AmpliSeq cDNA Synthesis for Illumina: A Complete Guide to Targeted RNA Sequencing Workflows
This comprehensive guide explores the AmpliSeq cDNA synthesis workflow for Illumina RNA sequencing, providing researchers and drug development professionals with foundational knowledge, methodological protocols, troubleshooting strategies, and performance validation data.
AmpliSeq cDNA Synthesis for Illumina: A Complete Guide to Targeted RNA Sequencing Workflows
Abstract
This comprehensive guide explores the AmpliSeq cDNA synthesis workflow for Illumina RNA sequencing, providing researchers and drug development professionals with foundational knowledge, methodological protocols, troubleshooting strategies, and performance validation data. The article covers the transformative role of this targeted RNA sequencing approach, which enables highly accurate gene expression quantification and fusion detection from minimal RNA input (as low as 1 ng), making it particularly valuable for precious clinical samples like FFPE tissues and blood. By comparing AmpliSeq to traditional RNA-seq methods and offering practical optimization techniques, this resource serves as an essential reference for implementing cost-effective, sensitive transcriptome analysis in biomedical research.
Understanding AmpliSeq Technology: Revolutionizing Targeted RNA Sequencing
The analysis of gene expression has been revolutionized by next-generation sequencing (NGS) technologies. While whole transcriptome sequencing (RNA-seq) provides a comprehensive view of all RNA molecules, it carries limitations including significant input RNA requirements, complex data analysis, and substantial data storage needs [1]. Targeted RNA sequencing approaches, such as those enabled by AmpliSeq for Illumina, overcome these challenges by focusing on a predefined set of genes of interest, enabling highly sensitive and cost-effective expression analysis.
The AmpliSeq for Illumina cDNA Synthesis kit serves as the critical first step in converting total RNA to cDNA for subsequent targeted amplification and sequencing [2] [3]. This targeted approach is particularly valuable for research applications where sample quantity is limited or when studying specific gene panels relevant to disease mechanisms, such as cancer research or inherited disorders [4]. By focusing sequencing power on specific targets, researchers can achieve greater depth of coverage while reducing costs and simplifying data analysis compared to whole transcriptome methods.
Technical Specifications and Workflow Components
Key Technical Specifications
The AmpliSeq for Illumina cDNA Synthesis kit is optimized for use with low-input RNA samples, making it suitable for challenging sample types common in research settings [2]. The table below summarizes the essential technical specifications for the cDNA synthesis step and associated library preparation workflow.
Table 1: Technical Specifications of AmpliSeq for Illumina cDNA Synthesis and Library Preparation
| Parameter | Specification | Application Notes |
|---|---|---|
| Input Quantity | 1-100 ng total RNA (10 ng per pool recommended) [2] | Suitable for limited and precious samples |
| Hands-On Time | <1.5 hours for library prep [2] | Streamlined workflow increases laboratory efficiency |
| Total Library Prep Time | ~5-7 hours [4] | Can be completed in a single workday |
| Species Compatibility | Human, Mouse, Rat; Compatible with any species [2] | Predefined genomes available for common model organisms |
| Panel Content Range | 12 to 12,288 amplicons [2] | From focused panels to comprehensive transcriptome coverage |
| Sample Type Compatibility | Blood, FFPE tissue [2] | Ideal for clinically relevant sample types |
Essential Research Reagent Solutions
Successful implementation of the AmpliSeq for Illumina RNA workflow requires several key components that work together in an integrated system. The following table outlines the essential research reagent solutions and their specific functions within the workflow.
Table 2: Research Reagent Solutions for AmpliSeq RNA Workflows
| Component | Function | Specific Examples |
|---|---|---|
| cDNA Synthesis Kit | Converts total RNA to cDNA for amplification | AmpliSeq for Illumina cDNA Synthesis (Cat. # 20022654) [2] [3] |
| Library Preparation Kit | Prepares sequencing-ready libraries from cDNA | AmpliSeq Library PLUS for Illumina (24, 96, or 384 reactions) [2] |
| Index Adapters | Enables sample multiplexing | AmpliSeq CD Indexes (Set A-D, 384 total indexes) [2] |
| Target Panels | Defines genes targeted for sequencing | Focus Panel (52 genes), Comprehensive Panel (20,000+ targets), or Custom panels [4] [1] |
| Library Normalization | Normalizes libraries for balanced sequencing | AmpliSeq Library Equalizer (bead-based normalization) [2] [3] |
| Specialized Sample Prep | Processes challenging sample types | AmpliSeq for Illumina Direct FFPE DNA (for tissue samples) [2] |
Experimental Workflow and Protocol
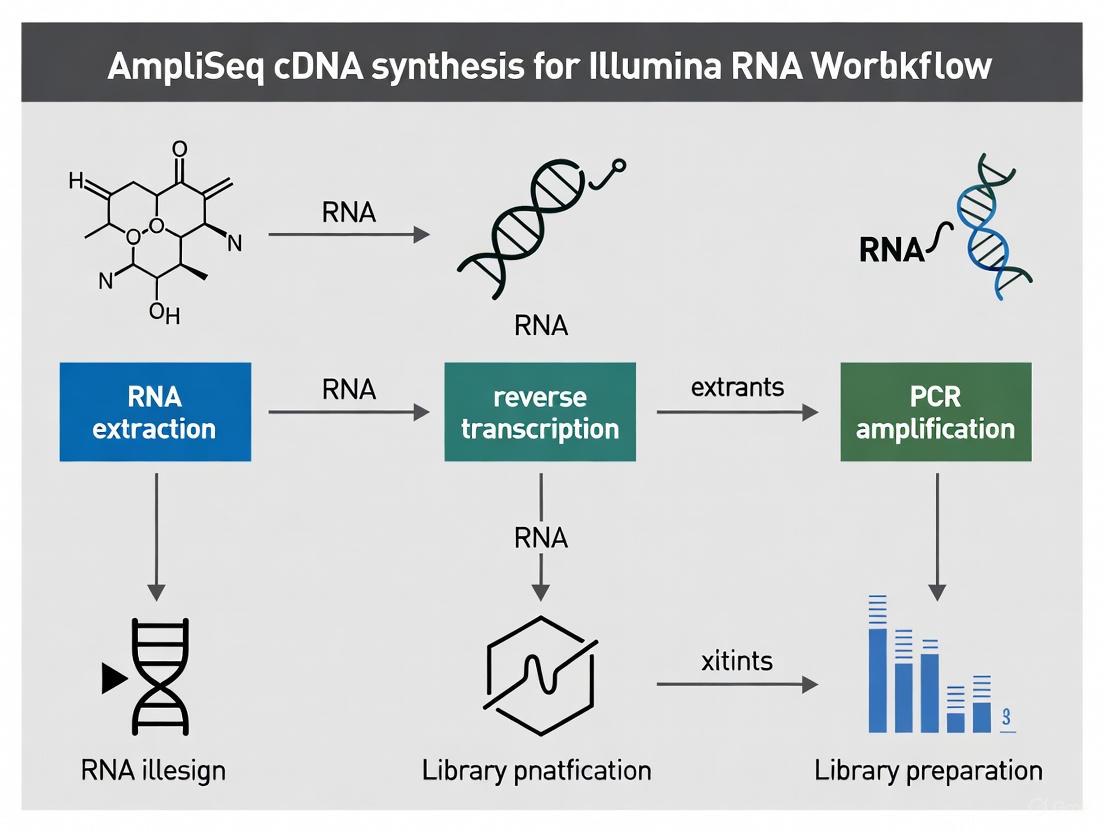
The AmpliSeq for Illumina RNA-to-results workflow integrates multiple steps from sample preparation through data analysis, with cDNA synthesis serving as the foundational molecular biology step that initiates the process.
Figure 1: The complete AmpliSeq for Illumina RNA-to-results workflow, highlighting the central role of cDNA synthesis in the targeted RNA sequencing process.
Detailed cDNA Synthesis and Library Preparation Protocol
The initial phase of the AmpliSeq for Illumina RNA workflow involves converting RNA to cDNA and preparing sequencing-ready libraries. This protocol ensures that the resulting libraries accurately represent the original transcriptome while maintaining the specificity required for targeted sequencing.
Step 1: RNA Quality Assessment and cDNA Synthesis
- Begin with total RNA extraction from your sample source (blood, FFPE tissue, or cell cultures).
- Use the AmpliSeq for Illumina cDNA Synthesis Kit (Component: SuperScript VILO cDNA Synthesis Mix) to reverse transcribe total RNA into first-strand cDNA [3].
- Critical Step: Use 10 ng of total RNA as input for optimal results, though the protocol supports 1-100 ng ranges for limited samples [2].
- Incubate reactions according to manufacturer specifications to ensure complete cDNA synthesis.
Step 2: Multiplex PCR Amplification
- Amplify targeted regions using predefined or custom AmpliSeq panels.
- The AmpliSeq for Illumina Focus Panel, for example, targets 52 genes with known relevance to solid tumors using a single pool of primers that generates 284 amplicons for RNA analysis [5].
- PCR conditions are optimized to maintain proportional representation of transcripts while achieving specific amplification of targeted genes.
Step 3: Primer Digestion and Library Construction
- Digest remaining primers to prevent interference with downstream steps.
- Incorporate AmpliSeq CD Indexes via ligation to enable sample multiplexing [2].
- Technical Note: The CD Indexes are available in sets (A-D) providing 384 unique indexes for large-scale studies [2].
Step 4: Library Normalization and Pooling
- Normalize libraries using the AmpliSeq Library Equalizer, which employs a bead-based method to ensure equimolar representation of samples [2] [3].
- Pool indexed libraries in preparation for sequencing.
- Validate library quality and quantity using appropriate methods such as fluorometry or capillary electrophoresis.
Performance Validation and Method Comparison
Analytical Performance Metrics
The performance of the AmpliSeq for Illumina workflow with cDNA synthesis has been rigorously validated in comparative studies. A comprehensive evaluation published in BMC Genomics demonstrated strong concordance between AmpliSeq and established RNA-seq methods [1].
Table 3: Performance Validation of AmpliSeq for Illumina Transcriptome Analysis
| Performance Metric | Result | Comparative Benchmark |
|---|---|---|
| Fold-Change Concordance | Pearson's r = 0.92 vs. Illumina HiSeq [1] | High correlation with established platforms |
| Sensitivity | Detection of 20,000+ distinct human RNA targets [1] | Comprehensive transcriptome coverage |
| Input RNA Requirement | 10 ng total RNA (as low as 1 ng supported) [2] | 10-100x less input than some RNA-seq methods [1] |
| Application Range | DEG analysis, clustering, PCA [1] | Suitable for various analysis types |
| Reproducibility | High intra- and inter-run consistency [1] | Reliable for longitudinal studies |
Comparison to Alternative Methods
When compared to whole transcriptome sequencing approaches, AmpliSeq for Illumina demonstrates particular advantages in several key areas. The targeted nature of AmpliSeq makes it especially suitable for studies where specific gene panels are of interest, or when sample input is limited [1]. A 2014 study comparing cDNA synthesis and library preparation methods found that method selection significantly impacts outcomes in gene expression studies, with targeted approaches like AmpliSeq providing more reliable results when working with limited RNA inputs [6].
The integration with qPCR represents another important consideration in method selection. As noted by Thermo Fisher Scientific, "we don't live in a 'real-time PCR vs. NGS' world; rather the two technologies are complementary" [7]. In practice, qPCR is often used both upstream of NGS to check cDNA integrity, and downstream to verify NGS results, creating a validated workflow for gene expression analysis [7].
For research applications requiring highly sensitive detection of specific transcripts across many samples, AmpliSeq for Illumina provides a robust solution that balances comprehensive coverage with practical considerations of cost, time, and data management [4] [1].
Applications in Research and Drug Development
The AmpliSeq for Illumina cDNA synthesis and RNA workflow supports diverse research applications across multiple scientific domains. In clinical cancer research, targeted panels like the AmpliSeq for Illumina Focus Panel enable investigators to profile solid tumors using specific gene signatures with known relevance to cancer biology [4] [5]. The workflow's compatibility with FFPE tissue specimens further enhances its utility in translational research where archived samples represent valuable resources [2].
For drug development professionals, the platform offers a streamlined approach to biomarker discovery and validation. The ability to design custom panels using the Illumina DesignStudio software allows researchers to create targeted content specific to their therapeutic area [4] [8]. The availability of community panels—predesigned sequencing panels containing content selected with input from leading disease researchers—provides additional resources for studying well-characterized disease mechanisms [4].
The platform's scalability across Illumina sequencing systems (from iSeq 100 to NextSeq 2000) makes it suitable for both small-scale pilot studies and larger validation cohorts, providing a consistent methodology throughout the drug discovery pipeline [2] [5].
Targeted RNA sequencing represents a significant evolution in transcriptome analysis, enabling researchers to focus on specific genes or pathways of interest with high efficiency. Unlike traditional whole-transcriptome RNA-seq, which sequences all RNA molecules present in a sample, targeted approaches like the AmpliSeq for Illumina platform use multiplex PCR to amplify and sequence a predefined set of gene targets [9]. This methodology offers two fundamental advantages that are particularly valuable for clinical research and drug development: significantly reduced input requirements and a highly focused, customizable design.
The AmpliSeq for Illumina workflow integrates PCR-based library preparation with Illumina's sequencing by synthesis (SBS) technology, creating an optimized system for analyzing dozens to thousands of targets simultaneously [10]. This technical approach enables researchers to overcome common challenges in genomic research, including limited sample availability and the need for cost-effective, high-throughput analysis of specific gene panels.
Key Advantages of AmpliSeq Technology
Low Input Requirements
The AmpliSeq for Illumina platform demonstrates exceptional performance with minimal starting material, a critical feature for precious or limited samples.
| Input Specification | AmpliSeq for Illumina Performance |
|---|---|
| Minimum Input Quantity | 1 ng of RNA [10] |
| Recommended Input Quantity | 1-100 ng RNA (10 ng recommended) [11] |
| Compatible Sample Types | Blood, FFPE tissue [10] [12] |
| Hands-on Time | < 1.5 hours [10] |
| Total Assay Time | 5.5-7.5 hours [10] |
This low input requirement is particularly advantageous for formalin-fixed paraffin-embedded (FFPE) tissue samples, which are often clinically valuable but yield limited quantities of degraded RNA [10] [12]. The ability to generate reliable gene expression data from just 1 ng of input RNA enables researchers to utilize archival samples that would be insufficient for traditional RNA-seq protocols, thereby expanding the scope of retrospective clinical studies.
Targeted Design Capabilities
The targeted design of AmpliSeq panels provides researchers with precise control over their genomic investigations, allowing focus on specific pathways, disease mechanisms, or gene sets of interest.
| Design Feature | Capability |
|---|---|
| Target Range | 12 to 1200 gene targets in a single assay [10] |
| Design Resource | >20,000 human RefSeq genes available in DesignStudio [10] |
| Application Flexibility | Gene expression, fusion detection, transcript variant analysis [12] |
| Multiplexing Capacity | Up to 384-plex [10] |
The customizable nature of these panels enables drug development professionals to create tailored solutions for specific research questions. For oncology applications, the AmpliSeq for Illumina Custom RNA Fusion Panel can detect fusion genes while simultaneously profiling expression of 12-1200 additional targets, providing comprehensive molecular characterization from a single assay [12]. This targeted approach conserves sequencing resources and data analysis time by eliminating irrelevant genomic content.
Comparative Performance Data
Advantage Over Traditional RNA-seq Methods
When compared to traditional RNA-seq methods, AmpliSeq technology demonstrates distinct advantages in several key performance metrics:
| Parameter | AmpliSeq for Illumina Transcriptome Panel | Traditional Whole Transcriptome Methods |
|---|---|---|
| Input Requirement | 1-100 ng RNA [11] | Typically 100-1000 ng for standard protocols [11] |
| Hands-on Time | < 1.5 hours [11] | 3-5.5 hours for comparable workflows [11] |
| Assay Time | ~6 hours (library prep only) [11] | 7-11.5 hours [11] |
| Content Specification | Targeted: >20,000 human RefSeq genes [11] | Comprehensive: coding + non-coding RNA [11] |
| FFPE Compatibility | Yes [10] [12] | Limited for many protocols [8] |
The targeted design of AmpliSeq panels enables exceptional data quality with minimal input. Experimental data demonstrates high concordance between replicates (R² = 0.997) and high read alignment rates, confirming the reliability of the method even with challenging sample types [10].
Application-Specific Advantages
In cancer research, the combination of low input requirements and targeted design enables comprehensive molecular profiling from clinically relevant samples. The platform's ability to work with FFPE tissues and blood samples makes it particularly valuable for translational research and biomarker discovery [12]. For drug development professionals, this facilitates pharmacodynamic biomarker assessment and patient stratification strategy implementation using sample types routinely available in clinical trials.
Experimental Protocols
AmpliSeq for Illumina RNA Workflow Protocol
The standard protocol for AmpliSeq-based targeted RNA sequencing involves a streamlined workflow that can be completed in less than 8 hours with minimal hands-on time:
Detailed Procedural Steps
cDNA Synthesis:
Library Preparation:
Indexing and Normalization:
Sequencing and Analysis:
- Sequence on compatible Illumina platforms (iSeq 100, MiSeq, NextSeq series) [10]
- Analyze data using Illumina-supported analysis tools specific to the RNA application workflow
Custom Panel Design Protocol
For researchers requiring customized target content, the panel design process utilizes Illumina's DesignStudio software:
The Scientist's Toolkit: Research Reagent Solutions
| Research Reagent | Function | Application Notes |
|---|---|---|
| AmpliSeq cDNA Synthesis for Illumina | Converts total RNA to cDNA | Required for all AmpliSeq RNA panels; number of reactions varies by panel type [10] |
| AmpliSeq Library PLUS | Library preparation reagents | Available in 24, 96, or 384 reactions; separate purchase from panels and indexes [10] |
| AmpliSeq UD Indexes | Sample multiplexing | 24 indexes for 24 samples; enables sample pooling and tracking [10] |
| AmpliSeq CD Indexes | High-plex sample multiplexing | 96 indexes per set (Sets A-D available); enables large-scale studies [10] |
| AmpliSeq Library Equalizer | Library normalization | Optional but recommended for normalizing libraries before sequencing [10] |
| AmpliSeq ERCC RNA Spike-In Mix | Process controls | External RNA controls for quantitating differential gene expression [10] |
Application in Drug Development and Clinical Research
The low input requirements and targeted design of AmpliSeq technology make it particularly valuable for drug development pipelines. In target identification and validation phases, customized panels can focus on specific pathways or disease mechanisms, generating high-quality data from limited preclinical samples. During clinical development, the platform's compatibility with FFPE tissues and blood enables comprehensive biomarker analysis from samples routinely collected in clinical trials [12].
For cancer research, the AmpliSeq for Illumina Custom RNA Fusion Panel demonstrates exceptional performance in detecting fusion genes, with experimental data showing 100% call rates for expected fusions in validation studies [12]. This reliability, combined with the ability to simultaneously profile gene expression, provides a comprehensive molecular profiling solution from minimal input material.
The targeted nature of AmpliSeq panels also streamlines data analysis and interpretation for drug development professionals. By focusing on predefined gene sets, bioinformatic analysis becomes more efficient, and regulatory submission packages can be more readily prepared with standardized, reproducible genomic data.
Targeted RNA sequencing enables researchers to focus on specific genes of interest, providing a cost-effective and efficient method for gene expression analysis. The AmpliSeq for Illumina platform offers a streamlined, multiplex polymerase chain reaction (PCR)-based workflow that is particularly advantageous for working with challenging sample types, such as formalin-fixed, paraffin-embedded (FFPE) tissues and blood [2]. This application note details the integrated workflow centered on three core components: the AmpliSeq Library PLUS kit, the cDNA Synthesis kit, and various Index Adapters. When used together, these components enable the preparation of high-quality sequencing libraries from minimal RNA input in under 8 hours, providing a robust solution for research in drug development and other biological fields [10].
The AmpliSeq for Illumina RNA workflow is a multi-stage process that converts RNA into sequence-ready libraries. The following diagram illustrates the key steps and the point at which the three core components are utilized.
Research Reagent Solutions
The following table catalogues the essential reagents required to execute the AmpliSeq for Illumina RNA workflow, along with their specific functions.
Table 1: Key Research Reagent Solutions for the AmpliSeq RNA Workflow
| Component Name | Core Function | Key Specifications | Compatible Illumina Systems |
|---|---|---|---|
| AmpliSeq Library PLUS [2] | Provides core reagents for library construction post-target amplification. | ~5 hr assay time; <1.5 hr hands-on time; 1-100 ng input DNA. | MiSeq, iSeq, NextSeq series, MiniSeq |
| AmpliSeq cDNA Synthesis Kit [10] | Converts total RNA to cDNA, which is the required starting material for RNA panels. | Required for all AmpliSeq RNA panels. | MiSeq, iSeq, NextSeq series, MiniSeq |
| AmpliSeq UD Indexes [2] | Uniquely labels each library for multiplexing; mitigates index hopping. | 24 indexes, sufficient for 24 samples. | All Illumina systems with 2- or 4-color chemistry |
| AmpliSeq CD Indexes [2] | Labels libraries using combinatorial dual indexing for multiplexing. | 96 indexes per set (Set A, B, C, D); can label 96 samples per set. | All Illumina systems with 2- or 4-color chemistry |
| AmpliSeq Custom RNA Panel [10] | Targeted panel for amplifying 12 to 1,200 gene targets of interest in a single assay. | 5.5-7.5 hr total assay time; 1 ng cDNA input. | MiSeq, iSeq, NextSeq series, MiniSeq |
| AmpliSeq Library Equalizer [2] | Optional beads and reagents for normalizing libraries before pooling and sequencing. | Simplifies and standardizes the library normalization process. | N/A |
Detailed Experimental Protocols
cDNA Synthesis from Total RNA
The workflow begins with the synthesis of complementary DNA (cDNA), as the AmpliSeq RNA panels require cDNA as their input material [10].
- Input Material: Total RNA.
- Primer Selection: The protocol can be adapted based on the primers used. For gene-specific primers, a denaturation step is recommended for targets with high secondary structure. A typical 20 µL reaction includes the RNA template, 4 µL of 5x RT Buffer, 1 µL of dNTP Mix (10 mM each), 0.5 µL of RNase Inhibitor, and 0.5 µL of Reverse Transcriptase [13].
- Thermal Cycling:
- Denaturation & Primer Annealing: If using a denaturation protocol, incubate the RNA and primer mix at 65-70°C for 5 minutes, then hold at room temperature or on ice.
- First-Strand Synthesis:
- For gene-specific primers: Incubate at 50°C for 30-60 minutes.
- For oligo-dT or random primers: Incubate at 42°C for 10 minutes, followed by 50°C for 30-60 minutes [13].
- Enzyme Inactivation: Heat the reaction to 70°C for 10 minutes [13].
- Output: The resulting first-strand cDNA is ready for the target amplification step and should be stored at -20°C if not used immediately.
Target Amplification and Library Construction
This stage uses the synthesized cDNA as input for a targeted, multiplex PCR reaction to create the sequencing library.
- Input Material: 1 ng of cDNA is recommended [10].
- Panel Configuration: The AmpliSeq Custom RNA Panel is designed to target 12 to 1,200 genes. Panels are designed using Illumina's DesignStudio Software from a menu of over 20,000 human RefSeq genes [10].
- Library Preparation with AmpliSeq Library PLUS:
- Target Amplification: The custom RNA panel primers are used in a multiplex PCR to amplify the specific gene targets from the cDNA pool.
- Library Construction: The AmpliSeq Library PLUS kit reagents are used to partially digest the primer sequences and ligate the required adapter sequences to the amplicons, creating the sequencing library [2].
Key Quantitative Data: Table 2: Performance Specifications of the Core Workflow Components
Component Assay Time Hands-on Time Input Quantity Key Metric cDNA Synthesis ~1 hour < 30 minutes 10 pg - 5 µg Total RNA [13] cDNA yield for 1-100 ng input [2] Library Prep (Library PLUS) ~5 hours [2] < 1.5 hours [2] 1-100 ng (10 ng recommended per pool) [2] 12 to 12,288 amplicons [2] Custom RNA Panel (Total Workflow) 5.5-7.5 hours [10] < 1.5 hours [10] 1 ng cDNA [10] Up to 1,200 targets [10]
Indexing, Normalization, and Pooling
To enable multiplexing, each library is tagged with a unique index sequence, normalized to ensure equal representation, and then pooled.
- Indexing Strategies:
- Unique Dual Indexes (UDI): This strategy uses a unique identifier on both ends of the DNA fragment. UDIs are highly recommended as they mitigate index hopping—a phenomenon where indexes are misassigned between samples—which is particularly important on patterned flow cell instruments like the NovaSeq 6000 system [14]. The AmpliSeq UD Indexes (24 Indexes) kit is suitable for experiments with up to 24 samples [2].
- Combinatorial Dual Indexes (CDI): This approach uses a limited set of indexes that are combined. While it allows for 96 unique combinations from fewer unique sequences, the indexes are not fully unique and share some sequences, which can make them more susceptible to index hopping compared to UDIs [14]. The AmpliSeq CD Indexes (e.g., Set A-D) are available for larger studies [2].
- Library Normalization and Pooling:
- Normalization: The AmpliSeq Library Equalizer is an optional but recommended kit that uses beads and reagents to normalize libraries, ensuring equimolar amounts of each library before pooling [2].
- Pooling: The normalized, indexed libraries are combined into a single tube.
Troubleshooting and Technical Notes
- Index Hopping: For the highest data accuracy, especially on high-output instruments like the NovaSeq 6000 system, the use of Unique Dual Indexes (UDI) is strongly recommended over Combinatorial Dual Indexes to minimize sample misassignment [14].
- Low Input RNA: The addition of an RNase Inhibitor (included in some cDNA synthesis kits) is recommended and may be essential when working with low amounts of starting RNA to prevent template degradation [13].
- Challenging RNA Templates: For RNA targets with a high degree of secondary structure, using the sample denaturation protocol (65-70°C for 5 minutes) during cDNA synthesis and increasing the reverse transcription incubation temperature to 55°C can improve yields [13].
The AmpliSeq for Illumina technology provides a targeted, multiplex PCR-based next-generation sequencing (NGS) workflow designed for robust performance with challenging sample types, including formalin-fixed, paraffin-embedded (FFPE) tissues. This integrated system enables researchers to investigate gene expression and detect critical biomarkers like gene fusions from low-input RNA samples. The workflow begins with the critical step of cDNA synthesis, converting RNA to cDNA, which is then amplified using sequence-specific primers for targets of interest. This approach is particularly valuable in clinical cancer research, where it facilitates the identification of therapeutic targets and diagnostic biomarkers from limited and degraded samples [10] [4].
A key advantage of the AmpliSeq method is its minimal hands-on time (under 1.5 hours) and rapid assay time (approximately 5.5 to 7.5 hours), allowing researchers to go from nucleic acid to sequencing-ready libraries in less than a day. The technology supports a wide range of input quantities, requiring as little as 1 ng of RNA, making it suitable for precious clinical specimens where material is limited. Its compatibility with various Illumina sequencing systems, from the iSeq 100 to the NextSeq 2000, provides scalability to meet different throughput needs [10] [2].
Technical Specifications and Workflow Components
Key Technical Specifications
The table below summarizes the core technical specifications of the AmpliSeq for Illumina RNA workflow, highlighting its flexibility and efficiency for targeted RNA sequencing applications.
Table 1: Technical Specifications of the AmpliSeq for Illumina RNA Workflow
| Parameter | Specification |
|---|---|
| Assay Time | 5.5 - 7.5 hours (total); ~5-6 hours (library prep only) |
| Hands-on Time | < 1.5 hours |
| Input Quantity | 1 - 100 ng RNA (10 ng recommended) |
| Multiplexing Capacity | Up to 384 samples using CD Indexes |
| Nucleic Acid Type | RNA (requires cDNA synthesis) |
| Specialized Sample Types | Blood, FFPE tissue |
| Compatible Instruments | iSeq 100, MiSeq, MiniSeq, NextSeq 500/550/1000/2000 |
| Method | Targeted RNA sequencing, Amplicon sequencing |
| Key Applications | Gene expression analysis, fusion detection, transcript variant analysis |
The Scientist's Toolkit: Essential Research Reagents
A successful AmpliSeq for Illumina experiment requires several key components. The table below details the essential reagents and their specific functions within the workflow.
