CCR5Δ32 Mutation: From Natural HIV Resistance to Therapeutic Gene Editing
This article provides a comprehensive analysis of the CCR5Δ32 mutation, a 32-base-pair deletion in the CCR5 gene that confers significant resistance to HIV-1 infection.
CCR5Δ32 Mutation: From Natural HIV Resistance to Therapeutic Gene Editing
Abstract
This article provides a comprehensive analysis of the CCR5Δ32 mutation, a 32-base-pair deletion in the CCR5 gene that confers significant resistance to HIV-1 infection. We explore the foundational mechanism by which this mutation prevents the CCR5 co-receptor's expression on the cell surface, thereby blocking the primary entry pathway for R5-tropic HIV strains. The discussion extends to methodological applications, including the development of CCR5 antagonists and pioneering gene-editing technologies like CRISPR/Cas9 that aim to mimic this natural resistance. The review also addresses key challenges such as viral tropism switching and optimization strategies involving multi-target gene editing. Finally, we examine the validation of this mechanism through population genetics, clinical case studies, and comparative analyses with other protective alleles, synthesizing the profound implications for future HIV cure strategies and therapeutic development.
The Genetic Shield: Deconstructing the CCR5Δ32 Mutation and Its HIV Blockade Mechanism
CCR5's Role as a Critical HIV-1 Co-receptor
The C-C chemokine receptor type 5 (CCR5) has been established as an essential co-receptor for human immunodeficiency virus type 1 (HIV-1) entry into host cells. As a member of the G-protein coupled receptor (GPCR) superfamily, this seven-transmembrane protein is expressed on various leukocytes including macrophages, dendritic cells, and CD4+ T-cells, where it normally functions in inflammatory signaling pathways [1] [2]. The seminal discovery that individuals carrying a homozygous 32-base pair deletion in the CCR5 gene (CCR5-Δ32) demonstrated natural resistance to HIV-1 infection transformed our understanding of viral entry mechanisms and created new avenues for therapeutic intervention [3] [4]. This technical guide comprehensively examines CCR5's pivotal role in HIV-1 pathogenesis, the protective mechanism of the Δ32 mutation, current experimental methodologies for studying coreceptor function, and emerging therapeutic strategies that target this critical viral entry pathway.
CCR5 Structure and Natural Function
CCR5 is a 352-amino-acid protein located on chromosome 3 at position 3p21.31 [4]. Its structure comprises seven transmembrane helices with an extracellular N-terminus and three extracellular loops (ECLs), particularly ECL2, which form critical interaction sites for both natural ligands and viral envelope proteins [5]. Under physiological conditions, CCR5 binds inflammatory β-chemokines including RANTES (CCL5), MIP-1α (CCL3), and MIP-1β (CCL4) [2]. This binding activates downstream signaling pathways through G-proteins, leading to coordinated immune cell migration, secretion of pro-inflammatory cytokines, and stimulation of both innate and adaptive immune responses [2]. The receptor's expression on memory CD4+ T-cells, macrophages, and dendritic cells highlights its fundamental role in immune coordination and inflammatory response management.
The Mechanism of HIV-1 Entry and Coreceptor Dependency
HIV-1 entry into target cells requires a meticulously coordinated sequence of interactions between viral envelope proteins and host cell receptors. The process initiates when the viral gp120 glycoprotein binds to the primary CD4 receptor on susceptible cells, inducing conformational changes that expose previously cryptic epitopes [6]. These structural rearrangements enable gp120 to engage with a coreceptor—predominantly CCR5 or CXCR4—with the specific V3 loop region of gp120 playing a decisive role in coreceptor selection [7] [8]. Following successful coreceptor binding, further conformational changes activate the gp41 fusion peptide, which facilitates viral and cellular membrane fusion, culminating in viral entry [6].
HIV-1 strains are categorized based on their coreceptor preference: R5-tropic viruses utilize CCR5, X4-tropic viruses utilize CXCR4, and dual-tropic viruses can utilize both coreceptors [6]. During early and chronic infection stages, R5-tropic strains overwhelmingly predominate, making CCR5 the most clinically relevant coreceptor for initial infection and transmission [3] [6]. The emergence of X4-tropic variants typically occurs later in disease progression and is associated with accelerated CD4+ T-cell decline and more rapid disease progression [7] [8].
Table 1: HIV-1 Tropism Classification and Characteristics
| Tropism Classification | Primary Coreceptor | Stage of Infection | Clinical Association |
|---|---|---|---|
| R5-tropic | CCR5 | Early/Chronic | Primary transmission, macrophage tropism |
| X4-tropic | CXCR4 | Late | Accelerated CD4+ decline, syncytium formation |
| Dual/Mixed-tropic | CCR5 and/or CXCR4 | Any stage | Variable clinical course |
The CCR5-Δ32 Mutation: A Natural Resistance Mechanism
The CCR5-Δ32 mutation represents a 32-base pair deletion in the CCR5 gene that results in a frameshift and premature translational termination [4]. The truncated protein product lacks three transmembrane domains, extracellular and intracellular loops, and consequently cannot embed in the cell membrane, remaining instead as a non-functional intracellular peptide [4]. This loss-of-function mutation provides remarkable HIV-1 resistance in a gene dosage-dependent manner: heterozygous individuals exhibit delayed disease progression, while homozygous individuals demonstrate near-complete resistance to R5-tropic HIV-1 infection [4].
The epidemiological distribution of the Δ32 allele reveals striking geographical patterns, with highest frequencies in Northern European populations (approximately 10% allele frequency, 1% homozygosity) and a pronounced north-to-south gradient across Europe [4]. Multiple evolutionary hypotheses have been proposed to explain this distribution, including selective pressure from historical pathogens such as Yersinia pestis (bubonic plague) or Variola major (smallpox), though conclusive evidence remains elusive [4]. The profound HIV resistance observed in CCR5-Δ32 homozygotes was definitively demonstrated in the cases of the "Berlin" and "London" patients—HIV-positive individuals who received allogeneic hematopoietic stem cell transplantation from CCR5-Δ32 homozygous donors and subsequently achieved long-term viral remission without antiretroviral therapy [3] [9].
Experimental Methods for Studying CCR5 Coreceptor Function
Tropism Determination Assays
Accurate determination of viral tropism is essential both for clinical management and research applications. Phenotypic assays directly measure coreceptor usage through recombinant virus entry assays but are limited by cost, complexity, and processing time [6]. Genotypic methods infer tropism from V3 loop sequence characteristics, with computational tools like coreceptor-specific weight matrices (CMs) achieving accuracies exceeding 95% by incorporating position-specific scoring and charge rules [7]. Ultra-deep pyrosequencing (UDPS) technologies enable detection of minor CXCR4-using variants at frequencies below 5%, providing critical sensitivity for identifying tropism transitions in viral quasispecies [8].
Conformational Analysis of CCR5 Populations
Advanced immunological techniques using monoclonal antibodies with distinct epitope specificities have revealed that CCR5 exists in multiple conformational states on the cell surface, with only specific subpopulations permitting HIV-1 entry [5]. Super-resolution microscopy demonstrates that antibodies targeting the N-terminal region (such as CTC8) most effectively block viral entry by recognizing and binding to the specific CCR5 conformations required for gp120 interaction [5]. Infection experiments show selective internalization of these permissive CCR5 subpopulations following HIV-1 exposure, confirming their essential role in viral entry [5].
Diagram 1: HIV-1 Entry Mechanism via CD4 and CCR5
CCR5-Targeted Therapeutic Strategies
CCR5 Antagonists
Maraviroc, a small molecule CCR5 antagonist, represents the first licensed antiretroviral drug targeting a host factor rather than viral enzymes [6]. It functions allosterically by binding to a transmembrane pocket of CCR5, inducing conformational changes that prevent gp120 interaction while preserving chemokine binding and signaling functions [1] [6]. Clinical application requires pretreatment tropism testing to confirm CCR5 dependence, as X4-tropic viruses are intrinsically resistant [6].
Gene Editing Approaches
Gene editing technologies represent a revolutionary approach to recreating the protective Δ32 phenotype in patient-derived cells. Multiple platforms have demonstrated efficacy in disrupting CCR5 expression, including zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the CRISPR/Cas9 system [3]. Each technology offers distinct advantages and limitations regarding specificity, efficiency, and delivery considerations.
Table 2: Gene Editing Technologies for CCR5 Disruption
| Technology | Mechanism of Action | Editing Efficiency | Clinical Status |
|---|---|---|---|
| ZFNs | Custom zinc finger proteins fused to FokI nuclease recognize and cleave specific DNA sequences | Moderate | Phase I/II trials demonstrated safety and virological benefit |
| TALENs | Modular transcription activator-like effector proteins fused to FokI nuclease for DNA cleavage | High with improved specificity | Preclinical studies show efficient CCR5 editing |
| CRISPR/Cas9 | Guide RNA directs Cas9 nuclease to specific genomic loci for targeted cleavage | High, enables multiplex editing | Early-phase clinical trials (e.g., NCT03164135) demonstrate feasibility |
Recent innovations combine CCR5 knockout with knock-in strategies for HIV-1 inhibiting antibodies, creating multilayered resistance in hematopoietic stem and progenitor cells (HSPCs) [9]. This approach simultaneously provides cell-intrinsic resistance through CCR5 disruption and cell-extrinsic protection via secretion of broadly neutralizing antibodies (bNAbs) such as 10-1074, PGDM1400, and Ibalizumab [9].
Diagram 2: Multilayered HIV Resistance through Gene Editing
Research Reagent Solutions
The following essential research reagents represent critical tools for investigating CCR5 biology and developing therapeutic interventions:
Table 3: Essential Research Reagents for CCR5 Investigation
| Reagent Category | Specific Examples | Research Application |
|---|---|---|
| CCR5 monoclonal antibodies | CTC8 (anti-N-terminal), CTC5, 2D7, 45523, 45531 | Distinguishing conformational subpopulations; neutralization studies |
| HIV-1 inhibiting antibodies | Ibalizumab, 10-1074, PGDM1400, CAP256V2LS, 3BNC117 | Neutralization assays; knock-in strategies for gene therapy |
| Gene editing systems | ZFNs (SB-728-T), TALENs, CRISPR/Cas9, Base editors | CCR5 disruption in HSPCs and T-cells; therapeutic development |
| Cell lines | JC10 (low CCR5), JC53 (high CCR5), U87.CD4, TZM-bl | Tropism assays; viral entry studies; neutralization assays |
| Chemokine ligands | RANTES/CCL5, MIP-1α/CCL3, MIP-1β/CCL4 | Competition binding studies; internalization assays |
CCR5 stands as a critically validated co-receptor for HIV-1 entry and an exemplary model of how host genetics can inform therapeutic development. The structural biology of CCR5, its precise role in viral entry mechanisms, and the remarkable resistance conferred by the Δ32 mutation collectively provide a robust foundation for multiple therapeutic strategies. Current approaches encompass small molecule antagonists, gene editing platforms, and combination therapies that leverage both cell-intrinsic and cell-extrinsic resistance mechanisms. Despite significant progress, challenges remain in ensuring complete safety of gene editing approaches, preventing coreceptor tropism switching, and developing globally accessible therapies. Future research directions will likely focus on multiplexed gene editing strategies targeting both CCR5 and CXCR4, personalized approaches accounting for clinical heterogeneity, and sophisticated delivery systems to enhance therapeutic efficacy while minimizing off-target effects. The continued investigation of CCR5 biology promises to yield increasingly potent interventions in the ongoing effort to achieve durable HIV-1 remission and eventual eradication.
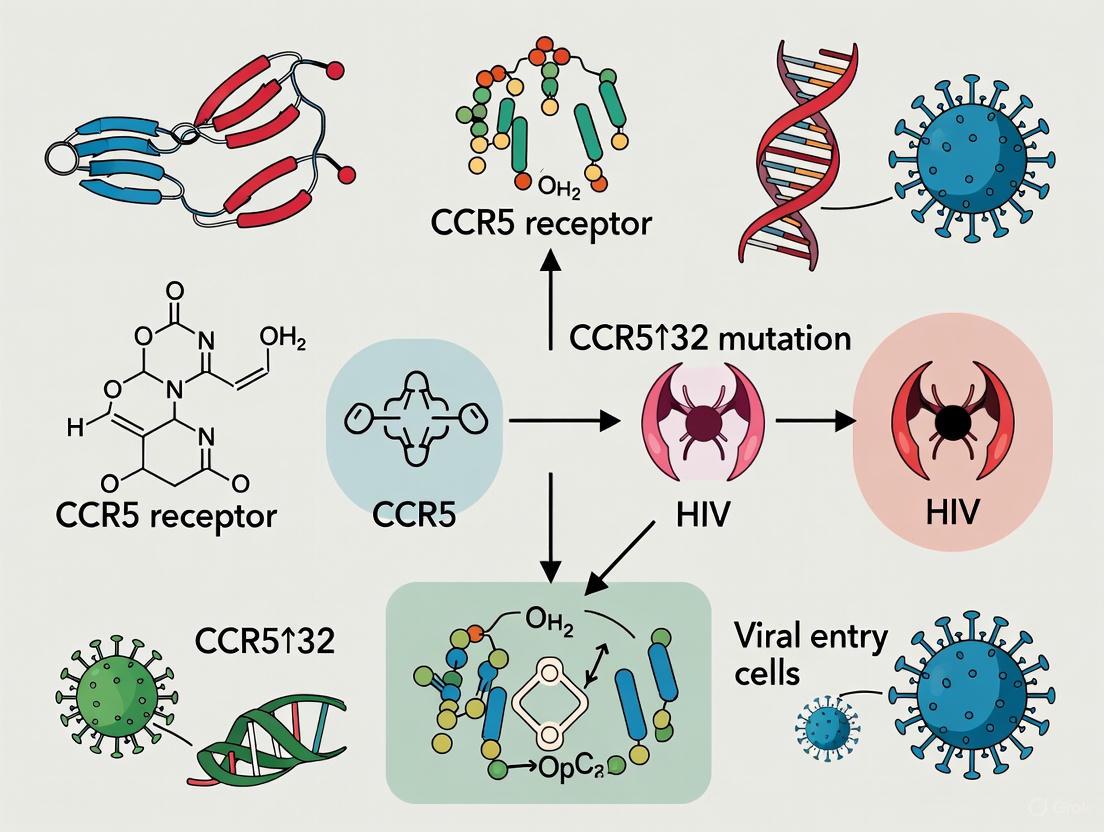
The Structural Consequence of the 32-Base-Pair Deletion
The C-C chemokine receptor type 5 (CCR5) serves as a critical co-receptor for human immunodeficiency virus (HIV) entry into host cells. A naturally occurring 32-base-pair deletion (CCR5-Δ32) in the CCR5 gene results in a truncated protein that confers resistance to HIV-1 infection in homozygous individuals. This whitepaper provides a comprehensive technical analysis of the structural alterations caused by the CCR5-Δ32 mutation, detailing the molecular mechanisms through which these changes disrupt CCR5 function and prevent viral entry. Within the broader context of HIV resistance research, we examine experimental methodologies for studying this mutation, visualize key signaling pathways and structural relationships, and catalog essential research tools for investigating CCR5 biology. The structural insights derived from CCR5-Δ32 research continue to inform novel therapeutic strategies, including CCR5 antagonists and gene editing approaches for HIV treatment.
CCR5 is a G-protein-coupled receptor (GPCR) containing seven transmembrane α-helices, three extracellular loops, and three intracellular loops [10]. It is expressed on various immune cells including T-lymphocytes, monocytes, macrophages, dendritic cells, and natural killer cells [11] [2]. As a chemokine receptor, CCR5 interacts primarily with CCL3 (MIP-1α), CCL4 (MIP-1β), and CCL5 (RANTES), mediating leukocyte migration and inflammatory responses [10] [12]. Beyond its natural immunological functions, CCR5 gained prominence as the major co-receptor for R5-tropic HIV-1 strains, working in concert with CD4 to facilitate viral entry into target cells [12] [13].
The CCR5-Δ32 mutation is a 32-base-pair deletion within the coding region of the CCR5 gene on chromosome 3 (3p21.31) [10] [14]. This deletion causes a frameshift during translation, resulting in a prematurely truncated protein that lacks normal structural and functional domains [14] [15]. Individuals homozygous for this mutation (Δ32/Δ32) demonstrate substantial resistance to HIV-1 infection due to the absence of functional CCR5 on cell surfaces, while heterozygous individuals (WT/Δ32) show delayed disease progression due to reduced receptor expression [10] [12] [16].
This technical guide examines the structural consequences of the CCR5-Δ32 deletion within the broader research context of HIV resistance mechanisms. We analyze how specific structural alterations disrupt receptor function, summarize key experimental approaches for studying this mutation, and provide visual representations of the structural and mechanistic relationships.
Structural Biology of Wild-Type CCR5 and Δ32 Variant
Wild-Type CCR5 Architecture
The wild-type CCR5 protein consists of 352 amino acids forming a characteristic GPCR structure [12] [14]. Key structural elements include:
- Seven transmembrane α-helices that traverse the plasma membrane
- Three extracellular loops responsible for ligand binding
- Three intracellular loops involved in G-protein coupling and signal transduction
- An extracellular N-terminus and intracellular C-terminus [10] [12]
The second extracellular loop (ECL2) contains the critical 2D7 epitope, which serves as the essential binding site for the HIV-1 envelope glycoprotein gp120 [15]. Helices 2 and 3 play fundamental roles in chemokine-induced CCR5 activation [10]. The receptor is palmitoylated at cysteine residues in its C-terminal domain, facilitating localization to plasma membrane lipid rafts where it clusters with CD4 to create optimal HIV-1 docking sites [12].
Structural Consequences of Δ32 Deletion
The 32-base-pair deletion produces a frameshift mutation beginning at amino acid position 185, introducing a premature stop codon that results in a severely truncated protein of only 215 amino acids [14] [15]. This truncation causes critical structural deficiencies:
Table 1: Structural Domains Affected by CCR5-Δ32 Mutation
| Structural Domain | Wild-Type CCR5 | CCR5-Δ32 Variant | Functional Consequence |
|---|---|---|---|
| Transmembrane Domains | 7 complete domains | Only 3-4 domains incorporated | Incomplete membrane integration |
| Second Extracellular Loop | Intact with 2D7 epitope | Completely absent | Eliminates HIV-1 gp120 binding site |
| C-terminal Region | Full intracellular domain | Severely truncated | Disrupted intracellular signaling |
| Cellular Localization | Cell surface expression | Retained intracellularly | Prevents viral docking |
The truncated Δ32 protein lacks the final three transmembrane domains and the entire second extracellular loop, which contains the vital 2D7 binding epitope required for HIV-1 interaction [15]. Without this critical structural element, HIV-1 cannot effectively bind to the receptor complex. Furthermore, the mutation removes palmitoylation sites in the C-terminal domain, disrupting proper trafficking and membrane localization [12]. The misfolded protein is retained in the endoplasmic reticulum and fails to reach the cell surface, preventing any potential interaction with HIV-1 [11].
Diagram 1: Structural Comparison of Wild-Type and Δ32 CCR5. The CCR5-Δ32 variant exhibits severe structural truncation affecting membrane integration, key functional domains, and cellular localization.
Quantitative Analysis of CCR5-Δ32 Distribution
The CCR5-Δ32 allele demonstrates significant geographic variation in its distribution, with highest frequencies observed in Northern European populations. This uneven distribution suggests historical selective pressures, possibly from pathogens such as smallpox or plague [14] [15].
