Assessing Reproducibility of the AmpliSeq Childhood Cancer Panel: A Technical and Clinical Validation for DNA and RNA Analysis in Pediatric Oncology
This article provides a comprehensive evaluation of the reproducibility and reliability of the AmpliSeq for Illumina Childhood Cancer Panel, a targeted NGS solution for pediatric and young adult cancers.
Assessing Reproducibility of the AmpliSeq Childhood Cancer Panel: A Technical and Clinical Validation for DNA and RNA Analysis in Pediatric Oncology
Abstract
This article provides a comprehensive evaluation of the reproducibility and reliability of the AmpliSeq for Illumina Childhood Cancer Panel, a targeted NGS solution for pediatric and young adult cancers. We synthesize data from technical validations and clinical implementation studies, detailing performance metrics for both DNA and RNA components. Covering foundational principles, methodological workflows, troubleshooting strategies, and comparative performance against other assays, this resource is designed to inform researchers, scientists, and drug development professionals on integrating this panel into robust, reproducible genomic profiling pipelines for precision medicine in childhood cancers.
Understanding the AmpliSeq Childhood Cancer Panel: Design, Content, and Importance of Reproducibility in Pediatric Genomics
The molecular landscape of pediatric cancers is distinct from that of adult cancers, necessitating specialized genomic tools for accurate diagnosis and treatment. Next-generation sequencing (NGS) panels designed specifically for childhood cancers are critical for detecting key somatic variants, including single nucleotide polymorphisms (SNPs), insertions-deletions (indels), gene fusions, and copy number variants (CNVs). The reproducibility of results generated by these panels is a cornerstone of their clinical utility, ensuring that findings are consistent, reliable, and translatable across different laboratories. This guide objectively compares the performance of the AmpliSeq for Illumina Childhood Cancer Panel with other available solutions, focusing on their design, content, and the experimental data that underpins their reliability in a research context.
AmpliSeq for Illumina Childhood Cancer Panel
The AmpliSeq Childhood Cancer Panel for Illumina is a targeted resequencing solution designed for the comprehensive evaluation of somatic variants associated with childhood and young adult cancers [1]. Its core design and specifications are summarized below.
- Targeted Genes: The panel interrogates 203 genes with established associations to pediatric and young adult cancers [1].
- Variant Detection: It is engineered to detect multiple variant classes, including SNPs, indels, CNVs, and gene fusions [1].
- Sample Compatibility: The panel is compatible with a range of sample types, including blood, bone marrow, and FFPE tissue, requiring only 10 ng of high-quality DNA or RNA as input [1].
- Workflow Integration: It is part of an integrated workflow that includes PCR-based library preparation and Illumina SBS sequencing technology, with a hands-on time of less than 1.5 hours for library preparation [1].
Comparative Panel Landscape
Other panels have been developed to address the need for pediatric cancer genomic profiling. The following table provides a high-level comparison of key panels.
- OncoKids Panel: An amplification-based NGS assay that covers the full coding regions of 44 cancer predisposition genes, mutation hotspots in 82 genes, amplification events in 24 genes, and 1,421 targeted gene fusions via its RNA content. It uses 20 ng of DNA and RNA input and is validated for FFPE tissue, frozen tissue, bone marrow, and peripheral blood [2].
- SJPedPanel: Developed by St. Jude Children's Research Hospital, this panel was designed from the ground up for pediatric cancers. It was created by concentrating genetic knowledge from the Pediatric Cancer Genome Project and is optimized for performance, providing coverage of approximately 90% of pediatric cancer driver genes, a significant improvement over other panels which were reported to be closer to 60% [3]. It is particularly effective in challenging scenarios such as samples with low tumor purity or post-bone marrow transplantation [3].
Table 1: Comparison of Pediatric Cancer Targeted Sequencing Panels
| Feature | AmpliSeq Childhood Cancer Panel | OncoKids Panel | SJPedPanel |
|---|---|---|---|
| Number of Targeted Genes | 203 genes [1] | 44 cancer predisposition genes + 82 genes (hotspots) + 24 genes (amplification) [2] | Information not specified in search results |
| RNA Fusion Targets | Included (exact number not specified) [1] | 1,421 targeted gene fusions [2] | Information not specified in search results |
| Input Requirement (DNA/RNA) | 10 ng [1] | 20 ng [2] | Information not specified in search results |
| Key Differentiator | Integrated Illumina workflow; low input requirement | Broad fusion detection; includes cancer predisposition loci | Designed specifically for pediatrics; high coverage of pediatric drivers (~90%) [3] |
Experimental Data and Performance Comparison
Independent validation studies provide critical data on the performance of these panels. The OncoKids panel was validated using a cohort of 192 unique clinical samples, demonstrating "robust performance was observed for analytical sensitivity, reproducibility, and limit of detection studies" [2]. This supports its use for routine clinical testing.
The SJPedPanel was benchmarked against six other commercially available panels [3]. Its iterative, knowledge-informed design allowed it to "outperform existing cancer gene panels," providing superior coverage of known pediatric cancer driver genes. Furthermore, in certain situations like low tumor purity samples, the panel can "outperform gold-standard whole genome sequencing" by enabling high-depth sampling of a focused genomic region, thus filling an important clinical gap [3].
Table 2: Experimental Performance Metrics from Validation Studies
| Panel | Validation Cohort | Key Performance Findings |
|---|---|---|
| OncoKids | 192 unique clinical samples [2] | Robust analytical sensitivity, reproducibility, and limit of detection [2]. |
| SJPedPanel | Compared against 6 other commercial panels; over 600 clinical samples [3] | Provides ~90% coverage of pediatric cancer driver genes (vs. ~60% for others); effective for low tumor purity samples where WGS fails [3]. |
The Reproducibility Framework in Pediatric Cancer Genomics
The reproducibility of NGS panel results is not solely a function of the wet-lab protocol. It is increasingly supported by open-science initiatives that provide harmonized datasets and reproducible analysis workflows. The Open Pediatric Cancer (OpenPedCan) Project is a key example, offering a harmonized, multi-omic dataset from over 6,000 pediatric cancer patients [4]. The project delivers "reproducible, dockerized workflows" for data processing, enabling researchers to validate findings and methodologies in a consistent computational environment. Such resources provide a framework for benchmarking the performance and output of targeted panels like AmpliSeq, OncoKids, and SJPedPanel, thereby reinforcing the reproducibility of research built upon them.
Methodologies: Experimental Protocols and Workflows
AmpliSeq for Illumina Library Preparation Workflow
The following diagram outlines the core experimental workflow for preparing sequencing libraries using the AmpliSeq for Illumina technology, which is central to the Childhood Cancer Panel.
OpenPedCan Data Harmonization and Analysis Workflow
For panels used in a research context, integration into larger analysis frameworks is crucial. The OpenPedCan project employs a sophisticated workflow to harmonize data from multiple sources, which can be used to analyze and validate output from different panels.
The Scientist's Toolkit: Essential Research Reagents and Solutions
The following table details key reagents and materials required to implement the AmpliSeq for Illumina Childhood Cancer Panel in a research setting.
Table 3: Key Research Reagent Solutions for the AmpliSeq Workflow
| Item | Function | Example Product (Illumina) |
|---|---|---|
| Library Preparation Kit | Provides core reagents for PCR-based library construction. | AmpliSeq Library PLUS [1] |
| Childhood Cancer Panel | The core primer pool targeting the 203 genes associated with pediatric cancers. | AmpliSeq for Illumina Childhood Cancer Panel [1] |
| Index Adapters | Unique nucleotide sequences ligated to each sample to allow multiplexing of multiple libraries in a single sequencing run. | AmpliSeq CD Indexes (e.g., Set A-D) [1] |
| Library Normalization Reagent | Simplifies and automates the process of balancing library concentrations prior to pooling for sequencing. | AmpliSeq Library Equalizer for Illumina [1] |
| cDNA Synthesis Kit | Converts input RNA into cDNA, a required step when using RNA with the panel. | AmpliSeq cDNA Synthesis for Illumina [1] |
| FFPE DNA Preparation Kit | Enables direct library construction from FFPE tissues without the need for deparaffinization or DNA purification. | AmpliSeq for Illumina Direct FFPE DNA [1] |
The AmpliSeq Childhood Cancer Panel represents a well-integrated, targeted solution for investigating pediatric cancers, with key advantages in workflow speed and low input requirements. However, the landscape of pediatric cancer genomics offers other robust options. The OncoKids panel provides extensive validation data and broad fusion detection, while the St. Jude SJPedPanel demonstrates how a purpose-built design can achieve superior coverage of pediatric-specific driver genes, particularly in diagnostically challenging low-purity samples. The reproducibility of research utilizing any of these panels is greatly enhanced by global, open-science initiatives like the OpenPedCan Project, which provide the harmonized data and computational frameworks necessary for independent verification and collaborative discovery.
Reproducibility is a critical challenge in next-generation sequencing (NGS), impacting the reliability of data used for clinical diagnostics and research. This guide objectively compares the performance of the AmpliSeq for Illumina Childhood Cancer Panel, a targeted solution for pediatric cancers, against broader NGS reproducibility findings, providing experimental data and methodologies.
Quantifying Reproducibility: Performance Data Comparison
The following tables summarize key quantitative data on reproducibility from a validation study of the AmpliSeq Childhood Cancer Panel and from broader NGS research, highlighting the panel's performance in a clinical context.
Table 1: Key Performance Metrics of the AmpliSeq Childhood Cancer Panel [5]
| Metric | DNA (SNVs & Indels) | RNA (Fusion Genes) |
|---|---|---|
| Sensitivity | 98.5% (at 5% VAF) | 94.4% |
| Specificity | 100% | 100% |
| Reproducibility | 100% | 89% |
| Limit of Detection | 5% Variant Allele Frequency (VAF) | Not Specified |
Table 2: Comparative Reproducibility Findings from Broader NGS Studies
| Study Focus | Key Concordance/Discordance Finding | Major Factor Identified |
|---|---|---|
| Inter-assay Variability [6] | 71.8% discordance between two different NGS panels using identical DNA. | Sample type (FFPE vs. fresh frozen) and panel analytical features. |
| Inherited Variants with WGS [7] | Bioinformatics pipelines (aligners & callers) had a larger impact on variant reproducibility than sequencing platform or library prep. | Variant class (SNVs more reproducible than indels) and genome context. |
| Impact of Sample Type [6] | Significantly higher discordance rate for FFPE samples compared to fresh frozen (FF) samples. | FFPE DNA quality and tumor heterogeneity. |
Experimental Protocols: Assessing Reproducibility
Protocol: Analytical Validation of the AmpliSeq Childhood Cancer Panel
This detailed methodology was used to generate the performance metrics in Table 1 [5].
1. Sample Selection and Controls:
- Commercial Controls: Used SeraSeq Tumor Mutation DNA Mix (for DNA variants) and SeraSeq Myeloid Fusion RNA Mix (for RNA fusions) as positive controls. Used the NA12878 cell line and IVS-0035 as negative controls.
- Patient Cohorts: Selected 76 pediatric patients with acute leukemia (BCP-ALL, T-ALL, AML), prioritizing samples with high-quality nucleic acids.
2. Nucleic Acid Extraction and QC:
- DNA Extraction: Performed using Qiagen kits (Gentra Puregene, QIAamp DNA Mini/Micro).
- RNA Extraction: Conducted via guanidine thiocyanate-phenol-chloroform method or column-based methods (e.g., Direct-zol RNA MiniPrep).
- Quality Control: Assessed purity (OD260/280 >1.8) via spectrophotometry and integrity via Labchip or TapeStation. Concentration was determined by fluorometric quantification (Qubit).
3. Library Preparation and Sequencing:
- Library Prep: Followed the manufacturer's protocol (Illumina). Used 100 ng of DNA and 100 ng of RNA (reverse-transcribed to cDNA) per sample to generate amplicon libraries.
- Pooling and Sequencing: DNA and RNA libraries were pooled at a 5:1 ratio, diluted to 17–20 pM, and sequenced on an Illumina MiSeq sequencer.
4. Data Analysis and Validation:
- Variant Calling: Variants were called using the panel's built-in analysis pipeline.
- Orthogonal Confirmation: Identified DNA variants and RNA fusions were confirmed using conventional methods like Sanger sequencing, PCR, and quantitative RT-PCR.
- Metric Calculation:
- Sensitivity: (True Positives / (True Positives + False Negatives)) * 100
- Specificity: (True Negatives / (True Negatives + False Positives)) * 100
- Reproducibility: Percentage of variants consistently identified in replicate experiments.
Protocol: Assessing Inter-Assay Variability in NGS
This methodology underpins the findings on inter-assay discordance summarized in Table 2 [6].
1. Sample and Panel Design:
- Samples: Utilized 30 patient-derived DNA samples (10 fresh frozen, 20 FFPE).
- Assays: Compared two different CLIA-certified laboratory-developed tests: a Tumor-Only (TO) panel (OncoPrime, 215 genes) and a paired Tumor-Normal (TN) panel (NCC Oncopanel v4, 114 genes).
2. Experimental Comparison:
- Primary Analysis: The same DNA sample from each patient was submitted to both the TO and TN panel assays.
- Supplementary Analysis: Additional slices from the same FFPE blocks (n=20) were submitted to a second, independent TN panel assay to assess variability within the same tumor block.
3. Data Analysis:
- Variant Comparison: Reported short variants (SNVs and indels up to 5 bp) from both panels were compared.
- Discordance Rate Calculation: Defined as 100% minus the concordance rate, where concordance rate = (number of variants found in both panels) / (number of variants found in both panels + all discordant variants).
Visualizing NGS Reproducibility Concepts
NGS Wet Lab to Dry Lab Workflow
This diagram illustrates the complete NGS workflow, highlighting stages where technical variance can be introduced. The wet lab phase (gold) involves sample and library preparation, where factors like sample type (FFPE vs. fresh frozen) and input quality significantly impact reproducibility [6] [8]. The dry lab phase (green) encompasses bioinformatics, where the choice of aligners and variant callers has been shown to have a major influence on variant reproducibility [7].
Factors Affecting NGS Reproducibility
This diagram categorizes the primary sources of technical variance in NGS. Bioinformatics pipelines (aligners and callers) have been identified as having a larger impact on reproducibility than the sequencing platform itself [7]. The sample type is another critical factor, with formalin-fixed paraffin-embedded (FFPE) samples showing significantly higher discordance rates compared to fresh frozen tissues [6]. Finally, the variant class matters, as single-nucleotide variants (SNVs) are generally more reproducible than insertions and deletions (indels), especially those longer than 5 base pairs [7].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for NGS Reproducibility Studies [5]
| Item | Function in Reproducibility Context |
|---|---|
| SeraSeq Tumor Mutation DNA Mix | Multiplex biosynthetic positive control with known variants at defined allele frequencies (e.g., 10% VAF). Essential for establishing sensitivity and limit of detection. |
| SeraSeq Fusion RNA Mix | Synthetic RNA positive control containing known fusion genes. Validates fusion detection sensitivity and specificity in the wet lab workflow. |
| NA12878 Cell Line DNA | Well-characterized reference genome from Coriell Institute. Serves as a critical negative control and benchmark for inherited variant calling. |
| Qubit dsDNA/RNA BR Assay Kits | Fluorometric quantification for accurate nucleic acid concentration measurement. Superior to spectrophotometry for library preparation input, crucial for reproducibility. |
| AmpliSeq for Illumina Childhood Cancer Panel | Targeted amplicon-based panel integrating library prep reagents for 203 genes. Standardizes the initial steps of the NGS workflow across samples. |
| AmpliSeq cDNA Synthesis for Illumina | Converts total RNA to cDNA for RNA-based fusion detection in the panel. Ensures high-quality input for RNA sequencing applications. |
| TapeStation System (Agilent) | Microfluidic capillary electrophoresis for assessing DNA and RNA integrity. Provides critical quality control (QC) data before library prep. |
The Critical Role of Reproducible Results in Diagnostic Refinement and Clinical Decision-Making
Reproducibility forms the cornerstone of reliable clinical genomics, ensuring that diagnostic results remain consistent across different laboratories, sequencing runs, and analysis methods. In the field of pediatric oncology, where treatment decisions hinge on precise molecular characterization, the ability to generate reproducible data becomes critical for diagnostic refinement and therapeutic decision-making. Next-generation sequencing (NGS) panels like the AmpliSeq for Illumina Childhood Cancer Panel have emerged as powerful tools for comprehensive genomic profiling of childhood cancers. This guide objectively evaluates the performance of this targeted panel, with a specific focus on its reproducibility in analyzing DNA and RNA variants, and examines how these characteristics support its role in clinical research and diagnostic refinement.
Performance Evaluation: Accuracy and Reproducibility Metrics
Rigorous technical validation studies demonstrate that the AmpliSeq Childhood Cancer Panel delivers highly reproducible results across key performance parameters essential for reliable clinical research.
Table 1: Key Performance Metrics of the AmpliSeq Childhood Cancer Panel
| Performance Parameter | DNA Analysis | RNA Analysis | Experimental Details |
|---|---|---|---|
| Sensitivity | 98.5% (for variants at 5% VAF) | 94.4% | Using commercial control materials [5] |
| Specificity | 100% | Information not specified in search results | Using commercial control materials [5] |
| Reproducibility | 100% | 89% | Measured across replicates [5] |
| Mean Read Depth | >1000x | Information not specified in search results | Ensures sufficient coverage for reliable variant calling [5] |
| Variant Types Detected | SNVs, InDels, CNVs | Gene fusions | Panel covers 203 genes, 97 fusions, 82 DNA variants, 24 CNVs [5] [1] |
The panel's high reproducibility for DNA variants (100%) ensures that single nucleotide variants (SNVs), insertions-deletions (InDels), and copy number variants (CNVs) can be consistently detected across repeated runs [5]. While slightly lower, the 89% reproducibility for RNA-based fusion detection still demonstrates substantial consistency for transcriptomic analysis. The high sensitivity down to 5% variant allele frequency (VAF) for DNA enables detection of low-level somatic mutations, which is crucial for identifying subclonal populations in heterogeneous tumor samples [5].
Experimental Protocols for Validation
The validation methodology followed standardized protocols to ensure rigorous assessment of the panel's capabilities.
Sample Selection and Preparation
- Control Materials: The validation utilized commercially available reference standards including SeraSeq Tumor Mutation DNA Mix and SeraSeq Myeloid Fusion RNA Mix to establish sensitivity, specificity, and limit of detection [5].