Table 2: Essential Research Reagents for the AmpliSeq Workflow
| Component | Function | Key Features |
|---|---|---|
| AmpliSeq cDNA Synthesis for Illumina | Converts total RNA to cDNA for subsequent library preparation. | Required for working with all AmpliSeq for Illumina RNA Panels. |
| AmpliSeq Custom RNA Panel | Targets 12 to 1,200 user-defined human RefSeq genes. | Designed via DesignStudio Software from over 20,000 genes. |
| AmpliSeq Transcriptome Human Gene Expression Panel | Ready-to-use panel measuring >20,000 human RefSeq genes. | Captures >95% of the human transcriptome in a single assay. |
| AmpliSeq Library PLUS | Contains reagents for preparing sequencing libraries from amplicons. | Available in 24, 96, and 384 reaction sizes. |
| AmpliSeq CD Indexes | Unique dual indexes for multiplexing samples prior to sequencing. | Enable pooling of up to 384 samples in a single run. |
| AmpliSeq Library Equalizer | Bead-based normalization reagent for streamlining library pooling. | Optional but recommended for simplifying workflow. |
| AmpliSeq ERCC RNA Spike-In Mix | External RNA controls for quantitating differential gene expression. | Requires separate ERCC RNA Companion Panel. |
Application Note: Enhancing Fusion Detection in Solid Tumors
Clinical Challenge and Experimental Rationale
Gene fusions are pivotal molecular biomarkers for tumor diagnosis, classification, and targeted therapy. However, their accurate detection in clinical samples, which are often FFPE-derived and contain degraded RNA, presents a significant challenge. While traditional methods like FISH and IHC are limited in their multiplexing capacity, standard NGS approaches that rely solely on either DNA or RNA can lead to false negatives. A 2025 study validated an integrated DNA- and RNA-based targeted sequencing assay to overcome these limitations, demonstrating that a combined approach maximizes detection sensitivity for fusion events in solid tumors [15].
Detailed Experimental Protocol
The following protocol outlines the key steps for detecting gene fusions using an integrated DNA and RNA approach with the AmpliSeq workflow.
Table 3: Protocol for Integrated DNA/RNA-Based Fusion Detection
| Step | Procedure | Critical Parameters |
|---|---|---|
| 1. Sample Preparation | Extract DNA and RNA from FFPE tumor samples or use commercial reference standards. | Input: 1-100 ng of RNA and 1-100 ng of DNA (10 ng recommended per pool). |
| 2. cDNA Synthesis | Convert total RNA to cDNA using the AmpliSeq cDNA Synthesis for Illumina kit. | Use reaction mix and enzyme blend per manufacturer's instructions. |
| 3. Library Preparation (DNA & RNA) | Prepare separate AmpliSeq libraries from DNA and cDNA using a custom panel. | Panel design: 16 genes related to targeted therapy. Use AmpliSeq Library PLUS and CD Indexes. |
| 4. Library Normalization & Pooling | Normalize libraries using AmpliSeq Library Equalizer. Pool equalized libraries. | Bead-based normalization ensures equimolar representation. |
| 5. Sequencing | Sequence pooled libraries on an Illumina MiSeq, iSeq, or NextSeq system. | Use MiSeq Reagent Kit v2 or v3; aim for high coverage (>421x). |
| 6. Data Analysis | Align reads and call fusions using DRAGEN RNA Amplicon pipeline or similar tools. | Cross-validate fusions called from DNA and RNA data streams. |
Key Performance Metrics and Validation Data
The integrated assay was rigorously validated for sensitivity, specificity, and reproducibility. The table below summarizes the key performance data from the validation study.
Table 4: Performance Metrics of the Integrated Fusion Detection Assay
| Metric | Result | Experimental Details |
|---|---|---|
| Analytical Sensitivity | 100% (28/28 known positive samples detected) | Validation cohort of 60 clinical FFPE samples. |
| Analytical Specificity | 96.9% (31/32), corrected to 100% after Sanger validation | One additional TPM3::NTRK1 fusion discovered. |
| Limit of Detection (DNA) | 5% mutational abundance | Determined via serial dilution of reference standards. |
| Limit of Detection (RNA) | 250-400 copies/100 ng RNA | Determined via serial dilution of reference standards. |
| Intra-Assay Reproducibility | 100% concordance across triplicate runs | Tested with samples containing multiple fusions. |
| Inter-Assay Reproducibility | 100% concordance across three different sequencing runs | Demonstrated robust performance across batches. |
The study demonstrated that the DNA and RNA results are complementary. The DNA assay alone showed 93.4% concordance with previous results, missing some fusions like ETV6::NTRK3, while the RNA assay alone showed 86.9% concordance, missing others like TRIM46::NTRK1. However, when results from both levels were integrated, the assay achieved 100% sensitivity and specificity, identifying all known fusions and even discovering a previously false-negative TPM3::NTRK1 fusion in a papillary thyroid carcinoma sample [15].
Diagram 1: Integrated DNA and RNA Fusion Detection Workflow. This workflow shows the parallel processing of DNA and RNA from a single FFPE sample, culminating in integrated analysis for comprehensive fusion detection.
Comparative Analysis with Other Methodologies
AmpliSeq vs. Whole-Transcriptome Methods
The AmpliSeq for Illumina Transcriptome Human Gene Expression Panel offers a targeted alternative to whole-transcriptome sequencing for specific applications. The table below compares key features of these approaches.
Table 5: Comparison of Targeted vs. Whole-Transcriptome RNA Sequencing
| Characteristic | AmpliSeq Transcriptome Panel | Illumina Stranded Total RNA Prep |
|---|---|---|
| Method | Targeted, multiplex PCR | Whole-transcriptome, ligation-based |
| Assay Time | 6 hours (library prep) | ~7 hours |
| Hands-on Time | < 1.5 hours | < 3 hours |
| Input Quantity | 1-100 ng RNA (10 ng recommended) | 1-1000 ng total RNA |
| Content | >20,000 human RefSeq genes | Coding + non-coding RNA |
| Best For | Focused gene expression, fusion detection in predefined targets | Novel transcript discovery, global transcriptome analysis |
Performance in FFPE vs. Fresh-Frozen Samples
The compatibility of the AmpliSeq workflow with FFPE samples is critical for clinical research. A 2024 study directly compared fusion detection by RNA sequencing in matched FFPE and freshly frozen (FF) colorectal cancer samples. The research found no statistically significant difference in the number of chimeric transcripts detected between the two sample types. This demonstrates the robustness of the method for degraded RNA typical of archival FFPE material. The study successfully identified a novel, potentially actionable LRRFIP2::ALK fusion in one patient, highlighting the clinical utility of the approach [16].
Diagram 2: Complementary Nature of DNA and RNA Fusion Detection. The integrated strategy overcomes the individual limitations of DNA-only or RNA-only approaches, providing a more comprehensive solution for clinical fusion detection.
The AmpliSeq for Illumina RNA workflow, anchored by its robust cDNA synthesis step, provides a versatile and efficient platform for a broad spectrum of research applications, from comprehensive gene expression profiling to sensitive fusion detection in human samples. The integration of DNA and RNA-based NGS, as demonstrated in recent clinical validations, represents a significant advancement for molecular pathology, enabling complementary detection that maximizes sensitivity and specificity. This approach is particularly powerful for profiling solid tumors from FFPE samples, where material is limited and RNA is often degraded. By leveraging the low input requirements, rapid turnaround time, and compatibility with multiple sequencing platforms offered by the AmpliSeq technology, researchers and drug development professionals can reliably identify therapeutically actionable biomarkers to advance precision medicine.
The AmpliSeq for Illumina technology represents a targeted, PCR-based RNA sequencing approach designed for robust gene expression analysis with exceptional efficiency. This methodology is engineered to deliver high-quality data from challenging samples, including those derived from formalin-fixed, paraffin-embedded (FFPE) tissues and blood, making it particularly valuable for clinical cancer research and drug development [4] [2]. The workflow integrates seamlessly with Illumina's sequencing-by-synthesis (SBS) technology and automated data analysis pipelines, enabling researchers to focus on biological interpretation rather than complex bioinformatics [4]. A core component of this system is the AmpliSeq cDNA Synthesis step, which converts total RNA into cDNA, serving as the template for subsequent highly multiplexed PCR amplification [10]. This document details the technical specifications, protocols, and key reagents that underpin the AmpliSeq for Illumina RNA workflow, providing a comprehensive guide for its application in research settings.
Technical Specifications and Performance Data
The AmpliSeq for Illumina RNA workflow is characterized by its low input requirements and time-efficient protocol. The key technical specifications for different RNA panel types are consolidated in the table below for direct comparison.
Table 1: Technical Specifications of AmpliSeq for Illumina RNA Panels
| Specification | Custom RNA Panel | Transcriptome Human Gene Expression Panel | Library Prep (General) |
|---|---|---|---|
| Input RNA Quantity | 1 ng [10] | 1-100 ng (10 ng recommended) [11] | 1-100 ng (10 ng recommended per pool) [2] |
| Hands-on Time | < 1.5 hours [10] | < 1.5 hours [11] | < 1.5 hours [2] |
| Total Assay Time (Library Prep) | 5.5 - 7.5 hours [10] | ~6 hours [11] | ~5 hours [2] |
| Number of Targets | Up to 1,200 genomic targets [10] | >20,000 human RefSeq genes [11] | 12 to 12,288 amplicons [2] |
| Key Application | Custom targeted gene expression [10] | Whole-transcriptome targeted expression [11] | Targeted RNA sequencing [2] |
A comparative study published in BMC Genomics underscores the reliability of this approach, demonstrating a strong concordance in log2 fold change for all genes when comparing the AmpliSeq Transcriptome panel to standard Illumina HiSeq RNA-seq (Pearson’s r = 0.92) [1]. The study concluded that AmpliSeq excels as a highly sensitive and cost-effective approach for very large-scale gene expression analysis and mRNA marker screening with high accuracy, particularly for genes with high abundance [1].
Detailed Experimental Protocol
cDNA Synthesis and Library Preparation Workflow
The following diagram outlines the core steps of the AmpliSeq for Illumina RNA workflow, from sample input to sequencing-ready libraries.
Diagram 1: AmpliSeq RNA-to-Library Workflow.
Step-by-Step Protocol:
- cDNA Synthesis: Using the AmpliSeq cDNA Synthesis for Illumina kit, convert total RNA (1-100 ng) into cDNA. This step is mandatory for all AmpliSeq for Illumina RNA panels and provides the template for targeted amplification [10] [11].
- Multiplex PCR Amplification: Combine the synthesized cDNA with a selected AmpliSeq Panel (e.g., Custom RNA, Transcriptome, or Focus Panel) and the AmpliSeq Library PLUS master mix. The panel contains thousands of primer pairs in a single pool that simultaneously amplify the targeted genomic regions of interest via multiplex PCR [4] [10].
- Primer Digestion: Following PCR amplification, any remaining primers are enzymatically digested to prevent interference with downstream library preparation steps [4].
- Library Assembly and Indexing: The amplified targets are then ligated to Illumina-specific adapters, including unique molecular barcodes (AmpliSeq CD Indexes or UD Indexes), using reagents from the Library PLUS kit. This step makes the amplicons compatible with Illumina sequencing platforms and allows for multiplexing of multiple samples in a single sequencing run [2].
- Library Normalization and Pooling (Optional but Recommended): Use the AmpliSeq Library Equalizer to normalize libraries, ensuring balanced representation before pooling. This bead-based normalization method is designed to streamline the process and improve data uniformity [10].
Sequencing and Data Analysis
After library preparation and quality control, pooled libraries are sequenced on a compatible Illumina benchtop sequencer, such as the iSeq 100, MiSeq, or NextSeq series [2] [10]. Data analysis can be performed efficiently without extensive bioinformatics resources. The primary analysis options are:
- Cloud-Based Analysis: Utilize the DRAGEN Amplicon pipeline on the BaseSpace Sequence Hub for secondary analysis. The DRAGEN RNA Amplicon app performs differential expression analysis and gene fusion calling [4].
- On-Instrument Analysis: Perform secondary analysis directly on the sequencing instrument using Local Run Manager software, which provides a user-friendly interface for amplicon analysis and variant calling [4].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of the AmpliSeq for Illumina RNA workflow requires a set of core and accessory reagents. The following table details these essential components and their functions.
Table 2: Key Research Reagent Solutions for the AmpliSeq Workflow
| Reagent Solution | Function | Key Features |
|---|---|---|
| AmpliSeq cDNA Synthesis Kit [10] | Converts total RNA to cDNA for subsequent PCR amplification. | Mandatory for RNA panels; compatible with low-input (from 1 ng) and degraded RNA from FFPE samples. |
| AmpliSeq Panels (e.g., Custom, Transcriptome, Focus) [4] [10] [11] | Target-specific primer pools for multiplex PCR amplification. | Available as ready-to-use or custom designs; targets from dozens to over 20,000 genes. |
| AmpliSeq Library PLUS Kit [2] | Master mix and reagents for PCR amplification and library construction. | Enables fast library prep (~5-7 hrs) with minimal hands-on time (<1.5 hrs). |
| AmpliSeq CD/UD Indexes [2] | Dual-index adapters for sample multiplexing. | Allows pooling of up to 384 unique samples in a single sequencing run. |
| AmpliSeq Library Equalizer [10] | Bead-based normalization reagent. | Optional but recommended for easy and efficient library normalization before pooling. |
| AmpliSeq for Illumina Direct FFPE DNA [2] | Prepares DNA directly from FFPE tissues. | Used for DNA-based panels; eliminates need for deparaffinization or DNA purification. |
The AmpliSeq for Illumina RNA workflow, anchored by its robust cDNA synthesis step, provides a streamlined and highly effective solution for targeted gene expression analysis. Its defining characteristics—low RNA input requirements (1-100 ng), minimal hands-on time (<1.5 hours), and rapid assay duration (approximately 5-7.5 hours)—make it an indispensable tool for researchers and drug development professionals. The availability of both pre-designed and custom panels offers unparalleled flexibility to focus on specific genes or pathways relevant to disease research. When combined with the integrated suite of reagents and simplified data analysis solutions, this technology empowers laboratories to generate high-quality, actionable transcriptomic data efficiently and reliably.
Implementing the AmpliSeq Workflow: Step-by-Step Protocol and Panel Design
Targeted RNA sequencing represents a powerful approach for focused gene expression analysis, enabling researchers to concentrate resources on specific genomic regions of interest. The AmpliSeq for Illumina technology provides a robust, multiplex PCR-based methodology for constructing sequencing-ready libraries from RNA samples. This workflow is particularly valuable in research areas where sample quantity is limited, as it requires only 1-100 ng of input RNA, with 10 ng recommended per pool [2]. The entire library preparation process requires approximately 5-7.5 hours of assay time with less than 1.5 hours of hands-on time, making it an efficient solution for researchers studying coding transcriptomes across various applications including oncology, infectious disease, and genetic disease research [17] [10].
A key advantage of the AmpliSeq approach lies in its targeted nature, which allows for deep sequencing of specific transcripts without the need for extensive bioinformatics analysis and data storage associated with whole transcriptome methods [1]. The technology demonstrates exceptional performance in gene expression quantification, showing strong concordance with established RNA-seq methods while excelling in limiting areas of traditional RNA-seq, such as situations with minimal starting material [1]. This workflow integrates seamlessly with Illumina sequencing systems, creating a complete solution from sample preparation to final analysis.
Experimental Design and Workflow Considerations
Sample Requirements and Quality Control
Successful implementation of the AmpliSeq RNA workflow begins with proper sample preparation and quality assessment. Input RNA can be derived from various sources including blood, FFPE tissue, and cell cultures [2]. For optimal results, RNA purity should be confirmed with OD260/280 ratio >1.8 using spectrophotometric methods, while integrity can be assessed through automated electrophoresis systems such as Agilent Bioanalyzer or TapeStation [18]. Fluorometric quantification using assays like Qubit RNA BR Assay Kit is recommended for accurate concentration measurements [18].
The AmpliSeq technology is compatible with a range of RNA input quantities (1-100 ng), enabling analysis of challenging sample types. For FFPE and low-quality samples, the stranded total RNA prep approach provides high coverage uniformity and reliability even from degraded material [17]. When working with limited samples, the technology's low input requirement makes it particularly advantageous compared to traditional RNA-seq methods that typically require significantly more starting material [1].
Panel Selection and Experimental Configuration
Researchers can select from various AmpliSeq panel options depending on their experimental needs:
- Custom RNA Panels: Target 12 to 1,200 genomic targets of interest, designed using Illumina's DesignStudio software from a menu of over 20,000 human RefSeq genes [10]
- Pre-designed Panels: Including options for whole transcriptome analysis, mRNA sequencing, and targeted enrichment [17]
- Specialized Panels: Such as the Childhood Cancer Panel that analyzes 203 genes associated with pediatric cancers, demonstrating high sensitivity (98.5% for DNA variants) and clinical utility [18]
The choice between panels depends on the research objectives, with targeted panels providing cost-effective solutions for focused experiments while whole transcriptome options offer comprehensive coverage.
Table 1: AmpliSeq Panel Options and Specifications
| Panel Type | Number of Targets | Primary Applications | Key Features |
|---|---|---|---|
| Custom RNA | 12-1,200 | Targeted gene expression | Design flexibility for specific gene sets |
| Whole Transcriptome | >20,000 | Comprehensive profiling | Broad coverage of human transcriptome |
| Childhood Cancer | 203 | Pediatric oncology research | Analyzes fusions, SNVs, InDels, CNVs |
| Stranded Total RNA | Whole transcriptome | Coding and noncoding RNA | Integrated enzymatic rRNA depletion |
Step-by-Step Protocol: From RNA to Sequencing-Ready Libraries
cDNA Synthesis from RNA Templates
The AmpliSeq RNA workflow begins with the conversion of total RNA to cDNA using the AmpliSeq cDNA Synthesis for Illumina kit [10]. This step requires:
- Input RNA: 10 ng of total RNA is typically used, though the protocol accommodates 1-100 ng inputs [2]
- Reverse Transcription: Using SuperScript VILO cDNA Synthesis Kit or similar systems to generate first-strand cDNA [18] [1]
- Quality Assessment: Verify cDNA synthesis success before proceeding to amplification
The cDNA synthesis step is critical for maintaining accurate representation of transcript abundances in the final library. The use of optimized reverse transcription conditions ensures high fidelity in converting RNA templates to amplifiable cDNA molecules.
Targeted Amplification and Library Construction
Following cDNA synthesis, targeted amplification proceeds using the AmpliSeq Library PLUS kit with specific primer panels:
- Multiplex PCR Amplification: Amplify targets of interest using pool-specific primers
- Amplicon Generation: Produce ~150 bp amplicons representing targeted genes [1]
- Partial Digestion: Utilize enzymatic digestion to prepare amplicons for adapter ligation
- Adapter Ligation: Incorporate Illumina-specific adapters and sample barcodes (indexes) using AmpliSeq CD or UD Indexes [2]
This PCR-based approach replaces nonspecific hybridization steps, resulting in high-specificity, high-uniformity amplified libraries [2]. The multiplexing capability allows multiple genes to be analyzed in a single assay, significantly reducing processing time and costs.
Library Normalization, Pooling, and Quality Control
The final steps prepare libraries for sequencing:
- Library Normalization: Use AmpliSeq Library Equalizer for Illumina to normalize libraries, an optional but recommended step that simplifies pooling [10]
- Quality Assessment: Evaluate library quality and quantity using Agilent Bioanalyzer High Sensitivity chip [18]
- Pooling: Dilute libraries to appropriate concentration (typically 100 pM) and pool multiple samples (up to 8 samples per pool recommended)
- Sequencing Ready: The pooled libraries are now ready for template preparation and sequencing on Illumina platforms
Table 2: AmpliSeq Library Preparation Specifications and Performance
| Parameter | Specification | Notes |
|---|---|---|
| Hands-on time | <1.5 hours | Minimal manual intervention required |
| Total assay time | 5.5-7.5 hours | Library preparation component only |
| Input quantity | 1-100 ng (10 ng recommended) | Suitable for limited samples |
| Multiplexing capacity | Up to 384-plex | Using unique dual indexes |
| Amplicon size | ~150 bp | Optimized for sequencing efficiency |
| Reproducibility | R² = 0.997 | Demonstrated with technical replicates |
| Compatibility | MiSeq, iSeq, NextSeq systems | Multiple Illumina sequencing platforms |
Downstream Processing and Data Analysis
Sequencing and Initial Data Processing
Following library preparation, pooled libraries undergo sequencing on Illumina platforms such as MiSeq, iSeq, or NextSeq systems [2]. The targeted nature of AmpliSeq libraries means that fewer raw reads are required for accurate gene expression quantification compared to traditional whole-transcriptome RNA sequencing [1]. This efficiency translates to reduced sequencing costs and faster analysis times.
Initial data processing involves:
- Demultiplexing: Separating sequenced reads by sample using the incorporated barcodes
- Quality Control: Assessing read quality using tools like FastQC or Falco to identify potential issues [19] [20]
- Read Alignment: Mapping reads to reference transcripts or genomes using alignment tools such as Bowtie, HISAT2, or STAR [19] [21]
Gene Expression Quantification and Differential Expression Analysis
The core analysis focuses on quantifying gene expression levels and identifying differentially expressed genes (DEGs) between experimental conditions:
- Read Counting: Using tools like featureCounts to assign reads to genes [19]
- Abundance Estimation: Employing statistical methods such as RSEM (RNA-Seq by Expectation Maximization) for accurate transcript quantification, particularly valuable for handling reads that map to multiple genes or isoforms [21]
- Differential Expression Analysis: Utilizing specialized packages like DESeq2 to identify statistically significant changes in gene expression between sample groups [19]
- Visualization: Creating plots such as heatmaps and volcano plots to represent genes and gene sets of interest [19]
The AmpliSeq approach demonstrates strong concordance with other gene expression quantification methods, with reported Pearson correlation coefficients of r = 0.92 when comparing log2 fold change to both Illumina HiSeq and Ion Torrent Proton platforms [1].
Technical Validation and Performance Metrics
Quality Assurance and Analytical Validation
Comprehensive technical validation of the AmpliSeq RNA workflow demonstrates robust performance characteristics:
- Sensitivity and Specificity: The AmpliSeq Childhood Cancer Panel showed 98.5% sensitivity for DNA variants with 5% variant allele frequency (VAF) and 94.4% sensitivity for RNA fusions, with 100% specificity for DNA [18]
- Reproducibility: High reproducibility rates are achieved, with 100% reproducibility for DNA and 89% for RNA in the Childhood Cancer Panel [18]
- Coverage Uniformity: Consistent coverage across targets, with mean read depth greater than 1000× reported in validation studies [18]
- Limit of Detection: Reliable detection of variants at low frequencies (5% VAF) [18]
These performance metrics make the technology suitable for both research and clinical applications where accuracy and reproducibility are paramount.
Comparison to Alternative Methodologies
When compared to whole transcriptome sequencing approaches, AmpliSeq demonstrates several advantages:
- Lower Input Requirements: Successful with as little as 1 ng total RNA versus typically 100-1000 ng for standard RNA-seq [2] [17]
- Reduced Sequencing Costs: Requires fewer reads per sample for equivalent gene-level quantification [1]
- Faster Turnaround: Streamlined workflow with less hands-on time [2]
- Superior Performance with Challenging Samples: Enhanced performance with degraded samples such as FFPE tissues [17]
Evaluation studies have demonstrated that AmpliSeq "excels in the limiting areas of RNA-seq for gene expression quantification analysis" and represents "a very sensitive and cost-effective approach for very large scale gene expression analysis" [1].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for AmpliSeq RNA Workflow
| Reagent/Kit | Function | Application Notes |
|---|---|---|
| AmpliSeq Library PLUS | Core library preparation reagents | Includes reagents for preparing 24, 96, or 384 libraries |
| AmpliSeq CD Indexes | Sample barcoding | 8 bp indexes for multiplexing up to 384 samples |
| AmpliSeq cDNA Synthesis | RNA to cDNA conversion | Required for working with RNA panels; includes reaction mix and enzyme blend |
| AmpliSeq Library Equalizer | Library normalization | Optional but recommended for normalizing libraries before pooling |
| AmpliSeq Custom RNA Panel | Targeted amplification | 12-1,200 gene targets designed via DesignStudio |
| AmpliSeq ERCC RNA Spike-In Mix | Process controls | External RNA controls for quantitating differential gene expression |
| AmpliSeq for Illumina Direct FFPE DNA | FFPE sample processing | Prepares DNA from unstained, slide-mounted FFPE tissues without deparaffinization |
Workflow Visualization
Diagram 1: AmpliSeq RNA Workflow from Extraction to Sequencing. This visualization outlines the key steps in preparing RNA samples for sequencing using the AmpliSeq for Illumina methodology, highlighting critical quality control points and processing stages.
Troubleshooting and Optimization Guidelines
Common Challenges and Solutions
Successful implementation of the AmpliSeq RNA workflow may require optimization in specific scenarios:
- Low Yield Libraries: Increase input RNA within recommended range (up to 100 ng) or verify RNA quality metrics prior to cDNA synthesis
- Uneven Coverage: Ensure proper library normalization using AmpliSeq Library Equalizer and verify primer pool performance [10]
- High Duplicate Rates: Reduce PCR cycle numbers during amplification or increase starting material to improve library complexity
- Failed Quality Control: Implement rigorous RNA quality assessment and consider using the AmpliSeq for Illumina Direct FFPE DNA kit for challenging sample types [2]
For specialized applications such as wastewater surveillance or other low-template samples, modified approaches like the ARTIC-Amp protocol (which combines targeted amplification with rolling circle amplification) have shown improved performance over standard methods [22].
Automation and High-Throughput Applications
The AmpliSeq RNA workflow is amenable to automation, with compatibility for liquid handling robots enabling higher throughput processing [17]. For large-scale studies:
- Batch Processing: Utilize 384-reaction kits for processing large sample numbers [2]
- Quality Monitoring: Implement automated electrophoresis systems (e.g., Labchip, TapeStation) for efficient quality control [18]
- Data Analysis Pipelines: Employ automated analysis solutions such as the DRAGEN RNA Pipeline for efficient secondary analysis [17]
The scalability of the AmpliSeq system makes it suitable for studies ranging from small pilot investigations to large-scale population analyses, with consistent performance across sample batches.
Within the framework of AmpliSeq cDNA synthesis for Illumina RNA workflow research, the conversion of total RNA to complementary DNA (cDNA) represents a critical initial step that determines the success of subsequent targeted sequencing applications. The AmpliSeq cDNA Synthesis for Illumina kit is specifically designed to integrate seamlessly with AmpliSeq for Illumina RNA Panels, enabling researchers to generate high-quality sequencing libraries from minimal RNA input [23]. This protocol is optimized for a broad spectrum of investigative contexts, from oncology research to immunology studies, providing the foundation for precise gene expression quantification and variant detection [2] [24].
The AmpliSeq technology employs a highly multiplexed PCR-based workflow that replaces nonspecific hybridization steps, resulting in libraries with high specificity and uniformity [2]. This targeted approach allows for the analysis of multiple genes in a single assay, significantly saving time and reducing overall sequencing costs while maintaining analytical sensitivity and accuracy comparable to traditional RNA-seq methods [25].
Materials and Equipment
Research Reagent Solutions
The following table details essential materials and their specific functions within the AmpliSeq cDNA synthesis workflow:
Table 1: Essential Research Reagents and Materials
| Item Name | Function/Application |
|---|---|
| AmpliSeq cDNA Synthesis for Illumina Kit [23] | Converts total RNA to cDNA for use with AmpliSeq for Illumina RNA Panels. |
| AmpliSeq for Illumina RNA Panel (e.g., Immune Response, Comprehensive Panel v3) [3] | Provides target-specific primer pools for multiplexed amplification of genes of interest. |
| AmpliSeq Library PLUS for Illumina [3] [2] | Contains reagents for preparing sequence-ready libraries from the synthesized cDNA. |
| AmpliSeq CD Indexes for Illumina [3] [2] | Enables sample multiplexing by adding unique barcode sequences to each library. |
| AmpliSeq Library Equalizer for Illumina [3] | Normalizes libraries using a bead-based method prior to sequencing. |
Technical Specifications and Sample Requirements
The AmpliSeq cDNA Synthesis kit is supplied in a 2-tube format, separating the reaction mix from the enzyme blend to ensure stability [23]. The number of reactions per kit varies depending on the specific AmpliSeq RNA panel used, which dictates planning for larger studies.
