Beyond the Static Snapshot: Integrating Crystal Structures and NMR Ensembles for Dynamic Drug Discovery
This article provides a comprehensive comparison of X-ray crystallography and NMR spectroscopy for protein structure determination, tailored for researchers and drug development professionals.
Beyond the Static Snapshot: Integrating Crystal Structures and NMR Ensembles for Dynamic Drug Discovery
Abstract
This article provides a comprehensive comparison of X-ray crystallography and NMR spectroscopy for protein structure determination, tailored for researchers and drug development professionals. It explores the foundational principles of each technique, highlighting how static crystal structures complement dynamic NMR ensembles. The content covers practical methodologies and applications in structure-based drug design, addresses common challenges and optimization strategies, and discusses advanced validation frameworks for integrative models. By synthesizing key takeaways, the article underscores the synergistic power of combining these techniques to illuminate protein flexibility and dynamics, ultimately guiding the development of more effective therapeutics.
Static Snapshots vs. Dynamic Movies: Core Principles of Crystallography and NMR
In structural biology, X-ray crystallography and Nuclear Magnetic Resonance (NMR) spectroscopy represent two foundational techniques for determining the three-dimensional structures of proteins and other biological macromolecules. While X-ray structures provide a detailed view of the molecular arrangement within a crystal lattice, they represent a static, time- and ensemble-averaged model of the dynamic reality within the crystal. The interpretation of electron density maps and the associated B-factors (atomic displacement parameters) is therefore crucial for understanding both the structure and inherent dynamics of biological molecules.
This guide objectively compares the interpretation of crystal structures, with a specific focus on electron density and B-factors, against the ensemble representations provided by solution-state NMR. This comparison is particularly relevant for researchers in drug development who rely on accurate structural information, as the choice of technique can significantly influence the interpretation of molecular flexibility, binding interactions, and ultimately, drug design strategies.
Comparative Analysis of Structural Techniques
Table 1: Core Comparison of X-ray Crystallography and NMR Spectroscopy
| Feature | X-ray Crystallography | NMR Spectroscopy |
|---|---|---|
| Primary Output | Single, time- and ensemble-averaged model [1] | Ensemble of models representing conformational diversity [2] [3] |
| Sample State | Solid crystal lattice | Solution (near-native conditions) |
| Key Metrics | Electron density fit, B-factors, Resolution | Root Mean Square Deviation (RMSD) within ensemble, restraint violations |
| Typical Backbone RMSD between NMR and X-ray structures | - | 1.5 Å to 2.5 Å [2] |
| Interpretation of Flexibility | Isotropic/Anisotropic B-factors [4] | Root Mean Square Fluctuation (RMSF) across the ensemble [5] |
| Handling of Disorder | High B-factors, omitted atoms; can be over-interpreted [6] | Intrinsically represented by conformational diversity in the ensemble |
| View of Hydrogen Bonding | Inferred from atomic proximity; H atoms largely invisible [7] | Directly probed via chemical shifts (e.g., downfield for H-bond donors) [7] |
| Limitations | Static snapshot, crystal packing effects, "invisible" atoms with high B-factors [6] [1] | Limited by molecular size, sparse restraints can lead to less reliable atom positions [5] [8] |
Table 2: Performance in Structure-Based Drug Design Context
| Aspect | X-ray Crystallography | NMR Spectroscopy |
|---|---|---|
| Throughput | High for established soaking systems; limited by crystallization success [7] | No crystallization needed; can screen directly in solution [7] |
| Ligand Binding Insights | Static snapshot of a single dominant binding mode [7] | Can reveal multiple bound states and dynamic interactions [7] |
| Observation of Water Networks | ~80% of bound waters are observable [7] | Can detect highly mobile bound waters not seen in X-ray [7] |
| Information on Molecular Interactions | Inferred from electron density and geometry [7] | Directly measured (e.g., chemical shifts report on H-bonds) [7] |
| Notable Finding for Membrane Proteins | Transmembrane regions are typically straighter and more tightly packed [5] | NMR ensembles show higher convergence in the membrane region [5] |
Experimental Protocols and Data Interpretation
Interpreting Electron Density and B-Factors in Crystallography
In X-ray crystallography, the atomic model is refined to fit the experimental electron density map. The B-factor, or atomic displacement parameter, quantifies the smearing of an atom's electron density due to thermal vibration, static disorder, or other factors. It is mathematically defined as ( B = 8π²u² ), where ( u² ) is the mean-square amplitude of atomic vibration [6] [4].
A critical challenge is that B-factors are influenced by many non-physiological factors, including crystal lattice defects, refinement artifacts, and the resolution of the data [4] [1]. Consequently, raw B-factors are not directly transferable between different structures. To enable meaningful comparisons, B-factors must be rescaled. A common method is Z-transformation: [ B{ri} = \frac{Bi - B{ave}}{B{std}} ] where ( B{ri} ) is the rescaled B-factor for atom *i*, ( B{ave} ) is the average B-factor of the structure, and ( B_{std} ) is the standard deviation [4].
Interpreting regions with high B-factors requires caution. Atoms with B-factors larger than 100 Ų make a negligible contribution to the calculated structure factors, and their positions are not supported by experimental evidence [6]. A strategy to define an upper limit for plausible B-factors (Bmax) involves extrapolating the relationship between a structure's average B-factor and its solvent content. This Bmax value is resolution-dependent, being approximately 25 Ų at very high resolution (<1.5 Å) and rising to about 80 Ų at low resolution (>3.3 Å). Structures with average B-factors exceeding B_max should be treated with caution [6].
It is also important to recognize that refined B-factors can significantly underestimate the true level of microscopic heterogeneity present in the crystal. Molecular dynamics simulations have shown that even at high resolution (1.0 Å), refined B-factors can underestimate the actual atomic fluctuations by up to sixfold for some well-resolved atoms [1].
Protocol for Validating a Crystal Structure against Electron Density
Objective: To assess the local fit of an atomic model to its experimental electron density map, identifying regions where the model may be poorly supported or over-interpreted.
Workflow:
Methodology:
- Data Acquisition: Download the structure file (PDB format) and its associated sigma-scaled 2Fo-Fc electron density map (CCP4 format) from resources like the Protein Data Bank in Europe (PDBe) [9].
- Electron Density Ratio Calculation: For each non-hydrogen atom i, sum the electron density values (ρm) of all significant voxels within a defined radius of the atom. Calculate the electron density ratio ( ri = \frac{\sum \rhom}{Zi} ), where ( Zi ) is the number of electrons for the atom (hydrogens are accounted for by adding their electrons to the bonded atom) [9].
- Normalization: Normalize the density ratio ( ri ) to account for the discrete voxel representation and its dependence on the atom's B-factor. This correction produces a normalized, chemically informative metric (( r{i-corrected} )) that is consistent across the entire structure and comparable between different structures [9].
- Aggregation and Analysis: Aggregate the corrected electron density values from the atomic level to the residue and chain levels. A residue or atom with a significantly low corrected electron density indicates a region where the model is not well-supported by experimental data and should be interpreted with caution [9].
Protocol for Comparing an NMR Ensemble to a Crystal Structure
Objective: To quantitatively assess the conformational differences between a protein's structure determined by X-ray crystallography and by NMR spectroscopy in solution.
Workflow:
Methodology:
- Data Curation: Select a non-redundant pair of structures for the same protein or a close homolog. For the NMR structure, use the entire ensemble of models [2] [5].
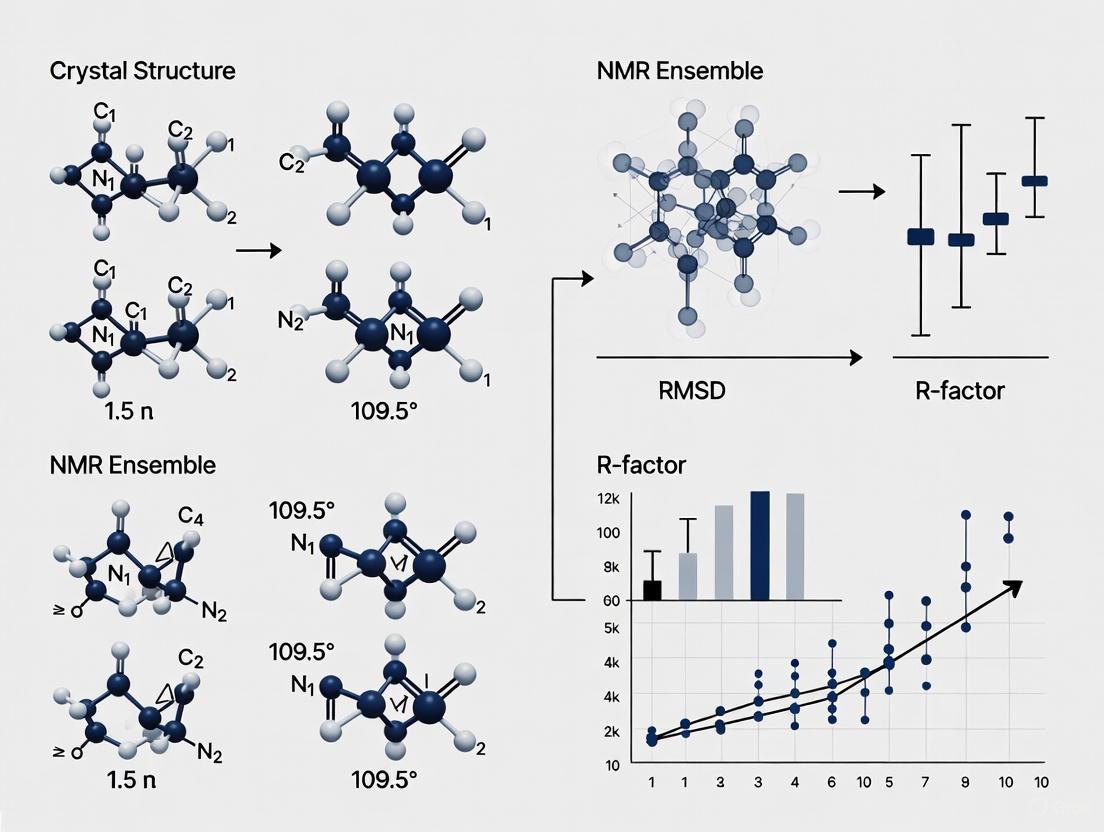
- Structural Alignment and Global Comparison: Perform a structural alignment of the NMR models to the crystal structure, typically using the backbone atoms of well-ordered regions. Calculate the global backbone Root Mean Square Deviation (RMSD) between each NMR model and the crystal structure. For a set of 109 proteins, the average RMSD values fall between 1.5 Å and 2.5 Å [2].
- Local Flexibility Analysis: Calculate the Root Mean Square Fluctuation (RMSF) for the backbone atoms of the NMR ensemble to identify flexible regions. Compare this to the B-factor profile of the crystal structure.
- Regional Analysis:
- Secondary Structure: Beta-strands typically show better agreement between NMR and crystal structures than helices and loops [2].
- Side Chains: Hydrophobic, buried side chains usually adopt very similar conformations in both states, while hydrophilic, surface-exposed side chains may differ more due to crystal packing or solvation effects [2].
- Loops and Binding Sites: Analyze conformational differences in loop regions and active/binding sites, as these can have functional implications independent of crystal contacts [2].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Software for Structural Studies
| Item | Function | Application Context |
|---|---|---|
| Detergents (e.g., DPC, DHPC) | Mimic the membrane environment for solubilizing membrane proteins [5] | NMR & Crystallography of membrane proteins |
| Lipidic Cubic Phase (LCP) | Membrane mimetic that promotes crystallization of membrane proteins [5] | Crystallography (e.g., Bacteriorhodopsin) |
| Isotope-labeled Amino Acids (e.g., ¹³C-Val, ¹⁵N-Leu) | Enable specific labeling for resolving NMR signals and probing interactions [7] | NMR spectroscopy (SBDD, assignment) |
| Molecular Replacement Software (e.g., Phaser) | Solves the crystallographic "phase problem" using a known homologous structure [4] | X-ray crystallography |
| Refinement Software (e.g., Phenix, TEMPy-ReFF) | Fits and refines an atomic model against experimental data (X-ray maps or cryo-EM maps) [10] | X-ray crystallography & Cryo-EM |
| Ensemble Analysis Software (e.g., EnsembleFlex) | Extracts, quantifies, and visualizes conformational heterogeneity from PDB ensembles [3] | NMR & multi-model analysis |
In structural biology, the shift from viewing proteins as static entities to understanding them as dynamic systems is crucial for unraveling their true function. While X-ray crystallography provides high-resolution snapshots, it often captures a single, rigid conformation. In contrast, solution-state Nuclear Magnetic Resonance (NMR) spectroscopy uniquely characterizes proteins in their native-like aqueous environment, revealing the dynamic conformational ensembles that are fundamental to biological activity. This guide objectively compares these techniques for researchers and drug development professionals.
The table below summarizes the core distinctions between the two methods, highlighting how their complementary strengths address different research questions.
Table 1: Core Method Comparison: NMR Spectroscopy vs. X-ray Crystallography
| Feature | X-ray Crystallography | Solution-State NMR Spectroscopy |
|---|---|---|
| Molecular Weight Limit | Effectively no limit [11] | Traditionally limited, though advancing for >80 kDa complexes [11] |
| Resolution | High (~1 Å) [11] | High (~1-2 Å) [11] |
| Sample State | Solid crystal | Solution (native-like conditions) |
| Conformational Dynamics | No [11] | Yes, across multiple timescales [11] |
| Hydrogen Atom Information | No (effectively "blind" to H) [11] | Yes (direct probe of H-bonding) [11] |
| Throughput Viability | Yes (high-throughput soaking systems) [11] | Yes, for specific applications [11] |
| Key Limitation | Requires high-quality crystals; infers molecular interactions [11] | Sparse data for large systems; complex data analysis [11] [5] |
Quantitative Comparisons in Structure Determination
Direct comparisons of structures solved by both X-ray crystallography and NMR reveal measurable differences that underscore the influence of method and environment.
Table 2: Empirical Data from Comparative Studies
| Protein/System Studied | Key Comparative Findings | Implication |
|---|---|---|
| General Soluble Proteins | Average backbone RMSD between crystal and NMR structures is 1.0-1.4 Å [5]. | NMR and crystal structures are largely congruent for core folded regions. |
| Membrane Proteins | RMSDs are below 5 Å in the membrane region; crystal structures often have straighter transmembrane helices and tighter packing [5]. | The membrane mimetic used (micelles, bicelles, etc.) significantly influences the observed structure. |
| Streptococcus pneumoniae PsrP | MD simulations started from an AlphaFold structure were validated with NMR relaxation data; only specific trajectory segments matched experiments, revealing flexible functional regions [12] [13]. | Integrative approaches are essential to identify biologically relevant dynamic states from computational models. |
| Intrinsically Disordered Proteins (IDPs) | NMR is the primary method for determining atomic-resolution conformational ensembles of IDPs, often integrated with MD simulations and SAXS [14]. | NMR is indispensable for studying proteins that lack a fixed structure. |
Experimental Protocols for Modern NMR Ensemble Determination
The following workflows represent cutting-edge methodologies that integrate NMR with computational modeling to determine accurate dynamic ensembles.
Workflow 1: Integrative NMR-MD for Dynamic Ensembles
This protocol, adapted from studies on folded proteins, uses molecular dynamics (MD) and NMR relaxation to derive time-resolved conformational ensembles [12] [13] [15].
Diagram Title: Integrative NMR-MD Workflow
Step-by-Step Protocol:
- Initial Model Generation: Obtain a starting 3D structure using AlphaFold [12] [13].
- Molecular Dynamics Simulation: Perform a long, unconstrained MD simulation in explicit solvent using a modern force field (e.g., CHARMM36m, a99SB-disp) [14] [13].
- NMR Data Acquisition: Collect experimental NMR relaxation data, including longitudinal (R1) and transverse (R2) rates, heteronuclear NOEs, and cross-correlated relaxation (ηxy) rates on the protein in solution [13].
- Back-Calculation: Extract backbone amide vector motions from the MD trajectory and use them to back-calculate the expected NMR relaxation parameters (R1, NOE, ηxy) for every segment of the simulation [12] [13].
- Trajectory Selection: Identify stable segments of the MD trajectory (RMSD plateaus) for which the back-calculated NMR parameters show strong agreement with the experimental data [12].
- Ensemble Validation: The selected segments collectively form the final 4D conformational ensemble, which is validated against the full set of experimental NMR data [13].
Workflow 2: Maximum Entropy Reweighting for IDP Ensembles
This method is specifically designed for Intrinsically Disordered Proteins (IDPs) and uses a maximum entropy principle to refine MD ensembles against sparse experimental data [14].
Diagram Title: MaxEnt Ensemble Refinement
Step-by-Step Protocol:
- Diverse Simulation: Run extensive, all-atom MD simulations of the IDP using multiple state-of-the-art force fields (e.g., a99SB-disp, CHARMM36m) [14].
- Multi-Technique Data Collection: Acquire ensemble-averaged experimental data, such as NMR chemical shifts (CS), paramagnetic relaxation enhancements (PREs), and Small-Angle X-ray Scattering (SAXS) profiles [14].
- Forward Modeling: Predict the values of the experimental measurements from every frame of the MD simulations using physical models [14].
- Automated Reweighting: Apply a maximum entropy reweighting algorithm. This procedure adjusts the statistical weights of the MD frames with the minimal change needed to achieve agreement with the entire experimental dataset. A key parameter is the Kish ratio (K), which is set to retain a sufficient number of effective conformations (e.g., K=0.10 keeps ~3000 structures from 30,000) to prevent overfitting [14].
- Convergence Check: The resulting ensemble should be robust, meaning reweighting simulations from different starting force fields converges to a highly similar conformational distribution, providing a force-field independent view of the IDP's dynamics [14].
Table 3: Key Research Reagent Solutions for NMR Ensemble Studies
| Tool / Resource | Function in Research |
|---|---|
| Selective Side-Chain Labeling | Uses (^{13}\mathrm{C})-labeled amino acid precursors to simplify NMR spectra and provide specific probes for protein-ligand interactions, enabling high-throughput applications in drug discovery [11]. |
| NMR Relaxation Measurements | Parameters like longitudinal (R1) and transverse (R2) relaxation rates and heteronuclear NOEs provide detailed insight into internal dynamics on picosecond-to-nanosecond timescales [13]. |
| Cross-Correlated Relaxation (ηxy) | An advanced NMR parameter that is less biased by slow exchange processes than R2, providing a cleaner readout of fast dynamics for validating MD ensembles [13]. |
| Advanced Force Fields | Modern molecular mechanics force fields (e.g., CHARMM36m, a99SB-disp) with improved water models provide more accurate physical models for MD simulations of both folded and disordered proteins [14] [13]. |
| Integrative Modeling Software | Software platforms like Rosetta and automated maximum entropy reweighting scripts are used to refine computational models against sparse experimental data [14] [5]. |
| Restraint-Assisted Structure Prediction (RASP) | A deep learning model derived from AlphaFold that can directly incorporate sparse distance restraints (e.g., from NMR) to improve structure prediction, especially for multi-domain and few-MSA proteins [16]. |
For decades, the primary goal of structural biology has been to determine the precise three-dimensional atomic coordinates of biological macromolecules. This pursuit has yielded over 200,000 structures in the Protein Data Bank, most representing single, static snapshots of proteins captured primarily through X-ray crystallography. The longstanding paradigm in molecular biology has been that each protein sequence folds into a single, averaged 3D structure under given conditions, an assumption that deeply influenced both experimental approaches and computational methods [15]. The recent revolutionary advances in artificial intelligence, particularly AlphaFold, have further cemented our ability to predict these static structures with remarkable accuracy, marking a transformative milestone in structural biology [17].
However, this static representation presents an incomplete picture of protein reality. Proteins are fundamentally dynamic entities that sample multiple conformational states to perform their biological functions. As one review notes, "protein function is not solely determined by static three-dimensional structures but is fundamentally governed by dynamic transitions between multiple conformational states" [17]. This limitation of static structures becomes particularly significant when considering that many pathological conditions, including Alzheimer's disease, Parkinson's disease, and other disorders, stem from protein misfolding or abnormal dynamic conformations [17]. The shift from static to multi-state representations is therefore crucial for understanding the mechanistic basis of protein function and regulation.
This guide provides a comprehensive comparison between static protein structures and NMR-derived ensembles, examining what each approach reveals about protein structure, what they miss, and how integrative methods are bridging the gap between these complementary techniques.
Comparative Analysis of Structural Biology Techniques
Technical Foundations and Limitations
Table 1: Fundamental characteristics of static structures versus NMR ensembles
| Characteristic | Static Structures (X-ray/cryo-EM) | NMR Ensembles | Integrated Approaches |
|---|---|---|---|
| Structural Representation | Single conformation | Multiple conformations (10-40 typically) | 4D conformational ensembles (3D space + time) |
| Timescale Resolution | Static snapshot | Picoseconds to seconds | Femtoseconds to milliseconds (MD) |
| Sample Environment | Crystalline state or frozen | Solution state, near-physiological conditions | Various, including in-cell |
| Key Limitations | packing effects, crystal artifacts | Molecular weight constraints, interpretation complexity | Computational cost, validation challenges |
| Dynamic Information | Indirect (B-factors) | Direct (relaxation, order parameters) | Direct from simulation and experiment |
| Functional Insights | Orthosteric sites, binding pockets | Allosteric pathways, conformational selection | Complete mechanistic picture |
Static structures obtained through X-ray crystallography and cryo-EM provide high-resolution snapshots that are invaluable for understanding overall protein architecture, active site geometry, and protein-ligand interactions. However, these techniques have inherent limitations. Crystallographic B-factors are affected by packing and other special features of the crystalline state, and elevated B-factors may not solely indicate macromolecular flexibility [18]. Furthermore, the crystallization process itself may select for specific conformations while excluding others that are functionally relevant.