- Patient Cohorts: The study included 76 pediatric patients diagnosed with B-cell precursor ALL (n=51), T-ALL (n=11), and AML (n=14) from multiple centers, with selection prioritizing samples with high DNA/RNA quality and those that could benefit from NGS after inconventional diagnostic results [5].
- Nucleic Acid Extraction: DNA was extracted using Qiagen kits (Gentra Puregene, QIAamp DNA Mini, or QIAamp DNA Micro), while RNA was extracted using either manual guanidine thiocyanate-phenol-chloroform method or column-based methods [5].
Library Preparation and Sequencing
- Input Requirements: The protocol requires only 10 ng of high-quality DNA or RNA, making it suitable for precious, limited pediatric samples [1].
- Library Construction: The panel uses a PCR-based approach to generate 3069 DNA amplicons and 1701 RNA amplicons, with RNA first reverse transcribed to cDNA [5].
- Sequencing: Libraries were pooled at a 5:1 DNA:RNA ratio and sequenced on Illumina MiSeq sequencers, with compatibility extending to NextSeq and MiniSeq systems [5] [1].
The following diagram illustrates the complete validation workflow, from sample preparation to clinical interpretation:
Diagram Title: Childhood Cancer Panel Validation Workflow
Research Reagent Solutions and Essential Materials
Successful implementation of the AmpliSeq Childhood Cancer Panel requires specific companion reagents and accessories.
Table 2: Essential Research Reagents and Materials
| Reagent/Material | Function/Purpose | Specifications |
|---|---|---|
| AmpliSeq Library PLUS | Library preparation reagents | Available in 24, 96, or 384 reactions [1] |
| AmpliSeq CD Indexes | Sample barcoding for multiplexing | 8 bp indexes in sets A-D (384 total indexes) [1] |
| AmpliSeq cDNA Synthesis | Converts RNA to cDNA for fusion detection | Required for RNA analysis [1] |
| AmpliSeq Library Equalizer | Normalizes libraries before sequencing | Streamlines workflow [1] |
| AmpliSeq for Illumina Direct FFPE DNA | Processes FFPE tissue without DNA purification | Enables analysis of archived specimens [1] |
Clinical Impact and Diagnostic Utility
The ultimate validation of any diagnostic tool lies in its ability to generate clinically actionable information. Studies demonstrate that the Childhood Cancer Panel identified clinically relevant results in 43% of patients tested in one cohort [5]. The clinical impact of detected variants was substantial:
- DNA Mutations: 49% of identified mutations were considered targetable, while 41% refined diagnosis [5]
- RNA Fusions: 97% of detected fusion genes had diagnostic impact, providing crucial diagnostic refinement [5]
This high clinical impact rate underscores how reproducible NGS testing can directly influence patient management by identifying targetable alterations and refining diagnostic classification beyond what conventional methodologies can achieve.
The Broader Context: Reproducibility Challenges in Genomic Analysis
The rigorous validation of targeted panels like the AmpliSeq Childhood Cancer Panel addresses significant reproducibility challenges in broader genomic analysis:
- Bioinformatics Variability: Bioinformatics tools can introduce both deterministic variations (algorithmic biases) and stochastic variations (intrinsic randomness) that affect result consistency [9]
- Technical Replicates: Consistency across technical replicates (same biological sample sequenced multiple times) is essential for establishing genomic reproducibility, though generating them increases costs and complexity [9]
- RNA-Seq Specific Challenges: Studies have identified technical biases in RNA-seq data, such as sample-specific length effects where gene length influences measured expression changes, potentially leading to false results if not properly corrected [10]
These contextual challenges highlight why standardized, validated panels with established reproducibility metrics provide significant value for clinical research applications where consistency across experiments and laboratories is paramount.
The AmpliSeq for Illumina Childhood Cancer Panel demonstrates strong performance characteristics for reproducible detection of DNA and RNA variants in pediatric cancer samples. With high sensitivity, specificity, and reproducibility metrics, combined with substantial clinical impact in diagnostic refinement and identification of targetable alterations, this targeted NGS approach provides researchers with a reliable tool for pediatric oncology genomics. The standardized protocols and defined performance parameters support its role in generating consistent, clinically relevant data across research settings, addressing fundamental reproducibility requirements in genomic medicine. As the field continues to emphasize reproducibility as a cornerstone of reliable diagnostics, such validated approaches will remain essential for advancing precision oncology in childhood cancers.
In the pursuit of precision medicine, the reproducibility of genomic results is a cornerstone of reliable biomarker discovery and clinical research [11]. This is particularly critical for targeted sequencing panels, such as the AmpliSeq Childhood Cancer Panel, which are designed to detect somatic variants across DNA and RNA from precious clinical samples [1]. The synergy between DNA and RNA workflow components, from initial amplicon distribution to final library preparation, directly influences the consistency and accuracy of downstream results. Genomic reproducibility, defined as the ability of bioinformatics tools and wet-lab protocols to maintain consistent results across technical replicates, is a fundamental metric often challenged by technical variability in sequencing and computational analysis [11]. This guide objectively compares the performance of different library preparation methodologies within the context of ensuring reproducible DNA and RNA results in childhood cancer research.
Experimental Protocols for Performance Comparison
To evaluate the reproducibility and performance of different library preparation workflows, we focus on two primary types of experimental data: validation studies of commercial panels and controlled in-silico simulations.
Validation of the Watchmaker RNA-Seq Workflow
A direct benchmark study was performed comparing the Watchmaker Genomics (WMG) RNA-sequencing workflow with a standard RNA capture method [12]. The experimental protocol was as follows:
- Samples: Universal human reference RNA (UHRR), whole blood (WB), a Horizon Discovery reference sample (HD200), and formalin-fixed paraffin-embedded (FFPE) samples.
- Library Preparation: The WMG RNA library prep with Polaris Depletion was executed, with a total hands-on time of approximately 4 hours. This was compared directly to a standard capture-based method requiring about 16 hours.
- Sequencing and Analysis: Standard Illumina sequencing was performed. Downstream bioinformatic analysis quantified duplication rates, mapping rates, rRNA and globin read counts, and the number of genes detected.
In-silico Simulation of Technical Noise
The GENOMICON-Seq simulation tool was used to model the impact of technical variation on low-frequency mutation detection, a key challenge in somatic variant calling from cancer samples [13]. The protocol involves:
- Ground Truth Mutation Insertion: User-defined mutations are inserted into a reference genome (e.g., HPV16 or human exome) using one of three modes: "deterministic mode" for controlled VAFs, "specific mutation rate mode" for random mutations, or "SBS-Mimicry mode" to replicate cancer mutational signatures from COSMIC.
- Workflow Simulation: For amplicon sequencing, the tool simulates PCR errors and amplification efficiency drops. For whole exome sequencing (WES), it simulates probe-capture enrichment biases.
- Read Generation and Analysis: A modified InSilicoSeq engine generates Illumina-style reads with platform-specific error models. The resulting FASTQ files allow researchers to track the fate of ground-truth mutations and benchmark variant callers against a known truth set.
Comparative Performance Data
The following tables summarize quantitative data from the cited experimental and simulation studies, providing a clear comparison of key performance metrics.
Table 1: Experimental RNA-Seq Workflow Performance Comparison (Watchmaker vs. Standard Method) [12]
| Performance Metric | Sample Type | Watchmaker Workflow | Standard Method |
|---|---|---|---|
| Assay Time | All | ~4 hours | ~16 hours |
| PCR Duplication Rate | UHRR | Significantly Reduced | Higher |
| Whole Blood | Significantly Reduced | Higher | |
| FFPE | Significantly Reduced | Higher | |
| Uniquely Mapped Reads | All | Significantly Increased | Lower |
| rRNA Reads | Whole Blood | Fewer | More |
| FFPE | Fewer | More | |
| Globin Reads | Whole Blood | Reduced | More |
| Genes Detected | All | ~30% More | Baseline |
Table 2: Simulated Impact of Technical Factors on Low-Frequency Mutation Detection (GENOMICON-Seq) [13]
| Simulation Factor | Study Case | Impact on Mutation Detection |
|---|---|---|
| Polymerase Error Rate | Amplicon (A1) | Higher error rates increase background noise, complicating true low-frequency variant identification. |
| Input Copy Number | Amplicon (A2) | Low viral/genome copy numbers reduce the probability of detecting true low-frequency mutations. |
| Sequencing Depth | Amplicon (A3) & WES (W1, W2) | Higher read depth improves the detection of alternative alleles, especially at lower frequencies. |
| Capture Bias (WES) | WES (W1-W3) | Probe-capture enrichment can lead to the loss of mutations if their fragments are undersampled. |
| Sequencing Bias | WES (W3) | Length-weighted sequencing bias can skew coverage, affecting variant allele frequency (VAF) accuracy. |
Workflow Visualization
The following diagram illustrates the synergistic DNA and RNA workflow for targeted amplicon sequencing, highlighting critical control points for ensuring genomic reproducibility. The process is aligned with the AmpliSeq methodology and incorporates principles for minimizing technical variation [11] [1].
Diagram 1: Targeted Amplicon Sequencing Workflow. This flowchart outlines the integrated DNA and RNA pathway for library preparation using the AmpliSeq technology, highlighting key reagent-dependent steps and the convergence point for data analysis [1].
The Scientist's Toolkit: Essential Research Reagents
Successful and reproducible library preparation relies on a suite of specialized reagents. The following table details key components for the AmpliSeq for Illumina workflow.
Table 3: Essential Research Reagent Solutions for the AmpliSeq Workflow [1]
| Research Reagent | Function |
|---|---|
| AmpliSeq for Illumina Childhood Cancer Panel | A ready-to-use primer pool for targeted amplification of 203 genes associated with childhood and young adult cancers. |
| AmpliSeq Library PLUS | Master mix containing enzymes and buffers for the PCR-based construction of sequencing libraries. |
| AmpliSeq CD Indexes | Unique nucleotide sequences (barcodes) used to label individual samples, enabling multiplexed sequencing. |
| AmpliSeq cDNA Synthesis for Illumina | Reagents for converting total RNA to cDNA, a mandatory step prior to library prep when using RNA samples. |
| AmpliSeq Library Equalizer for Illumina | A bead-based solution for normalizing library concentrations, ensuring balanced representation of samples in a sequencing run. |
| AmpliSeq for Illumina Direct FFPE DNA | Enables DNA preparation from FFPE tissues within the AmpliSeq protocol, bypassing the need for deparaffinization or DNA purification. |
Discussion
The drive for genomic reproducibility necessitates rigorous evaluation of every step in the sequencing workflow, from sample input to computational analysis [11]. As the performance data indicates, modern library preparation methods like the Watchmaker workflow offer significant gains in speed and data quality, which directly contribute to more consistent results by reducing technical artifacts like high duplication rates and inefficient rRNA depletion [12]. Furthermore, the AmpliSeq panel's integrated system, when used with its specified reagent toolkit (Table 3), provides a standardized path to minimize inter-experimental variation.
A critical, often overlooked, aspect of reproducibility is the computational analysis. Bioinformatics tools can both remove and introduce unwanted variation. For instance, the consistency of read alignment tools like BWA-MEM can be affected by the order of reads, and variant callers may produce different results on technical replicates, especially in complex genomic regions [11]. This underscores the importance of using simulation tools like GENOMICON-Seq to benchmark bioinformatics pipelines against a known ground truth before applying them to real clinical data [13]. By understanding the impact of parameters such as polymerase error, input copy number, and sequencing depth (Table 2), researchers can proactively design experiments and analytical thresholds that enhance the reliability of their findings.
In conclusion, achieving reproducible DNA and RNA results in childhood cancer research is a multi-faceted challenge. It requires the synergistic combination of optimized wet-lab protocols, robust and integrated reagent systems, and a rigorous, simulation-informed bioinformatic approach.
Implementing the Panel: Standardized Protocols for Reproducible DNA and RNA Library Preparation and Sequencing
A critical factor in the success of next-generation sequencing (NGS) is the quality and quantity of nucleic acid input. This guide objectively compares the performance of the AmpliSeq for Illumina Childhood Cancer Panel, which specifies 10 ng of high-quality DNA or RNA, against other common targeted sequencing and whole transcriptome methods. The data presented herein, framed within the broader thesis of ensuring reproducible research results, provides scientists with the evidence needed to select the appropriate methodology for their sample type and research goals.
Input Specifications and Performance Comparison
The table below summarizes the key input specifications and performance characteristics of the AmpliSeq Childhood Cancer Panel alongside other commonly used methods.
Table 1: Comparison of Input Specifications and Performance Across Methods
| Method | Recommended Input | Hands-On Time | Assay Time | Key Performance Characteristics | Best for Reproducibility When: |
|---|---|---|---|---|---|
| AmpliSeq Childhood Cancer Panel [1] | 10 ng DNA or RNA | < 1.5 hours | 5-6 hours | Constant gene detection across inputs (100-100K cells); high alignment rates (81-92%) [14]. | Working with limited, low-input, or FFPE samples and require consistent target coverage. |
| AmpliSeq Custom DNA Panel [15] | 1–100 ng (10 ng recommended) | 1.5 hours | ~5 hours | Flexible, targeted design for specific genes or regions. | Studying non-standard gene sets or species with constrained sample material. |
| SMARTer Ultra-Low Input RNA-Seq [14] | Varies by cell count | Not Specified | Not Specified | Decreasing detected genes with lower input; higher PCR duplication rates at low inputs [14]. | RNA quantity is not a limiting factor and detection of non-coding genes is required. |
| Illumina DNA Prep [16] | 100-500 ng (Large Genomes) | ~2 hours | 3-4 hours | Robust whole-genome or whole-exome sequencing. | High-quality, abundant DNA is available for broad genomic applications. |
| Illumina Nextera XT [16] | 1 ng | Not Specified | 5.5 hours | Very low input DNA requirement for WGS. | Dealing with extremely low DNA amounts for de novo assembly or WGS. |
Experimental Protocols for Key Comparisons
Protocol: Evaluating Ultra-Low Input RNA Sequencing Methods
A 2019 study directly compared the performance of AmpliSeq technology with SMARTer-based methods at progressively lower cell inputs, providing critical data on reproducibility at the limits of detection [14].
- Sample Preparation: Primary human naïve CD4 T cells were purified from healthy donors and activated. Cells were serially diluted to achieve inputs of 100,000, 5,000, 1,000, and 100 cells [14].
- RNA Extraction: RNA was extracted using the Qiagen RNeasy micro kit, which was validated to provide the lowest CT values and highest consistency across donors, especially at the 100-cell input [14].
- Library Preparation:
- SMART Protocol: Libraries were prepared using SMART-Seq v4 (Clontech) with two different Illumina-compatible library prep methods: Nextera (SMARTNxt) and Clontech's own low-input protocol (SMARTCC).
- AmpliSeq Protocol: Libraries were prepared using the targeted AmpliSeq transcriptome approach.
- Sequencing and Analysis: Libraries were sequenced. For a fair comparison, an equal number of total aligned reads (~17 million) was used per sample. The number of detected genes (count > 0), alignment rates, and PCR duplication rates were calculated and compared across inputs and methods [14].
Protocol: Comprehensive Evaluation of AmpliSeq Transcriptome
A 2015 study performed a comprehensive comparison of the AmpliSeq whole-transcriptome method against traditional RNA-seq on Illumina HiSeq and Ion Torrent Proton platforms [17].
- Sample Types:
- Reference RNAs: Agilent Universal Human Reference RNA (UHRR) and Ambion Human Brain Reference RNA (HBRR).
- Biological Samples: RNA from human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs).
- Library Preparation:
- AmpliSeq: 10 ng of total RNA was converted to cDNA, followed by targeted amplification of over 20,000 human RNA targets using a single primer pool.
- RNA-seq: Methods used were poly-A enrichment for Illumina and ribosomal RNA depletion for Proton.
- Data Analysis: The log2 fold change of expression between UHRR and HBRR was compared across methods using Pearson correlation. Overall performance was assessed using ROC curves, Matthew’s correlation coefficient, and RMSD. Global expression patterns in hiPSC-CMs were evaluated with clustering and principal component analysis (PCA) [17].
Experimental Workflow and Logical Pathway
The following diagram illustrates the logical pathway and key decision points for optimizing nucleic acid input in targeted sequencing, based on the experimental data.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents for Nucleic Acid Input Optimization and Library Preparation
| Reagent Solution | Core Function | Role in Ensuring Reproducibility |
|---|---|---|
| AmpliSeq Library PLUS Kit [1] [15] | Provides core reagents for PCR-based library construction. | Standardizes the library prep process across samples, minimizing technical variation. |
| AmpliSeq CD Indexes [1] [15] | Unique barcodes to label individual samples pre-pooling. | Enables high-plex multiplexing, reduces batch effects, and allows precise sample tracking. |
| AmpliSeq for Illumina Direct FFPE DNA [1] | Prepares DNA from FFPE tissues without deparaffinization or purification. | Recovers reproducible data from challenging, clinically relevant sample types. |
| AmpliSeq cDNA Synthesis for Illumina [1] | Converts total RNA to cDNA for use with RNA panels. | Provides a standardized, efficient starting point for RNA-based targeted sequencing. |
| AmpliSeq Library Equalizer for Illumina [1] | Normalizes libraries post-preparation. | Ensures balanced representation of each library in the final pool, preventing read-depth bias. |
| Qiagen RNeasy Micro Kit [14] | Column-based purification of high-quality RNA from limited samples. | Delivers consistent RNA yield and purity from low inputs, as validated in performance studies. |
| Quantitative PCR (qPCR) Assay [18] [19] | Accurately quantifies amplifiable library fragments and determines optimal PCR cycles. | Prevents over- or under-cycling during library amplification, minimizing artifacts and duplicates. |
The choice of library preparation method and adherence to its input specifications are fundamental to achieving reproducible results. The AmpliSeq Childhood Cancer Panel, optimized for 10 ng of input, demonstrates a key advantage in low-input scenarios where other methods falter. The targeted amplicon approach maintains a consistent number of detected genes even down to 100-cell inputs, whereas whole-transcriptome methods like SMARTer show a significant drop in detected genes and a dramatic increase in PCR duplication rates [14]. Furthermore, in a comprehensive evaluation, the AmpliSeq whole-transcriptome method showed a strong correlation (Pearson’s r = 0.92) with traditional RNA-seq in differential gene expression analysis, confirming its accuracy and reliability [17].