Table 2: Kit Configuration and Sample Requirements
| Parameter | Specification |
|---|---|
| Kit Configuration | 2-tube format (Reaction Mix & Enzyme Blend) [23] |
| Reactions per Kit | 100 reactions for Immune Response, Focus, Comprehensive Panel v3, and Custom panels [23] |
| Reactions per Kit | 200 reactions for Transcriptome Human Gene Expression, Myeloid, Immune Repertoire Plus, and TCR beta Panels [23] |
| Input Quantity | 1-100 ng total RNA (10 ng per pool recommended) [2] |
| Hands-On Time | < 1.5 hours for library prep [2] |
| Total Assay Time | ~5 hours (library prep only) [2] |
Methodologies
The complete experimental pathway from sample to sequencer is illustrated below, highlighting the role of cDNA synthesis within the broader context:
Detailed cDNA Synthesis Protocol
Procedure
Initiate the cDNA synthesis reaction by combining the isolated total RNA with the provided reaction mix and enzyme blend [23]. The specific thermal cycling conditions should follow the manufacturer's recommendations for the particular AmpliSeq RNA panel in use. This step efficiently converts the RNA template into stable cDNA, which serves as the input for the subsequent targeted amplification.
Library Preparation and Sequencing
Following cDNA synthesis, the workflow proceeds through a series of integrated steps:
- Targeted Amplification: The synthesized cDNA is amplified using the selected AmpliSeq for Illumina panel (e.g., Comprehensive Panel v3), which contains primer pools designed to generate thousands of specific amplicons from targeted genes [24].
- Library Construction: Amplification products are used to prepare sequence-ready libraries using the AmpliSeq Library PLUS for Illumina kit [3] [2].
- Indexing: Unique sample identifiers (indexes) are ligated using the AmpliSeq CD Indexes kit, enabling multiplexing of multiple samples in a single sequencing run [2].
- Sequencing: Final libraries are sequenced on supported Illumina platforms, such as the MiSeq, iSeq, or NextSeq systems [2] [24].
Application Example: Comprehensive Panel v3 Workflow
The Comprehensive Panel v3 exemplifies a typical application, requiring coordinated use of multiple kits. The following diagram and table detail the specific reagent requirements for different study scales.
Table 3: Reagent Planning for Comprehensive Panel v3 Workflow (per sample)
| Component | Function | Required per 24 Samples |
|---|---|---|
| AmpliSeq for Illumina Comprehensive Panel v3 | Provides targeted primer pools for DNA and RNA | 1 panel [24] |
| AmpliSeq Library PLUS for Illumina | Prepares sequence-ready libraries | 2 x 24-reaction kits [24] |
| AmpliSeq CD Indexes Set A | Provides unique barcodes for sample multiplexing | 1 set (96 indexes) [24] |
| AmpliSeq cDNA Synthesis for Illumina | Converts total RNA to cDNA for the RNA portion | 1 kit [24] |
Performance and Validation
Technical Validation
Independent research has demonstrated that the AmpliSeq targeted approach excels in gene expression quantification analysis. A comprehensive performance comparison revealed a strong concordance between AmpliSeq and traditional RNA-seq methods, with Pearson correlation coefficients of r = 0.92 for log2 fold change comparisons using standard reference RNA samples [25]. This high level of accuracy, combined with the method's sensitivity, makes AmpliSeq a robust and cost-effective approach for large-scale gene expression analysis and mRNA marker screening [25].
The technology is particularly well-suited for profiling limited or challenging sample types, including formalin-fixed, paraffin-embedded (FFPE) tissue, while maintaining flexibility in input RNA requirements from 1 to 100 ng [2]. Specialized accessories like the AmpliSeq for Illumina Direct FFPE DNA further enhance compatibility with archived clinical samples [3].
Discussion
Integrating the cDNA synthesis protocol within the complete AmpliSeq for Illumina RNA workflow provides researchers with a streamlined solution for targeted transcriptome studies. The combination of the AmpliSeq cDNA Synthesis kit with tailored RNA panels creates a standardized pipeline that minimizes hands-on time while maximizing data quality and reproducibility [2].
This methodology is particularly valuable in drug development contexts where reproducible, high-throughput screening of gene expression patterns in response to therapeutic interventions is required. The availability of predefined panels focusing on key biological pathways—such as oncology (Comprehensive Panel, Myeloid), immunology (Immune Response), and others—enables researchers to quickly implement targeted sequencing without the need for extensive custom panel design [3] [24].
The AmpliSeq platform's compatibility with multiple Illumina sequencing systems, from the benchtop MiniSeq to the higher-throughput NextSeq series, offers scalability to match varying project needs [24]. This flexibility, combined with the demonstrated performance characteristics, establishes this cDNA synthesis and library preparation protocol as a robust foundation for advanced transcriptome research in both basic and translational science.
Within the framework of AmpliSeq cDNA synthesis for Illumina RNA workflow research, the AmpliSeq Library PLUS kit provides a foundational and efficient method for constructing sequencing-ready libraries. This targeted RNA sequencing approach leverages a highly multiplexed polymerase chain reaction (PCR)-based workflow, enabling researchers to simultaneously amplify hundreds to thousands of specific gene targets from cDNA derived from RNA samples [2] [10]. The protocol is optimized for compatibility with a range of Illumina benchtop sequencers, including the MiSeq, iSeq 100, and NextSeq series systems, facilitating a seamless transition from library preparation to sequencing and analysis [2] [10] [11].
The key advantage of this methodology lies in its ability to generate high-quality, specific data from challenging sample types, such as formalin-fixed, paraffin-embedded (FFPE) tissue and blood, even with low input quantities down to 1 ng of RNA (with 10 ng recommended) [10] [11]. By replacing nonspecific hybridization steps with a targeted PCR amplification, the workflow achieves high specificity and uniformity, making it an indispensable tool for researchers and drug development professionals focused on gene expression and transcript variant analysis [2] [4].
Key Specifications and Performance Data
The table below summarizes the core performance characteristics and technical specifications of the AmpliSeq for Illumina library preparation workflow, which are critical for experimental planning.
Table 1: Key Specifications of the AmpliSeq for Illumina Workflow
| Parameter | Specification |
|---|---|
| Total Assay Time | ~5–7.5 hours (library prep only; excludes quantification, normalization, and pooling) [2] [10] [11] |
| Hands-On Time | < 1.5 hours [2] [10] [11] |
| Input Quantity | 1–100 ng RNA (10 ng recommended per pool) [10] [11] |
| Mechanism of Action | Multiplex PCR [2] [10] [11] |
| Number of Amplicons | 12 to 12,288 for DNA; up to 1,200 for custom RNA panels [2] [10] [26] |
| Specialized Sample Types | Blood, FFPE tissue [2] [10] [11] |
| Key Supported Variant Classes | Transcript variants, Gene fusions, SNPs, Indels [2] [11] |
This streamlined workflow demonstrates robust performance, as evidenced by experimental data showing a high concordance (R² = 0.997) between technical replicates when using custom RNA panels, ensuring reliable and reproducible results [10].
Detailed Experimental Protocol
The entire process, from cDNA to sequencing-ready libraries, can be completed in less than a day. The following diagram illustrates the key stages of the AmpliSeq for Illumina library preparation workflow.
Step-by-Step Methodology
cDNA Synthesis (Pre-requisite): For RNA workflows, total RNA must first be converted to cDNA using the AmpliSeq cDNA Synthesis for Illumina kit. This step provides the single-stranded cDNA template required for the subsequent multiplex PCR amplification [10] [11].
Multiplex PCR Amplification:
- Reaction Setup: Combine the synthesized cDNA (1-100 ng, 10 ng recommended) with the chosen AmpliSeq panel (e.g., Custom RNA Panel, Transcriptome Human Gene Expression Panel) and the AmpliSeq Library PLUS master mix [10] [11].
- Amplicon Generation: The panel consists of a highly multiplexed primer pool designed to simultaneously amplify the targeted genomic regions of interest. This PCR-based enrichment replaces nonspecific hybridization, resulting in a library with high specificity and uniformity [2] [4].
Post-PCR Cleanup:
- Primer Digestion: Following PCR amplification, the remaining primers are partially digested using a specific enzyme blend included in the Library PLUS kit. This prepares the amplicon ends for the subsequent adapter ligation step [4].
Adapter Ligation and Barcoding:
- Indexing: Adapter sequences, which include Illumina sequencing motifs and sample-specific barcodes (indexes), are ligated to the ends of the amplicons. This step is crucial for multiplexing, as it allows multiple libraries to be pooled and sequenced together on the same flow cell. Common index options include the AmpliSeq UD Indexes (24 indexes) or AmpliSeq CD Indexes (96 indexes per set) [2] [10].
Library Normalization and Pooling:
- Quantification and Normalization: The final libraries are quantified. The AmpliSeq Library Equalizer for Illumina is an optional but recommended accessory that provides a bead-based method for normalizing libraries, ensuring balanced representation in the final pool [2] [10].
- Pooling: The normalized, barcoded libraries are combined into a single pool for a single sequencing run [2].
The Scientist's Toolkit: Essential Research Reagents
Successful execution of the AmpliSeq for Illumina workflow requires several key components. The table below lists the essential reagents and their specific functions within the protocol.
Table 2: Essential Research Reagent Solutions for the AmpliSeq Workflow
| Component | Function | Example Catalog Number |
|---|---|---|
| AmpliSeq Library PLUS Kit | Core library prep reagents for PCR, primer digestion, and adapter ligation. Available in 24, 96, and 384 reactions. [2] | 20019101 (24-rxn) |
| AmpliSeq Panel | Targeted primer pool for multiplex PCR (e.g., Custom RNA, Transcriptome, Focus Panel). Defines the genomic content of your library. [10] [11] | 20020496 (Custom RNA) |
| AmpliSeq cDNA Synthesis Kit | Converts total RNA to cDNA; required for all AmpliSeq for Illumina RNA panels. [10] [11] | 20022654 |
| AmpliSeq Index Adapters | Contains unique barcodes for sample multiplexing. UD Indexes (24) or CD Indexes (96 per set). [2] [10] | 20019104 (UD Indexes) |
| AmpliSeq Library Equalizer | Bead-based normalization kit; optional but recommended for simplifying library pooling. [2] [10] | 20019171 |
| AmpliSeq for Illumina Direct FFPE DNA | For direct library construction from slide-mounted FFPE tissues without DNA purification. [2] | 20023378 |
The AmpliSeq Library PLUS kit, integrated with cDNA synthesis, provides a robust and streamlined solution for targeted RNA sequencing. Its fast, multiplexed PCR-based workflow, combined with low input requirements and compatibility with degraded samples from FFPE tissues, makes it particularly valuable for research and drug development in oncology and other fields where sample material is limited [2] [26] [4]. The availability of both ready-to-use and custom panels via the DesignStudio tool allows researchers to tailor their genomic investigations with high precision, efficiency, and data quality.
In the realm of precision medicine and oncological research, targeted RNA sequencing has emerged as a powerful methodology that balances comprehensive transcriptome analysis with cost-effective sequencing. Researchers face a critical decision point when selecting between custom RNA panels and specialized RNA fusion panels, each designed to address distinct research questions within a broad target range of 12 to 1200 genes. Within the context of the AmpliSeq cDNA synthesis workflow for Illumina RNA sequencing, this selection becomes increasingly significant, as it directly impacts library preparation efficiency, data quality, and analytical outcomes.
RNA fusion panels provide a targeted approach for detecting known and novel fusion transcripts—hybrid genes formed through chromosomal rearrangements that serve as critical diagnostic biomarkers and therapeutic targets in cancer [27]. These panels employ specialized chemistries like anchored multiplex PCR (AMP) to capture breakpoints without prior knowledge of partner genes, enabling discovery of novel fusions alongside known oncogenic drivers [27]. In contrast, custom RNA panels offer researchers the flexibility to design targeted sequencing approaches around specific research interests, whether focusing on particular pathways, disease mechanisms, or functional gene groupings [28] [29]. The decision between these approaches hinges on multiple factors, including research objectives, sample types, and analytical requirements.
Technology Comparison: Strategic Advantages and Applications
RNA Fusion Panels: Specialized Detection of Structural Variants
RNA fusion panels are optimized for sensitive detection of gene fusions, which are particularly prevalent in hematological malignancies and solid tumors. These panels target specific exon regions of genes known to be involved in oncogenic fusions, enabling comprehensive profiling of both known and novel fusion events.
Table 1: Commercial RNA Fusion Panel Specifications
| Panel Name | Target Genes | Target Range | Input Requirements | Specialized Applications |
|---|---|---|---|---|
| TruSight RNA Fusion Panel [30] | 507 | Fixed | 10 ng total RNA; 20–100 ng FFPE RNA | Pan-cancer fusion detection in hematologic and solid tumors |
| iGeneTech Hema Tumor Fusion RNA Panel [31] | 141 | Fixed | Optimized for FFPE and low-input samples | Hematologic malignancies with focus on drug resistance and prognosis |
| Archer FusionPlex Custom Panel [27] | 17 (customizable) | 12+ | 20–50 ng FFPE RNA | Targeted fusion detection with AMP technology for known and novel partners |
The TruSight RNA Fusion Panel exemplifies a comprehensive solution, covering 507 fusion-associated genes related to cancer in a single assay, with specialized optimization for challenging sample types like formalin-fixed paraffin-embedded (FFPE) tissues [30]. This panel employs a capture-based chemistry that provides a simple, streamlined method for isolating targeted regions of interest from total RNA, making it particularly valuable for clinical research settings where sample quality and quantity may be limiting factors.
Validation studies of RNA fusion panels have demonstrated exceptional performance characteristics. One systematic validation of a 17-gene fusion panel reported accuracy, reproducibility, and precision above 99%, with a limit of detection for most RNA fusion transcripts of approximately 50 copies [27]. The implementation of such panels as reflex tests following DNA sequencing has been shown to increase diagnostic yield by 10% in tumor samples lacking DNA driver mutations, significantly expanding the therapeutic landscape without substantial increases in processing time or cost [27].
Custom RNA Panels: Flexible, Focused Assay Design
Custom RNA panels empower researchers to design targeted sequencing approaches tailored to specific experimental needs, providing the flexibility to focus on genes of interest while maintaining the benefits of targeted sequencing's cost efficiency and simplified data analysis.
Table 2: Custom RNA Panel Design Considerations
| Design Factor | Options | Impact on Panel Performance |
|---|---|---|
| Target Region | Full transcript coverage, Specific exons, Non-coding regions | Determines ability to detect variants, splicing events, and novel transcripts |
| Primer Design | Automated platforms (e.g., ampPD), Manual curation | Affects amplification efficiency, coverage uniformity, and specificity |
| Sample Input | Standard (100 ng), Low-input (1-10 ng), Degraded (FFPE) | Influences sensitivity, reproducibility, and success rate |
| Multiplexing Capacity | 12-1200 targets | Impacts cost efficiency and experimental scale |
The SLIMamp technology from Pillar Biosciences exemplifies advancements in custom panel design, enabling efficient sequencing from as little as 2.5 ng of input DNA and improving coverage uniformity through elimination of unwanted amplicons [28]. This approach is particularly valuable for precious or limited samples where maximum information recovery is essential. Custom panels also excel in simplifying bioinformatics workflows by excluding irrelevant genomic regions, thereby streamlining variant calling and interpretation [28].
Custom panels provide distinct advantages in oncology research, where they can be designed to sequence beyond traditional gene "hot spots" to include entire coding sequences or non-coding regions with clinical significance [28]. The AmpliSeq for Illumina platform supports this custom approach, offering researchers the ability to design panels focusing on specific pathways or disease mechanisms with as few as 12 or as many as 1200 targets, seamlessly integrating with the AmpliSeq cDNA synthesis workflow.
Comparative Analysis: Fusion vs. Custom Panel Applications
Table 3: Method Selection Guide: RNA Fusion Panels vs. Custom RNA Panels
| Feature | RNA Fusion Panels | Custom RNA Panels |
|---|---|---|
| Primary Application | Fusion transcript detection in cancer research | Focused gene expression and variant analysis across diseases |
| Target Flexibility | Fixed or limited customization around fusion genes | Highly flexible; researcher-defined targets |
| Novel Discovery Capability | Detects novel fusion partners through breakpoint mapping | Limited to pre-selected targets; no novel fusion detection |
| Sample Requirements | Optimized for FFPE and low-quality samples (10-100 ng) | Varies by design; can be optimized for low input (1-100 ng) |
| Hands-on Time | ~11 hours (library prep to sequencing) [30] | Variable; typically less than broad transcriptome approaches |
| Bioinformatics Complexity | Moderate; requires specialized fusion calling algorithms | Simplified through targeted analysis |
| Ideal Research Context | Oncology biomarker discovery, clinical assay development | Pathway-focused studies, validation cohorts, translational research |
Panel Selection Framework: Matching Technology to Research Needs
Selecting between custom RNA and RNA fusion panels requires systematic evaluation of research objectives, sample characteristics, and practical constraints. The following decision workflow provides a structured approach to this selection process:
Decision Workflow for Panel Selection
Key Selection Criteria
Research Objectives: RNA fusion panels are indispensable for studies focused specifically on oncogenic fusion events, particularly in clinical oncology settings where comprehensive fusion profiling directly impacts therapeutic decisions [27]. Custom RNA panels excel in pathway-focused studies, biomarker validation, and focused mechanistic investigations where predefined gene sets are already established [32] [28].
Sample Characteristics: Both panel types can be optimized for challenging samples, but RNA fusion panels often include specific validation for FFPE tissues and low-input samples, making them preferable for clinical archives and biopsies [30] [27]. Custom panels offer flexibility in adapting to various sample types through tailored primer design and optimization.
Target Flexibility vs. Standardization: Fixed-content RNA fusion panels provide standardized workflows and established validation metrics, advantageous for clinical applications and multi-institutional studies [27]. Custom panels enable investigator-driven target selection, crucial for emerging research fields or specialized biological pathways not covered by commercial offerings [29].
Multiplexing and Throughput Requirements: Recent technological advances enable both panel types to support moderate multiplexing. The Ion AmpliSeq Transcriptome Human Gene Expression Kit demonstrates the capacity to target 20,802 genes while maintaining compatibility with limited RNA input (as little as 10 ng), though with constraints on sample multiplexing (approximately 16 samples per run) [33]. Alternative technologies like the MERCURIUS family of solutions enable multiplexing of up to 384 samples, significantly enhancing throughput for large-scale studies [33].
Integrated Experimental Protocol: AmpliSeq Workflow for Fusion Detection
This section outlines a comprehensive protocol for implementing targeted RNA sequencing within the AmpliSeq framework, incorporating best practices for reliable results across custom and fusion panel applications.
Sample Preparation and Quality Control
RNA Extraction and QC: Extract total RNA using validated kits (e.g., Qiagen AllPrep DNA/RNA FFPE kit) [27]. For FFPE samples, use 5-10 unstained slides with 5-μm sections, with macrodissection guided by H&E staining to ensure ≥20% tumor cellularity [27]. Quantify RNA using fluorometric methods (e.g., Qubit) and assess integrity. Input requirements typically range from 10-100 ng for standard samples, with FFPE samples requiring 20-100 ng [30] [27].
cDNA Synthesis: Convert RNA to cDNA using the SuperScript VILO cDNA Synthesis Kit within the AmpliSeq workflow [33]. For comprehensive transcript coverage, employ a combination of anchored oligo(dT) primers (ensuring 3' end capture) and random hexamer primers (providing whole-transcriptome representation) [34]. This dual-priming strategy minimizes 3' bias and ensures accurate representation of gene expression across transcript lengths, particularly critical for fusion detection where breakpoints may occur in 5' regions.
Library Preparation and Target Enrichment
Target Amplification: For custom panels, utilize automated primer design platforms (e.g., Pillar's ampPD) to optimize primer specificity and coverage uniformity [28]. For fusion panels, employ anchored multiplex PCR (AMP) chemistry which uses molecular barcodes and universal primer binding sites to ligate with double-stranded cDNA, enabling selection of fragments with known and novel fusion partners [27].
Library QC and Normalization: Perform pre-sequence quality check using qPCR (e.g., KAPA SYBR FAST qPCR Master Mix) to assess library quality and quantity [27]. Normalize libraries to ensure equal representation before pooling. For custom panels, implement PiVAT software or similar bioinformatics tools to filter reads without proper amplicon structure, enabling variant calling with frequencies as low as 1% [28].
Sequencing and Data Analysis
Sequencing Configuration: Sequence on appropriate Illumina platforms (MiSeq, NextSeq 500/550, or MiniSeq) using recommended read lengths (e.g., 2×76 bp for fusion detection) [30]. For custom panels, adjust sequencing depth based on target numbers—higher depth (≥500x) for variant detection, moderate depth (≥100x) for expression quantification.
Fusion Detection Analysis: Process FastQ files through specialized algorithms (e.g., Archer Analysis, STAR-Fusion) to identify fusion events supported by both spanning reads and junction reads [31]. Establish confidence thresholds based on fusion scores (typically >0.6 for high-confidence calls) and orthogonal validation [30].
Research Reagent Solutions: Essential Materials for Targeted RNA Sequencing
Table 4: Essential Research Reagents for Targeted RNA Sequencing Workflows
| Reagent Category | Specific Products | Function in Workflow | Key Features |
|---|---|---|---|
| RNA Extraction | Qiagen AllPrep DNA/RNA FFPE kit [27], Meridian RNA Isolation Kits [34] | Nucleic acid purification from various sample types | Optimized for FFPE tissues, high yield, inhibitor-free |
| cDNA Synthesis | SuperScript VILO cDNA Synthesis Kit [33], SensiFAST cDNA Synthesis Kit [34] | Reverse transcription of RNA to cDNA | Combination of oligo(dT) and random hexamer primers, low-input capability |
| Library Preparation | Archer FusionPlex Reagents [27], Ion AmpliSeq Transcriptome reagents [33] | Target amplification and library construction | Molecular barcoding, AMP technology for fusion detection |
| Target Enrichment | TruSight RNA Fusion Oligo Panel [30], iGeneTech AIdesign Hema Panel [31] | Hybrid capture or amplicon-based enrichment | 3X tiling strategy, exon-junction coverage |
| QC & Quantification | KAPA SYBR FAST qPCR Master Mix [27], Qubit fluorometric systems [27] | Library quality assessment and quantification | Sensitive detection, accurate normalization |
Case Studies: Application in Oncological Research
Implementing a Reflex Testing Algorithm in Solid Tumors
A clinical validation study demonstrated the utility of implementing an RNA fusion panel as a reflex test following DNA variant analysis [27]. In this approach, 450 tumor samples lacking DNA driver mutations were subjected to additional RNA fusion testing using a 17-gene custom fusion panel. This sequential testing algorithm resulted in a 10% increase in diagnostic yield with minimal additional processing time and cost [27]. The study highlights the practical advantage of targeted RNA fusion panels in clinical settings, where comprehensive molecular profiling directly impacts therapeutic decisions. The validated panel demonstrated >99% accuracy, reproducibility, and precision across 44 FFPE and fresh-frozen specimens representing NSCLC, thyroid cancer, glioblastomas, and gastrointestinal tumors [27].
Combining Short-Read and Long-Read Sequencing for Comprehensive Fusion Detection
Emerging methodologies combine the strengths of different sequencing technologies to overcome limitations of individual approaches. A recent study developed a workflow that integrated targeted panel-based sequencing with whole-transcriptome long-read sequencing to enhance fusion detection in glioma samples [35]. Researchers first adapted libraries from the short-read CHOP Cancer Fusion Panel (targeting 119 oncogenes) for Oxford Nanopore Technologies' long-read platform, confirming compatibility and reducing turnaround times. Most significantly, when they applied whole-transcriptome long-read sequencing to 24 glioma samples with negative short-read fusion panel results, they identified 20 candidate novel fusions that were subsequently experimentally validated [35]. This approach demonstrates how hybrid strategies can maximize detection sensitivity while leveraging the benefits of targeted panels.
The selection between custom RNA panels and specialized RNA fusion panels represents a critical decision point in targeted transcriptome analysis. RNA fusion panels offer specialized, optimized solutions for comprehensive fusion detection in oncology research, particularly valuable in clinical settings where sensitivity and reproducibility are paramount. Custom RNA panels provide unparalleled flexibility for focused research applications, enabling investigators to concentrate resources on biologically or clinically relevant targets.
Within the AmpliSeq cDNA synthesis workflow for Illumina platforms, both panel types benefit from standardized, robust library preparation methods that maintain compatibility with challenging sample types, including FFPE tissues. The emerging trend of combining targeted approaches with whole-transcriptome sequencing demonstrates the potential for hybrid strategies to maximize detection capabilities while maintaining cost efficiency.
As targeted sequencing technologies continue to evolve, researchers should consider not only current needs but also emerging applications, such as long-read sequencing integration and automated bioinformatics solutions, that may influence panel design and selection criteria. By aligning panel choice with specific research objectives, sample characteristics, and analytical requirements, investigators can optimize their transcriptional profiling strategies to generate robust, actionable data across diverse research contexts.
The ability to generate reliable transcriptomic data from challenging sample types is a cornerstone of advanced clinical and translational research. Formalin-fixed paraffin-embedded (FFPE) tissues and whole blood specimens represent invaluable resources for studying disease mechanisms, particularly in cancer and rare genetic disorders. However, these samples present significant technical hurdles: RNA from FFPE tissues is often fragmented and chemically modified, while blood-derived RNA contains abundant globin and ribosomal RNA (rRNA) that can dominate sequencing libraries and obscure biological signals [36] [37] [38]. The AmpliSeq for Illumina platform addresses these challenges through a targeted, PCR-based workflow that maintains robust performance with low-input and degraded samples [4] [2] [39]. This application note details optimized protocols and experimental considerations for successful RNA sequencing from these valuable but demanding sample types, enabling researchers to unlock molecular insights from archival and minimally-invasive specimens.
Sample-Type-Specific Challenges and Solutions
FFPE Tissue Specimens
Archival FFPE tissues represent an immense resource for biomedical research, particularly for studies requiring long-term clinical follow-up. However, the formalin fixation process fragments RNA and introduces chemical modifications, while long-term storage can further degrade nucleic acids [36] [37]. Successful RNA-seq from these specimens requires specialized approaches from extraction through data analysis.
RNA extraction from FFPE tissues can be optimized through pathologist-assisted macrodissection to enrich for regions of interest. One study reported average RNA yields of 402 ng/cm² from oropharyngeal squamous carcinoma specimens stored for up to 20 years, demonstrating that even decades-old blocks can yield sufficient material for sequencing [37]. The DV200 value (percentage of RNA fragments >200 nucleotides) serves as a more reliable quality metric than RIN for FFPE RNA, with samples having DV200 >30% generally producing usable sequencing data [36].
For library preparation, the TaKaRa SMARTer Stranded Total RNA-Seq Kit v2 and Illumina Stranded Total RNA Prep with Ribo-Zero Plus have both demonstrated effectiveness with FFPE samples. A comparative analysis revealed that the TaKaRa kit achieved comparable gene expression quantification to the Illumina kit while requiring 20-fold less RNA input, a crucial advantage for limited samples [36]. Both kits generated data with high concordance in differential expression and pathway analyses, despite differences in rRNA depletion efficiency and alignment rates [36].
Blood-Derived Specimens
Blood samples offer a minimally-invasive alternative for transcriptomic studies, particularly valuable for rare diseases and longitudinal monitoring. However, blood-derived RNA presents unique challenges including high levels of globin mRNA and rRNA that can consume sequencing depth, and the limited expression of tissue-specific genes in peripheral blood mononuclear cells (PBMCs) [40] [38].