In contrast, NMR spectroscopy captures protein behavior in solution under conditions closer to the physiological environment. The NMR-based structure determination process typically generates multiple models (typically 10-40), collections that are called "ensembles" [18]. As one methodology paper explains, "In conjunction with the recognition of the functional role of internal dynamics of proteins at various timescales, there is an emerging use of dynamic structural ensembles instead of individual conformers" [19]. These ensembles are usually substantially more diverse than conventional NMR ensembles and eliminate the expectation that a single conformer should fulfill all NMR parameters originating from 10^16 - 10^17 molecules in the sample tube [19].
Quantitative Comparison of Structural and Dynamic Information
Table 2: Quantitative assessment of information content across methods
| Parameter | X-ray Crystallography | NMR Spectroscopy | Molecular Dynamics | Integrated Approaches |
|---|---|---|---|---|
| Atomic Coordinates | Precise (0.5-2.5 Å resolution) | Well-defined backbone, variable side chains | Atomic detail (0.1 Å precision) | Atomic detail with uncertainty estimates |
| Backbone Flexibility | B-factors (temperature factor) | S² order parameters (0-1 scale) | Root mean square fluctuations | Combined experimental/theoretical metrics |
| Timescale Coverage | None | ps-ns (S²), μs-ms (Rex), slower (exchange) | fs-μs (enhanced sampling extends this) | Comprehensive coverage across timescales |
| Conformational Diversity | Limited to crystal contacts | Explicitly represented in ensemble | Sampled through simulation | Validated diversity through experimental agreement |
| Allosteric Communication | Indirect through comparison of structures | PRE, RDC, chemical exchange | Interaction networks, correlation analysis | Mechanistic models with experimental validation |
Recent advances have enabled more direct comparisons between these techniques. A 2021 study compared patterns in protein flexibility between crystallographic B-factors and NMR ensembles, finding that "coordinate uncertainties in an NMR-derived 'ensemble' of structures are highly correlated to coordinate variances across MD trajectories" [18]. Interestingly, the study identified a persistent pattern in backbone heavy atom coordinate uncertainties in NMR ensembles that also exists in MD simulations but not in crystallographic B-factors, suggesting that MD trajectories and NMR ensembles capture motional behavior of peptide bond units not captured by B-factors [18].
A particularly insightful application of comparative structural analysis comes from a 2025 study on protein phosphorylation effects. This research, which systematically analyzed how phosphorylation affects backbone conformation, protein dynamics, and mechanical strain, found that "phosphorylation commonly induces small, stabilizing conformational changes through conformational selection and frequently modulates local residue fluctuations, influencing overall protein motion" [20]. Notably, the study found that phosphorylation was significantly linked to global changes in backbone conformation, though most changes tend to be small (median backbone RMSD 1.14 ± 3.13 Å), with only 28.14% of phosphorylation events associated to changes ≥ 2 Å [20].
Experimental Protocols for Ensemble Determination
NMR-Based Ensemble Generation
The determination of dynamic conformational ensembles by NMR employs distinct experimental protocols compared to single-structure determination:
Sample Preparation: Proteins are uniformly labeled with ^15N and ^13C, with specific labeling strategies for larger proteins. For in-cell NMR studies, proteins are introduced into living cells (E. coli, yeast, or mammalian) through electroporation or other methods, and the cells are maintained in specially designed bioreactors that supply fresh medium to prolong viability during data acquisition [21].
Data Collection: A combination of experiments is required to obtain structural and dynamic information:
- NOESY: Provides distance restraints through nuclear Overhauser effects
- RDC: Residual dipolar couplings provide orientational restraints
- Relaxation Measurements: R1, R2, and heteronuclear NOE provide dynamics on ps-ns timescales
- Paramagnetic Effects: PRE (paramagnetic relaxation enhancement) and PCS (pseudocontact shifts) provide long-range distance restraints
Structure Calculation: Unlike conventional single-conformer refinement, ensemble methods use specialized protocols such as:
- DER (Dynamic Ensemble Refinement): Generates ensembles reflecting internal dynamics
- MUMO (Minimal Under-restraining Minimal Over-restraining): Addresses different restraint averaging across ensemble sizes
- EROS (Ensemble Refinement with Orientational Restraints): Incorporates RDCs and other orientational restraints
- ISD (Inferential Structure Determination): Bayesian approach avoiding inherent errors in conventional refinement
The CoNSEnsX (Consistency of NMR-derived Structural Ensembles with eXperimental data) web server provides a standardized approach for evaluating dynamic conformational ensembles against experimental NMR data [19]. This approach gives a complete evaluation of these ensembles by assessing correspondence with diverse independent NMR parameters.
Integrative Approaches Combining Multiple Methods
A promising integrative methodology was demonstrated in a 2025 study that combined AlphaFold, molecular dynamics, and NMR relaxation data [15]. The protocol involves:
Initial Structure Generation: Using AlphaFold to generate a starting structural model, recognizing that "AlphaFold-generated structural ensembles are considered promising starting points for MD simulations, as they may effectively explore a broad range of local and global energy minima" [15].
Molecular Dynamics Sampling: Performing extensive MD simulations (often hundreds of nanoseconds to microseconds) using improved force fields such as AMBER99SB or OPLS. The simulations sample conformational space without experimental restraints initially.
Experimental Validation: Measuring NMR relaxation parameters including longitudinal (R1) and transverse (R2) relaxation rates, heteronuclear NOE, and cross-correlated relaxation (ηxy) rates.
Ensemble Selection: Identifying trajectory segments consistent with experimental observables through back-calculation of NMR parameters from the MD trajectory and selection of regions with stable RMSD that best match experimental data.
This approach addresses a key challenge in structural biology: "Obtaining a reliable 4D model (defined as a three-dimensional spatial structure evolving over time) of the most energetically favourable, and therefore most populated, region of conformational space offers a more realistic and comprehensive understanding of protein function in living systems" [15].
Table 3: Key research reagents and computational resources for structural ensemble studies
| Resource Category | Specific Tools/Reagents | Function/Application | Key Features |
|---|---|---|---|
| NMR Structure Calculation | CYANA, XPLOR-NIH, CNS, CS-RosettaCM | Conversion of NMR data to 3D structures | Traditional single-conformer refinement |
| Ensemble Generation | DER, MUMO, EROS, ISD, ABSURDer | Dynamic ensemble refinement | Ensemble-aware restraint handling |
| Validation Tools | CoNSEnsX, PRIDE-NMR | Ensemble validation against experimental data | Web server availability for accessibility |
| Molecular Dynamics | GROMACS, AMBER, OpenMM, CHARMM | Sampling conformational space | Improved force fields for accuracy |
| Specialized Databases | ATLAS, GPCRmd, MemProtMD | MD trajectories for specific protein classes | Community resources for validation |
| In-Cell NMR Tools | Bioreactor systems, isotope labeling schemes | Structural studies in cellular environments | Maintains cell viability during experiments |
The toolkit for studying dynamic conformational ensembles has expanded significantly, with both experimental and computational resources becoming more accessible. For NMR studies, specialized databases have emerged to support dynamic conformation research, including ATLAS (comprising simulations of approximately 2000 representative proteins), GPCRmd (focusing on G protein-coupled receptors), and SARS-CoV-2 protein databases [17]. These resources provide essential reference data and trajectories for method development and validation.
For in-cell applications, specialized bioreactor systems have been developed that continuously supply fresh medium into the NMR tube to prolong the lifetime of cells (at least 24 hours), enabling the acquisition of 3D NMR data that requires longer measurement times [21]. Additionally, paramagnetic probes and ^19F labeling strategies have expanded the applicability of NMR for atomic-level characterization of protein structure in mammalian cells, overcoming challenges associated with line broadening in cellular environments [21].
Functional Implications for Drug Discovery
The limitations of static structures have direct consequences for drug discovery and development. Static structures primarily reveal orthosteric binding sites - the primary functional sites where substrates or inhibitors bind directly. However, they often miss allosteric regulation mechanisms, conformational selection processes, and the dynamic interplay between different functional states.
A telling example comes from the analysis of phosphorylation effects: "Notably, a small but significant subset of phosphosites shows mechanical coupling with functional sites, aligning with the domino model of allosteric signal transduction" [20]. This finding has profound implications for drug design, suggesting that targeting allosteric networks influenced by phosphorylation may provide new therapeutic opportunities.
The integration of static and dynamic approaches is particularly valuable for understanding protein-protein interactions and signaling networks. In vivo cross-linking mass spectrometry (XL-MS) enables analysis of protein structure and interaction at the cellular proteomic level, providing complementary information to NMR-based approaches [21]. As one perspective notes, "In vivo XL-MS method is crucial for unraveling the native conformation of protein complexes directly within the complex and dynamic microenvironments of cells and tissues" [21].
For drug development professionals, these insights translate into practical considerations:
- Target Identification: Dynamic ensembles may reveal cryptic binding pockets not apparent in static structures
- Mechanism Understanding: Allosteric networks illuminated by dynamics studies suggest alternative modulation strategies
- Specificity Optimization: Understanding conformational heterogeneity helps design selective inhibitors that distinguish between similar binding sites
- Polypharmacology: Ensemble information enables rational design of compounds that affect multiple conformational states
The field of structural biology is undergoing a fundamental paradigm shift from static structures to dynamic ensemble representations. As one review observes, "In the post-AlphaFold era, driven by breakthrough advancements in static protein structures, the paradigm of protein research is gradually shifting from static structures to dynamic conformations" [17]. This transition requires the development of novel, conceptually distinct computational methods and experimental tools [15].
The integration of multiple approaches - static structures from crystallography and cryo-EM, dynamic information from NMR, computational sampling from MD simulations, and AI-based structure prediction - provides the most comprehensive understanding of protein function. As one study demonstrates, the combination of AlphaFold, MD, and NMR relaxation allows researchers to "identify biologically relevant holistic time-resolved 4D conformational ensembles" that capture the complete dynamic picture of backbone and side chains [15].
For researchers and drug development professionals, the key insight is that static structures and dynamic ensembles provide complementary rather than competing information. Static structures reveal the architectural framework and precise atomic coordinates, while dynamic ensembles illuminate the functional motions, allosteric pathways, and conformational heterogeneity essential for biological activity. The most effective strategies will leverage both approaches to overcome their individual limitations and provide a more complete understanding of the relationship between protein structure, dynamics, and function.
High-resolution three-dimensional structures are fundamental to modern biology, yet the dominant techniques for obtaining them—X-ray crystallography and solution-state Nuclear Magnetic Resonance (NMR) spectroscopy—provide fundamentally different views of the protein universe. X-ray crystallography produces a precise, static snapshot of a protein's most stable conformation, trapped within a crystal lattice. In contrast, NMR spectroscopy yields an ensemble of structures, offering a dynamic view of the protein's conformational landscape in a near-native solution environment. This guide provides an objective comparison of these two powerful techniques, framing them not as competitors, but as complementary tools for elucidating the critical link between protein dynamics and biological function.
Quantitative Comparison: Crystallography vs. NMR
A systematic analysis of proteins studied by both X-ray crystallography and NMR reveals consistent, quantifiable differences in the resulting structural models. The table below summarizes key comparative metrics derived from large-scale studies of these matched protein pairs.
Table 1: Overall Structural Comparison between Crystal and NMR Structures
| Comparison Metric | X-ray Crystallography | Solution NMR Spectroscopy | Key Findings from Matched Protein Pairs |
|---|---|---|---|
| Global Backbone RMSD | Baseline | 1.0 - 2.5 Å [2] | Average backbone RMSD for soluble proteins is 1.0-1.4 Å over core residues [5]. |
| Membrane Protein RMSD | Baseline | < 5.0 Å [5] | In membrane regions, RMSDs are below 5 Å, with higher NMR convergence in this area [5]. |
| Secondary Structure Agreement | Baseline | β-strands match best [2] | β-strands show better agreement than helices or loops; loop differences are independent of crystal packing [2]. |
| Side-chain Conformations | Baseline | Hydrophobic residues more similar [2] | Buried hydrophobic side chains show higher similarity; different rotamers for buried side chains are rare [2]. |
| Stereo-chemical Quality | Higher [5] | Lower (pre-refinement) [5] | Crystal structures typically exhibit higher stereochemical correctness and tighter packing [5]. |
| Structural Convergence | Single model | Ensemble-dependent | For 76% of pairs, RMSD between methods is larger than the spread within the NMR ensemble itself [5]. |
Further analysis reveals that the degree of divergence is not random but is influenced by the local structural and chemical environment.
Table 2: Correlation of Structural Differences with Protein Features
| Protein Feature | Correlation with NMR-Crystal RMSD | Interpretation |
|---|---|---|
| Residue Type | Hydrophobic residues are more similar than hydrophilic ones [2]. | Hydrophobic residues are often well-packed in the protein core, restricting conformational variability. |
| Solvent Accessibility | Modest correlation (correlation coefficient = 0.462) [2]. | Solvent-exposed residues have greater freedom of movement, especially in solution. |
| Location in Membrane Proteins | Higher convergence in membrane region than in soluble domains [5]. | The membrane environment imposes physical constraints on transmembrane helices. |
| Impact of Crystal Packing | Minimal influence on conformational differences in loops [2]. | Crystal contacts do not appear to be the primary driver of differences in flexible regions. |
Experimental Protocols for Structure Determination
X-ray Crystallography Workflow
The process of structure determination by X-ray crystallography involves several standardized steps, from protein production to model refinement.
- Protein Production and Crystallization: The target protein is expressed and purified to homogeneity. It is then concentrated and subjected to crystallization trials, where thousands of conditions are screened to find the optimal mix of precipants, buffers, and salts that promote the formation of well-ordered, three-dimensional crystals. For membrane proteins, this requires the use of membrane mimetics such as detergent micelles, lipidic cubic phases (LCP), or bicelles to mimic the native lipid environment [5].
- Data Collection and Phasing: A single crystal is exposed to a high-intensity X-ray beam. The resulting diffraction patterns are collected and processed to determine the crystal's symmetry and unit cell parameters. The "phase problem" is then solved using methods like Molecular Replacement (if a homologous structure exists) or Experimental Phasing (e.g., via selenomethionine incorporation).
- Model Building and Refinement: An atomic model is built into the experimental electron density map. The model is iteratively refined against the diffraction data to improve its agreement with the experimental observations (R-factors) while maintaining proper stereochemistry.
Solution NMR Spectroscopy Workflow
NMR structure determination relies on extracting distance and angle constraints from the protein's nuclear spins.
- Sample Preparation for NMR: The protein is uniformly labeled with stable isotopes (15N and 13C). This is essential for the multidimensional NMR experiments required to resolve and assign signals. For large proteins (>25 kDa), more sophisticated labeling schemes (e.g., deuteration, methyl-specific labeling) are employed. Membrane proteins are studied in membrane mimetics such as detergent micelles (e.g., DPC, DHPC) or nanodiscs [5] [7].
- NMR Data Acquisition and Assignment: A suite of multi-dimensional NMR experiments (e.g., HSQC, NOESY, TROSY) is performed to correlate the spins of neighboring nuclei. The first critical step is resonance assignment, which links each NMR signal to a specific atom in the protein sequence.
- Constraint-Based Structure Calculation: Nuclear Overhauser Effect (NOE) signals are used to generate a list of interatomic distance restraints. Additional restraints from residual dipolar couplings (RDCs) and scalar couplings (J-couplings) define backbone and side-chain torsion angles. Structures are calculated by using simulated annealing or molecular dynamics to find conformations that satisfy all experimental constraints, resulting in an ensemble of models.
Diagram 1: Comparative structural biology workflows.
Capturing Functional Dynamics: Beyond the Static Structure
Biological activity is inherently dynamic, and both methodologies are evolving to capture this reality.
Time-Resolved and Dynamic Crystallography
Traditional crystallography captures a thermodynamically stable state. However, new techniques are making it possible to observe protein motions within the crystal. Electric-field stimulated time-resolved X-ray crystallography (EFX) applies an external electric field to initiate synchronized protein motions (e.g., ion conduction through a channel) and uses rapid, pulsed X-rays to capture a molecular movie of the action [22]. Concurrently, advanced Molecular Dynamics (MD) simulations are now being performed in explicitly modeled crystal environments, accounting for crystal contacts and solvent composition. These simulations can reach millisecond timescales and help interpret time-resolved data by providing an ensemble view of the conformational heterogeneity present even within a crystal [23].
NMR as a Native Probe for Dynamics and Interactions
NMR spectroscopy is uniquely suited to study dynamics and interactions directly in solution, under physiological conditions. It provides atomistic information on hydrogen bonding and other non-covalent interactions by measuring chemical shifts, which is crucial for understanding the enthalpic contributions to ligand binding [7]. Furthermore, NMR relaxation experiments can quantify motions on timescales from picoseconds to seconds, directly linking dynamics to function. This makes NMR particularly powerful for studying weak interactions, conformational entropy, and the role of water networks in binding—factors that are largely invisible to crystallography [7].
Diagram 2: Multi-technique approach to protein dynamics.
Research Reagent Solutions for Structural Biology
Successful structure determination, particularly for challenging targets like membrane proteins, relies on specialized reagents and methodologies.
Table 3: Essential Research Reagents and Methodologies
| Reagent / Methodology | Function in Research | Application Context |
|---|---|---|
| Membrane Mimetics (e.g., DPC Micelles, LCP, Bicelles, Nanodiscs) | Replicate the native lipid bilayer to solubilize and stabilize membrane proteins for structural studies [5]. | Used in both crystallography and NMR for studying membrane proteins. The choice of mimetic can influence the observed structure. |
| Isotope Labeling (15N, 13C, 2H) | Incorporates NMR-active nuclei into the protein, enabling signal detection and assignment in multi-dimensional NMR experiments [7]. | Essential for all solution NMR structure determination. Specific labeling schemes (e.g., methyl-labeled, perdeuterated) overcome size limitations. |
| Crystallization Screens (Sparse Matrix) | Pre-formulated sets of conditions that systematically vary precipitants, salts, and pH to identify initial crystallization hits. | A standard first step in any crystallography project. |
| Advanced Force Fields (e.g., Amber ff14SB, CHARMM36m) | Mathematical models of interatomic interactions used in MD simulations to accurately predict protein dynamics and energetics [23]. | Critical for running MD simulations in solution or crystal environments. Force field choice impacts the accuracy of simulated dynamics. |
| TROSY-based NMR Experiments | Reduces signal line-widths in large molecules by suppressing relaxation effects, effectively extending the molecular weight limit for NMR [7]. | Essential for studying large proteins and complexes, often in combination with deuteration. |
| Electric Field Stimulation Cells | Experimental apparatus to apply precise electric fields to protein crystals, initiating synchronous conformational changes for time-resolved studies [22]. | Used specifically in EFX experiments to trigger and observe functional motions. |
The choice between X-ray crystallography and NMR spectroscopy is not a matter of selecting the superior technique, but of choosing the right tool for the biological question. Crystallography provides unparalleled resolution and detail for stable states, while NMR offers a unique window into flexibility, dynamics, and solution-state heterogeneity. The future of structural biology lies in integrating these complementary views. The combination of time-resolved crystallography, advanced NMR, and molecular dynamics simulations is creating a powerful new paradigm—one that moves beyond static snapshots to deliver a dynamic, mechanistic understanding of how proteins function as sophisticated molecular machines. This integrated approach is essential for tackling complex problems in structural biology and rational drug design.
From Structures to Drugs: Practical Applications in Discovery and Design
Structure-based drug design (SBDD) represents a cornerstone of modern pharmaceutical research, providing a rational framework for transforming initial hits into optimized drug candidates by leveraging detailed 3D structural information [7]. For decades, X-ray crystallography has dominated this field, enabling researchers to visualize protein-ligand complexes at atomic resolution. However, this technique captures a single, static snapshot and faces inherent limitations, including low success rates in crystallization and an inability to observe hydrogen atoms or dynamic behaviors [7] [24]. These shortcomings are particularly problematic for studying complex biological systems where molecular flexibility and transient interactions are critical for function.
Solution-state Nuclear Magnetic Resonance (NMR) spectroscopy has emerged as a powerful alternative that complements and extends the capabilities of crystallographic methods. NMR-SBDD provides detailed information about protein-ligand complexes directly in solution, capturing their dynamic nature and revealing molecular interactions that are often invisible to other techniques [7] [25]. With continuous advancements in NMR hardware, isotopic labeling strategies, and computational workflows—including the integration of artificial intelligence—NMR is overcoming traditional limitations and establishing itself as an indispensable tool for modern drug discovery pipelines [7] [26].
Table 1: Core Techniques in Structure-Based Drug Design
| Technique | Key Applications in SBDD | Key Limitations |
|---|---|---|
| X-ray Crystallography | High-resolution static structures; workhorse for SBDD [27] | Requires high-quality crystals; cannot study dynamics; "blind" to hydrogen atoms [7] |
| Cryo-EM | Structural analysis of large complexes and membrane proteins [24] | Limited resolution for small proteins; specialized equipment required [7] [24] |
| Solution-State NMR | Studying dynamics, weak interactions, and protein-ligand ensembles in solution [7] [25] | Molecular weight limitations; requires isotope labeling; complex data analysis [7] [24] |
Comparative Analysis: NMR-SBDD Versus Traditional Structural Methods
Fundamental Advantages of the NMR Approach
Solution-state NMR spectroscopy offers distinctive capabilities that make it particularly valuable for drug discovery:
Direct Observation of Molecular Interactions: NMR provides direct access to atomistic information, particularly through ¹H chemical shifts that report on hydrogen-bonding interactions. Protons with large downfield chemical shift values typically act as hydrogen bond donors, while upfield shifts indicate interactions with aromatic systems [7]. This direct observation contrasts with crystallography, where interactions are inferred from atomic proximity.