For researchers focused on reproducibility in childhood cancer research or any field with limited sample material, the evidence strongly supports the use of targeted AmpliSeq panels. Its robust performance with low and challenging sample types, combined with a fast, streamlined workflow, makes it a superior choice for generating reliable and comparable data across experiments and laboratories.
In targeted next-generation sequencing (NGS), the journey from nucleic acids to a sequenced library is a critical determinant of data quality and reproducibility. For research applications such as the AmpliSeq Childhood Cancer Panel, a meticulously optimized and consistent library preparation workflow is paramount for generating reliable, comparable results across experiments and laboratories. This guide details the comprehensive library preparation process, from cDNA synthesis to final library pooling, while objectively comparing the performance of the AmpliSeq method against alternative approaches. Framed within the broader context of reproducibility in cancer research, we provide the experimental protocols, quantitative data, and key insights necessary for researchers and drug development professionals to make informed decisions.
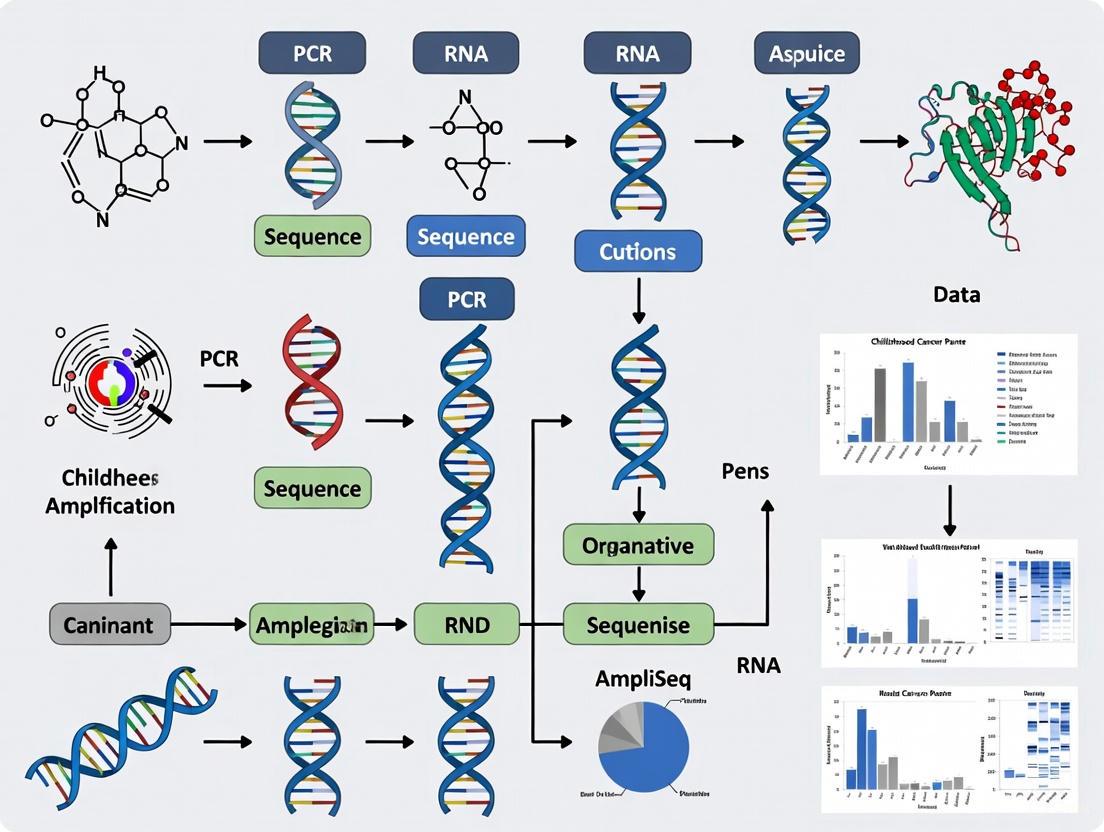
The Library Preparation Workflow: A Step-by-Step Guide
The process of creating a sequencing-ready library involves a series of precise molecular biology steps. The following diagram outlines the complete workflow for the AmpliSeq Childhood Cancer Panel, which processes DNA and RNA in parallel from a single sample.
Workflow Overview: The process begins with a paired sample, from which 10 ng of DNA and 10 ng of RNA are used as input [1]. The RNA undergoes reverse transcription to complementary DNA (cDNA) using the AmpliSeq cDNA Synthesis kit, a critical step for converting RNA targets into an amplifiable DNA format [20] [1]. Both the DNA and the synthesized cDNA then enter the targeted amplification step, where a multiplexed polymerase chain reaction (PCR) uses numerous primer pairs to simultaneously amplify the 203 genes of interest, generating thousands of amplicons [1]. The subsequent step involves a partial enzymatic digestion to cleave the primer sequences from the amplicons. This is followed by the ligation of Illumina P5 and P7 flow cell attachment sequences and the addition of unique index sequences (barcodes) to each sample, enabling multiplexing [20]. Finally, libraries are purified, quantified, normalized to ensure equimolar representation, and pooled in a recommended 5:1 DNA-to-RNA volume ratio before sequencing [20].
Technology Comparison and Performance Data
Selecting an appropriate library prep method is fundamental to experimental success. The table below compares the AmpliSeq for Illumina Custom DNA Panel with two other common Illumina methods, highlighting key specifications that impact reproducibility and practical application.
| Feature | AmpliSeq for Illumina | Nextera Rapid Capture | Nextera XT |
|---|---|---|---|
| Description | Targeted study of genes/regions with high accuracy [21] | Custom workflow for enrichment of targeted content [21] | Prepares libraries for amplicons with minimal hands-on time [21] |
| Method | Amplicon [21] | Enrichment [21] | Amplicon [21] |
| Input Amount | 1–100 ng (10ng per pool recommended) [21] | 50 ng DNA [21] | 1 ng DNA [21] |
| FFPE Compatible | Yes [21] | No [21] | No [21] |
| Multiplexing | Up to 96-plex [21] | Up to 96-plex [21] | Up to 96-plex [21] |
| Hands-On Time | < 1.5 hours [1] | Not Specified | ~15 minutes [21] |
Performance Analysis and Key Differentiators
- Input Material and Sample Type: AmpliSeq's compatibility with formalin-fixed, paraffin-embedded (FFPE) tissue is a significant advantage in clinical cancer research, where such archived samples are a primary source of material [21]. Furthermore, its ability to work with both DNA and RNA from the same low input (10 ng) streamlines the analysis of paired samples [1].
- Workflow Efficiency: The AmpliSeq Childhood Cancer Panel boasts a hands-on time of less than 1.5 hours, significantly less than many traditional methods [1]. This streamlined process reduces opportunities for manual error, directly enhancing reproducibility.
- Targeted Accuracy: As an amplicon-based method, AmpliSeq demonstrates high sensitivity for detecting variants, including SNPs, indels, CNVs, and gene fusions, from minimal input [1]. This contrasts with enrichment-based methods like Nextera Rapid Capture, which may have different performance characteristics for specific variant types.
Reproducibility in Focus: Experimental Evidence
Reproducibility—the ability of a bioinformatics tool or experimental method to maintain consistent results across technical replicates—is a cornerstone of reliable genomics [9]. Factors such as library storage time and input quantity are potential sources of variation.
Impact of Library Storage and Input Quantity
A foundational study investigated the impact of several sample preparation factors on RNA-seq results. The key findings are summarized below.
Experimental Protocol: This study used the mRNA TruSeq v.2 kit (Illumina) to prepare libraries from RNA isolated from human primary B and CD4+ cells [22]. To test the effect of input RNA, titrations of 1 μg, 500 ng, 250 ng, and 100 ng of the same sample RNA were used to construct cDNA libraries [22]. For storage time, original cDNA libraries were compared to the same libraries after three years of storage at -80°C [22]. Bioinformatics analysis involved aligning reads to the GRCh38 genome with HISAT2 and performing differential expression analysis with edgeR [22].
Conclusion: The study found that variations in input RNA quantity and extended library storage time did not significantly alter overall gene transcriptional expression profiles [22]. This evidence strongly supports the robustness of well-standardized NGS library prep protocols against these technical variables, a principle that extends to targeted panels like AmpliSeq when protocols are rigorously followed.
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful library preparation relies on a suite of specialized reagents. The following table details the key components required for the AmpliSeq Childhood Cancer Panel workflow.
| Item Name | Function | Specifications |
|---|---|---|
| AmpliSeq Childhood Cancer Panel | Ready-to-use primer pool for amplifying 203 target genes associated with pediatric cancers [1]. | 24 reactions per kit [1]. |
| AmpliSeq Library PLUS for Illumina | Core library preparation reagents for amplification, digestion, ligation, and purification [1]. | Available in 24-, 96-, and 384-reaction configurations [20]. |
| AmpliSeq CD Indexes | Unique nucleotide barcodes (indexes) added to each sample for multiplexing [1]. | Sold in sets (A, B, C, D); each set contains 96 unique 8 bp indexes [1]. |
| AmpliSeq cDNA Synthesis for Illumina | Converts total RNA to cDNA for subsequent amplification in RNA panels [1]. | Required for RNA input; number of reactions varies by panel [1]. |
| AmpliSeq Library Equalizer | Bead-based normalization solution to ensure equimolar library pooling [1]. | Simplifies and standardizes the final, critical step before sequencing [1]. |
The path from cDNA synthesis to a pooled, indexed library is a finely tuned sequence of molecular events that forms the foundation of any robust NGS study. The AmpliSeq for Illumina Childhood Cancer Panel exemplifies a modern targeted approach, offering a streamlined workflow, low input requirements, and compatibility with challenging but clinically vital sample types like FFPE. As the experimental evidence demonstrates, the reproducibility of results—even across variables like input quantity and storage time—is achievable with standardized, kit-based methods. For researchers pursuing discoveries in childhood cancers, a deep understanding of this process is not merely technical; it is a prerequisite for generating the high-quality, reliable data that drives scientific progress and drug development forward.
Within the context of reproducibility research for the AmpliSeq for Illumina Childhood Cancer Panel, selecting an appropriate sequencing platform is a critical methodological consideration. This pan-cancer targeted panel analyzes 203 genes associated with childhood and young adult cancers, detecting single nucleotide variants (SNVs), insertions-deletions (indels), copy number variants (CNVs), and gene fusions from DNA and RNA in a single assay [1] [5]. The reproducibility of its results is foundational to its clinical utility in refining diagnosis, prognosis, and therapeutic strategies for pediatric acute leukemia [5]. This guide objectively compares the performance of the MiSeq, NextSeq, and MiniSeq systems—three commonly used Illumina benchtop sequencers—for this specific application, providing supporting experimental data and structured protocols to inform platform selection and ensure reliable, repeatable outcomes.
Sequencing Platform Comparison and Performance Specifications
Key performance metrics for the MiSeq, NextSeq, and MiniSeq systems differ significantly, directly impacting throughput, run time, and project scalability. These differences must be aligned with the specific data and sample throughput needs of a research or clinical study.
Table 1: Key Performance Metrics for Benchtop Sequencing Systems [23] [24] [25]
| Specification | MiSeq Series | MiSeqDx | NextSeq 550 System | MiniSeq System |
|---|---|---|---|---|
| Maximum Output | 0.3-15 Gb | 0.3-15 Gb | 20-120 Gb | 1.65-7.5 Gb |
| Maximum Reads per Run | 1-25 million | 1-25 million | 130-400 million | 8-25 million |
| Maximum Read Length | 2 × 300 bp | 2 × 300 bp | 2 × 150 bp | 2 × 150 bp |
| Typical Run Time | 5-55 hours | 4-55 hours | 11-29 hours | 4-24 hours |
| Recommended Application Throughput | Low to mid | Low to mid | Mid to high | Low |
Table 2: Supported Applications and Key Differentiating Factors [23] [1] [24]
| Feature | MiSeq Series | NextSeq 550 System | MiniSeq System |
|---|---|---|---|
| Officially Supported for Childhood Cancer Panel? | Yes (MiSeq, MiSeqDx in Research Mode) [1] | Yes [1] | Yes [1] |
| Chemistry | 4-color SBS [26] | 2-color SBS [26] | 2-color SBS [27] |
| Typical Use Case | Targeted gene sequencing, small genome sequencing, amplicon sequencing [24] | Exome sequencing, transcriptome sequencing, large targeted panels [24] | Targeted gene sequencing [25] |
| Key Consideration for Reproducibility | The 4-color chemistry is considered the "gold standard" and may introduce fewer batch effects compared to 2-color systems when mixing data [26]. | Data from 2-color chemistry should not be naively combined with 4-color data in a single analysis without batch effect correction [26]. | Ideal for low-plex targeted studies but is scheduled to be obsolete, with orders ending in 2025 [25]. |
Experimental Protocols for Platform Validation and Reproducibility
A critical study validating the AmpliSeq Childhood Cancer Panel on the MiSeq system provides a robust experimental framework for assessing platform performance and ensuring reproducible results [5]. The following detailed methodology can be adapted as a template for qualifying any of the three platforms for this specific panel.
Sample Selection and Nucleic Acid Preparation
- Commercial Controls: Utilize commercially available reference standards to establish baseline performance. The validation study used:
- SeraSeq Tumor Mutation DNA Mix: A multiplex biosynthetic mixture of DNA variants at approximately 10% variant allele frequency (VAF) to assess DNA sensitivity and specificity.
- SeraSeq Myeloid Fusion RNA Mix: A mixture of synthetic RNA fusions (e.g., ETV6::ABL1, RUNX1::RUNX1T1) to assess RNA fusion detection capability.
- Negative controls: DNA from NA12878 and RNA from IVS-0035 [5].
- Patient Samples: Select patient samples with high-quality nucleic acids. The validation study used 76 pediatric patients with acute leukemia, prioritizing samples with high DNA and RNA quality [5].
- Nucleic Acid QC: Integrity and purity are paramount.
- Purity: Determine using spectrophotometry (e.g., NanoDrop); all samples should have an OD260/280 ratio >1.8 [5] [28].
- Integrity: Assess using automated electrophoresis systems (e.g., Agilent Bioanalyzer or TapeStation). For RNA, a RNA Integrity Number (RIN) > 7 is recommended [5] [28]. Intact total RNA should show sharp 28S and 18S ribosomal bands with a 2:1 intensity ratio [28].
- Quantification: Use fluorometric methods (e.g., Qubit Fluorimeter) for accurate concentration measurement, as spectrophotometry can be influenced by contaminants [5] [28].
Library Preparation and Sequencing
The following protocol is adapted from the panel's manufacturer and the cited validation study [1] [5].
- Library Preparation: Perform using the AmpliSeq for Illumina Childhood Cancer Panel kit per manufacturer's instructions.
- Input: Use 100 ng of DNA and 100 ng of RNA (converted to cDNA) per sample.
- Amplicon Generation: The panel generates 3,069 DNA amplicons and 1,701 RNA amplicons.
- Indexing: Use unique barcodes for each sample to enable multiplexing.
- Library Pooling and Loading: After individual library QC, pool DNA and RNA libraries at a 5:1 ratio (DNA:RNA). The final pool is diluted to an appropriate loading concentration (e.g., 17–20 pM) for sequencing [5].
- Sequencing: Load the normalized pool onto the chosen platform (MiSeq, NextSeq, or MiniSeq) using a flow cell and reagent kit compatible with the desired output and read length. The validation study used a MiSeq system [5].
Data Analysis and Performance Qualification
- Sequencing Metrics: The validation study established the following key performance metrics for the panel, which can be used as benchmarks [5]:
- Mean Read Depth: > 1000x.
- Sensitivity: 98.5% for DNA variants at 5% VAF; 94.4% for RNA fusions.
- Specificity: 100% for both DNA and RNA.
- Reproducibility: 100% for DNA and 89% for RNA.
- Performance Qualification (PQ): For ongoing quality assurance, Illumina offers Performance Qualification services. These services run comprehensive, audit-ready protocols to verify that each system functions according to pre-set performance specifications, which is crucial for maintaining reproducibility after major repairs or at regular intervals [29].
Figure 1: Experimental workflow for reproducible sequencing with the AmpliSeq Childhood Cancer Panel, highlighting critical quality control checkpoints.
The Scientist's Toolkit: Essential Reagents for the Childhood Cancer Panel Workflow
Table 3: Key Research Reagent Solutions for the AmpliSeq Childhood Cancer Panel Workflow [1]
| Item | Function | Catalog ID Example |
|---|---|---|
| AmpliSeq for Illumina Childhood Cancer Panel | Core panel for investigating 203 genes; sufficient for 24 samples. | 20028446 |
| AmpliSeq Library PLUS | Reagents for preparing sequencing libraries; sold in 24, 96, or 384 reactions. | 20019101 |
| AmpliSeq CD Indexes | Unique barcodes for multiplexing samples; multiple sets (A-D) are available. | 20019105 |
| AmpliSeq cDNA Synthesis for Illumina | Converts total RNA to cDNA, required for RNA input into the panel. | 20022654 |
| AmpliSeq for Illumina Direct FFPE DNA | Prepares DNA from FFPE tissues without need for deparaffinization or purification. | 20023378 |
| AmpliSeq Library Equalizer for Illumina | Beads and reagents for normalizing libraries prior to pooling and sequencing. | 20019171 |
The choice between MiSeq, NextSeq, and MiniSeq systems for running the AmpliSeq Childhood Cancer Panel involves a direct trade-off between throughput, runtime, and data compatibility. The MiSeq system, with its 4-color chemistry and proven track record in targeted sequencing, is often the preferred platform for ensuring maximum reproducibility, particularly for studies where data may be combined from multiple runs or sites [5] [26]. The NextSeq 550 system offers a powerful solution for higher-throughput laboratories but requires careful attention to potential batch effects if combining its 2-color data with MiSeq data [26]. Researchers should note that the MiniSeq system is scheduled for obsolescence, making it a less future-proof investment despite its suitability for low-plex targeted studies [25].
For research focused on the reproducibility of AmpliSeq Childhood Cancer Panel results, the following recommendations are critical:
- Standardize the Platform: For a single study, consistently use one platform and reagent kit version to minimize technical variation.
- Implement Rigorous QC: Adhere to strict nucleic acid quality controls (RIN > 7, fluorometric quantification) as detailed in the experimental protocol [5] [28].
- Use Reference Materials: Incorporate well-characterized positive and negative controls in every run to monitor assay performance and validate sensitivity and specificity metrics [5].