Research demonstrates that PBMCs express approximately 69.4% of genes from large disease panels, with particularly strong performance for neurodevelopmental disorders (79.7% of intellectual disability and epilepsy genes expressed) [40]. For rare disease diagnostics, short-term cultured PBMCs treated with cycloheximide (CHX) to inhibit nonsense-mediated decay (NMD) have proven effective for capturing aberrant transcripts resulting from pathogenic variants [40]. The effectiveness of CHX treatment can be monitored using SRSF2 transcripts as an internal control, with studies showing increased expression of NMD-sensitive SRSF2 isoforms from 4.55% to 8.58% after treatment [40].
For whole blood studies, specialized depletion methods are essential. The REALLY hg-rNONE kit simultaneously depletes rRNA and hemoglobin transcripts, significantly improving mapping rates and detection of informative genes [41]. Without such depletion, hemoglobin transcripts can constitute up to 40% of sequencing reads from blood samples, drastically reducing study power [41].
Low-Quality and Low-Input RNA Samples
Degraded RNA samples with low integrity numbers (RIN < 3) require modified approaches that do not depend on intact RNA. Random primed cDNA synthesis rather than poly-A selection is recommended for these samples, as it does not exhibit the 3' bias associated with degraded RNA [38]. The REALLY rNONE workflow has demonstrated success with highly degraded FFPE RNA (RIN < 3) with inputs as low as 10ng, generating libraries with high mapping quality where double-stranded methods fail even with increased PCR cycles [41].
For amplification-based methods like AmpliSeq, the extremely high efficiency of targeted PCR enables robust performance with minute RNA quantities. The AmpliSeq for Illumina workflow officially recommends 10ng input per pool but can function with as little as 1ng of input material, making it suitable for precious biopsies with limited yield [2] [39].
Quantitative Comparison of RNA-Seq Methods for Challenging Samples
Table 1: Performance comparison of RNA-seq methods and kits for different sample types
| Method/Kits | Recommended Input | FFPE Performance | Blood Performance | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| AmpliSeq for Illumina | 1-100 ng (10 ng recommended) | Excellent with specialized panels [39] | Excellent with cDNA synthesis [2] | Fast workflow (5-6 hr), minimal hands-on time (<1.5 hr), simple data analysis [2] [39] | Targeted approach limits discovery potential |
| TaKaRa SMARTer Stranded Total RNA-Seq v2 | Low input (20-fold less than conventional) | High-quality data, comparable to Illumina kit [36] | Information not available in search results | Ultra-low input capability, stranded libraries | Higher rRNA content (17.45%), increased duplication rate [36] |
| Illumina Stranded Total RNA with Ribo-Zero Plus | Conventional input | High-quality data, better alignment rates [36] | Information not available in search results | Efficient rRNA depletion (0.1% rRNA), low duplication rate [36] | Higher input requirements |
| REALLY rNONE/hg-rNONE | 10-250 ng | Works with highly degraded RNA (RIN < 3) [41] | hg-rNONE depletes hemoglobin transcripts [41] | Simple workflow, directional libraries without 2nd strand synthesis, handles degraded samples [41] | Newer technology with less established track record |
| snRandom-seq | Single nucleus | Designed specifically for FFPE [42] | Information not available in search results | Single-nucleus resolution, broad RNA coverage including non-coding RNAs [42] | Complex protocol (4 days), specialized equipment required |
Table 2: Key quality metrics for RNA-seq from challenging samples
| Quality Metric | Target Values for FFPE | Target Values for Blood | Interpretation |
|---|---|---|---|
| DV200 | >30% [36] | Not critical | Percentage of RNA fragments >200 nucleotides; key for FFPE quality assessment |
| RIN | Not reliable for FFPE | >7 for high-quality blood RNA [38] | RNA Integrity Number; less informative for FFPE samples |
| rRNA Content | <10% ideal [36] | <5% with depletion [41] | Percentage of ribosomal reads; indicates depletion efficiency |
| Mapping Rate | >80% [36] | >70% with hemoglobin depletion [41] | Percentage of reads aligning to reference genome |
| Genes Detected | >13,000 protein-coding genes [37] | Varies by cell type and panel | Measure of library complexity and coverage |
| Globin/ Hemoglobin Reads | Not applicable | <5% with depletion [41] | Critical metric for blood samples; should be minimized through depletion |
Detailed Experimental Protocols
AmpliSeq for Illumina cDNA Synthesis and Library Preparation Protocol
The AmpliSeq for Illumina system provides a streamlined workflow for targeted RNA sequencing from challenging samples. The complete protocol requires 5-6 hours for library preparation with less than 1.5 hours of hands-on time [2] [39].
cDNA Synthesis from Total RNA
Input Requirements: Use 1-100 ng of total RNA (10 ng recommended per pool). For FFPE samples, ensure DV200 >30% [36]. For blood samples, consider prior globin RNA depletion if using whole blood [38] [41].
Reverse Transcription: Utilize the AmpliSeq cDNA Synthesis Module to convert total RNA to cDNA. The kit includes reaction mix and enzyme blend optimized for low-input and challenging samples [2].
Quality Assessment: Verify cDNA synthesis success through fluorometric quantification. Proceed immediately to library preparation to minimize degradation.
Library Preparation via Multiplex PCR
Target Amplification: Combine cDNA with the appropriate AmpliSeq panel (e.g., Comprehensive Panel v3, Focus Panel, or custom designs). The Comprehensive Panel v3 targets 161 oncogenes through 4,648 amplicons divided into four pools [39].
PCR Conditions: Perform multiplex PCR amplification following manufacturer's recommendations. The highly efficient PCR enables robust amplification even from degraded templates.
Primer Digestion: Digest remaining PCR primers to prevent interference with downstream steps.
Adapter Ligation: Add Illumina-compatible adapters and sample-specific indexes using AmpliSeq CD Indexes or UD Indexes. The system supports multiplexing of up to 96 samples in a single run [2].
Library Normalization: Use AmpliSeq Library Equalizer for efficient normalization before pooling libraries. Purify final library pool using SPRI beads.
Sequencing and Data Analysis
Sequencing Configuration: Most AmpliSeq panels require 2 × 150 bp reads. For the Comprehensive Panel v3, sequence 16 samples on a NextSeq 550 Mid Output flow cell or 48 samples on a High Output flow cell to achieve minimum coverage of 500× [39].
Data Analysis: Utilize the DRAGEN RNA Amplicon pipeline for secondary analysis, including differential expression and gene fusion calling. The pipeline is available via BaseSpace Sequence Hub or Local Run Manager [4].
Specialized Protocol for FFPE-Derived RNA
For optimal results with FFPE samples, specific modifications to the standard protocol are recommended:
Pathologist-Assisted Macrodissection: Prior to nucleic acid extraction, employ precise macrodissection to enrich for regions of interest while excluding non-relevant tissue areas [36]. This is particularly crucial for tumor microenvironment studies where contamination from adjacent normal tissue can confound results.
Simultaneous DNA/RNA Extraction: Use the AllPrep DNA/RNA FFPE kit (Qiagen) following manufacturer's instructions with modifications. Begin with 6 μm thick sections, with average total RNA yield of approximately 3629 ng from ~9 slides (2 cm² each) [37].
RNA Demodification and Fragmentation: For older FFPE blocks, implement RNA demodification protocols to reverse formalin-induced modifications. For highly fragmented RNA, additional fragmentation may be unnecessary [43].
Quality Control: Use DV200 rather than RIN for quality assessment. Samples with DV200 values ranging from 37% to 70% have been successfully sequenced despite extensive fragmentation [36].
Library Preparation Selection: Choose FFPE-optimized kits such as the TaKaRa SMARTer Stranded Total RNA-Seq Kit v2 for limited samples or Illumina Stranded Total RNA Prep with Ribo-Zero Plus for conventional inputs [36].
Specialized Protocol for Blood-Derived RNA
For transcriptomic analysis from blood specimens:
Sample Collection and Preservation: Collect blood in PAXgene tubes or RNAlater for stabilization. These preservation methods require specific extraction protocols downstream [38].
PBMC Isolation and Culture: Isolate PBMCs using density gradient centrifugation. For rare disease diagnostics involving potential NMD substrates, culture cells for short-term with cycloheximide (CHX) treatment to inhibit NMD [40].
RNA Extraction: Use column-based methods that retain small RNAs if studying microRNAs. For standard mRNA sequencing, most commercial kits provide sufficient quality.
Globin and rRNA Depletion: Employ depletion strategies for whole blood RNA. The REALLY hg-rNONE kit simultaneously depletes rRNA and hemoglobin transcripts, significantly improving unique mapping rates [41]. Alternatively, the Epicentre Ribo-Zero rRNA depletion kit followed by random-primed cDNA synthesis can be used [38].
Quality Assessment: Verify RNA quality through bioanalyzer and fluorometric quantification. For NMD inhibition studies, confirm efficacy by assessing increased expression of NMD-sensitive transcripts such as SRSF2 [40].
Workflow Visualization and Decision Framework
Diagram 1: Sample processing workflow for FFPE and blood specimens. This decision framework guides researchers to appropriate methods based on sample type and quality.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key reagents and kits for RNA sequencing from challenging samples
| Reagent/Kits | Primary Application | Key Features | Sample Compatibility |
|---|---|---|---|
| AmpliSeq for Illumina Library PLUS | Library preparation | Fast workflow (5-6 hr), minimal hands-on time (<1.5 hr), supports 1-100 ng input [2] | FFPE tissue, blood, low-quality RNA |
| AmpliSeq cDNA Synthesis for Illumina | cDNA synthesis | Converts total RNA to cDNA optimized for AmpliSeq panels [2] | All sample types |
| AmpliSeq for Illumina Comprehensive Panel v3 | Targeted sequencing | Investigates 161 cancer-associated genes, detects SNVs, Indels, CNVs, fusions [39] | FFPE tissue |
| REALLY hg-rNONE | rRNA and hemoglobin depletion | Simultaneously depletes rRNA and hemoglobin transcripts, directional libraries [41] | Whole blood, other samples with hemoglobin |
| AllPrep DNA/RNA FFPE Kit | Nucleic acid extraction | Simultaneous DNA/RNA extraction from FFPE tissues [37] | FFPE tissue |
| TaKaRa SMARTer Stranded Total RNA-Seq Kit v2 | Whole transcriptome | Ultra-low input capability, works with degraded RNA [36] | FFPE tissue, low-input samples |
| Illumina Stranded Total RNA Prep with Ribo-Zero Plus | Whole transcriptome | Efficient rRNA depletion (0.1% rRNA), high alignment rates [36] | FFPE tissue, standard quality samples |
Data Analysis and Quality Control Considerations
Robust data processing pipelines are essential for deriving reliable biological insights from challenging samples. For FFPE-derived data, specific normalization approaches address technical artifacts:
Filtering and Normalization: Remove non-protein coding genes and exclude zero counts. Calculate 75th percentile read values for each sample, then normalize counts by both sample-specific upper quartile and gene size [37].
Technical Outlier Detection: Implement median absolute deviation (MAD) approaches to identify and remove technical outliers that could skew results [37].
Data Transformation and Scaling: Perform log2 transformation after adding a small constant (0.01) to all values. Rescale data to a global median (e.g., 7.0) to enable consistent threshold application across experiments with varying sequencing depths [37].
Pathway Analysis Validation: Verify biological consistency through pathway enrichment analysis. Studies show 80% concordance in enriched pathways between different library preparation methods when analyzing the same biological samples [36].
For targeted approaches like AmpliSeq, the integrated DRAGEN RNA Amplicon pipeline provides optimized variant calling, differential expression analysis, and fusion detection specifically designed for amplicon-based data [4].
The methodologies detailed in this application note demonstrate that robust RNA sequencing from challenging sample types is now achievable through careful protocol selection and optimization. The AmpliSeq for Illumina platform provides a particularly streamlined solution for targeted RNA sequencing from FFPE tissues and blood specimens, enabling researchers to leverage these valuable resources for biomarker discovery, diagnostic development, and mechanistic studies. As single-nucleus technologies like snRandom-seq mature and become more accessible [42], and depletion methods continue to improve [41], the research community can increasingly overcome the historical limitations of these precious clinical samples, opening new avenues for understanding disease biology and advancing personalized medicine.
Within the context of AmpliSeq for Illumina RNA workflow research, the reproducibility and technical performance of gene expression experiments are critical for generating reliable data. This application note details the roles of both required and optional components in the workflow, with a specific focus on the AmpliSeq Library Equalizer and ERCC Spike-In Controls. The Library Equalizer is an integral part of the library preparation process, while the ERCC Spike-In Controls provide a powerful external standard for quality control and data normalization, enabling researchers to control for technical variability and accurately interpret biological results [44] [45].
Core Components of the AmpliSeq for Illumina RNA Workflow
The AmpliSeq for Illumina RNA workflow is a targeted, multiplex PCR-based approach designed for next-generation sequencing (NGS) on Illumina platforms. The following diagram illustrates the key stages of the complete workflow, integrating both required and optional components.
Required Research Reagent Solutions
The following table details the essential components for constructing an AmpliSeq for Illumina cDNA library.
Table 1: Required Components for the AmpliSeq for Illumina RNA Workflow
| Component Name | Catalog Number Example | Function in the Workflow |
|---|---|---|
| AmpliSeq cDNA Synthesis for Illumina | 20022654 | Converts total RNA to cDNA for subsequent PCR amplification in RNA panels [46] [2]. |
| AmpliSeq Library PLUS for Illumina | 20019101 (24 rxns) | Contains the core reagents for library amplification, including DNA Ligase and FuPa Reagent [46] [2]. |
| AmpliSeq CD/UD Indexes | 20019105 (Set A) | Unique oligonucleotide sequences ligated to amplicons to enable multiplexing of samples [2] [47]. |
| AmpliSeq RNA Panel | 20019164 (Focus Panel) | A ready-to-use or custom set of primers targeting specific genes of interest for amplification [4] [47]. |
The Library Equalizer: An Essential Workflow Component
The AmpliSeq Library Equalizer for Illumina (Catalog No. 20019171) is a critical, though sometimes considered optional, reagent for normalizing libraries prior to sequencing.
- Function: It automates the normalization and pooling of amplified libraries, ensuring a balanced representation of each sample in the final sequencing pool. This process improves data quality and reduces manual hands-on time [46] [2].
- Kit Contents: The kit contains Equalizer Beads, Equalizer Capture solution, Elution Buffer, Wash Buffer, and Equalizer Primer [46].
- Storage: The components are stored between 2°C to 8°C, except for the Wash Buffer, which is stable at room temperature (15°C to 30°C) [46].
ERCC Spike-In Controls: An Optional Tool for Rigorous QC
The ERCC (External RNA Controls Consortium) RNA Spike-In Mix is a set of external controls developed by a consortium including NIST to address challenges in technical variability and data reproducibility in gene expression analysis [44] [45].
Composition and Rationale
The controls consist of 92 synthetic, unlabeled, polyadenylated transcripts that mimic natural eukaryotic mRNAs, with lengths ranging from 250 to 2,000 nucleotides [44]. These transcripts are formulated into defined blends with known concentrations. By spiking a constant amount of these controls into an RNA sample after isolation but before library preparation, researchers can create an internal standard curve for their experiment [44].
Key Applications and Configurations
The primary applications of ERCC controls and the available kit configurations are summarized in the table below.
Table 2: ERCC Spike-In Control Kits and Their Applications
| Kit Name | Catalog Number | Contents | Primary Application |
|---|---|---|---|
| ERCC RNA Spike-In Mix | 4456740 | 10 µL of Spike-In Mix 1, 1.75 mL Nuclease-free Water | Assess platform dynamic range and limit of detection [44]. |
| ERCC ExFold RNA Spike-In Mixes | 4456739 | 10 µL each of Spike-In Mix 1 & 2, 1.75 mL Nuclease-free Water | Assess the accuracy of differential gene expression measurements [44]. |
The ExFold kit is specifically designed for fold-change analysis, as the two mixes contain the same 92 transcripts but in different, highly concordant abundance ratios across four subgroups. This allows for precise calculation of expression fold-changes with a high degree of confidence [44].
Integrated Experimental Protocols
Protocol 1: Incorporating ERCC Spike-In Controls into the AmpliSeq Workflow
This protocol describes the key steps for using ERCC controls to monitor technical performance.
- Step 1: Spike-In Addition. After total RNA isolation and quality assessment, add the recommended volume of the appropriate ERCC Spike-In Mix (e.g., 10 µL of Mix 1 from Catalog No. 4456740) directly to your RNA sample. This must be performed before the cDNA synthesis step [44].
- Step 2: Proceed with Standard Workflow. Continue with the standard AmpliSeq for Illumina RNA protocol, which includes: cDNA synthesis using the AmpliSeq cDNA Synthesis for Illumina kit (20022654), followed by multiplexed PCR amplification with your chosen AmpliSeq panel and the Library PLUS kit [2] [47].
- Step 3: Library Normalization. Use the AmpliSeq Library Equalizer for Illumina (20019171) to normalize the final libraries before pooling and sequencing on an Illumina platform [46].
- Step 4: Data Analysis with ERCC Metrics. Following sequencing, analyze the data using the
erccdashboardsoftware package available from NIST. This tool uses the known input amounts of the spike-ins to generate performance metrics, including dynamic range, limit of detection, and ratio measurement bias, providing an independent assessment of the experiment's technical quality [45].
Protocol 2: Validation of Gene Expression Measurements
A systematic comparison of RNA-seq procedures underscores the importance of technical validation.
- qRT-PCR Validation: As performed in scientific studies, select a set of constitutively expressed genes (housekeeping genes) and genes of interest from your RNA-seq data. Validate their expression levels using TaqMan qRT-PCR assays on the same cDNA samples [48].
- Data Normalization for qRT-PCR: For the qRT-PCR data, use a robust normalization method such as the global median normalization approach, which calculates a normalization factor using the median Ct value of genes with Ct < 35 for each sample. This helps correct for potential biases, such as under-expression of common housekeeping genes (e.g., GAPDH, ACTB) under certain treatment conditions [48].
- Correlation Analysis: Compare the normalized expression values from the AmpliSeq workflow (with and without ERCC-based normalization) to the qRT-PCR results. This validation provides a measure of the accuracy and precision of the gene expression quantification from your primary method [48].
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Quality-Driven AmpliSeq Research
| Item | Function |
|---|---|
| AmpliSeq cDNA Synthesis for Illumina (20022654) | Converts input RNA into template-ready cDNA for amplification [2]. |
| AmpliSeq Library Equalizer for Illumina (20019171) | Reagents for automated normalization of libraries to ensure even sequencing coverage [46]. |
| ERCC RNA Spike-In Mix (4456740) | External RNA controls to assess technical performance and dynamic range [44]. |
| ERCC ExFold Spike-In Mixes (4456739) | Two mixes of controls for validating differential gene expression measurements [44]. |
erccdashboard R Package |
Software for analyzing data from ERCC controls and generating performance metrics [45]. |
Optimizing Results: Troubleshooting Common cDNA Synthesis and Library Prep Issues
In targeted RNA sequencing workflows, such as the AmpliSeq for Illumina platform, the consistent generation of high-quality complementary DNA (cDNA) is a critical prerequisite for reliable gene expression data. The AmpliSeq for Illumina RNA workflow requires cDNA synthesis as an essential first step, enabling the analysis of a broad range of input materials (1-100 ng RNA, with 10 ng recommended) from various sample types, including challenging formalin-fixed, paraffin-embedded (FFPE) tissues and blood [10] [11] [47]. However, many researchers encounter suboptimal cDNA yields that compromise downstream library preparation, sequencing efficiency, and ultimately, experimental outcomes.
Low cDNA yield directly impacts the sensitivity and accuracy of transcript detection, particularly for low-abundance genes critical in regulatory processes and biomarker studies [49] [50]. This application note systematically addresses the primary factors contributing to inefficient cDNA synthesis—RNA integrity and reverse transcription optimization—within the context of the AmpliSeq workflow. By implementing the detailed protocols and troubleshooting strategies outlined herein, researchers can significantly improve cDNA yield, quality, and reproducibility, thereby enhancing the robustness of their transcriptional analyses in drug development and clinical research applications.
RNA Quality Assessment and Degradation Prevention
The integrity of starting RNA material represents the most fundamental variable influencing cDNA synthesis efficiency. Degraded RNA templates cannot support complete reverse transcription, leading to truncated cDNA fragments, biased transcript representation, and ultimately, low yields that compromise downstream AmpliSeq library preparation.
RNA Integrity Monitoring
- Spectrophotometric and Fluorometric Assessment: Utilize multiple quantification methods (e.g., Nanodrop, Qubit, BioAnalyzer) to obtain comprehensive RNA quality metrics [51]. While UV spectrophotometry provides information about protein contamination (260/280 ratio) and solvent contamination (260/230 ratio), fluorometric methods offer greater accuracy for quantifying nucleic acid concentration specifically.
- RNA Integrity Number (RIN): Employ microfluidic capillary electrophoresis platforms (e.g., Agilent BioAnalyzer, Bio-Rad Experion) to assign RIN values, which numerically represent RNA degradation levels [51]. Samples with RIN values below 7.0 may exhibit compromised cDNA synthesis efficiency and should be used with caution.
Practical RNA Preservation Techniques
- Inhibition of RNases: Implement rigorous RNase-free techniques throughout sample processing, including wearing gloves, using aerosol-barrier pipette tips, and regularly decontaminating work surfaces with appropriate RNase deactivating solutions [52].
- Optimal Storage Conditions: Store purified RNA at -80°C in nuclease-free buffers with minimal freeze-thaw cycles. Aliquot RNA samples to avoid repeated freezing and thawing, which accelerates degradation.
- Inhibitor Removal: Ensure complete removal of common reverse transcription inhibitors during RNA purification, including salts, metal ions, ethanol, phenol, and heparin [52]. Specific purification methods should be selected based on starting materials (e.g., blood, tissues, cells) to maximize inhibitor removal while maintaining RNA integrity.
Table 1: Troubleshooting RNA Quality Issues
| Problem | Potential Causes | Solutions |
|---|---|---|
| Low RIN values | Improper tissue preservation, RNase contamination, prolonged room temperature exposure | Flash-freeze samples in LN₂, use RNase inhibitors, minimize thaw time |
| Spectral ratio deviations | Protein or solvent contamination | Add purification steps (e.g., column cleanup, reprecipitation) |
| Discrepant concentration readings | Method-specific limitations or contaminants | Cross-validate with multiple quantification methods [51] |
| Degraded RNA in FFPE samples | Fixation time, storage duration, extraction method | Optimize fixation protocols, use specialized FFPE RNA extraction kits |
Reverse Transcription Optimization Strategies
The reverse transcription step converts RNA templates to cDNA through the action of reverse transcriptase (RT) enzymes. Multiple factors in this process significantly impact cDNA yield, including enzyme selection, reaction composition, and cycling conditions.
Reverse Transcriptase Selection
Reverse transcriptases vary considerably in their properties, which directly influence cDNA yield and quality. While the AmpliSeq for Illumina workflow includes a dedicated cDNA Synthesis Kit [3] [2], understanding enzyme characteristics remains valuable for troubleshooting:
- Enzyme Origins and Characteristics: Most reverse transcriptases derive from either Avian Myeloblastosis Virus (AMV) or Moloney Murine Leukemia Virus (MMLV). AMV RT exhibits high RNase H activity and thermal stability but produces shorter cDNA fragments (<5 kb). MMLV RT has lower RNase H activity and can generate longer cDNAs (<7 kb) [52].
- Engineered Reverse Transcriptases: Modern engineered MMLV derivatives (e.g., SuperScript IV, Maxima H Minus) feature mutated RNase H domains that enhance processivity, thermostability (up to 55°C), and cDNA yields, particularly with challenging or suboptimal RNA templates [52] [49].
- Performance in Low-Input Applications: Studies comparing RT enzymes for ultralow RNA input (0.5-5 pg) demonstrated that Maxima H Minus Reverse Transcriptase provided superior sensitivity for detecting low-abundance genes and higher cDNA yields at minimal input amounts compared to other enzymes [49].
Reaction Component Optimization
The precise formulation of the reverse transcription reaction mixture profoundly impacts efficiency:
Primer Selection: The choice of priming strategy significantly influences cDNA yield and representation:
- Oligo(dT) Primers: Prime specifically from the poly-A tails of mRNA, providing targeted amplification of protein-coding transcripts but potentially missing non-polyadenylated RNAs and exhibiting 3' bias [51].
- Random Hexamers: Prime throughout the RNA sequence, enabling detection of non-polyadenylated RNAs and better coverage of transcript 5' regions, though may increase ribosomal cDNA background [52].
- Gene-Specific Primers: Provide highly specific priming for particular targets of interest, offering superior sensitivity for low-expression genes but requiring multiple reactions for analyzing multiple targets [50].
- Mixed Priming Approaches: Combining oligo(dT) and random hexamers can capitalize on the advantages of both methods, particularly for whole-transcriptome analyses [51].
Reaction Buffer Composition: Optimize buffer components according to manufacturer recommendations, paying particular attention to:
- DTT Concentration: Use fresh DTT solutions to maintain reducing conditions necessary for enzyme stability.
- Magnesium and Potassium Levels: These cations influence enzyme processivity and fidelity; deviations from recommended concentrations can dramatically impact yield.
- dNTP Quality: Use high-quality, freshly prepared dNTPs at recommended concentrations (typically 0.5-1 mM each) to ensure efficient incorporation [52].
Thermal Cycling Parameters
Optimized thermal cycling conditions enhance reverse transcription efficiency, particularly for structured RNA templates:
- Annealing Conditions: When using random hexamers, include a preliminary 10-minute incubation at room temperature (~25°C) to promote primer annealing before temperature elevation [52].
- Elevated Reaction Temperatures: For GC-rich templates or RNAs with significant secondary structure, employ thermostable reverse transcriptases at higher temperatures (50-55°C) to melt stable structures that would otherwise impede reverse transcription [52].
- Reaction Duration: Polymerization time should be optimized based on the processivity of the specific reverse transcriptase used. Engineered enzymes with high processivity may require only 10-30 minutes, while wild-type MMLV often needs >60 minutes for complete cDNA synthesis [52].
AmpliSeq-Specific Workflow Considerations
The AmpliSeq for Illumina platform employs a targeted, multiplex PCR-based approach that places specific demands on the cDNA synthesis step to ensure optimal performance.
cDNA Synthesis for AmpliSeq Panels
The AmpliSeq workflow requires conversion of total RNA to cDNA using the dedicated AmpliSeq cDNA Synthesis for Illumina kit prior to library preparation [3] [11] [2]. This step is essential for all AmpliSeq RNA panels, including:
- Transcriptome Human Gene Expression Panel (>20,000 human RefSeq genes)
- Focus Panel (52 genes with relevance to solid tumors)
- Custom RNA Panels (12-1200 user-defined targets)
- Comprehensive Cancer Panels (e.g., Comprehensive Panel v3)
The cDNA synthesis kit includes a specialized reaction mix and enzyme blend optimized for compatibility with downstream AmpliSeq library preparation, ensuring uniform coverage across targets of varying abundance and sequence context [11] [47].
Input RNA Recommendations
While AmpliSeq for Illumina RNA panels support a broad input range (1-100 ng total RNA), consistent results are achieved with 10 ng input RNA [11] [47]. Input quantities below this recommendation may require special considerations:
- Ultralow Input Protocols: For precious samples yielding less than 10 ng RNA, incorporate carrier RNA or employ specialized ultralow input protocols that enhance reaction efficiency through modified reverse transcriptases and template-switching oligonucleotides [49].
- RNA Quantification Accuracy: Consistently use the same quantification method throughout an experiment to maintain input RNA consistency, as different measurement platforms (Nanodrop, Qubit, BioAnalyzer) can yield varying concentration estimates [51].
Genomic DNA Removal
Trace genomic DNA (gDNA) contamination can lead to false-positive signals and reduced assay specificity. Effective gDNA removal strategies include:
- DNase I Treatment: Traditional DNase I digestion effectively removes gDNA but requires careful inactivation before cDNA synthesis to prevent degradation of cDNA products.
- Thermolabile Double-Strand-Specific DNases: Enzymes such as ezDNase offer streamlined workflows with mild temperature inactivation (55°C) that doesn't compromise RNA integrity or interfere with subsequent cDNA synthesis [52].
Advanced Methodologies for Challenging Samples
Certain sample types present unique challenges for cDNA synthesis that require specialized approaches beyond standard protocols.