Table 2: Global Distribution of CCR5-Δ32 Allele Frequency
| Population | Δ32 Allele Frequency | Homozygous Frequency | Research Context |
|---|---|---|---|
| Northern European | 16% (Norway) | ~1% | Highest global frequency [10] |
| Central European | 11% (Germany) | ~1% | Intermediate frequency [10] [17] |
| Southern European | 4-6% (Italy, Greece) | <0.5% | Declining north-south gradient [18] [14] |
| African & Asian | ~0% | ~0% | Virtually absent [17] [14] |
| Admixed Latin American | 4-5% (Brazil), 12% (Chile) | Variable | Reflects European admixture [10] [18] |
| Peruvian | 2.7% (heterozygous only) | 0% | Low prevalence in study cohort [16] |
The CCR5-Δ32 mutation follows a pronounced north-to-south gradient in Europe, with highest frequencies in Scandinavian and Baltic populations (approximately 16%) and decreasing frequencies in Southern European populations (4-6%) [10] [14]. The mutation is virtually absent in African, Asian, and indigenous American populations, though variable frequencies occur in admixed Latin American populations due to European gene flow [10] [18] [16]. In Peru, recent research found a 2.7% prevalence of heterozygous individuals and no homozygous cases among the study population [16].
Methodological Approaches for CCR5-Δ32 Research
Genotyping Techniques
Endpoint PCR with Gel Electrophoresis
- Principle: Amplification of CCR5 gene region flanking the Δ32 deletion using specific primers, followed by size separation on agarose gels [16].
- Protocol Details:
- Primers: Forward: 5′-ACCAGATCTCTCAAAAAGAAGGTCT-3′, Reverse: 5′-CATGATGGTGAAGATAAGCCTCCACA-3′ [16]
- Amplification Conditions: Initial denaturation at 98°C for 30 seconds; 35 cycles of 98°C for 30 seconds, 60°C for 30 seconds, 72°C for 15 seconds; final extension at 72°C for 3 minutes [16]
- Product Analysis: Wild-type allele produces 225bp fragment; Δ32 allele produces 193bp fragment; heterozygotes show both bands [16]
- Applications: Initial screening, population studies, clinical genotyping [16].
Real-Time PCR with Probe Detection
- Principle: Quantitative amplification with allele-specific fluorescent probes for high-throughput genotyping [16].
- Advantages: Higher throughput, reduced contamination risk, potential for quantification [16].
DNA Sequencing
- Principle: Direct determination of nucleotide sequence across the CCR5 gene region to confirm deletion and identify potential additional variations [16].
- Methodology: Sanger sequencing of PCR products using Big Dye Terminator chemistry followed by capillary electrophoresis [16].
- Applications: Validation of PCR results, identification of rare variants, quality control [16].
Functional Assays
Cell Surface Expression Analysis
- Flow Cytometry: Uses CCR5-specific antibodies (e.g., recognizing 2D7 epitope) to quantify receptor expression on peripheral blood mononuclear cells (PBMCs) or transfected cell lines [12].
- Immunofluorescence Microscopy: Visualizes subcellular localization of CCR5 in permeabilized vs. non-permeabilized cells to demonstrate intracellular retention of Δ32 variant [11].
Viral Entry Assays
- Pseudovirus Infection: Engineered HIV-1 pseudotypes with envelope proteins from R5-tropic strains used to infect cells with different CCR5 genotypes [12] [13].
- Cell-Cell Fusion Assays: Measures fusion between cells expressing HIV-1 envelope and target cells expressing CD4/CCR5 [12].
Gene Expression Profiling
- Microarray Analysis: Global gene expression comparison between CCR5 wild-type and Δ32 carrier cells to identify differentially expressed genes [17].
- Methodology: CD34+ hematopoietic progenitor cells from genotyped donors analyzed using Affymetrix HG-U133plus 2.0 arrays [17].
Diagram 2: Experimental Workflow for CCR5-Δ32 Research. The methodology encompasses sample collection, genotyping, functional validation, and data analysis to comprehensively characterize the mutation.
Gene Editing Approaches
CRISPR-Cas9 Systems
- Application: Precise genome editing to introduce CCR5 disruptions in hematopoietic stem cells (HSCs) and CD4+ T-cells [15] [13].
- Protocol: CD34+ HSPCs transfected with CRISPR-Cas9 constructs targeting CCR5; edited cells transplanted into immunodeficient mice to assess HIV-1 resistance in vivo [15].
- Outcome: Long-term CCR5 ablation and sustained HIV-1 resistance in humanized mouse models [15] [13].
Zinc Finger Nucleases (ZFNs) and TALENs
- Application: Earlier gene editing platforms for CCR5 disruption, with clinical trials demonstrating safety and feasibility [13] [2].
- Clinical Data: ZFN-modified CD4+ T-cells (SB-728) showed enhanced resistance to HIV-1 infection and potential viral load reductions in patients [2].
Research Reagent Solutions
Table 3: Essential Research Tools for CCR5-Δ32 Investigations
| Reagent/Category | Specific Examples | Research Application | Technical Function |
|---|---|---|---|
| Genotyping Primers | CCR5 DELTA1/DELTA2 [16] | Mutation screening | Amplify wild-type (225bp) and Δ32 (193bp) alleles |
| Antibodies | Anti-CCR5 (2D7 epitope) [12] | Surface expression analysis | Flow cytometry and immunofluorescence detection |
| Cell Lines | PM1, HEK-293T, TZM-bl | Viral entry assays | Model systems for HIV-1 infectivity studies |
| Gene Editing Tools | CRISPR-Cas9, ZFNs, TALENs [15] [13] | Functional validation | Targeted CCR5 disruption in primary cells |
| Chemokine Ligands | Recombinant CCL3, CCL4, CCL5 [10] [12] | Signaling studies | CCR5 activation and internalization assays |
| Animal Models | Humanized mouse models (NSG) | In vivo studies | HIV-1 challenge of edited human cells |
Implications for HIV-1 Therapeutic Development
The structural insights from CCR5-Δ32 research have directly informed multiple therapeutic strategies for HIV-1 infection:
Small Molecule Antagonists
- Maraviroc, an allosteric CCR5 inhibitor approved for clinical use in 2007, mimics the protective effect of Δ32 by blocking HIV-1 engagement without triggering signaling pathways [10] [12].
Gene Editing Therapies
- The "Berlin" and "London" patients, cured of HIV-1 after CCR5-Δ32 stem cell transplantation, provide proof-of-concept for CCR5-targeted therapies [18] [13] [2].
- CRISPR-Cas9 approaches to disrupt CCR5 in hematopoietic stem cells represent promising functional cure strategies currently in clinical trials (NCT03164135) [15] [13].
Combination Approaches
- Multiplex gene editing targeting both CCR5 and CXCR4 co-receptors prevents viral tropism switching [13].
- Integration of CCR5 editing with broadly neutralizing antibodies or latency-reversing agents addresses both viral entry and reservoir persistence [13] [2].
The structural characterization of CCR5-Δ32 continues to guide rational drug design and gene therapy development, highlighting the fundamental importance of understanding structure-function relationships in biomedical research.
The C-C chemokine receptor type 5 (CCR5) serves as a critical coreceptor for human immunodeficiency virus (HIV) entry into target cells. A naturally occurring 32-base pair deletion in the CCR5 gene (CCR5Δ32) confers profound resistance to HIV-1 infection in homozygous individuals. This technical review delineates the molecular and cellular mechanisms through which the CCR5Δ32 mutation prevents functional receptor expression on the cell membrane. We examine how the truncated protein product undergoes misprocessing, intracellular retention, and accelerated degradation, ultimately resulting in the absence of CCR5 from the cell surface. Furthermore, we explore secondary effects including receptor dimerization and altered chemokine receptor homeostasis. Within the broader context of HIV resistance research, understanding these mechanisms provides the foundational knowledge for developing CCR5-targeted therapeutic interventions.
CCR5 is a seven-transmembrane G-protein-coupled receptor (GPCR) that normally facilitates immune cell migration and inflammatory responses by binding chemokines including CCL3 (MIP-1α), CCL4 (MIP-1β), and CCL5 (RANTES) [12] [19]. As a primary coreceptor for R5-tropic HIV-1 strains, CCR5 enables viral entry into CD4+ T lymphocytes upon binding to the viral envelope glycoprotein gp120 [20] [21]. The CCR5Δ32 mutation, a 32-base pair deletion in the coding region of the CCR5 gene, represents a loss-of-function mutation that substantially reduces susceptibility to HIV-1 infection in heterozygous individuals and provides near-complete protection in homozygous carriers [4] [21].
The CCR5Δ32 allele occurs with approximately 10% frequency in European populations, with a north-to-south clinal distribution, but is virtually absent in native African, Asian, and Indigenous American populations [18] [4] [21]. This mutation results in a frameshift during translation and generates a prematurely truncated protein that lacks normal structural domains essential for proper membrane localization and function [20] [4]. This review systematically examines the molecular journey of the mutant CCR5Δ32 protein from synthesis to degradation, explaining the mechanistic basis for its absence from the plasma membrane.
Molecular Consequences of the Δ32 Deletion
Structural Domains Affected
The CCR5Δ32 mutation occurs in the region encoding the second extracellular loop of the receptor, resulting in a frameshift that produces a severely truncated protein [4]. The table below summarizes the key structural deficiencies in the mutant protein compared to the wild-type CCR5.
Table 1: Structural Comparison of Wild-Type CCR5 and CCR5Δ32 Mutant Protein
| Structural Feature | Wild-Type CCR5 | CCR5Δ32 Mutant | Functional Consequence |
|---|---|---|---|
| Amino Acid Length | 352 amino acids | Truncated (approximately 215 amino acids) | Premature termination [4] |
| Transmembrane Domains | 7 domains | Only 3-4 domains | Incomplete membrane integration [4] |
| Extracellular Loops | 3 complete loops | Disrupted second loop | Impaired ligand binding [4] |
| Intracellular Loops | 3 complete loops | Truncated | Compromised signaling [4] |
| C-terminal Domain | Complete phosphorylation sites | Severely truncated | Altered trafficking & internalization [19] |
Biosynthetic Processing and Intracellular Trafficking
Following translation, the mutant CCR5Δ32 protein exhibits fundamental defects in its biosynthetic processing pathway. Unlike the wild-type receptor, which undergoes proper folding, post-translational modifications, and trafficking through the Golgi apparatus to the plasma membrane, the truncated protein fails to progress beyond the endoplasmic reticulum (ER) [20] [22].
The diagram below illustrates the comparative trafficking pathways of wild-type CCR5 versus the CCR5Δ32 mutant protein.
The CCR5Δ32 protein accumulates in the endoplasmic reticulum and fails to undergo complete glycosylation and other maturation processes essential for forward trafficking [20]. Pulse-chase experiments demonstrate significantly accelerated degradation kinetics of the CCR5Δ32 protein compared to wild-type CCR5, with rapid disappearance of the mutant protein during the chase period [20]. This ER retention and premature degradation constitute the primary mechanism preventing cell surface expression.
Experimental Evidence and Methodologies
Protein Detection and Localization Studies
Multiple experimental approaches have demonstrated the absence of functional CCR5Δ32 protein at the plasma membrane. Western blot analyses of peripheral blood mononuclear cell (PBMC) lysates from CCR5Δ32 homozygous individuals show either complete absence or dramatically reduced levels of the CCR5 protein compared to wild-type individuals [20]. In one comprehensive study, endogenous CCR5Δ32 protein was detected in all HIV-negative CCR5-/- PBMC samples (25/25) but was absent in four of six unrelated HIV-positive CCR5-/- PBMC samples, with only low levels detected in the remaining two samples [20].
Immunofluorescence and confocal microscopy studies provide visual evidence of the divergent subcellular localization. While wild-type CCR5 localizes predominantly to the plasma membrane, the CCR5Δ32 mutant protein remains trapped in intracellular compartments, showing minimal co-localization with plasma membrane markers [22]. These studies further demonstrated that in the steady state, wild-type and truncated CCR5 proteins segregate into non-overlapping subcellular compartments, challenging earlier hypotheses about heterodimer-induced retention [22].
Table 2: Key Experimental Evidence Supporting CCR5Δ32 Absence from Membrane
| Experimental Method | Key Findings | References |
|---|---|---|
| Western Blot Analysis | Absent or dramatically reduced CCR5 protein in CCR5Δ32 homozygotes | [20] |
| Flow Cytometry | No detectable CCR5 on cell surface of PBMCs from CCR5Δ32 homozygotes | [20] [22] |
| Confocal Microscopy | Intracellular retention of CCR5Δ32; absence from plasma membrane | [22] |
| Pulse-Chase Experiments | Accelerated degradation of CCR5Δ32 protein compared to wild-type | [20] |
| HIV Entry Assays | Resistance to R5-tropic HIV infection in CCR5Δ32 cells | [20] [12] |
Functional Assays of Coreceptor Activity
Functional assessments further corroborate the absence of CCR5 from the cell surface. HIV-1 envelope glycoprotein-mediated fusion assays demonstrate that CD4+ T lymphocytes isolated from CCR5Δ32 homozygous individuals show significantly reduced fusion with cells expressing R5-tropic HIV-1 Env proteins [20]. Viral infection experiments confirm that PBMCs from CCR5Δ32 homozygous individuals are highly resistant to infection by R5-tropic HIV-1 strains, while remaining susceptible to X4-tropic viruses that utilize CXCR4 as their coreceptor [20] [12].
Notably, when researchers expressed adenovirus-encoded CCR5Δ32 protein in PBMCs from HIV-positive CCR5-/- individuals, the protective effect was restored in some cases, but not in others, suggesting that protein stability and potential degradation mechanisms may vary between individuals [20]. In samples where the protective effect was not restored, pulse-chase analyses demonstrated disappearance of the adenovirus-encoded CCR5Δ32 protein and accumulation of wild-type CCR5 during chase periods [20].
Research Reagent Solutions
The following table compiles essential reagents and methodologies utilized in CCR5Δ32 research, providing a toolkit for investigators in this field.
Table 3: Essential Research Reagents for CCR5Δ32 Studies
| Reagent/Technique | Specific Example | Research Application | Key References |
|---|---|---|---|
| Anti-CCR5 Antibodies | PE-conjugated anti-CCR5 (Clone 556042) | Flow cytometry detection of surface CCR5 | [20] |
| Adenovirus Vectors | Ad5/Δ32 encoding CCR5Δ32 | Recombinant protein expression in PBMCs | [20] |
| PBMC Isolation | Phytohemagglutinin-activated PBMCs | Primary cell model for HIV infection studies | [20] |
| Gene Editing Tools | CRISPR/Cas9, ZFNs, TALENs | Targeted CCR5 disruption for therapeutic applications | [3] |
| HIV Entry Assays | Env-mediated cell fusion assay | Functional assessment of coreceptor activity | [20] |
Broader Implications for HIV Resistance Research
The elucidation of CCR5Δ32 mechanisms has directly inspired therapeutic strategies for HIV treatment and cure. The most prominent example involves hematopoietic stem cell transplantation from CCR5Δ32 homozygous donors to HIV-positive patients, which has resulted in sustained viral remission in several reported cases (the "Berlin," "London," and "Düsseldorf" patients) [3] [18]. This approach functionally recapitulates the natural resistance mechanism by reconstituting the immune system with CCR5-deficient CD4+ T cells that are resistant to HIV entry.
Furthermore, gene editing technologies specifically targeting CCR5 have emerged as promising therapeutic modalities. Clinical trials utilizing zinc finger nucleases (SB-728-T), CRISPR/Cas9 systems (NCT03164135), and other gene editing platforms aim to disrupt CCR5 expression in autologous T cells or hematopoietic stem cells for subsequent transplantation [3]. These approaches represent translational applications of the fundamental knowledge gained from studying the natural CCR5Δ32 variant.
The diagram below illustrates how research on the natural CCR5Δ32 mutation has informed the development of CCR5-targeted therapies for HIV.
Beyond HIV therapy, understanding CCR5Δ32 mechanisms has implications for various physiological and pathological processes. CCR5 plays roles in inflammatory diseases, cancer metastasis, and cognitive function, suggesting that modulation of CCR5 expression may have therapeutic value beyond HIV treatment [4] [21]. However, the pleiotropic functions of CCR5 also highlight potential unintended consequences of CCR5-targeted interventions, emphasizing the need for precise therapeutic control.
The CCR5Δ32 mutation prevents membrane expression through a multi-step process involving protein truncation, misfolding, endoplasmic reticulum retention, and accelerated degradation. The resultant absence of functional CCR5 from the cell surface presents a formidable barrier to R5-tropic HIV-1 entry, explaining the profound resistance observed in homozygous individuals. This natural protective mechanism has provided fundamental insights into CCR5 biology and HIV pathogenesis while inspiring innovative therapeutic modalities including CCR5-targeted gene editing and stem cell transplantation. Future research directions include optimizing gene editing efficiency, understanding potential compensatory mechanisms by alternative coreceptors, and elucidating the long-term consequences of CCR5 ablation in diverse human populations.
The CCR5Δ32 mutation, a 32-base-pair deletion in the gene encoding the C-C chemokine receptor type 5 (CCR5), exhibits a profound gene dosage effect that confers varying degrees of resistance to human immunodeficiency virus (HIV) infection. Individuals homozygous for this mutation possess near-complete resistance to R5-tropic HIV-1 strains, whereas heterozygous individuals display partial resistance characterized by reduced susceptibility and improved disease outcomes. This whitepaper delineates the molecular and cellular mechanisms underlying this gene dosage effect, synthesizing quantitative data on its impact on viral transmission, receptor expression, and clinical progression. Furthermore, it explores the translational application of these principles in novel therapeutic strategies, including gene editing and stem cell transplantation, framing the discussion within the broader context of CCR5-targeted HIV cure research. The content is structured to provide researchers, scientists, and drug development professionals with a comprehensive technical guide to this critical area of study.
The CC chemokine receptor 5 (CCR5) is a G-protein-coupled receptor (GPCR) with seven transmembrane domains, constitutively expressed on the surface of immune cells including T lymphocytes, macrophages, and dendritic cells [12]. Its primary physiological role involves mediating leukocyte migration to sites of inflammation through interaction with chemokines such as CCL3, CCL4, and CCL5 [12] [23]. Crucially, CCR5 serves as the principal co-receptor for R5-tropic HIV-1 strains, which dominate during the initial and chronic phases of infection [12]. Viral entry requires the sequential binding of HIV gp120 to CD4 and then to the CCR5 co-receptor, initiating fusion and infection [13].
The CCR5Δ32 mutation (rs333) is a 32-base-pair deletion within the coding region of the CCR5 gene on chromosome 3 (3p21.31) [14]. This deletion induces a frameshift, resulting in a truncated, non-functional receptor that is not expressed on the cell surface due to degradation during protein synthesis [12] [14]. The genotypic distribution of this allele follows a Mendelian inheritance pattern, producing three distinct phenotypes with significant implications for HIV susceptibility. The protective effect of this mutation demonstrates a clear gene dosage effect, making it a paradigm for understanding the relationship between host genetics and infectious disease susceptibility.
Molecular Mechanism of the CCR5Δ32 Gene Dosage Effect
The gene dosage effect of the CCR5Δ32 mutation manifests primarily at the protein level, directly influencing the surface density of CCR5 receptors available for HIV-1 entry.