Solution-State Environment: NMR studies proteins in conditions closer to their native physiological state, avoiding potential artifacts induced by crystallization [7]. This is especially valuable for intrinsically disordered proteins, flexible linkers, and membrane-associated systems that often resist crystallization [7] [24].
Dynamic Information: Unlike the static snapshots provided by crystallography, NMR can capture the dynamic behavior of protein-ligand complexes, including multiple bound states, conformational entropy, and differential hydration effects [7]. This provides critical insights into the subtle interplay between enthalpy and entropy that governs binding affinity.
Hydrogen Atom Resolution: NMR is uniquely capable of detecting hydrogen atoms and their interactions, including hydrogen bonds and non-classical interactions, which are essentially invisible to X-ray crystallography [7]. This information is crucial for understanding the precise geometry of binding interactions.
Technical Comparisons Across Structural Biology Techniques
Table 2: Quantitative Comparison of Structural Biology Techniques in Drug Discovery
| Parameter | X-ray Crystallography | Cryo-EM | Solution-State NMR |
|---|---|---|---|
| Typical Resolution | Atomic (0.5-2.5 Å) [27] | Near-atomic to atomic (1.5-4 Å) [24] | Atomic (0.5-3 Å) [7] |
| Sample Requirements | High-quality single crystals | Vitreous ice (no crystals) | Solution in appropriate buffer |
| Protein Size Range | No strict upper limit | >50 kDa optimal [7] | Typically <50 kDa [24] |
| Success Rate (from cloning) | ~25% yield crystals [7] | Varies widely | Higher for challenging targets [7] |
| Throughput | High with established soaking systems [7] | Medium | Medium to high [7] |
| Observation of Hydrogens | No [7] | No | Yes [7] |
| Dynamic Information | No [7] | Limited | Yes [7] |
Experimental Protocols and Workflows in NMR-SBDD
Core Methodologies for Protein-Ligand Complex Characterization
NMR-SBDD employs diverse experimental approaches to elucidate protein-ligand interactions:
Chemical Shift Perturbation: This method monitors changes in NMR chemical shifts when a ligand binds to a protein, providing information about the binding interface and affinity [25]. The technique is particularly valuable for mapping interaction surfaces and studying weak interactions that might be missed by other methods.
NOESY (Nuclear Overhauser Effect Spectroscopy): NOESY experiments measure through-space dipolar couplings between nuclei, providing distance restraints critical for determining 3D structures of protein-ligand complexes [28]. Recent advancements have integrated AI-assisted peak assignment, dramatically reducing analysis time from months to hours [28].
19F-NMR Screening: Fluorine NMR has emerged as a powerful screening tool due to the high sensitivity of ¹⁹F chemical shifts to environmental changes. This approach enables efficient fragment-based screening and can probe protein interactions both in vitro and in cellular environments [25] [29].
TROSY (Transverse Relaxation-Optimized Spectroscopy): For larger proteins (>50 kDa), TROSY-based experiments overcome traditional size limitations by optimizing relaxation properties, extending the molecular weight range accessible to NMR [7].
AI-Enhanced Workflows for Accelerated Analysis
Recent advances have integrated artificial intelligence to address traditional bottlenecks in NMR data analysis:
AI-NMR Workflow Integration - Figure 1: Modern NMR-SBDD integrates AI-based conformer generation with experimental validation.
The FAAST (iterative Folding Assisted peak ASsignmenT) pipeline represents a breakthrough in NMR analysis, combining experimental data with the RASP (Restraints Assisted Structure Predictor) model to assign NOESY peaks and generate structural ensembles in hours rather than months [28]. This approach demonstrates particularly strong performance for multi-domain proteins and those with limited sequence homologs, where traditional methods often struggle [28].
Successful implementation of NMR-SBDD requires specialized reagents and computational resources:
Table 3: Key Research Reagent Solutions for NMR-SBDD
| Reagent/Resource | Function/Purpose | Application Example |
|---|---|---|
| ¹³C-labeled Amino Acid Precursors | Selective side-chain labeling for reduced spectral complexity [7] | Studying large proteins and specific molecular interactions |
| 19F-Labeling Probes | Sensitive environmental reporters for protein studies [25] [29] | Fragment screening and in-cell applications |
| Cryogenic NMR Probes | Enhanced sensitivity for studying low-concentration samples [29] | High-throughput screening and unstable proteins |
| Hyperpolarization Agents | Signal enhancement for low-abundance species [29] | Transient states and metabolic studies |
| RASP Software | AI-driven structure prediction with experimental restraints [28] | Multi-domain protein structure determination |
| FAAST Pipeline | Automated NOESY assignment and structure generation [28] | Rapid structural analysis of protein-ligand complexes |
Case Studies and Experimental Data
Application to Challenging Drug Targets
NMR-SBDD has demonstrated particular value for target classes that resist characterization by traditional methods:
Intrinsically Disordered Proteins: NMR has directly detected NH-π hydrogen bonds on the surface of an intrinsically disordered peptide, illustrating its unique capability to study systems that are inaccessible to crystallography [30]. Such interactions are crucial for understanding molecular recognition in these challenging targets.
Multi-Domain Proteins: For the multi-domain protein 6XMV, where both AlphaFold2 and MEGA-Fold incorrectly predicted relative domain positions, the incorporation of NMR-derived restraints corrected inter-domain positioning, improving the TM-score from 0.51 to 0.79 [28]. This highlights NMR's value in validating and correcting computational models.
Proteins with Limited Sequence Homology: For viral protein 7NBV, which has only three sequences in its multiple sequence alignment, the addition of NMR restraints progressively improved structure quality, increasing the TM-score from 0.43 to 0.77 with just 50 restraints [28].
Quantitative Performance Assessment
Table 4: Experimental Validation Data for NMR-SBDD Applications
| Target System | Traditional Method Result | NMR-SBDD Enhancement | Validated Improvement |
|---|---|---|---|
| Multi-domain Protein (6XMV) | AF2 TM-score: 0.51 [28] | +55% accuracy | TM-score: 0.79 with restraints [28] |
| Few-MSA Protein (7NBV) | Baseline TM-score: 0.43 [28] | +79% accuracy | TM-score: 0.77 with 50 restraints [28] |
| NOESY Assignment | Conventional time: months [28] | 90%+ time reduction | FAAST pipeline: hours [28] |
Integrated Structural Biology: The Future of Drug Discovery
The most powerful applications of structural biology in drug discovery emerge from integrating multiple techniques rather than relying on any single method. NMR spectroscopy provides unique insights into dynamic processes and hydrogen-bonding networks that complement high-resolution static structures from crystallography [7]. Meanwhile, cryo-EM offers capabilities for studying large complexes that may challenge both crystallography and NMR [24]. The emerging paradigm involves using computational frameworks, particularly AI-based systems like AlphaFold-NMR, to harmonize data from these diverse experimental sources [31].
This integrative approach enables researchers to select conformational states from AI-generated ensembles that best explain experimental NMR data, revealing previously hidden structural states that provide novel insights into protein structure-dynamic-function relationships [31]. As these technologies continue to mature, the drug discovery pipeline will increasingly leverage the complementary strengths of multiple structural biology techniques, computational prediction, and experimental validation to tackle increasingly challenging therapeutic targets.
Leveraging Crystallography for High-Throughput Soaking and Screening
In modern drug discovery, determining the three-dimensional structure of target proteins and their complexes with potential drug molecules provides an invaluable blueprint for rational drug design. Among the techniques available, X-ray crystallography stands as the dominant method, accounting for approximately 84% of all structures deposited in the Protein Data Bank (PDB), while NMR spectroscopy and cryo-electron microscopy contribute the remainder [32]. The technique of high-throughput crystallographic soaking has emerged as a particularly powerful methodology for rapidly screening countless potential drug compounds against crystalline protein targets. This approach involves immersing pre-formed protein crystals in solutions containing small molecule ligands or fragments, allowing these compounds to diffuse through the crystal lattice and bind to their target sites. When performed at scale with automation, this method enables the structural characterization of dozens to hundreds of protein-ligand interactions in a time-efficient manner.
Framed within the broader thesis of comparing crystal structures with NMR ensembles research, this guide objectively examines the performance, capabilities, and limitations of high-throughput crystallographic soaking and screening against its NMR-based counterparts. While crystallography provides high-resolution structural snapshots, NMR ensembles capture dynamic conformational states in solution—a complementary perspective that is crucial for understanding protein-ligand interactions in more physiologically relevant conditions [7]. The following sections provide a detailed comparison of these techniques, supported by experimental data and methodological protocols to guide researchers in selecting the optimal approach for their structural biology challenges.
Technical Comparison: Crystallography Versus NMR for Screening
Table 1: Key Characteristics of Structural Biology Techniques for Drug Discovery
| Parameter | X-ray Crystallography | NMR Spectroscopy |
|---|---|---|
| Throughput Capability | High (especially with fragment soaking) [33] [34] | Medium (requires individual samples or mixtures) [35] |
| Sample Requirement | 5 mg at ~10 mg/mL; highly pure, crystallizable protein [32] | >200 μM in 250-500 μL; isotope labeling often required [32] |
| Structure Type | Static snapshot | Dynamic ensemble in solution |
| Molecular Weight Range | Essentially unlimited [32] | Typically <50 kDa (with technical advancements expanding this) [7] |
| Hydrogen Atom Detection | Poor (hydrogen atoms largely invisible) [7] | Excellent (direct detection of hydrogen bonds and protonation states) [7] |
| Ligand Binding Information | Inferred from electron density | Directly measured through chemical shifts and relaxation [36] |
| Dynamic Behavior | Limited information | Comprehensive data on kinetics and dynamics [7] |
| Water Molecule Detection | ~80% of bound waters observable [7] | Full hydration networks detectable |
| Typical Screening Application | Fragment screening via soaking [32] | Hit identification and validation [35] |
Table 2: Performance Metrics for High-Throughput Soaking Technologies
| Metric | Traditional Soaking | Advanced Platforms (e.g., SmartSoak) | NMR-Based Screening |
|---|---|---|---|
| Setup Time | Weeks to months [33] | As little as 2 weeks [33] | Days to weeks [35] |
| Success Rate | Industry standard | ≥50% higher than conventional [33] | High for initial hit identification [35] |
| Ligand Concentration | Limited by crystal damage | Supports higher concentrations [33] | Limited by solubility and signal |
| Target Flexibility | Requires stable crystals | Improved for challenging targets [33] | Handles flexible systems well [7] |
| Data Completeness | High for well-diffracting crystals | Comprehensive with QC package [33] | Partial for large systems |
Experimental Protocols and Methodologies
High-Throughput Crystallographic Soaking Workflow
Protein Crystallization Preparation The process begins with the generation of reproducible, high-quality protein crystals. Using purified protein samples at concentrations typically around 10 mg/mL, initial crystallization conditions are identified through sparse matrix or statistically designed screening approaches [37]. Robotic liquid handling systems dispense nanoliter volumes of protein and screening solutions, with incubation occurring at controlled temperatures (commonly 4°C and 20°C) [34]. For membrane proteins, specialized mimetics such as lipidic cubic phases (LCP) are often employed to create a more native-like environment [32]. The HTX Lab at EMBL Grenoble exemplifies this automated approach, using crystal farms with capacity to handle hundreds of crystallization experiments simultaneously [34].
Soaking System Establishment Once reproducible crystals are obtained, establishing a robust soaking system is crucial. The proprietary SmartSoak technology exemplifies an optimized approach that systematizes this traditionally trial-and-error process [33]. Key considerations include:
- Identification of cryoprotectants compatible with the crystal system
- Determination of optimal ligand concentration ranges
- Optimization of soaking duration to balance between ligand penetration and crystal integrity
- Development of harvesting protocols that minimize crystal damage
Advanced facilities employ automated CrystalDirect harvesters capable of processing and freezing up to 400 crystals per operation cycle [34].
Ligand Soaking and Data Collection The actual soaking process involves transferring crystals to solutions containing the ligand of interest, typically using acoustic dispensing systems for precise volume control [34]. Soaking times range from minutes to hours, depending on ligand properties and crystal characteristics. After soaking, crystals are cryocooled in liquid nitrogen to preserve their structure during data collection. Synchrotron sources provide the high-intensity X-rays needed for rapid data collection, with facilities like the ESRF-EMBL Joint Structural Biology Group offering dedicated beamlines for high-throughput structural biology [34].
Data Processing and Structure Determination Diffraction data processing involves indexing, integrating, and scaling the collected images to produce structure factor amplitudes. Molecular replacement typically serves as the primary phasing method when similar structures are available. For fragment screening campaigns, specialized software like PanDDA (Pan-Dataset Density Analysis) helps identify weak ligand density across multiple datasets [32]. The final structural models are refined iteratively, balancing agreement with experimental data and proper stereochemistry.
NMR-Based Screening Workflow
Sample Preparation and Isotope Labeling NMR screening requires highly soluble, stable protein samples at concentrations typically above 200 μM in volumes of 250-500 μL [32]. For proteins larger than 5 kDa, isotopic labeling with ¹⁵N and ¹³C is necessary, most commonly achieved through recombinant expression in E. coli grown in defined media containing these isotopes as the sole nitrogen and carbon sources [32]. Specific labeling strategies, such as selective side-chain labeling, can simplify spectra and focus on specific interaction sites [7].
Ligand-Observed Screening (1D Techniques) Initial fragment screening typically employs one-dimensional ligand-observed methods that don't require protein isotopic labeling:
- Saturation Transfer Difference (STD): Identifies compounds that bind weakly by selectively saturating protein resonances and detecting magnetization transfer to bound ligands [36] [35].
- Water-LOGSY (Water-Ligand Observed via Gradient Spectroscopy): Exploits water-mediated nuclear Overhauser effects (NOEs) to detect binding, particularly effective for identifying fragments with poor solubility [36] [35].
- T₁ρ relaxation measurements: Binding enhances ligand relaxation rates; this method enables affinity estimates and identification of interacting functional groups [36].
Protein-Observed Screening (2D Techniques) For hit validation and binding site identification, two-dimensional protein-observed methods are employed:
- Chemical Shift Perturbation (CSP): Monitors changes in chemical shifts of protein resonances upon ligand binding, identifying interaction sites [36] [35].
- SAR by NMR: Systematically uses chemical shift perturbations to guide fragment linking and optimization [36].
Structure Calculation of Protein-Ligand Complexes For successful hits, more extensive NMR experiments are conducted to determine the three-dimensional structure of the complex. NOE-based distance restraints, residual dipolar couplings, and chemical shift-derived torsion angles are used in computational structure calculation protocols to generate structural ensembles representing the solution state of the complex [7].
Essential Research Reagent Solutions
Table 3: Key Research Reagents and Materials for Structural Biology Screening
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Crystallization Screens | Sparse matrix or statistical sampling of chemical space to identify initial crystallization conditions [37] | Commercial screens available (e.g., from Hampton Research, Molecular Dimensions) |
| Lipidic Cubic Phase (LCP) Materials | Membrane mimetic for crystallizing membrane proteins [32] | Monolein-based systems commonly used for GPCRs and transporters |
| Detergents | Solubilize and stabilize membrane proteins in aqueous solutions [5] | Vary in properties (e.g., DDM, OG, LDAO); selection is empirical |
| Isotope-Labeled Nutrients | ¹⁵N-ammonium salts and ¹³C-glucose for producing labeled proteins for NMR [32] | Required for protein-observed NMR studies of proteins >5 kDa |
| Cryoprotectants | Prevent ice formation during crystal cryocooling [34] | Glycerol, ethylene glycol, or various sugars commonly used |
| Fragment Libraries | Collections of 500-15,000 small molecules (<300 Da) for initial screening [35] | Designed following "Rule of Three" for optimal physicochemical properties |
Comparative Analysis and Strategic Implementation
Method-Specific Limitations and Considerations
Crystallography-Specific Challenges The primary bottleneck in crystallography remains obtaining well-diffracting crystals, with statistics indicating that only approximately 25% of proteins that are successfully cloned, expressed, and purified yield crystals suitable for structure determination [7]. This challenge is particularly acute for membrane proteins and highly flexible targets. Additionally, the molecular interactions observed in crystal structures are inferred from electron density maps rather than directly measured, and hydrogen bonding information must be deduced indirectly since hydrogen atoms are not directly visualized in most X-ray structures [7]. Crystallography also provides limited information about the dynamic behavior of protein-ligand complexes, capturing primarily a single, static snapshot of the bound state.
NMR-Specific Limitations NMR spectroscopy faces inherent sensitivity limitations, typically requiring protein concentrations in the hundreds of micromolar range, which can be challenging to achieve for some targets [32]. The technique also has practical size limitations, with traditional solution-state NMR becoming increasingly challenging for proteins larger than 50 kDa, though technical advancements like TROSY-based experiments are continually pushing this boundary [7]. NMR structure determination remains more time-consuming than crystallography for high-throughput applications, with data collection and analysis requiring days to weeks compared to hours for routine crystal structures [35].
Synergistic Applications in Drug Discovery
The most effective drug discovery pipelines strategically leverage both crystallography and NMR to overcome their individual limitations. Crystallography excels at providing high-resolution structural models that efficiently guide medicinal chemistry optimization, while NMR provides critical information about dynamics, hydration, and allosteric effects that complement the static crystallographic snapshots [7]. This synergy is particularly powerful in fragment-based drug discovery (FBDD), where NMR often identifies initial fragment hits and characterizes their binding mode, followed by crystallography to provide detailed structural information for optimization [35]. Statistical evidence demonstrates this complementarity, with NMR being used in approximately 80% of clinical compounds for initial hit identification, while crystallography becomes increasingly dominant in later optimization stages [35].
For challenging targets where crystallization proves difficult, NMR-driven structure-based drug design (NMR-SBDD) provides an alternative approach that combines selective isotope labeling with advanced computational workflows to generate protein-ligand ensembles suitable for guiding optimization [7]. This approach is particularly valuable for studying proteins with intrinsic flexibility or those that undergo conformational changes upon ligand binding—scenarios that are often difficult to capture using traditional crystallographic approaches.
High-throughput crystallographic soaking represents a powerful methodology for accelerating structure-based drug discovery, particularly when implemented with advanced automation and processing pipelines. The technology enables rapid structural characterization of dozens to hundreds of protein-ligand interactions, providing invaluable insights for medicinal chemistry optimization. When evaluated against NMR-based approaches, crystallography offers superior throughput and resolution for well-behaved targets that form quality crystals, while NMR provides unique capabilities for studying dynamics, solvent interactions, and challenging targets that resist crystallization.
The most successful structural biology programs strategically integrate both techniques, leveraging their complementary strengths to build a more comprehensive understanding of protein-ligand interactions. As both technologies continue to advance—with improvements in automation, data collection, and computational analysis—their synergistic application will undoubtedly continue to drive innovations in drug discovery and development.
Fragment-based drug discovery (FBDD) has evolved into a mainstream strategy for identifying novel therapeutic compounds, particularly against challenging biological targets. This approach involves screening small, low molecular weight compounds (fragments) and optimizing them into potent drug leads [38] [39]. Two principal techniques have emerged as powerful tools in FBDD campaigns: Nuclear Magnetic Resonance (NMR) spectroscopy, renowned for its solution-based detection of weak affinity interactions, and X-ray crystallography, valued for its atomic-resolution structural visualization of binding modes [35] [40]. While each technique possesses distinct strengths and limitations, their integration creates a synergistic pipeline that accelerates lead discovery. This guide objectively compares the performance of these methodologies within the context of FBDD, examining how their combined application provides a more complete understanding of fragment binding than either technique could deliver independently, ultimately advancing the broader thesis of comparing crystal structures with NMR ensembles in research.
Table 1: Core Technique Profiles in FBDD
| Feature | NMR Spectroscopy | X-ray Crystallography |
|---|---|---|
| Primary Role in FBDD | Detection of weak binding events and affinity measurement [38] | High-resolution visualization of binding modes and sites [40] |
| Typical Affinity Detection Range | Millimolar to high micromolar [38] [39] | Not a direct affinity method; infers affinity from occupancy [41] |
| Sample State | Solution (native-like conditions) [42] | Crystalline solid state [43] |
| Key Advantage | Detects binding without prior functional knowledge [38] | Provides atomic-level structural data for optimization [32] |
| Major Technical Hurdle | Protein size limitations for target-detected methods [32] | Requirement for robust, reproducible crystallization [43] [42] |
Experimental Protocols and Workflows
NMR-Based Fragment Screening Methodologies
NMR screening employs two principal experimental paradigms: ligand-detected and target-detected methods. Ligand-detected NMR methods, including Saturation Transfer Difference (STD) and Water-Ligand Observed via Gradient Spectroscopy (Water-LOGSY), monitor changes in the fragment's NMR signals upon binding to the protein target [35]. These techniques are particularly valuable because they do not require isotopic labeling of the protein, can be performed with small amounts of protein (nanomoles), and are amenable to high-throughput screening of fragment mixtures [38] [35]. Conversely, target-detected NMR (e.g., 2D (^{1}H)-(^{15}N) HSQC) monitors perturbations in the protein's NMR signals upon fragment binding. This approach requires (^{15}N)-labeled (and sometimes (^{13}C)-labeled) protein but provides crucial information about the binding site and can validate binding events [35]. The protein size limit for this method is typically around 25-30 kDa, though advanced techniques can extend this range [32].