Ultralow RNA Input Applications
For minute RNA inputs (<100 pg), as encountered in single-cell analyses, laser-capture microdissected samples, or circulating tumor cells, specialized methodologies dramatically improve cDNA yield:
- Template-Switching Mechanisms: Methods like Ordered Two-Template Relay (OTTR) and SMART-based protocols capture end-to-end sequences of input molecules while appending sequencing adapters in the same reverse transcription step, significantly improving sensitivity for short or fragmented RNAs [53] [49].
- Enhanced Sensitivity Modifications: Incorporation of modified template-switching oligonucleotides (rN-modified TSO) and optimized reverse transcriptases (e.g., truncated Bombyx mori R2 RT) improve capture precision and reduce nonproductive sequencing reads [53].
- Reaction Miniaturization: Reducing reaction volumes (to 5-10 µL) increases effective reactant concentrations, enhancing molecular interaction frequencies and yields from limited input materials.
Degraded RNA Samples
FFPE tissues frequently yield degraded RNA that challenges conventional cDNA synthesis protocols:
- RNA Integrity Preservation: Optimize FFPE processing protocols to minimize RNA degradation, including reduced fixation times, proper tissue processing, and specialized storage conditions.
- Targeted Amplification Approaches: AmpliSeq panels perform robustly with partially degraded RNA because they target shorter amplicons (typically <175 bp) that are more likely to be preserved in degraded samples [11] [47].
- 3'-Bias Utilization: For severely degraded samples, employ detection methods focused on the 3' ends of transcripts, as these regions are preferentially preserved in degradation processes.
Experimental Protocols
Protocol 1: Optimized cDNA Synthesis for Standard RNA Inputs (10-100 ng)
This protocol is adapted for use with the AmpliSeq cDNA Synthesis for Illumina kit and follows manufacturer recommendations with optimized parameters for improved yield [52] [3].
Materials:
- AmpliSeq cDNA Synthesis for Illumina (Catalog #20022654)
- High-quality RNA samples (RIN > 8.0 recommended)
- Nuclease-free water
- Thermal cycler
Procedure:
- Genomic DNA Removal:
- Combine 1-100 ng RNA (10 ng recommended) with 2 µL of gDNA Removal Buffer in a 0.2 mL nuclease-free tube.
- Incubate at 50°C for 5 minutes, then place immediately on ice.
Reverse Transcription Master Mix Preparation:
- Prepare the following mixture for each reaction:
- 4 µL 5× cDNA Synthesis Buffer
- 1 µL cDNA Synthesis Enzyme
- 1 µL cDNA Synthesis Primer Mix
- Nuclease-free water to a final volume of 20 µL
- Prepare the following mixture for each reaction:
Reaction Assembly and Incubation:
- Combine the gDNA-treated RNA with the master mix.
- Mix thoroughly by pipetting and centrifuge briefly.
- Incubate in a thermal cycler using the following conditions:
- 25°C for 10 minutes (primer annealing)
- 50°C for 30 minutes (cDNA synthesis)
- 70°C for 10 minutes (enzyme inactivation)
- Hold at 4°C
Product Storage:
- Use cDNA immediately for AmpliSeq library preparation or store at -20°C for up to one week.
Protocol 2: Enhanced cDNA Synthesis for Limited or Degraded RNA (<10 ng)
This protocol incorporates modifications to maximize yield from challenging samples, integrating principles from ultralow input RNA sequencing methods [49].
Materials:
- Maxima H Minus Reverse Transcriptase or equivalent engineered enzyme
- Modified template-switching oligonucleotides (rN-TSO)
- RNase inhibitors
- Betaine (5M stock)
- dNTPs (100mM)
Procedure:
- RNA Denaturation and Primer Annealing:
- Combine limited RNA input (0.5 pg-10 ng) with 1 µL of 50 µM oligo(dT) primer and 1 µL of 10 µM template-switching oligonucleotide.
- Incubate at 72°C for 3 minutes, then immediately transfer to ice.
Reverse Transcription Reaction Assembly:
- Prepare the following master mix for each reaction:
- 2 µL 5× RT buffer
- 0.5 µL RNase inhibitor (40 U/µL)
- 0.5 µL 100 mM dNTPs
- 0.5 µL 5M betaine
- 1 µL Maxima H Minus Reverse Transcriptase (200 U/µL)
- Nuclease-free water to 10 µL final volume
- Prepare the following master mix for each reaction:
Template-Switching Reaction:
- Combine the master mix with the denatured RNA-primer mixture.
- Incubate in a thermal cycler with the following program:
- 42°C for 90 minutes
- 10 cycles of (50°C for 2 minutes, 42°C for 2 minutes)
- 70°C for 15 minutes
- Hold at 4°C
Product Purification:
- Purify cDNA using solid-phase reversible immobilization (SPRI) beads according to manufacturer instructions.
- Elute in 10-15 µL nuclease-free water.
Research Reagent Solutions
Table 2: Essential Reagents for Optimized cDNA Synthesis
| Reagent Category | Specific Products | Function & Features |
|---|---|---|
| Reverse Transcriptases | AmpliSeq cDNA Synthesis for Illumina, SuperScript IV, Maxima H Minus | Converts RNA to cDNA; engineered enzymes offer higher thermostability, processivity, and lower RNase H activity [52] [49] |
| RNA Protection Reagents | RNase inhibitors, TRIzol, RNAlater | Preserves RNA integrity during storage and processing; inhibits ubiquitous RNases [52] [50] |
| gDNA Removal Kits | ezDNase Enzyme, Turbo DNA-free Kit, DNase I | Eliminates genomic DNA contamination without damaging RNA; thermolabile versions simplify inactivation [52] [50] |
| Priming Systems | Oligo(dT)₁₈, random hexamers, gene-specific primers, template-switching oligonucleotides | Initiates cDNA synthesis; choice affects transcript coverage and bias [49] [51] |
| Specialized Buffers | Betaine, DTT, magnesium solutions, K⁺-acetate | Enhances reverse transcription efficiency through destabilization of secondary structures and optimal cation concentrations [49] |
Optimizing cDNA synthesis represents a critical step in ensuring the success of AmpliSeq for Illumina RNA workflows, particularly when working with limited or challenging sample types. Through systematic attention to RNA quality assessment, reverse transcriptase selection, reaction component optimization, and specialized protocols for difficult samples, researchers can significantly improve cDNA yield and quality. The methodologies outlined in this application note provide a comprehensive framework for addressing low cDNA yield challenges, enabling more robust and reproducible gene expression data in drug development and clinical research applications.
Implementing these evidence-based approaches—from basic RNA handling practices to advanced template-switching protocols—will empower researchers to maximize the value of precious samples and generate high-quality sequencing data that faithfully represents the transcriptional landscape of their experimental systems.
Figure 1. cDNA Synthesis Optimization Workflow
The synthesis of complementary DNA (cDNA) is a foundational step in the Illumina RNA workflow, enabling the conversion of RNA transcripts into sequenceable DNA libraries. However researchers frequently encounter amplification issues that compromise data quality and experimental outcomes. Within the context of AmpliSeq cDNA synthesis for Illumina RNA workflows, two predominant technical challenges account for most amplification failures: RNA secondary structures that impede reverse transcriptase processivity, and suboptimal priming strategies that yield incomplete or biased cDNA representation. These issues manifest practically as low or no amplification in downstream applications, truncated cDNA fragments, and poor coverage of transcriptomes.
Secondary structures form when single-stranded RNA molecules fold onto themselves via intramolecular base pairing, creating stable hairpins, stem-loops, and other complex conformations. These structures present physical barriers to reverse transcriptase enzymes, causing them to dissociate from the template and resulting in incomplete cDNA synthesis. Simultaneously, the choice of priming strategy fundamentally determines which RNA species are reverse transcribed and with what efficiency. Oligo(dT) primers, random hexamers, and gene-specific primers each exhibit distinct strengths and limitations in different experimental contexts. This application note provides detailed methodologies for diagnosing, troubleshooting, and overcoming these amplification challenges within Illumina RNA workflows, supported by structured experimental data and optimized protocols.
Understanding and Disrupting RNA Secondary Structures
The Impact of Secondary Structures on Reverse Transcription
RNA secondary structures represent one of the most significant obstacles to efficient cDNA synthesis. Thermostable reverse transcriptases that withstand elevated temperatures are particularly effective for GC-rich templates, as they remain enzymatically active at temperatures that melt these stable structures [54]. When reverse transcriptase encounters these barriers, it may pause, dissociate from the RNA template, or produce truncated cDNA fragments, ultimately compromising downstream applications including library preparation and sequencing [54].
Experimental Protocols for Disrupting Secondary Structures
Protocol: Thermal Denaturation of RNA Secondary Structures
- Sample Preparation: Dilute purified RNA to working concentration in nuclease-free water or appropriate buffer [54].
- Denaturation: Heat RNA sample at 65°C for approximately 5 minutes in a thermal cycler or heat block [54].
- Rapid Cooling: Immediately transfer samples to ice and hold for at least 2 minutes to prevent reformation of secondary structures [54].
- Immediate Use: Proceed directly with reverse transcription reaction after this pretreatment.
Protocol: Elevated Temperature Reverse Transcription
- Select Appropriate Enzyme: Choose a thermostable reverse transcriptase capable of functioning efficiently at elevated temperatures (e.g., 50°C or higher) [54].
- Modify Reaction Conditions: Set up reverse transcription reaction according to manufacturer instructions with adjusted temperature parameters.
- Incubate: Perform cDNA synthesis at the optimized higher temperature (typically 50-60°C depending on enzyme specifications) [54].
- Validate: Assess cDNA yield and fragment size distribution using bioanalyzer or similar quality control method.
Table 1: Comparison of Secondary Structure Disruption Methods
| Method | Optimal Conditions | Advantages | Limitations |
|---|---|---|---|
| Thermal Denaturation | 65°C for ~5 min, rapid cooling on ice | Simple, requires no specialized reagents, effective for most structures | Effects temporary, structures may reform during cooling |
| Elevated Temperature RT | 50-60°C during reverse transcription | Continuous structure disruption throughout elongation, higher processivity | Requires thermostable enzyme, may reduce enzyme longevity |
| Denaturant Addition | Varies by denaturant type | Can be combined with thermal methods, effective for extreme structures | May inhibit reverse transcriptase, requires optimization |
Optimizing Priming Strategies for Comprehensive cDNA Coverage
Primer Selection Guidelines for Different RNA Templates
The choice of reverse transcription primer fundamentally determines cDNA representation and library complexity. Each priming strategy exhibits distinct strengths and limitations that must be matched to experimental goals and RNA template characteristics.
Oligo(dT) Primers specifically target the 3' poly(A) tails of eukaryotic messenger RNAs, providing selective amplification of protein-coding transcripts while excluding structural and non-polyadenylated RNAs. However, this approach introduces significant 3' bias, as reverse transcriptase may not fully process along lengthy transcripts, particularly when RNA integrity is compromised [55]. This limitation becomes critically important when designing PCR primers that anneal to the 5' end of the target sequence, as they may lack complementary template, resulting in false-negative results [55].
Random Hexamers anneal nonspecifically throughout the RNA population, theoretically providing uniform coverage across all RNA species regardless of polyadenylation status. In practice, however, this approach preferentially reverse transcribes ribosomal RNA due to its overwhelming abundance in total RNA extracts, potentially drowning out signal from low-abundance transcripts [55]. For weakly expressed genes, the proportion converted to cDNA may fall beneath detection thresholds in subsequent qPCR or sequencing applications.
Gene-Specific Primers offer maximal specificity for targeted applications but are impractical for whole-transcriptome approaches. These primers are ideal for one-step RT-PCR workflows focusing on particular genes of interest [55].
Experimental Protocol: Primer Optimization and Validation
Protocol: Systematic Primer Evaluation
- RNA Quality Assessment: Verify RNA integrity using agarose gel electrophoresis or microfluidics-based systems (e.g., Bioanalyzer). Intact eukaryotic RNA should display sharp ribosomal bands (28S and 18S in mammalian RNA) without smearing [54].
- Parallel Reverse Transcription: Set up identical reverse transcription reactions differing only in primer strategy:
- Tube A: Oligo(dT) primer (0.5 μg/reaction)
- Tube B: Random hexamers (50-250 ng/reaction)
- Tube C: Combination of both (0.25 μg oligo(dT) + 25-125 ng random hexamers)
- cDNA Synthesis: Perform reverse transcription according to enzyme manufacturer's protocol.
- Downstream Validation: Assess cDNA quality using:
- qPCR with primers spanning different regions of reference genes (5', middle, and 3')
- Bioanalyzer profile to visualize cDNA size distribution
- Spike-in controls to quantify amplification efficiency
Protocol: Primer Switching for Problematic Templates
- Identify Amplification Failure: Note when specific targets fail to amplify despite high-quality RNA template.
- Redesign PCR Primers: If using oligo(dT) priming, redesign PCR primers to anneal nearer the 3' end of the transcript [55].
- Switch Priming Strategy:
- From oligo(dT) to random hexamers: When detecting transcripts without poly(A) tails (e.g., viral or histone RNAs) or when dealing with degraded samples [54].
- From random hexamers to oligo(dT): When signal from rRNA overwhelms mRNA detection or when specific enrichment of polyadenylated transcripts is desired [55].
- Validate with Controls: Include appropriate positive controls (known expressed genes) and negative controls (no-template and no-RT) with each priming strategy.
Table 2: Primer Selection Guide for Challenging RNA Templates
| RNA Template Type | Recommended Primer | Rationale | Modifications |
|---|---|---|---|
| Bacterial RNA | Random primers | Bacterial mRNA lacks poly(A) tails, making oligo(dT) ineffective [54] | Use longer random primers (8-9 mers) for enhanced coverage |
| Degraded RNA | Random primers | Fragmented RNA may lack 3' ends where oligo(dT) binds [54] | Increase primer concentration to ensure binding to low-complexity fragments |
| RNA with High GC Content | Random primers + elevated temperature | Reduces secondary structure interference during priming | Combine with thermal denaturation protocol |
| Low Abundance Transcripts | Oligo(dT) or gene-specific | Reduces background from abundant rRNA species [55] | Use sequence-specific primers for ultimate specificity |
| Full-Length cDNA Generation | Oligo(dT) | Promotes complete transcription from natural 3' start site | Use reverse transcriptase with high processivity |
Integrated Workflow for Troubleshooting Amplification Issues
Diagnostic Framework for Amplification Failures
When facing amplification issues in cDNA synthesis, a systematic diagnostic approach efficiently identifies the root cause. Begin by assessing RNA integrity through gel electrophoresis or microfluidics; degraded RNA appears as a smear rather than discrete ribosomal bands [55]. Next, perform a no-reverse transcriptase control (-RT) to detect genomic DNA contamination, which manifests as amplification in the absence of reverse transcriptase [54]. If these preliminary checks pass, proceed to evaluate secondary structures and priming strategies using the protocols outlined below.
Comprehensive Troubleshooting Protocol
Protocol: Sequential Troubleshooting of Amplification Problems
Verify RNA Quality and Purity
- Assess RNA integrity by agarose gel electrophoresis or microfluidics
- Check RNA purity by UV spectroscopy (A260/A280 ratio ~2.0, A260/A230 >2.0)
- Address degradation issues by minimizing freeze-thaw cycles, using RNase inhibitors, and ensuring nuclease-free working conditions [54]
Eliminate Genomic DNA Contamination
- Treat RNA samples with DNase I prior to reverse transcription
- Include a no-RT control in downstream amplification assays
- Use PCR primers spanning exon-exon junctions to distinguish cDNA from gDNA amplification [54]
Address Secondary Structure Interference
- Implement thermal denaturation protocol (Section 2.2)
- Switch to thermostable reverse transcriptase
- Perform reverse transcription at elevated temperatures (50°C) [54]
Optimize Priming Strategy
- Match primer type to RNA template (refer to Table 2)
- Test combination approaches (oligo(dT) + random hexamers)
- For problematic specific targets, redesign PCR primers to anneal nearer the 3' end [55]
Evaluate and Modify Reaction Components
- Ensure appropriate dNTP concentration (0.5 mM or less) [56]
- Verify reaction component freshness and proper storage
- Confirm magnesium concentration optimization
Research Reagent Solutions
Table 3: Essential Reagents for Overcoming Amplification Challenges
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Thermostable Reverse Transcriptases | SuperScript IV, ThermoScript | Synthesizes cDNA at elevated temperatures to melt secondary structures | Essential for GC-rich templates; enables reactions at 50-60°C [54] |
| RNase Inhibitors | Recombinant RNaseOUT, Protector RNase Inhibitor | Protects RNA templates from degradation during handling | Critical for long transcripts and low-abundance targets [54] |
| Priming Systems | Oligo(dT)18-20, Random Hexamers, Gene-Specific Primers | Initiates cDNA synthesis at specific or random sites | Selection depends on RNA type and experimental goals [54] [55] |
| DNA Removal Reagents | DNase I (RNase-free), TURBO DNase | Eliminates genomic DNA contamination | Essential for accurate gene expression quantification [54] |
| RNA Integrity Tools | Bioanalyzer RNA kits, Fragment Analyzer | Assesses RNA quality before cDNA synthesis | Identifies degradation issues before committing to library prep [54] |
Workflow Visualization
Diagram 1: Systematic troubleshooting workflow for cDNA amplification issues
Successful cDNA synthesis within the Illumina RNA workflow requires meticulous attention to two fundamental challenges: RNA secondary structures that impede reverse transcriptase processivity, and priming strategies that must be carefully matched to experimental requirements. The protocols and guidelines presented in this application note provide a systematic framework for diagnosing and resolving amplification issues, enabling researchers to obtain comprehensive cDNA coverage across diverse RNA template types. By implementing thermal denaturation protocols for structured regions, selecting appropriate priming strategies based on RNA characteristics, and utilizing thermostable reverse transcriptases where needed, researchers can significantly improve cDNA yield, representation, and overall sequencing data quality. These optimized approaches ensure that AmpliSeq cDNA synthesis serves as a robust foundation rather than a limiting factor in Illumina RNA workflows, ultimately generating more reliable and comprehensive transcriptome data for drug development and basic research applications.
Within the framework of research utilizing the AmpliSeq for Illumina RNA workflow, stringent quality control (QC) is paramount to success. This targeted RNA sequencing approach, which involves converting RNA to cDNA followed by multiplex PCR-based library construction, is highly dependent on the quality of the starting material and the accuracy of the final library preparation [2]. Failures in RNA integrity or library quantification can lead to wasted resources, inconclusive results, and failed sequencing runs. This application note provides detailed protocols and structured data summaries for the two most critical QC checkpoints: initial RNA integrity assessment and final library quantification, specifically contextualized for the AmpliSeq workflow.
RNA Integrity Assessment
The integrity of RNA is a fundamental prerequisite for successful downstream applications, as degradation can severely skew gene expression measurements [57] [58]. For AmpliSeq cDNA synthesis, ensuring that the input RNA is intact is the first and one of the most critical steps.
Methods and Instrumentation
The most reliable method for assessing RNA integrity is capillary electrophoresis, which provides an objective and quantitative measurement.
- Principle of Operation: This method uses microfluidics to separate RNA fragments by size. The RNA is passed through fine capillaries containing a sieving polymer matrix. As fragments migrate, they pass a detector that measures fluorescence from an intercalating dye. The result is an electropherogram, a plot of fluorescence intensity over time (or fragment size) [59] [58].
- Key Output - RNA Integrity Number (RIN): The RIN is an algorithm-developed metric that assigns RNA quality a value from 1 (completely degraded) to 10 (perfectly intact). The algorithm considers the entire electrophoretic trace, including the ratio of 28S to 18S ribosomal RNA peaks, the presence of degradation products, and other features, providing a robust and reproducible metric [57] [60].
- Common Platforms: The Agilent 2100 Bioanalyzer is a widely adopted system for this analysis, used in conjunction with RNA-specific chips (e.g., RNA 6000 Nano/Pico kits). Equivalent systems include the Fragment Analyzer and the TapeStation [59] [61].
Protocol: RNA Integrity Analysis Using a Bioanalyzer
This protocol outlines the key steps for assessing RNA integrity using the Agilent 2100 Bioanalyzer.
Equipment and Reagent Setup:
- Agilent 2100 Bioanalyzer instrument.
- RNA 6000 Nano or Pico Kit (choice depends on expected RNA concentration).
- RNase-free pipette tips and tubes.
- A thermal block or water bath set to 70°C.
Sample Preparation:
- Thaw the RNA ladder (included in the kit) and all samples on ice.
- Denature the ladder and samples by heating at 70°C for 2 minutes, then immediately cool on ice.
Chip Priming and Loading:
- Prepare the gel-dye mix according to the kit instructions.
- Load the gel-dye mix into the appropriate well on the microfluidic chip. Use a pipette with a plunger to prime the chip.
- Pipette 5 µL of the RNA marker into the ladder well and each sample well.
- Add 1 µL of the denatured RNA ladder to its designated well.
- Add 1 µL of each denatured sample to the respective sample wells.
Vortexing and Run:
- Place the chip in the vortexer and vortex for 1 minute at the specified speed.
- Insert the chip into the Bioanalyzer and start the run using the appropriate assay settings (e.g., "RNA 6000 Nano" assay).
Data Interpretation:
- After the run, the software will generate an electropherogram and a gel-like image for each sample.
- The software automatically calculates the RIN value. Visually inspect the electropherogram for distinct 18S and 28S rRNA peaks (for eukaryotic samples) and a flat baseline. A low RIN and a smeared electropherogram indicate degradation.
Acceptable RIN Values for AmpliSeq Workflow
The required RIN threshold can vary based on the sample type and specific research goals. However, general guidelines for RNA-Seq applications, including AmpliSeq, are stringent.
Table 1: Interpretation of RNA Integrity Number (RIN) Scores
| RIN Score | Interpretation | Suitability for AmpliSeq RNA Workflow |
|---|---|---|
| 9 - 10 | Excellent/Intact | Ideal. Highly recommended for optimal results. |
| 8 | Good | Good. Generally acceptable for most applications. |
| 7 | Moderate | Conditionally Acceptable. May be used but can introduce bias; results require careful validation. |
| < 7 | Degraded/Poor | Not Recommended. High risk of failure and unreliable data; should not be used [60]. |
For formalin-fixed, paraffin-embedded (FFPE) samples, which are inherently degraded, the RIN metric is less reliable, and alternative QC metrics or specialized kits like the Illumina RNA Prep with Enrichment may be more appropriate [17].
The following workflow diagram illustrates the logical progression of the RNA QC process and its critical link to the downstream cDNA synthesis and library prep steps.
Library Quantification and QC
Following successful cDNA synthesis and AmpliSeq library construction, accurate quantification of the final sequencing library is a non-negotiable step for achieving optimal sequencing performance. Inaccurate quantification is the most common cause of over- or under-clustering on the flow cell, leading to poor data quality, reduced yield, or even run failure [62].
Quantification Methods
Different quantification methods provide different types of information, and their use cases vary significantly.
Fluorometric Methods (e.g., Qubit):
- Principle: Use fluorescent dyes that selectively bind to double-stranded DNA (dsDNA). The fluorescence intensity is proportional to the mass of dsDNA present [62] [58].
- Advantages: Highly specific for dsDNA; not affected by contaminants like salts, free nucleotides, or single-stranded RNA. Essential for measuring sample concentration in ng/µL before normalization.
- Disadvantage: Cannot distinguish between functional library molecules with intact adapters and non-functional by-products like adapter dimers or primer dimers. This can lead to an overestimation of the usable library concentration [62] [61].
qPCR-Based Methods (e.g., KAPA Biosystems kits):
- Principle: Uses primers that bind to the P5 and P7 adapter sequences added during library construction. Only fragments that possess both adapters—a requirement for cluster amplification on the Illumina flow cell—are amplified and quantified [62].
- Advantages: Quantifies only "amplifiable" library fragments. This is the gold-standard method for obtaining a molar concentration (nM) for accurate pooling and loading.
- Disadvantage: Does not provide information about library size distribution or the presence of by-products [62] [61].
Microfluidics-Based Analysis (e.g., Bioanalyzer, TapeStation):
- Principle: As described for RNA analysis, this method separates DNA fragments by size, providing a visual profile of the library.
- Primary Role: Quality Control, not primary quantification. It is used to confirm the expected size distribution of the library, assess the average fragment size (critical for converting mass to molarity), and detect contaminants like adapter dimers or primer dimers (seen as a peak around ~100-150 bp) [62] [61].
Protocol: Library QC and Quantification Workflow
A robust QC strategy combines the strengths of multiple methods.
Post-Library Construction Analysis:
- Run 1 µL of the final library on a Bioanalyzer High Sensitivity DNA chip or equivalent.
- Inspect the trace: Confirm the library profile is a single, clean peak in the expected size range. The absence of a large peak in the ~100-150 bp region indicates low adapter-dimer contamination. Note the average fragment size.
Fluorometric Quantification:
- Using a Qubit dsDNA HS Assay, measure the concentration of the library in ng/µL. This confirms sufficient mass for sequencing.
qPCR Quantification:
- Perform qPCR using a kit designed for Illumina libraries (e.g., KAPA Library Quantification Kit).
- Use a previously sequenced library of known concentration as a positive control.
- Run samples and standards in triplicate across at least two dilutions (e.g., 1:10,000 and 1:20,000).
- Calculate the library concentration in nM based on the standard curve and the average fragment size obtained from the Bioanalyzer [62].
Methods Comparison and Best Practices
A clear understanding of the strengths and limitations of each method is key to proper library quantification.
Table 2: Comparison of Library Quantification and QC Methods
| Method | What It Measures | Primary Use | Key Advantage | Key Limitation |
|---|---|---|---|---|
| Fluorometry (Qubit) | Mass of all dsDNA | Initial quantification & mass check | Specific for dsDNA; unaffected by contaminants | Overestimates functional library by counting non-functional products |
| qPCR | Concentration of amplifiable fragments | Final quantification for pooling | Measures only fragments capable of forming clusters | Does not provide size information |
| Microfluidics (Bioanalyzer) | Size distribution of all nucleic acids | Quality control & size determination | Visualizes library profile and contaminants | Not optimal for accurate quantification of broad libraries [62] |
For AmpliSeq libraries, which are amplicon-based and typically have a relatively narrow size distribution, quantification by Bioanalyzer can be considered, but qPCR remains the most accurate method for determining molarity [62] [2].
Avoid UV Spectrophotometry: Methods like NanoDrop should be avoided for final library quantification because they detect free nucleotides, single-stranded RNA, and other contaminants, leading to a significant overestimation of usable library concentration [62] [58].
The following workflow synthesizes the library QC process into a logical sequence of steps.
The Scientist's Toolkit: Essential Reagents and Materials
The following table details key research reagent solutions essential for implementing the quality control checkpoints described in this document.
Table 3: Essential Reagents and Kits for RNA and Library QC
| Item | Function/Application | Example Products |
|---|---|---|
| Microfluidics RNA Analysis Kit | Assesses RNA integrity and calculates the RIN number. | Agilent RNA 6000 Nano/Pico Kit [59] |
| Microfluidics DNA Analysis Kit | Analyzes the size distribution and quality of final NGS libraries. | Agilent High Sensitivity DNA Kit [61] |
| dsDNA Fluorescent Dye | Precisely quantifies the mass concentration of dsDNA libraries, unaffected by common contaminants. | Qubit dsDNA HS Assay Kit [62] [61] |
| Library Quantification Kit | Accurately determines the molar concentration of "amplifiable" library fragments via qPCR. | KAPA Library Quantification Kit for Illumina [62] |
| AmpliSeq Library Prep Kit | Multiplex PCR-based kit for targeted RNA library preparation from cDNA. | AmpliSeq Library PLUS for Illumina [2] |
| cDNA Synthesis Kit | Converts total RNA to cDNA for use with AmpliSeq RNA panels. | AmpliSeq cDNA Synthesis for Illumina [2] |
The success of sophisticated downstream methodologies, such as the AmpliSeq for Illumina RNA workflow, is fundamentally contingent upon the quality and integrity of the starting RNA material. This application note details the essential best practices for RNA handling, emphasizing RNase-free techniques and immediate processing. Within the context of AmpliSeq cDNA synthesis for Illumina, these procedures are not merely preliminary steps but are integral to generating biologically relevant and reproducible gene expression data. The highly multiplexed, PCR-based nature of AmpliSeq technology is designed for sensitivity and specificity, goals that can only be realized with high-quality, intact RNA input. Proper sample stabilization and RNA isolation ensure that the subsequent synthesis of cDNA—a required step for AmpliSeq RNA panels—accurantly reflects the in vivo transcriptome, thereby safeguarding the investment in sequencing and the validity of the resulting data [10] [11].