Protein Expression and Truncation
The wild-type CCR5 protein is a 352-amino-acid polypeptide embedded in the cell membrane. The Δ32 variant produces a mutant RNA that is translated into a severely truncated peptide lacking the final three transmembrane domains, extracellular loops, and the C-terminal tail [14]. This aberrant structure cannot traffic to the cell membrane and is retained intracellularly, where it is degraded [12]. Consequently, the mutation exhibits a gene-dosage effect: heterozygous individuals (CCR5/CCR5Δ32) exhibit reduced CCR5 expression, while homozygous individuals (CCR5Δ32/CCR5Δ32) show a near-total absence of surface CCR5 [23].
Table 1: Impact of CCR5Δ32 Genotype on Receptor Expression and HIV-1 Susceptibility
| Genotype | CCR5 Surface Expression | Phenotypic Consequence for R5-tropic HIV-1 |
|---|---|---|
| CCR5/CCR5 (Wild-type Homozygous) | Normal, high | Fully susceptible to infection |
| CCR5/CCR5Δ32 (Heterozygous) | Reduced (~50% or less) | Partially resistant; slower disease progression |
| CCR5Δ32/CCR5Δ32 (Mutant Homozygous) | Absent or non-functional | Highly resistant to infection; largely protected |
The following diagram illustrates the molecular consequence of the Δ32 mutation and its gene dosage effect on CCR5 expression and HIV entry.
Impact on Viral Entry and Infectivity
The reduced receptor density in heterozygous individuals directly lowers the probability of successful viral attachment and entry. Mathematical models of HIV transmission dynamics incorporate this effect, assigning lower per-contact transmission probabilities for partnerships involving heterozygous individuals [24]. For homozygous individuals, the absence of the CCR5 receptor creates an insurmountable barrier for R5-tropic viruses, preventing the initial establishment of infection in the vast majority of documented cases [25] [26]. It is critical to note that this protection is specific to R5-tropic HIV-1; rare cases of infection in CCR5Δ32 homozygotes have been attributed to exposure to viral strains that utilize alternative co-receptors, such as CXCR4 (X4-tropic) [12].
Quantitative Clinical and Epidemiological Evidence
The gene dosage effect is corroborated by extensive clinical and epidemiological studies that quantify its impact on transmission risk and disease course.
HIV Susceptibility and Transmission
Cohort studies of high-risk, HIV-seronegative individuals have provided robust evidence for the protective effect of both homozygous and heterozygous genotypes.
Table 2: Epidemiological and Clinical Outcomes by CCR5Δ32 Genotype
| Parameter | CCR5/CCR5 (Wild-type) | CCR5/CCR5Δ32 (Heterozygous) | CCR5Δ32/CCR5Δ32 (Homozygous) |
|---|---|---|---|
| Relative Risk of HIV Infection | 1.0 (Reference) | Significantly Reduced (RR = 0.30, CI: 0.08-0.97) [25] | Near-complete protection [25] |
| Pre-AIDS Viral Load | High | Lower than wild-type [24] | Not applicable (protected from infection) |
| Progression to AIDS | Standard rate | Delayed by ~2 years [24] | Not applicable |
| Per-act Transmission Probability (Asymptomatic Stage, M to F) | 0.0005 [24] | 0.0005 (Similar to wild-type, but infectiousness is reduced due to lower viral load) [24] | Effectively 0 [24] |
A longitudinal cohort study of 2,996 high-risk individuals found that heterozygous Caucasian men who have sex with men (MSM) had a significantly reduced relative risk of HIV seroconversion (RR = 0.30) compared to wild-type individuals [25]. Cross-sectional analyses further supported this, showing a higher prevalence of the heterozygous genotype among MSM who reported unprotected receptive anal intercourse, suggesting a survival advantage in this high-exposure group [25]. The prevalence of the homozygous genotype was also positively associated with age in high-HIV-prevalence cities, indicating that these resistant individuals persist in the population despite long-term high-risk exposure [25].
Corroboration from Interventional Studies: CCR5Δ32 HSCT
The most compelling evidence for a cure-like outcome comes from allogeneic hematopoietic stem cell transplantation (HSCT) from CCR5Δ32/CCR5Δ32 donors to HIV-infected patients with hematological malignancies. The "Berlin," "London," and "Düsseldorf" patients achieved sustained HIV remission after transplantation, allowing for analytical treatment interruption (ATI) without viral rebound [13] [26]. In-depth virological and immunological characterization of one such patient revealed no replication-competent virus despite sporadic traces of HIV DNA, and waning HIV-specific immune responses, indicating a lack of ongoing antigenic stimulation [26]. This intervention effectively converts the recipient's immune system to a homozygous CCR5Δ32 phenotype, confirming the sufficiency of this genotype for achieving HIV cure.
Experimental Approaches and Research Methodologies
Research into the CCR5Δ32 gene dosage effect relies on a suite of well-established experimental protocols.
Key Experimental Protocols
1. Genotyping and Population Genetics:
- Methodology: DNA is typically extracted from peripheral blood mononuclear cells (PBMCs). The CCR5Δ32 variant is identified using polymerase chain reaction (PCR) with primers flanking the deletion, followed by gel electrophoresis. Wild-type alleles produce a 332-bp band, while Δ32 alleles produce a 300-bp band. Heterozygous individuals show both bands [14] [23].
- Application: This foundational technique is used in cohort studies to correlate genotype with HIV status, viral load, and disease progression [25]. It is also essential for screening potential donors for HSCT and gene therapy trials.
2. Viral Outgrowth Assays and Reservoir Quantification:
- Methodology: To assess the presence of replication-competent virus in patients post-HSCT or in therapy trials, quantitative viral outgrowth assays (qVOA) are performed. PBMCs from the patient are co-cultured with healthy donor CD4+ T cells stimulated to maximize viral induction. The culture supernatants are then tested for p24 antigen or HIV RNA to confirm viral growth [26].
- Application: This method was critical in demonstrating the absence of replication-competent virus in the cured "Düsseldorf" patient, despite the detection of fragmented viral DNA [26].
3. In Vivo Outgrowth Assays using Humanized Mice:
- Methodology: Immunodeficient mice (e.g., NSG mice) are engrafted with human immune cells to create "humanized" models. Patient-derived cells or tissues are transplanted into these mice, which are then monitored for the emergence of HIV viremia over time, in the absence of ART [26].
- Application: This highly sensitive in vivo method provides a more robust assessment of the residual reservoir than in vitro assays and was used to validate the cure in several HSCT cases [26].
The following diagram visualizes the multi-faceted experimental workflow for validating HIV cure in a CCR5Δ32/Δ32 HSCT patient.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents and Materials for CCR5Δ32 and HIV Cure Research
| Research Reagent / Tool | Function and Application | Key Details |
|---|---|---|
| CCR5Δ32 Genotyping Primers | Amplification of wild-type and mutant CCR5 alleles for PCR-based genotyping. | Specific primers yield 332-bp (WT) and 300-bp (Δ32) products for gel analysis [25]. |
| CCR5-Specific Monoclonal Antibodies | Flow cytometric quantification of CCR5 surface expression on CD4+ T cells. | Critical for confirming reduced/absent receptor expression in heterozygous/homozygous individuals [23]. |
| Humanized Mouse Models (e.g., NSG) | In vivo assessment of latent HIV reservoirs and efficacy of curative interventions. | Provide a robust in vivo system to test for replication-competent virus post-therapy [26]. |
| CRISPR/Cas9 Gene Editing Systems | Precision genome editing for CCR5 knockout in hematopoietic stem cells or T cells. | Creates a CCR5-null phenotype analogous to homozygosity; used in therapeutic development (e.g., NCT03164135) [13]. |
| Broadly Neutralizing Antibodies (bNAbs) | Passive immunization to target circulating virus and suppress viremia. | Used in combination with other therapies to control viral replication during treatment interruptions [13]. |
Therapeutic Applications and Future Directions
The elucidation of the CCR5Δ32 gene dosage effect has directly inspired multiple therapeutic avenues aimed at mimicking this natural resistance.
CCR5 Blockade and Gene Editing
The development of the CCR5 antagonist maraviroc provided pharmacological proof-of-concept for blocking this co-receptor. However, the goal of achieving a permanent cure has shifted toward gene editing strategies. Technologies like CRISPR/Cas9 are being employed to disrupt the CCR5 locus in patient-derived hematopoietic stem and progenitor cells (HSPCs) or T cells, with the aim of generating a continuous supply of HIV-resistant immune cells [13]. Early-phase clinical trials (e.g., NCT03164135) have demonstrated the feasibility and safety of this approach [13].
Overcoming Limitations: Multi-Target Strategies
A significant challenge in CCR5-targeted therapy is the potential for viral evolution toward CXCR4-tropic strains, which could circumvent the blockade. To address this, next-generation strategies involve multiplex gene editing, simultaneously targeting CCR5, CXCR4, and even integrated HIV proviral DNA (e.g., the Long Terminal Repeat - LTR) to create a comprehensive viral blockade [13]. Furthermore, the synergistic integration of gene editing with immunotherapy (e.g., anti-HIV CAR-T cells or immune checkpoint inhibitors) is being explored to enhance the clearance of infected cells and achieve a functional cure [13].
The CCR5Δ32 mutation stands as a seminal example of a host genetic variant with a clear gene dosage effect conferring differential resistance to HIV infection. The homozygous state provides near-absolute protection against R5-tropic HIV, while heterozygosity confers partial resistance through delayed disease progression and reduced transmission risk. The molecular basis—graded reduction in CCR5 surface expression—has been unequivocally validated by the success of CCR5Δ32/Δ32 HSCT in achieving HIV cure. This foundational knowledge has catalyzed the development of a robust pipeline of CCR5-targeted therapies, from small molecule antagonists to advanced gene editing platforms. Future research must focus on optimizing the efficiency and safety of these interventions, combating viral tropism switching, and integrating them with complementary immunotherapies to translate the protective principle of the CCR5Δ32 gene dosage effect into a scalable and accessible cure for HIV.
The C-C chemokine receptor type 5 (CCR5) serves roles far beyond its well-characterized function as an HIV-1 co-receptor. This review synthesizes current understanding of CCR5's integral functions in immune regulation, the profound evolutionary implications of the CCR5-Δ32 mutation, and the cutting-edge therapeutic strategies emerging from this knowledge. Framed within the context of a broader thesis on the mechanisms of CCR5Δ32-mediated HIV resistance, we detail how this natural mutation confers protection against infection and how researchers are leveraging this insight through advanced gene-editing technologies. For scientists and drug development professionals, this whitepaper provides a comprehensive technical guide, complete with structured data, experimental protocols, and visualizations of key pathways and methodologies.
CCR5 is a seven-transmembrane, G protein-coupled receptor (GPCR) that is primarily involved in immune surveillance and inflammatory response [21]. Its expression is found on a wide array of bone-marrow-derived cells, including lymphocytes, monocyte/macrophages, granulocytes, T cells, and specialized immune cells such as natural killer (NK) cells and regulatory T (Treg) cells [10] [21]. The receptor is activated by its natural agonist ligands—CCL3 (MIP-1α), CCL4 (MIP-1β), and CCL5 (RANTES)—which stimulate cell migration and mediate inflammatory responses [21]. The lifecycle of the CCR5 receptor involves continuous internalization and recycling; upon ligand binding and activation, it undergoes rapid phosphorylation, internalizes via clathrin-coated pits, and is then recycled back to the plasma membrane [10] [21].
The discovery of a 32-base-pair deletion (Δ32) in the CCR5 gene, which results in a truncated protein that is not expressed on the cell surface, revealed a direct link between CCR5 and HIV-1 resistance [10] [4]. Homozygous carriers (Δ32/Δ32) exhibit virtually total protection against infection with CCR5 (R5)-tropic HIV-1 strains, the dominant form transmitted between humans [4] [27]. This protective mechanism, stemming from the absence of the critical co-receptor required for viral entry, provides a powerful natural model of immunity and forms the cornerstone for developing CCR5-targeted therapies.
Mechanism of CCR5 in HIV-1 Entry and Δ32-Mediated Resistance
HIV-1 Entry Mechanism
The process by which HIV-1 infects a host cell is a multi-step sequence that requires coreceptor engagement:
- CD4 Binding: The viral envelope glycoprotein (gp120) binds to cellular CD4 receptors.
- Conformational Change: This binding induces a structural shift in gp120, exposing the V3 loop.
- Coreceptor Engagement: The exposed V3 loop interacts with and binds to a coreceptor, primarily CCR5 or CXCR4.
- Membrane Fusion: Coreceptor binding triggers a further conformational change that unmasks gp41, facilitating fusion of the viral envelope with the host cell membrane and subsequent viral entry [27].
R5-tropic HIV-1 strains, which utilize CCR5, are predominantly involved in viral transmission and are the principal circulating strain in early infection [27]. The high density of CCR5 expression on cells in the genital mucosa is believed to be a key factor in this preferential transmission [27].
The CCR5-Δ32 Mutation: A Loss-of-Function Variant
The CCR5-Δ32 variant is a 32-base-pair deletion in the CCR5 coding region, causing a frameshift during translation. This results in a defective, truncated peptide that lacks three transmembrane domains and cannot embed itself in the cell membrane, remaining in the cytoplasm instead [4]. The phenotypic effects are genotype-dependent:
- Wild-type (CCR5/CCR5): Normal CCR5 expression; full susceptibility to R5-tropic HIV-1.
- Heterozygous (CCR5/Δ32): Reduced CCR5 expression on the cell surface; leads to decreased susceptibility to HIV-1 infection and slower disease progression.
- Homozygous (Δ32/Δ32): Absence of CCR5 surface expression; confers near-complete resistance to infection by R5-tropic HIV-1 strains [10] [4] [28].
Table 1: Impact of CCR5-Δ32 Genotype on HIV-1 Infection
| Genotype | CCR5 Surface Expression | Susceptibility to R5-tropic HIV-1 | Clinical Outcome |
|---|---|---|---|
| Wild-type (CCR5/CCR5) | Normal | High | Standard infection and progression |
| Heterozygous (CCR5/Δ32) | Reduced | Moderate | Delayed disease progression |
| Homozygous (Δ32/Δ32) | Absent | Very Low / None | High resistance to infection |
The following diagram illustrates the mechanism of HIV-1 cellular entry and how the Δ32 mutation confers resistance at the molecular level.
Broader Immunological Functions and Pathogenic Roles of CCR5
Beyond its role in HIV entry, CCR5 is a critical regulator of immune cell trafficking and function. It mediates the chemotaxis of T lymphocytes, macrophages, and dendritic cells to sites of inflammation and infection [21]. It also controls the action of specific cell types, including NK cells and Treg cells, and is expressed by tissue-resident memory T cells, which are crucial for barrier immunity [10]. This broad immunological role means that CCR5 dysregulation or its genetic ablation can have significant consequences in various viral infections.
Table 2: Impact of CCR5 and CCR5-Δ32 on Selected Viral Infections Beyond HIV
| Viral Pathogen | Role/Impact of Functional CCR5 | Effect of CCR5-Δ32 / Deficiency |
|---|---|---|
| West Nile Virus (WNV) | Facilitates leukocyte trafficking to the brain for viral control. | Increased risk of symptomatic WNV infection and fatal outcome [10] [29]. |
| Influenza Virus | Not fully defined. | Associated with a higher frequency of fatal outcome (mouse model and human study) [4]. |
| Tick-Borne Encephalitis Virus | Recruits memory T cells to the brain. | Inadequate immune response in the brain; impaired viral clearance [4]. |
| Hepatitis B & C (HBV, HCV) | Modulates antiviral immune responses. | Conflicting reports; some studies associate Δ32 with modified disease progression or treatment response [29]. |
The pleiotropic nature of CCR5 signaling means that therapeutic strategies targeting this receptor, while promising for HIV, must be carefully evaluated for potential off-target effects on other immune functions.
Evolutionary Insights: Origin and Selection of the CCR5-Δ32 Allele
The global distribution of the CCR5-Δ32 allele is highly distinctive. It is found principally in European and Western Asian populations, with an average frequency of approximately 10%, and is virtually absent in Sub-Saharan African, Asian, and Native American populations [4] [21]. Within Europe, a strong north-south cline exists, with the highest frequencies in Nordic and Baltic regions (up to 16%) and the lowest in Southern Europe (e.g., 4-7%) [21] [30].
This geographic pattern, combined with the allele's relatively young estimated age (between 700 and 3500 years, though some evidence suggests it could be older), provides strong evidence that it has been under intense historical positive selection [4] [31] [30]. The selective advantage for heterozygous carriers in the past has been estimated to be as high as 5-35% [30]. Since HIV-1 is a recent human pathogen, it cannot account for this historical selection, leading to several competing hypotheses.
Table 3: Prominent Evolutionary Theories for CCR5-Δ32 Selection
| Hypothesis | Proposed Selective Agent | Key Evidence & Arguments | Status & Challenges |
|---|---|---|---|
| Bubonic Plague | Yersinia pestis (bacteria) | Correlates with the timing and devastation of the Black Death in Europe. | Largely refuted; CCR5 is not a primary receptor for Y. pestis; limited protective effect in models [4]. |
| Smallpox | Variola major (virus) | Smallpox was a major human pathogen for centuries with high fatality. | A leading candidate; smallpox was a strong selective force, but direct mechanistic evidence is limited [4]. |
| Hemorrhagic Fevers | Unknown ancient virus | Suggests plagues were viral, not bacterial, based on symptoms and transmission patterns. | Highly speculative; lacks identification of a specific pathogen [4]. |
| Viking Dispersal | N/A (Non-selective) | Spread of the allele from a Northern European origin via Viking raids and migration. | Explains geographic distribution via dispersal, but does not identify the initial selective pressure [4] [30]. |
Quantitative modeling suggests that with modest gradients in selection intensity, the allele's origin could be outside Northern Europe, and that its current distribution is likely the result of both strong selection and long-range dispersal events [30].
Experimental and Therapeutic Applications
Research Reagent Solutions
The following table details key reagents and technologies essential for research in CCR5 biology and therapeutic development.
Table 4: Essential Research Reagents and Tools for CCR5 Investigation
| Reagent / Technology | Function / Mechanism | Application in Research |
|---|---|---|
| Zinc Finger Nucleases (ZFNs) | Engineered nucleases that induce DNA double-strand breaks at specific CCR5 loci. | One of the earliest tools for CCR5 gene editing in clinical trials (e.g., SB-728-T) [3]. |
| TALENs | Modular DNA-binding domains fused to FokI nuclease for targeted CCR5 cleavage. | Provides improved specificity over ZFNs; automated production systems developed for clinical-scale cell production [3]. |
| CRISPR/Cas9 System | sgRNA directs Cas9 nuclease to create site-specific breaks in the CCR5 gene. | Highly efficient CCR5 editing; allows for multiplexed gene targeting; used in clinical trials (NCT03164135) [3] [32]. |
| Base Editors | Fusion of Cas proteins with deaminases enabling precise single-nucleotide changes without double-strand breaks. | Emerging technology for precise CCR5 mutation; reduces risks associated with DNA breaks [3]. |
| CCR5 Antagonists (e.g., Maraviroc) | Small molecule allosteric inhibitors that bind CCR5, preventing gp120 recognition. | Approved HIV-1 treatment; used to study the consequences of CCR5 pharmacological blockade [27]. |
| Monoclonal Antibodies (e.g., PRO 140) | Bind to the extracellular domain of CCR5, blocking gp120 interaction. | Investigational therapeutic; tool for studying receptor occupancy and immune-mediated inhibition [27]. |
| C46 Fusion Inhibitor | A membrane-anchored peptide that inhibits viral fusion, independent of coreceptor use. | Used in combination with CCR5 editing to protect against both R5- and X4-tropic HIV-1 [32]. |
Detailed Experimental Protocol: CRISPR/Cas9-Mediated CCR5 Knockout
The following methodology, adapted from a 2024 study, outlines the steps for generating a cellular model resistant to HIV-1 via CCR5 knockout [32].