Crystallographic Fragment Screening Protocols
Crystallographic fragment screening involves soaking protein crystals in solutions containing high concentrations of fragments [44]. The process requires a reliable supply of high-quality, reproducible crystals that can withstand handling and soaking in DMSO-containing fragment solutions [40]. Modern high-throughput platforms, such as the XChem facility at Diamond Light Source or the FragMAX platform at MAX IV Laboratory, have dramatically increased the throughput of this method, enabling the collection and processing of hundreds of datasets per day [40]. A key requirement is that the crystal packing must allow access to the ligand-binding site. Crystals with different packing (space groups) can be beneficial as they may reduce the incidence of false negatives by providing alternative access routes [40]. The final output is an electron density map into which fragment hits can be modeled, directly revealing their binding geometry and protein interactions.
Performance Comparison and Experimental Data
Quantitative Technique Comparison
The selection between NMR and crystallography for FBDD is often dictated by project-specific goals, target properties, and resource availability. Objective performance data reveals distinct operational profiles for each technique.
Table 2: Performance Metrics and Operational Characteristics
| Parameter | NMR Spectroscopy | X-ray Crystallography |
|---|---|---|
| Typical Hit Rate | Varies; generally higher than HTS [39] | Reported 13-16% for focused libraries [40] |
| Sample Consumption | ~100-500 μg per data point (ligand-detected) [32] | Single crystal per fragment/mixture [40] |
| Throughput | Minutes per sample (1D ligand-detected) [35] | Hundreds of datasets per day (automated) [40] |
| Affinity Information | Direct measurement (K~d~) possible [38] [35] | Indirect; inferred from occupancy [41] |
| Binding Site Info | Yes (via CSP mapping for labeled proteins) [35] | Direct visualization [44] [40] |
| Structure Requirement | Not required for ligand-detected screening [38] | Essential; high-quality crystals mandatory [43] |
Analysis of Complementary Strengths and Limitations
The quantitative data reveals a fundamental complementarity. NMR spectroscopy excels as a primary screening tool, especially for novel targets where binding sites are unknown. Its ability to detect very weak interactions (K~d~ in the millimolar range) and directly quantify binding affinity without requiring crystallization makes it invaluable for the initial identification of fragment hits [38] [39]. Furthermore, NMR is less prone to the false-positive results that can plague other screening techniques because it directly observes the binding event [38]. A significant limitation, however, is that for target-detected methods, the protein molecular weight is a constraint, and the technique does not automatically provide the detailed three-dimensional structural picture needed for efficient chemical optimization.
X-ray crystallography, in contrast, provides an unambiguous, atomic-resolution snapshot of the protein-fragment complex [40]. This detailed structural information is paramount for guiding medicinal chemistry efforts, as it reveals precise atom-atom interactions, solvation patterns, and protein conformational changes induced by binding. The main limitation is the stringent requirement for a robust, high-throughput crystallization system that produces crystals tolerant to soaking and diffract to a resolution sufficient to identify small fragments [44] [42]. For some targets, particularly membrane proteins, this can be a major bottleneck. Additionally, crystallography does not directly measure binding affinity.
Integrated Screening Strategy: A Case-Driven Pipeline
The most successful FBDD campaigns leverage the strengths of both techniques in a coordinated pipeline. The synergistic workflow typically begins with a broad screen using ligand-detected NMR to identify bona fide binders from a large library under native solution conditions [38] [35]. Hits are then validated and their binding sites roughly mapped using target-detected NMR if feasible. These validated, site-assigned fragments subsequently serve as candidates for crystallographic screening. Focusing crystallographic efforts on a pre-validated subset of fragments maximizes the use of precious crystal resources and beamtime.
Once a crystal structure of a protein-fragment complex is obtained, it creates a powerful feedback loop. The structure explains the chemical basis for the binding affinity measured by NMR and reveals vectors for fragment growth. As chemists synthesize elaborated compounds, NMR can rapidly assess whether the new analogs maintain binding and quantify improvements in affinity, while iterative crystallography provides structural validation and guides further optimization [35] [40]. This cycle of design (informed by structure), synthesis, and biophysical validation (by NMR and crystallography) efficiently progresses a weak fragment into a potent lead candidate.
Table 3: Essential Research Reagent Solutions
| Reagent / Material | Function in FBDD |
|---|---|
| Rule-of-Three Compliant Fragment Library | A curated collection of small molecules (MW <300, ClogP ≤3, HBD/HBA ≤3) designed to efficiently explore chemical space [38] [40]. |
| Isotope-Labeled Protein (¹⁵N, ¹³C) | Essential for target-detected NMR studies (e.g., 2D (^{1}H)-(^{15}N) HSQC) to map fragment binding sites [35] [32]. |
| Crystallization Plates & Reagents | Tools and chemical screens for obtaining high-quality, reproducible protein crystals suitable for high-throughput soaking [40]. |
| Synchrotron Beamline Access | High-flux X-ray source enabling rapid data collection from hundreds of crystals, a cornerstone of modern fragment screening [43] [40]. |
| DMSO-d⁶ | Deuterated dimethyl sulfoxide; a common solvent for preparing fragment stock solutions for both NMR and crystallography experiments [40]. |
NMR affinity screening and crystallographic detection are not competing but fundamentally complementary technologies in the FBDD arsenal. NMR serves as a sensitive, solution-based tool for identifying and validating weak fragment binders under physiologically relevant conditions, while X-ray crystallography provides the high-resolution structural blueprint essential for rational chemical optimization. The integration of these techniques into a cohesive pipeline—where NMR identifies initial hits and monitors affinity, and crystallography visualizes binding modes—creates a powerful engine for drug discovery. This synergy is particularly critical for challenging targets where conventional methods fail. As both technologies continue to advance, with improvements in NMR sensitivity, cryo-EM, and computational methods, their combined application will remain a cornerstone of structural biology-driven drug design, offering a comprehensive approach to understanding both the structure and dynamics of protein-ligand interactions.
Understanding allostery—the process by which a binding event at one site of a protein influences activity at a distant, functional site—requires a detailed map of molecular interactions, including hydrogen bonding and hydration networks. Two primary experimental techniques, X-ray crystallography and Nuclear Magnetic Resonance (NMR) spectroscopy, are at the forefront of providing these atomic-resolution insights. While crystallography has delivered the majority of high-resolution static snapshots found in the Protein Data Bank, NMR spectroscopy is unequaled in its ability to detect structural and dynamical changes in biomolecules under native solution conditions [45]. This guide provides an objective comparison of these two powerful techniques, focusing on their application in mapping the intricate interplay of hydrogen bonds, hydration, and allosteric pathways. The choice between them is not a matter of which is superior, but which is most appropriate for the specific biological question at hand, particularly when studying dynamic allosteric regulation.
Fundamental Principles and Comparative Capabilities
At their core, X-ray crystallography and NMR spectroscopy rely on fundamentally different physical principles to extract structural information. X-ray crystallography infers atomic positions by measuring the diffraction pattern produced when X-rays interact with a crystalline lattice of the protein. The resulting electron density map is interpreted to build an atomic model. This process excels at providing high-resolution, static snapshots but traditionally obscures the dynamic ensemble nature of proteins, especially when crystals are cryo-cooled, a process that can alter conformational distributions [46].
In contrast, NMR spectroscopy probes the local magnetic environments of atomic nuclei (e.g., 1H, 15N, 13C) in solution. Parameters such as chemical shift, scalar coupling, and relaxation rates provide a wealth of information on bond distances, angles, dynamics, and interactions. NMR is uniquely capable of characterizing equilibrium dynamics and conformational fluctuations on timescales from picoseconds to seconds and beyond, making it ideal for studying the dynamic nature of allostery [47].
Table 1: Core Methodological Principles and Outputs
| Feature | X-ray Crystallography | Solution NMR Spectroscopy |
|---|---|---|
| Fundamental Principle | Measurement of X-ray diffraction from a crystal lattice | Detection of nuclear spin transitions in a magnetic field |
| Sample State | Static, crystalline solid | Dynamic, solution (or occasionally solid) state |
| Primary Output | Single, time-averaged electron density map; atomic coordinates | Ensemble of structures consistent with experimental restraints |
| Key Measurables | Structure factor amplitudes (& phases), B-factors | Chemical shifts, J-couplings, NOEs, relaxation rates (R₁, R₂, NOE), residual dipolar couplings |
| Inherent Dynamics | Limited; inferred from B-factors or disorder | A core strength; directly probed across wide time scales |
The most significant distinction lies in their treatment of protein dynamics and heterogeneity. Crystallography typically produces a single model that represents a time- and space-averaged conformation. While multi-conformer models and tools like qFit can extract heterogeneity information, this is most accurate at room temperature [46]. NMR, by its nature, defines an ensemble of structures and can quantitatively probe the kinetics and thermodynamics of conformational exchange processes that are fundamental to allosteric mechanisms [47].
Technical Performance and Application in Allostery Research
The practical application of these techniques reveals distinct strengths and limitations. The quality of a crystallographic model is critically dependent on resolution, which determines the clarity of the electron density map. High-resolution (<1.5 Å) data are required to unambiguously define hydrogen atom positions and precise water networks. However, a significant technical challenge is X-ray radiation damage. At cryogenic temperatures, damage can introduce artifactual conformational heterogeneity, complicating structural interpretation. Room-temperature crystallography provides more accurate ensemble information but is more susceptible to radiation-induced decay [46].
NMR's strength is its site-specific resolution of dynamics. Experiments like Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersion can quantitatively characterize "invisible" excited states that are populated as low as 0.5%, revealing how allosteric ligands redistribute conformational ensembles on micro- to millisecond timescales [47]. For example, studies of the catabolite activator protein (CAP) showed that binding of the first cAMP ligand enhances dynamics across the protein, facilitating cooperative binding of the second ligand [47].
Table 2: Mapping Allostery and Hydration: A Technical Comparison
| Aspect | X-ray Crystallography | Solution NMR Spectroscopy |
|---|---|---|
| Hydrogen Bond Detection | Indirect, from atomic distances and angles; direct H-atom location requires ~1.2 Å resolution or better | Direct, via scalar J-couplings (hydrogen bonds); through-space correlations (NOEs) |
| Hydration Site Mapping | Excellent for identifying ordered, long-lived water molecules in high-resolution structures | Probes dynamics and residence times of water; detects protein-water NOEs for bound water |
| Allosteric Pathway Identification | Inferred from structural comparisons of different states (e.g., apo/holo); can miss dynamic contributions | Directly probes dynamic couplings and allosteric networks through relaxation and chemical shift perturbations |
| Sensitivity to Dynamics | Indirect (B-factors, multi-conformer models); can be limited by crystal packing and cryo-cooling | Direct and quantitative across picosecond-second timescales |
| Sample Requirements | Requires high-quality, well-diffracting crystals | Requires soluble, isotopically labeled (15N, 13C) protein; can be challenging for large complexes |
Hydration plays a critical and often allosteric role in protein function. Crystallography excels at pinpointing the locations of ordered water molecules within a structure. Computational studies leveraging these structures have shown that hydration sites are spatially fragmented and nonuniform, and that ligand binding can alter hydration at remote, allosteric sites [48]. NMR provides a complementary dynamic perspective. Techniques like water-NOE (wNOE) and water-ROE (wROE) experiments allow for site-specific detection of hydration dynamics near the protein surface. For instance, in the metalloregulator CzrA, zinc binding was shown to be entropically driven, a phenomenon attributed in part to the release of water molecules from the protein surface, an effect detectable by NMR [49].
Experimental Protocols for Key Assays
NMR Relaxation Dispersion to Probe Allosteric Dynamics
Objective: To characterize low-populated, energetically excited states and quantify conformational exchange processes on the micro- to millisecond timescale that are crucial for allosteric communication.
Protocol Summary:
- Sample Preparation: Prepare a uniformly 15N-labeled protein sample in a suitable buffer. For larger proteins (>25 kDa), perdeuteration is often necessary.
- Data Collection: Record a series of 1H-15N heteronuclear single quantum coherence (HSQC) spectra or TROSY-based variants for large proteins, using a CPMG pulse sequence. The experiment is performed at multiple magnetic field strengths (e.g., 600, 800 MHz) and with a varying frequency (νCPMG) of the applied refocusing pulses.
- Data Analysis: For each resolved backbone amide peak, the measured transverse relaxation rate (R2) is plotted against νCPMG to generate a relaxation dispersion profile.
- Quantitative Modeling: Profiles from multiple residues and field strengths are globally fitted to a kinetic model (e.g., a two-state exchange process A ⇌ B) to extract the exchange rate constant (kex), the population of the minor state (pB), and the chemical shift difference (Δω) between states for each nucleus [47].
Crystallographic Workflow for Conformational Ensemble Analysis
Objective: To extract information on conformational heterogeneity and alternative side-chain conformations from a single crystal at room temperature, providing accurate ensemble information.
Protocol Summary:
- Data Collection: Collect a complete, high-resolution (<1.5 Å) X-ray diffraction data set from a single protein crystal at room temperature (277-298 K), using a minimal crystal rotation range to mitigate radiation damage.
- Damage Monitoring: To assess damage, collect sequential data sets from the same crystal and monitor the decay of the overall diffraction intensity.
- Model Building and Refinement: Build an initial atomic model into the electron density map. Subsequently, use the software qFit to automatically build a multi-conformer model that represents the ensemble of states present in the crystal.
- Heterogeneity Quantification: Analyze the resulting multi-conformer model to calculate conformational order parameters (S2), which quantify the heterogeneity at each atomic position, integrating both harmonic (B-factors) and anharmonic (alternative conformers) motions [46].
Visualization of Experimental Workflows and Allosteric Concepts
The following diagrams illustrate the core workflows for the two techniques and a conceptual model of dynamic allostery.
Diagram 1: Comparative Structural Biology Workflows. This diagram contrasts the sequential steps of solution NMR spectroscopy, which culminates in a conformational ensemble, with those of X-ray crystallography, which typically produces a single, static model.
Diagram 2: Integrated View of Dynamic Allostery. This diagram illustrates how an allosteric effector binding event is communicated via a dynamic network, involving changes in protein dynamics and surface hydration, to regulate the active site.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful structural biology studies depend on high-quality materials and specialized reagents. The following table details key solutions used in the experiments cited throughout this guide.
Table 3: Key Research Reagent Solutions
| Reagent / Solution | Function / Description | Experimental Context |
|---|---|---|
| Isotopically Labeled Proteins | Proteins enriched with 15N and/or 13C for NMR detection; essential for multi-dimensional NMR experiments. | Required for backbone assignment, relaxation (CPMG) studies, and observing large proteins [47]. |
| Crystallization Screening Kits | Sparse matrix solutions (e.g., from Hampton Research, Jena Bioscience) to identify initial crystal growth conditions. | Fundamental first step for obtaining protein crystals for X-ray studies [50]. |
| Liquid Injection Systems | Microfluidic devices (e.g., GDVN, VF) for delivering a stream of microcrystals in serial crystallography. | Enables data collection at XFELs and synchrotrons with minimal sample consumption [50]. |
| Paramagnetic Probes (e.g., TEMPOL) | Small, stable nitroxide radicals used in NMR to probe surface accessibility and hydrophobicity. | Used to identify protein "hot spots" by measuring paramagnetic relaxation enhancement (PRE) [51]. |
| Software: qFit | Computational tool for automated modeling of multi-conformer structures into crystallographic electron density. | Critical for extracting accurate conformational ensemble information from room-temperature X-ray data [46]. |
| Software for RD Analysis | Programs (e.g., CATIA, ChemEx) for global fitting of relaxation dispersion data to extract kinetic/thermodynamic parameters. | Essential for quantifying micro- to millisecond dynamics from CPMG experiments [47]. |
Navigating Technical Challenges and Optimizing Multi-Method Workflows
Overcoming Crystallization Hurdles and NMR Assignment Bottlenecks
Structural biology provides the fundamental blueprint for modern drug discovery, with X-ray crystallography and Nuclear Magnetic Resonance (NMR) spectroscopy serving as two pivotal techniques for determining three-dimensional molecular structures. While X-ray crystallography has been the dominant workhorse, generating the majority of structures in the Protein Data Bank, it faces significant crystallization hurdles that limit its application to many biologically important targets. Simultaneously, NMR spectroscopy offers a powerful solution-state alternative but has traditionally been hampered by assignment bottlenecks and molecular weight limitations. This guide provides an objective comparison of these complementary techniques, focusing on their specific challenges and the modern solutions advancing structural-based drug discovery.
Technical Comparison: Crystallography vs. NMR in Drug Discovery
Table 1: Fundamental comparison of X-ray crystallography and NMR spectroscopy for structure determination
| Parameter | X-ray Crystallography | Solution-State NMR |
|---|---|---|
| Sample Requirement | High-quality crystals | Soluble, isotopically labeled protein |
| Sample State | Crystal lattice | Solution environment |
| Structural Output | Single static model | Ensemble of conformations |
| Hydrogen Atom Detection | Essentially invisible | Directly observable |
| Dynamic Information | Limited | Comprehensive |
| Typical Throughput | High (especially with soaking) | Medium |
| Membrane Protein Success | Challenging, requires special mimetics [5] | Challenging, requires membrane mimetics [5] |
| Molecular Weight Range | Virtually unlimited | Traditionally limited (~50 kDa), expanding with new methods [7] |
| Key Bottlenecks | Crystallization, crystal quality | Signal assignment, sensitivity, data analysis |
Table 2: Quantitative comparison of structural features between crystal and NMR structures of membrane proteins [5]
| Structural Characteristic | Crystal Structures | NMR Structures |
|---|---|---|
| RMSD in Membrane Region | Reference | Typically <5 Å compared to crystal structures |
| Transmembrane Region | Straighter helices | More structural variability |
| Stereo-chemical Correctness | Higher | Variable, can be improved with refinement |
| Packing Density | Tighter packing | Looser packing |
| Ensemble Convergence | Single model | Higher convergence in membrane regions |
Overcoming Crystallization Challenges in X-ray Crystallography
Fundamental Limitations and Practical Hurdles
The process of obtaining high-quality crystals represents the most significant bottleneck in X-ray crystallography. Statistics from a Human Proteome Structural Genomics project reveal that only 25% of successfully cloned, expressed, and purified proteins yield crystals suitable for X-ray structure determination [7]. This challenge is particularly acute for membrane proteins, which constitute important drug targets but require specialized membrane mimetics such as detergents or lipidic cubic phases [5] [52].
The fundamental assumptions in structure-guided drug design often overlook crystallography's limitations: protein structures are assumed to be completely correct, ligand interactions are presumed accurately modeled, and the determined structures are considered biologically relevant [53]. In practice, these assumptions frequently prove problematic, with examples of retracted structures due to fundamental errors in interpretation [53].
Experimental Protocols for Crystallization Optimization
Protein Engineering for Enhanced Crystallization
- In situ proteolysis: Limited digestion to remove flexible termini that inhibit crystallization [52]
- Reductive methylation: Chemical modification of lysine residues to enhance crystal contacts [52]
- Thermostabilizing mutations: Introduction of mutations to reduce flexibility, particularly for membrane proteins [5]
High-Throughput Crystallization Screening
- Automated screening with 96-well plates containing 0.2 μL protein solution per drop [52]
- Fluorescence-based thermal shift assays to identify optimal constructs and conditions [52]
- Dynamic light scattering to assess sample homogeneity and monodispersity [52]
Advanced Crystallization Techniques
- Lipid cubic phase crystallization for membrane proteins [5]
- Microseeding to improve crystal quality
- Co-crystallization with binding partners to stabilize specific conformations
Crystallization Workflow: This diagram illustrates the iterative process of protein crystallization, with the red arrow highlighting the common bottleneck requiring sample reengineering.
Addressing NMR Assignment Bottlenecks
Traditional Limitations and Modern Solutions
NMR spectroscopy provides unparalleled insights into protein dynamics and molecular interactions in solution but faces distinct challenges in signal assignment and data interpretation. Traditional NMR assignment bottlenecks include limited sensitivity, molecular weight constraints, and the time-intensive nature of spectral analysis [7].
Modern approaches have significantly alleviated these constraints through technological and methodological advances. Isotope labeling strategies, particularly selective side-chain labeling with 13C-labeled amino acid precursors, have streamlined the assignment process [7]. Hardware improvements including cryogenic probes and higher field magnets have enhanced sensitivity, while novel experiments such as TROSY-based techniques have extended the applicable molecular weight range [7].
Experimental Protocols for Streamlined NMR Analysis
Sample Preparation and Labeling Strategies
- Uniform 15N/13C labeling for backbone assignment
- Selective side-chain labeling with 13C-amino acid precursors to simplify spectra [7]
- Specific protonation patterns to reduce signal overlap
Advanced NMR Experiments
- TROSY-based experiments for large proteins (>50 kDa) [7]
- Long-lived coherences to extend relaxation times [7]
- Dynamic nuclear polarization for sensitivity enhancement [7]
Computational and Automation Approaches
- Automated peak picking and assignment algorithms [54] [55]
- Integration of artificial intelligence for spectral analysis [7]
- Computer-assisted structure elucidation (CASE) systems [55]
- Automated structure verification (ASV) systems [55]
NMR Structure Determination: This workflow highlights how software solutions (yellow ellipse) integrate at critical bottleneck points to automate assignment and structure calculation.