The following sections provide a detailed protocol, from sample collection to RNA storage, designed to maximize RNA yield and integrity for seamless integration with the AmpliSeq for Illumina library preparation workflow.
Establishing an RNase-Free Environment
Ribonucleases (RNases) are ubiquitous, extraordinarily stable enzymes that do not require cofactors to function, making them a persistent threat to RNA integrity. A dedicated RNase-free workspace is the first and most critical defense [63].
Personal Protective Equipment and Laboratory Hygiene
- Gloves: Wear disposable gloves at all times and change them frequently, especially after touching surfaces, skin, or hair, to prevent the transfer of RNases from the body to samples [63] [64] [65].
- Lab Coats: Dedicate a clean lab coat for RNA work to minimize contamination from clothing or the general laboratory environment [64].
Decontamination of Workspaces and Equipment
- Surfaces and Equipment: Before starting, decontaminate all work surfaces, pipettes, and other equipment with an RNase-inactivating solution, such as RNaseZap or RNase-X, followed by a wipe with nuclease-free water or 70% ethanol [64] [65].
- Glassware and Plasticware: Use sterile, disposable plasticware whenever possible, as it is manufactured to be RNase-free. For reusable glassware, bake at 180°C for at least 4 hours. Treat reusable plasticware by soaking in 0.1 M NaOH/1 mM EDTA, followed by thorough rinsing with nuclease-free water [63] [65].
Reagents and Solutions
- Source: Use only reagents certified as nuclease-free.
- Water and Buffers: Utilize nuclease-free, ultra-filtered water for all solutions. Note that Tris-based buffers cannot be treated with DEPC (diethyl pyrocarbonate) due to chemical reactivity; instead, dedicate a bottle of Tris salts for RNA work and prepare solutions with DEPC-treated, autoclaved water [64] [65].
Sample Collection, Stabilization, and Immediate Processing
RNA degradation begins immediately upon sample collection, primarily due to endogenous RNases. Therefore, the moments following collection are the most critical for preserving RNA integrity [66] [63].
Sample Collection and Rapid Stabilization
The chosen stabilization method must instantly inactivate RNases at the moment of cell or tissue disruption.
- Liquid Nitrogen Flash-Freezing: Snap-freezing small tissue pieces (≤0.5 cm) in liquid nitrogen is an effective method. The frozen samples can be pulverized and then processed or stored at -70°C to -80°C for later use [66] [67].
- Stabilization Solutions: Commercial reagents like RNAlater or RNAprotect offer flexibility. Tissues or cells are simply immersed in the solution, which rapidly permeates to stabilize RNA. This allows samples to be stored for days or even months at 4°C or -20°C (depending on the solution) before processing, without sacrificing RNA quality [66] [63]. These solutions are compatible with most subsequent RNA extraction procedures [66].
- Specialized Blood Collection: For whole blood, which is exceptionally rich in RNases, use dedicated collection systems like PAXgene Blood RNA Tubes or Tempus Blood RNA Tubes. These contain proprietary reagents that immediately stabilize RNA upon venipuncture, ensuring an accurate gene expression profile from the moment of draw [68].
Homogenization and Lysis
Once stabilized, samples must be homogenized in a denaturing lysis buffer.
- Lysis Buffer: Homogenize samples immediately in a lysis buffer containing strong denaturants, such as guanidinium isothiocyanate or TRIzol (phenol/guanidine isothiocyanate), which denature proteins and inactivate RNases on contact [66] [67].
- Method Selection: The choice of homogenization method (e.g., Dounce homogenizer, bead mill, Polytron) should be appropriate for the sample type to ensure complete disruption and efficient contact with the lysis buffer [66] [63].
Table 1: Sample Stabilization Methods and Their Applications
| Stabilization Method | Mechanism of Action | Recommended Sample Types | Compatibility with AmpliSeq Workflow |
|---|---|---|---|
| Liquid Nitrogen Flash-Freezing | Instant freezing halts all enzymatic activity. | Tissues (must be small), cell pellets. | High; frozen powder can be lysed directly. |
| RNAlater / RNAprotect | Chemical solution permeates cells to stabilize RNA. | Most tissues, cell cultures, body fluids. | High; tissue is simply removed from solution for lysis. |
| PAXgene / Tempus Tubes | Proprietary reagents in blood collection tubes. | Whole blood. | High; RNA is extracted directly from the tube. |
RNA Isolation Techniques and Considerations
Selecting the appropriate RNA isolation method is crucial for balancing yield, purity, and compatibility with downstream AmpliSeq library preparation.
- Organic Extraction (e.g., TRIzol): Considered the gold standard for RNA preparation, especially for difficult samples (high in nucleases, lipids, or carbohydrates). It involves phenol-chloroform extraction and alcohol precipitation. While highly effective, it is labor-intensive and involves hazardous chemicals [66] [64].
- Filter-Based Spin Basket Formats: These silica-membrane columns offer convenience and ease of use. They are amenable to single-sample and 96-well processing. However, they can be prone to clogging with particulate matter [66] [67].
- Magnetic Particle Methods: These use paramagnetic beads to bind RNA and are ideal for automation and high-throughput workflows. They avoid filter clogging and have efficient solution-based binding kinetics [66] [67].
Genomic DNA Removal
Carry-over genomic DNA can cause significant issues in downstream applications like RT-qPCR and RNA sequencing. On-column DNase digestion during the purification process is the recommended and most efficient method for removing residual DNA, as it is easier and allows for higher RNA recovery than post-purification treatments [67].
Table 2: Comparison of Total RNA Isolation Methods
| Purification Method | Key Principle | Benefits | Drawbacks | Suitability for AmpliSeq |
|---|---|---|---|---|
| Organic Extraction | Phenol-chloroform phase separation. | Rapid nuclease denaturation; scalable; handles difficult samples. | Use of hazardous chemicals; laborious; difficult to automate. | Excellent for challenging samples. |
| Spin Basket Column | RNA binding to silica membrane. | Convenient; easy to use; amenable to 96-well format. | Potential for filter clogging; fixed binding capacity. | Good for most sample types. |
| Magnetic Beads | RNA binding to paramagnetic particles. | Easy automation; no filter clogging; high throughput. | Potential bead carry-over; requires magnetic rack. | Ideal for automated, high-throughput labs. |
RNA Storage, Quantitation, and Quality Assessment
Optimal RNA Storage Conditions
Purified RNA remains susceptible to degradation via hydrolysis and must be stored properly.
- Aliquoting: Divide RNA into single-use aliquots to avoid repeated freeze-thaw cycles [63] [67].
- Storage Buffer: Resuspend RNA in a certified RNase-free buffer, such as THE RNA Storage Solution or nuclease-free water. The presence of EDTA (in TE buffer, pH 7.5) is beneficial as it chelates divalent cations like Mg²⁺ that catalyze RNA hydrolysis [64] [67].
- Temperature: For short-term storage (up to a few weeks), -20°C is acceptable. For long-term storage, -70°C to -80°C is mandatory to preserve RNA integrity for months or years [63] [65] [67].
Quantitation and Quality Control
Accurate assessment of RNA concentration and quality is a non-negotiable step prior to library preparation for sequencing.
- UV Spectrophotometry: Measures absorbance at 260 nm (A260) for concentration and the A260/A280 ratio for purity. An A260/A280 ratio of 1.8–2.1 indicates highly purified RNA. Traditional spectrophotometers require dilution in a specific buffer, while microvolume systems (e.g., NanoDrop) use only 1-2 µL of sample [66] [67].
- Fluorometric Methods (e.g., Qubit): These assays use RNA-binding fluorescent dyes and are more sensitive and specific than UV spectroscopy, providing accurate concentration even with dilute samples or in the presence of contaminants [67].
- Capillary Electrophoresis (e.g., Agilent Bioanalyzer): This is the gold standard for assessing RNA integrity. It provides an RNA Integrity Number (RIN), where a value of 10 represents perfectly intact RNA. While some applications like qRT-PCR can tolerate lower RINs, a minimum RIN of 7 is generally recommended for sequencing workflows, including AmpliSeq [66] [67].
Integrated Workflow for AmpliSeq for Illumina RNA Library Preparation
The following diagram and protocol outline the integrated workflow from sample to sequencer, highlighting how the preceding RNA handling practices feed into the AmpliSeq technology.
Detailed Protocol Steps
- Input RNA: Begin with high-quality total RNA (1-100 ng; 10 ng recommended) that has passed QC checks as described in Section 5.2 [10] [11].
- cDNA Synthesis: Convert the total RNA to cDNA using the AmpliSeq cDNA Synthesis for Illumina kit. This step is mandatory for all AmpliSeq for Illumina RNA Panels [10] [2] [11].
- Library Preparation (AmpliSeq Library PLUS): The cDNA is used as input into a highly multiplexed PCR reaction using the AmpliSeq Library PLUS kit and a specific RNA panel (e.g., Custom RNA Panel or Transcriptome Human Gene Expression Panel). This generates a library of sequence-specific amplicons [10] [2].
- Indexing and Normalization: Unique index adapters (e.g., AmpliSeq CD Indexes) are ligated to the amplicons to allow for sample multiplexing. The AmpliSeq Library Equalizer is recommended for easy and efficient normalization of libraries before pooling [10] [2].
- Sequencing: The pooled, normalized libraries are sequenced on a compatible Illumina sequencer (e.g., iSeq 100, MiSeq, NextSeq 1000/2000 systems) [10] [11].
Research Reagent Solutions for the AmpliSeq Workflow
Table 3: Essential Reagents and Kits for the AmpliSeq RNA Workflow
| Product Name | Function | Role in the Workflow |
|---|---|---|
| RNAlater Stabilization Solution | Stabilizes and protects cellular RNA in tissues and cells. | Preserves RNA integrity from the moment of sample collection until RNA isolation. |
| TRIzol Reagent | Monophasic solution of phenol and guanidine isothiocyanate. | Effective RNA isolation from difficult samples (high in nucleases, fats, or polysaccharides). |
| PureLink RNA Mini Kit | Spin-column based total RNA purification. | Efficient and easy isolation of high-quality RNA from most common sample types. |
| AmpliSeq cDNA Synthesis for Illumina | Converts total RNA to cDNA. | Mandatory first step to prepare RNA sample for any AmpliSeq for Illumina RNA Panel. |
| AmpliSeq for Illumina Transcriptome Human Gene Expression Panel | Ready-to-use primer pool for >20,000 human RefSeq genes. | Targeted panel for measuring whole-transcriptome gene expression. |
| AmpliSeq Library PLUS for Illumina | Reagents for preparing sequencing libraries from amplicons. | Generates the sequencing-ready library from the cDNA and panel amplicons. |
| AmpliSeq CD Indexes for Illumina | Unique nucleotide sequences for sample multiplexing. | Allows pooling of multiple libraries by tagging each with a unique index. |
| AmpliSeq Library Equalizer for Illumina | Beads and reagents for library normalization. | Simplifies and standardizes the process of normalizing libraries before pooling. |
The journey to robust and reliable RNA sequencing data begins long before the sample is loaded onto the sequencer. Meticulous adherence to RNase-free techniques and immediate sample stabilization are the foundational pillars supporting the entire AmpliSeq for Illumina RNA workflow. By implementing the best practices and detailed protocols outlined in this application note—from establishing a contamination-free workspace to selecting the appropriate isolation method and performing rigorous QC—researchers can ensure that their input RNA is of the highest possible quality. This, in turn, guarantees that the sophisticated AmpliSeq technology performs as designed, generating accurate gene expression data that truly reflects the biological system under investigation.
Within the research workflow for AmpliSeq cDNA synthesis for Illumina, the creation of a custom RNA panel is a critical step that determines the success and accuracy of subsequent gene expression and fusion analysis. The AmpliSeq for Illumina Custom RNA Panel enables researchers to design targeted panels investigating 12 to 1200 genomic targets of interest, using a menu of over 20,000 human RefSeq genes via the DesignStudio web-based assay design tool [10]. For fusion detection, the AmpliSeq for Illumina Custom RNA Fusion Panel extends this capability to include well-annotated fusion genes while maintaining gene expression profiling capabilities [12]. This application note details the optimized methodologies and considerations for achieving high coverage uniformity and reliable performance in custom panel design, providing researchers with a framework for maximizing data quality from limited RNA samples, including those from challenging sources like FFPE tissue and blood.
Custom Design Optimization Strategies
DesignStudio Assay Design Tool Utilization
The DesignStudio Assay Design Tool serves as the cornerstone of custom panel creation, offering researchers a free, user-friendly web interface for developing personalized panel content tailored to specific study requirements [4]. For researchers whose genes of interest are not covered by ready-to-use panels, DesignStudio facilitates the submission of target regions of interest and returns optimized panel content [4]. The tool allows for the design of both Custom RNA Panels (for gene expression) and Custom RNA Fusion Panels (for fusion detection and gene expression) from extensive menus of human RefSeq genes and well-annotated fusion genes [10] [12]. For complex designs or non-human targets, Illumina provides complimentary concierge design services through their sales team to assist researchers with challenging design scenarios [69].
Content Specification and Flexibility
Custom RNA panels offer researchers remarkable flexibility in content specification, with the ability to target from 12 up to 1200 gene targets in a single assay [10]. The AmpliSeq for Illumina Custom RNA Fusion Panel specifically enables the design of panels containing 12 to 1200 gene targets, incorporating both fusion detection and gene expression profiling capabilities [12]. For comprehensive transcriptome-wide analysis, the ready-to-use AmpliSeq for Illumina Transcriptome Human Gene Expression Panel targets 20,802 human RefSeq genes (covering >95% of human RefSeq genes) in a single assay, eliminating the need for custom design while providing extensive coverage [11]. This panel efficiently captures the human transcriptome using sequence-specific amplicons arranged in a single pool for streamlined processing [11].
Table 1: AmpliSeq Custom and Ready-to-Use RNA Panel Options
| Panel Type | Target Capacity | Primary Application | Key Features |
|---|---|---|---|
| Custom RNA Panel | 12-1200 gene targets [10] | Gene expression profiling | Design from >20,000 human RefSeq genes [10] |
| Custom RNA Fusion Panel | 12-1200 gene targets [12] | Fusion detection & gene expression | Menu of >1000 well-annotated fusion genes [12] |
| Transcriptome Human Gene Expression Panel | 20,802 genes [11] | Whole-transcriptome analysis | >95% of human RefSeq genes; ready-to-use [11] |
Coverage Uniformity Optimization
Technical Parameters Affecting Uniformity
Coverage uniformity across targeted regions is influenced by multiple technical parameters in the AmpliSeq workflow. The multiplex PCR-based mechanism employed by AmpliSeq panels demonstrates robust performance with various sample types, including challenging FFPE tissues [10] [12] [26]. The AmpliSeq for Illumina library preparation workflow requires minimal hands-on time (<1.5 hours) and can be completed in approximately 5.5-7.5 hours total assay time [10]. Library preparation itself takes approximately 5-7 hours with only 1.5 hours of hands-on time, making it an efficient workflow for research settings [4]. The system operates effectively with low input quantities, requiring as little as 1 ng of input RNA (with 10 ng recommended), which is particularly beneficial for limited clinical samples [10] [11].
Panel Performance and Validation
The integrated AmpliSeq workflow, which combines PCR-based library preparation with Illumina sequencing by synthesis (SBS) chemistry and automated analysis, demonstrates high concordance between replicates (R² = 0.997) and high read alignment rates [10]. For fusion detection, the Custom RNA Fusion Panel has demonstrated 100% call rates for multiple known fusions including EML4-ALK, KIF5B-RET, CD74-ROS1, and TPM3-NTRK1 in validation studies [12]. The focused panel approach consistently delivers high coverage uniformity and on-target alignment, even with DNA extracted from FFPE tissues of varying quality [47]. The AmpliSeq for Illumina Focus Panel, which investigates 52 genes with known relevance to solid tumors, has shown 100% concordance between expected and detected variant frequency in validation studies [47].
Essential Research Reagent Solutions
A successful AmpliSeq for Illumina custom RNA workflow requires several key components beyond the custom panel itself. The required products include the panel, library prep reagents (Library PLUS), and indexes, with additional accessory products available for specialized applications [10] [12].
Table 2: Essential Research Reagent Solutions for AmpliSeq Custom RNA Workflow
| Component | Function | Key Specifications |
|---|---|---|
| AmpliSeq cDNA Synthesis for Illumina | Converts total RNA to cDNA for RNA panels | Required for all AmpliSeq for Illumina RNA panels [10] [12] |
| AmpliSeq Library PLUS for Illumina | Library preparation reagents | Available in 24, 96, or 384 reactions [2] |
| AmpliSeq UD Indexes or CD Indexes | Sample multiplexing | UD Indexes: 24 indexes; CD Indexes: 96 indexes per set [10] [2] |
| AmpliSeq Library Equalizer for Illumina | Library normalization | Optional but recommended for normalizing libraries [10] |
| ERCC RNA Spike-In Mix & Companion Panel | RNA controls for quantitating differential gene expression | External RNA controls developed by ERCC [10] |
Experimental Protocol for Custom Panel Validation
Sample Preparation and Quality Control
Begin with RNA extraction from your sample source (blood, FFPE tissue, or other relevant biological material). Assess RNA quality and quantity using appropriate methods such as spectrophotometry or fluorometry. For FFPE samples, additional quality assessment is recommended. Convert total RNA to cDNA using the AmpliSeq cDNA Synthesis for Illumina kit, following manufacturer recommendations [10] [12]. This step is required for all AmpliSeq for Illumina RNA panels. The cDNA synthesis reaction includes reaction mix and enzyme blend specifically formulated for compatibility with AmpliSeq RNA panels [2]. For low-input samples, consider incorporating the ERCC RNA Spike-In Mix at this stage to monitor technical performance throughout the workflow, using the companion panel for subsequent analysis [10].
Library Preparation and Sequencing
The library preparation process follows these key steps:
Multiplex PCR Amplification: Combine the synthesized cDNA with your custom designed panel and AmpliSeq Library PLUS reagents. The multiplex PCR amplifies all targeted regions simultaneously in a highly specific reaction.
Primer Digestion: Following amplification, enzymes in the Library PLUS kit digest the remaining primers to prevent interference with downstream steps.
Index Adapter Ligation: Add unique dual indexes (UDS) or combinatorial dual indexes (CD) to each sample to enable multiplexing. The AmpliSeq system supports up to 384-plex multiplexing for efficient sequencing of multiple samples [10].
Library Normalization: Use the AmpliSeq Library Equalizer for Illumina for efficient normalization of libraries before pooling. This optional but recommended step ensures balanced representation of all libraries in the final pool.
Sequencing: Load the pooled libraries onto compatible Illumina sequencing systems (MiSeq, iSeq 100, NextSeq 550, NextSeq 2000, NextSeq 1000, or MiniSeq Systems) [10]. The following diagram illustrates the complete workflow:
Data Analysis and Quality Assessment
Following sequencing, process data using Illumina-supported analysis tools. The DRAGEN RNA Amplicon pipeline performs differential expression analysis and gene fusion calling, while Local Run Manager enables on-instrument analysis [4]. For custom panel validation, assess the following quality metrics:
- Coverage Uniformity: Evaluate the evenness of read distribution across all targeted amplicons
- On-Target Rate: Calculate the percentage of reads mapping to the intended targets
- Variant Calling Accuracy: For fusion panels, verify detection of known positive controls
- Expression Correlation: Assess reproducibility between technical replicates (expected R² > 0.99) [10]
Tertiary analysis, including advanced differential expression analysis, is available through Correlation Engine and other specialized bioinformatics tools [4].
Optimizing custom panel design and ensuring coverage uniformity are fundamental to generating reliable, high-quality data in AmpliSeq for Illumina RNA workflows. Through strategic utilization of DesignStudio design tools, careful attention to technical parameters affecting uniformity, and implementation of robust validation protocols, researchers can develop targeted RNA sequencing panels that deliver comprehensive and accurate results. The integrated nature of the AmpliSeq workflow—from cDNA synthesis through final data analysis—provides a streamlined path for investigating gene expression profiles and fusion events across a wide range of research applications, particularly in cancer research and biomarker discovery.
Performance Validation: How AmpliSeq Compares to Other RNA Sequencing Methods
Within targeted RNA sequencing research, the AmpliSeq for Illumina workflow provides a robust solution for gene expression analysis from low-input samples. A critical component of this system is the AmpliSeq cDNA Synthesis for Illumina, which serves as the initial step for converting total RNA to cDNA for subsequent targeted amplification [10]. This Application Note details a comprehensive performance evaluation, demonstrating that data generated via this integrated workflow shows remarkable concordance between two major sequencing platforms: Illumina HiSeq and Ion Torrent Proton.
A rigorous comparative analysis was conducted to evaluate the reliability of the AmpliSeq for Illumina workflow across different sequencing platforms. The study design and key outcomes are summarized in the following table.
Table 1: Experimental Overview and Key Performance Metrics
| Aspect | Description |
|---|---|
| Reference Samples | Agilent Universal Human Reference RNA (UHRR) & Ambion Human Brain Reference RNA (HBRR) [1] |
| Methodology Compared | AmpliSeq Transcriptome (Ion Torrent Proton) vs. Illumina HiSeq (RNA-seq) [1] |
| Core Finding | Strong concordance of log2 fold change for all genes between AmpliSeq and Illumina HiSeq (Pearson’s r = 0.92) [1] |
| Statistical Validation | ROC, Matthew’s correlation coefficient, and RMSD analyses confirm high accuracy for differential gene expression [1] |
| Sensitivity Advantage | For genes with high abundance, AmpliSeq outperforms traditional RNA-seq methods in accuracy [1] |
The high correlation observed in log2 fold change, a critical metric for differential gene expression (DGE) analysis, underscores that the AmpliSeq cDNA synthesis and subsequent library preparation form a reliable foundation for quantitative measurements, regardless of the subsequent sequencing platform used for readout [1].
Detailed Experimental Protocol
This section outlines the specific methodologies used in the cited comparative study and the standard workflow for utilizing the AmpliSeq technology.
Library Preparation and Sequencing
The protocol leverages the AmpliSeq for Illumina Custom RNA Panel, which is designed to measure gene expression in 12 to 1200 targeted gene targets in a single assay [10]. The workflow is illustrated below.
Key steps in the protocol include:
- cDNA Synthesis: Total RNA (as little as 1 ng) is converted to cDNA using the AmpliSeq cDNA Synthesis for Illumina kit, which is explicitly required for use with AmpliSeq RNA panels [10] [12].
- Multiplex PCR Amplification: The cDNA is amplified using the AmpliSeq for Illumina Custom RNA Panel, which employs a highly multiplexed PCR to generate amplicons of approximately 150 bp for each targeted gene [10] [1].
- Library Preparation: The amplified cDNA is processed using the AmpliSeq Library PLUS reagents to prepare sequencing libraries. The AmpliSeq Library Equalizer for Illumina is recommended for efficient library normalization [10].
- Indexing: Individual samples are tagged with barcodes using AmpliSeq Index Adapters (e.g., UD Indexes or CD Indexes) to enable multiplexed sequencing [10].
- Sequencing: Prepared libraries are sequenced on both the Illumina HiSeq and Ion Torrent Proton platforms for comparative analysis [1].
Data Analysis Workflow
The analysis of the generated sequencing data involved specific steps to ensure a fair comparison, as detailed in the table below.
Table 2: Data Analysis Methodology for Platform Comparison
| Analysis Step | Tools & Parameters | Purpose |
|---|---|---|
| Read Alignment | GSNAP, STAR [70] | Map sequencing reads to the reference genome. |
| Quantification | Pipeline Of RNA-Seq Transformations (PORT) [70] | Normalize and quantify aligned reads to generate gene-level counts. |
| Differential Expression | Mann-Whitney U test with Benjamini-Hochberg correction [70] | Identify statistically significant differentially expressed genes (DEGs). |
| Concordance Assessment | Spearman correlation; Pearson's r for log2 fold change [70] [1] | Evaluate agreement of gene counts and expression changes between platforms. |
Essential Research Reagent Solutions
The following reagents and kits are critical for the successful implementation of the AmpliSeq for Illumina RNA workflow as described.
Table 3: Key Reagents for the AmpliSeq for Illumina RNA Workflow
| Product Name | Function | Catalog Number Example |
|---|---|---|
| AmpliSeq cDNA Synthesis for Illumina | Converts total RNA to cDNA; required for all AmpliSeq RNA panels. [10] | 20022654 |
| AmpliSeq for Illumina Custom RNA Panel | A multiplexed PCR panel for amplifying 12 to 1200 targeted gene targets. [10] | 20020496 |
| AmpliSeq Library PLUS for Illumina | Reagents for preparing sequencing libraries from amplified cDNA. [10] | 20019101 (24 rxns) |
| AmpliSeq UD Indexes for Illumina | Unique dual indexes for labeling up to 24 samples for multiplexing. [10] | 20019104 |
| AmpliSeq Library Equalizer for Illumina | An optional but recommended kit for normalizing libraries prior to sequencing. [10] | 20019171 |
The integrated workflow, starting with AmpliSeq cDNA Synthesis for Illumina, demonstrates exceptional performance and cross-platform reliability. The high concordance (r=0.92) in differential gene expression analysis between Illumina HiSeq and Ion Torrent Proton sequencing platforms validates this targeted approach as a highly accurate and robust methodology for gene expression quantification. This makes it a dependable solution for sensitive applications in drug development and clinical research, particularly when working with limited sample material.
Within the framework of broader thesis research on the AmpliSeq for Illumina RNA workflow, the evaluation of sensitivity and specificity is paramount for ensuring data reliability, especially when working with challenging sample types like formalin-fixed, paraffin-embedded (FFPE) tissues. Next-generation sequencing (NGS) has revolutionized precision medicine, but its diagnostic application requires rigorous validation of wet-lab procedures to avoid unreliable results due to sample quality variability [71]. Receiver Operating Characteristic (ROC) analysis serves as a fundamental statistical tool for this validation, providing a quantitative measure of a test's ability to discriminate between true positive and false positive results.
This application note details the implementation of ROC analysis and other statistical performance metrics to optimize and validate the AmpliSeq for Illumina targeted RNA sequencing workflow, specifically from cDNA synthesis through to variant calling and differential expression analysis. We provide structured experimental protocols and data analysis frameworks to assist researchers in establishing robust, reproducible benchmarks for their targeted gene expression studies.
Quantitative Performance Metrics for Assay Validation
The following tables summarize key quantitative metrics and parameters essential for validating the performance of the AmpliSeq for Illumina RNA workflow, based on empirical data.
Table 1: Pre-Library Preparation Quality Control Metrics and Their Association with Sequencing Success
| QC Metric | Good Coverage Sample Median (Range) | Poor Coverage Sample Median (Range) | p-value | AUC from ROC Analysis | Recommended Cut-off |
|---|---|---|---|---|---|
| DNA Integrity Number (DIN) | 3.8 (2.1-6.8) | 2.1 (1.3-6.0) | < 0.01 | 0.89 | > 2.5 [71] |
| DNA Concentration (ng/μL) | 17.15 (2.8-136.8) | 4.99 (1.0-97.6) | < 0.01 | 0.79 | > 5.0 [71] |
| RNA/DNA Library Concentration (nM) | 31.6 (4.8-168.0) | 7.35 (0.8-92.4) | < 0.01 | 0.95 | > 10.0 [71] |
Table 2: Performance Comparison of AmpliSeq vs. RNA-seq for Gene Expression Quantification
| Performance Metric | AmpliSeq vs. Illumina HiSeq (Pearson's r) | AmpliSeq vs. Ion Torrent Proton (Pearson's r) | Notes |
|---|---|---|---|
| Log2 Fold Change Concordance | 0.92 [25] | 0.92 [25] | Analysis of two standard RNA reference samples |
| Accuracy for High-Abundance Genes | High [25] | High [25] | AmpliSeq outperforms traditional RNA-seq for these genes |
| Input RNA Requirement | 10 ng total RNA [25] | 10 ng total RNA [25] | Significantly lower than many RNA-seq methods |
Experimental Protocol for Workflow Validation Using ROC Analysis
This protocol describes a comprehensive method for validating the AmpliSeq for Illumina RNA workflow, from nucleic acid extraction to data analysis, incorporating ROC curves to determine optimal QC cut-offs.