- Perform Regular Qualification: Utilize services like Illumina Performance Qualification (PQ) to ensure the sequencing instrument itself continues to perform to specifications over time, which is a cornerstone of reproducible science [29].
Reproducibility forms the cornerstone of reliable scientific research, particularly in clinical genomics where diagnostic and treatment decisions hinge on consistent results. For researchers using targeted sequencing panels like the AmpliSeq for Illumina Childhood Cancer Panel, achieving reproducibility requires precise optimization of library preparation parameters, specifically the DNA:RNA pooling ratios and sequencing depth. This guide examines the experimental data supporting specific protocol configurations that ensure optimal coverage and reproducible detection of somatic variants, gene fusions, and other clinically relevant alterations in pediatric cancer samples. The integration of both DNA and RNA analysis in a single workflow presents unique challenges for standardization, making the establishment of validated protocols particularly critical for multi-center studies and clinical implementation.
Technical Specifications of the AmpliSeq Childhood Cancer Panel
The AmpliSeq for Illumina Childhood Cancer Panel is a targeted resequencing solution designed for comprehensive evaluation of pediatric and young adult cancers. Its technical profile supports the simultaneous analysis of multiple variant types from minimal input material, making it suitable for diverse sample types commonly encountered in pediatric oncology research.
Table 1: AmpliSeq Childhood Cancer Panel Technical Specifications
| Parameter | Specification | Relevance to Reproducibility |
|---|---|---|
| Genes Targeted | 203 genes associated with childhood cancer [1] | Standardized target region enables consistent coverage across runs |
| Input Requirement | 10 ng high-quality DNA or RNA [1] | Minimizes sample quality issues that affect reproducibility |
| Assay Time | 5-6 hours (library prep only) [1] | Streamlined workflow reduces technical variability |
| Variant Types Detected | SNPs, indels, CNVs, gene fusions, somatic variants [1] | Comprehensive profiling with standardized methodologies |
| Compatible Systems | MiSeq, NextSeq 500/1000/2000, MiniSeq [1] | Flexibility across Illumina platforms maintains result consistency |
The panel employs a PCR-based amplification approach that generates 3,069 DNA amplicons and 1,701 RNA amplicons, with average sizes of 114 bp and 122 bp respectively [5]. This targeted design is particularly suited for pediatric leukemias, which characteristically have a low mutational burden but clinically relevant alterations [5].
Optimized DNA:RNA Pooling Ratios for Integrated Analysis
The ratio at which DNA and RNA libraries are pooled prior to sequencing significantly impacts the balance of genomic and transcriptomic information obtained. Experimental validation studies have identified optimal ranges for this critical parameter.
Empirically Validated Pooling Ratio
A comprehensive validation study of the AmpliSeq Childhood Cancer Panel established a 5:1 DNA:RNA pooling ratio as optimal for balanced variant detection [5]. In this protocol, final libraries were diluted to 2 nM, after which DNA and RNA libraries were pooled at this specific ratio before sequencing on a MiSeq instrument [5]. This ratio prioritizes genomic coverage while maintaining sufficient transcriptomic data for fusion detection, reflecting the panel's design emphasis on both DNA mutations and RNA fusions relevant to pediatric cancers.
Impact on Assay Performance
The 5:1 ratio demonstrated high sensitivity in validation studies, achieving 98.5% for DNA variants (at 5% variant allele frequency) and 94.4% for RNA fusions [5]. The balance also supported robust reproducibility, with 100% reproducibility for DNA and 89% for RNA findings [5]. This ratio effectively accommodates the typically lower representation of RNA fragments in combined library preparations while ensuring adequate coverage for fusion detection.
Recommended Sequencing Depth for Comprehensive Coverage
Sequencing depth fundamentally determines the confidence of variant calls and the comprehensiveness of genomic coverage. The specialized requirements of pediatric cancer research necessitate specific depth considerations.
Depth Requirements for Variant Detection
Validation studies for the Childhood Cancer Panel utilized a mean read depth greater than 1000×, which proved sufficient for reliable detection of diverse variant types [5]. This depth exceeds typical whole-genome sequencing recommendations (30-100×) due to the targeted nature of the panel and the need to detect low-frequency variants in heterogeneous cancer samples.
Table 2: Recommended Sequencing Depth by Variant Type
| Variant Type | Recommended Depth | Rationale | Supporting Evidence |
|---|---|---|---|
| SNVs/Indels | >1000× mean depth | Enables detection of variants with 5% VAF [5] | 98.5% sensitivity achieved in validation [5] |
| Gene Fusions | Sufficient coverage at fusion junctions | Critical for detecting low-expression fusions [5] | 94.4% sensitivity for fusion detection [5] |
| Copy Number Variants | Consistent coverage across targets | Reduces false-positive CNV calls [2] | Robust CNV detection in pediatric tumors [2] |
Coverage Uniformity and Quality Metrics
Beyond raw depth, coverage uniformity across targeted regions is equally critical for reproducibility. The AmpliSeq panel validation demonstrated that the obtained depth provided 95.6% concordance for single nucleotide variants (SNVs) compared to orthogonal methods, indicating excellent coverage uniformity [5]. For clinical applications, the panel achieved 100% specificity, confirming that the combination of depth and uniformity minimizes false positives [5].
Experimental Protocols for Reproducible Results
Standardized experimental protocols are essential for maintaining reproducibility across different laboratories and sample batches. The following methodologies are supported by empirical validation data.
Library Preparation Workflow
The recommended protocol begins with 100 ng of input DNA and 100 ng of input RNA, which is reverse transcribed to cDNA using the AmpliSeq cDNA Synthesis kit [5]. Amplicon libraries are generated through consecutive PCRs with sample-specific barcodes. After quality control checks, libraries are cleaned up and quantified, then diluted to 2 nM before employing the critical 5:1 DNA:RNA pooling ratio [5]. The final pool is diluted to 17-20 pM for sequencing on MiSeq or compatible Illumina platforms [5].
Quality Control Checkpoints
Rigorous quality control is embedded throughout the workflow to ensure reproducible outcomes. DNA and RNA purity should be verified with OD260/280 ratio >1.8, while integrity is assessed via Labchip or TapeStation [5]. Post-sequencing, quality metrics include mapping rates (70-90% expected for human genome), read distribution across genomic features, and coverage uniformity [30]. For the Childhood Cancer Panel, validation studies established a 98.5% sensitivity threshold for DNA variants at 5% VAF as a key quality benchmark [5].
Performance Comparison with Alternative Approaches
Understanding how the optimized AmpliSeq protocol compares to other NGS approaches provides context for its reproducibility advantages in pediatric cancer research.
Comparison with Other NGS Methods
The AmpliSeq Childhood Cancer Panel's performance can be contrasted with other NGS approaches used in pediatric oncology:
Table 3: Performance Comparison with Alternative NGS Methods
| Method | Sensitivity for DNA Variants | Sensitivity for RNA Fusions | Reproducibility | Input Requirements |
|---|---|---|---|---|
| AmpliSeq Childhood Cancer Panel (with 5:1 pooling) | 98.5% (5% VAF) [5] | 94.4% [5] | 100% DNA, 89% RNA [5] | 10 ng DNA/RNA [1] |
| OncoKids Panel | Not specified in results | Not specified in results | Robust performance [2] | 20 ng DNA/RNA [2] |
| Simul-seq Method (WGS/WTS) | 95.6% SNV concordance [31] | High-quality transcriptome data [31] | Comparable to biological replicates [31] | 50 ng total nucleic acid [31] |
| Standard RNA-seq | Not primary focus | Dependent on depth and coverage [30] | Varies with protocol [30] | Varies by protocol |
Impact on Clinical Utility
The reproducibility of the optimized AmpliSeq protocol directly translates to clinical impact. In validation studies, 49% of mutations and 97% of fusions identified had demonstrable clinical impact, with 41% of mutations refining diagnosis and 49% considered targetable [5]. Overall, the panel produced clinically relevant results in 43% of patients tested in the validation cohort [5], demonstrating how standardized protocols enhance clinical utility.
Essential Research Reagent Solutions
Implementing reproducible NGS library preparation requires specific reagent systems designed to maintain consistency throughout the workflow.
Table 4: Essential Research Reagents for Library Preparation
| Reagent Solution | Function | Role in Reproducibility |
|---|---|---|
| AmpliSeq Library PLUS | PCR-based library preparation | Standardized amplification across samples [1] |
| AmpliSeq CD Indexes | Sample multiplexing | Enables batch processing without cross-sample contamination [1] |
| AmpliSeq Library Equalizer | Library normalization | Ensures balanced representation in pooled libraries [1] |
| AmpliSeq cDNA Synthesis | RNA to cDNA conversion | Maintains transcript representation integrity [1] |
| AmpliSeq Direct FFPE DNA | DNA from FFPE tissues | Standardizes challenging sample types [1] |
The experimental data supporting a 5:1 DNA:RNA pooling ratio combined with >1000× mean read depth establishes a validated standard for reproducible research using the AmpliSeq Childhood Cancer Panel. This optimized protocol demonstrates that precise technical configurations directly enable high sensitivity (98.5% for DNA variants), specificity (100%), and clinical utility (43% of patients) in pediatric cancer genomics [5]. As the field moves toward increasingly integrated genomic and transcriptomic profiling, such standardized approaches will be essential for multi-center research collaborations and the translation of NGS findings into clinical practice. The ongoing development of automated library preparation systems [32] [33] [34] promises to further enhance reproducibility by reducing manual intervention and variability, ultimately advancing the precision oncology paradigm for childhood cancers.
Optimizing Performance and Troubleshooting Common Challenges in DNA and RNA Analysis
In genomic research, particularly in oncology, the ability to accurately detect low-frequency variants and gene fusions is paramount for understanding cancer heterogeneity, minimal residual disease, and early treatment response. The challenge intensifies when working with degraded samples from formalin-fixed, paraffin-embedded (FFPE) tissue or limited biopsy material, where nucleic acid quantity and quality are suboptimal. Within this context, genomic reproducibility—defined as the ability of bioinformatics tools to maintain consistent results across technical replicates—becomes a critical benchmark for evaluating any sensitive detection method [11].
Targeted sequencing approaches, such as the AmpliSeq for Illumina Childhood Cancer Panel, offer a balanced solution for comprehensive genomic evaluation of pediatric and young adult cancers. This panel targets 203 genes associated with childhood cancers while requiring only 10 ng of input DNA or RNA and featuring less than 1.5 hours of hands-on time [1]. However, achieving reliable detection of variants at low variant allele frequencies (VAFs) down to 5% and accurate fusion calling requires careful consideration of both wet-lab and computational methodologies. This guide objectively compares the performance of various approaches within the critical framework of experimental reproducibility.
Wet-Lab Methodologies: Foundation of Sensitive Detection
Targeted Amplicon Sequencing for Limited Samples
Amplicon-based next-generation sequencing (NGS) methods provide a robust approach for mutation detection in samples with limited quantity, a common scenario in clinical practice. The principal advantage of this technology is its minimal input requirement—as little as 10 ng of nucleic acid—enabling analysis of over 95% of samples compared to higher-input methods that may fail in 20-30% of cases due to quantity not sufficient (QNS) status [35].
The AmpliSeq Childhood Cancer Panel employs multiplex PCR for library preparation, generating amplicons that cover genes associated with leukemias, brain tumors, sarcomas, and other pediatric cancers [1]. This targeted approach demonstrates particular strength for known fusion detection, with reported positive predictive value (PPV) of 100% for intergenic fusions across thousands of cases [35]. The tradeoff, however, is limited capability to detect novel fusion partners not explicitly targeted by the panel design.
Unique Molecular Identifiers for Ultra-Sensitive Detection
For detection of variants below 1% VAF, Unique Molecular Identifier (UMI) technologies provide enhanced error correction capabilities. UMIs are short random oligonucleotide sequences that label individual DNA molecules before amplification, enabling bioinformatic distinction between true variants and artifacts introduced during PCR or sequencing [36] [37].
Table 1: Comparison of UMI-Based vs. Raw-Reads-Based Variant Calling
| Feature | UMI-Based Methods | Raw-Reads-Based Methods |
|---|---|---|
| Theoretical Detection Limit | 0.025% VAF [37] | 0.05%-1% VAF [37] |
| Error Correction Mechanism | Molecular barcoding with consensus building | Statistical modeling of sequencing errors |
| Input Requirements | Typically higher due to UMI incorporation | Lower, more flexible |
| Best-Performing Tools | DeepSNVMiner, UMI-VarCal [37] | LoFreq, Pisces [37] |
| Sensitivity/Precision at 0.1% VAF | 88%/100% (DeepSNVMiner) [37] | <50% with high false positives [37] |
The enhanced sensitivity of UMI-based approaches comes with increased complexity and cost. However, for applications requiring detection of ultra-rare variants, such as monitoring clonal evolution or early resistance mutations, this investment is justified.
Bioinformatics Strategies: From Raw Data to High-Confidence Calls
Low-Frequency Variant Calling Tools
Multiple bioinformatic tools have been developed specifically for low-frequency variant detection, each employing distinct statistical approaches to distinguish true biological variants from technical artifacts.
Table 2: Performance Comparison of Low-Frequency Variant Callers
| Variant Caller | Type | Detection Limit | Sensitivity at 0.1% VAF | Precision at 0.1% VAF | Key Algorithm |
|---|---|---|---|---|---|
| DeepSNVMiner | UMI-based | 0.025% | 88% | 100% | UMI family consensus with strand bias filter [37] |
| UMI-VarCal | UMI-based | 0.1% | 84% | 100% | Poisson statistical test with position-specific errors [37] |
| LoFreq | Raw-reads | 0.05% | <50% | Moderate | Bernoulli trial with base quality integration [37] |
| Pisces | Raw-reads | 0.05% | <50% | Moderate | Q-score based on Poisson model [37] |
| MAGERI | UMI-based | 0.1% | Low | High | Beta-binomial modeling of UMI groups [37] |
| smCounter2 | UMI-based | 0.5% | Low | High | Beta-binomial distribution for non-reference UMIs [37] |
UMI-based callers generally outperform raw-reads-based callers, particularly at VAFs below 1%. However, factors beyond sheer sensitivity must be considered, including computational resources, analysis time, and compatibility with existing workflows.
Fusion Detection and Validation Pipeline
Gene fusions represent critical driver events in many childhood cancers, requiring specialized detection approaches. A robust fusion validation pipeline integrates evidence from both RNA and DNA sequencing data to maximize confidence in fusion calls [38].
Diagram 1: Integrated RNA-DNA Fusion Validation Pipeline. This workflow combines the transcriptomic evidence from RNA-Seq with genomic breakpoint validation in WGS data to identify high-confidence fusion events [38].
The fusion validation approach depicted in Diagram 1 demonstrates how leveraging matched whole-genome sequencing (WGS) data can confirm fusion transcripts identified through RNA-Seq. This method focuses computational resources on specific genomic regions of interest, significantly improving both speed and sensitivity compared to genome-wide structural variant detection tools like Manta and BreakDancer [38].
Experimental Protocols for Robust Detection
Library Preparation for Low-Input Samples
The AmpliSeq for Illumina Childhood Cancer Panel protocol requires 5-6 hours for library preparation (excluding quantification and normalization), with less than 1.5 hours of hands-on time [1]. For optimal performance with low-input samples:
Input Quantity: Use 10 ng of high-quality DNA or RNA as standard; the panel can work with inputs as low as 1 ng when necessary [1] [39]
FFPE Samples: Employ AmpliSeq for Illumina Direct FFPE DNA to prepare DNA from unstained, slide-mounted FFPE tissues without deparaffinization or DNA purification [1]
RNA Considerations: When working with RNA targets, use AmpliSeq cDNA Synthesis for Illumina to convert total RNA to cDNA before library preparation [1]
Library Normalization: Utilize AmpliSeq Library Equalizer for consistent library normalization, critical for reproducible results across sequencing runs [1]
Sequencing Configuration for Sensitivity
Achieving 5% VAF detection requires sufficient sequencing depth to ensure statistical confidence in variant calls:
Coverage Depth: Target minimum 500× coverage for reliable detection of variants at 5% VAF [39]
Instrument Selection: The panel is compatible with MiSeq, NextSeq, and MiniSeq systems; for larger studies, NextSeq 550/1000/2000 systems enable 48 samples per run at 500× coverage [1] [39]
Quality Control: Implement rigorous QC metrics including sample-to-sample contamination checks using the AmpliSeq for Illumina Sample ID Panel, which targets validated SNPs [1]
Bioinformatics Parameters for Low VAF Detection
When analyzing sequencing data for low-frequency variants:
Variant Filtering: For UMI-based data, apply strand bias filters and homopolymer region filters to reduce false positives [37]
VAF Thresholds: Set appropriate VAF thresholds based on validated limits of detection for your specific variant caller; for 5% VAF detection, most tools perform excellently, but precision decreases significantly below 0.5% for raw-reads-based callers [37]
Visual Validation: Implement integrative genomics viewers for manual inspection of putative low-frequency variants, particularly those near known problematic genomic regions
Research Reagent Solutions
Table 3: Essential Research Reagents for Sensitive Detection
| Reagent / Product | Function | Application in Sensitive Detection |
|---|---|---|
| AmpliSeq Childhood Cancer Panel [1] | Targeted primer panel | Investigates 203 genes associated with childhood cancers with minimal input requirements |
| AmpliSeq Library PLUS [1] | Library preparation reagents | Provides enzymes and buffers for PCR-based library construction |
| AmpliSeq CD Indexes [1] | Sample barcoding | Enables multiplexing of up to 384 samples, reducing batch effects |
| AmpliSeq cDNA Synthesis for Illumina [1] | RNA to cDNA conversion | Essential for RNA-based fusion detection from low-quality inputs |
| AmpliSeq Direct FFPE DNA [1] | DNA preparation from FFPE | Enables analysis of archival specimens without DNA purification |
| AmpliSeq Library Equalizer [1] | Library normalization | Ensures balanced representation in pooled libraries |
Performance Comparison and Reproducibility Considerations
Technology Tradeoffs in Clinical Context
Each detection technology presents distinct tradeoffs. Amplicon-based approaches like the AmpliSeq Childhood Cancer Panel offer robust detection of known targets with minimal input, but have limitations in detecting novel fusion partners [35]. In comparison, hybrid capture-based methods provide broader coverage but require substantially more input material (typically 100 ng), resulting in higher QNS rates [35].