Objective: To disrupt the CCR5 gene in the MT4CCR5 cell line using a ribonucleoprotein (RNP) complex delivery method for protection against R5-tropic HIV-1.
Materials:
- Cell Line: MT4CCR5 (CD4+ T-cell line expressing CCR5)
- Proteins: Recombinant S. pyogenes Cas9 protein
- Nucleic Acids: Two synthetic single-guide RNAs (sgRNAs) targeting the first exon of the human CCR5 gene, specifically designed at the Δ32 mutation site for high efficiency and low off-target effects.
- Equipment: Nucleofector device, flow cytometer, SDS-PAGE and Western blot apparatus, T7 Endonuclease I assay kit.
Procedure:
- RNP Complex Formation:
- Prepare two doses of the RNP complex:
- Dose 1: Combine 6 µg of Cas9 protein with 2 µg of each sgRNA (total 4 µg sgRNA).
- Dose 2: Combine 10 µg of Cas9 protein with 4 µg of each sgRNA (total 8 µg sgRNA).
- Incubate the complexes at room temperature for 10-20 minutes to allow formation.
- Prepare two doses of the RNP complex:
Cell Nucleofection:
- Harvest and wash MT4CCR5 cells. Resuspend the cell pellet in an appropriate nucleofection solution.
- Mix the cell suspension with the pre-formed RNP complex.
- Transfer the cell-RNP mixture to a nucleofection cuvette and electroporate using a pre-optimized nucleofection program for human T-cell lines.
Post-Nucleofection Culture:
- Immediately transfer the cells to pre-warmed culture medium.
- Incubate at 37°C with 5% CO₂ for 3 days to allow for gene editing and protein turnover.
Efficiency Assessment:
- Cleavage Efficiency (Day 3): Harvest a subset of cells. Extract genomic DNA and perform a T7 Endonuclease I (T7E1) assay on the amplified CCR5 target region. Cleaved PCR products indicate successful genome editing.
- Protein Knockdown (Day 3):
- Flow Cytometry: Stain cells with a fluorescently labeled anti-CCR5 antibody. Analyze by flow cytometry to quantify the percentage of CCR5-negative cells. Dose 2 typically achieves >97% reduction in CCR5 expression [32].
- Western Blotting: Lyse another subset of cells. Perform SDS-PAGE and Western blotting using an anti-CCR5 antibody to confirm the loss of CCR5 protein.
Functional Validation:
- Challenge the CCR5-knockout cells and control cells with an R5-tropic HIV-1 strain.
- Monitor for resistance by measuring parameters such as cell viability (via 7AAD staining), p24 antigen production, and viral reverse transcriptase activity over 7-14 days.
The workflow for this protocol, from preparation to validation, is summarized in the following diagram.
Synergistic Therapeutic Strategies
Current research is moving beyond single-target approaches. A prominent strategy involves combining CCR5 editing with other anti-HIV transgenes, such as the C46 fusion inhibitor, to create cells resistant to both R5- and X4-tropic HIV-1, thereby preventing viral escape via tropism switching [3] [32]. Furthermore, the integration of gene editing with immunotherapies—such as checkpoint blockade (e.g., anti-PD-1) or engineered CAR-T cells—is being explored to enhance the clearance of latent viral reservoirs, representing the next frontier in the quest for an HIV cure [3].
The study of CCR5 provides a profound example of how a deep understanding of a fundamental immunological receptor—from its role in health and disease to the evolutionary history of its protective variant—can catalyze revolutionary biomedical advances. The CCR5-Δ32 mutation is a natural blueprint for HIV resistance, demonstrating the power of human genetics to inform therapeutic development. For researchers and drug developers, the continued investigation of CCR5 is paramount. Future work must focus on refining the safety and specificity of gene-editing platforms, understanding the long-term immunological consequences of CCR5 modulation, and developing accessible, scalable curative strategies. The journey from evolutionary insight to clinical application for CCR5 is well underway, offering a robust framework for tackling one of modern medicine's most persistent challenges.
Translating Natural Resistance into Therapeutic Innovation
{# The 'Berlin' and 'London' Patient Case Studies}
| Characteristic | Berlin Patient (Timothy Ray Brown) | London Patient (Adam Castillejo) |
|---|---|---|
| Medical Condition | Acute Myeloid Leukemia (AML) [33] | Hodgkin's Lymphoma [33] |
| Transplant Type | Two allogeneic HSCT procedures [33] | Single allogeneic HSCT [33] |
| Conditioning Regimen | Total body irradiation with each transplant [33] | Reduced-intensity (LACE chemotherapy & Alemtuzumab), no irradiation [33] |
| Graft-versus-Host Disease | Experienced [34] | Mild gut GvHD [33] |
| Reported Remission Duration | >13 years (until passing) [34] | 30 months post-ATI (as of 2020 report) [35] |
| Key Virological Findings | No replication-competent virus detected in extensive testing [34] | No replication-competent virus in blood, CSF, gut, or lymphoid tissue; undetectable plasma RNA (<1 copy/mL) [35] |
| Key Immunological Findings | Loss of HIV-specific antibodies and T-cell responses [33] | Absent HIV-specific T-cell responses; declining, low-avidity Env antibodies [35] |
The concept of a cure for Human Immunodeficiency Virus type 1 (HIV-1) was, for decades, a theoretical pursuit until the landmark case of the "Berlin Patient" [33]. This case, followed by the "London Patient," provided the first tangible proof that sterilizing cure—the complete eradication of replication-competent virus from the body—is achievable in humans. Both individuals received allogeneic hematopoietic stem cell transplantation (allo-HSCT) from donors with a homozygous mutation in the CCR5 gene (CCR5Δ32/Δ32) to treat life-threatening hematological malignancies [33] [26]. The sustained remission of HIV-1 after the discontinuation of antiretroviral therapy (ART) in these cases offers critical insights into the mechanisms of viral persistence and clearance. Furthermore, a third case, the "Düsseldorf Patient," has since been reported with similar outcomes, reinforcing the concept [26] [36]. These cases collectively form a foundational proof of concept, demonstrating that the CCR5 coreceptor is indispensable for viral rebound and that its absence, coupled with a reconstituted immune system, can lead to a cure [33] [35].
Clinical Case Profiles and Experimental Methodologies
The Berlin Patient: A Pioneering Case
The Berlin Patient, Timothy Ray Brown, was diagnosed with HIV-1 in 1995 and later with acute myeloid leukemia (AML) [34]. To treat his leukemia, he underwent two allogeneic HSCT procedures in 2007 and 2008. Critically, his physician selected a donor who was homozygous for the CCR5Δ32 mutation [33] [34]. His conditioning regimen included total body irradiation with each transplant, and he experienced graft-versus-host disease (GvHD) [33]. ART was discontinued at the time of the first transplant. Despite extensive and repeated testing over more than a decade, no replication-competent HIV-1 was ever detected in his blood, gut, or other tissues, and he showed a loss of HIV-specific immune responses, consistent with a cure [34]. He remained free of HIV until his passing in 2020 from recurrent leukemia.
The London Patient: Validation with a Less Intensive Approach
The London Patient, Adam Castillejo, was diagnosed with HIV-1 in 2003 and with Hodgkin's Lymphoma in 2012 [33]. After chemotherapy failed, he received a single allo-HSCT in 2016 from a CCR5Δ32/Δ32 donor. His conditioning regimen used reduced-intensity chemotherapy (LACE) and anti-thymocyte globulin (Alemtuzumab), but no irradiation [33]. He experienced only mild gut GvHD. ART was maintained after transplant and then intentionally interrupted 16 months post-transplant in a controlled analytical treatment interruption (ATI) [33]. As of the 2020 report, he had been in remission for 30 months post-ATI, with no detectable replication-competent virus in blood, cerebrospinal fluid, lymph node, or gut tissue [35]. His HIV-1-specific T-cell responses became undetectable, and his antibody levels declined, indicating a lack of antigenic stimulation [33] [35].
Core Experimental Protocols for Validating Cure
The extensive virological and immunological profiling conducted on these patients followed rigorous experimental protocols, which are now considered benchmarks for cure research.
- Quantitative Viral Outgrowth Assay (QVOA): This is the gold-standard assay for quantifying the replication-competent latent reservoir. Resting CD4+ T cells from the patient are maximally activated ex vivo to induce virus production, which is then measured. For the London Patient, QVOA performed on a total of 24 million resting CD4+ T cells showed no reactivatable virus, yielding an reservoir estimate of <0.029 infectious units per million (IUPM) cells [33] [37].
- Droplet Digital PCR (ddPCR): This ultrasensitive nucleic acid detection technique was used to quantify total HIV-1 DNA (both intact and defective) in patient cells and tissues. While sporadic, very low-level positive signals for HIV-1 DNA were occasionally detected, repeated ddPCR assays on the London Patient's gut and lymph node tissues were largely negative [26] [35].
- Viral Tropism Assay: To confirm that the patient's pre-transplant virus was susceptible to CCR5 disruption, the viral tropism was determined. This involves sequencing the V3 loop of the HIV-1 envelope gene and using computational algorithms (e.g., PSSM, geno2pheno) to predict coreceptor usage. For both the Berlin and London Patients, the pre-existing virus populations were predicted to be exclusively CCR5-tropic [33] [37].
- In Vivo Outgrowth Assays: To provide even more sensitive evidence, researchers used humanized mouse models. Immune cells from the Düsseldorf Patient were transplanted into immunodeficient mice, and no virus was recovered, confirming the absence of replication-competent virus in the tested samples [26].
[HIV Entry Block by CCR5Δ32]
The CCR5Δ32 Mutation: Mechanism of HIV Resistance
The CCR5 coreceptor, a G-protein-coupled receptor expressed on the surface of macrophages and CD4+ T cells, is the primary portal of entry for HIV-1 during the initial and chronic stages of infection [13] [10]. The virus first binds to the CD4 receptor, then interacts with CCR5, which triggers fusion and allows the viral genome to enter the host cell [4].
The CCR5Δ32 mutation is a 32-base-pair deletion in the coding region of the CCR5 gene. This deletion causes a frameshift during translation, resulting in a severely truncated and non-functional protein that is not expressed on the cell surface [10] [4]. In individuals who are homozygous (Δ32/Δ32) for this mutation, CD4+ T cells lack the CCR5 coreceptor entirely. Since the vast majority of HIV-1 strains require CCR5 for entry, these cells are highly resistant to infection [10]. This natural resistance is the foundational mechanism behind the cures in the Berlin and London Patients. Their new, donor-derived immune systems were composed of CD4+ T cells that were genetically resistant to infection by the patients' pre-existing CCR5-tropic HIV-1 reservoirs, preventing viral rebound after ART cessation [33].
Critical Research Reagents and Methodologies
The following toolkit summarizes key reagents and assays essential for replicating and advancing this research.
| Research Tool | Function / Explanation |
|---|---|
| CCR5Δ32/Δ32 Donor Cells | Source of hematopoietic stem cells that give rise to an HIV-1-resistant immune system upon transplantation [33] [26]. |
| Ultra-Sensitive Viral Load Assays | PCR-based assays (e.g., with LOD of <1 copy RNA/mL) to monitor for viral rebound during ATI with maximum sensitivity [33] [35]. |
| Droplet Digital PCR (ddPCR) | An absolute nucleic acid quantification method used to precisely measure levels of HIV-1 DNA in tissue and cell samples, distinguishing between intact and defective provinces [26] [35]. |
| Quantitative Viral Outgrowth Assay (QVOA) | The gold-standard assay for quantifying the inducible, replication-competent latent HIV-1 reservoir in resting CD4+ T cells [33] [37]. |
| Humanized Mouse Models | In vivo model (e.g., NOG mice) used to test for the presence of replication-competent virus by implanting patient-derived cells and monitoring for viremia [26]. |
| Intracellular Cytokine Staining (ICS) | Flow cytometry-based method to detect and characterize virus-specific T-cell responses (e.g., to HIV-1 Gag, Pol, Nef) [26]. |
[Reservoir Measurement Assays]
Broader Implications and Future Directions
The cases of the Berlin and London Patients are not treatments for the general HIV-positive population due to the high mortality risk and scarcity of matched CCR5Δ32/Δ32 donors [13]. However, they serve as a critical proof of concept, directly informing the development of safer, scalable cure strategies.
The primary direction is the development of gene editing therapies to mimic the CCR5Δ32 phenotype. The CRISPR/Cas9 system has been used in a clinical trial (NCT03164135) to edit the CCR5 gene in hematopoietic stem cells from a patient with HIV and acute lymphoblastic leukemia, demonstrating feasibility and safety [13] [38]. This approach aims to create a patient's own CCR5-modified immune system, eliminating the need for a donor and the risk of GvHD.
Furthermore, these cases highlight the need for multi-target strategies. Because HIV can switch to use the CXCR4 coreceptor if CCR5 is unavailable, future therapies may involve simultaneous knockout of both CCR5 and CXCR4, or targeting the integrated viral DNA itself (e.g., the HIV LTR promoter) to achieve a more robust and comprehensive cure [13]. The successful cure of the "Geneva Patient," who received a transplant from a wild-type (non-CCR5Δ32) donor, and the "New Berlin Patient," who received cells from a heterozygous donor, suggests that factors beyond CCR5 ablation—such as graft-versus-reservoir effects—may also contribute to reservoir reduction, opening additional avenues for research [34].
The C-C chemokine receptor type 5 (CCR5) serves as a critical coreceptor for human immunodeficiency virus (HIV) entry into host cells, playing an indispensable role in the initial stages of viral infection and dissemination [12]. The discovery that a natural 32-base pair deletion in the CCR5 gene (CCR5-Δ32) confers strong resistance to HIV-1 infection in homozygous individuals provided a foundational insight that has shaped therapeutic development [39] [40]. This genetic variant produces a nonfunctional receptor that prevents R5-tropic HIV strains from entering target cells, effectively mimicking a natural form of immunity [41]. CCR5 antagonist drugs represent a direct pharmacological strategy to replicate this protective mechanism, offering a host-targeted approach to prevent viral entry and slow disease progression [12] [42].
The CCR5-Δ32 mutation is notably prevalent in European populations, with heterozygote frequency of approximately 9% and homozygote frequency of about 1% [41]. Individuals homozygous for this mutation do not express functional CCR5 receptors on their cell surfaces and demonstrate significant resistance to HIV-1 infection despite multiple high-risk exposures [40] [41]. Heterozygous carriers exhibit intermediate protection, with approximately 50% reduction in functional CCR5 receptors due to dimerization between mutant and wild-type receptors that interferes with proper cellular transport [41]. This gene-dose effect provides a compelling natural model for pharmacological intervention [12].
Biological Mechanism of CCR5 in HIV Entry
Structural and Functional Basis of CCR5
CCR5 belongs to the large family of G-protein coupled receptors (GPCRs), characterized by seven transmembrane domains that traverse the cell membrane [12]. This receptor is expressed on various immune cells including T cells, macrophages, dendritic cells, and microglial cells, where it normally functions as a receptor for chemokines such as RANTES (CCL5), MIP-1α (CCL3), and MIP-1β (CCL4) [12] [42]. These chemokines are involved in coordinating immune responses by directing leukocyte trafficking to inflamed or damaged tissues [42].
In the context of HIV infection, the viral envelope glycoprotein gp120 first binds to the primary CD4 receptor on target cells, inducing conformational changes that expose previously hidden epitopes, allowing subsequent interaction with CCR5 as a coreceptor [42] [43]. This sequential binding triggers further structural rearrangements in the viral envelope that ultimately facilitate fusion between viral and cellular membranes, enabling viral entry [12].
Table 1: CCR5 Expression and Ligands
| Cell Types Expressing CCR5 | Natural Ligands (Chemokines) | Role in HIV Infection |
|---|---|---|
| CD4+ T cells (Th0, Th1, memory) | CCL3 (MIP-1α), CCL4 (MIP-1β) | Primary coreceptor for R5-tropic HIV |
| Monocytes, macrophages | CCL5 (RANTES) | Facilitates viral entry and cell-to-cell spread |
| Dendritic cells | CCL8 (MCP-2), CCL13 (MCP-4) | Antigen presentation and viral dissemination |
| Microglial cells | CCL3L1, CCL4L1 (variant chemokines) | Enables CNS infection and viral reservoirs |
Molecular Consequences of the Δ32 Mutation
The CCR5-Δ32 mutation is characterized by a 32-base pair deletion in the coding region of the CCR5 gene, which produces a frameshift during translation and introduces a premature stop codon [41]. This genetic alteration results in a truncated protein of only 215 amino acids instead of the normal 352, which fails to localize to the cell surface due to improper folding and retention within intracellular compartments [41]. The absence of functional CCR5 receptors on the cell surface effectively blocks the entry of R5-tropic HIV strains, which represent the predominantly transmitted variants [39] [40].
The protective effect of this mutation follows a gene-dose pattern: homozygous individuals (Δ32/Δ32) demonstrate near-complete resistance to R5-tropic HIV infection, while heterozygous individuals (+/Δ32) typically show delayed disease progression and reduced viral loads compared to those with wild-type CCR5 [41]. This graded protection has important implications for therapeutic strategies aiming to partially or fully recapitulate this natural resistance mechanism [12].
Diagram 1: Molecular consequences of CCR5-Δ32 mutation. The 32-base pair deletion produces a truncated, non-functional receptor that prevents HIV entry.
CCR5 Antagonist Drugs: Development and Mechanisms
First-Generation CCR5 Antagonists
The development of CCR5 antagonists has focused on creating compounds that allosterically inhibit the receptor's interaction with HIV gp120, effectively mimicking the protective effect of the Δ32 mutation through pharmacological means [42]. Maraviroc, approved by the FDA in 2007, represents the first successful drug in this class and remains the only CCR5 antagonist approved for clinical use against HIV [42]. As a small molecule antagonist, maraviroc binds to a hydrophobic cavity within the transmembrane domains of CCR5, inducing conformational changes that prevent gp120 binding while sparing natural chemokine signaling to a significant extent [44] [42].
Early clinical trials demonstrated maraviroc's efficacy in treatment-experienced patients with R5-tropic HIV, but also revealed limitations including hepatotoxicity concerns and the necessity for tropism testing prior to initiation [42]. The MOTIVATE studies showed significant viral load reduction in HAART-experienced patients, but also highlighted that virological failure often resulted from the emergence of pre-existing CXCR4-using virus populations not detected by screening assays [42]. This underscored the importance of reliable tropism testing and the potential for viral escape through coreceptor switching [42].
Second-Generation and Dual-Target Antagonists
Next-generation CCR5 antagonists have sought to improve upon the limitations of earlier compounds through enhanced pharmacokinetic profiles and expanded therapeutic targets [42]. Cenicriviroc represents a significant advancement as a dual CCR5/CCR2 antagonist that allows for once-daily dosing and possesses additional anti-inflammatory properties [42]. This dual mechanism offers potential benefits for addressing HIV-associated inflammation and comorbidities, particularly in the context of nonalcoholic steatohepatitis (NASH) where CCR2-mediated monocyte recruitment plays a pathogenic role [44] [42].
Novel compounds such as GRL-117C demonstrate exceptionally potent anti-HIV activity with sub-nanomolar IC50 values against wild-type R5-HIV-1, comparable to maraviroc in experimental models [44]. Structural modeling indicates that these compounds bind in the hydrophobic cavity of CCR5 beneath the second extracellular loop, with amino acid interactions largely overlapping with those of maraviroc, explaining observed cross-resistance patterns [44]. The continuous development of CCR5 inhibitors with unique binding profiles and immunomodulating properties remains an active area of investigation [44].