Integrated Structural Biology: Combining Complementary Approaches
The limitations of both techniques have spurred the development of integrated approaches that leverage their complementary strengths. NMR-driven structure-based drug design (NMR-SBDD) combines selective labeling and computational workflows to generate protein-ligand ensembles that capture dynamic information missing from static crystal structures [7].
For membrane proteins, comparative studies reveal that while crystal structures typically show higher stereochemical quality and tighter packing, NMR ensembles better represent structural flexibility and provide higher convergence in membrane regions [5]. Computational refinement using programs like Rosetta can improve NMR structure quality, reducing differences between techniques [5].
The emergence of AlphaFold predictions has added another dimension to structural biology, with studies indicating that AlphaFold predictions are typically more accurate than NMR structures, except in cases involving local dynamics where NMR may be superior [56]. This suggests a role for NMR in validating and refining computational predictions where necessary.
Essential Research Reagent Solutions
Table 3: Key research reagents and solutions for structural biology applications
| Reagent/Solution | Function | Application Context |
|---|---|---|
| Lipidic Cubic Phase Matrices | Membrane mimetic for crystallization | Membrane protein crystallography [5] |
| Detergents (DDM, DHPC, OG) | Solubilize membrane proteins | Both NMR and crystallography of membrane proteins [5] |
| 13C/15N-labeled Amino Acids | Isotopic labeling for NMR | Protein sample preparation for NMR [7] |
| Crystallization Screens | Matrix of conditions for initial crystal hits | High-throughput crystallography [52] |
| Cryoprotectants | Protect crystals during freezing | X-ray data collection at cryogenic temperatures |
| NMR Processing Software | Automated data analysis | Overcoming assignment bottlenecks [54] [55] |
Both crystallography and NMR spectroscopy continue to evolve, addressing their respective bottlenecks through methodological and technological innovations. Crystallography benefits from advanced automation, crystal engineering, and improved detection systems, while NMR advances through sophisticated labeling strategies, sensitivity enhancements, and artificial intelligence-driven analysis. The most powerful structural biology approaches strategically combine these complementary techniques, leveraging their respective strengths to provide comprehensive insights into molecular structure and function that drive rational drug design forward. As both fields continue to advance, the integration of computational predictions with experimental validation represents the next frontier in overcoming persistent challenges in structure determination.
Addressing Molecular Weight Limitations and Spectral Overlap in NMR
Nuclear Magnetic Resonance (NMR) spectroscopy is a powerful technique for determining the three-dimensional structures and dynamics of proteins and other biomolecules at atomic resolution. However, two persistent challenges have historically limited its application to larger biological systems: molecular weight limitations and spectral overlap. As molecular weight increases, slower tumbling rates in solution lead to broader resonance lines and signal loss, while the growing number of signals creates spectral overlap that complicates assignment and interpretation. This guide compares contemporary strategies and technological advancements that collectively address these challenges, enabling researchers to study increasingly complex systems with unprecedented detail.
Technical Comparison of Resolution-Enhancing NMR Methods
The table below summarizes the core methodologies developed to overcome molecular weight limitations and spectral overlap in NMR spectroscopy, with their respective experimental approaches and performance characteristics.
Table 1: Comparison of NMR Methods for Addressing Molecular Weight and Spectral Overlap Challenges
| Method | Technical Approach | Molecular Weight Applicability | Key Performance Metrics | Limitations |
|---|---|---|---|---|
| Ultrahigh Field (UHF) NMR [57] | 1.1-1.3 GHz spectrometers with HTS magnets; external (^2)H lock for field stability | Up to 144 kDa demonstrated [57] | 0.1-0.3 ppm linewidths in solids; 500+ backbone amides resolved in 2D spectra [57] | Instrument cost; field instability in new magnets; specialized probe design needed |
| FRI-NMR [58] | Finite-rate-of-innovation sampling; autocorrelation-based parameter estimation | Validated on 8.6 kDa ubiquitin and 23 kDa p50-NTD [58] | Resolves peaks separated by ≤10 Hz (below Fourier limit); accurate with 40% fewer data points [58] | Performance depends on signal-to-noise; validation needed for diverse systems |
| TROSY-based Methods [7] | Cross-correlated relaxation optimization; selective labeling schemes | Effective for systems >50 kDa [7] | Signal intensity preservation in large complexes; reduced overlap through selective observation [7] | Requires specific isotopic labeling schemes; pulse sequence optimization |
| Pure Shift NMR [59] | Homonuclear decoupling to collapse multiplet structure | Broad applicability across sizes | Converts multiplets to singlets; resolves overlap in crowded regions [59] | Sensitivity reduction; specialized pulse sequences required |
| Restraint-Assisted AI Prediction (RASP) [16] | Deep learning with experimental restraints integrated via MSA and IPA biases | Improved performance for multi-domain and few-MSA proteins [16] | TM-score improvement from 0.51 to 0.79 for problematic targets [16] | Dependent on restraint quality; training data limitations |
Experimental Protocols and Methodological Implementation
Ultrahigh-Field Solid-State NMR with External Deuterium Lock
Purpose: Achieve ultrahigh resolution (0.1-0.3 ppm) for high molecular-weight proteins in solid state. [57]
Sample Requirements: Microcrystalline protein samples or other solid-state preparations (e.g., membrane proteins in liposomes). For external lock capability, a D(_2)O-filled capillary is incorporated in the probe design. [57]
Equipment: 1.1 GHz or higher NMR spectrometer with HTS magnet; SSNMR probe with external (^2)H lock coil; magic angle spinning capability. [57]
Step-by-Step Workflow:
- Magnetic Field Stabilization: Implement external (^2)H lock system with D(_2)O-filled capillary positioned within magnetic field sweet spot. [57]
- Consensus Shimming: Simultaneously optimize magnetic field homogeneity for both sample coil ((^{13})C signal from adamantane) and lock coil ((^{2})H signal from D(_2)O). [57]
- Data Acquisition: Apply LOW-BASHD (Long-Observation-Window Band-Selective Homonuclear Decoupling) during acquisition to suppress (^{13})C-(^{13})C scalar couplings. [57]
- Processing: Process with optimized parameters to maintain resolution gains achieved experimentally. [57]
Critical Parameters:
- Lock coil must achieve (^2)H linewidth ≤10 Hz for optimal stability. [57]
- Magnetic field sweet spot in UHF magnets is approximately 35 mm, requiring compact probe design. [57]
- Sample temperature must be controlled to minimize field fluctuations. [57]
FRI-NMR for Super-Resolved Spectroscopy
Purpose: Resolve peaks separated below the Fourier resolution limit using parametric signal processing. [58]
Sample Requirements: Standard solution NMR samples; method particularly beneficial for rapidly relaxing signals or limited acquisition time. [58]
Equipment: Conventional NMR spectrometer; standard processing software with FRI-NMR implementation.
Step-by-Step Workflow:
- Data Acquisition: Acquire FID with appropriate number of points based on desired resolution enhancement. [58]
- Signal Modeling: Model FID as sum of exponentially damped sinusoids: (\tilde{f}(nTs) = \sum{l=1}^{L} al e^{(-\alphal + j\omegal)nTs} + w(n)). [58]
- Autocorrelation Analysis: Compute autocorrelation function to concentrate noise at zero-lag while preserving sum-of-exponential property. [58]
- Parameter Estimation: Extract frequencies (chemical shifts), amplitudes, and relaxation rates using FRI estimation techniques. [58]
- Spectral Reconstruction: Generate spectrum with Dirac impulses at estimated frequencies with corresponding amplitudes. [58]
Critical Parameters:
- For two closely spaced peaks (ΔF = 10 Hz), FRI-NMR requires approximately 40% fewer points than Fourier transform. [58]
- Effective even at low S/N (7.5-15 demonstrated). [58]
- Performance depends on accurate modeling of signal damping characteristics. [58]
AI-Assisted Structure Prediction with Experimental Restraints
Purpose: Improve structure prediction accuracy, particularly for multi-domain and few-MSA proteins, using experimental NMR restraints. [16]
Sample Requirements: NMR-derived distance restraints (NOE) and chemical shift data.
Equipment: RASP software implementation; conventional NMR data for restraint generation.
Step-by-Step Workflow:
- Restraint Preparation: Convert experimental NMR data (NOE peaks, chemical shifts) to distance restraints. [16]
- Model Input: Feed sequence information and restraints into RASP model as MSA and IPA biases. [16]
- Iterative Refinement: Use FAAST pipeline for iterative NOE assignment and structure generation. [16]
- Validation: Assess model quality via pLDDT scores and restraint compliance. [16]
Critical Parameters:
- Restraint quality more important than quantity; information content crucial. [16]
- pLDDT scores correlate with accuracy (r=0.71 with TM-score). [16]
- Model sensitive to inconsistent restraints; can identify problematic constraints. [16]
Workflow Visualization: Integrating Advanced NMR Methods
Diagram 1: Integrated NMR Workflow for High Molecular Weight Systems. This workflow shows how contemporary methods combine to address molecular weight and resolution challenges in structural biology.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagents and Materials for Advanced NMR Applications
| Reagent/Material | Function | Application Notes |
|---|---|---|
| High-Temperature Superconducting (HTS) Magnets [57] | Generate ultrahigh fields (1.1-1.3 GHz) for enhanced resolution | Essential for UHF NMR; requires specialized infrastructure [57] |
| External ²H Lock System [57] | Magnetic field stabilization via D₂O reference | Critical for UHF NMR stability; implemented in specialized probes [57] |
| Selective ¹³C-Labeled Amino Acids [7] | Specific side-chain labeling for spectral simplification | Enables targeted observation in complex systems [7] |
| Magic Angle Spinning (MAS) Probes [57] | Anisotropy averaging for resolution enhancement in solids | Required for SSNMR of membrane proteins, fibrils [57] |
| Paramagnetic Tags [7] | Generate long-range distance restraints via PRE | Extends observable distance range for large complexes [7] |
| Alignment Media [7] | Enable RDC measurement for orientation restraints | Provides global structural information [7] |
The methods compared in this guide demonstrate that molecular weight limitations and spectral overlap in NMR spectroscopy are no longer absolute barriers. Ultrahigh-field NMR provides the physical foundation for enhanced resolution, while sophisticated processing techniques like FRI-NMR extract more information from acquired data. AI-assisted approaches leverage experimental restraints to guide structure prediction, particularly for challenging targets. The optimal approach depends on the specific system: UHF SSNMR excels with very large, insoluble complexes; TROSY methods remain valuable for solution studies of large proteins; and FRI-NMR offers resolution enhancement when hardware limitations exist. As these technologies continue to mature and integrate, NMR spectroscopy is poised to expand its transformative impact on structural biology and drug discovery for increasingly complex biological systems.
Molecular dynamics (MD) simulations provide an atomistic view of biomolecular motion, but their predictive power is intrinsically linked to the accuracy of the force fields employed. This guide objectively compares the performance of contemporary biomolecular force fields by examining quantitative data from studies validated against nuclear magnetic resonance (NMR) spectroscopy and crystallography. We find that no single force field universally outperforms others across all systems and properties. Key lessons emerge: the treatment of electrostatics and the careful selection of validation metrics are critical, and simulations are susceptible to initial conditions and sampling limitations. This analysis provides researchers with a framework for selecting force fields and underscores the importance of continued refinement driven by experimental data.
Molecular dynamics simulations have matured into an indispensable tool for investigating biological processes, from protein folding and conformational changes to drug binding. The validity of these simulations, however, is ultimately determined by the accuracy of the underlying force fields that describe the forces between all atoms [60]. Force fields are mathematical models parameterized against experimental and quantum mechanical data, and their imperfections can lead to significant discrepancies between simulation outcomes and real-world behavior.
The need to validate and refine force fields is particularly acute when simulations are used to interpret data from experimental structural biology techniques like X-ray crystallography and NMR spectroscopy. These techniques provide complementary insights—crystallography offers high-resolution static snapshots, while NMR yields ensemble-averaged structural and dynamic information in solution. Discrepancies often arise when MD simulations fail to reproduce the dynamic profiles or conformational equilibria observed in NMR experiments, or when they drift excessively from starting crystal structures. Analyzing these discrepancies provides a powerful pathway for force field improvement. This guide compares the performance of popular force fields, summarizes supporting experimental data, and details the protocols used for their validation, all within the critical context of bridging computational models with experimental reality.
Comparative Performance of Biomolecular Force Fields
Systematic evaluations against experimental data, particularly from NMR, reveal that force field performance is not uniform but depends heavily on the biomolecular system and the property being measured.
Performance with Globular Proteins
Microsecond MD simulations of globular proteins like ubiquitin and the GB3 domain have been scrutinized against NMR data, including residual dipolar couplings (RDCs) and J-couplings across hydrogen bonds [61].
Table 1: Comparison of Force Field Performance for Proteins
| Force Field | Electrostatics Method | Protein Tested | Key Performance Metric | Result |
|---|---|---|---|---|
| AMBER99sb | Particle-Mesh Ewald (PME) | Ubiquitin & GB3 | RDC agreement (RRDC) | Best overall performance [61] |
| AMBER99sb | Cut-off/Reaction Field | Ubiquitin & GB3 | RDC agreement (RRDC) | Poor performance [61] |
| AMBER03 | PME | Ubiquitin & GB3 | RDC agreement (RRDC) | Moderate performance [61] |
| CHARMM22 | PME | Ubiquitin & GB3 | RDC agreement (RRDC) | Variable performance [61] |
| OPLS/AA | PME | Ubiquitin & GB3 | RDC agreement (RRDC) | Variable performance [61] |
| GROMOS96-43a1 | PME | Ubiquitin & GB3 | RDC agreement (RRDC) | Variable performance [61] |
| ff99 | Explicit Solvent (TIP3P) | HEWL, Ubiquitin | Methyl order parameter (O²ₐₓᵢₛ) correlation | R² ~0.46 [62] |
| CHARMM27 | Explicit Solvent (TIP3P) | HEWL, Ubiquitin | Methyl order parameter (O²ₐₓᵢₛ) correlation | R² ~0.45 (nominally better) [62] |
| ff99SB | Explicit Solvent (TIP3P) | Hen Egg White Lysozyme | Backbone N-H S² order parameters | Improved vs. ff99; depends on starting structure [63] |
A critical finding is that the method for treating long-range electrostatics profoundly influences quality. The Particle-Mesh Ewald (PME) method consistently outperforms cut-off or reaction-field approaches, even for force fields originally parameterized with the latter [61]. Furthermore, the reproduction of J-couplings across hydrogen bonds suggests deficiencies in how most modern force fields describe these crucial interactions [61].
Performance with Nucleic Acids
Nucleic acids present a distinct challenge, with early force fields like CHARMM22 known to over-stabilize the A-form of DNA relative to the B-form. This was addressed in the CHARMM27 reparameterization [64].
Table 2: Comparison of Force Field Performance for Nucleic Acids
| Force Field | System Tested | Key Finding | Experimental Validation |
|---|---|---|---|
| CHARMM22 | DNA Dodecamer | Over-stabilization of the A-form of DNA [64] | Comparison to crystal structures & solution NMR |
| CHARMM27 | DNA Dodecamer | Improved equilibrium between A and B DNA forms [64] | X-ray structures & solution data |
| AMBER 4.1 | DNA Decamer | Good structural stability with PME [64] | MD-derived vs. experimental helicoidal parameters |
| BMS | DNA Decamer | Performance compared to CHARMM & AMBER [64] | Sequence-dependent structural variations |
| OL-force fields | DNA Double Helix | Arguably among the best for B-DNA description [60] | Multiple structural benchmarks |
| Tumuc1 | DNA Double Helix | Arguably among the best for B-DNA description [60] | Multiple structural benchmarks |
Despite progress, challenges remain. The description of sugar puckering is a recognized problem, and electrostatic parameters are considered deprecated, potentially explaining some ongoing deficiencies [60]. The OL-force fields and Tumuc1 are currently considered top performers for describing the DNA double helix [60].
Performance with Membranes
Force field evaluation extends to complex systems like lipid bilayers. A 2023 pre-print systematically evaluated force fields for binary mixtures of phosphatidylcholine and cholesterol against C-H bond order parameters and lateral diffusion from NMR, and form factors from X-ray scattering [65]. The study concluded that none of the tested force fields clearly outperformed the others across all properties, though the Slipids parameters provided the best overall performance. This highlights the difficulty in creating a universally accurate force field, even for fundamental building blocks of cellular membranes [65].
Experimental Protocols for Force Field Validation
The credibility of force field comparisons rests on robust and reproducible experimental protocols. The following methodologies are commonly employed in benchmarking studies.
Molecular Dynamics Simulation Protocol
A typical MD validation study follows a standardized workflow to ensure comparability:
- System Setup: A crystal or NMR structure is solvated in a box of explicit water molecules (e.g., TIP3P model). The system is neutralized with counterions (e.g., Na⁺, Cl⁻).
- Energy Minimization: The system is minimized to remove steric clashes.
- Equilibration: Short simulations are run with position restraints on solute atoms to equilibrate the solvent and ions around the biomolecule. These restraints are gradually released.
- Production Simulation: Unrestrained MD is performed for a defined time, often ranging from tens of nanoseconds to microseconds. Multiple independent replicates may be run to improve sampling. Studies cited herein used simulation lengths from 30 ns to 1 μs [61] [62].
- Trajectory Analysis: Snapshots from the production trajectory are saved at regular intervals (e.g., every 1-100 ps) for subsequent analysis.
The treatment of electrostatics is a critical parameter. The Particle-Mesh Ewald (PME) method is the current standard for handling long-range electrostatic interactions [64]. Simulations are commonly performed in the NVT (constant Number of particles, Volume, and Temperature) or NPT (constant Number, Pressure, and Temperature) ensembles using periodic boundary conditions.
Calculation of NMR Observables from MD Trajectories
To directly compare simulation results with experiment, NMR observables are back-calculated from the MD ensemble.
- Residual Dipolar Couplings (RDCs): RDCs are computed from the averaged orientations of inter-nuclear vectors (e.g., N-H) relative to the alignment tensor. The agreement is often quantified using an R-factor (e.g., R_RDC), with lower values indicating better agreement [61].
- J-Couplings: Scalar couplings, especially across hydrogen bonds (h³J_NC'), are computed based on empirical relationships with H-bond geometry (e.g., distance and angle) [61].
- Order Parameters (S²): Generalized order parameters for N-H bonds or methyl group symmetry axes (O²_axis) are calculated to quantify ps-ns timescale motion. The isotropic reorientational eigenmode dynamics (iRED) approach or the long-time limit approximation are commonly used [63] [62]. The correlation (R²) between simulated and experimental values is a key metric.
- Nuclear Overhauser Effects (NOEs): Effective interproton distances for NOEs are calculated as ensemble and time averages. Violations of experimental NOE distance bounds are summed to evaluate structural agreement [61].
Figure 1: The force field validation workflow. An experimental structure is used to set up and run an MD simulation. The resulting trajectory is used to calculate experimental observables like NMR parameters, which are then quantitatively compared back to real experimental data to validate or refine the force field.
Critical Considerations in Force Field Validation
The Critical Role of the Starting Structure
A crucial, often overlooked factor is the dependence of simulation outcomes on the initial coordinates. Research on hen egg white lysozyme demonstrated that different experimental starting structures can lead to significant differences in MD-derived S² order parameters [63]. In flexible loop regions, these differences were even larger than those caused by changing the force field itself. This implies that an apparent agreement with experiment could be fortuitous if it depends on a specific starting structure.
Mitigation Strategies:
- Evaluate trajectories started from multiple, conformationally diverse experimental structures in parallel.
- Ensure adequate sampling of flexible regions, which may require simulation times of ~100 ns or longer.
- Calculate S² parameters by averaging over short time windows (~1-5 ns) rather than the entire trajectory, which helps obtain consistent results irrespective of the starting coordinates [63].
Limitations of Fixed-Charge Force Fields
Most widely used force fields employ a fixed-charge model for electrostatics, which does not account for polarization effects—the redistribution of electron density in response to the changing chemical environment [66]. This can lead to inaccuracies in modeling molecular interactions. The development of polarizable force fields (e.g., AMOEBA+) aims to address this by allowing charges to respond to their environment, though at a significantly higher computational cost [66].
The Sampling Challenge vs. Force Field Accuracy
Longer simulations are often assumed to be better. However, with current force fields, simulations beyond hundreds of nanoseconds "run an increased risk of undergoing transitions to nonnative conformational states or will persist within states of high free energy for too long, thus skewing the obtained population frequencies" [61]. This indicates a complex interplay where improved sampling can sometimes reveal force field inaccuracies more starkly. For some properties, multiple short (50 ns) simulations can yield better agreement with NMR data than a single, longer 1 μs simulation, likely due to more diverse sampling of native-like states [61].