The missed detection rate for novel fusions with targeted amplicon approaches is estimated at less than 1% of all solid tumor cases, and as low as 0.1% for lung cancers where panel design is most optimized [35]. This must be balanced against the 20-30% of samples that would be inadequate for higher-input methods.
Reproducibility Framework
Within the context of genomic reproducibility, several factors impact the consistency of low VAF and fusion detection:
Technical Replicates: Sequence the same biological sample across multiple library preparations and sequencing runs to assess technical variability [11]
Bioinformatic Consistency: Select tools with deterministic algorithms; stochastic methods can introduce unwanted variation even with identical input data [11]
Background Error Profiling: Characterize platform-specific error rates using control samples to establish baseline expectations [36]
Reproducibility challenges are particularly pronounced for fusion detection, where different algorithms show limited overlap in results [38]. Integrating evidence from multiple callers or orthogonal validation provides the most reliable approach for clinical or research applications.
Achieving sensitive detection of low VAF variants (down to 5%) and gene fusions requires integrated methodological excellence across wet-lab and computational domains. Targeted amplicon sequencing approaches like the AmpliSeq Childhood Cancer Panel provide a robust foundation for known targets with limited samples, while UMI-based methods extend detection limits for ultra-rare variants. Crucially, performance evaluation must occur within a reproducibility framework that acknowledges the inherent tradeoffs between detection breadth, input requirements, and analytical sensitivity. By implementing the strategies and comparisons outlined in this guide, researchers can optimize their experimental and computational pipelines for confident detection of biologically significant low-frequency genomic events.
Mitigating Contamination and Managing Low-Diversity Amplicon Libraries
The reproducibility of research using targeted sequencing panels, such as the AmpliSeq Childhood Cancer Panel, hinges on effectively managing contamination and analyzing low-diversity amplicon libraries. These challenges become particularly acute in low-biomass environments or when target input is limited, where contaminating DNA can constitute a substantial proportion of sequencing data and dramatically impact analytical outcomes [40] [41]. In clinical genomics, where the AmpliSeq panel identifies diagnostic, prognostic, and therapeutic markers in pediatric acute leukemia, failure to mitigate contamination risks false-positive variant calls or obscured true signals, directly affecting patient management decisions [5].
Contamination concerns extend beyond human DNA to include microbial sources in microbiome studies and cross-sample contamination in high-throughput sequencing workflows. Even following best-practice guidelines that reduce contamination by over 90%, residual contaminants can still influence results, particularly in differential abundance analyses [40]. The research community has responded with stringent guidelines focusing on every study stage, from initial sample collection through data analysis and reporting, especially for low-biomass systems [41]. This guide objectively compares contemporary methodologies for contamination mitigation, providing experimental data and protocols to enhance the reproducibility and reliability of amplicon sequencing in critical research applications.
Comparative Analysis of Contamination Mitigation Approaches
Methodologies and Performance Metrics
We evaluated three primary contamination mitigation strategies—preventative wet-lab practices, bioinformatic subtraction, and CRISPR-based depletion—focusing on their impact on data integrity, practical implementation, and suitability for different research contexts. The table below summarizes the comparative performance of these approaches based on published validations and experimental data.
Table 1: Performance Comparison of Contamination Mitigation Methods
| Method Category | Key Examples | Efficiency (% Contaminant Removal) | Impact on True Signals | Implementation Complexity | Best-Suited Applications |
|---|---|---|---|---|---|
| Preventative Wet-Lab Practices | Sterile technique, UV sterilization, Bleach treatment, PPE usage [41] | >90% reduction achievable [40] | Minimal risk of true signal loss | Moderate (requires rigorous discipline) | All low-biomass studies; essential foundation |
| Bioinformatic Subtraction | Decontam, MicroDecon, background subtraction using negative controls [40] [41] | Varies with tool and parameters; can be highly effective | Risk of false negatives if overly aggressive [40] | Low to Moderate (computational) | Post-hoc correction; studies with appropriate controls |
| CRISPR-Based Depletion | Cas-16S-seq (for host depletion in plant studies) [42] | 63.2% to 2.9% in root samples; 99.4% to 11.6% in phyllosphere [42] | Minimal off-target when gRNAs are specific [42] | High (requires protocol optimization) | Studies with abundant predictable contaminants (e.g., host DNA) |
Impact on Statistical Outcomes and Data Interpretation
The influence of contamination on downstream statistical analyses varies significantly across methodological approaches. Quantitative assessments demonstrate that while contamination has minimal impact on weighted beta diversity metrics, it substantially alters the number of differentially abundant taxa when at least 10 contaminant sequences are present [40]. The effect increases with contamination levels and affects choice of differential abundance tool performance, with DESeq2 outperforming ANCOM-BC under stochastically distributed contamination [40].
Notably, the primary drivers of statistical outcomes in microbiome studies remain group dissimilarity and the number of unique taxa, with contamination playing a secondary role primarily affecting the magnitude rather than direction of findings [40]. When validated protocols with internal negative controls are implemented, residual contamination rarely determines whether microbiome differences are detected between groups, though it may affect the number of differentially abundant taxa identified [40].
Experimental Protocols for Contamination Control
Standardized Preventive Protocol for Low-Biomass Samples
Based on consensus guidelines for low-biomass microbiome studies, the following protocol establishes minimal standards for preventing contamination during sample processing [41]:
Sample Collection and Handling:
- Decontaminate all equipment, tools, vessels, and gloves with 80% ethanol (to kill microorganisms) followed by a nucleic acid degrading solution (e.g., sodium hypochlorite, UV-C exposure, or commercial DNA removal solutions) to eliminate residual DNA [41].
- Use single-use DNA-free consumables whenever possible. For reusable equipment, employ sterilization methods such as autoclaving followed by DNA removal treatments.
- Implement appropriate personal protective equipment (PPE) including gloves, face masks, and clean suits to limit human-derived contamination from skin cells, aerosols, or hair [41].
Negative Control Implementation:
- Process multiple negative controls alongside experimental samples, including:
- Extraction controls (blank extraction reagents)
- Amplification controls (water blanks)
- Sampling controls (empty collection vessels, air swabs) [41]
- Sequence these controls in the same run as experimental samples to identify contaminant sequences for bioinformatic removal.
Laboratory Workflow:
- Maintain physical separation of pre- and post-amplification activities to prevent amplicon contamination.
- Use dedicated equipment and reagents for low-biomass samples, particularly when working with clinical specimens requiring high sensitivity like the AmpliSeq Childhood Cancer Panel [5].
CRISPR/Cas9-Mediated Host DNA Depletion (Cas-16S-seq)
For studies plagued by abundant host DNA contamination (e.g., plant microbiota, human tissue samples), the Cas-16S-seq method provides targeted depletion [42]:
gRNA Design and Validation:
- Establish a bioinformatics pipeline to design guide RNAs (gRNAs) targeting host 16S rRNA genes (e.g., mitochondrial or chloroplast) without bacterial off-targets.
- Screen candidate gRNAs against comprehensive 16S rRNA databases (e.g., RDP, SILVA, GreenGenes) to ensure specificity.
- Select gRNAs targeting regions containing PAM sites (5'-NGG-3') unique to host sequences.
Wet-Lab Implementation:
- Perform first-round PCR amplification of 16S rRNA genes using universal primers with appropriate adaptors.
- Treat PCR products with Cas9 nuclease complexed with host-specific gRNAs to cleave host-derived amplicons.
- Conduct second-round index PCR to amplify remaining (bacterial) amplicons for sequencing.
- Validate method effectiveness using artificially mixed communities and experimental samples, comparing to standard protocols.
Performance Assessment:
- For rice microbiota profiling, Cas-16S-seq reduced host sequences from 63.2% to 2.9% in root samples and from 99.4% to 11.6% in phyllosphere samples while increasing bacterial species detection without introducing amplification bias [42].
Visualizing Contamination Mitigation Workflows
Integrated Strategy for Contamination Management
The following diagram illustrates the comprehensive workflow for mitigating contamination across experimental phases, from sample collection to data interpretation:
Integrated Contamination Management Workflow
CRISPR/Cas9 Depletion Mechanism
The Cas-16S-seq method specifically targets host-derived contamination while preserving bacterial signals, as visualized below:
CRISPR/Cas9 Host DNA Depletion Mechanism
Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for Contamination Mitigation
| Reagent/Material | Function | Implementation Example | Considerations for Reproducibility |
|---|---|---|---|
| DNA Decontamination Solutions | Degrades contaminating DNA on surfaces and equipment | Sodium hypochlorite (bleach), commercial DNA removal solutions [41] | Effectiveness varies by formulation; validate concentration and exposure time |
| Ultra-Clean Consumables | Pre-introduction of contaminants during sample processing | DNA-free tubes, filters, and collection vessels [41] | Lot-to-lot variability requires verification with negative controls |
| Personal Protective Equipment (PPE) | Reduces human-derived contamination | Gloves, masks, clean suits, hair nets [41] | Proper donning procedures critical; minimize skin and aerosol exposure |
| Negative Control Materials | Identifies contamination sources and levels | Blank extraction reagents, sterile swabs, air exposure plates [41] | Must mirror sample processing exactly; multiple controls recommended |
| CRISPR/Cas9 Reagents | Targeted depletion of specific contaminating sequences | Cas9 nuclease, host-specific gRNAs [42] | gRNA specificity must be validated against target database to prevent off-target effects |
| Amplification Reagents | PCR-based target enrichment | AmpliSeq Childhood Cancer Panel primers [5] | Low DNA input (20 ng) requires optimized master mixes to maintain sensitivity |
Effective contamination management requires integrated strategies spanning preventive measures, wet-lab depletion technologies, and bioinformatic corrections. The reproducibility of AmpliSeq Childhood Cancer Panel results and similar targeted sequencing applications depends on recognizing that while contamination cannot be entirely eliminated, its impacts can be minimized and accurately accounted for in data interpretation [40] [5]. Quantitative evidence confirms that with appropriate controls and validated protocols, residual contamination rarely determines whether significant differences are detected between experimental groups, though it may affect the number of differentially abundant taxa identified [40].
The research community's move toward standardized reporting of contamination control measures, as outlined in recent consensus statements [41], will enhance cross-study comparability and methodological transparency. As novel approaches like CRISPR-based depletion mature [42], they offer promising avenues for further improving signal-to-noise ratios in challenging samples. By implementing the compared methodologies with appropriate consideration of their strengths and limitations, researchers can significantly enhance the reliability and reproducibility of their amplicon sequencing data, ultimately strengthening conclusions in critical research areas such as pediatric cancer diagnostics.
The reproducibility of research findings, particularly in molecular diagnostics using targeted panels like the AmpliSeq for Illumina Childhood Cancer Panel, is fundamentally dependent on sample quality. Next-generation sequencing (NGS) has redefined diagnostic and therapeutic strategies for cancers, including pediatric leukemias, by allowing parallel analysis of numerous genes and alteration types [5]. However, the inherent challenges of common specimen types—Formalin-Fixed Paraffin-Embedded (FFPE) tissues, bone marrow, and blood—can introduce pre-analytical variables that jeopardize data integrity. This guide objectively compares best practices for these sample types, providing structured experimental data and protocols to uphold the reproducibility of NGS results.
Understanding Sample-Specific Challenges and Quality Control
Each sample type presents unique biochemical and physical challenges that can degrade nucleic acid quality and impact downstream sequencing.
- FFPE Tissues: The formalin fixation process causes RNA fragmentation and cross-linking, while long-term storage can lead to nucleic acid degradation [43]. The paraffin embedding process also introduces contaminants that can inhibit enzymatic reactions during library preparation.
- Bone Marrow Aspirates (BMA): The quality of BMA is highly technique-dependent. A "dry tap" or a "bloody tap" (dilute aspirate) can yield insufficient material for analysis. Specimens are highly susceptible to clotting if not immediately treated with appropriate anticoagulants like EDTA or sodium heparin [44]. Morphological assessment becomes unreliable if EDTA-fixed aspirates are processed beyond 2 hours [44].
- Blood Specimens: Blood contains high concentrations of ribonucleases (RNases) that rapidly degrade RNA upon collection; 99% of free RNA can be degraded within 15 seconds of exposure to plasma [45]. Furthermore, globin mRNA and ribosomal RNA (rRNA) can constitute up to 80% and 90% of total RNA respectively, consuming valuable sequencing reads and reducing gene detection sensitivity [45].
Quantitative Quality Control (QC) Thresholds
The table below summarizes the minimum recommended QC metrics for each specimen type to ensure successful sequencing with the AmpliSeq Childhood Cancer Panel.
Table 1: Quality Control Thresholds for Different Specimen Types
| Specimen Type | Key QC Metric | Minimum Recommended Threshold | Impact on Sequencing |
|---|---|---|---|
| FFPE | RNA Concentration [43] | 25 ng/µL | Library preparation failure |
| Pre-capture Library Qubit [43] | 1.7 ng/µL | Inadequate sequencing data | |
| Bone Marrow | Aspirate Morphology [44] | Process within 2 hours of collection | Artificial dysplastic features |
| Clot Specimen [44] | Fixed in formalin | Suitable for IHC; not for flow cytometry | |
| Whole Blood | RNA Stabilization [45] | Use PAXgene or Tempus tubes | Prevents massive RNA degradation |
| Globin Depletion [45] | Use globin mRNA removal protocols | Increases gene detection rates by >30% |
Best Practices for Sample Processing and Workflows
Adherence to standardized protocols from collection to nucleic acid extraction is critical for maintaining sample integrity and ensuring reproducible NGS data.
FFPE Specimen Protocols
For FFPE samples, the choice of library preparation protocol significantly influences data quality. A comparative study evaluated two common methods:
- TruSeq RNA Exome (Illumina): This protocol involves library preparation without fragmentation, followed by exome capture. It is designed to be compatible with degraded RNA from FFPE samples.
- NEBNext rRNA Depletion (New England Biolabs): This protocol uses ribosomal RNA depletion to enrich for mRNA and may include fragmentation based on the RNA Integrity Number (RIN).
Table 2: Comparison of FFPE RNA-Seq Library Prep Protocols
| Protocol | Key Feature | Performance in FFPE |
|---|---|---|
| TruSeq RNA Exome | Exome capture post-library prep | Demonstrated better performance in alignment rates, SNP concordance, and sample-wise correlation [43]. |
| NEBNext rRNA Depletion | Ribosomal RNA removal | An alternative method; performance relative to exome capture may vary with FFPE RNA degradation levels [43]. |
Methodology: In a pilot study, seven paired FFPE and fresh frozen (FFzn) samples from benign breast disease patients were processed using both protocols. Bioinformatics metrics, including alignment statistics, SNP concordance with whole exome sequencing (WES) data, junction coverage, and sample-wise correlation, were used for evaluation [43].
Bone Marrow and Blood Workflows
For bone marrow and blood, the immediate stabilization of nucleic acids is the most critical step.
Bone Marrow Laboratory Evaluation [44]:
- Site: Posterior iliac crest is preferred.
- Aspiration (BMA): Perform first to avoid a bloody tap. Prepare smears immediately at the bedside.
- Biopsy (BMB): Obtain a solid core after aspiration from the same entry site.
- Anticoagulants:
- Sodium Heparin: For aspirates destined for flow cytometry.
- EDTA: For molecular studies and aspirate smears for morphological analysis.
Whole Blood RNA-Seq Best Practice [45] [46]:
- Collection: Draw blood directly into RNA-stabilizing tubes (e.g., PAXgene or Tempus).
- RNA Extraction: Use silica-membrane columns or acidic phenol/chloroform.
- DNase Treatment: Strongly recommended due to high DNA content in blood cells.
- RNA Pre-treatment: Deplete globin mRNA and rRNAs to significantly increase gene detection rates and free up sequencing space.
Impact of Sample Quality on Experimental Reproducibility
Variations in sample handling introduce significant technical noise, which can obscure biological signals and compromise the reproducibility of the AmpliSeq Childhood Cancer Panel results.
Temporal Stability of Blood Specimens
Research demonstrates that blood sample storage time directly impacts transcriptome profiles. One study found that the number of differentially expressed genes (DEGs) increased with longer storage times of peripheral blood mononuclear cells (PBMCs) at room temperature [46]. While the total number of detected genes may not change significantly, the expression levels of specific genes can shift, potentially mimicking disease-associated signatures if not controlled for.
Methodology: PBMCs from healthy donors were stored at room temperature for 0, 1, 4, and 8 hours post-collection before RNA extraction. Whole blood was also stored at 4°C for 0, 4, 8, 24, and 32 hours. All RNA samples underwent high-throughput sequencing, and DEG analysis was performed comparing each time point to the baseline (0h) [46].
Performance of the AmpliSeq Childhood Cancer Panel
The validated performance of a sequencing panel under optimal conditions establishes a benchmark for assessing the impact of sample quality.
Methodology: The AmpliSeq Childhood Cancer Panel was technically validated using commercial control samples. The panel targets 203 genes, analyzing gene fusions, SNVs, InDels, and CNVs. Key performance metrics were assessed [5]:
- Sensitivity/Specificity: Evaluated using commercial positive controls (SeraSeq Tumor Mutation DNA Mix and Myeloid Fusion RNA Mix) and negative controls.
- Limit of Detection (LOD): Determined by testing variants at different allele frequencies.
- Reproducibility: Assessed via replicate experiments.