Table 2: Characteristics of Major CCR5 Antagonist Drugs
| Compound | Development Status | Key Properties | IC50 Against R5-HIV-1 | Dosing Schedule |
|---|---|---|---|---|
| Maraviroc | FDA-approved (2007) | Selective CCR5 antagonist | 0.6-0.7 nM (MAGI assay) [44] | Twice daily [42] |
| Cenicriviroc | Phase II clinical trials | Dual CCR5/CCR2 antagonist | Not specified | Once daily [42] |
| Vicriviroc | Clinical trials discontinued | Second-generation antagonist | Less potent than Maraviroc [44] | Not specified |
| GRL-117C | Preclinical research | Novel small molecule inhibitor | 0.6 nM (MAGI assay) [44] | Not determined |
| OB-002/5P12-RANTES | Phase I for HIV prevention | CCL5 analog, potent antagonist | Not specified | Topical formulation [45] |
Quantitative Analysis of Antagonist Efficacy
Antiviral Potency Assessments
The efficacy of CCR5 antagonists is quantitatively evaluated through multiple assay systems, each providing complementary data on antiviral activity. The MAGI assay (Multinuclear Activation of a Galactosidase Indicator) typically yields lower IC50 values compared to p24 production assays in peripheral blood mononuclear cells (PBMCs), reflecting differences in experimental sensitivity and cellular context [44]. For instance, GRL-117C demonstrates an IC50 of 0.6 nM in MAGI assays compared to 8.1 nM in PBMC-based p24 assays when tested against HIV-1Ba-L, a pattern consistently observed across this drug class [44].
These compounds also show potent activity against transmitter/founder (T/F) viruses, which are almost exclusively R5-tropic and responsible for establishing new infections [44]. Against a panel of four T/F infectious clones, GRL-117C exhibited IC50 values ranging from 1.9-3.2 nM, comparable to maraviroc (1.7-2.3 nM), confirming their potential effectiveness in preventing initial viral transmission [44].
Resistance Profiles and Cross-Resistance Patterns
The emergence of resistant viral strains presents a significant challenge for CCR5 antagonist therapy. In vitro selection experiments have generated HIV-1 variants capable of utilizing CCR5 even in the presence of inhibitors like AD101 and vicriviroc, without switching to CXCR4 usage [44]. These resistant variants typically contain mutations in the V3 loop of gp120 that enable recognition of the drug-bound conformation of CCR5 [44].
Cross-resistance profiles vary among CCR5 antagonists. Vicriviroc-resistant viruses (D1/85.16) demonstrate high-level resistance to SCH-C (68-fold) but remain susceptible to other drugs in the class, with fold-resistance values ranging from 3.6 to 12.5 [44]. Similarly, AD101-resistant viruses (CC101.19) show significant resistance to SCH-C (>150-fold) but more modest resistance to maraviroc, APL, and GRL compounds (2.6 to 15-fold) [44]. These patterns reflect overlapping but distinct binding interactions with the CCR5 receptor.
Table 3: Efficacy of CCR5 Inhibitors Against Resistant HIV-1 Strains
| Compound | Activity Against Wild-type (IC50 nM) | Fold Resistance (AD101-resistant) | Fold Resistance (Vicriviroc-resistant) | Cross-resistance Profile |
|---|---|---|---|---|
| SCH-C | 5.2-6.1 nM (CC1/85) [44] | >150-fold [44] | 68-fold [44] | High cross-resistance |
| Maraviroc | 0.7 nM (MAGI assay) [44] | 15-fold [44] | 12.5-fold [44] | Moderate cross-resistance |
| GRL-117C | 0.6 nM (MAGI assay) [44] | 9.3-fold [44] | 8.5-fold [44] | Moderate cross-resistance |
| APL | 0.2 nM (MAGI assay) [44] | 2.6-fold [44] | 3.6-fold [44] | Low cross-resistance |
Experimental Protocols for CCR5 Antagonist Evaluation
Standardized Antiviral Potency Assays
The evaluation of CCR5 antagonist efficacy employs standardized laboratory protocols to quantitatively assess antiviral activity:
MAGI Assay Protocol:
- Seed MAGI/CCR5 cells (HeLa cells expressing CD4 and CCR5 with an HIV LTR-driven β-galactosidase reporter) in 96-well plates at 1×10⁴ cells/well and incubate for 24 hours at 37°C [44].
- Prepare serial dilutions of CCR5 antagonists in culture medium (typically ranging from 0.1 nM to 1000 nM).
- Incubate cells with compound dilutions for 30 minutes prior to infection with R5-tropic HIV-1 (e.g., HIV-1Ba-L) at approximately 100 TCID50 per well.
- After 48 hours incubation, fix cells and stain with X-gal solution to detect β-galactosidase activity.
- Quantify blue foci formation microscopically; calculate IC50 values using non-linear regression analysis of dose-response curves [44].
PBMC-based p24 Assay Protocol:
- Isplicate human PBMCs from healthy donors and stimulate with phytohemagglutinin (PHA) for 48-72 hours [44].
- Pre-incubate cells with CCR5 antagonist dilutions for 60 minutes before infection with R5-tropic HIV-1.
- Maintain cultures for 7 days with appropriate compound concentrations.
- Measure p24 antigen levels in culture supernatants using commercial ELISA kits.
- Calculate IC50 values from dose-response curves of p24 inhibition [44].
Binding Competition Assays
Radioligand binding studies provide critical data on receptor interaction mechanisms:
[¹²⁵I]-CCL3 Binding Assay Protocol:
- Prepare membranes from CCR5-expressing cells (e.g., B300-19 murine pre-B cells stably transfected with human CCR5) [46].
- Incubate membrane preparations (10-20 μg protein) with fixed concentration of [¹²⁵I]-CCL3 (0.1 nM) and varying concentrations of unlabeled competitors (CCR5 antagonists or natural ligands) in binding buffer.
- Conduct incubation for 60-90 minutes at 27°C to reach equilibrium.
- Separate bound from free radioligand by rapid filtration through GF/B filters presoaked in 0.3% polyethyleneimine.
- Measure filter-bound radioactivity using a gamma counter; calculate Ki values using the Cheng-Prusoff equation to determine binding affinity [46].
Diagram 2: Standardized workflow for evaluating CCR5 antagonist efficacy in antiviral assays.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for CCR5 Antagonist Studies
| Reagent/Cell Line | Specific Function | Application in CCR5 Research |
|---|---|---|
| MAGI/CCR5 cells | HeLa-derived cells expressing CD4, CCR5, and β-galactosidase reporter under HIV LTR | Quantitative high-throughput screening of antiviral activity [44] |
| PM-1/CCR5 cells | T-cell line expressing CD4 and CCR5 | Studies of viral entry inhibition and replication kinetics |
| B300-19 transfectants | Murine pre-B cells stably expressing human CCR5 | Radioligand binding studies and species selectivity experiments [46] |
| [¹²⁵I]-CCL3 | Radiolabeled chemokine ligand | Competitive binding assays to determine antagonist affinity [46] |
| R5-tropic HIV-1 Ba-L | Macrophage-tropic HIV strain | Standard challenge virus for antiviral assays [44] |
| Transmitter/Founder (T/F) clones | HIV strains representing initial transmission variants | Evaluation of prevention efficacy against relevant viruses [44] |
| CCR5 inhibitor-resistant variants | HIV with V3 loop mutations (e.g., CC101.19) | Cross-resistance profiling and resistance mechanism studies [44] |
| Anti-CCR5 monoclonal antibodies (e.g., 2D7) | Receptor detection and neutralization | Flow cytometry, receptor mapping, and functional blocking studies |
CCR5 antagonist drugs represent a successful example of pharmacological mimicry of a protective genetic variant, translating natural resistance mechanisms into therapeutic interventions. The development of these compounds has validated CCR5 as a viable drug target and provided important insights into viral entry mechanisms, resistance development, and the complex interplay between host genetics and infectious disease susceptibility [12] [42].
Future directions in this field include the optimization of dual-target antagonists with enhanced anti-inflammatory properties, strategies to overcome or prevent resistance, and exploration of CCR5 inhibition in contexts beyond HIV, such as in COVID-19 and other inflammatory conditions [44] [45]. Gene editing approaches that directly target the CCR5 locus represent another frontier, potentially offering a one-time curative strategy that more permanently recapitulates the Δ32 mutation [39] [47]. As research continues to elucidate the intricate biology of CCR5 and its role in disease, the pharmacological mimicry of protective mutations remains a powerful paradigm for therapeutic development.
The discovery that a 32-base pair deletion (CCR5Δ32) in the C-C chemokine receptor 5 (CCR5) gene confers natural resistance to HIV-1 infection provided a foundational rationale for therapeutic gene editing [13] [48]. Individuals homozygous for the CCR5Δ32 mutation express a truncated, non-functional CCR5 protein that is not localized to the cell surface, thereby preventing entry of the most common CCR5 (R5)-tropic HIV-1 strains [49] [28]. The documented cures of the "Berlin," "London," and "Düsseldorf" patients following allogeneic hematopoietic stem cell transplantation (HSCT) from CCR5Δ32/Δ32 homozygous donors validated CCR5 disruption as a viable path to an HIV cure [13] [50]. However, the rarity of the homozygous CCR5Δ32 genotype (~1% in the Caucasian population) and the challenges of allogeneic HSCT severely limit this approach [48] [32].
Gene editing technologies overcome these limitations by enabling precise genetic modification of a patient's own cells to mimic the CCR5Δ32 phenotype. These platforms facilitate the creation of double-strand breaks (DSBs) at specific genomic loci, which are then repaired by the cell's endogenous non-homologous end joining (NHEJ) pathway. This error-prone repair results in insertions or deletions (indels) that disrupt the target gene [48]. This whitepaper provides a technical comparison of the three primary nuclease platforms—Zinc Finger Nucleases (ZFNs), Transcription Activator-Like Effector Nucleases (TALENs), and CRISPR/Cas9—for CCR5 disruption, detailing their mechanisms, experimental protocols, and applications within HIV cure research.
Comparative Analysis of Gene Editing Platforms
The three main nuclease platforms share the common goal of creating targeted DSBs but differ fundamentally in their design, mechanism, and practical application.
Table 1: Core Characteristics of Gene Editing Platforms for CCR5 Disruption
| Feature | ZFNs | TALENs | CRISPR/Cas9 |
|---|---|---|---|
| Mechanism of Action | Custom zinc finger proteins fuse to FokI nuclease; requires a pair binding opposite DNA strands [48] [51] | Custom TALE proteins fuse to FokI nuclease; requires a pair binding opposite DNA strands [48] [51] | gRNA guides Cas9 nuclease to complementary DNA sequence; single nuclease suffices [48] [51] |
| Targeting Specificity | DNA triplet sequence (~18-36 bp per pair) [51] | Single nucleotide sequence (~30-40 bp per pair) [51] | 20-nucleotide gRNA sequence + PAM (NGG for SpCas9) [48] |
| Key Advantages | First platform in clinical trials; well-validated for CCR5 [3] [52] | High specificity; modular DNA-binding domain design [3] [51] | Simple gRNA design; highly efficient; allows multiplexed editing [13] [51] |
| Primary Limitations | Complex protein engineering; high cost; potential off-target effects [13] [3] | Large gene size complicates delivery; labor-intensive assembly [3] [51] | Off-target effects; PAM sequence dependency; potential immune responses [13] [3] |
| Clinical Trial Status (for CCR5) | Multiple completed/ongoing trials (e.g., SB-728-T) [3] [52] | Preclinical stage | Early-phase trials (e.g., NCT03164135) [13] [32] |
Table 2: Quantitative Performance Metrics in CCR5 Editing
| Metric | ZFNs | TALENs | CRISPR/Cas9 |
|---|---|---|---|
| Typical Editing Efficiency (in T cells or HSPCs) | Demonstrated clinical efficacy [52] | Efficient editing demonstrated [3] | 52%-97% (dose-dependent) [32] [50] |
| Off-Target Risk (Theoretical) | Moderate [3] | Low to Moderate [3] [51] | Moderate to High (mitigable with high-fidelity Cas9) [3] [51] |
| Multiplexing Capacity | Low (complex to engineer) [51] | Low (complex to engineer) [51] | High (easy to design multiple gRNAs) [13] [51] |
| Reported HIV Protection In Vitro | Resistance to R5-tropic HIV [49] | Resistance to R5-tropic HIV [3] | Resistance to R5, X4, and dual-tropic HIV [49] [32] |
Diagram 1: Gene editing platform workflows for CCR5 disruption.
Detailed Experimental Protocols for CCR5 Editing
CRISPR/Cas9-Mediated CCR5 Knockout in Hematopoietic Stem/Progenitor Cells (HSPCs)
The following protocol, adapted from recent preclinical studies, achieves high-frequency CCR5 editing in HSPCs using Cas9 ribonucleoprotein (RNP) complexes [49] [50].
1. Guide RNA (gRNA) Design and Selection:
- Target Region: Focus on exon 3 of the human CCR5 gene, which corresponds to the region of the natural CCR5Δ32 mutation [32] [50].
- In Silico Screening: Use prediction software (e.g., from Broad Institute or commercial vendors) to identify 4-6 candidate gRNAs with high on-target scores.
- Off-Target Prediction: Analyze the human genome for sequences with ≤3 base pair mismatches to exclude gRNAs with high-risk off-target profiles [50].
- Validation: Synthesize gRNAs and test editing efficiency and CCR5 protein knockdown in human cell lines (e.g., CEM.NKR-CCR5+ or K562) before moving to primary cells.
2. RNP Complex Formation and Delivery:
- RNP Complex Assembly: Complex chemically synthesized, modified gRNAs with purified, high-concentration SpCas9 protein. A representative recipe is:
- Cell Preparation: Isolate and activate human CD34+ HSPCs from mobilized peripheral blood or cord blood. Maintain cells in serum-free medium supplemented with cytokines (SCF, TPO, FLT3-L).
- Electroporation: Use a specialized nucleofector device and a CD34+ cell-specific kit. Resuspend 1x10^5 to 1x10^6 cells in the provided solution, mix with the pre-formed RNP complex, and electroporate using the recommended program [50].
3. Post-Editing Analysis and Validation:
- Editing Efficiency: At 48-72 hours post-electroporation, extract genomic DNA. Use the T7 Endonuclease I (T7E1) assay or, for higher precision, deep sequencing (NGS) to quantify indel frequency at the CCR5 locus. Efficiencies of >90% are achievable [50].
- Protein Knockdown Validation: 5-7 days post-editing, analyze cells by flow cytometry using anti-CCR5 antibodies to confirm reduction of surface CCR5 expression. Western blot can provide complementary data [32].
- Functional HIV Challenge: Differentiate edited HSPCs into macrophages or T cells in vitro. Challenge these cells with CCR5-tropic HIV-1 (e.g., HIV-1BaL). Monitor viral replication over 2-4 weeks by measuring p24 antigen levels in the supernatant via ELISA. Successful editing results in significant suppression of p24 levels compared to control cells [49].
Protocol for Simultaneous CCR5 and CXCR4 Disruption
To prevent viral escape via coreceptor switching (to CXCR4-tropic strains), a multi-target editing strategy is employed [49] [13].
- Multiplexed gRNA Design: Select high-efficiency gRNAs for both CCR5 and CXCR4 genes. CRISPR/Cas9 is uniquely suited for this, as multiple gRNAs can be co-delivered with a single Cas9 protein [49] [13].
- Co-delivery: Formulate an RNP complex containing Cas9 protein with a cocktail of CCR5- and CXCR4-targeting gRNAs. Electroporate into primary CD4+ T cells or HSPCs as described above.
- Validation: Use flow cytometry to confirm concurrent reduction of CCR5 and CXCR4 surface expression. Challenge the dual-edited cells with R5-tropic, X4-tropic, and dual-tropic HIV-1 strains to demonstrate broad-spectrum resistance [49].
- Caution: Studies note that combined CCR5/CXCR4 knockout in CD4+ T cells may lead to poor engraftment in the bone marrow of humanized mouse models, a critical safety consideration for translational work [49].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for CCR5 Gene Editing Experiments
| Reagent / Solution | Function / Purpose | Specific Examples / Notes |
|---|---|---|
| SpCas9 Nuclease | The "scissors" that create the double-strand break in DNA. | Recombinantly expressed and purified protein; available from commercial vendors (e.g., Sigma, Thermo Fisher, IDT). |
| CRISPR gRNAs | Guides the Cas9 nuclease to the specific target site in the CCR5 gene. | Chemically synthesized, modified (e.g., 2'-O-methyl) gRNAs with 3' protospacers for enhanced stability [50]. |
| Nucleofector System | Enables highly efficient delivery of RNP complexes into hard-to-transfect primary cells. | Lonza 4D-Nucleofector with specific kits for CD34+ cells or T cells is the industry standard [50]. |
| Cytokine Cocktails | Maintains viability and stemness of HSPCs during and after the editing process. | Typically includes Stem Cell Factor (SCF), Thrombopoietin (TPO), and FMS-like tyrosine kinase 3 ligand (Flt3-L) [50]. |
| T7 Endonuclease I Assay | A fast and cost-effective method for initial quantification of editing efficiency. | Detects DNA mismatches in PCR products caused by indels. Less sensitive than NGS [32]. |
| Next-Generation Sequencing (NGS) | The gold standard for precise quantification of editing efficiency and profiling of indel patterns. | Provides deep sequencing data for on-target and potential off-target sites [49] [50]. |
| Anti-CCR5 Antibody | Validates successful knockdown of the CCR5 protein at the cell surface. | Fluorescently conjugated antibody for flow cytometry analysis (e.g., clone 2D7/CCR5) [49] [32]. |
ZFNs, TALENs, and CRISPR/Cas9 have each demonstrated the capability to disrupt CCR5 and confer resistance to HIV-1 in preclinical models. The choice of platform involves a trade-off between simplicity and multiplexing capacity (favoring CRISPR/Cas9) and established clinical history (favoring ZFNs). The field is evolving toward combinatorial strategies that simultaneously target CCR5, CXCR4, and even the HIV provirus itself to construct a comprehensive barrier against viral escape [13] [3]. Furthermore, the integration of gene editing with immunotherapy, such as engineering HIV-resistant CAR-T cells, represents the next frontier in the development of a durable, scalable cure for HIV [13]. As delivery methods improve and safety profiles are refined in ongoing clinical trials, autologous transplantation with gene-edited HSPCs stands as a promising strategy for replicating the success of the Berlin patient on a broader scale.
The mechanism of the CCR5Δ32 mutation is a cornerstone of HIV resistance research. This natural polymorphism involves a 32-base-pair deletion in the CCR5 gene, encoding a defective protein that cannot embed in the cell membrane. The CCR5 co-receptor is normally essential for R5-tropic HIV entry into CD4+ immune cells. In homozygous individuals (approximately 1% of European populations), this mutation confers near-complete resistance to HIV infection, while heterozygotes experience slower disease progression [14]. This natural resistance mechanism has inspired two primary therapeutic approaches: allogeneic hematopoietic stem cell transplantation (HSCT) from CCR5Δ32 donors and autologous CAR-T cells engineered for HIV resistance.