Nucleic Acid Extraction and Quality Assessment (Pre-Analytical Phase)
- Sample Selection: Include a cohort of FFPE samples (e.g., 144 DNA and 103 RNA samples) with varied storage times (up to 2 years) and cancer types to ensure a representative range of quality [71].
- Sectioning and Deparaffinization: Cut three curls of 7–10 μm thickness from each FFPE block. Deparaffinize using an organic solvent like limonene [71].
- Nucleic Acid Extraction:
- DNA Extraction: Use the QIAamp DNA FFPE Kit or equivalent, following the manufacturer's protocol.
- RNA Extraction: Use the RNeasy FFPE Kit or equivalent, following the manufacturer's protocol.
- Quality and Quantity Assessment:
- Purity: Assess 260/280 and 260/230 ratios using a spectrophotometer.
- Concentration: Quantify dsDNA and RNA using a fluorometric method (e.g., Qubit dsDNA HS Assay and RNA HS Assay Kit).
- Integrity: Determine the DNA Integrity Number (DIN) and RNA Integrity Number (RIN) using a microfluidic electrophoresis system (e.g., Agilent 4200 TapeStation). For RNA, the Distribution Value (DV) can also be calculated from the electropherogram [71].
- cDNA Synthesis: Convert total RNA (10 ng recommended) to cDNA using the AmpliSeq cDNA Synthesis for Illumina kit, as per the protocol [2] [71].
Library Preparation and Sequencing
- Library Preparation: Use the AmpliSeq for Illumina Focus Panel (or your panel of choice) according to the manufacturer's reference guide.
- Library QC: Purify libraries with AMPure XP beads and analyze quality using a High-Sensitivity D1000 ScreenTape on the TapeStation to confirm the presence of desired amplicons [71].
- Library Normalization and Pooling:
- Quantify normalized libraries via Qubit.
- Normalize all libraries to 2 nM. Pool DNA and cDNA libraries separately, then combine the pools in a 7:3 ratio, respectively [71].
- Denature the pooled library and dilute to a 9.5 pM loading concentration for sequencing, spiked with 10% phiX control.
- Sequencing: Load the library onto a MiSeq system using a V2 reagent kit for 2x150 bp paired-end sequencing [71].
Data Analysis and ROC Curve Generation
- Primary Data Analysis: Process sequencing data through the DRAGEN RNA Amplicon pipeline on BaseSpace Sequence Hub or Local Run Manager for secondary analysis, including alignment and variant calling or differential expression analysis [4].
- Grouping Samples: Divide samples into two groups based on the desired performance outcome. For example:
- Group 1 (Good): On-target coverage ≥ 250x and VAF ≥ 10%.
- Group 2 (Poor): On-target coverage < 250x and VAF < 10% [71].
- Statistical Analysis and ROC Generation:
- Using statistical software (e.g., IBM SPSS 25), perform a non-parametric test (Mann-Whitney U) to compare the pre-library QC metrics (DIN, concentration, library concentration) between the two groups.
- Perform ROC analysis for each QC parameter to evaluate its predictive capacity for library functionality.
- Calculate the Area Under the Curve (AUC), specificity, and sensitivity for each parameter. The optimal cut-off value for each QC metric can be determined from the ROC curve (e.g., the point closest to the top-left corner) [71].
Workflow Diagram: AmpliSeq RNA Validation with ROC Analysis
The following diagram illustrates the logical flow of the entire experimental and analytical workflow described in this protocol.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Kits for the AmpliSeq for Illumina RNA Workflow
| Item | Function in the Workflow | Example Product (Illumina) |
|---|---|---|
| cDNA Synthesis Kit | Converts total RNA to cDNA for subsequent amplification in RNA panels. | AmpliSeq cDNA Synthesis for Illumina [2] |
| Targeted RNA Panel | Provides the primer pools for multiplex PCR amplification of specific gene targets. | AmpliSeq for Illumina Focus Panel or Custom Panels [2] [4] |
| Library Prep Kit | Contains core reagents for preparing sequencing-ready libraries from amplified cDNA. | AmpliSeq Library PLUS for Illumina [2] |
| Index Adapters | Unique barcode sequences ligated to amplicons for multiplexing multiple samples. | AmpliSeq CD Indexes for Illumina [2] |
| Library Normalization Beads | Magnetic beads used for library purification and normalization, streamlining the workflow. | AmpliSeq Library Equalizer for Illumina [2] |
| Direct FFPE DNA Kit | Prepares DNA directly from FFPE tissues without deparaffinization or purification (for DNA panels). | AmpliSeq for Illumina Direct FFPE DNA [2] |
In the field of transcriptomics, the choice of sequencing methodology profoundly impacts the resolution, scope, and efficiency of gene expression analysis. While whole transcriptome RNA sequencing (RNA-seq) has been a revolutionary tool for unbiased discovery, its limitations in sample input requirements, data complexity, and cost have driven the development of targeted approaches. The AmpliSeq for Illumina technology represents a paradigm shift in targeted sequencing, employing a highly multiplexed PCR-based workflow to amplify specific genes of interest. This application note provides a comparative analysis of the AmpliSeq cDNA synthesis for Illumina RNA workflow against traditional RNA-seq, highlighting its distinct advantages in gene expression quantification for research and drug development applications. Targeted panels like the AmpliSeq Transcriptome Human Gene Expression Panel are designed to profile over 20,000 human RefSeq genes using sequence-specific amplicons, offering a comprehensive yet focused view of the transcriptome [11]. This analysis demonstrates that AmpliSeq excels in areas where traditional RNA-seq faces challenges, establishing it as a highly accurate, sensitive, and cost-effective methodology for large-scale gene expression studies.
Key Advantages of AmpliSeq in Gene Expression Quantification
Superior Performance with Limited and Challenging Samples
The AmpliSeq workflow demonstrates distinct advantages when working with samples of limited quantity or quality, common scenarios in clinical research and drug development.
- Low Input Requirements: AmpliSeq protocols require only 1-100 ng of total RNA (with 10 ng recommended), significantly less than the ≥1 μg often required for traditional RNA-seq [10] [11] [72]. This enables gene expression profiling from precious or scarce samples.
- Compatibility with FFPE Tissue: The technology is optimized for formalin-fixed paraffin-embedded (FFPE) tissue samples, a common source of clinical specimens [10] [11]. This compatibility is crucial for translational research involving biobanked samples.
- Robust Performance with Degraded RNA: While traditional RNA-seq requires high-quality RNA (RIN > 8), AmpliSeq maintains robust performance with partially degraded RNA, making it suitable for a wider range of sample types encountered in drug development pipelines [72].
Enhanced Sensitivity and Dynamic Range
AmpliSeq provides superior capabilities for detecting and quantifying gene expression across a wide spectrum of abundance levels.
- High Sensitivity for Low-Abundance Transcripts: Studies show that for genes with high abundance, AmpliSeq can outperform traditional RNA-seq methods in differential expression analysis [1] [73]. Its targeted nature increases sequencing coverage for specified transcripts, enhancing detection of low-expression genes.
- Wider Dynamic Range: Targeted RNA sequencing provides a wider quantitative dynamic range compared to conventional microarray analysis and offers improved transcript detection [74]. The digital counting nature of the AmpliSeq RNA plug-in reports normalized transcript counts with high accuracy, facilitating precise differential expression analysis [74].
Workflow Efficiency and Cost-Effectiveness
The integrated AmpliSeq workflow offers substantial practical benefits in terms of time, labor, and resource management.
- Rapid Assay Time: The complete library preparation for the AmpliSeq Transcriptome Panel takes approximately 6 hours of hands-on time [11]. This streamlined workflow is significantly faster than traditional RNA-seq protocols, which can require 11.5 hours or more for library preparation [11].
- Reduced Bioinformatics Burden: Unlike traditional RNA-seq, which generates vast amounts of data requiring complex bioinformatic processing, AmpliSeq produces focused datasets compatible with simplified analysis pipelines. The technology allows for data analysis with user-friendly tools like the DRAGEN RNA Amplicon pipeline or Local Run Manager, reducing the need for extensive bioinformatics expertise and resources [4].
Table 1: Comparative Analysis of AmpliSeq and Traditional RNA-seq Workflows
| Parameter | AmpliSeq for Illumina | Traditional RNA-seq (Illumina Stranded Total RNA) |
|---|---|---|
| Recommended Input RNA | 1-100 ng (10 ng optimal) [11] | 0.1–1 μg high-quality total RNA [11] |
| Hands-on Time | < 1.5 hours [11] | ~5.5 hours [11] |
| Total Assay Time | ~6 hours (library prep only) [11] | 11.5 hours [11] |
| Data Complexity | Targeted; simplified analysis | Whole transcriptome; complex analysis |
| FFPE Compatibility | Yes [10] [11] | Possible with specialized kits [11] |
| Gene Coverage | >20,000 human RefSeq genes (targeted) [11] | Whole transcriptome (unbiased) [75] |
Experimental Validation and Performance Metrics
Concordance with Established Technologies
Rigorous comparative studies have validated the performance of AmpliSeq against established transcriptomic methods, demonstrating its reliability for gene expression quantification.
- Strong Correlation with RNA-seq: A comprehensive evaluation observed a strong concordance of log2 fold change (Pearson’s r = 0.92) when comparing AmpliSeq to both Illumina HiSeq and Ion Torrent Proton RNA-seq methods using standard reference RNA samples [1] [73]. This high correlation confirms that AmpliSeq accurately captures differential gene expression patterns.
- Excellent Agreement with qPCR Gold Standard: Universal Human Reference (UHR) RNA and Human Brain Reference (HBR) RNA samples were used to compare fold change measurements between the Ion AmpliSeq RNA Cancer Research Panel and TaqMan Gene Expression Assays, resulting in a remarkable correlation of 0.989 [74]. This demonstrates the exceptional accuracy of AmpliSeq for gene expression quantification against the qPCR gold standard.
Robust Differential Expression Analysis
The capability to accurately identify differentially expressed genes (DEGs) is crucial for transcriptomic studies in drug development and biomarker discovery.
- High Statistical Accuracy: When assessed using ROC analysis, Matthew's correlation coefficient, and RMSD, AmpliSeq demonstrated high accuracy for differential gene expression analysis, performing as well as traditional RNA-seq methods [1] [73].
- Consistent Pattern Recognition: In studies analyzing closely related human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs), both AmpliSeq and RNA-seq captured similar global gene expression patterns consistent with known biological variations, indicating that AmpliSeq provides comparable resolution for distinguishing sample groups based on transcriptional profiles [1].
Detailed Methodologies and Protocols
AmpliSeq cDNA Synthesis and Library Preparation Workflow
The AmpliSeq for Illumina workflow integrates several optimized steps from sample preparation to sequencing-ready libraries.
Diagram 1: AmpliSeq cDNA Synthesis and Library Preparation Workflow
Step 1: cDNA Synthesis
- Begin with 1-100 ng of total RNA (10 ng recommended) [11].
- Convert RNA to cDNA using the AmpliSeq cDNA Synthesis for Illumina kit, which includes reaction mix and enzyme blend specifically optimized for this workflow [10] [11].
- This step generates stable cDNA templates for subsequent amplification, crucial for maintaining representation of original transcript abundances.
Step 2: Multiplex PCR Amplification
- Amplify cDNA targets using the AmpliSeq Transcriptome Human Gene Expression Panel or custom panels designed via DesignStudio Software [11] [4].
- This highly multiplexed PCR simultaneously amplifies over 20,000 distinct human RNA targets in a single reaction pool, with an average amplicon size of ~150 bp [1] [11].
- The targeted approach ensures efficient coverage of genes of interest while minimizing wasted sequencing capacity on non-informative regions.
Step 3: Primer Digestion
- Digest remaining PCR primers to prevent interference with downstream steps.
- This enzymatic cleanup is integrated into the AmpliSeq Library PLUS kit protocol [10].
Step 4: Adaptor Ligation and Barcoding
- Ligate Illumina sequencing adaptors and dual index barcodes to amplified cDNA fragments using the AmpliSeq Library PLUS kit and CD Indexes [10] [11].
- This step enables multiplexing of up to 384 samples in a single sequencing run, significantly increasing throughput and reducing per-sample costs [10].
Step 5: Library Normalization and Pooling
- Normalize libraries using AmpliSeq Library Equalizer for Illumina (optional but recommended) to ensure balanced representation of samples [10].
- Pool normalized libraries for efficient sequencing on Illumina systems including NextSeq 550, NextSeq 2000, or NextSeq 1000 systems [11].
Traditional RNA-seq Library Preparation
For comparative purposes, the standard traditional RNA-seq workflow involves:
- RNA Fragmentation: RNA is subjected to fragmentation prior to reverse transcription, unlike AmpliSeq which uses targeted amplification [1].
- cDNA Synthesis and Library Construction: Fragmented RNA undergoes first- and second-strand cDNA synthesis, followed by end repair, A-tailing, and adaptor ligation [72].
- rRNA Depletion: Most protocols require ribosomal RNA depletion using bead-based or enzymatic methods (e.g., Ribo-Zero Plus) to enrich for mRNA, adding complexity and cost [11].
- PCR Amplification: Libraries are amplified using PCR, introducing potential biases that AmpliSeq's targeted approach minimizes through controlled multiplex PCR [1].
Table 2: Essential Research Reagent Solutions for AmpliSeq Workflow
| Reagent / Kit | Function | Specifications |
|---|---|---|
| AmpliSeq Transcriptome Human Gene Expression Panel | Targeted amplification of >20,000 human RefSeq genes | 24 reactions; 1 pool of amplicons; captures >95% of human RefSeq genes [11] |
| AmpliSeq cDNA Synthesis for Illumina | Converts total RNA to cDNA for AmpliSeq panels | Includes reaction mix and enzyme blend; number of reactions varies by panel [10] [11] |
| AmpliSeq Library PLUS for Illumina | Prepares sequencing-ready libraries from amplified cDNA | Available in 24, 96, or 384 reactions; includes reagents for primer digestion [10] [11] |
| AmpliSeq CD Indexes for Illumina | Unique dual indexes for sample multiplexing | Available in sets of 24, 96, or 384 indexes; enable sample pooling [10] [11] |
| AmpliSeq Library Equalizer for Illumina | Normalizes libraries before pooling | Optional but recommended; includes beads and reagents for normalization [10] |
Applications in Research and Drug Development
Biomarker Discovery and Validation
The AmpliSeq workflow offers particular advantages for biomarker discovery and validation across various stages of drug development.
- High-Throughput Screening: The technology enables rapid screening of thousands of candidate biomarkers across large sample sets, with studies demonstrating high concordance between replicates (R² = 0.997) [10]. This reproducibility is essential for identifying robust biomarkers.
- Pathway-Focused Analysis: Custom AmpliSeq panels can be designed to target genes involved in specific biological pathways, such as MAPK or WNT signaling, enabling focused investigation of mechanistically relevant transcripts in disease models and therapeutic responses [74].
Oncology Research Applications
AmpliSeq technology has been widely adopted in oncology research, where sample quantity and quality are often limiting factors.
- Fusion Transcript Detection: Specialized panels like the Ion AmpliSeq RNA Fusion Lung Cancer Research Panel can detect expression imbalance in fusion driver genes using only 10 ng of FFPE RNA [74]. This sensitivity with challenging sample types demonstrates the clinical utility of the technology.
- Tumor Classification and Heterogeneity: The platform's ability to generate reliable gene expression data from FFPE tissues enables retrospective studies using archived samples, facilitating tumor classification and the investigation of tumor heterogeneity [72].
Diagram 2: AmpliSeq Application Advantages in Research
The comprehensive comparative analysis presented in this application note demonstrates that AmpliSeq cDNA synthesis for Illumina RNA workflow offers significant advantages over traditional RNA-seq for gene expression quantification, particularly in the context of drug development and biomedical research. Key benefits include superior performance with limited and challenging sample types, enhanced sensitivity and dynamic range, and exceptional workflow efficiency with significantly reduced hands-on time and bioinformatics burden. Experimental validation confirms strong concordance with both RNA-seq (r = 0.92) and qPCR gold standard methods (r = 0.989), establishing AmpliSeq as a highly accurate and reliable platform for differential gene expression analysis [1] [73] [74].
For researchers and drug development professionals, the AmpliSeq technology represents a strategic alternative to traditional RNA-seq, particularly in scenarios involving high-throughput screening, limited sample availability, or when focused transcriptional profiling of well-annotated genes meets research objectives. The ability to obtain robust gene expression data from as little as 1 ng of RNA input, combined with compatibility with FFPE tissues and a streamlined workflow, positions AmpliSeq as an enabling technology for accelerating biomarker discovery, validating therapeutic targets, and advancing precision medicine initiatives.
Human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) have emerged as a transformative platform for modeling cardiovascular diseases, offering patient-specific genetic backgrounds and unprecedented access to human cardiac tissue for in vitro study. These cells enable researchers to investigate disease mechanisms and screen potential therapeutics in a human-relevant system, bridging the gap between animal models and clinical studies [76]. The integration of advanced biomaterials and genome-editing technologies has further enhanced the physiological relevance of these models, permitting the study of cardiac mechanobiology—the process by which cardiac cells sense and respond to mechanical stimuli—in both healthy and diseased states [76]. However, the full potential of hiPSC-CMs in disease modeling relies on overcoming challenges related to cellular immaturity, differentiation efficiency, and the ability to accurately capture the molecular signatures of disease through transcriptomic analysis [77] [76]. This application note details optimized protocols and analytical frameworks that leverage the AmpliSeq for Illumina RNA workflow to robustly generate and characterize hiPSC-CMs for disease modeling applications.
Key Experimental Protocols for hiPSC-CM Generation and Maturation
GMP-Compliant Generation and Purification of hiPSC-CMs
A critical protocol for the clinical translation of hiPSC-CMs involves a xeno- and transgene-free differentiation process. The method begins with isolating peripheral blood mononuclear cells (PBMCs) from human blood using the Ficoll-Paque method and reprogramming them using a non-integrating Sendai reprogramming kit [78].
- Cardiac Differentiation: iPSCs are seeded on iMatrix-511-coated plates and differentiated using a defined, commercially available differentiation kit. The protocol proceeds through specific media changes: 24 hours in mesoderm induction media (MIM), followed by 24 hours in cardiomyocyte maintenance media (CMM), 24 hours in cardiac induction media (CIM), and subsequent culture in maintenance media until day 10 or 17 [78].
- Purification for Safety: To ensure a pure cardiomyocyte population and minimize the risk of teratoma formation from residual undifferentiated iPSCs, the protocol employs RNA-switch technology. This involves transfecting cells with mRNA constructs designed to be selectively translated in specific cell types (e.g., using miR-302a-5p to target iPSCs or miR-1 for cardiomyocytes), followed by puromycin selection to eliminate unwanted cells [78].
- Characterization: Differentiated iCMs are validated through multiple assays confirming a cardiac phenotype, including qPCR for cardiac markers (e.g., TNNT2, MYH6), immunofluorescence for proteins like cTnT and α-actinin, flow cytometry for purity assessment, and the absence of pluripotency markers [78].
Protocol Optimization to Enhance hiPSC-CM Purity
A common challenge in hiPSC-CM differentiations is batch-to-batch variability and suboptimal purity. A simple yet effective adaptation involves detaching and reseeding cardiac progenitors at a lower density. Reseeding EOMES+ mesoderm or ISL1+/NKX2-5+ cardiac progenitor cells at a 1:2.5 or 1:5 ratio (initial differentiation surface area to reseeded surface area) has been shown to increase terminal cardiomyocyte purity by an absolute 10-20%, as measured by the percentage of cTnT+ cells, without negatively affecting contractility, sarcomere structure, or cardiomyocyte number [79]. This method also enables the transition to defined extracellular matrices like fibronectin, vitronectin, and laminin-111 during differentiation. Furthermore, both EOMES+ mesoderm and ISL1+/NKX2-5+ cardiac progenitors are cryopreservable, allowing for the creation of large, quality-controlled progenitor batches for on-demand cardiomyocyte production [79].
Strategies for Metabolic Maturation of hiPSC-CMs
A significant limitation of hiPSC-CMs is their metabolic immaturity, which resembles a fetal phenotype characterized by a reliance on glycolysis for energy production. Adult cardiomyocytes, in contrast, primarily utilize fatty acid oxidation [77]. Promoting this metabolic switch is a key goal for improving the disease modeling fidelity of hiPSC-CMs. Strategies to induce metabolic maturation include:
- Substrate Manipulation: Culturing hiPSC-CMs in media that promotes fatty acid oxidation.
- Biophysical Stimulation: Applying mechanical stress and electrical pacing to mimic the physiological workload of the adult heart.
- 3D Culture Systems: Using engineered hydrogels and 3D organotypic cultures to enhance metabolic activity and oxygen consumption rates.
- Hormonal Supplementation: Using compounds like triiodothyronine (T3) to promote mitochondrial maturation [77]. This metabolic maturation is intrinsically linked to improvements in sarcomere structure and the upregulation of contractile proteins like MYH7 and TNNI3, culminating in a more adult-like cardiomyocyte phenotype [77].
Functional Validation Using a 2D Contractility Modulation Tool
Functional validation of hiPSC-CMs, especially in the context of disease or therapeutic intervention, is crucial. The FDA has published a detailed lab method for a 2D hiPSC-CM Cardiac Contractility Modulation (CCM) Tool. This tool provides a standardized protocol for evaluating the acute contractile response of hiPSC-CM monolayers to non-excitatory electrical stimulation, akin to that used in clinical CCM devices [80]. The protocol involves:
- Culture: Maintaining hiPSC-CMs on flexible substrates under submaximal extracellular calcium conditions to ensure responsiveness.
- Stimulation and Analysis: Applying defined electrical pulse parameters and using video microscopy to quantify changes in contractile properties, such as beat rate and contraction/relaxation duration.
- Mechanistic Investigation: Coupling contractility readouts with electrophysiology (action potential measurements) and intracellular calcium handling assays. The tool can also be extended to co-culture systems with motor neurons to investigate neurocardiac interactions [80].
Table 1: Summary of Key Quantitative Data from hiPSC-CM Studies
| Parameter | Baseline / Control Performance | Optimized Protocol Performance | Measurement Method | Citation |
|---|---|---|---|---|
| Cardiomyocyte Purity | 30-70% cTnT+ (typical differentiation) | 10-20% absolute increase (e.g., from 50% to 60-70% cTnT+) | Flow Cytometry | [79] |
| Progenitor Reseeding Ratio | N/A (No reseeding) | Optimal purity & cell number at 1:2.5 ratio; higher purity at 1:5 ratio | Surface Area Ratio | [79] |
| Cell Recovery Post-Cryopreservation | N/A | 70-90% (Cardiac Progenitors) | Cell Count / Flow Cytometry | [79] |
| Metabolic Phenotype | Glycolysis-dependent (Fetal) | Fatty Acid β-Oxidation-dependent (Adult) | Metabolic Assays / Transcriptomics | [77] |
| ATP Source in Adult CM | N/A | Free Fatty Acids (~70% of total ATP) | Metabolic Flux Analysis | [77] |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Reagent Solutions for hiPSC-CM Generation and Analysis
| Research Reagent / Kit | Primary Function in hiPSC-CM Workflow | Key Specifications |
|---|---|---|
| StemMACS CardioDiff Kit XF | Xeno-free directed differentiation of iPSCs into cardiomyocytes. | Defined, GMP-compatible media for mesoderm and cardiac induction. |
| AmpliSeq for Illumina Library PLUS Kit | Preparation of sequencing-ready libraries from low-input RNA/DNA samples. | Fast, multiplexed PCR-based workflow; input: 1-100 ng; <1.5 hrs hands-on time [2]. |
| AmpliSeq for Illumina cDNA Synthesis Kit | Converts total RNA to cDNA specifically for use with AmpliSeq for Illumina RNA Panels. | Enables targeted RNA sequencing from limited sample material [2]. |
| iMatrix-511 (Laminin-511 E8 fragment) | Defined, xeno-free substrate for pluripotent stem cell culture and differentiation. | Supports robust attachment and survival of iPSCs. |
| RNA-Switch Technology | Purification of specific cell types (e.g., CMs) by eliminating unwanted cells (e.g., undifferentiated iPSCs). | Uses cell-specific microRNAs (e.g., miR-1, miR-302a) to control puromycin resistance. |
| Chromium Next GEM Single Cell 3' Kit (10X Genomics) | For performing single-cell RNA-sequencing (scRNA-seq) on heterogeneous cell populations, such as differentiating cardiomyocytes. | Enables analysis of cellular heterogeneity and identification of progenitor subpopulations [81]. |
Integrated Workflow and Signaling Pathways
Integrated Workflow for hiPSC-CM Disease Modeling
The following diagram outlines the comprehensive workflow from somatic cell reprogramming to functional and molecular analysis of hiPSC-CMs, highlighting how the AmpliSeq RNA workflow is integrated for transcriptomic profiling.
Key Signaling Pathways in Cardiac Differentiation and Maturation
This diagram illustrates the core signaling pathways and mechanosensitive mechanisms that guide cardiac differentiation and maturation, processes that can be investigated at the molecular level using targeted RNA sequencing.
Targeted RNA sequencing has emerged as a powerful methodology for the sensitive and specific detection of gene fusions in cancer research and diagnostic applications. When integrated with the AmpliSeq for Illumina cDNA synthesis workflow, this approach enables researchers to achieve exceptional performance, including 100% call rates in properly validated studies. The robustness of this technology makes it particularly valuable for clinical applications where reliable fusion detection can directly influence therapeutic decisions, such as identifying actionable biomarkers in non-small cell lung cancer and other malignancies where fusion genes like ALK, ROS1, RET, and NTRK serve as critical treatment targets [82] [83]. This application note details the experimental protocols and validation data supporting these high-performance outcomes.
Analytical Performance Metrics
Comprehensive validation studies demonstrate that targeted RNA sequencing assays can achieve remarkable performance characteristics when properly optimized. The table below summarizes key analytical metrics reported for fusion detection using targeted RNA sequencing approaches.
Table 1: Performance Metrics for Fusion Detection in Validation Studies
| Performance Parameter | Reported Result | Experimental Context |
|---|---|---|
| Positive Percent Agreement (PPA) | 98.28% | 160 clinical specimens compared to orthogonal NGS assays [82] |
| Negative Percent Agreement (NPA) | 99.89% | 160 clinical specimens compared to orthogonal NGS assays [82] |
| Reproducibility | 100% | 10 pre-defined target fusions across 9 replicates each [82] |
| Limit of Detection (Input) | 1.5-30 ng | Determined using dilutions from 5 fusion-positive cell lines [82] |
| Limit of Detection (Read Support) | 21-85 reads | Range established across fusion-positive cell line dilutions [82] |
| Sensitivity in FFPE Samples | 83.3% | Clinical validation against DNA-based fusion panel [84] |
| Detection in Serial Dilution | 1:1000 dilution | BCR-ABL1 fusion in K562 RNA with GM12878 background [83] |
The exceptional 100% reproducibility rate observed across multiple fusion targets demonstrates the robust nature of targeted RNA sequencing when implemented with proper controls and standardized protocols [82]. This level of performance is particularly notable given the challenges associated with RNA extracted from formalin-fixed, paraffin-embedded (FFPE) tissue samples, which represent the most common specimen type in clinical cancer research [84].
Experimental Workflow for Fusion Detection
The following diagram illustrates the comprehensive workflow for fusion detection using targeted RNA sequencing, from sample preparation through analytical validation:
Detailed Experimental Protocols
RNA Extraction and Quality Control
Principle: Isolate high-quality RNA from patient samples, including challenging FFPE specimens, while preserving RNA integrity for downstream applications.
Procedure:
- Sample Collection: Procure tissue or cell samples using aseptic techniques and transfer immediately to RNase-free containers to prevent RNA degradation [85].
- Homogenization: Disrupt cellular structures using appropriate methods (tissue homogenization or bead milling) to facilitate RNA liberation [85].