Table 3: Validation Metrics of the AmpliSeq Childhood Cancer Panel
| Metric | DNA (SNVs/InDels) | RNA (Fusions) |
|---|---|---|
| Mean Read Depth | >1000x [5] | >1000x [5] |
| Sensitivity | 98.5% (at 5% VAF) [5] | 94.4% [5] |
| Specificity | 100% [5] | 100% [5] |
| Reproducibility | 100% [5] | 89% [5] |
| Clinical Utility | 49% of mutations had clinical impact [5] | 97% of fusions had clinical impact [5] |
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 4: Key Reagents for Quality NGS Sample Preparation
| Reagent / Kit | Function | Application / Note |
|---|---|---|
| PAXgene Blood RNA Tube | Stabilizes RNA at collection | Inactivates RNases in whole blood; critical for accurate transcriptomics [45]. |
| TruSeq RNA Library Prep for Enrichment | FFPE RNA-Seq library prep | Optimized for degraded RNA; used with exome capture [43]. |
| RiboCop HMR+Globin / Globin Block | Depletes globin mRNA & rRNA | Frees sequencing space; significantly boosts gene detection in blood RNA-Seq [45]. |
| AmpliSeq for Illumina Childhood Cancer Panel | Targeted NGS sequencing | Analyzes 203 genes for fusions, SNVs, InDels, CNVs; validated for low input (20ng DNA/RNA) [5]. |
| Qubit Fluorometer & RNA HS Assay | Accurate RNA quantification | Preferable for library prep over UV-spectrophotometry due to higher accuracy with degraded samples. |
The journey to reproducible AmpliSeq Childhood Cancer Panel results begins the moment a sample is collected. There is no universal "best" sample type; rather, the optimal choice is guided by the clinical question, followed by the rigorous application of specimen-specific best practices. For FFPE samples, this means adhering to strict RNA input concentrations and choosing the right library protocol. For bone marrow, it requires meticulous attention to collection order and timing. For blood, immediate RNase inactivation and globin depletion are non-negotiable. By integrating the standardized protocols, quality thresholds, and essential tools outlined in this guide, researchers and clinicians can significantly reduce pre-analytical variability, thereby ensuring that the powerful genetic data generated translates into reliable diagnoses, prognoses, and therapeutic strategies for patients.
Utilizing Automated Library Preparation and Normalization Kits for Enhanced Reproducibility
Reproducibility forms the cornerstone of reliable scientific research, particularly in clinical genomics where diagnostic and treatment decisions depend on accurate, repeatable results. Technical variability in next-generation sequencing (NGS) workflows—from library preparation through data analysis—poses significant challenges for cross-laboratory consistency. This comparison guide objectively evaluates automated library preparation and normalization technologies designed to enhance reproducibility, with specific application to the AmpliSeq for Illumina Childhood Cancer Panel. We focus on performance metrics, experimental methodologies, and integrated workflows that help researchers and drug development professionals achieve more reliable, consistent genomic data in pediatric cancer research.
Library Preparation Kit Performance Comparison
Comprehensive Performance Metrics for Library Preparation Kits
Table 1: Comparative performance of library preparation kits for low-input and standard RNA/DNA applications
| Kit Name | Input Range | Hands-on Time | Total Workflow Time | Key Performance Metrics | Reproducibility Assessment |
|---|---|---|---|---|---|
| AmpliSeq for Illumina Childhood Cancer Panel | 10 ng DNA/RNA | <1.5 hours | 5-6 hours (library prep only) | Sensitivity: DNA 98.5% (5% VAF), RNA 94.4%; Specificity: 100% [5] | High reproducibility for DNA; 89% for RNA fusion detection [5] |
| Illumina TruSeq Stranded mRNA | 50-500 ng total RNA | ~3-4 hours | 9 hours | Pearson correlation >0.97 with reference methods [47] | High inter-laboratory consistency for protein-coding genes [48] |
| Swift RNA Library Prep | 10-100 ng total RNA | ~1.5 hours | 4.5 hours | Fewest DEGs attributable to input amount; >80% uniquely mapped reads [47] | High agreement with reference datasets (correlation >0.97) [47] |
| Swift Rapid RNA Library Prep | 50-200 ng total RNA | ~1 hour | 3.5 hours | Equivalent library complexity to TruSeq; uniform coverage [47] | Consistent performance across input amounts [47] |
| SMART-Seq v4 Ultra Low Input RNA | 250 pg-4 ng total RNA | ~2 hours | ~6 hours | Spearman correlation >0.8 with TruSeq standards [49] | Suitable for RiboTag-IP samples with intronic read retention [49] |
Normalization Kit Performance and Specifications
Table 2: Comparison of library normalization technologies for NGS workflows
| Normalization Kit | Technology Principle | Input Compatibility | Recovery Output | Hands-on Time | Integration with Automation |
|---|---|---|---|---|---|
| Auto-Mag DNA Normalization Kit | Magnetic bead limited binding capacity | gDNA, PCR products, NGS libraries | ~400 ng (standard), ~200 ng (alternate) | ~30 minutes | Full compatibility [50] |
| QIAseq Universal Normalizer Kit | Modified primers with magnetic bead chemistry | All Illumina libraries with intact P5/P7 | 4 nmol/L | ~30 minutes | Designed for automated workflows [51] |
| AmpliSeq Library Equalizer for Illumina | Bead-based normalization | AmpliSeq libraries | Sequencing-ready pools | <30 minutes | Optimized for AmpliSeq workflows [1] |
| Illumina DNA Prep Built-in Normalization | Bead-linked transposome saturation | gDNA for WGS and targeted | Consistent coverage | Minimal additional time | Fully integrated [52] |
Experimental Protocols and Methodologies
Validation Protocol for the AmpliSeq Childhood Cancer Panel
The technical validation and clinical utility study of the AmpliSeq for Illumina Childhood Cancer Panel provides a comprehensive methodological framework for assessing reproducibility [5]. The experimental protocol encompasses:
Sample Selection and Controls:
- Commercial controls: SeraSeq Tumor Mutation DNA Mix (v2 AF10 HC) and SeraSeq Myeloid Fusion RNA Mix
- Negative controls: NA12878 (DNA) and IVS-0035 (RNA)
- Patient samples: 76 pediatric patients with BCP-ALL (n=51), T-ALL (n=11), and AML (n=14)
Library Preparation Methodology:
- Input: 100 ng DNA for DNA libraries, 100 ng RNA for RNA libraries (converted to cDNA)
- Amplicon generation: 3,069 amplicons per DNA sample, 1,701 amplicons per RNA sample
- Library pooling: DNA and RNA libraries pooled at 5:1 ratio
- Sequencing: MiSeq platform with 17-20 pM final concentration
Quality Metrics and Analysis:
- Sensitivity and specificity calculations using commercial controls
- Limit of detection (LOD) establishment for variant allele frequencies
- Reproducibility assessment through replicate testing
- Clinical impact evaluation by expert review
Low-Input RNA-Seq Comparative Analysis Protocol
The systematic comparison of strand-specific RNA-seq library preparation methods for low input samples provides a robust experimental design for evaluating performance across kits [47]. Key methodological aspects include:
Sample Design:
- Universal Human Reference RNA (UHRR) from 10 human cancer cell lines
- Input amounts spanning manufacturer recommendations: 10-500 ng total RNA
- Replication: 5 samples per condition for statistical power
Library Preparation Conditions:
- Three methods tested: Illumina TruSeq stranded mRNA, Swift RNA, Swift Rapid RNA
- Strand-specificity mechanisms: dUTP labeling (TruSeq) vs. Adaptase technology (Swift kits)
- mRNA enrichment: oligo(dT) selection for all kits
- Sequencing depth: 20 million reads per library, subsampled to 10 million for analysis
Quality Assessment Metrics:
- Mapping efficiency (>80% threshold)
- Gene detection counts (12,000 protein-coding genes expected)
- Ribosomal RNA contamination (<1% threshold)
- Strand specificity (>90% correct strand mapping)
- Library complexity measurements
Diagram 1: NGS workflow with key reproducibility factors. Experimental variability arises from multiple technical aspects including input amount, library preparation method, and normalization chemistry, impacting final analytical reproducibility.
Reproducibility Assessment Frameworks
Multi-Center Benchmarking Approaches
Large-scale consortium studies have developed comprehensive frameworks for assessing reproducibility across laboratories. The Quartet project and MAQC consortium provide robust methodologies for evaluating technical performance [48]:
Reference Materials Design:
- Quartet RNA samples: Four related cell lines with subtle biological differences
- MAQC samples: Large biological differences (cancer cell lines vs. brain tissue)
- ERCC spike-in controls: 92 synthetic RNAs with known concentrations
- Defined mixture samples: T1 (3:1) and T2 (1:3) for ratio-based assessment
Performance Metrics Suite:
- Signal-to-Noise Ratio (SNR): Based on principal component analysis
- Absolute expression accuracy: Correlation with TaqMan reference datasets
- Differential expression accuracy: Detection of known DEGs
- Technical reproducibility: Inter- and intra-laboratory concordance
Experimental Diversity:
- 45 independent laboratories
- 26 different experimental processes
- 140 bioinformatics pipelines
- 1080 RNA-seq libraries total
Key Reproducibility Findings from Multi-Center Studies
The Quartet project revealed critical insights about reproducibility challenges in real-world settings [48]:
Inter-laboratory Variability:
- SNR values varied widely: 0.3-37.6 for Quartet samples vs. 11.2-45.2 for MAQC samples
- Greater inter-laboratory variations in detecting subtle differential expressions
- 17 out of 45 laboratories had SNR values <12 for Quartet samples, indicating quality issues
Major Variability Sources:
- Experimental factors: mRNA enrichment method and strandedness protocols
- Bioinformatics factors: Gene annotation sources, alignment tools, and normalization methods
- Batch effects: Significant impact when samples processed separately
Research Reagent Solutions Toolkit
Table 3: Essential research reagents and their functions in automated library preparation workflows
| Reagent/Kits | Primary Function | Compatibility | Key Features for Reproducibility |
|---|---|---|---|
| AmpliSeq Library PLUS | PCR-based library construction | AmpliSeq panels | Consistent amplicon generation across samples [1] |
| AmpliSeq CD Indexes | Sample multiplexing | Illumina systems | Unique dual indexes to reduce index hopping [1] |
| Auto-Mag DNA Normalization Beads | Magnetic bead normalization | Various DNA types | Limited binding capacity for consistent recovery [50] |
| QIAseq Normalizer Primer Mix | Library modification for normalization | Illumina libraries | Modified primers for bead-based quantification [51] |
| AmpliSeq cDNA Synthesis for Illumina | RNA to cDNA conversion | AmpliSeq RNA panels | High-efficiency reverse transcription [1] |
| AmpliSeq Direct FFPE DNA | DNA from FFPE tissues | AmpliSeq panels | Bypasses deparaffinization and purification [1] |
| Illumina DNA/RNA UD Indexes | Unique dual indexing | Illumina DNA Prep | Complete removal of index hopping effects [52] |
| ERCC RNA Spike-In Mix | Process controls | RNA-seq workflows | 92 synthetic RNAs for technical monitoring [48] |
Integrated Workflow for Enhanced Reproducibility
Diagram 2: Integrated automated workflow for the AmpliSeq Childhood Cancer Panel. This unified approach incorporates simultaneous processing of DNA and RNA samples with integrated normalization to minimize technical variability and enhance reproducibility across experimental batches.
Based on the comprehensive comparison of automated library preparation and normalization technologies, several best practices emerge for enhancing reproducibility in genomic research, particularly with the AmpliSeq Childhood Cancer Panel:
Experimental Design Recommendations:
- Implement reference materials (Quartet, MAQC, or ERCC spike-ins) in each batch
- Use unique dual indexes to minimize index hopping and sample cross-talk
- Maintain consistent input quantities within study cohorts when possible
- Process cases and controls simultaneously to reduce batch effects
Technology Selection Guidelines:
- For low-input samples (<10 ng), consider SMART-Seq v4 or Swift RNA kits
- For standardized clinical applications, TruSeq and AmpliSeq provide highest reproducibility
- Integrate bead-based normalization for consistent library representation
- Utilize stranded protocols to resolve overlapping gene expression
Quality Control Metrics:
- Establish minimum thresholds for sensitivity (>95%) and specificity (>99%)
- Require >80% mapping efficiency for RNA-seq experiments
- Monitor ribosomal RNA contamination (<1% for polyA-selected libraries)
- Verify inter-laboratory correlation coefficients >0.9 for reference materials
The integration of automated library preparation systems with bead-based normalization technologies significantly enhances reproducibility across sequencing batches and laboratories. For the AmpliSeq Childhood Cancer Panel specifically, the incorporated validation framework demonstrates clinical-grade reproducibility with 98.5% sensitivity for DNA variants and 94.4% for RNA fusions, establishing a robust foundation for pediatric cancer diagnostics and research applications.
Technical Validation and Comparative Analysis: Performance Metrics Against Established Standards and Alternative Panels
The integration of Next-Generation Sequencing (NGS) into clinical practice has revolutionized the molecular diagnosis of pediatric cancers, which possess distinct genetic landscapes compared to adult malignancies [5]. Analytical validation is a critical prerequisite for clinical implementation, establishing the performance metrics of a diagnostic test. This guide objectively compares the analytical validation results of the AmpliSeq for Illumina Childhood Cancer Panel against other commercially available panels, situating the findings within the broader thesis of reproducible research in genomic assay development [5] [2] [53].
Performance Comparison of Pediatric Cancer NGS Panels
The table below summarizes key analytical validation metrics for three targeted NGS panels designed for pediatric cancers.
| Parameter | AmpliSeq for Illumina Childhood Cancer Panel [5] | OncoKids Panel [2] | CANSeqTMKids Panel [53] |
|---|---|---|---|
| DNA Sensitivity | 98.5% (at 5% VAF) | Robust performance reported | >99% (at 5% Allele Fraction) |
| RNA Sensitivity | 94.4% | Robust performance reported | >99% |
| Specificity | 100% (DNA & RNA) | 100% reported | >99% |
| Reproducibility | 100% (DNA), 89% (RNA) | High reproducibility reported | >99% |
| Limit of Detection (LoD) | 5% VAF for DNA variants | Low input (20 ng DNA/RNA) | 5% AF for SNVs/INDELs |
| Target Coverage | Mean depth >1000x | 44 genes (full coding), 82 hotspots, 24 CNVs | 130 genes (SNV/INDEL), 91 fusions |
| Sample Input | 100 ng DNA & RNA | 20 ng DNA & RNA | As low as 5 ng nucleic acid |
Key Insights from Comparative Data
- The AmpliSeq panel demonstrates high sensitivity and perfect specificity, with its validation study detailing performance against specific variant allele frequencies [5].
- The CANSeqTMKids panel also reports >99% sensitivity and specificity, achieving this with a lower required nucleic acid input, which can be crucial for precious pediatric samples [53].
- All three panels are designed to target the unique genomic features of childhood cancers, supporting their use in comprehensive molecular profiling [5] [2] [53].
Experimental Protocols for Key Validation Experiments
The high performance claims for these panels are underpinned by rigorous experimental validation. The following workflow details the key steps in the analytical validation process for the AmpliSeq panel [5].
Detailed Methodologies
The validation of the AmpliSeq panel followed a structured approach [5]:
- Sample Selection and Controls: The study used 76 pediatric patient samples (B-ALL, T-ALL, AML) alongside commercial positive and negative controls (SeraSeq Tumor Mutation DNA Mix and Fusion RNA Mix) to assess sensitivity, specificity, and limit of detection [5].
- Library Preparation and Sequencing: Libraries were prepared from 100 ng of DNA and RNA using the AmpliSeq for Illumina Childhood Cancer Panel kit. The pooled libraries were sequenced on an Illumina MiSeq platform, achieving a mean read depth of greater than 1000x [5].
- Orthogonal Confirmation: Results from the NGS panel were confirmed using established conventional techniques, including labeled-PCR for
FLT3-ITDandNPM1, Sanger sequencing forcKITandGATA1, and quantitative RT-PCR for fusion genes [5].
Research Reagent Solutions for NGS Panel Validation
A successful validation study relies on specific, high-quality reagents and materials. The following table details key solutions used in the featured AmpliSeq validation study [5].
| Research Reagent | Function / Purpose | Example Product / Source |
|---|---|---|
| Nucleic Acid Extraction Kits | Isolate high-quality DNA and RNA from diverse sample types. | Gentra Puregene kit (Qiagen), QIAamp DNA Mini Kit, TriPure (Roche) [5] |
| Quantification & QC Instruments | Precisely measure nucleic acid concentration, purity, and integrity. | Qubit 4.0 Fluorometer, Quawell Q5000 UV-Vis, Labchip, TapeStation [5] |
| Targeted NGS Panel | Simultaneously interrogate multiple gene targets for variants and fusions. | AmpliSeq for Illumina Childhood Cancer Panel (203 genes) [5] |
| Commercial Reference Standards | Act as positive controls to establish sensitivity, specificity, and LoD. | SeraSeq Tumor Mutation DNA Mix, SeraSeq Myeloid Fusion RNA Mix [5] |
| Library Prep Chemistry | Generate barcoded, sequencing-ready libraries from input nucleic acids. | AmpliSeq cDNA Synthesis kit, IonCode Barcode Adapters [5] |
| Sequencing Platform | Perform high-throughput sequencing of prepared libraries. | Illumina MiSeq Sequencer [5] |
The analytical validation of the AmpliSeq for Illumina Childhood Cancer Panel demonstrates that it is a highly sensitive, specific, and reproducible tool for clinical genomic profiling in pediatric leukemia [5]. When compared to similar panels like OncoKids and CANSeqTMKids, it holds its own with excellent performance metrics [2] [53].
These validation studies underscore a critical principle in modern science: the verifiability of research claims is paramount [54]. The rigorous, documented methodology behind these panels—from sample preparation and library construction to data analysis—provides a transparent framework that allows other scientists to assess, reproduce, and build upon this work. This commitment to transparency and reproducibility ensures that such advanced genomic tools can be reliably integrated into clinical practice, ultimately guiding personalized treatment decisions for pediatric cancer patients [5] [55].
Reproducibility is a cornerstone of reliable molecular diagnostics, ensuring that sequencing assays deliver consistent results across repeated experiments. For comprehensive genomic profiling in pediatric cancers, where treatment decisions hinge on accurate detection of diverse alterations, demonstrating robust inter-run and intra-run concordance is particularly critical. Targeted next-generation sequencing (NGS) panels must maintain high precision across multiple variant types to guide clinical applications. This guide objectively compares the reproducibility performance of the AmpliSeq for Illumina Childhood Cancer Panel with other DNA and RNA assays, providing researchers with experimental data to inform their selection of appropriate genomic profiling tools.
Comparative Reproducibility Performance of Targeted NGS Panels
The table below summarizes key reproducibility metrics from validation studies of several targeted NGS panels, including the AmpliSeq Childhood Cancer Panel.