The CCR5Δ32 allele demonstrates a north-to-south frequency gradient across Europe, with highest prevalence in Scandinavian and Baltic populations (~10-16% allele frequency), declining to lower frequencies in southern regions. The mutation is almost absent in Asian, African, and indigenous American populations, though gene flow has created local peaks in admixed populations like Chile (12%) and Ashkenazi Jewish populations (11-20%) [14]. This geographic distribution reflects historical selective pressures, potentially from pathogens like smallpox or hemorrhagic fevers, though the exact evolutionary driver remains debated [14].
Hematopoietic Stem Cell Transplantation with CCR5Δ32 Donors
Mechanism and Clinical Evidence
Allogeneic HSCT from CCR5Δ32/Δ32 homozygous donors aims to replace a patient's HIV-susceptible immune system with genetically resistant CCR5-deficient cells. This approach was pioneered with the Berlin, London, and Düsseldorf patients – individuals with HIV and hematological malignancies who received CCR5Δ32/Δ32 allogeneic stem cell transplants and achieved sustained ART-free HIV remission [53] [54]. The therapeutic effect derives from both the CCR5 deficiency preventing HIV entry and graft-versus-host disease contributing to elimination of residual HIV reservoirs through alloreactive responses [53].
Table 1: Clinical Outcomes of CCR5Δ32/Δ32 HSCT for HIV Infection
| Patient Case/Study | Underlying Condition | Conditioning Regimen | GVHD | ART Cessation | Remission Duration |
|---|---|---|---|---|---|
| Berlin Patient [54] | Acute Myeloid Leukemia | Chemotherapy + TBI | Mild-Moderate | Yes | >5 years |
| London Patient [53] | Hodgkin's Lymphoma | Chemotherapy + TBI | Mild | Yes | >2 years |
| Düsseldorf Patient [53] | Acute Myeloid Leukemia | Chemotherapy + TBI | Moderate | Yes | >6 years |
| Systematic Review Cases [55] | Various Hematologic Malignancies | Varied (Chemo ± TBI) | Mild-Severe | Yes | 60-80% achieved ≥12 months remission |
A recent systematic review of five case studies reported that 60-80% of patients receiving CCR5Δ32/Δ32 HSCT achieved at least 12 months of ART-free HIV remission, with a subset maintaining long-term remission beyond 5 years [55]. The approach demonstrates greatest efficacy when complete donor chimerism is achieved, wherein over 95% of immune cells derive from the CCR5-deficient donor [54].
Experimental Protocol: CCR5Δ32/Δ32 HSCT
Patient Selection and Conditioning
- Identify HIV+ patients with hematological malignancies requiring HSCT
- Perform HLA typing and CCR5Δ32 screening of potential donors
- Administer myeloablative conditioning (e.g., cyclophosphamide + total body irradiation) to eliminate residual bone marrow and reduce HIV reservoirs
- Continue antiretroviral therapy throughout the procedure when possible [54]
Transplantation and Engraftment Monitoring
- Infuse CCR5Δ32/Δ32 allogeneic hematopoietic stem cells (~4-8×10^6 CD34+ cells/kg)
- Monitor engraftment through daily complete blood counts
- Assess donor chimerism weekly using PCR-based methods on peripheral blood and bone marrow
- Administer immunosuppressants (cyclosporine, methotrexate) for GVHD prophylaxis [54] [55]
Post-Transplant Evaluation
- Quantitative HIV DNA PCR in peripheral blood mononuclear cells
- Viral load monitoring after ART interruption (typically 3-6 months post-engraftment)
- HIV antibody titers and T-cell reactivity assessments
- Reservoir quantification via quantitative viral outgrowth assays [54]
Figure 1: CCR5Δ32/Δ32 HSCT Clinical Workflow
CAR-T Cell Engineering for HIV Resistance
CAR-T Cell Design and Generational Evolution
Chimeric antigen receptor T-cell (CAR-T) therapy for HIV involves engineering autologous T-cells to express synthetic receptors targeting HIV antigens, primarily envelope glycoproteins. These CAR-T cells combine the HIV-specificity of antibodies with the cytotoxic potency of T-cells, operating independently of MHC presentation – a critical advantage against HIV which downregulates MHC class I [53].
Table 2: Generational Evolution of Anti-HIV CAR-T Cells
| Generation | Components | Advantages | Limitations |
|---|---|---|---|
| First Generation [53] | CD4 extracellular domain + CD3ζ intracellular domain | MHC-independent recognition, specific gp120 targeting | Limited persistence, no costimulation, poor in vivo efficacy |
| Second Generation [53] | CD4 or bNAb extracellular + CD28 or 4-1BB + CD3ζ | Enhanced persistence, improved expansion, better viral control | Susceptible to HIV infection, potential viral escape |
| Third Generation [53] | Multiple costimulatory domains (CD28+4-1BB+CD3ζ) | Stronger cytokine production, enhanced killing capacity | Increased complexity of manufacturing |
| Fourth Generation (TRUCK) [53] | CAR + inducible cytokine (IL-12) | Recruitment of innate immunity, enhanced tumor microenvironment penetration | Cytokine-related potential toxicity |
| Advanced Configurations [56] | bNAb-based multispecific CARs + CCR5 knockout | HIV resistance, broad variant recognition, reduced viral escape | Technically challenging manufacturing process |
Current approaches combine CAR engineering with CCR5 knockout using gene editing technologies like zinc-finger nucleases (ZFNs) or CRISPR-Cas9, creating HIV-resistant CAR-T cells with enhanced durability [57]. The dual-targeting "duoCAR" designs incorporate two distinct HIV-specificities to prevent viral escape [53].
Experimental Protocol: Engineering HIV-Resistant CAR-T Cells
CAR Construct Design and Vector Production
- Select targeting domains: CD4 for gp120 or broadly neutralizing antibodies (bNAbs) for conserved epitopes
- Clone CAR construct into lentiviral vector with CD8α hinge/transmembrane domain
- Incorporate 4-1BB and CD3ζ signaling domains (second generation)
- Produce lentiviral vectors via transfection of HEK293T cells [53] [57]
T-Cell Isolation and Engineering
- Isolate peripheral blood mononuclear cells from HIV+ donor via apheresis and density centrifugation
- Activate CD8+ and CD4+ T-cells with anti-CD3/anti-CD28 beads
- Transduce with lentiviral CAR vectors (MOI 5-20) via spinoculation
- Edit CCR5 locus using ZFNs or CRISPR-Cas9 via electroporation
- Expand cells in IL-2 and IL-15 media for 10-14 days [57]
Quality Control and Potency Assessment
- Flow cytometry for CAR expression (detected via Fab anti-IgG)
- CCR5 knockout efficiency measurement by sequencing or antibody staining
- In vitro cytolysis assay against HIV-infected cells
- Viral inhibition assay measuring p24 reduction in supernatant
- Cytokine release profiling upon HIV antigen exposure [57]
Figure 2: HIV-Resistant CAR-T Cell Manufacturing
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Engineering HIV-Resistant Immune Cells
| Reagent/Category | Specific Examples | Research Function | Technical Notes |
|---|---|---|---|
| CAR Constructs [53] [57] | CD4ζ, bNAb-based CARs, duoCAR | Target HIV-infected cells | CD4ζ first generation; bNAbs target conserved epitopes |
| Gene Editing Tools [54] [57] | ZFNs, CRISPR-Cas9, TALENs | Disrupt CCR5 expression | CRISPR-Cas9 offers high efficiency and multiplexing |
| Viral Vectors [53] [57] | Lentiviral, Retroviral vectors | Deliver CAR constructs | Lentiviral preferred for T-cell transduction |
| Cell Culture Media [57] | IL-2, IL-15, Anti-CD3/CD28 beads | T-cell activation and expansion | IL-15 enhances memory T-cell persistence |
| Flow Cytometry Antibodies [57] | Anti-CAR detection antibodies, CCR5 antibodies | Validate CAR expression and CCR5 knockout | Fab fragments for CAR detection without Fc binding |
| HIV Detection Assays [54] | p24 ELISA, Quantitative PCR, Viral outgrowth assays | Measure HIV reservoir and viral inhibition | QVOA gold standard for latent reservoir quantification |
| Animal Models [57] | Humanized mice, Primate models | Preclinical efficacy testing | BLT mice for human immune system reconstitution |
Comparative Analysis and Future Directions
Therapeutic Comparison
HSCT from CCR5Δ32 donors represents a complete immune system replacement strategy, suitable primarily for HIV patients with coincident hematological malignancies due to transplant-associated mortality risks (15-20%) and GVHD incidence [54] [55]. In contrast, CAR-T cell approaches offer a targeted immunotherapy with potentially better safety, though they face challenges of viral escape, limited tissue penetration, and CAR-T cell persistence [53].
Recent clinical trials demonstrate progress in overcoming these limitations. A Phase I trial of bNAb-based CAR-T showed long-term in vivo persistence without safety concerns [53]. The duoCAR-T approach targeting multiple HIV epitopes simultaneously has entered Phase I/II trials (NCT04648046) to address viral escape variants [53].
Integration with Complementary Approaches
The future of HIV cure research likely involves combination strategies that pair engineered cell therapies with other interventions:
- Latency reversal agents to expose reservoir cells for elimination by CAR-T cells [53]
- Broadly neutralizing antibodies to neutralize free virions and synergize with cellular therapies [56]
- Immune checkpoint inhibitors to overcome T-cell exhaustion in persistent HIV infection [53]
Notably, research initiated for HIV directly informed cancer CAR-T therapies, with the first anti-HIV CAR-T clinical trials predating cancer applications [58]. This reverse translation exemplifies how HIV research continues to drive innovation in cellular immunotherapy.
Engineering HIV-resistant immune cells through CCR5Δ32 HSCT or CAR-T technology represents a promising frontier in HIV cure research. The CCR5Δ32 mutation provides a natural blueprint for resistance, which can be replicated through gene editing or leveraged through transplantation. While HSCT offers proof-of-concept for HIV eradication, its utility is limited by donor availability and procedure-related risks. CAR-T approaches provide a more scalable strategy, particularly as gene editing technologies improve in efficiency and safety. The integration of these approaches with complementary strategies targeting the latent reservoir offers the most promising path toward a sustainable HIV remission.
The pursuit of a cure for Human Immunodeficiency Virus (HIV) has evolved from viral suppression to eradication strategies. Despite the transformative success of antiretroviral therapy (ART), which effectively controls viral replication, it fails to eliminate latent viral reservoirs and necessitates lifelong medication, presenting challenges such as cumulative drug toxicity and the emergence of resistant strains [13] [59]. The discovery that a homozygous mutation in the C-C chemokine receptor 5 (CCR5) gene, known as CCR5-Δ32, confers natural resistance to R5-tropic HIV infection provided a foundational insight for curative approaches [13] [60]. This natural resistance mechanism was spectacularly validated by the cases of the "Berlin," "London," and a third patient, who were functionally cured of HIV following hematopoietic stem-cell transplantation (HSCT) from donors with a homozygous CCR5-Δ32 mutation [32] [60].
However, the scarcity of naturally compatible CCR5-Δ32 donors and the medical risks of allogeneic HSCT limit its broad application [32]. This has catalyzed the development of novel strategies to mimic this natural resistance artificially. Two of the most promising fields are gene editing, which aims to permanently disrupt the CCR5 gene or excise integrated provirus, and immunotherapy, which seeks to enhance the immune system's ability to clear infected cells [13] [59]. Individually, each approach has shown distinct limitations: single-target CCR5 editing leaves cells vulnerable to CXCR4-tropic (X4) viral strains, while immunotherapy alone may lack the persistence to eradicate a deeply latent reservoir [13] [32]. Consequently, the next frontier in HIV cure research involves synergistic integration of these modalities, creating a multi-pronged defense that leverages the long-term protection of gene editing and the potent clearance capabilities of engineered immunity [13]. This whitepaper provides a technical guide to the mechanisms, methodologies, and emerging evidence supporting this combined strategy.
Scientific Foundation: Core Concepts and Mechanisms
The CCR5-Δ32 Mutation and HIV Resistance
The CCR5 protein is a seven-transmembrane G-protein-coupled receptor that serves as a primary co-receptor for HIV-1 entry into CD4+ T-cells and macrophages [61]. The virus initially binds to the CD4 receptor, inducing a conformational change that allows its gp120 envelope protein to engage with CCR5, a step critical for viral fusion and entry [61]. The CCR5-Δ32 mutation is a naturally occurring 32-base-pair deletion in the CCR5 gene's coding region. This deletion creates a frameshift and a premature stop codon, resulting in a truncated, non-functional protein that is not expressed on the cell surface [61] [60]. Consequently, individuals who are homozygous for this mutation (CCR5-Δ32/Δ32) lack CCR5 co-receptors and exhibit high-level resistance to infection by R5-tropic HIV strains, which dominate during the early and chronic phases of infection [13] [60]. The foundational proof-of-concept for targeting CCR5 was established by the cure of HIV in patients who received HSCT from CCR5-Δ32/Δ32 donors, demonstrating that a CCR5-deficient immune system can confer long-term viral remission without ART [32] [60].
Gene Editing Platforms for CCR5 Disruption
Gene editing technologies enable precise, permanent disruption of the CCR5 gene in human cells, aiming to recapitulate the protective effect of the Δ32 mutation. The primary platforms are compared in Table 1.
Table 1: Comparative Characteristics of Major Gene Editing Technologies for CCR5-Targeted HIV Therapy [13]
| Technology | Mechanism of Action | Key Features | Therapeutic Potential for HIV |
|---|---|---|---|
| Zinc Finger Nucleases (ZFNs) | Fusion of zinc finger DNA-binding domain to FokI endonuclease. | First platform used in clinical trials for CCR5; high specificity but complex design. | Clinical trials (e.g., NCT01252641, NCT01543152) show safety and feasibility of infusing ZFN-modified autologous CD4+ T-cells. |
| TALENs | Fusion of TALE DNA-binding domain to FokI endonuclease. | Simpler design rules than ZFNs; modular protein engineering. | Demonstrated efficient CCR5 editing; can be paired with ZFNs for multi-locus editing [13]. |
| CRISPR/Cas9 | RNA-guided DNA cleavage using Cas9 nuclease and sgRNA. | Simple design, high efficiency, multiplexibility, and low cost. | Progressed to early-phase clinical trials (NCT03164135) for CCR5-edited HSCs; suitable for multi-target strategies [13] [32]. |
Among these, the CRISPR/Cas9 system has emerged as the most versatile tool due to its simplicity and efficiency. It can be programmed with multiple single-guide RNAs (sgRNAs) to target several genomic loci simultaneously, a capability critical for addressing viral escape mechanisms [13].
Synergistic Immunotherapy Approaches
Immunotherapy aims to empower the host immune system to recognize and eliminate HIV-infected cells. Key strategies include:
- HIV-Specific CAR-T Cells: T-cells are engineered to express a Chimeric Antigen Receptor (CAR) that recognizes HIV envelope proteins on infected cells, leading to their targeted destruction. Second-generation CAR-T cells can be further engineered for enhanced persistence and function [13] [59].
- Immune Checkpoint Inhibition: Chronic HIV infection leads to T-cell exhaustion, characterized by upregulated checkpoint molecules like PD-1. Blocking these pathways with antibodies can reinvigorate HIV-specific T-cell responses and potentially facilitate latent reservoir clearance [13].
- Broadly Neutralizing Antibodies (bNAbs): These antibodies target conserved regions of the HIV envelope, neutralizing a broad spectrum of viral strains and mediating antibody-dependent cellular cytotoxicity (ADCC) to eliminate infected cells [59].
The synergy emerges when gene-edited, HIV-resistant cells are combined with these potent immune effectors, creating a population of target cells that are both difficult to infect and capable of aggressive viral clearance.
Quantitative Data and Multi-Target Editing Strategies
Relying solely on CCR5 disruption is insufficient for a comprehensive HIV cure. A key challenge is viral tropism switching, where selective pressure from CCR5 knockout can favor the emergence of CXCR4-tropic (X4) viruses, which can then infect cells via the intact CXCR4 co-receptor [13]. Furthermore, for cells already harboring integrated provirus, simply blocking entry does not address the latent reservoir, which can reactivate via the Long Terminal Repeat (LTR) region [13].
Multi-Target Gene Editing
To construct a comprehensive viral barrier, advanced strategies employ multiplexed gene editing. The CRISPR/Cas system is particularly adept at this, allowing for the simultaneous knockout of host genes and disruption of viral DNA [13] [32]. Quantitative data on the efficiency of such approaches are summarized in Table 2.
Table 2: Quantitative On-Target Efficiency and Off-Target Profiles of Gene Editing Platforms [13]
| Editing Platform | Target Loci | Reported On-Target Efficiency | Key Off-Target Risks |
|---|---|---|---|
| TALENs | CCR5 | High (Schwarze et al., 2021) [13] | Lower off-target risk compared to CRISPR/Cas9. |
| CRISPR/Cas9 | CCR5, CXCR4, HIV LTR | Superior efficacy in inhibiting viral replication and transmission [13]. | Higher off-target risk; requires careful sgRNA design and validation. |
| CRISPR/Cas12a (Cpf1) | CCR5, CXCR4, HIV LTR | Well-suited for simultaneous targeting of diverse loci via crRNA arrays [13]. | Unique cleavage profile (sticky ends) may influence repair outcomes. |
The rationale for a multi-target attack is clear:
- Knockout of CCR5 and CXCR4 prevents de novo infection by both R5- and X4-tropic viruses, effectively closing the door to most circulating strains [13] [32].
- Editing the HIV LTR suppresses transcriptional activation, helping to lock the provirus in a latent state and preventing reactivation ("block and lock" strategy) [13].
- Targeting viral structural genes (e.g., Gag, Pol) can directly disrupt the production of functional viral particles [13].
A 2024 study demonstrated the power of combining CCR5 knockout with an additional anti-HIV transgene. Researchers used CRISPR/Cas9 to ablate CCR5 in a cell line and concurrently introduced a lentiviral vector expressing C46, a membrane-anchored HIV-1 fusion inhibitor. This combined approach provided robust protection against both R5- and X4-tropic HIV-1 challenges, outperforming either single method [32].
The Gene-Immune Synergy Workflow
The conceptual and technical workflow for integrating these therapies involves a coordinated sequence of ex vivo and in vivo steps, designed to establish a durable, HIV-resistant immune system.
Diagram 1: Combined therapy workflow for creating an HIV-resistant immune system by engineering and infusing patient cells.
Detailed Experimental Protocols
This section outlines a core methodology for implementing a combined gene editing and immunotherapy strategy, based on a 2024 study [32].
Protocol: Combining CRISPR/Cas9-Mediated CCR5 Knockout with C46 Fusion Inhibitor Expression
Objective: To generate a population of MT4CCR5 cells (or primary CD4+ T-cells) resistant to both R5- and X4-tropic HIV-1 through simultaneous CCR5 gene knockout and stable expression of the C46 fusion inhibitor.
Key Research Reagent Solutions:
- In-house Cas9 Protein: Purified Cas9 nuclease for ribonucleoprotein (RNP) complex formation.
- Single-Guide RNAs (sgRNAs): Two sgRNAs designed to target the first exon of the human CCR5 gene, specifically at the Δ32 mutation site (sequences as validated in [32]).
- Lentiviral Vector pCDH-EF1a-C46-Puro: Construct for expression of the C46 HIV-1 fusion inhibitor, driven by an EF1a promoter, with a puromycin resistance gene for selection.
- MT4CCR5 Cell Line: A model cell line expressing both CD4 and CCR5, susceptible to HIV-1 infection.
- Nucleofector System: For high-efficiency delivery of RNP complexes into cells.