Table 3: Key Resources for Force Field Research
| Category | Item | Function in Research |
|---|---|---|
| Software & Tools | GROMACS, AMBER, NAMD, CHARMM | MD simulation packages used to run and analyze simulations [61] [64]. |
| Particle-Mesh Ewald (PME) | The standard algorithm for accurate treatment of long-range electrostatic interactions in periodic systems [61] [64]. | |
| Force Fields | AMBER (e.g., ff99SB, ff19SB) | Family of force fields for proteins and nucleic acids; often show high accuracy for RDCs [61] [66]. |
| CHARMM (e.g., CHARMM27, CHARMM36) | Family of force fields; often optimized for agreement with thermodynamic and NMR data [64] [62]. | |
| GROMOS, OPLS-AA | Force fields parameterized with a focus on accurate thermodynamic properties of liquids [66]. | |
| SLIPIDS | Specialized force field for lipid membranes, showing good performance for lipid-cholesterol mixtures [65]. | |
| Experimental Data | NMR Order Parameters (S²) | Site-specific measure of ps-ns backbone (N-H) and side-chain (methyl) dynamics for validation [63] [62]. |
| Residual Dipolar Couplings (RDCs) | NMR observables sensitive to the average orientation of internuclear vectors, used to validate structural ensembles [61]. | |
| Scalar J-Couplings | NMR observables reporting on torsion angles and hydrogen-bond geometry [61]. |
The systematic comparison of MD force fields against NMR and crystallographic data reveals a nuanced landscape. While modern force fields like AMBER99sb (for proteins) and CHARMM27/OL (for nucleic acids) demonstrate strong performance, no single force field is flawless or universally superior. The community has moved towards force-field-specific recommendations, acknowledging that the optimal choice depends on the target system and the properties of interest.
Key lessons for practitioners include:
- The PME method is non-negotiable for accurate electrostatic treatment.
- Validation must be multi-faceted, leveraging diverse NMR parameters (RDCs, S², J-couplings) beyond simple structural metrics.
- Starting structure bias must be considered, especially for flexible regions.
- Implicit solvent models generally perform poorly in reproducing side-chain dynamics compared to explicit water [62].
Future development will focus on incorporating more physical realism, such as polarizability and charge flux, to overcome the limitations of fixed-charge models [66]. Furthermore, the integration of new computational approaches, including machine learning and automated parameter optimization against increasingly large and diverse experimental datasets, promises to usher in a new generation of more accurate and reliable force fields. This ongoing refinement, firmly grounded in experimental discrepancy, will expand the boundaries of what is possible with biomolecular simulation.
Integrating Computational Docking with Experimental NMR Affinity Screens
The process of drug discovery is a complex and costly endeavor, with a significant number of candidates failing during clinical phases [67]. In this challenging landscape, structure-based drug design has emerged as a powerful approach to improve efficiency. Two techniques in particular—computational molecular docking and experimental NMR affinity screening—have demonstrated remarkable synergy when integrated [67]. Molecular docking provides an extremely rapid method to evaluate likely binders from large chemical libraries with minimal cost, while NMR screens can directly detect protein-ligand interactions and measure corresponding dissociation constants [67]. When combined, these methods create a powerful pipeline that leverages the strengths of both in silico and empirical approaches, offering researchers a robust toolkit for identifying and validating potential therapeutic compounds within the context of comparing structural biology methods.
Molecular Docking Fundamentals
Molecular docking is a computational technique that predicts the binding affinity and orientation of ligands when bound to their receptor proteins [68]. The process primarily involves two key steps: sampling algorithm and scoring function [68]. The sampling algorithm explores possible conformations and orientations of the ligand within the protein's binding site, while the scoring function ranks these poses based on estimated binding affinity [68].
Table 1: Common Molecular Docking Search Algorithms
| Algorithm Type | Subtypes | Key Characteristics | Example Software |
|---|---|---|---|
| Systematic | Conformational Search | Gradually changes torsional, translational, and rotational degrees of freedom | FlexX, DOCK |
| Fragmentation | Docks multiple fragments that are connected or built outward from initial bound fragment | LUDI | |
| Database Search | Pre-generates reasonable conformations from molecular databases | FLOG | |
| Stochastic | Monte Carlo | Randomly places ligands, scores them, then generates new configurations | MCDOCK, ICM |
| Genetic Algorithm | Uses "genes" to describe configuration, with score as "fitness" for generating subsequent generations | GOLD, AutoDock | |
| Tabu Search | Avoids previously examined areas of conformational space | PRO LEADS, Molegro Virtual Docker |
The accuracy of docking predictions varies, with successful redocking of compounds into known protein-ligand structures achieving more than 70% accuracy within 2 Å root mean square deviation (RMSD) of the actual ligand pose [67]. However, limitations include potentially inaccurate scoring functions, use of rigid protein models, and simplified solvation models [67].
NMR Affinity Screening Techniques
NMR spectroscopy offers versatile approaches for detecting protein-ligand interactions through either ligand-based or target-based methods [67]. These techniques detect binding events by monitoring changes in NMR parameters between bound and unbound states.
Table 2: Key NMR Screening Methods and Applications
| Method Category | Specific Techniques | Detected Parameter | Key Applications | Throughput |
|---|---|---|---|---|
| Ligand-based NMR | STD NMR, WaterLOGSY, SLAPSTIC, TINS | Saturation transfer, diffusion coefficients, relaxation | Primary screening of compound mixtures, KD measurement | High (100s-1000s of compounds/day) |
| Target-based NMR | 2D 1H-15N HSQC | Chemical shift perturbations | Binding site identification, binding site validation, co-structure generation | Medium (10s of compounds/day) |
Ligand-based methods typically utilize one-dimensional 1H-NMR experiments that are fast (2-5 minutes) and can screen compound mixtures without deconvolution, requiring minimal protein concentration (<10 μM) and no isotopic labeling [67]. In contrast, target-based screens employ two-dimensional 1H-15N/13C HSQC experiments that require 15N/13C-labeled protein and longer experiment times (>10 minutes) but provide residue-specific interaction data and binding site information [67].
Integrated Workflow: Combining Computational and Experimental Approaches
The powerful synergy between molecular docking and NMR screening emerges when these techniques are combined in a logical workflow that leverages their complementary strengths. The integrated approach follows a cyclical process of computational prediction and experimental validation that progressively refines lead compounds.
This workflow demonstrates how the integration creates a powerful feedback loop. Molecular docking rapidly narrows large virtual libraries to a manageable number of candidates for experimental testing [67]. NMR then validates these hits and provides critical structural information about the binding interaction [67] [11]. This experimental data can subsequently refine the computational models, leading to more accurate predictions in subsequent cycles [69].
Experimental Protocols: Detailed Methodologies for Key Techniques
Molecular Docking and Virtual Screening Protocol
The computational docking process follows a standardized workflow:
Protein Preparation: Obtain the three-dimensional structure of the target protein from experimental methods (X-ray crystallography, NMR, or cryo-EM) or homology modeling. Remove water molecules and co-crystallized ligands, then add hydrogen atoms and assign partial charges using appropriate force fields [68].
Ligand Library Preparation: Collect structures of potential ligands in standardized formats (e.g., SDF, MOL2). Generate three-dimensional conformations and optimize geometries using energy minimization. Prepare ligand files with appropriate charges and atom types for the docking software [68].
Docking Execution: Define the binding site coordinates based on known ligand positions or predicted active sites. Select appropriate search algorithms (systematic, stochastic, or deterministic methods) and scoring functions (force field-based, empirical, knowledge-based, or consensus) [68]. Run docking simulations with sufficient sampling to ensure comprehensive coverage of possible binding modes.
Analysis and Prioritization: Rank compounds based on docking scores and binding poses. Cluster similar binding modes and visually inspect top-ranked complexes for favorable interactions. Select top candidates (typically 20-100 compounds) for experimental validation [67].
NMR Affinity Screening Protocol
The experimental NMR screening process involves these key steps:
Sample Preparation: Prepare protein sample in appropriate buffer (typically 10-50 μM concentration for ligand-observed methods). For target-observed methods, prepare 15N-labeled protein (50-200 μM). Prepare ligand stocks in DMSO-d6 or buffer matching protein conditions [67].
Ligand-Observed Screening (Primary Screen):
- Acquire 1H NMR reference spectra of individual compounds or mixtures.
- Collect STD-NMR spectra by selectively saturating protein resonances (typically at -1 to -2 ppm) and comparing with off-resonance saturation (50-100 ppm).
- Calculate STD amplification factors by (I0 - Isat)/I0, where I0 is the off-resonance intensity and Isat is the on-resonance intensity.
- Identify hits showing significant STD effects (>10-20% attenuation) [69].
Target-Observed Mapping (Secondary Screen):
- Acquire 2D 1H-15N HSQC spectrum of apo protein.
- Titrate hits from primary screen and collect HSQC spectra at each concentration.
- Monitor chemical shift perturbations using the formula: Δδ = √(ΔδH2 + (αΔδN)2), where α is a scaling factor (typically 0.1-0.2).
- Identify residues showing significant perturbations (>mean + 1 standard deviation) to map binding epitope [67].
Comparative Analysis: Strengths and Limitations in Structural Biology Context
When evaluating computational docking and NMR screens within the broader context of structural biology techniques, distinct advantages and limitations emerge for each method.
Table 3: Technique Comparison in Structural Biology Research
| Parameter | Molecular Docking | NMR Affinity Screens | X-ray Crystallography |
|---|---|---|---|
| Throughput | Very High (1000s-100,000s compounds/day) | High (100s-1000s compounds/day) | Low (1 complex at a time) |
| Cost per Compound | Very Low | Moderate | High |
| Experimental Requirements | Protein structure | Protein sample (<10 μM) | Protein crystals |
| Binding Site Information | Predicted | Experimental mapping (HSQC) | Experimental atomic resolution |
| Dynamic Information | Limited | Yes (kinetics, dynamics) | No (static snapshot) |
| Hydrogen Bond Detection | Infered from geometry | Direct (via chemical shifts) | Indirect (inferred) |
| False Positive Rate | High (requires validation) | Low | Very Low |
The integration of docking with NMR specifically addresses the fundamental limitations of crystallography-driven approaches, which include inability to capture dynamic behavior, challenges with crystallization, and being "blind" to hydrogen information [11]. NMR provides direct experimental observation of hydrogen bonds through chemical shift information and captures the dynamic nature of protein-ligand interactions that static crystal structures cannot reveal [11].
Case Study: Practical Application in Carbohydrate Antibody Research
A compelling example of this integrated approach comes from research on the sialyl-Tn carbohydrate antigen, a tumor-associated epitope. The study combined multiple computational and experimental techniques to elucidate antibody-glycan interactions that are notoriously difficult to characterize due to challenges in crystallizing antibody-carbohydrate complexes [69].
The research employed this multi-technique workflow:
- Computational Modeling: Generated antibody homology models and performed molecular docking of the glycan antigen [69].
- Experimental Specificity Profiling: Used quantitative glycan microarray screening to determine apparent KD values [69].
- Binding Site Analysis: Conducted alanine scanning mutagenesis to identify key residues in the antibody combining site [69].
- Structural Validation: Employed saturation transfer difference NMR (STD-NMR) to define the glycan-antigen contact surface [69].
- Model Selection: Used the experimental data as metrics for selecting the optimal 3D-model from thousands of docking-generated options [69].
This case study exemplifies how the integration of computational and experimental approaches enables researchers to overcome the limitations of individual techniques and obtain high-quality structural information for challenging systems.
Research Reagent Solutions: Essential Materials and Tools
Successful implementation of integrated docking and NMR screening requires specific reagents and computational tools.
Table 4: Essential Research Reagents and Computational Tools
| Category | Specific Items | Function/Purpose | Examples |
|---|---|---|---|
| Computational Tools | Docking Software | Predict ligand binding poses and affinity | AutoDock Vina, Glide, GOLD, DOCK [68] |
| Molecular Visualization | Analyze and visualize docking results | UCSF Chimera, PyMOL | |
| Structure Preparation | Prepare protein and ligand structures for docking | Schrödinger Protein Prep Wizard, OpenBabel | |
| NMR Reagents | Isotopically Labeled Proteins | For target-based NMR screening | 15N, 13C-labeled proteins |
| NMR Screening Libraries | Diverse compound collections for screening | Fragment libraries, lead-like compounds | |
| Buffer Components | Maintain protein stability during NMR | Deuterated buffers, stabilizing additives | |
| Experimental Validation | Reference Ligands | Positive controls for binding | Known binders with measured KD |
| Binding Assay Reagents | Orthogonal validation of hits | ITC, SPR, fluorescence-based assays |
Advanced Applications and Future Directions
The continuing evolution of both computational and NMR-based approaches promises enhanced integration in structural biology research. Recent advances include the development of deep learning algorithms for improved pose selection in docking studies, addressing one of the fundamental limitations of traditional scoring functions [70]. Simultaneously, NMR methodology has advanced with new restraint-assisted structure prediction tools like RASP (Restraint Assisted Structure Predictor) that directly incorporate experimental data to improve AI-based structure prediction, particularly for multi-domain proteins and those with limited sequence homologs [16].
The emerging paradigm of NMR-driven structure-based drug design (NMR-SBDD) combines selective side-chain labeling strategies with computational workflows to generate protein-ligand ensembles that capture dynamic information not accessible through crystallography alone [11]. This approach is particularly valuable for studying flexible systems, transient interactions, and allosteric binding sites that constitute challenging targets for traditional structure-based drug design.
These technological advancements collectively point toward a future with tighter integration between computational predictions and experimental validation, enabling more efficient drug discovery pipelines and expanding the range of target classes amenable to structure-based approaches.
Benchmarking and Validation: Ensuring Accuracy in Integrative Models
Cross-validation in structural biology serves as a critical methodology for assessing the quality and reliability of macromolecular structures, particularly when comparing models determined by different techniques. In the context of comparing crystal structures with NMR ensembles, cross-validation employs independent experimental parameters—Residual Dipolar Couplings (RDCs), relaxation data, and chemical shifts—to evaluate structural accuracy without the risk of overfitting. This approach is fundamentally important because it tests a model's predictive power against data not used in the structure calculation process, providing an unbiased assessment of structural quality [71].
The theoretical foundation of this methodology lies in the fact that different types of NMR data provide complementary structural information. RDCs offer long-range orientational constraints, relaxation data probe dynamics on various timescales, and chemical shifts provide sensitive indicators of local conformation. When used in combination for cross-validation, these parameters can reveal inaccuracies in structural models that might otherwise remain undetected. This is especially valuable when comparing crystal structures, which represent a static conformation in a crystalline environment, with NMR ensembles, which capture the dynamic behavior of proteins in solution [2] [5].
Recent assessments, such as the CASP16 Conformational Ensembles Experiment, have highlighted both the necessity and challenges of this approach. In this evaluation, computational methods struggled to accurately predict the distribution of relative orientations of protein domains connected by flexible linkers when validated against combined NMR RDC and SAXS data. None of the submitted predictions provided close fits to the experimental data, underscoring the critical importance of rigorous cross-validation with independent parameters [71].
Experimental Parameters for Cross-Validation
Residual Dipolar Couplings (RDCs)
Residual Dipolar Couplings provide global orientational information that is highly sensitive to molecular architecture and dynamics. RDCs arise from the partial alignment of molecules in a magnetic field, which creates a small but measurable anisotropic component to normally isotropic nuclear interactions. The resulting dipolar couplings contain information about the angle between internuclear vectors and the alignment tensor, offering long-range structural restraints that are particularly valuable for validating domain orientations and detecting conformational changes [72].
The measurement of RDCs has been significantly advanced by methods like ARTSY (Analysis of TROSY Spectra), which enables precise determination of 1H-15N and 1H-13C RDCs even in larger systems. This technique analyzes intensity ratios in TROSY-HSQC spectra recorded with different dephasing delays, minimizing problems associated with resonance overlap. The precision of RDC measurements is directly related to the signal-to-noise ratio, approximately 30/(S/N) Hz for 15N-1H and 65/(S/N) Hz for 13C-1H couplings [72]. This precision makes RDCs exceptionally valuable for cross-validation, as they can detect subtle discrepancies between structural models and actual molecular conformations.
Relaxation Parameters
Relaxation parameters provide direct insights into molecular dynamics across various timescales, offering a complementary validation metric to structural data. NMR relaxation measurements, including T1, T2, and heteronuclear NOE, probe motions from picoseconds to nanoseconds, while techniques like CPMG (Carr-Purcell-Meiboom-Gill) relaxation dispersion and CEST (Chemical Exchange Saturation Transfer) can detect and characterize conformational exchanges on microsecond to millisecond timescales [73].
These dynamic parameters serve as excellent cross-validation tools because they are sensitive to both local flexibility and global conformational changes. For instance, regions showing elevated flexibility in NMR ensembles should correlate with higher B-factors in crystal structures, while discrepancies might indicate crystal packing artifacts or limitations in the NMR ensemble. The combination of relaxation data with structural information allows researchers to build more accurate models of protein energy landscapes and functionally relevant motions [73].
Chemical Shifts
Chemical shifts serve as extremely sensitive probes of local electronic environment, making them ideal for validating secondary structure and local conformation. The chemical shift of a nucleus is influenced by numerous factors including backbone torsion angles, hydrogen bonding, and sidechain orientation. This sensitivity has led to the development of powerful validation tools such as chemical shift prediction programs (e.g., SHIFTX2) and the ability to back-calculate expected chemical shifts from structural models [2].
In cross-validation protocols, chemical shifts provide a particularly valuable metric because they can be measured quickly and accurately, even for large proteins. Significant deviations between experimentally observed chemical shifts and those back-calculated from structural models often indicate local structural inaccuracies. Furthermore, chemical shifts can be used to validate the presence of specific secondary structure elements identified in crystal structures when compared to NMR data [2].
Methodologies for Cross-Validation
Experimental Protocols
RDC Measurement Protocol
The measurement of RDCs using the ARTSY method follows a standardized protocol that ensures accuracy and reproducibility. For a typical protein sample, the procedure begins with the preparation of an alignment medium, such as Pf1 phage, added to the protein solution to induce partial alignment. The sample concentration typically ranges from 0.7-0.8 mM in appropriate buffer conditions, with the alignment strength monitored by measuring the 2H quadrupole splitting of the solvent (e.g., 10.1 Hz for a 10 mg/mL Pf1 concentration) [72].
The core of the ARTSY experiment involves recording two 1H-15N TROSY-HSQC spectra with different dephasing delays (typically 22 ms and 44 ms). These spectra are acquired with water-selective excitation schemes that leave water magnetization unperturbed, enabling rapid repetition rates and enhanced signal-to-noise through transfer of magnetization from bulk water. The RDC values are then extracted by analyzing the intensity ratios of cross-peaks in the two spectra, with the coupling size calculated using the formula: RDC = (1/πτ) × arccos(2 × (I₁/I₂ - 1)), where τ is the dephasing delay and I₁/I₂ is the intensity ratio [72].
Table 1: Key Parameters for RDC Measurement Using ARTSY Method
| Parameter | Typical Value | Purpose |
|---|---|---|
| Protein Concentration | 0.7-0.8 mM | Optimal signal-to-noise |
| Pf1 Phage Concentration | 7-10 mg/mL | Induces partial alignment |
| ²H Quadrupole Splitting | 6.9-10.1 Hz | Monitors alignment strength |
| Dephasing Delays | 22 ms, 44 ms | Enables RDC calculation from intensity ratios |
| Temperature | 25°C | Standard measurement condition |
| Acquisition Time | 2-3 days per sample | Typical for high-quality data |
Relaxation Measurements Protocol
Relaxation measurements follow well-established protocols that have been optimized for accuracy and precision. For T1 and T2 measurements, a series of spectra are acquired with increasing relaxation delays, typically spanning a range from 10-1000 ms for T1 and 10-300 ms for T2. The resulting peak intensities are fitted to exponential decay curves to extract relaxation rates. Heteronuclear NOE values are determined from the ratio of peak intensities in spectra acquired with and without proton saturation [73].
For dynamics occurring on microsecond-millisecond timescales, CPMG relaxation dispersion experiments measure R₂ eff as a function of CPMG field strength, while CEST experiments monitor signal attenuation as a function of saturation frequency and power. These experiments require careful optimization of sampling schemes and fitting procedures to extract accurate parameters for conformational exchange, including rates, populations, and chemical shift differences between states [73].
Data Analysis and Validation Workflows
The cross-validation process follows a systematic workflow that integrates multiple types of experimental data to assess structural quality. The fundamental principle is to use each type of data—RDCs, relaxation, and chemical shifts—to validate structures determined using other types of constraints. This approach prevents overfitting and provides a more realistic assessment of model accuracy [71].
A typical cross-validation workflow begins with the division of experimental restraints into training and test sets. The structure is calculated using only the training set restraints, and then the quality of the structure is assessed by its ability to predict the test set data. This process is repeated multiple times with different divisions of the data to ensure robust validation. For RDCs, the validation typically involves comparing experimental RDCs with those back-calculated from the structural model using the determined alignment tensor. Significant discrepancies often indicate domain misorientation or the presence of conformational dynamics not captured in the model [71] [72].
Table 2: Cross-Validation Metrics for Different NMR Parameters
| Parameter | Validation Metric | Typical Threshold | Structural Interpretation |
|---|---|---|---|
| RDCs | Q-factor | <0.3 indicates good fit | Validates global fold and domain orientation |
| Chemical Shifts | RMSD to predicted | <0.3 ppm for ¹H, <3 ppm for ¹⁵N | Validates local geometry and secondary structure |
| Relaxation (R₂/R₁) | Reduced χ² | <1.5 | Validates overall tumbling and internal dynamics |
| NOE Violations | Number >0.5 Å | <5% of total restraints | Validates interatomic distances |
Diagram Title: Cross-Validation Workflow for NMR Structures
Comparative Analysis: Crystal Structures vs. NMR Ensembles
Systematic Comparisons from Large-Scale Studies
Large-scale comparative analyses have revealed consistent patterns in the similarities and differences between crystal structures and NMR ensembles. A comprehensive study of 109 protein pairs, each with structures determined by both X-ray crystallography and NMR spectroscopy, found that root-mean-square deviations (RMSDs) between the two forms typically range from approximately 1.5 Å to 2.5 Å. This analysis revealed several important trends: hydrophobic amino acids show greater similarity between crystal and NMR structures than hydrophilic residues, beta strands generally match better than helices or loops, and variations in buried side chain conformations are relatively rare [2].