Table 1: Reproducibility Metrics for DNA and RNA Targeted Sequencing Panels
| Assay Name | Targeted Variants | Intra-run Reproducibility | Inter-run Reproducibility | Key Findings |
|---|---|---|---|---|
| AmpliSeq for Illumina Childhood Cancer Panel [56] | DNA: SNVs, InDels; RNA: Fusions | DNA: 100%; RNA: 89% | 100% for DNA variants | Demonstrated high sensitivity for DNA (98.5% for variants with 5% VAF) and RNA (94.4%) |
| FoundationOneRNA [57] | RNA: Fusions (318 genes), Gene expression (1521 genes) | 100% for 10 pre-defined fusions | Not explicitly stated | High reproducibility observed across 9 replicates per sample over 3 different days |
| Integrated DNA/RNA Solid Tumor Assay [58] | DNA and RNA: Fusions (16 genes) | 100% for all fusion-positive samples | 100% concordance across three different sequencing runs | CV of FFPM values in RNA assay showed consistent results in repeated experiments |
| OncoKids [2] | DNA: 44 genes (full coding), 82 genes (hotspots), 24 genes (amplifications); RNA: 1421 fusions | Robust performance in reproducibility studies | Robust performance in reproducibility studies | Compatible with low DNA/RNA input (20 ng each); validated with 192 clinical samples |
| Rapid Pan-Heme (RPPH) Assay [59] | DNA: >400 genes (SNVs, InDels, fusions) | Meets NYS CLEP standards | Meets NYS CLEP standards | Achieves stringent analytical sensitivity and reproducibility criteria for regulatory compliance |
Experimental Protocols for Assessing Reproducibility
AmpliSeq Childhood Cancer Panel Validation Protocol
The reproducibility of the AmpliSeq Childhood Cancer Panel was assessed through a rigorous validation protocol [56]. Library preparation was performed using the manufacturer's instructions with 100 ng of DNA and 100 ng of RNA per sample. The DNA component generated 3069 amplicons covering coding regions of multiple genes, while the RNA component targeted 1701 amplicons for fusion detection.
For precision assessment, the validation included:
- Inter-run reproducibility: Testing across multiple sequencing runs
- Intra-run reproducibility: Testing within the same sequencing run
- Sample types: Commercial controls including SeraSeq Tumor Mutation DNA Mix and SeraSeq Myeloid Fusion RNA Mix
- Analysis focus: Fusion genes, single nucleotide variants (SNVs), and insertions/deletions (InDels) in leukemia-related genes
The panel demonstrated 100% reproducibility for DNA variants and 89% reproducibility for RNA fusions, with high sensitivity for DNA (98.5% for variants with 5% variant allele frequency) and RNA (94.4%) [56].
FoundationOneRNA Precision Study Design
The FoundationOneRNA assay employed a comprehensive approach to assess precision [57]:
- Sample preparation: Ten FFPE samples harboring 10 fusions were processed on 3 different days
- Replication scheme: Three replicates per day, for a total of 9 replicates per source sample
- Quality control: All replicates (9 replicates × 10 source samples) passed quality control steps
- Result: 100% reproducibility for all 10 pre-defined target fusions across all replicates
This study design demonstrated the robust reproducibility of fusion detection in the FoundationOneRNA assay across multiple runs and days [57].
Integrated DNA/RNA Solid Tumor Assay Precision Testing
The integrated DNA/RNA solid tumor assay implemented a precision validation protocol to assess both intra-run and inter-run reproducibility [58]:
- Sample selection: Three samples containing (1) a standard FFPE sample with six NTRK fusions, (2) a clinical FFPE sample with EML4::ALK, and (3) a clinical negative FFPE sample
- Intra-run assessment: Performed in triplicate within one sequencing run
- Inter-run assessment: Performed in triplicate across three different sequencing runs
- Quantitative analysis: Calculated coefficient of variation (CV) for allele frequency (DNA) and fusion fragment per million (FFPM) values (RNA)
- Result: Complete concordance of gene fusion results for all samples across different runs with consistent CV values [58]
Research Reagent Solutions for Reproducibility Studies
Table 2: Essential Research Reagents for NGS Reproducibility Studies
| Reagent/Control Type | Specific Examples | Function in Reproducibility Studies |
|---|---|---|
| DNA Reference Standards | SeraSeq Tumor Mutation DNA Mix (v2 AF10 HC) [56] | Provides known DNA variants at specific allele frequencies for sensitivity and reproducibility testing |
| RNA Fusion Controls | SeraSeq Myeloid Fusion RNA Mix [56] | Contains synthetic RNA fusions combined with reference RNA for fusion detection reproducibility |
| Negative Controls | NA12878 (DNA), IVS-0035 (RNA) [56] | Establishes baseline for false positive rates and assay specificity |
| Fusion-Positive Cell Lines | Custom cell lines with known fusions [57] | Enables limit of detection studies and precision assessment across dilution series |
| Library Preparation Kits | AmpliSeq for Illumina Childhood Cancer Panel kit [56] | Standardized reagents for target amplification and library preparation |
| Sequencing Platforms | Illumina HiSeq4000 [57], other Illumina systems | Provides the sequencing engine with consistent output quality |
Workflow Diagrams for Reproducibility Assessment
NGS Reproducibility Validation Workflow
DNA vs. RNA Concordance Relationship
Discussion and Clinical Implications
The reproducibility data presented demonstrates that modern targeted NGS panels can achieve high inter-run and intra-run concordance when properly validated. The AmpliSeq Childhood Cancer Panel shows robust performance for DNA variants (100% reproducibility) with slightly lower but still substantial reproducibility for RNA fusions (89%) [56]. This pattern of high DNA reproducibility with somewhat reduced RNA consistency is observed across multiple platforms, likely reflecting the greater instability of RNA as an analyte and the technical challenges of fusion detection.
The integrated DNA/RNA approach exemplified by the solid tumor assay [58] demonstrates how combining both analytes can achieve 100% reproducibility while compensating for the limitations of each individual method. This complementary approach aligns with expert recommendations that advocate for unified DNA and RNA NGS strategies to maximize detection sensitivity for fusion genes in clinical practice [58].
For pediatric cancer applications, where sample material is often limited, the demonstrated ability of these assays to maintain reproducibility with low input amounts (20-100 ng) [56] [2] is particularly significant. Furthermore, achieving reproducibility metrics that meet stringent regulatory standards such as those from the New York State Department of Health's Clinical Laboratory Evaluation Program [59] underscores the growing maturity of NGS technologies for clinical diagnostics.
When selecting appropriate assays for research or clinical applications, scientists should consider both the demonstrated reproducibility metrics and the validation study designs that support them. The experimental protocols outlined herein provide templates for rigorous assessment of assay precision, enabling informed decisions about technology implementation in precision oncology contexts.
The molecular landscape of pediatric cancers is distinct from adult malignancies, characterized by a lower mutational burden but a higher prevalence of clinically significant structural variants, such as gene fusions [60]. Next-generation sequencing (NGS) panels have become indispensable tools for delineating this landscape, providing critical information for diagnosis, prognosis, and therapeutic targeting. However, the true value of these panels in both research and clinical translation is fundamentally dependent on the reproducibility of their DNA and RNA results. This guide provides a objective comparison of the performance and technical characteristics of two prominent pediatric cancer panels—the AmpliSeq for Illumina Childhood Cancer Panel and Children's Hospital Los Angeles's OncoKids—within the critical context of analytical reproducibility. It is important to note that despite a comprehensive search, no performance or validation data for the "CANSeqTMKids" panel was available in the public domain for inclusion in this comparison.
This section details the fundamental design and technical profiles of the two comparable panels.
AmpliSeq for Illumina Childhood Cancer Panel
The AmpliSeq Childhood Cancer Panel is a targeted resequencing solution from Illumina designed for the comprehensive evaluation of somatic variants associated with childhood and young adult cancers [1]. It is a PCR-based amplicon sequencing assay that simultaneously analyzes 203 genes. Its key technical specifications are summarized in the table below and its integrated workflow is designed to streamline the process from library preparation to analysis [1].
OncoKids
The OncoKids panel was developed at Children's Hospital Los Angeles (CHLA) to address the specific genomic profile of pediatric cancers, which could not be adequately covered by simply modifying adult cancer panels [60]. It is an amplification-based NGS assay that combines DNA and RNA analysis to detect a full spectrum of alterations across pediatric malignancies, including leukemias, sarcomas, and brain tumors [61] [60]. The panel uses the Ion Torrent S5 sequencing platform and is optimized for low input amounts, making it suitable for a variety of sample types, including retrospective analysis of formalin-fixed, paraffin-embedded (FFPE) tissue [61] [60].
Table 1: Core Technical Specifications of Pediatric Cancer NGS Panels
| Feature | AmpliSeq for Illumina Childhood Cancer Panel | OncoKids |
|---|---|---|
| Target Genes | 203 genes [1] | 44 genes with full exon coverage; 82 mutation hotspots; 24 genes for CNVs [61] |
| Variant Types Detected | SNPs, Indels, CNVs, Gene Fusions, Somatic Variants [1] | Mutations, Gene Amplifications, Gene Fusions [61] |
| RNA Fusion Targets | 97 gene fusions [5] [56] | 1,421 targeted gene fusions [61] |
| Nucleic Acid Input | 10 ng DNA or RNA [1] | 20 ng DNA and 20 ng RNA [61] |
| Sample Types | Blood, Bone Marrow, FFPE Tissue [1] | Fresh, Frozen, or FFPE Tissue; Bone Marrow; Peripheral Blood [61] |
| Sequencing Platform | Illumina MiSeq, NextSeq, MiniSeq Systems [1] | Ion Torrent S5 [60] |
| Library Prep Method | PCR-based amplicon [1] | Amplification-based (Ion AmpliSeq) [60] |
Experimental Protocols and Workflow Diagrams
Understanding the detailed experimental protocols is essential for assessing the potential sources of technical variability and ensuring reproducibility.
AmpliSeq Childhood Cancer Panel Workflow
The following diagram and protocol describe the standard operating procedure for the AmpliSeq panel, as utilized in a key validation study [5] [56].
Detailed Methodology [5] [56]:
- Nucleic Acid Extraction & Quantification: DNA and RNA are extracted using kits (e.g., QIAamp DNA Mini Kit, TriPure reagent). Quality control is performed via spectrophotometry (OD260/280 >1.8) and integrity assessment (e.g., Agilent TapeStation). Fluorometric quantification (Qubit Fluorometer) is used for accurate concentration measurement.
- Library Preparation: For DNA, 100 ng is used to generate 3,069 amplicons. For RNA, 100 ng is first reverse-transcribed to cDNA using the AmpliSeq cDNA Synthesis kit, targeting 1,701 amplicons for fusion detection. Amplicon libraries are constructed via consecutive PCRs with sample-specific barcodes.
- Sequencing: Barcoded DNA and RNA libraries are pooled at a 5:1 ratio, diluted to 17–20 pM, and sequenced on an Illumina MiSeq sequencer.
OncoKids Panel Workflow
The OncoKids workflow is designed to be a consolidated test, replacing multiple single-analyte assays.
Key Workflow Characteristics [61] [60]: The OncoKids assay is notable for its very low nucleic acid input requirement (20 ng each of DNA and RNA), making it suitable for limited samples. It uses the Ion AmpliSeq technology on the Ion Torrent S5 platform. A distinctive feature of the OncoKids program is the integrated access to clinical experts for pathology consultations and guidance on further testing, including germline mutation analysis [60].
Comparative Performance and Reproducibility Data
Independent validation studies provide the critical data necessary to objectively compare the analytical performance of these panels.
Quantitative Performance Metrics
Table 2: Analytical Performance Metrics from Validation Studies
| Performance Metric | AmpliSeq for Illumina Childhood Cancer Panel | OncoKids |
|---|---|---|
| Mean Read Depth | >1000x [5] [62] | Not explicitly stated |
| DNA Sensitivity (at 5% VAF) | 98.5% [5] [62] | Robust performance per validation [61] |
| RNA Sensitivity | 94.4% [5] [62] | Robust performance per validation [61] |
| Specificity | 100% (DNA) [5] [62] | Robust performance per validation [61] |
| Reproducibility | 100% (DNA), 89% (RNA) [5] [62] | High reproducibility reported [61] |
| Limit of Detection (LOD) | High sensitivity for variants at 5% VAF [5] | Validated with low input amounts (20 ng) [61] |
| Clinical Impact in Cohort | 43% of patients had clinically relevant findings [5] [62] | Designed to guide diagnosis and treatment [60] |
Analysis of Reproducibility and Real-World Performance
The data in Table 2 highlights several key points regarding reproducibility and utility:
- The AmpliSeq panel has been rigorously validated, demonstrating exceptionally high sensitivity and specificity. The 89% reproducibility for RNA fusion detection, while high, indicates a known area where technical variability can be introduced, potentially during the reverse transcription or cDNA amplification steps. This underscores the importance of strict RNA quality control in the workflow [5] [62].
- A study of 76 pediatric acute leukemia patients using the AmpliSeq panel found that 97% of the identified fusions and 49% of the mutations had a direct clinical impact, refining diagnosis or revealing targetable alterations [5] [56]. This demonstrates a high positive predictive value for clinically actionable results.
- While specific sensitivity/specificity values for OncoKids are not detailed in the available public summaries, its validation involved a large cohort of 192 unique clinical samples across a wide range of pediatric tumor types, confirming its "robust performance" for sensitivity, reproducibility, and limit of detection [61]. Its design as a combined DNA/RNA assay that conserves tissue is a significant practical advantage for the pediatric setting [60].
The Scientist's Toolkit: Essential Research Reagent Solutions
Implementing and validating these NGS panels requires a suite of specialized reagents and controls.
Table 3: Key Reagents and Materials for Panel Implementation
| Item | Function | Example Product |
|---|---|---|
| Positive Control DNA | Assesses assay sensitivity and variant calling accuracy for DNA variants. | SeraSeq Tumor Mutation DNA Mix [5] [56] |
| Positive Control RNA | Validates the entire RNA workflow, from cDNA synthesis to fusion detection. | SeraSeq Myeloid Fusion RNA Mix [5] [56] |
| Negative Control | Identifies background noise, contamination, or false-positive calls. | NA12878 (DNA), IVS-0035 (RNA) [5] [56] |
| Library Prep Kit | Contains reagents for generating sequencing libraries from amplicons. | AmpliSeq Library PLUS for Illumina [1] |
| Index Adapters | Unique barcodes for multiplexing multiple samples in a single sequencing run. | AmpliSeq CD Indexes for Illumina [1] |
| cDNA Synthesis Kit | Converts input RNA to cDNA for subsequent amplification in RNA fusion assays. | AmpliSeq cDNA Synthesis for Illumina [1] |
| FFPE DNA Solution | Enables library construction directly from FFPE tissues without separate DNA purification. | AmpliSeq for Illumina Direct FFPE DNA [1] |
| Fusion Caller Software | Bioinformatics tools for identifying gene fusions from RNAseq data. | Arriba, FusionCatcher, STAR-Fusion, Dragen [63] |
Both the AmpliSeq for Illumina Childhood Cancer Panel and the OncoKids panel demonstrate strong performance as comprehensive tools for the genomic characterization of pediatric cancers. The choice between them may depend on several factors:
- Platform Preference: The decision may be influenced by an institution's existing sequencing infrastructure (Illumina vs. Ion Torrent).
- Evidence Level: Researchers requiring detailed, published validation metrics for reproducibility in leukemia may lean on the AmpliSeq panel, whereas others may value the extensive pediatric focus and integrated clinical consultation offered with OncoKids.
- Workflow Needs: The very low input requirement of OncoKids is advantageous for precious pediatric samples, while the AmpliSeq panel's hands-on time of <1.5 hours offers efficiency [1].
A key future direction is the move beyond targeted panels to genome-wide approaches. Researchers at CHLA have developed an exome capture-based RNA-sequencing assay that integrates four fusion callers to identify novel or unexpected fusions missed by targeted panels, addressing a critical gap where initial targeted testing is non-informative [63]. This highlights that while targeted panels like AmpliSeq and OncoKids offer reproducible and efficient profiling of known targets, the field is advancing towards more agnostic methods to fully capture the complex genomic landscape of childhood cancers.
The integration of next-generation sequencing (NGS) into pediatric oncology represents a paradigm shift from traditional histopathological diagnosis to molecularly-driven classification. The AmpliSeq for Illumina Childhood Cancer Panel is a targeted sequencing solution designed to comprehensively evaluate somatic variants in 203 genes associated with childhood and young adult cancers, including leukemias, brain tumors, and sarcomas [1]. This assessment evaluates the panel's clinical utility within the broader context of reproducibility research, examining its impact on diagnostic refinement, prognostic stratification, and identification of targetable therapies.
The clinical utility of genomic tests is defined by their ability to inform treatment decisions that positively change patient outcomes [64]. For pediatric cancers, which have a relatively low mutational burden but clinically relevant alterations, targeted panels like AmpliSeq offer a practical approach to precision medicine by focusing on genes with established significance [5]. This analysis compares the performance of the AmpliSeq Childhood Cancer Panel against alternative approaches, supported by experimental data from validation studies and clinical applications.
Performance Comparison with Alternative Methodologies
Technical Specifications and Workflow Comparison
The AmpliSeq Childhood Cancer Panel employs a PCR-based library preparation method that analyzes 3069 DNA amplicons and 1701 RNA amplicons, requiring only 10 ng of input DNA or RNA [1]. This low input requirement makes it particularly suitable for pediatric cases where sample material is often limited. The panel detects multiple variant types including single nucleotide variants (SNVs), insertions-deletions (indels), copy number variants (CNVs), and gene fusions simultaneously [1] [5].
Table 1: Comparison of Technical Specifications for Pediatric Cancer NGS Panels
| Parameter | AmpliSeq Childhood Cancer Panel | OncoKids Panel | Conventional Methods (FISH, Karyotyping, PCR) |
|---|---|---|---|
| Genes Covered | 203 genes | 206 genes (44 cancer predisposition loci, 82 mutation hotspots, 24 amplification genes) | Targeted to specific alterations |
| Input Requirements | 10 ng DNA or RNA | 20 ng DNA and 20 ng RNA | Varies by method, typically higher |
| Hands-on Time | <1.5 hours | Not specified | Extensive for multiple tests |
| Assay Time | 5-6 hours (library prep only) | Not specified | Days to weeks for complete profiling |
| Variant Types Detected | SNVs, Indels, CNVs, fusions | SNVs, Indels, CNVs, fusions | Limited to specific alteration types per test |
| Sample Compatibility | Blood, bone marrow, FFPE tissue | FFPE tissue, frozen tissue, bone marrow, peripheral blood | Varies by method |
Compared to traditional methods like fluorescent in situ hybridization (FISH), karyotyping, and polymerase chain reaction (PCR), which test specific alterations in separate assays, the AmpliSeq panel provides comprehensive profiling in a single workflow [5]. The OncoKids panel, another pediatric-focused NGS assay, shows similar capabilities but requires slightly higher input amounts (20 ng each of DNA and RNA) [2].