Methodology:
- CRISPR/Cas9 RNP Complex Formation: Combine 10 µg of in-house Cas9 protein with 4 µg of each sgRNA (total 8 µg sgRNA). Incubate at room temperature for 10-20 minutes to form the RNP complex [32].
- Cell Nucleofection: Harvest and resuspend MT4CCR5 cells in an appropriate nucleofection solution. Mix the cell suspension with the pre-formed RNP complex and electroporate using a optimized nucleofection program. Include controls transfected with Cas9 or sgRNAs alone [32].
- Assessment of Editing Efficiency (3 days post-nucleofection):
- T7 Endonuclease I (T7E1) Assay: Extract genomic DNA and PCR-amplify the CCR5 target region. Digest the PCR product with T7E1, which cleaves mismatched DNA heteroduplexes. Analyze cleavage fragments by gel electrophoresis to estimate indel frequency [32].
- Flow Cytometry: Stain live cells with a fluorescently labeled anti-CCR5 antibody. Analyze using flow cytometry to determine the percentage of CCR5-negative cells. The cited study achieved a 97.89% reduction in CCR5 surface expression with this protocol [32].
- Western Blot: Confirm reduction of CCR5 protein expression levels in the edited cell population.
- Lentiviral Transduction for C46 Expression: Package the pCDH-EF1a-C46-Puro vector into lentiviral particles. Transduce the CCR5-edited cells with the viral supernatant. 48 hours post-transduction, begin selection with puromycin (e.g., 1-2 µg/mL) for 1-2 weeks to generate a pure population of cells expressing the C46 fusion inhibitor [32].
- Functional HIV Challenge Assay: Challenge the dual-modified cells (CCR5-KO + C46), control cells (wild-type, CCR5-KO only, C46 only) with R5-tropic (e.g., Bal.) and X4-tropic (e.g., NL4-3) HIV-1 strains.
- Metrics: Monitor and compare:
- Cell viability (e.g., via 7AAD staining) over 7-14 days.
- Viral replication by measuring p24 antigen levels in the culture supernatant via ELISA.
- Expected Outcome: The combined therapy group should demonstrate significantly superior protection, evidenced by higher cell viability and lower p24 levels, against both viral tropisms compared to any single-therapy group [32].
- Metrics: Monitor and compare:
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Combined Gene Editing and Immunotherapy Studies
| Reagent / Tool | Function / Purpose | Example Application |
|---|---|---|
| CRISPR/Cas9 System | Precise genomic DNA cleavage for gene knockout (CCR5, CXCR4) or proviral excision. | Creating HIV co-receptor resistant hematopoietic stem cells (HSCs) and T-cells [13] [32]. |
| Lentiviral Vectors | Stable delivery of transgenes (e.g., C46, CAR constructs, bNAbs) into target cells. | Engineering T-cells to express anti-HIV CARs or fusion inhibitors [32] [59]. |
| Zinc Finger Nucleases (ZFNs) | Alternative gene editing platform for site-specific gene disruption. | Used in early clinical trials (Sangamo BioSciences) to create CCR5-modified CD4+ T-cells [62] [60]. |
| Broadly Neutralizing Antibodies (bNAbs) | Neutralize diverse HIV strains and recruit immune effector functions. | Administered in vivo to clear circulating virus and opsonize infected cells post cell-therapy [59]. |
| Immune Checkpoint Inhibitors | Block inhibitory receptors (e.g., PD-1) to reverse T-cell exhaustion. | Used in combination with CAR-T cells to enhance their cytotoxic activity and persistence [13]. |
| Acute Phase Protein & Cytokine Panels | Quantify inflammatory biomarkers (e.g., IL-6, CRP, sCD14) to monitor immune activation and therapy safety. | Assessing systemic inflammation and immune reconstitution in pre-clinical and clinical studies [63] [64]. |
Integrated Therapeutic Workflows and Future Directions
The ultimate application of these strategies involves a multi-step process that begins with harvesting a patient's own cells, engineering them ex vivo, and reinfusing them to rebuild an HIV-resistant immune system, potentially supported by subsequent in vivo treatments.
Diagram 2: Defense mechanisms of synergistic multi-target gene editing and immunotherapy.
Future work must focus on optimizing delivery systems, enhancing the safety profile of gene editors to minimize off-target effects, and developing personalized combination regimens based on patient-specific factors such as viral tropism and genetic background [13]. The path forward requires a collaborative, multi-disciplinary effort to translate these powerful synergistic strategies from the laboratory to the clinic, offering hope for a definitive cure for HIV.
Navigating Scientific Hurdles: Tropism Switching, Safety, and Enhanced Efficacy
The phenomenon of coreceptor switching from CCR5 (R5) to CXCR4 (X4) represents a critical juncture in HIV-1 pathogenesis, presenting substantial challenges for therapeutic strategies built upon CCR5 blockade. HIV-1 entry into host cells requires interaction with the CD4 receptor and one of two principal coreceptors, CCR5 or CXCR4 [65]. The CCR5Δ32 mutation, a well-characterized 32-base-pair deletion in the CCR5 gene, confers significant resistance to R5-tropic HIV-1 infection. Homozygous carriers produce a truncated protein that fails to embed in the cell membrane, effectively blocking the primary entry portal for most transmitted viruses [14] [66]. This natural resistance mechanism has inspired therapeutic approaches targeting CCR5. However, the adaptive capacity of HIV-1 enables viral escape through the emergence of X4-tropic variants, which utilize the CXCR4 coreceptor and are frequently associated with accelerated CD4+ T-cell decline and disease progression [67] [68].
The prevalence of X4-tropic viruses increases from approximately 0-20% during early infection to over 50% in late-stage disease among subtype B infections [67]. While X4 variants were historically considered less efficient at establishing new infections, a groundbreaking 2023 study identified a strictly CXCR4-tropic transmitted/founder (T/F) virus in an individual with wild-type CCR5, demonstrating unequivocally that X4-tropic HIV-1 can traverse mucosal barriers and establish clinical infection [69]. This patient experienced dramatically accelerated CD4+ T-cell depletion at a rate of 1.6 cells/μL/day compared to 0.27 cells/μL/day in R5-infected controls, highlighting the enhanced pathogenicity of X4 variants [69]. This evidence fundamentally challenges the paradigm that CXCR4 use is exclusively a late-stage adaptation, underscoring the imperative to address X4 tropism in comprehensive HIV therapeutic strategies.
Biological Mechanisms of CXCR4 Tropism and Viral Pathogenesis
Molecular Determinants of Coreceptor Usage
The genetic basis for coreceptor specificity primarily resides in the V3 loop of the HIV-1 envelope glycoprotein gp120. X4-tropic viruses typically exhibit an increased positive charge in the V3 loop, enhancing interaction with the negatively charged extracellular domains of CXCR4 [67]. However, coreceptor specificity is not exclusively determined by V3; mutations in C2, V4/V5, and gp41 regions also contribute to coreceptor affinity and entry efficiency [67]. The transition from R5 to X4 usage often occurs through intermediate R5X4 variants that can utilize both coreceptors, with progressive evolution toward exclusive CXCR4 dependence [67] [68].
Coreceptor usage fundamentally determines cellular tropism and pathogenic impact. CXCR4 is broadly expressed on naïve and central memory CD4+ T-cells, while CCR5 expression is predominantly on effector memory and transitional memory subsets [69]. This expression pattern underlies the distinct pathogenic profiles of R5 and X4 viruses. The broader cellular targeting of X4 variants enables infection of naïve T-cells, which constitute a larger reservoir of potential target cells and may explain the accelerated CD4+ decline observed with X4 tropism [69].
Table 1: Coreceptor Expression Patterns on CD4+ T-Cell Subsets
| CD4+ T-Cell Subset | CCR5 Expression | CXCR4 Expression | Primary Viral Tropism |
|---|---|---|---|
| Naïve | Undetectable | High | X4-tropic |
| Central Memory (CM) | Low | High | X4-tropic |
| Transitional Memory (TM) | High | Moderate | R5-tropic |
| Effector Memory (EM) | High | Low | R5-tropic |
Pathogenic Consequences of X4 Tropism
The expanded target cell pool available to X4-tropic viruses underlies their enhanced pathogenicity. The case study of patient 40700, infected with a transmitted X4-tropic virus, demonstrated preferential depletion of naïve and central memory CD4+ subsets, with decline rates of 0.97 cells/μL/day and 0.42 cells/μL/day, respectively [69]. This contrasts with significantly slower decline in effector and transitional memory subsets (0.17 and 0.07 cells/μL/day), consistent with the CXCR4 expression gradient across T-cell populations [69].
Notably, all CD4+ subsets, including naïve cells, showed evidence of productive infection in this case, with the naïve subset exhibiting a 9.1-fold disparity between total and integrated HIV DNA [69]. This suggests that while X4 viruses efficiently enter naïve T-cells, the resting state of these cells may impair viral integration, potentially explaining the high ratio of unintegrated to integrated DNA. The robust viral replication of X4 variants, with viral loads sustained above 10⁵ copies/mL, coupled with increased immune activation, creates a perfect storm for rapid immunological decline [69].
Figure 1: X4-tropic HIV Pathogenesis Pathway
Experimental Models and Methodologies for Studying X4 Tropism
Coreceptor Usage Assays
Determining HIV-1 coreceptor tropism is essential for both clinical management and research applications. The TZM-bl cell line provides a standardized platform for tropism determination through pseudovirus entry assays. In this system, entry activity of HIV-1 pseudoviruses is measured in the presence of specific coreceptor inhibitors. AMD3100 (a CXCR4 inhibitor) and Maraviroc (a CCR5 inhibitor) enable pharmacological distinction of coreceptor preference; X4-tropic viruses show complete inhibition by AMD3100 but not Maraviroc [69].
The NP-2 cell line panel, engineered to express individual coreceptors, offers complementary tropism determination. X4-tropic viruses demonstrate high-efficiency infection in NP-2/CXCR4 cells while failing to infect NP-2/CCR5 cells [69]. This system also facilitates investigation of alternative coreceptor usage, with some X4-tropic viruses showing low-efficiency usage of CCR3, APJ, and FPRL1 [69]. For comprehensive characterization, primary CD4+ T-cells from multiple donors provide the most physiologically relevant replication platform, confirming that X4-tropic transmitted/founder viruses are fully replication-competent with kinetics comparable to laboratory-adapted X4 strains like NL4-3 [69].
Table 2: Experimental Models for X4 Tropism Determination
| Assay System | Key Components | Readout | Applications |
|---|---|---|---|
| TZM-bl Inhibition | CD4/CCR5/CXCR4+ cells, coreceptor inhibitors | Luminescence from reporter gene | Phenotypic tropism, inhibitor sensitivity |
| NP-2 Panel | CD4+ cells expressing single coreceptors | Viral p24 production or reporter signal | Coreceptor specificity, alternative coreceptor use |
| Primary CD4+ T-Cells | Purified CD4+ cells from healthy donors | Viral replication kinetics (p24) | Physiological replication capacity, cytopathicity |
| Trofile Assay | Proprietary cell line with reporter constructs | Luciferase activity | Clinical tropism testing, CCR5 inhibitor eligibility |
Genotypic Prediction and Evolution Analysis
Genotypic prediction algorithms based on V3 loop sequences provide a rapid, cost-effective alternative to phenotypic tropism assays. Tools like Geno2Pheno employ statistical models to predict coreceptor usage, with the false positive rate (FPR) indicating the probability of CXCR4 use; the X4-tropic T/F virus from patient 40700 had an FPR of 0.1%, indicating high confidence CXCR4 usage [69]. However, comprehensive genotypic analysis requires examination of the entire envelope sequence, as mutations outside V3 in C2, V4/V5, and gp41 regions significantly influence coreceptor affinity and entry efficiency [67].
Single-genome amplification (SGA) and phylogenetic analysis enable detailed reconstruction of viral evolution and identification of transmitted/founder viruses. Application of Poisson-Fitter tools to SGA-derived sequences allows estimation of time since transmission based on viral diversification rates [69]. Next-generation sequencing approaches now permit high-resolution analysis of coreceptor gene variations and their association with disease progression, identifying specific polymorphisms in CXCR4 (e.g., rs2680880:A>T) that influence CD4+ T-cell recovery during antiretroviral therapy [70].
Figure 2: Coreceptor Tropism Determination Workflow
Therapeutic Strategies and Research Reagents
Targeting CXCR4 and Dual Antagonism Approaches
The clinical development of CXCR4 antagonists has progressed cautiously due to concerns about disrupting the physiological SDF-1/CXCR4 axis, which is crucial for hematopoietic stem cell homing and immune cell trafficking. Plerixafor (AMD3100), the most extensively characterized CXCR4 antagonist, demonstrates potent anti-X4 activity at nanomolar concentrations but causes side effects that have limited its approval for HIV treatment [65]. Its primary clinical application is for hematopoietic stem cell mobilization in oncology [65].
Given the limitations of single-coreceptor targeting, dual CCR5/CXCR4 antagonists represent the next frontier in entry inhibition. AMD3451 was identified as the first dual antagonist, though its moderate potency hindered clinical development [65]. Subsequent efforts have explored diverse chemical scaffolds, including pyrazolo-piperidine derivatives (e.g., compound 11 with IC₅₀ values of 3.8 µM against R5 and 0.8 µM against X4 viruses) and ingenol diterpene esters that downregulate both coreceptors and CD4 [65]. Peptide triazoles like KR21 represent another innovative approach, simultaneously targeting gp120 interactions with both CD4 and coreceptors [65].
Gene Editing and Immunotherapeutic Approaches
Gene editing technologies present a transformative approach to conferring permanent resistance to HIV infection. The cases of the "Berlin" and "London" patients, who achieved sustained HIV remission after hematopoietic stem cell transplantation from CCR5Δ32 homozygous donors, provide proof-of-concept for CCR5 disruption as a therapeutic strategy [13]. CRISPR/Cas9 systems now enable precise targeting of CCR5, CXCR4, and viral LTR regions in hematopoietic stem cells, with early-phase clinical trials (NCT03164135) demonstrating feasibility and safety [13].
To prevent viral escape through tropism switching, multiplex gene editing strategies simultaneously target multiple host and viral loci. Knocking out both CCR5 and CXCR4 creates a population of CD4+ T-cells resistant to both R5 and X4 viruses, while editing the HIV LTR can suppress viral reactivation from latency [13]. The CRISPR/Cas12a system offers advantages for multiplexing through its crRNA array, which can generate multiple mature crRNAs from a single transcript [13]. Combining gene editing with immunotherapeutic approaches such as HIV-specific CAR-T cells and immune checkpoint inhibitors creates synergistic strategies that couple durable cellular resistance with enhanced viral clearance capacity [13].
Table 3: Research Reagent Solutions for X4 Tropism Research
| Reagent/Cell Line | Specific Application | Key Features | Experimental Use |
|---|---|---|---|
| TZM-bl Cells | Viral entry and tropism determination | Engineered to express CD4/CCR5/CXCR4 with reporter genes | Coreceptor inhibition assays with AMD3100/Maraviroc |
| NP-2 Cell Panel | Coreceptor specificity profiling | CD4+ cells expressing individual coreceptors | Determination of coreceptor usage spectrum |
| AMD3100 | CXCR4-specific inhibition | Selective CXCR4 antagonist, IC₅₀ in nM range | Phenotypic confirmation of X4 tropism, control for specificity |
| Maraviroc | CCR5-specific inhibition | FDA-approved CCR5 antagonist | Exclusion of CCR5 usage, dual-tropism characterization |
| Geno2Pheno Algorithm | Genotypic tropism prediction | Statistical prediction based on V3 loop sequences | Initial tropism screening from envelope sequences |
| CRISPR/Cas9 Systems | Gene editing for coreceptor knockout | Targeted disruption of CCR5/CXCR4 genes | Generation of virus-resistant cell populations, therapeutic development |
The challenge of CXCR4-tropic HIV strains represents a critical frontier in the quest for comprehensive HIV therapeutics. The identification of transmitted X4-tropic viruses fundamentally alters our understanding of HIV transmission biology and underscores the limitations of CCR5-focused interventions [69]. The enhanced pathogenicity of X4 variants, mediated through their broader tropism for naïve and central memory CD4+ T-cells, necessitates therapeutic strategies that address both major coreceptor pathways [69] [68].
Future progress will likely emerge from integrated therapeutic approaches that combine dual coreceptor antagonists with immunotherapeutic modalities and strategic gene editing [65] [13]. The personalization of HIV therapy based on viral tropism, host genetics, and immune environment will be essential, particularly as next-generation sequencing reveals polymorphisms in CXCR4 and related genes that influence disease progression and treatment response [70]. While significant challenges remain—including potential toxicity concerns with CXCR4 blockade and the evolutionary capacity of HIV to exploit alternative entry pathways—the scientific foundation is being laid for a new generation of interventions capable of addressing the full spectrum of HIV-1 coreceptor usage.
The discovery that a small proportion of individuals show partial or complete innate resistance to HIV, largely due to a mutation in the CCR5 gene known as CCR5-Δ32, represents a cornerstone of HIV cure research [47]. Individuals homozygous for this 32-base-pair deletion are highly resistant to R5-tropic HIV-1 infection, as the mutation results in a non-functional CCR5 co-receptor that the virus cannot use for cellular entry [71] [72]. This natural genetic defense, observed in rare individuals such as the "Berlin" and "London" patients who were cured of HIV after receiving CCR5-Δ32 homozygous stem cell transplants, provides a powerful proof-of-concept for therapeutic strategies aimed at disrupting HIV co-receptors [3]. However, reliance on single-target approaches creates vulnerability to viral escape mechanisms, particularly through tropism switching to strains that utilize the CXCR4 co-receptor [3] [73]. This biological reality necessitates a comprehensive multi-target strategy that concurrently addresses both major HIV co-receptors and the integrated viral reservoir itself.
The limitations of current antiretroviral therapy (ART), including lifelong medication requirements, cumulative drug toxicity, and an inability to eradicate latent viral reservoirs, further underscore the need for transformative curative approaches [3] [74]. The latent HIV reservoir, consisting of resting CD4+ T-cells with integrated proviral DNA, represents the primary barrier to eradication, as cessation of ART inevitably leads to viral rebound [74]. Multi-target gene editing represents a paradigm shift from viral suppression to potential eradication by creating cells with intrinsic resistance to HIV infection while simultaneously targeting the integrated provirus. By concurrently disrupting CCR5, CXCR4, and the HIV Long Terminal Repeat (LTR), this approach aims to establish a comprehensive defense system that blocks viral entry at multiple points and prevents reactivation from latency, thereby addressing the fundamental challenges that have hindered cure efforts to date.
Core Concepts and Biological Rationale
CCR5-Δ32: A Natural Blueprint for Resistance
The CCR5-Δ32 mutation provides the strongest genetic evidence for targeting CCR5 therapeutically. A 2018 meta-analysis of 24 case-control studies demonstrated that individuals with the homozygous CCR5-Δ32 genotype (delta32/delta32) had significantly reduced susceptibility to HIV-1 infection (OR=0.25, 95%CI=0.09-0.68) [71]. At the molecular level, this 32-base-pair deletion in the CCR5 gene coding region creates a truncated protein that fails to reach the cell surface, thereby denying R5-tropic HIV strains their primary entry co-receptor [71] [47]. Heterozygous carriers also show some protective benefit, with studies of viremic nonprogressors (VNPs) - individuals who maintain stable CD4+ T-cell counts despite viral replication - revealing that many possess the CCR5-Δ32 mutation in a heterozygous state, resulting in lower CCR5 receptor density on target cells and consequently reduced viral infectivity [72].