The observed differences arise from fundamental methodological distinctions. Crystal structures represent a single, often thermodynamically favored conformation stabilized by crystal packing forces, while NMR ensembles capture a sampling of the conformational space accessible in solution. This distinction becomes particularly important for flexible regions, where crystal structures may show disorder or static conformations influenced by packing contacts, while NMR ensembles can better represent the dynamic nature of these regions [2].
Membrane Protein Specific Considerations
Membrane proteins present unique challenges for structural biology that further complicate comparisons between crystallographic and NMR-derived models. A specialized analysis of 14 membrane proteins with structures determined by both methods revealed that RMSDs in the membrane region are typically below 5 Å. The study identified several systematic differences: NMR ensembles show higher convergence in membrane regions, crystal structures generally have straighter transmembrane helices, and crystal structures tend to exhibit better stereochemical quality and tighter packing [5].
These differences can be attributed to the distinct membrane mimetics used in each method. Crystallographers often employ micelles or lipidic cubic phases optimized for crystal formation, while NMR spectroscopists use detergents or bicelles that maintain protein solubility but may differ in their physicochemical properties from native membranes. The biological relevance of structures determined in these artificial environments remains an active area of investigation, highlighting the importance of cross-validation approaches that can assess structural quality independent of the determination method [5].
Table 3: Crystal vs. NMR Structure Comparison Across Protein Types
| Comparison Metric | Soluble Proteins | Membrane Proteins | Primary Cause of Differences |
|---|---|---|---|
| Global Backbone RMSD | 1.5-2.5 Å | Up to 5.0 Å | Crystal packing vs. solution dynamics |
| Secondary Structure Variation | β-strands most similar | Helices show straightening | Membrane mimetics environment |
| Side Chain Conformations | Buried residues most similar | Variable in lipid-facing regions | Lipid vs. crystal environment |
| Dynamic Regions | Loops show greatest variation | Extramembranous loops most variable | Differential flexibility representation |
| Validation with RDCs | Q-factor <0.3 typically achievable | Often higher due to dynamics | Incomplete sampling of motions |
Case Studies and Applications
Staphylococcal Protein A (SpA) Domain Orientation
The CASP16 Conformational Ensembles Experiment provided a rigorous test case for evaluating cross-validation with independent parameters. This challenge involved predicting the interdomain pose distribution of a Staphylococcal protein A (SpA) construct in which two domains were connected by either a wild-type linker or an all-glycine linker. Twenty-five research groups submitted predicted conformational distributions, which were validated against experimental NMR RDC and SAXS data [71].
The results were revealing: although predictions spanned a wide range of accuracy, none provided close fits to the combined NMR and SAXS data. Particularly telling was the failure of all methods to recapitulate the observed differences between wild-type and Gly6 linker proteins evident in the SAXS data. This case study highlights both the strengths and limitations of current computational approaches and underscores the critical importance of cross-validation with multiple independent data types [71].
RNA Riboswitch Structure Validation
RDC measurements have proven particularly valuable for validating nucleic acid structures, as demonstrated in a study of a 71-nucleotide adenine riboswitch. In this case, 1H-15N and 1H-13C RDCs measured using the ARTSY method provided validation of the global architecture and specific orientation of helical elements. The RDC data were compatible with nucleotide-specifically modeled, idealized A-form geometry and a static orientation of helix 1 relative to the helix 2/3 pair, differing by approximately 6° from the orientation observed in the X-ray structure of the native riboswitch [72].
This example illustrates how RDCs can detect subtle but biologically relevant structural differences that might be obscured by crystal packing effects or limited resolution. The ability to validate global architecture independent of local distance restraints makes RDCs particularly powerful for cross-validation of complex RNA structures, which often exhibit conformational flexibility that is difficult to capture in crystal structures [72].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful cross-validation studies require carefully selected reagents and materials optimized for both NMR and crystallographic approaches. The following table details essential components for these investigations:
Table 4: Essential Research Reagents for Cross-Validation Studies
| Reagent/Material | Specifications | Function in Research |
|---|---|---|
| Alignment Media | Pf1 phage, 7-10 mg/mL | Induces partial alignment for RDC measurements |
| NMR Tubes | Shigemi microcells, 330 μL | Maximizes signal-to-noise for limited samples |
| Isotope Labeling | ¹⁵N, ¹³C, ²H-enriched | Enables multidimensional NMR experiments |
| Detergents | DPC, DHPC, DDM micelles | Membrane mimetics for soluble state NMR |
| Crystallization Reagents | Lipid cubic phases (LCP) | Membrane protein crystallization |
| Buffer Components | Potassium phosphate, MgCl₂, KCl | Maintains physiological conditions and stability |
| Protease Inhibitors | Broad-spectrum cocktails | Prevents sample degradation during data collection |
| Reducing Agents | DTT, TCEP | Maintains cysteine residues in reduced state |
Cross-validation with independent parameters represents a powerful paradigm for assessing the accuracy and biological relevance of macromolecular structures. The integration of RDCs, relaxation data, and chemical shifts provides a comprehensive framework for identifying limitations in both crystal structures and NMR ensembles, particularly for flexible systems and complex biomolecular assemblies. As structural biology continues to tackle increasingly challenging systems, from intrinsically disordered proteins to large macromolecular machines, these cross-validation approaches will become increasingly essential for distinguishing structural features from methodological artifacts.
The case studies and methodologies outlined in this guide demonstrate that while significant progress has been made in cross-validation protocols, important challenges remain. The inability of current computational methods to accurately predict the conformational distributions of multidomain proteins, as evidenced in the CASP16 assessment, highlights the need for continued development of both experimental and theoretical approaches. For researchers in structural biology and drug development, rigorous cross-validation with multiple independent parameters provides the most reliable path toward biologically meaningful structural models that can effectively guide therapeutic design.
Proteins are inherently dynamic molecules, and their functions are often underpinned by conformational dynamics rather than single, static structures. The recognition that internal dynamics at various timescales play a functional role has led to the emerging use of dynamic structural ensembles instead of individual conformers [74] [75]. These ensembles are usually substantially more diverse than conventional Nuclear Magnetic Resonance (NMR) ensembles and eliminate the expectation that a single conformer should fulfill all NMR parameters originating from 10^16 - 10^17 molecules in the sample tube [74]. This paradigm shift necessitates specialized computational tools that can generate, analyze, and validate these structural ensembles, particularly when comparing results from different experimental methods like X-ray crystallography and NMR spectroscopy.
Systematic comparisons have revealed that while crystal and NMR structures of the same protein generally show good agreement, with RMSD values typically ranging from about 1.5 Å to 2.5 Å, significant conformational differences can exist, especially in loops and solvent-exposed residues [2]. Beta strands on average match better between NMR and crystal structures than helices and loops, and hydrophobic amino acids are more similar in crystal and NMR structures than hydrophilic amino acids [2]. These observations highlight the importance of specialized tools that can handle the unique challenges of ensemble analysis and comparison. Within this landscape, CoNSEnsX and EnsembleFlex have emerged as complementary solutions addressing different aspects of ensemble analysis, validation, and application in structural biology and drug design.
CoNSEnsX (Consistency of NMR-derived Structural Ensembles with eXperimental data) is a specialized web application designed specifically for validating NMR-derived structural ensembles against experimental NMR parameters [74] [75]. It operates on the fundamental principle that NMR observables should be treated as ensemble properties rather than as stemming from a single conformer. The tool allows fast, simple, and convenient assessment of the correspondence of the ensemble as a whole with diverse independent NMR parameters [74]. Its web-based nature makes it particularly accessible to researchers who may not have extensive computational expertise but require robust validation of their NMR ensembles.
EnsembleFlex represents a more comprehensive computational suite designed to extract, quantify, and visualize conformational heterogeneity from experimentally determined structure ensembles, including those from X-ray crystallography, NMR, and cryo-electron microscopy (cryo-EM) [3] [76]. Its primary design goal is to enable both computational and experimental scientists to gain actionable insights into protein dynamics, ligand interactions, and drug-design applications. Unlike CoNSEnsX's specialized focus on NMR validation, EnsembleFlex adopts a broader approach to ensemble analysis, incorporating flexibility analysis, dimension reduction, clustering, and binding-site characterization for heterogeneous structural datasets [3].
Table 1: Core Characteristics of CoNSEnsX and EnsembleFlex
| Feature | CoNSEnsX+ | EnsembleFlex |
|---|---|---|
| Primary Focus | Validation of NMR ensembles | General analysis of conformational heterogeneity |
| Interface | Web server [77] | Graphical user interface (GUI) and scriptable pipelines [78] |
| Input Formats | Multi-model PDB file, BMRB format NOE distance restraints, BMRB restraint file [77] | Heterogeneous PDB ensembles (X-ray, NMR, cryo-EM) [3] |
| Key Methodologies | Ensemble-averaged analysis of experimental parameters, greedy selection approach [77] | Dual-scale flexibility analysis, dimension reduction, clustering [3] |
| Experimental Validation | Direct comparison with NMR parameters (NOEs, S², RDCs, chemical shifts) [75] | Optional integration with elastic network models [3] |
| Ideal Use Cases | Cross-validation of NMR ensembles, quality assessment of dynamic conformational ensembles | Drug-design applications, binding site analysis, high-throughput structural analysis |
Methodological Comparison: Analytical Approaches and Workflows
The fundamental difference between CoNSEnsX and EnsembleFlex lies in their analytical philosophies and workflows. CoNSEnsX is built around the concept of validating structural ensembles against experimental NMR data, employing ensemble-averaged analysis of all experimental parameters recognized in the input [77]. It evaluates the correspondence of NMR-derived parameters with those back-calculated from a protein structural ensemble, providing a crucial quality assessment specifically for dynamically relevant structural ensembles [75]. This approach is particularly valuable given that dynamic protein ensembles often reproduce even parameters not used for their calculations better than conformer sets obtained with single-structure refinement [75].
EnsembleFlex employs a more diverse methodological toolkit focused on characterizing conformational heterogeneity. It performs dual-scale flexibility analysis (backbone and side-chain) via optimized superposition, utilizing RMSD (Root Mean Square Deviation) and RMSF (Root Mean Square Fluctuation) calculations [3]. The tool incorporates dimension reduction techniques including both linear (Principal Component Analysis - PCA) and non-linear (Uniform Manifold Approximation and Projection - UMAP) methods, along with clustering for state identification [3] [78]. Additionally, it features automated ligand-site variability mapping and conserved-water identification, making it particularly valuable for drug discovery applications [3].
The following workflow diagrams illustrate the distinct methodological approaches of each tool:
CoNSEnsX+ Workflow for NMR Ensemble Validation
EnsembleFlex Workflow for Conformational Heterogeneity Analysis
Experimental Applications and Case Studies
Both tools have been validated through extensive case studies demonstrating their utility in practical research scenarios. CoNSEnsX has been comprehensively tested on multiple proteins, including human ubiquitin as a well-characterized and relatively rigid protein, a 35-residue protease inhibitor as a small flexible protein, and a disordered subunit of cGMP phosphodiesterase 5/6 representing intrinsically disordered proteins [74] [75]. In the ubiquitin analysis, researchers utilized as many as 16 different structural ensembles taken from publicly available databases such as the PDB and RECOORD, plus three additional structurally generated ensembles specifically for methodological evaluation [75]. This extensive testing demonstrated CoNSEnsX's capability to handle diverse types of structural ensembles and provide meaningful validation across different protein flexibility regimes.
EnsembleFlex has demonstrated its scalability and utility through case studies including adenylate kinase, hexokinase-1, interleukin-1β (IL-1β) fragment screens, and SARS-CoV-2 main protease ensembles [3] [76]. These applications highlight the tool's versatility in addressing biologically and pharmacologically relevant questions. The adenylate kinase and hexokinase case studies typically focus on characterizing large-scale domain motions critical to their catalytic mechanisms, while the interleukin-1β fragment screens and SARS-CoV-2 main protease analyses demonstrate applications in drug discovery, where understanding conformational heterogeneity can inform inhibitor design [76].
Table 2: Experimental Applications and Validation Approaches
| Tool | Validation Proteins/Systems | Experimental Parameters | Key Outcomes |
|---|---|---|---|
| CoNSEnsX+ | Human ubiquitin, Protease inhibitor, Disordered PDE subunit [75] | NOE restraints, S² order parameters, Chemical shifts, RDCs [75] | Comprehensive evaluation of ensemble consistency with experimental data [74] |
| EnsembleFlex | Adenylate kinase, Hexokinase-1, IL-1β fragment screens, SARS-CoV-2 main protease [76] | Backbone/side-chain RMSD/RMSF, PCA/UMAP projections, Binding site statistics [3] | Actionable insights into protein dynamics and drug-design applications [76] |
Technical Implementation and Accessibility
The technical implementation and accessibility profiles of CoNSEnsX and EnsembleFlex differ significantly, reflecting their distinct target user bases and analytical goals. CoNSEnsX is available as a web service through the CoNSEnsX+ server (http://consensx.itk.ppke.hu), providing a streamlined, platform-independent interface that requires no local installation [77]. This web-based architecture lowers the barrier to entry for experimental researchers who need to validate NMR ensembles but may lack extensive computational infrastructure or expertise. The server is specifically designed for structural ensembles generated to reflect the internal dynamics of proteins at a given timescale, typically produced by restrained molecular dynamics methods or selection-based approaches [77].
EnsembleFlex offers multiple installation options to accommodate different user preferences and technical environments. The most reproducible environment setup uses Docker, which provides containerization with all dependencies included, though this requires users to move input PDB files into mounted folders [78]. For Conda or Mamba users, the tool provides environment configuration files (environment.yml or environment_versioned.yml) to create compatible Python/R environments [78]. The software can be executed through a browser-based graphical user interface built with Streamlit or via command-line scripts, offering flexibility for both interactive exploration and batch processing [78]. This multi-faceted approach accommodates users with varying computational expertise, from structural biologists preferring point-and-click interfaces to computational researchers developing automated analysis pipelines.
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Resources for Ensemble Analysis
| Resource Type | Specific Tools/Formats | Function in Ensemble Analysis |
|---|---|---|
| Structural Input | Multi-model PDB files, X-ray structures, NMR ensembles, cryo-EM maps [3] [77] | Raw structural data for ensemble generation and analysis |
| Experimental Restraints | BMRB format NOE distance restraints, NMR constraint files [77] [75] | Experimental data for validation and refinement of ensembles |
| Analysis Packages | Bio3D (R), ProDy (Python), BioPython, UMAP, clustering algorithms [78] | Core analytical algorithms for flexibility analysis and dimension reduction |
| Validation Tools | SHIFTX, PALES, PRIDE-NMR algorithms [75] | Back-calculation of NMR parameters for experimental validation |
| Visualization | PyMol scripts, py3Dmol, Streamlit interface [78] | Visualization of ensemble properties and analytical results |
CoNSEnsX and EnsembleFlex represent complementary approaches to the challenging task of protein ensemble analysis, each excelling in different aspects of this rapidly evolving field. CoNSEnsX provides specialized, rigorous validation of NMR-derived ensembles against experimental data, ensuring that dynamic conformational ensembles accurately represent the experimental observations they aim to model [74] [75]. Its web-based accessibility makes it particularly valuable for experimental groups focused on NMR structure determination and validation. EnsembleFlex offers a comprehensive toolkit for extracting biologically and pharmacologically relevant insights from heterogeneous structural ensembles, with particular strength in characterizing conformational landscapes relevant to drug design [3] [76]. Its flexible deployment options and user-friendly interface make it accessible to both computational and experimental scientists.
The choice between these tools depends fundamentally on the research question at hand. For researchers focused on validating NMR ensembles or assessing the consistency of structural ensembles with experimental NMR parameters, CoNSEnsX provides targeted, methodologically rigorous solutions. For investigations aimed at understanding conformational heterogeneity across multiple experimental methods (X-ray, NMR, cryo-EM) and connecting this heterogeneity to biological function or drug discovery, EnsembleFlex offers a more comprehensive analytical framework. As structural biology continues to recognize the fundamental importance of dynamics in protein function, both tools represent valuable additions to the methodological toolkit of researchers studying protein structure, dynamics, and their biological implications.
Proteins are dynamic molecular machines whose biological functions are intrinsically linked to their flexibility [79] [80]. Understanding atomic-scale mobility is therefore crucial in structural biology and drug development. Three principal techniques provide complementary insights into protein dynamics: X-ray crystallography (via B-factors), Nuclear Magnetic Resonance (NMR) spectroscopy (via structural ensembles), and Molecular Dynamics (MD) simulations (via trajectories) [79] [80]. Each method captures flexibility through different parameters and under different experimental or computational conditions.
This guide objectively compares the performance of these techniques in characterizing protein flexibility, with a specific focus on a recently identified pattern in backbone atom mobility. The analysis is framed within the broader context of comparing crystal structures with NMR ensembles, highlighting where these methods converge and diverge in their dynamic assessments.
The table below defines the key parameters and their physical significances for each method.
Table 1: Key Parameters for Assessing Protein Flexibility
| Method | Primary Flexibility Parameter | Physical Significance | Sample Environment |
|---|---|---|---|
| X-ray Crystallography | Crystallographic B-factor (Debye-Waller factor) | Mean square displacement of atoms from vibrational motion and static disorder [79]. | Crystalline solid state. |
| NMR Spectroscopy | Coordinate variance/uncertainty across an ensemble | Lack of convergence in atomic coordinates, indicating local mobility and paucity of structural restraints [79] [80]. | Solution state (near-native conditions). |
| Molecular Dynamics (MD) | Coordinate variance across a simulation trajectory | Temporal fluctuation of atomic positions based on the employed force field [79]. | In silico simulation (solution or vacuum). |
Quantitative Comparison of Flexibility Patterns
A seminal 2021 study systematically compared these techniques, revealing a distinct and persistent pattern in backbone heavy atom flexibility [79] [80] [81]. The analysis applied Friedman's test to rank the uncertainties of backbone atoms (N, Cα, C', O) on a per-residue basis across numerous structures.
Table 2: Summary of Statistical Findings on Backbone Atom Flexibility [79]
| Method | Highest Average Rank (Most Flexible) | Lowest Average Rank (Least Flexible) | Statistical Significance of Pattern |
|---|---|---|---|
| NMR Ensembles | Carbonyl Oxygen (O) | Amide Nitrogen (N) & Carbonyl Carbon (C') | Significant in almost all ensembles. |
| MD Trajectories | Carbonyl Oxygen (O) | Amide Nitrogen (N) & Carbonyl Carbon (C') | Significant in most trajectories, persistent across force fields. |
| Crystallographic B-factors | No consistent pattern | No consistent pattern | Only a few structures showed significant differences. |
The data shows that NMR and MD consistently identify a pattern where the carbonyl oxygen is the most flexible backbone atom, while the amide nitrogen and carbonyl carbon are the most rigid. This pattern suggests a motional mode where the peptide plane pivots around the N and C' atoms [79]. In contrast, crystallographic B-factors do not show this systematic variation, implying the pattern may be specific to solution-state dynamics or not captured by crystalline lattice interactions.
Detailed Experimental Protocols
To ensure reproducibility, this section outlines the standard methodologies employed in the comparative studies.
NMR Ensemble Generation and Analysis
- Ensemble Calculation: Typically, 10-40 structural models are generated using simulated annealing restrained by experimental data, primarily from NOESY (Nuclear Overhauser Effect Spectroscopy) experiments, which provide interatomic distance constraints [79] [80].
- Flexibility Assessment: The primary source of flexibility information is the local lack of convergence in atomic coordinates across the ensemble. Flexible regions, like loops, have fewer long-range "contacts" or restraints, leading to higher coordinate uncertainty [79].
- Superimposition: Calculating meaningful coordinate variances requires optimal superimposition of the ensemble, often limited to a core atom set to avoid bias from flexible regions. Methods like FindCore (which uses an interatomic variance matrix) or THESEUS (which assumes a multivariate Gaussian distribution) are used for this purpose [79] [80].
Molecular Dynamics (MD) Simulation Workflow
- Seed Structure: Simulations are initiated from an experimentally determined structure (e.g., from XRD or NMR) [79].
- Force Field Application: The atomic interactions are governed by a mathematical force field (e.g., AMBER99SB or OPLS). The quality of the simulation is dependent on the force field's accuracy [79] [82].
- Trajectory Production: The system is equilibrated and then a production run is performed, often in the microcanonical (NVE) or canonical (NVT) ensemble, generating a trajectory of atomic coordinates over time [79] [82].
- Variance Calculation: Similar to NMR analysis, MD trajectories are superimposed, and coordinate variances are calculated for each atom over the simulated time course [79].
Crystallographic B-Factor Determination
- Data Collection: X-ray diffraction data is collected from a protein crystal, measuring the intensities of reflections.
- Model Refinement: An atomic model is refined against the diffraction data. The B-factor for each atom is refined to account for the fact that atomic scattering factors fall off more rapidly with angle than those for stationary atoms. This accounts for atomic displacement due to vibration or disorder [79].
- Interpretation: While indicative of flexibility, elevated B-factors can also arise from static disorder, crystal packing defects, or other factors that reduce reflection intensities [79] [80].
The following diagram visualizes this comparative analysis workflow.
Visualizing the Key Pattern in Peptide Bond Flexibility
The core finding from the comparative studies is a specific pattern of peptide bond mobility consistently observed in NMR and MD data but absent in crystallographic B-factors. The following diagram illustrates this motional model.
Research Reagent Solutions
The following table lists key computational and data resources essential for research in this field.