Analytical Performance and Reproducibility Data
Technical validation studies demonstrate that the AmpliSeq Childhood Cancer Panel achieves robust performance metrics. A 2022 study reported a mean read depth greater than 1000×, with high sensitivity for both DNA (98.5% for variants with 5% variant allele frequency) and RNA (94.4%), along with 100% specificity and reproducibility for DNA and 89% reproducibility for RNA [5].
Table 2: Analytical Performance Metrics from Validation Studies
| Performance Metric | DNA Analysis | RNA Analysis | Method of Validation |
|---|---|---|---|
| Sensitivity | 98.5% (for variants with 5% VAF) | 94.4% | Serial dilutions of positive controls |
| Specificity | 100% | 100% | Comparison with orthogonal methods |
| Reproducibility | 100% | 89% | Inter-run and intra-run replicates |
| Limit of Detection | 5% VAF | Not specified | Dilution series with commercial controls |
| Coverage Uniformity | Mean >1000× | Not specified | Sequencing metrics across amplicons |
These performance characteristics establish the technical reproducibility of the AmpliSeq panel, a critical foundation for its clinical application. The high sensitivity at low variant allele frequencies is particularly important for detecting subclonal populations in heterogeneous tumor samples.
Impact on Diagnostic and Prognostic Refinement
Diagnostic Reclassification Through Comprehensive Profiling
The AmpliSeq Childhood Cancer Panel demonstrates significant impact on diagnostic refinement in pediatric leukemia. In a study of 76 pediatric patients with acute leukemia, the panel identified clinically relevant results in 43% of patients, with 49% of mutations and 97% of fusions refining diagnosis [5]. These findings enabled more precise molecular classification beyond conventional diagnostic approaches.
The identification of specific genetic alterations through reproducible NGS testing directly informs prognostic stratification. For instance, the detection of KMT2A rearrangements, ETV6::RUNX1 fusion, or TP53 mutations carries significant prognostic implications that guide risk-adapted therapy intensification or de-escalation [5] [65]. The comprehensive nature of the panel allows for simultaneous assessment of multiple prognostic markers that would otherwise require separate tests.
Comparison with Alternative Genomic Approaches
Large precision medicine initiatives have employed various genomic approaches beyond targeted panels. The ZERO Childhood Cancer PRISM trial utilizes whole-genome sequencing (WGS), RNA-seq, and DNA methylation arrays [65], while the INFORM registry employs WES, low-coverage WGS, DNA methylation analysis, and RNAseq [65]. These comprehensive approaches may identify novel alterations beyond known targets but require more complex analytical pipelines and longer turnaround times.
Targeted panels like AmpliSeq offer advantages in clinical settings where rapid turnaround, ease of interpretation, and cost-effectiveness are priorities. The focused gene content facilitates reproducible variant interpretation and direct clinical actionability without the challenges of interpreting variants of unknown significance often encountered with WGS or WES.
Identification of Targetable Therapies and Clinical Impact
Actionable Alterations and Therapeutic Matching
The clinical utility of the AmpliSeq Childhood Cancer Panel extends significantly to therapy selection. In validation studies, 49% of mutations identified were considered targetable, enabling precision-guided treatment approaches [5]. The panel detects alterations in genes with available targeted therapies, such as FLT3, JAK2, BRAF, and NTRK fusions, facilitating enrollment in clinical trials or off-label use of targeted agents.
Major precision oncology platforms like MAPPYACTS and GAIN have demonstrated that molecularly guided therapies can produce meaningful clinical responses, particularly when based on high-level evidence [65]. The INFORM registry reported that patients receiving matched targeted therapies based on genomic profiling, including ALK, BRAF, and NTRK inhibitors, showed statistically significant improvement in progression-free survival and overall survival compared to those with similar alterations who did not receive targeted therapy [65].
Comparison of Clinical Utility Across Platforms
Table 3: Clinical Outcome Comparison Across Precision Medicine Platforms
| Precision Medicine Platform | PGT Uptake Rate | Objective Response Rate | Overall Clinical Benefit | Evidence Level |
|---|---|---|---|---|
| MAPPYACTS | 30% | 17% (all PGT) 38% ("ready for routine use") | Not specified | Tiered recommendations |
| GAIN/iCat2 | 12% | 17% | 24% | Observational study |
| INFORM | 28% | PFS improvement for specific targets | OS improvement for specific targets | 7-scale prioritization |
| ZERO Childhood Cancer | 43% | Not specified | Significant survival benefit in high-risk patients | WGS + RNAseq |
The relatively low uptake of precision-guided therapies (PGT) across platforms (10-33%) highlights implementation challenges, including drug access, clinical trial eligibility, and physician familiarity with targeted agents [65]. However, when administered, particularly early in the disease course based on high-level evidence, PGT demonstrates meaningful clinical benefit [65].
Experimental Protocols and Methodologies
Library Preparation and Sequencing Workflow
The experimental protocol for the AmpliSeq Childhood Cancer Panel follows a standardized workflow:
Nucleic Acid Extraction: DNA and RNA are extracted using validated methods, with quality assessment through spectrophotometry (OD260/280 ratio >1.8) and integrity measurement via Labchip or TapeStation [5].
Library Preparation: A total of 100 ng of DNA is used to generate 3069 amplicons, while 100 ng of RNA is reverse transcribed to cDNA using the AmpliSeq cDNA Synthesis kit before generating 1701 amplicons [5]. Amplicon libraries are prepared with sample-specific barcodes through consecutive PCRs.
Library Pooling and Normalization: DNA and RNA libraries are pooled at a 5:1 ratio, normalized using the AmpliSeq Library Equalizer, and sequenced on Illumina platforms (MiSeq, NextSeq series, or MiniSeq) [1] [5].
Data Analysis: Sequencing data are processed through bioinformatics pipelines for variant calling, annotation, and interpretation, with results reviewed by multidisciplinary molecular tumor boards.
Figure 1: AmpliSeq Childhood Cancer Panel Experimental Workflow
Orthogonal Validation Methods
To ensure reproducibility and accuracy, variants identified by the AmpliSeq panel are often confirmed by orthogonal methods:
- FLT3-ITD and NPM1 mutations: Validated by labeled-PCR amplification [5]
- FLT3 tyrosine kinase domain, cKIT, and GATA1 mutations: Confirmed by Sanger sequencing [5]
- Fusion genes: Verified by quantitative RT-PCR with specific primers and probes [5]
- Copy number variations: Validated by multiplex ligation-dependent probe amplification or array comparative genomic hybridization
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Research Reagents for AmpliSeq Panel Implementation
| Reagent/Component | Function | Specifications | Catalog Example |
|---|---|---|---|
| AmpliSeq Library PLUS | Library preparation reagents | Includes reagents for 24, 96, or 384 libraries | 20019101, 20019102, 20019103 [1] |
| AmpliSeq CD Indexes | Sample multiplexing | 8 bp indexes for labeling 96 samples per set | Sets A-D (20019105, 20019106, 20019107, 20019167) [1] |
| AmpliSeq cDNA Synthesis | RNA to cDNA conversion | Converts total RNA to cDNA for RNA panels | 20022654 [1] |
| AmpliSeq Library Equalizer | Library normalization | Normalizes libraries for sequencing | 20019171 [1] |
| AmpliSeq Direct FFPE DNA | DNA from FFPE tissue | Prepares DNA from FFPE tissues without deparaffinization | 20023378 [1] |
| SeraSeq Tumor Mutation DNA Mix | Positive control for DNA | Multiplex biosynthetic mixture with known variants | SeraSeq Tumor Mutation DNA Mix (v2 AF10 HC) [5] |
| SeraSeq Myeloid Fusion RNA Mix | Positive control for RNA | Synthetic RNA fusions with reference line RNA | SeraSeq Myeloid Fusion RNA Mix [5] |
Clinical Decision-Making Pathway
The clinical utility of genomic information depends on its effective integration into patient care pathways. The AmpliSeq panel results feed into a structured decision-making process:
Figure 2: Clinical Decision Pathway Following Genomic Testing
Molecular tumor boards play a crucial role in interpreting AmpliSeq results and making therapy recommendations. These multidisciplinary teams review the clinical relevance of genomic alterations, considering available evidence for targeted agents, clinical trial options, and practical feasibility [65]. The tiered evidence system used by platforms like MAPPYACTS categorizes recommendations as "ready for routine use," "investigational," or "hypothetical" based on the strength of supporting evidence [65].
The AmpliSeq Childhood Cancer Panel demonstrates substantial clinical utility through diagnostic refinement, prognostic stratification, and identification of targetable therapies in pediatric oncology. Its reproducible performance characteristics, with high sensitivity, specificity, and reproducibility, make it a reliable tool for clinical implementation. While comprehensive genomic approaches like WGS may identify more novel alterations, targeted panels offer practical advantages in turnaround time, cost-effectiveness, and interpretability.
Future directions should focus on expanding the evidence base for precision-guided therapies through collaborative trials, addressing barriers to targeted therapy access, and integrating non-genomic assays to provide a more comprehensive view of tumor biology. As precision medicine continues to evolve in pediatric oncology, reproducible genomic testing with demonstrated clinical utility will increasingly become standard of care for all children with cancer.
Conclusion
The AmpliSeq Childhood Cancer Panel demonstrates high reproducibility and robust performance for both DNA and RNA analysis, establishing it as a reliable tool for molecular profiling in pediatric oncology. Validation studies confirm exceptional sensitivity and specificity, enabling the detection of clinically actionable variants that refine diagnosis and inform treatment strategies in a significant proportion of patients. For the future, widespread adoption of this standardized panel promises to enhance consistency across research datasets, accelerate drug development for pediatric cancers, and solidify the role of precision medicine in clinical practice. Ongoing efforts should focus on integrating automated workflows to further improve reproducibility and expanding panel content to encompass emerging biomarkers.
References
- 1. AmpliSeq for Illumina Childhood Cancer Panel [https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/ampliseq-childhood-cancer-panel.html]
- 2. OncoKids: A Comprehensive Next-Generation Sequencing ... [https://www.sciencedirect.com/science/article/pii/S1525157818301028]
- 3. Gene panel for paediatric cancers increases access to high ... [https://ecancer.org/en/news/25106-gene-panel-for-paediatric-cancers-increases-access-to-high-quality-testing]
- 4. The Open Pediatric Cancer Project - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC11257555/]
- 5. Technical Validation and Clinical Utility of an NGS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9021638/]
- 6. Inter-assay variability of next-generation sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9013031/]
- 7. Assessing reproducibility of inherited variants detected with ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02569-8]
- 8. Guidelines for Validation of Next-Generation Sequencing– ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6941185/]
- 9. Genomic reproducibility in the bioinformatics era [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03343-2]
- 10. RNA sequencing: as accurate as once believed? [https://www.biotechniques.com/news/rna-sequencing-as-accurate-as-once-believed/]
- 11. Genomic reproducibility in the bioinformatics era - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11312195/]
- 12. Improving RNA-Seq Library Preparation for Accuracy [https://cellcarta.com/science-hub/rna-seq-library-preparation-watchmaker-workflow/]
- 13. GENOMICON-Seq enables realistic simulation of amplicon ... [https://www.nature.com/articles/s41598-025-05267-8]
- 14. Evaluation of ultra-low input RNA sequencing for the study ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6559993/]
- 15. AmpliSeq for Illumina Custom DNA Panel [https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/ampliseq-custom-dna-panel.html]
- 16. Which NGS DNA Library Prep Kit should you choose? [https://frontlinegenomics.com/which-ngs-dna-library-prep-kit-should-you-choose/]
- 17. Comprehensive evaluation of AmpliSeq transcriptome, a ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-015-2270-1]
- 18. Choosing the Right Method for Nucleic Acid Quantitation [https://worldwide.promega.com/resources/pubhub/choosing-the-right-method-for-nucleic-acid-quantitation/]
- 19. PCR Cycle Number in RNA-Seq [https://www.lexogen.com/blog/amplification-of-rna-seq-libraries-the-correct-pcr-cycle-number/]
- 20. AmpliSeq for Illumina Childhood Cancer Panel [https://support.illumina.com/sequencing/sequencing_kits/ampliseq-for-illumina-childhood-cancer/product-compatibility.html]
- 21. Comparison Table [https://emea.illumina.com/library-prep-array-kit-selector/research-use-only/dna/targeted/custom-content/comparison-table.html]
- 22. Exploring the effect of library preparation on RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6551333/]
- 23. Sequencing Platform Comparison Tool [https://emea.illumina.com/systems/sequencing-platforms/comparison-tool_msm_moved_msm_moved.html]
- 24. Sequencing Platforms | Illumina NGS platforms [https://www.illumina.com/systems/sequencing-platforms.html]
- 25. MiniSeq System | Small, affordable sequencing system [https://www.illumina.com/systems/sequencing-platforms/miniseq.html]
- 26. Nextseq and Miseq data output difference? [https://www.biostars.org/p/365296/]
- 27. MiniSeq System specifications | Key performance parameters [https://www.illumina.com/systems/sequencing-platforms/miniseq/specifications.html]
- 28. Getting Started with RNA-Sequencing (RNA-Seq) [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/getting-started-with-rna-seq?srsltid=AfmBOor9m-cLybSZoFkSNGTqgu5Pd9LPyi0iPpnVT51Voui3JN5LtFLN]
- 29. Performance Qualification (PQ) Services [https://www.illumina.com/products/by-type/service-products/performance-qualifications.html]
- 30. A survey of best practices for RNA-seq data analysis [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0881-8]
- 31. Simul-seq: combined DNA and RNA sequencing for whole- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5734913/]
- 32. Automated NGS Library Preparation System [https://www.datainsightsmarket.com/reports/automated-ngs-library-preparation-system-194529]
- 33. Next Generation Sequencing Library Preparation Market to [https://www.globenewswire.com/news-release/2025/11/17/3189266/0/en/Next-Generation-Sequencing-Library-Preparation-Market-to-Reach-USD-4-83-Billion-by-2032-as-Demand-for-Precision-Genomics-Accelerates-SNS-Insider.html]
- 34. Next-generation Sequencing Library Preparation Market ... [https://www.precedenceresearch.com/next-generation-sequencing-library-preparation-market]
- 35. Fusion Detection in Precision Oncology, Tradeoff ... [https://www.thermofisher.com/us/en/home/clinical/preclinical-companion-diagnostic-development/oncomine-oncology/education/fusion-detection-vail-interview.html]
- 36. Next-Generation Sequencing Methodologies To Detect ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10843083/]
- 37. Evaluating the performance of low-frequency variant ... [https://www.nature.com/articles/s41598-023-47135-3]
- 38. Fast and sensitive validation of fusion transcripts in whole- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10518092/]
- 39. AmpliSeq for Illumina Comprehensive Panel v3 [https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/ampliseq-comprehensive-panel.html]
- 40. Impact of study design, contamination, and data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12456016/]
- 41. Guidelines for preventing and reporting contamination in ... [https://www.nature.com/articles/s41564-025-02035-2]
- 42. Engineering CRISPR/Cas9 to mitigate abundant host ... [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-020-00859-0]
- 43. Quality control recommendations for RNASeq using FFPE ... [https://bmcmedgenomics.biomedcentral.com/articles/10.1186/s12920-022-01355-0]
- 44. Laboratory Evaluation of Bone Marrow - StatPearls - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK603716/]
- 45. Whole Blood RNA-Seq: Best Practice [https://www.lexogen.com/blog/whole-blood-rna-seq-best-practice/]
- 46. The Impact of Blood Sample Processing on Ribonucleic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11050547/]
- 47. Systematic comparative analysis of strand-specific RNA -seq library ... [https://www.nature.com/articles/s41598-021-04583-z?error=cookies_not_supported&code=2db5364a-9eef-45ee-9155-bd9079b8678b]
- 48. A real-world multi-center RNA-seq benchmarking study ... [https://www.nature.com/articles/s41467-024-50420-y]
- 49. A comparative analysis of library approaches for prep ... sequencing [https://pubmed.ncbi.nlm.nih.gov/30241496/]
- 50. Auto-Mag® DNA Normalization Kit [https://www.amdbiotech.com/products/auto-mag-dna-normalization-kit]
- 51. QIAseq Normalizer Kits [https://www.qiagen.com/us/products/discovery-and-translational-research/next-generation-sequencing/qiaseq-library-normalization]
- 52. Illumina DNA Prep product line | Fast, flexible library prep [https://sapac.illumina.com/products/by-brand/dna-prep-portfolio.html]
- 53. Analytical validation and implementation of a pan cancer ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2023.1067457/full]
- 54. TOP Guidelines [https://www.cos.io/initiatives/top-guidelines]
- 55. Yale Experts Lead Sessions on Research Integrity and Best ... [https://isps.yale.edu/news/blog/2025/10/reproducibility-matters-yale-experts-lead-sessions-on-research-integrity-and-best]
- 56. Technical Validation and Clinical Utility of an NGS ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2022.854098/full]
- 57. Analytical validation (accuracy, reproducibility, limit of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12431237/]
- 58. An accurate DNA and RNA based targeted sequencing ... [https://www.nature.com/articles/s41598-025-91640-6]
- 59. A Comprehensive Guide to Achieving New York State ... [https://www.sciencedirect.com/science/article/pii/S1525157825000674]
- 60. CHLA launches OncoKidsSM - a comprehensive DNA and RNA pediatric cancer panel [https://www.eurekalert.org/news-releases/776947]
- 61. OncoKids: A Comprehensive Next-Generation Sequencing Panel for Pediatric Malignancies - PubMed [https://pubmed.ncbi.nlm.nih.gov/30138724/]
- 62. Technical Validation and Clinical Utility of an NGS ... [https://pubmed.ncbi.nlm.nih.gov/35463953/]
- 63. Assay identifies clinically relevant gene fusions in pediatric ... [https://www.elsevier.com/about/press-releases/new-assay-identifies-clinically-relevant-gene-fusions-in-pediatric-tumors]
- 64. Clinical Utility: Informing Treatment Decisions by Changing ... [https://nam.edu/perspectives/clinical-utility-informing-treatment-decisions-by-changing-the-paradigm/]
- 65. The Growing Role of Precision Medicine in Paediatric ... [https://link.springer.com/article/10.1007/s11523-025-01178-w]