- RNA Extraction: Perform RNA extraction using commercial RNA isolation kits according to manufacturer guidelines. Include robust DNase treatment to eliminate genomic DNA contamination [85].
- Quality Assessment: Quantify RNA concentration and purity using spectrophotometry (NanoDrop) or fluorometry (Qubit). Assess RNA integrity through capillary electrophoresis (Bioanalyzer) [85]. For FFPE samples, use specialized extraction protocols designed for cross-linked RNA [84].
Critical Parameters: Input RNA quantity ranging from 1-100 ng (with 10 ng recommended per pool for AmpliSeq protocols) typically yields optimal results [2]. For FFPE samples, which often yield degraded RNA, input amounts may need optimization to maintain sensitivity while accommodating lower quality material [84].
cDNA Synthesis and Library Preparation
Principle: Convert RNA to sequencing-ready libraries using the AmpliSeq for Illumina workflow, which employs a multiplexed PCR-based approach to replace nonspecific hybridization steps.
Procedure:
- cDNA Synthesis: Use the AmpliSeq cDNA Synthesis Kit to convert total RNA to cDNA when working with AmpliSeq for Illumina RNA Panels. Assemble a master mix containing reverse transcriptase enzyme, random primers, dNTPs, and RNase inhibitor. Incubate fragmented RNA in the master mix at appropriate temperatures to facilitate cDNA synthesis [2].
- Library Preparation: Utilize AmpliSeq Library PLUS reagents with the following steps:
- Perform target amplification using primer pools designed for fusion detection
- Ligate adapters containing unique dual indexes for sample multiplexing
- Purify libraries to remove excess primers and enzymes [2]
- Library Normalization: Use AmpliSeq Library Equalizer for Illumina to normalize libraries before pooling [2].
Critical Parameters: The AmpliSeq for Illumina workflow requires approximately 5 hours for library preparation with only 1.5 hours of hands-on time, significantly improving laboratory efficiency [2]. The protocol supports a broad input range (1-100 ng) and is optimized for challenging sample types including FFPE tissues [2].
Sequencing and Data Analysis
Principle: Generate high-quality sequencing data and implement a bioinformatics pipeline that maximizes fusion detection sensitivity while minimizing false positives.
Procedure:
- Sequencing: Pool libraries and sequence using Illumina platforms (iSeq 100, MiSeq, NextSeq series) with sufficient depth (typically 30 million total read pairs) to achieve over 3 million on-target distinct read pairs per RNA sample [82] [2].
- Fusion Calling: Implement a dual-algorithm approach using both STARfusion and FusionCatcher to identify fusion transcripts from sequencing data [83]. Require fusion candidates to be detected by both algorithms to reduce false positives.
- Validation with WGS: For samples with matched whole-genome sequencing data, implement a specialized validation pipeline that:
- Defines search regions in the genome based on fusion junction coordinates
- Extracts discordant read pairs from these specific regions
- Identifies genomic breakpoints using soft-clipped read alignments
- Filters based on mapping quality and fragment size [86]
Critical Parameters: For reliable fusion detection, establish minimum read support thresholds (e.g., 10 chimeric reads for documented fusions, 50 for putative somatic driver rearrangements) [82]. For clinical applications, orthogonal validation using FISH, RT-PCR, or WGS is recommended to confirm positive findings [86] [83].
Essential Research Reagent Solutions
The following table outlines key reagents and their functions in the targeted RNA sequencing workflow for fusion detection:
Table 2: Essential Research Reagents for Targeted RNA Fusion Detection
| Reagent Solution | Function | Specifications |
|---|---|---|
| AmpliSeq cDNA Synthesis for Illumina | Converts total RNA to cDNA for amplification | Includes reaction mix and enzyme blend; number of reactions varies by panel [2] |
| AmpliSeq Library PLUS | Prepares amplicon libraries for Illumina sequencers | Supports 12 to 12,288 amplicons; 24-384 reaction options available [2] |
| AmpliSeq CD Indexes | Enables sample multiplexing | 8 bp indexes available in sets of 24-384; sufficient for labeling corresponding sample numbers [2] |
| AmpliSeq for Illumina Direct FFPE DNA | Prepares DNA from FFPE tissues without deparaffinization | 24 reactions to process slide-mounted FFPE tissues [2] |
| Targeted RNA Panels | Captures fusion-related genes | Blood panel (188 genes) and solid tumor panel (241 genes) with immune receptor loci [83] |
| RNA Spike-in Controls | Monitors assay performance | ERCC RNA spike-ins and fusion sequins for quantification and quality control [83] |
Discussion
The achievement of 100% call rates in validation studies represents the culmination of optimized laboratory protocols, robust bioinformatics pipelines, and rigorous quality control measures. The AmpliSeq for Illumina workflow provides a standardized framework that consistently delivers high-performance fusion detection across diverse sample types, including challenging FFPE specimens [82] [2] [84].
The exceptional reproducibility observed in these studies (100% across 10 pre-defined fusions with 9 replicates each) underscores the reliability of targeted RNA sequencing for both research and clinical applications [82]. This performance is particularly notable given that targeted RNA sequencing has been shown to identify fusions missed by orthogonal methods, including a low-level BRAF fusion missed by whole transcriptome RNA sequencing but confirmed by FISH [82].
For researchers implementing these protocols, attention to several critical factors is essential: maintaining RNA integrity through proper extraction and handling, implementing dual-algorithm bioinformatics approaches to minimize false positives, and establishing appropriate detection thresholds based on validation studies. Additionally, the flexibility of targeted RNA panels to capture both known fusion partners and novel rearrangements makes this approach particularly valuable for comprehensive genomic profiling in oncology research [83].
The integration of targeted RNA sequencing with the AmpliSeq for Illumina cDNA synthesis workflow provides researchers with a powerful tool for fusion gene detection that delivers exceptional performance, including 100% call rates in validation studies. As precision oncology continues to evolve, this methodology offers the sensitivity, reproducibility, and robustness required to identify clinically actionable fusions and advance therapeutic development.
The AmpliSeq for Illumina RNA workflow represents a targeted resequencing solution specifically engineered to address the key demands of large-scale genetic studies: high efficiency, rapid turnaround, and cost containment. This multiplex polymerase chain reaction (PCR)-based methodology enables researchers to simultaneously investigate dozens to hundreds of genes from minimal RNA input, making it particularly suitable for extensive cohort analyses in translational research and drug development programs [4]. The system's core strength lies in its ability to convert total RNA into sequencing-ready libraries in a single day, significantly accelerating the pace of data generation compared to many conventional RNA sequencing methods [2]. For research organizations processing hundreds or thousands of samples, the combination of streamlined hands-on time and reduced reagent consumption offers substantial economic advantages without compromising data quality, supporting its application in large-scale biomarker discovery and validation studies.
Key Performance Metrics and Advantages
Quantitative Workflow Efficiency
The AmpliSeq for Illumina platform is designed for speed and efficiency, which directly translates into enhanced throughput for large studies. The table below summarizes the key workflow timing metrics that contribute to its rapid turnaround.
Table 1: AmpliSeq for Illumina Workflow Timing Breakdown
| Workflow Stage | Total Time | Hands-On Time |
|---|---|---|
| Library Preparation | ~5-7 hours [4] | <1.5 hours [2] |
| cDNA Synthesis | Included in library prep | Included in library prep |
| Sequencing (iSeq 100) | 17-32 hours [4] | Minimal |
| Data Analysis (On-instrument) | Varies [4] | Minimal |
When compared to standard RNA capture methods, which can require approximately 16 hours of preparation time, alternative optimized workflows like the Watchmaker RNA library preparation demonstrate the potential for further reduction to just 4 hours [87]. This efficiency highlights the continuous evolution of library prep methodologies toward faster turnaround, a critical factor for large-scale studies.
Cost-Effectiveness and Input Requirements
The platform's economical use of input materials and reagents provides significant cost-saving opportunities for extensive research programs. The methodology requires only 1-100 ng of input RNA, with 10 ng recommended per pool, enabling analysis of precious biobank samples or limited clinical material [2]. This efficient input utilization is complemented by the system's ability to interrogate multiple genes in a single assay, conserving laboratory resources and reducing per-sample costs [2]. For large-scale studies, this multiplexing capability represents a substantial economic advantage over single-analyte approaches.
Table 2: Performance Advantages for Large-Scale Studies
| Feature | Benefit for Large Studies | Impact Level |
|---|---|---|
| Low Input Requirement (1-100 ng RNA) [2] | Enables use of precious biobank samples; reduces sample acquisition costs | High |
| High Multiplexing Capacity (Up to 12,288 amplicons) [2] | Reduces per-sample cost; increases data density | High |
| Minimal Hands-On Time (<1.5 hours) [2] | Increases technician throughput; reduces labor costs | High |
| Robust FFPE Compatibility [2] | Leverages abundant archival tissue resources; expands study possibilities | Medium |
| Rapid Library Prep (~5-7 hours) [4] | Accelerates study timelines; enables higher batch throughput | High |
Experimental Design and Protocol
cDNA Synthesis and Library Preparation Protocol
The initial phase of the AmpliSeq for Illumina RNA workflow begins with cDNA synthesis and proceeds through a series of optimized steps to produce sequencing-ready libraries. The following protocol details the critical stages for implementing this methodology in large-scale studies.
Required Materials:
- AmpliSeq cDNA Synthesis for Illumina Kit (20022654) [2]
- AmpliSeq for Illumina Library PLUS (24, 96, or 384 reactions) [2]
- AmpliSeq for Illumina CD Indexes (Set A-D, depending on multiplexing needs) [2]
- AmpliSeq Library Equalizer for normalization [2]
- RNA samples (1-100 ng input, 10 ng recommended per pool) [2]
- Nuclease-free water and standard laboratory equipment
Procedure:
cDNA Synthesis (Day 1)
Multiplex PCR Amplification (Day 1)
- Combine cDNA with the selected AmpliSeq for Illumina Panel (Ready-to-Use or Custom) and AmpliSeq Library PLUS master mix [4] [2].
- Perform multiplex PCR amplification using the following cycling conditions:
- Initial Denaturation: 99°C for 2 minutes
- Cycling: 99°C for 15 seconds, 60°C for 4 minutes (repeat for specified cycles based on panel)
- Hold: 10°C [4]
- This step simultaneously amplifies hundreds to thousands of target regions in a single reaction [4].
Primer Digestion and Partial Digestion (Day 1)
Ligation of Adapter Sequences (Day 1)
- Prepare ligation mix containing Illumina-specific adapter sequences and DNA ligase.
- Add mix to digested amplicons and incubate at 22°C for 30 minutes, followed by 68°C for 10 minutes [4].
- This step attaches platform-compatible adapters for sequencing.
Library Purification and Normalization (Day 1)
Figure 1: AmpliSeq for Illumina RNA Workflow from cDNA to Library. The process transforms RNA to sequencing-ready libraries in approximately 5-7 hours with less than 1.5 hours hands-on time [4] [2].
Sequencing and Data Analysis
Following library preparation, the process continues with sequencing and computational analysis to generate biological insights.
Sequencing (Day 2)
Data Analysis (Day 2-3)
- Perform secondary analysis using DRAGEN Amplicon pipeline on BaseSpace Sequence Hub or on-instrument with Local Run Manager [4].
- For RNA applications, DRAGEN RNA Amplicon performs differential expression analysis and gene fusion calling [4].
- Execute tertiary analysis as needed using platforms like Correlation Engine for biological interpretation [4].
Research Reagent Solutions
Successful implementation of the AmpliSeq for Illumina workflow in large-scale studies requires specific reagent systems designed for compatibility and scalability.
Table 3: Essential Research Reagents for AmpliSeq RNA Workflows
| Reagent Solution | Catalog Number Options | Primary Function | Considerations for Large Studies |
|---|---|---|---|
| Library Preparation | 20019101 (24-rxn)20019102 (96-rxn)20019103 (384-rxn) [2] | Core reagents for library construction | Bulk 384-reaction format offers best per-sample value for high-throughput applications |
| Index Adapters | 20019104 (24 indexes)20031676 (384 indexes) [2] | Sample multiplexing and identification | CD Indexes Set A-D provides 384 unique combinations for extensive multiplexing |
| cDNA Synthesis Kit | 20022654 [2] | Converts total RNA to cDNA | Reaction count varies by panel; verify capacity based on specific panel requirements |
| Library Normalization | 20019171 [2] | Normalizes libraries for balanced representation | Critical for maintaining sequencing efficiency across large sample batches |
| Direct FFPE DNA | 20023378 [2] | Processes FFPE tissue without DNA purification | Enables utilization of archival clinical resources; 24 reactions per kit |
Technical Performance and Quality Assessment
Analytical Performance Metrics
The AmpliSeq for Illumina RNA workflow delivers robust analytical performance that meets the stringent requirements of large-scale studies. Independent validation studies comparing alternative optimized RNA-seq methods have demonstrated significant improvements in key quality metrics, including reduction in PCR duplication rates and increases in uniquely mapped reads [87]. These technical advantages translate directly into more efficient sequencing utilization and higher data quality for extensive genomic investigations.
In comparative platform assessments, targeted amplicon sequencing approaches have demonstrated excellent performance characteristics, with one study reporting success rates of 94-96% for complete genome coverage using similar amplification-based methodologies [88]. The consistency of these results across multiple samples and batches underscores the reliability of the approach for large-scale applications where technical reproducibility is paramount.
Application-Specific Considerations
The flexibility of the AmpliSeq system enables researchers to tailor their approach based on specific study requirements. For investigations requiring specialized content beyond standard gene panels, the platform offers multiple design options through the AmpliSeq Designer tool, including Custom Panels and Community Panels developed with research consortia input [4] [89]. Recent updates to the design pipeline have enhanced performance for challenging samples by prioritizing shorter amplicons (75-175bp), which improves target amplification efficiency from low-quality input materials such as FFPE specimens [89].
For studies where comprehensive transcriptome information is required, recent advances in ultra-deep RNA sequencing demonstrate that increasing sequencing depth to approximately 1 billion reads can significantly enhance gene and isoform detection in clinically accessible tissues [90]. While standard AmpliSeq panels provide sufficient depth for targeted applications, these findings highlight the potential for specialized approaches when investigating low-abundance transcripts.
The AmpliSeq for Illumina RNA workflow represents a strategically advantageous solution for large-scale genetic studies where cost-effectiveness and rapid turnaround time are critical success factors. By combining minimal hands-on requirements (<1.5 hours) with rapid library construction (~5-7 hours) and efficient multiplexing capabilities, the system enables research organizations to accelerate study timelines while maintaining analytical rigor [4] [2]. The platform's compatibility with degraded sample types, including FFPE tissues, further enhances its utility for retrospective cohort studies that leverage existing biobank resources [2]. For research consortia and drug development programs requiring standardized, reproducible genetic analysis across hundreds or thousands of samples, this targeted RNA sequencing approach delivers the practical benefits necessary to advance genomic discovery while effectively managing resource constraints.
Conclusion
The AmpliSeq cDNA synthesis workflow for Illumina represents a significant advancement in targeted RNA sequencing, combining the sensitivity to work with minimal input RNA (as low as 1 ng) with the accuracy required for reliable differential gene expression analysis and fusion detection. Validation studies demonstrate exceptional concordance with traditional RNA-seq methods while offering practical advantages including reduced hands-on time, lower sequencing depth requirements, and cost-effectiveness for large-scale studies. As targeted sequencing continues to evolve, AmpliSeq technology is poised to play an increasingly vital role in translational research, clinical diagnostics, and drug development, particularly for precious sample types like FFPE tissues and limited clinical specimens. Future directions will likely expand its applications in single-cell analysis, liquid biopsies, and personalized medicine approaches where sample limitations traditionally posed significant barriers.
References
- 1. Comprehensive evaluation of AmpliSeq transcriptome, a ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-015-2270-1]
- 2. AmpliSeq for Illumina Library Prep, Indexes, and Accessories [https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/ampliseq-library-plus-adapters-accessories.html]
- 3. AmpliSeq Library PLUS for Illumina Compatible Products [https://support.illumina.com/sequencing/sequencing_kits/ampliseq-library-plus-for-illumina/compatibility.html]
- 4. AmpliSeq for Illumina Sequencing Solution [https://www.illumina.com/products/by-brand/ampliseq.html]
- 5. AmpliSeq for Illumina Focus Panel Compatible Products [https://support.illumina.com/sequencing/sequencing_kits/ampliseq-for-illumina-focus-panel/product-compatibility.html]
- 6. Comparison of library preparation methods reveals their ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4213505/]
- 7. Real-Time PCR (qPCR) and Whole-Transcriptome NGS [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/ngs-comparison.html]
- 8. Comparison Table [https://emea.illumina.com/library-prep-array-kit-selector/research-use-only/dna/targeted/custom-content/comparison-table.html]
- 9. Targeted RNA Sequencing | Focus on specific transcripts ... [https://www.illumina.com/techniques/sequencing/rna-sequencing/targeted-rna-seq.html]
- 10. AmpliSeq for Illumina Custom RNA Panel [https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/ampliseq-custom-rna-panel.html]
- 11. AmpliSeq for Illumina Transcriptome Human Gene ... [https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/ampliseq-transcriptome-gene-expression-panel.html]
- 12. AmpliSeq for Illumina Custom RNA Fusion Panel [https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/ampliseq-custom-rna-fusion-panel.html]
- 13. SCRIPT cDNA Synthesis Kit, Components and Classics [https://www.jenabioscience.com/molecular-biology/reverse-transcription-rt-pcr/components-and-classics/pcr-511-script-cdna-synthesis-kit]
- 14. Dual Index Sequencing | Reduce Per-Sample Costs ... [https://www.illumina.com/techniques/sequencing/ngs-library-prep/multiplexing/unique-dual-indexes.html]
- 15. An accurate DNA and RNA based targeted sequencing ... [https://www.nature.com/articles/s41598-025-91640-6]
- 16. Detection of fusion events by RNA sequencing in FFPE ... [https://pubmed.ncbi.nlm.nih.gov/39906487/]
- 17. RNA Library Preparation [https://www.illumina.com/techniques/sequencing/ngs-library-prep/rna.html]
- 18. Technical Validation and Clinical Utility of an NGS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9021638/]
- 19. A Guide to Basic RNA Sequencing Data Processing and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12067304/]
- 20. Reference-based RNA-Seq data analysis [https://training.galaxyproject.org/training-material/topics/transcriptomics/tutorials/ref-based/tutorial.html]
- 21. RSEM: accurate transcript quantification from RNA-Seq data ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-12-323]
- 22. A Benchmark of methods for SARS-CoV-2 whole genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12394145/]
- 23. Ampliseq™ cDNA Synthesis for Illumina [https://www.govsci.com/product-detail/Illumina-----Inc/20022654/EA/?srsltid=AfmBOopREY-ZrX5Lk0F63ohfuGj5iDCk4LJ2vB5Sirch2qjztNJ6WMVs]
- 24. AmpliSeq for Illumina Comprehensive Panel v3 [https://support.illumina.com/sequencing/sequencing_kits/ampliseq-for-illumina-comprehensive-panel-v3/product-compatibility.html]
- 25. Comprehensive evaluation of AmpliSeq transcriptome, a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4681149/]
- 26. AmpliSeq for Illumina Custom DNA Panel [https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/ampliseq-custom-dna-panel.html]
- 27. Key considerations for comprehensive validation of an RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7322345/]
- 28. 5 reasons to go custom with your next sequencing panel [https://www.pillarbiosci.com/blog/5-reasons-to-go-custom-with-your-next-sequencing-panel/]
- 29. Revolutionizing Genomic Research: Custom NGS Panels [https://www.igenetech.com/blog/revolutionizing-genomic-research-custom-ngs-panels.html]
- 30. TruSight RNA Fusion Panel [https://www.illumina.com/products/by-type/clinical-research-products/trusight-rna-fusion.html]
- 31. iGeneTech Hematologic Tumor Fusion Gene RNA Panel [https://www.igenetech.com/igenetech-hematologic-tumor-fusion-gene-rna-panel.html]
- 32. Genome-wide RNA-Seq vs. NanoString nCounter® ... [https://blog.crownbio.com/choosing-the-right-rna-analysis-methods]
- 33. What is Ion AmpliSeq ™ Targeted Sequencing Technology? [https://alitheagenomics.com/blog/what-is-ion-ampliseq-targeted-sequencing-technology]
- 34. Practical Tips for Reliable cDNA in qPCR Synthesis Workflows [https://www.mscience.com.au/cdna-synthesis-qpcr-tips/]
- 35. Article Combining panel-based and whole-transcriptome ... [https://www.sciencedirect.com/science/article/pii/S266723752500147X]
- 36. Comparative analysis of library preparation approaches for ... [https://www.nature.com/articles/s41598-025-12992-7]
- 37. Reliable RNA-seq analysis from FFPE specimens as a means ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0321631]
- 38. Success in RNA-Seq – Sample Prep and Workflow ... [https://www.rna-seqblog.com/success-in-rna-seq-sample-prep-and-workflow-considerations/]
- 39. AmpliSeq for Illumina Comprehensive Panel v3 [https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/ampliseq-comprehensive-panel.html]
- 40. Cracking rare disorders: a new minimally invasive RNA- ... [https://www.nature.com/articles/s41525-025-00502-7]
- 41. REALLY TECHNOLOGY [http://www.claretbio.com/really-technology]
- 42. Single-nucleus total RNA sequencing of formalin-fixed ... [https://www.rna-seqblog.com/single-nucleus-total-rna-sequencing-of-formalin-fixed-paraffin-embedded-samples-using-snrandom-seq/]
- 43. FFPE compatible RNA sequencing [https://bio-protocol.org/exchange/minidetail?id=9890846&type=30]
- 44. ERCC RNA Spike-In Mix 1 Kit | Buy Online | Invitrogen™ [https://www.thermofisher.com/order/catalog/product/4456740]
- 45. External RNA Controls Consortium | NIST [https://www.nist.gov/programs-projects/external-rna-controls-consortium]
- 46. AmpliSeq Library PLUS for Illumina Contents & Storage [https://support.illumina.com/sequencing/sequencing_kits/ampliseq-library-plus-for-illumina/contents-storage.html]
- 47. AmpliSeq for Illumina Focus Panel | Combined DNA and ... [https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/ampliseq-focus-panel.html]
- 48. Systematic comparison and assessment of RNA-seq ... [https://www.nature.com/articles/s41598-020-76881-x]
- 49. Optimization of library preparation based on SMART for ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-021-08132-w]
- 50. Optimization of quantitative reverse transcription PCR ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2022.954976/full]
- 51. Reverse Transcription [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/gene-expression-and-silencing/reverse-transcription?srsltid=AfmBOooy9RB3KfsEH-kAN-9Ho5pNw_fa5B7wkryMFJ25FuDJk79D6LkO]
- 52. Five Steps to Optimal cDNA Synthesis [https://www.thermofisher.com/us/en/home/life-science/pcr/reverse-transcription/5steps-cDNA.html]
- 53. Improved precision, sensitivity, and adaptability of ordered two ... [https://pubmed.ncbi.nlm.nih.gov/39626888/]
- 54. Reverse transcription troubleshooting guide [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/rt-education/reverse-transcription-troubleshooting.html]
- 55. Reverse Transcription: The Most Common Pitfalls! [https://bitesizebio.com/32253/the-most-common-issues-with-reverse-transcription/]
- 56. cDNA Synthesis Troubleshooting | Blog [https://go.zageno.com/blog/cdna-synthesis-troubleshooting]
- 57. RNA integrity number [https://en.wikipedia.org/wiki/RNA_integrity_number]
- 58. Methods of RNA Quality Assessment [https://worldwide.promega.com/resources/pubhub/methods-of-rna-quality-assessment/]
- 59. Is Your RNA Intact? Methods to Check RNA Integrity [https://www.thermofisher.com/tr/en/home/references/ambion-tech-support/rna-isolation/tech-notes/is-your-rna-intact.html]
- 60. What is the RNA Integrity Number (RIN)? [https://www.cd-genomics.com/longseq/resource-rna-integrity-number.html]
- 61. Library Preparation Quality Control and Quantification [https://www.lexogen.com/blog/rna-lexicon-library-preparation-quality-control-and-quantification/]
- 62. Best practices for library quantification [https://knowledge.illumina.com/library-preparation/general/library-preparation-general-reference_material-list/000003750]
- 63. Best practices for RNA storage and sample handling [https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/rna/introduction/general-remarks-on-rna-handling-storage-and-stabilization]
- 64. Tech Tip: Tips & Best Practices for Working with RNA [https://biotium.com/tech-tips-protocols/tips-best-practices-for-working-with-rna/]
- 65. Precautions for Handling of RNA [https://lifescience.roche.com/global/en/article-listing/article/precautions-for-handling-of-rna.html]
- 66. The Basics: RNA Isolation [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rna-isolation/general-articles/the-basics-rna-isolation.html]
- 67. Top Ten Ways to Improve Your RNA Isolation [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rna-isolation/general-articles/ten-ways-to-improve-your-rna-isolation.html]
- 68. Whole Blood RNA-Seq: Best Practice [https://www.lexogen.com/blog/whole-blood-rna-seq-best-practice/]
- 69. Illumina Custom Enrichment Panel v2 [https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/custom-enrichment-panels.html]
- 70. A comparison of Illumina and Ion Torrent sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5553782/]
- 71. Comprehensive Development and Implementation of Good ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9141409/]
- 72. Genome-Wide RNA-seq and Array-Based NanoString ... [https://blog.crownbio.com/genome-wide-rna-seq-and-array-based-nanostring-transcriptomic-technologies]
- 73. Comprehensive evaluation of AmpliSeq transcriptome, a novel... [https://pubmed.ncbi.nlm.nih.gov/26673413/]
- 74. Targeted RNA Sequencing for Gene... | Thermo Fisher Scientific - KR [https://www.thermofisher.com/kr/ko/home/life-science/sequencing/rna-sequencing/targeted-rna-sequencing-ion-torrent-next-generation-sequencing.html]
- 75. - RNA Microarrays | Seq technologies vs Compare [https://www.illumina.com/science/technology/next-generation-sequencing/beginners/advantages/rna-seq-vs-arrays.html]
- 76. Frontiers | Cardiac disease mechanobiology: advances using hiPSC-CMs [https://www.frontiersin.org/journals/cardiovascular-medicine/articles/10.3389/fcvm.2025.1642931/full]
- 77. Metabolic Maturation in hiPSC-Derived Cardiomyocytes: Emerging Strategies for Inducing the Adult Cardiac Phenotype [https://www.mdpi.com/1424-8247/18/8/1133]
- 78. Generation and purification of iPSC-derived cardiomyocytes for clinical applications | Stem Cell Research & Therapy | Full Text [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-025-04319-0]
- 79. Enhancing human pluripotent stem cell differentiation to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12124670/]
- 80. 2D Human Induced Pluripotent Stem Cell-derived Cardiomyocyte Cardiac Contractility Modulation Tool: 2D hiPSC-CM CCM Tool | Center for Devices and Radiological Health [https://cdrh-rst.fda.gov/2d-human-induced-pluripotent-stem-cell-derived-cardiomyocyte-cardiac-contractility-modulation-tool]
- 81. Optimizing single cell RNA sequencing of stem ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1590889/full]
- 82. Analytical validation (accuracy, reproducibility, limit of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12431237/]
- 83. Diagnosis of fusion genes using targeted RNA sequencing [https://www.nature.com/articles/s41467-019-09374-9]
- 84. Development and Validation of an RNA Sequencing Assay ... [https://www.sciencedirect.com/science/article/pii/S1525157820305791]
- 85. Deciphering RNA-seq Library Preparation: From Principles ... [https://www.cd-genomics.com/resouce-rna-seq-library-preparation-principles-protocol.html]
- 86. Fast and sensitive validation of fusion transcripts in whole ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05489-5]
- 87. Improving RNA-Seq Library Preparation for Accuracy [https://cellcarta.com/science-hub/rna-seq-library-preparation-watchmaker-workflow/]
- 88. Reflective Evaluation of Next-Generation Sequencing Data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12494147/]
- 89. Release Notes [https://www.ampliseq.com/displayChangeLog.action]
- 90. The utility of ultra-deep RNA sequencing in Mendelian ... [https://www.sciencedirect.com/science/article/pii/S0002929725003696]