Table 3: Essential Research Tools for Protein Flexibility Studies
| Tool Name | Type | Primary Function | Relevance to Flexibility Studies |
|---|---|---|---|
| FindCore / Expanded FindCore [79] | Software Algorithm | Identifies a core set of well-converged atoms for optimal superimposition of NMR ensembles. | Critical for obtaining reliable coordinate uncertainties by avoiding bias from flexible regions. |
| THESEUS [79] [80] | Software Program | Performs statistical superimposition of macromolecular structures assuming multivariate Gaussian coordinate uncertainties. | An alternative method for superimposing ensembles and analyzing coordinate variances in NMR and MD data. |
| AMBER99SB & OPLS [79] | Force Field | Mathematical functions and parameters defining atomic interactions in MD simulations. | The persistence of the flexibility pattern across different force fields underscores its robustness or indicates a shared parameterization challenge. |
| GAFF2 [82] | Force Field (Generalized Amber Force Field 2) | A force field for simulating small organic molecules and drugs. | Used in high-throughput MD simulations to generate IR and NMR spectral datasets [82]. |
| NESG NMR/X-ray Pairs [79] [80] | Structural Dataset | A collection of over 40 protein structures solved by both NMR and X-ray crystallography. | Provides the essential paired experimental data required for direct, structure-by-structure comparisons of flexibility. |
| USPTO-Spectra Dataset [82] | Computational Dataset | A synthetic dataset of anharmonic IR and DFT-based NMR spectra for 177K organic molecules. | Supports benchmarking of computational methodologies and development of AI models for spectral analysis and property prediction. |
The paradigm of structural biology is undergoing a fundamental shift, moving from the study of single, static protein structures toward the characterization of dynamic conformational ensembles. This transition recognizes that conformational heterogeneity is not merely a structural nuance but is essential for protein function [15]. Traditional single-structure approaches, including those powered by artificial intelligence like AlphaFold, provide unprecedented accuracy for predicting stable conformations but often miss the full spectrum of biologically relevant states [83]. The integration of computational predictions with experimental validation has emerged as a critical pathway for capturing this complexity. This guide examines the integrative approach of combining AlphaFold-predicted structures, molecular dynamics (MD) simulations, and NMR relaxation data to generate accurate, time-resolved 4D conformational ensembles, framed within the context of comparing the static nature of crystal structures with the dynamic reality captured by NMR ensembles.
The Limitations of Isolated Methods
The Static Picture from AlphaFold and Crystallography
AlphaFold 2 has revolutionized protein structure prediction, achieving atomic accuracy in many cases [84]. However, systematic evaluations reveal its limitations in capturing protein dynamics and multiple conformational states. A comprehensive analysis of nuclear receptor structures found that while AlphaFold achieves high accuracy for stable conformations, it systematically underestimates ligand-binding pocket volumes by 8.4% on average and misses functionally important asymmetry in homodimeric receptors [83]. These limitations stem from AlphaFold's training on static experimental structures from X-ray crystallography and cryo-EM, which inherently represent proteins in fixed states, often at cryogenic temperatures or in crystalline environments [85].
The predicted local distance difference test (pLDDT) score provided by AlphaFold reliably indicates model confidence but offers only a binary "order/disorder" distinction without capturing gradations in dynamics [85]. Large-scale comparisons show that while high pLDDT residues correlate well with rigid, well-folded regions, the metric fails to represent the nuanced dynamics observed in solution for flexible protein regions [85].
The Dynamic but Unvalidated View from Molecular Dynamics
Molecular dynamics simulations model protein motion by numerically solving Newton's equations of motion, theoretically providing a complete picture of conformational sampling. However, MD faces significant challenges, particularly the starting structure dependence of simulated dynamics [63]. Studies on hen egg white lysozyme demonstrated that different experimental starting structures could lead to even larger differences in MD-derived order parameters (S²) than those caused by using different force fields [63]. Without experimental validation, it remains difficult to judge whether an MD simulation accurately represents a protein's true dynamics or reflects force field inaccuracies and sampling limitations.
Experimental Dynamics from NMR Relaxation
Solution-state NMR spectroscopy is a powerful tool for studying conformational ensembles as it inherently captures the physical properties of biomolecules averaged across multiple conformations [15]. Relaxation measurements, including longitudinal (R1), transverse (R2) rates, and heteronuclear NOE, provide detailed insights into dynamic structural ensembles [15]. The model-free analysis of this data yields the generalized order parameter (S²), which quantifies the spatial restriction of internal motions on pico- to nanosecond timescales [15]. While exceptionally sensitive to dynamics, interpreting NMR relaxation data in structural terms remains challenging without complementary computational models [15].
Table 1: Key Limitations of Individual Structural Biology Methods
| Method | Key Strengths | Principal Limitations |
|---|---|---|
| AlphaFold | High accuracy for stable folds; Fast prediction; Good stereochemistry | Misses conformational diversity; Underestimates binding pocket volumes (8.4% on average); Cannot capture functional asymmetry [83] |
| X-ray Crystallography | Atomic resolution; Provides static snapshot | Crystalline environment artifacts; Limited visibility of flexible regions; Low temperatures [85] |
| Molecular Dynamics (MD) | Models full atomic motion; Physical force field | Starting structure dependence; Sampling limitations; Force field inaccuracies [63] |
| NMR Relaxation | Probes dynamics at physiological conditions; Multiple timescales | Challenging to translate to structural ensembles; Limited to smaller proteins; Complex interpretation [15] |
The Integrative Approach: Methodology and Workflow
The core integrative methodology combines the strengths of prediction, simulation, and experimental validation to overcome the limitations of each individual method [15]. This approach uses AlphaFold-predicted structures as high-quality starting points for free MD simulations, then selects trajectory segments consistent with experimental NMR relaxation data to identify biologically relevant conformational ensembles [15].
Experimental Protocols and Methodologies
AlphaFold Structure Prediction
The protocol begins with generating a structural model using AlphaFold. The network takes the primary amino acid sequence and aligned sequences of homologues as inputs, directly predicting the 3D coordinates of all heavy atoms through its Evoformer and structure module architecture [84]. The resulting model provides the initial coordinates for MD simulations, though researchers should be aware of its potential limitations in capturing flexible regions and binding pockets [83].
Molecular Dynamics Simulation Setup
For the extracellular region of Streptococcus pneumoniae PsrP, researchers performed free MD simulations starting from the AlphaFold-generated structure using modern force fields [15]. Key considerations include:
- Using periodic boundary conditions with explicit water models (e.g., TIP3P)
- Employing thermostats (e.g., Langevin thermostat) for temperature control
- Running equilibration phases (e.g., 25 ps) followed by extended production runs (e.g., 100+ ns)
- Ensuring adequate sampling of flexible regions, which may require ∼100 ns simulations [63]
NMR Relaxation Data Collection
The experimental backbone employs ¹⁵N-labeled proteins in solution. Key measurements include:
- Longitudinal (R1) and transverse (R2) relaxation rates
- ¹⁵N{¹H} heteronuclear NOE measurements
- Cross-correlated relaxation (ηxy) rates, which are less biased by slow conformational exchange than R₂ [15] Data collection requires optimized pulse programs and careful processing to ensure accurate relaxation parameters for comparison with MD trajectories [15].
Ensemble Validation and Selection
The critical integration step involves comparing back-calculated relaxation parameters from MD trajectories with experimental NMR data. Rather than reweighting the entire trajectory, the approach selects trajectory segments (RMSD plateaus) that show consistency with experimental observables [15]. For PsrP, only specific segments of long MD trajectories aligned well with experimental data, and these selected ensembles revealed functionally important flexible regions [15].
Diagram 1: Workflow for Integrative Modeling combining AlphaFold, MD, and NMR.
Comparative Performance Analysis
Quantitative Assessment of Method Capabilities
Table 2: Quantitative Performance Comparison of Structural Methods
| Performance Metric | AlphaFold Alone | MD Alone | NMR Alone | Integrated Approach |
|---|---|---|---|---|
| Static Structure Accuracy | High (0.96Å backbone RMSD) [84] | Variable (force field dependent) | Moderate (ensemble averaging) | High (AF starting point) [15] |
| Ligand Pocket Volume | Underestimated by 8.4% [83] | Accurate with validation | Not directly measured | Accurate (validated by MD/NMR) |
| Dynamic Timescales Captured | None (static model) | Picoseconds to microseconds | Picoseconds to milliseconds | Picoseconds to milliseconds |
| Loop Region Accuracy | Low (high flexibility) [85] | Variable (starting structure dependent) [63] | High (solution conditions) | High (experimentally validated) [15] |
| Functional Asymmetry Detection | Missed in homodimers [83] | Possible but requires validation | Detectable | Detectable and validated |
Case Study: Streptococcus pneumoniae PsrP
The application to the extracellular region of Streptococcus pneumoniae PsrP demonstrated the power of the integrative approach [15]. The method revealed two regions with increased flexibility that had important functional roles, which might have been missed by any single method. Only specific segments of the long MD trajectory aligned well with experimental NMR relaxation data, highlighting the importance of the selection process rather than taking the entire trajectory as biologically relevant [15].
Essential Research Reagents and Computational Tools
Table 3: Research Reagent Solutions for Integrative Modeling
| Tool/Reagent | Type | Primary Function | Key Features |
|---|---|---|---|
| AlphaFold | Software | Protein structure prediction | End-to-end deep learning; Evoformer architecture; pLDDT confidence score [84] |
| AMBER | Software | Molecular dynamics simulation | Force fields (ff99SB); Periodic boundary conditions; TIP3P water model [63] |
| GAFF | Software | Force field parameterization | Generalized Amber Force Field for small molecules [82] |
| CPMD | Software | First-principles dynamics | Density functional theory; Wannier function analysis [82] |
| ¹⁵N-labeled proteins | Biochemical reagent | NMR spectroscopy substrate | Enables ¹⁵N(¹H) NMR relaxation measurements [15] |
| LAMMPS | Software | Molecular dynamics | Classical MD simulations; Multiple force fields [82] |
The integration of AlphaFold, molecular dynamics, and NMR relaxation data represents a significant advancement in structural biology, moving beyond the limitations of single-method approaches. This integrative framework leverages the predictive power of AI, the temporal resolution of MD, and the experimental validation of NMR to construct accurate, dynamic conformational ensembles that more faithfully represent protein behavior in solution. For researchers comparing crystal structures with NMR ensembles, this approach provides a pathway to reconcile the static beauty of crystalline states with the dynamic reality of biological function. As methods for generating conformational ensembles continue to evolve—including AlphaFold-generated ensembles and database-derived models—the integration with experimental validation will remain crucial for capturing the full spectrum of protein structural heterogeneity.
Conclusion
The synergy between X-ray crystallography and NMR spectroscopy is paramount for a modern, dynamic understanding of protein structure. Crystallography provides high-resolution architectural blueprints, while NMR reveals the essential conformational flexibility and dynamics in solution that are critical for function. For drug discovery, this integration is transformative; it allows researchers to move beyond static binding sites to target specific conformational states and allosteric networks, understand enthalpy-entropy compensation, and design superior therapeutics. Future directions will be dominated by integrative approaches that combine these experimental techniques with computational powerhouses like AlphaFold and molecular dynamics. These hybrid methods, validated by robust tools like CoNSEnsX, are paving the way for the accurate prediction of holistic, time-resolved conformational ensembles, ultimately enabling the precise targeting of complex diseases with next-generation drugs.
References
- 1. X-ray refinement significantly underestimates the level of ... [https://www.nature.com/articles/ncomms4220]
- 2. Systematic Comparison of Crystal and NMR Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3032220/]
- 3. EnsembleFlex: Protein structure ensemble analysis made ... [https://www.sciencedirect.com/science/article/pii/S0969212625002497]
- 4. B-Factor Rescaling for Protein Crystal Structure Analyses [https://www.mdpi.com/2073-4352/14/5/443]
- 5. Comparison of NMR and crystal structures of membrane ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5790426/]
- 6. How large B-factors can be in protein crystal structures [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-018-2083-8]
- 7. NMR-driven structure-based drug discovery by unveiling ... [https://www.nature.com/articles/s42004-025-01542-x]
- 8. Article Free-Energy Calculations Highlight Differences in ... [https://www.sciencedirect.com/science/article/pii/S0969212601006608]
- 9. A chemical interpretation of protein electron density maps in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7423092/]
- 10. Cryo-EM structure and B-factor refinement with ensemble ... [https://www.nature.com/articles/s41467-023-44593-1]
- 11. NMR-driven structure-based drug discovery by unveiling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12126535/]
- 12. Combining AlphaFold, MD, and Amide 15N(1H) NMR ... [https://pubmed.ncbi.nlm.nih.gov/41009484/]
- 13. Accurate Protein Dynamic Conformational Ensembles [https://pmc.ncbi.nlm.nih.gov/articles/PMC12469643/]
- 14. Determining accurate conformational ensembles of ... [https://www.nature.com/articles/s41467-025-64098-3]
- 15. Accurate Protein Dynamic Conformational Ensembles [https://www.mdpi.com/1422-0067/26/18/8917]
- 16. Assisting and accelerating NMR assignment with ... [https://www.nature.com/articles/s42003-025-08466-1]
- 17. Beyond static structures: protein dynamic conformations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12262120/]
- 18. Patterns in Protein Flexibility: A Comparison of NMR “ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7967184/]
- 19. CoNSEnsX: an ensemble view of protein structures and NMR ... [https://bmcstructbiol.biomedcentral.com/articles/10.1186/1472-6807-10-39]
- 20. Global comparative structural analysis of responses to ... [https://www.nature.com/articles/s41467-025-64116-4]
- 21. Progress, Challenges and Opportunities of NMR and XL-MS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10900494/]
- 22. New x-ray technology captures proteins in motion [https://biologicalsciences.uchicago.edu/news/new-x-ray-technology-captures-proteins-motion]
- 23. Functional protein dynamics in a crystal [https://www.nature.com/articles/s41467-024-47473-4]
- 24. Comparing Analytical Techniques for Structural Biology [https://www.nanoimagingservices.com/about/blog/unraveling-the-mysteries-of-the-molecular-world-structural-biology-techniques]
- 25. Current NMR Techniques for Structure-Based Drug ... [https://pubmed.ncbi.nlm.nih.gov/29329228/]
- 26. "NMR-driven structure-based drug discovery by unveiling ... [https://www.linkedin.com/pulse/nmr-driven-structure-based-drug-discovery-unveiling-ken-wasserman-ju2ve]
- 27. Structure-Based Drug Design - Rowan Scientific [https://www.rowansci.com/technologies/structure-based-drug-design]
- 28. Assisting and accelerating NMR assignment with restrained ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12274491/]
- 29. NMR spectroscopy method development [https://www.nature.com/collections/figjjdbjgi]
- 30. Solution-state NMR - Latest research and news [https://www.nature.com/subjects/solution-state-nmr]
- 31. October 10, 2025 - Seminar - Hidden Structural States of ... [https://clas.ucdenver.edu/chemistry/2025/10/06/october-10-2025-seminar-hidden-structural-states-proteins-revealed-conformer-selection]
- 32. Comparison Of Structural Techniques: X-ray, NMR, Cryo-EM [https://peakproteins.com/a-comparison-of-the-structural-techniques-used-at-sygnature-discovery-x-ray-crystallography-nmr-and-cryo-em/]
- 33. SmartSoak® Technology — High-Throughput Crystal ... [https://www.crystalsfirst.com/smartsoak/]
- 34. High-throughput crystallisation and ligand screening [https://www.embl.org/services-facilities/grenoble/high-throughput-crystallisation/]
- 35. Fragment-Based Drug Discovery by NMR. Where Are the ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2022.834453/full]
- 36. Perspectives on NMR in drug discovery: a technique comes of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2891904/]
- 37. Lessons from high-throughput protein crystallization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3637395/]
- 38. Fragment-Based Drug Discovery Using NMR Spectroscopy [https://pmc.ncbi.nlm.nih.gov/articles/PMC3699969/]
- 39. NMR-Fragment Based Virtual Screening: A Brief Overview [https://pmc.ncbi.nlm.nih.gov/articles/PMC6017141/]
- 40. Crystallographic Fragment Screening in Drug Design [https://www.saromics.com/crystallographic-fragment-screening/]
- 41. Accelerating fragment-based drug discovery using grand ... [https://www.nature.com/articles/s41467-025-60561-3]
- 42. X-Ray Crystallography vs. NMR Spectroscopy [https://www.news-medical.net/life-sciences/X-Ray-Crystallography-vs-NMR-Spectroscopy.aspx]
- 43. Comparison of X-ray Crystallography, NMR and EM [https://www.creative-biostructure.com/comparison-of-crystallography-nmr-and-em_6.htm?srsltid=AfmBOoqUv7qzAS63XQokdvcEJajtW3OKWBwwRIQZYS26PX5MrFragI__]
- 44. Crystallographic fragment screening [https://pubmed.ncbi.nlm.nih.gov/22222452/]
- 45. Solution NMR Spectroscopy for the Study of Enzyme Allostery [https://pmc.ncbi.nlm.nih.gov/articles/PMC4937494/]
- 46. Evaluating the impact of X-ray damage on conformational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9344472/]
- 47. NMR Methods to Study Dynamic Allostery - Research journals [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1004620]
- 48. Heterogeneous and Allosteric Role of Surface Hydration for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10848206/]
- 49. Introducing NMR strategies to define water molecules that ... [https://www.sciencedirect.com/science/article/pii/S2666441023000225]
- 50. Sample delivery methods for protein X-ray crystallography ... [https://www.nature.com/articles/s41467-025-65173-5]
- 51. NMR Studies of Protein Hydration and TEMPOL Accessibility [https://www.sciencedirect.com/science/article/abs/pii/S0022283603008520]
- 52. X-ray crystallography over the past decade for novel drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4655606/]
- 53. Limitations and lessons in the use of X-ray structural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7185550/]
- 54. Mnova NMR Software for 1D and 2D NMR Data [https://mestrelab.com/main-product/nmr]
- 55. NMR Software | Processing, Prediction, and Assignment [https://www.acdlabs.com/solutions/nmr-spectroscopy/]
- 56. The accuracy of protein structures in solution determined ... [https://www.sciencedirect.com/science/article/pii/S0969212622001320]
- 57. Ultrahigh-resolution solid-state NMR for high–molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12285714/]
- 58. Super-Resolved Nuclear Magnetic Resonance Spectroscopy [https://www.nature.com/articles/s41598-017-09884-w]
- 59. qNMR of mixtures: what is the best solution to signal ... [https://mestrelab.com/articles/qnmr-of-mixtures-what-is-the-best-solution-to-signal-overlap.html]
- 60. Review The development of nucleic acids force fields [https://www.sciencedirect.com/science/article/pii/S0006349522039327]
- 61. Scrutinizing Molecular Mechanics Force Fields on the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2905107/]
- 62. On the ability of molecular dynamics force fields to recapitulate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4941777/]
- 63. Starting Structure Dependence of NMR Order Parameters ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2426661/]
- 64. A Comparison of Force Fields for Nucleic Acids - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1302718/]
- 65. Quantitative Comparison Against Experiments Reveals ... [https://chemrxiv.org/engage/chemrxiv/article-details/648acb0de64f843f41d0183a]
- 66. Supplemental: Overview of the Common Force Fields [https://computecanada.github.io/molmodsim-md-theory-lesson-novice/S01-Force_Fields_Overview/index.html]
- 67. Application of NMR and Molecular Docking in Structure- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6628936/]
- 68. Molecular docking as a tool for the discovery ... [https://www.nature.com/articles/s41598-023-40160-2]
- 69. A combined computational-experimental approach to ... [https://www.nature.com/articles/s41598-018-29209-9]
- 70. Addressing docking pose selection with structure-based ... [https://www.sciencedirect.com/science/article/pii/S2001037024001727]
- 71. Predicting Pose Distribution of Protein Domains Connected by ... [https://pubmed.ncbi.nlm.nih.gov/41102979/]
- 72. Measurement of 1H-15N and 1H-13C residual dipolar ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3184849/]
- 73. NMR methods for investigating functionally relevant ... [https://www.sciencedirect.com/science/article/pii/S2772516225000208]
- 74. CoNSEnsX: an ensemble view of protein structures and ... [https://pubmed.ncbi.nlm.nih.gov/21034466/]
- 75. an ensemble view of protein structures and NMR-derived ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2987814/]
- 76. EnsembleFlex: Protein structure ensemble analysis made ... [https://pubmed.ncbi.nlm.nih.gov/40713968/]
- 77. CoNSEnsX + [https://consensx.itk.ppke.hu/]
- 78. EnsembleFlex - Flexibility Analysis of Structure Ensembles [https://github.com/chembl/EnsembleFlex]
- 79. Patterns in Protein Flexibility: A Comparison of NMR “Ensembles”, MD Trajectories, and Crystallographic B-Factors - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7967184/]
- 80. Patterns in Protein Flexibility: A Comparison of NMR ... [https://www.mdpi.com/1420-3049/26/5/1484]
- 81. Patterns in Protein Flexibility: A Comparison of NMR ... [https://pubmed.ncbi.nlm.nih.gov/33803249/]
- 82. IR-NMR multimodal computational spectra dataset for ... [https://www.nature.com/articles/s41597-025-05729-8]
- 83. Bridging prediction and reality: Comprehensive analysis of ... [https://www.sciencedirect.com/science/article/pii/S2001037025001734]
- 84. Highly accurate protein structure prediction with AlphaFold [https://www.nature.com/articles/s41586-021-03819-2]
- 85. Gradations in protein dynamics captured by experimental ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283624005308]