The Imperative for Multi-Target Approaches
Single-target strategies, particularly those focusing exclusively on CCR5, face significant limitations due to HIV's evolutionary capacity. Following effective CCR5 disruption, selective pressure can favor the emergence of X4-tropic strains that utilize CXCR4 as an alternative co-receptor [3] [73]. The X4-tropic HIV-1 strains are present in approximately half of late-stage infections and are associated with more rapid disease progression [73]. Furthermore, even successful dual co-receptor disruption cannot address the established latent reservoir, where integrated provirus remains capable of reactivation via the Long Terminal Repeat (LTR) region, which contains strong promoter and enhancer elements that drive viral transcription once activated [3]. The LTR promotes Gag expression, enhances viral particle assembly, facilitates reverse transcription, and ultimately drives viral replication, making it a critical target for preventing reservoir reactivation [3].
Table 1: HIV Targets for Multi-Gene Editing Strategies
| Target | Type | Function | Rationale for Editing | Consequence of Disruption |
|---|---|---|---|---|
| CCR5 | Host gene | HIV-1 co-receptor (R5-tropic) | Primary entry point for most infections; natural Δ32 mutation confers resistance | Renders cells resistant to R5-tropic HIV entry; mimics natural immunity |
| CXCR4 | Host gene | HIV-1 co-receptor (X4-tropic) | Alternative entry pathway; emerges after CCR5 blockade | Prevents viral escape via tropism switching; blocks X4-tropic infection |
| HIV LTR | Viral DNA | Regulatory region for viral transcription | Controls viral reactivation from latency; critical for reservoir persistence | Disrupts viral replication cycle; prevents reactivation from latent reservoirs |
Molecular Mechanisms of HIV Entry and Latency
HIV entry into CD4+ T-cells occurs through a sequential process involving attachment to the CD4 receptor followed by engagement of either the CCR5 or CXCR4 co-receptor, triggering fusion and viral entry [3] [73]. The LTR region functions as the master regulatory switch for HIV replication, controlling transcription initiation and serving as a binding site for both viral and host transcription factors that can reactivate latent virus [3]. During latency, the integrated provirus remains transcriptionally silent but intact, poised for reactivation when appropriate signals are received through the LTR. This complex biology necessitates a multi-pronged editing approach that simultaneously disrupts both entry pathways and permanently disables the integrated provirus to achieve comprehensive viral control.
Quantitative Assessment of Gene Editing Outcomes
The efficacy of gene editing strategies is quantified through multiple parameters, including indel efficiency, protein disruption rates, and functional protection from HIV infection. Recent studies provide compelling data on the feasibility of concurrently targeting CCR5, CXCR4, and HIV LTR.
Table 2: Quantitative Outcomes of Gene Editing Approaches
| Target | Editing System | Cell Type | Efficiency (Indels) | Protein Reduction | HIV Protection | Citation |
|---|---|---|---|---|---|---|
| CXCR4 | CRISPR/Cas9 (gRNA #6) | Ghost X4 cells | 39.22% | 76.5% (FACS) | Complete resistance to X4-tropic HIV | [73] |
| CXCR4 | CRISPR/Cas9 (gRNA #7) | Ghost X4 cells | 39.22% | 70.5% (FACS) | Complete resistance to X4-tropic HIV | [73] |
| CXCR4 | CRISPR/Cas9 (gRNA #6) | Jurkat T cells | 45.03% | Significant decrease (Western blot) | Reduced p24 antigen levels | [73] |
| CXCR4 | CRISPR/Cas9 (gRNA #7) | Jurkat T cells | 44.31% | Significant decrease (Western blot) | Reduced p24 antigen levels | [73] |
| CCR5 | ZFNs (SB-728-T) | Primary T cells | Clinical trial data | N/A | Virological/immunological benefits | [3] |
| CCR5-Δ32/Δ32 | Natural mutation | Human population | N/A | 100% CCR5 disruption | >90% protection from HIV infection | [71] |
The data demonstrate that CRISPR/Cas9-mediated ablation of CXCR4 achieves high specificity with negligible off-target effects while maintaining normal cell division and propagation [73]. The complete resistance to HIV infection in edited Ghost X4 cells, as evidenced by undetectable GFP expression and significantly reduced viral titers, validates the therapeutic potential of co-receptor disruption [73]. Furthermore, the natural CCR5-Δ32 homozygote data provides a long-term safety profile for CCR5 disruption, with no obvious phenotypic consequences beyond HIV resistance except for potential increased susceptibility to certain pathogens [73] [47].
Experimental Protocols and Methodologies
CRISPR/Cas9-Mediated CXCR4 Disruption Protocol
The following detailed protocol for CXCR4 gene disruption in human primary CD4+ T-cells has been adapted from established methodologies with demonstrated efficacy [73]:
Phase 1: gRNA Design and Vector Construction
- Design 10-12 gRNAs targeting conserved sites in the first coding exon of human CXCR4 gene (Reference sequence: NC_000023.11)
- Select gRNAs with high on-target efficiency scores and minimal off-target potential using computational tools (e.g., CRISPRscan, MIT CRISPR Design)
- Clone selected gRNAs into a modular lentiviral sgRNA:Cas9 vector system under U6 promoter control
- Validate constructs through Sanger sequencing and restriction digest analysis
Phase 2: Lentiviral Production and Transduction
- Package lentiviral vectors in HEK-293T cells using standard third-generation packaging systems
- Concentrate viral supernatants via ultracentrifugation (25,000 RPM for 2 hours)
- Determine viral titer by p24 ELISA or quantitative PCR
- Transduce target cells (Ghost-CXCR4, Jurkat, or primary CD4+ T-cells) at MOI 40-60 in the presence of 8μg/mL polybrene
- Centrifuge plates at 800-1000 × g for 30-60 minutes (spinoculation) to enhance transduction efficiency
- Incubate at 37°C, 5% CO2 for 72-96 hours post-transduction
Phase 3: Efficiency Validation and Cell Sorting
- Harvest transduced cells and extract genomic DNA for T7 Endonuclease I (T7EN1) assay
- Perform PCR amplification of CXCR4 target region (primers: F-5'-CAGTCAGTATGCAGGAGAT-3', R-5'-CAGGAAATGACAGCCAAGC-3')
- Digest heteroduplex DNA with T7EN1 enzyme and analyze by agarose gel electrophoresis
- Calculate indel efficiency using band intensity quantification
- For protein-level validation, stain cells with anti-CXCR4-PE antibody and analyze by flow cytometry
- Sort CXCR4-negative populations using FACS Aria II or equivalent cell sorter
Phase 4: Functional HIV Protection Assay
- Infect sorted CXCR4-edited cells with HIV-1NL4-3 (X4-tropic) at MOI 1.0
- Monitor infection over 5-7 days using multiple readouts:
- GFP expression (for Ghost-CXCR4 reporter cells)
- p24 antigen ELISA at days 1, 3, 5, and 7 post-infection
- Viral RNA copy number by quantitative RT-PCR
- Intracellular HIV Gag staining and flow cytometry
- Include untransduced cells and non-targeting gRNA controls in all experiments
Multi-Target Editing Workflow
Advanced protocols now enable simultaneous editing of CCR5, CXCR4, and HIV LTR through multiplexed CRISPR approaches:
- Vector Design: Utilize either single vector systems with multiple gRNA expression cassettes or parallel transduction with separate lentiviral vectors
- gRNA Coordination: Employ distinct RNA polymerase III promoters (U6, H1, 7SK) to minimize recombination
- Delivery Optimization: Implement staggered transduction protocols with 24-hour intervals between different viral vectors to reduce competition
- Efficiency Assessment: Design locus-specific PCR amplicons for each target to quantify individual editing efficiencies
- Off-target Screening: Perform whole-genome sequencing or GUIDE-seq analysis to identify and verify specificity
Diagram 1: Multi-Target Gene Editing Workflow. This flowchart illustrates the comprehensive experimental pipeline for concurrent editing of CCR5, CXCR4, and HIV LTR, from initial gRNA design through final functional validation.
Research Reagent Solutions
Successful implementation of multi-target gene editing requires carefully selected reagents and systems optimized for efficient delivery and precise genomic modification.
Table 3: Essential Research Reagents for Multi-Target Gene Editing
| Reagent Category | Specific Product/System | Function | Key Considerations |
|---|---|---|---|
| Editing Systems | CRISPR/Cas9 (Streptococcus pyogenes) | RNA-guided DNA cleavage | High efficiency; enables multiplexing; various delivery formats available |
| ZFNs (SB-728-T clinical candidate) | Protein-based DNA recognition and cleavage | Early clinical validation; complex design but established safety profile | |
| TALENs | Protein-based DNA recognition and cleavage | High specificity; larger size challenges viral packaging | |
| Delivery Vectors | Lentiviral vectors (VSV-G pseudotyped) | Stable gene delivery to dividing and non-dividing cells | High transduction efficiency; insertional mutagenesis risk |
| Adenovirus-associated vectors (AAV) | Transient expression in non-dividing cells | Lower immunogenicity; limited packaging capacity | |
| Electroporation (mRNA/protein) | Direct delivery of editing components | Transient expression; reduced off-target risk; high efficiency in primary cells | |
| gRNA/Targeting | CXCR4 gRNA #6: 5'-GACTGACCAGAGAGCCAAGC-3' | Targets CXCR4 exon 1 | 45.03% efficiency in Jurkat cells [73] |
| CXCR4 gRNA #7: 5'-GCCAACCGTCAGTCTGCTAC-3' | Targets CXCR4 exon 1 | 44.31% efficiency in Jurkat cells [73] | |
| CCR5 gRNAs targeting Δ32 region | Mimics natural CCR5-Δ32 mutation | Various validated designs available | |
| HIV LTR gRNAs targeting NF-κB/Sp1 sites | Disrupts viral transcription factor binding | Multiple targets across U3/R regions | |
| Analytical Tools | T7 Endonuclease I (T7E1) assay | Detection of indel mutations | Rapid screening; semi-quantitative |
| Next-generation sequencing (NGS) | Comprehensive editing and off-target analysis | Gold standard for specificity assessment | |
| Flow cytometry (anti-CXCR4/CCR5 antibodies) | Protein-level validation of editing | Direct functional consequence measurement | |
| Cell Systems | Ghost-CXCR4 reporter cells | HIV infection and tropism studies | GFP expression under LTR control |
| Primary human CD4+ T-cells | Physiologically relevant target cells | Require activation for efficient editing | |
| Jurkat T-cell line | Model for mechanistic studies | Easy to culture; high editing efficiency |
Strategic Integration with Immunotherapy
The combination of multi-target gene editing with immunotherapy represents a promising frontier for achieving functional HIV cure. Current research indicates several synergistic approaches:
CAR-T Cell Engineering: Chimeric Antigen Receptor (CAR) T-cells specific for HIV envelope proteins can be further enhanced through CCR5 and CXCR4 gene editing, creating HIV-resistant immune effector cells with potent antiviral activity [3]. Studies demonstrate that allogeneic HIV-specific CAR-T cells engineered to secrete PD-1-blocking scFv show increased cytotoxicity against HIV Env+ cells, highlighting the potential of combined approaches [3].
Immune Checkpoint Modulation: The persistent expression of immune checkpoint molecules like PD-1 contributes to T-cell exhaustion in chronic HIV infection [3]. Gene editing technologies, including base editors, have been successfully employed to disrupt PD-1 expression, potentially reversing HIV-specific T-cell exhaustion and enhancing viral clearance [3]. Jia et al. (2025) successfully edited the PD-1 gene using CE-8e-SpRY mRNA base editors delivered via LVLPs under a "Gag-only" packaging strategy [3].
Broadly Neutralizing Antibody Combinations: Emerging clinical data support the combination of gene editing with passive immunization using broadly neutralizing antibodies (bNAbs). Week 52 results from a Phase 2 trial demonstrated that twice-yearly lenacapavir in combination with bNAbs (teropavimab and zinlirvimab) maintained viral suppression in people with HIV with susceptible viruses and was generally well tolerated [75]. This investigational combination has the potential to be the first twice-yearly complete regimen and is now progressing to Phase 3 clinical development [75].
Diagram 2: Gene Editing and Immunotherapy Synergy. This diagram illustrates the complementary mechanisms by which multi-target gene editing enhances various immunotherapeutic approaches, leading to improved outcomes across multiple dimensions of HIV control.
Multi-target gene editing represents a paradigm shift in HIV cure research, moving beyond viral suppression to potential eradication. The concurrent targeting of CCR5, CXCR4, and HIV LTR addresses the fundamental biological challenges that have hindered previous approaches: viral entry through multiple co-receptors and persistence in latent reservoirs. The natural resistance conferred by the CCR5-Δ32 mutation provides both validation for this strategy and a blueprint for therapeutic intervention.
Significant challenges remain in optimizing delivery efficiency, ensuring long-term safety, and addressing potential immune responses to editing components. The clinical translation of these approaches will require careful balance between therapeutic efficacy and risk management. However, the rapid advancement of gene editing technologies, combined with synergistic immunotherapies, creates an unprecedented opportunity to develop transformative interventions for HIV. As these technologies mature, personalized approaches considering individual viral tropism, reservoir characteristics, and host genetics will likely maximize therapeutic benefit while minimizing potential risks.
The integration of multi-target gene editing with other emerging modalities, including long-acting antiretrovirals and therapeutic vaccines, may ultimately provide the comprehensive strategy needed to achieve durable HIV remission or cure. With multiple clinical trials already underway and an expanding toolkit of precise gene editing technologies, the field is poised to translate these sophisticated scientific approaches into tangible benefits for people living with HIV.
The discovery that the CCR5Δ32 mutation confers natural resistance to HIV infection by obliterating the CCR5 chemokine receptor on cell surfaces has established a compelling therapeutic paradigm [12] [76]. This natural resistance mechanism, observed in homozygous CCR5Δ32 individuals and dramatically demonstrated by the "Berlin" and "London" HIV cure cases, has catalyzed the development of gene editing therapies designed to mimic this protective effect [3] [13]. Gene editing technologies—including CRISPR/Cas9, zinc finger nucleases (ZFNs), and transcription activator-like effector nucleases (TALENs)—are being leveraged to disrupt the CCR5 gene in hematopoietic stem cells and T cells to create HIV-resistant immune populations [3].
However, the therapeutic promise of CCR5 gene editing is tempered by significant safety concerns, primarily the risk of off-target effects and long-term genomic consequences. Off-target effects refer to unintended modifications at genomic sites with sequence similarity to the intended target, which can potentially lead to detrimental outcomes such as oncogenesis or disruption of essential genes [3] [13]. As research progresses toward clinical applications, establishing robust methodologies for assessing these risks becomes paramount for translational success. This technical guide provides a comprehensive framework for evaluating off-target effects and long-term safety in CCR5-targeted gene editing protocols, specifically contextualized within HIV resistance research.
Biological Foundation: CCR5 Structure, Function, and the Δ32 Mutation
The C-C chemokine receptor type 5 (CCR5) is a seven-transmembrane G-protein coupled receptor expressed predominantly on immature (Th0) and memory T-cells, macrophages, and dendritic cells [12]. Its natural ligands include pro-inflammatory chemokines CCL3 (MIP-1α), CCL4 (MIP-1β), and CCL5 (RANTES) [12]. For HIV-1 entry, the viral envelope glycoprotein gp120 initially binds to CD4, inducing conformational changes that expose its coreceptor binding site, which subsequently engages CCR5, facilitating viral fusion and entry [12] [3].
The CCR5Δ32 mutation is a 32-base pair deletion in the CCR5 gene that results in a frameshift and premature termination codon, preventing functional receptor expression on the cell surface [12] [76]. Individuals homozygous for this mutation demonstrate substantial resistance to R5-tropic HIV strains, which account for the majority of initial infections, without apparent immunodeficiency or other pathological phenotypes, though some studies suggest potential subtle immune alterations [12] [76]. This favorable safety profile makes CCR5 an attractive target for therapeutic gene editing.
Table 1: Biological Characteristics of CCR5 and the Δ32 Mutation
| Feature | Wild-Type CCR5 | CCR5Δ32 Mutant |
|---|---|---|
| Gene Structure | 352 amino acids; full-length coding sequence | 32-bp deletion; frameshift and premature stop codon |
| Receptor Expression | Present on cell surface (CD4+ T cells, macrophages) | Truncated protein; not expressed on cell surface |
| HIV Infection | Permissive to R5-tropic HIV entry | Highly resistant to R5-tropic HIV infection |
| Immune Function | Receptor for chemokines MIP-1α, MIP-1β, RANTES | No apparent immunodeficiency in homozygous individuals |
| Population Frequency | Global variation; Δ32 allele ~10% in Northern Europe | Highest frequency in European populations |
The following diagram illustrates the structural difference between wild-type CCR5 and the Δ32 mutant, and the consequential HIV resistance mechanism:
Gene Editing Platforms for CCR5 Targeting: Efficacy and Safety Profiles
Multiple gene editing platforms have been developed to disrupt the CCR5 locus, each with distinct mechanisms and safety considerations. The comparative characteristics of these platforms are detailed below:
Table 2: Gene Editing Platforms for CCR5 Targeting: Mechanisms and Safety Profiles
| Technology | Mechanism of Action | Editing Efficiency | Reported Off-Target Risks | Advantages for HIV Therapy |
|---|---|---|---|---|
| ZFNs | Custom zinc finger proteins fused to FokI nuclease dimer for DNA cleavage | Moderate (30-50% in clinical trials) | Higher risk of off-target effects; potential immunogenicity | Early clinical data (SB-728-T); established safety profile [3] |
| TALENs | Modular TALE proteins fused to FokI nuclease for specific DNA cleavage | High (>60% in preclinical studies) | Improved specificity over ZFNs; reduced off-target activity | High specificity; modular design facilitates targeting [3] |
| CRISPR/Cas9 | sgRNA directs Cas9 nuclease to genomic loci for double-strand breaks | Very High (80-95% in vitro) | Main concern is off-target effects at sites with seed sequence similarity | Enables multiplex editing (CCR5, CXCR4, LTR); easy design [3] [13] |
| Base Editors | Cas9 nickase fused to deaminase enables precise single-nucleotide changes | Variable (15-70% depending on system) | Potential for off-target base editing (DNA and RNA); limited editing window | Avoids double-strand breaks; precise nucleotide conversion [3] |
The workflow for targeting CCR5 using these platforms involves multiple coordinated steps:
Comprehensive Methodologies for Off-Target Detection
Robust assessment of off-target effects requires a multi-modal approach employing complementary biochemical, computational, and cellular methods. The most current and effective techniques are summarized below:
Biochemical and Cell-Based Assays
GUIDE-seq (Genome-wide Unbiased Identification of DSBs Enabled by sequencing): This method utilizes double-stranded oligodeoxynucleotides that integrate into double-strand breaks (DSBs) via non-homologous end joining. After editing, genome-wide sequencing identifies off-target integration sites, providing a comprehensive landscape of CRISPR/Cas9-induced DSBs with nucleotide-level resolution [3] [13].
CIRCLE-seq (Circularization for In vitro Reporting of Cleavage Effects by sequencing): An in vitro method where genomic DNA is fragmented, circularized, and amplified as a circular library. After incubation with CRISPR/Cas9 ribonucleoproteins, cleaved fragments are linearized and sequenced. CIRCLE-seq offers high sensitivity without cellular context constraints and can be performed prior to clinical application [13].
SITE-seq (Selective Enrichment and Identification of Tagged Genomic DNA Ends by sequencing): This approach biochemically identifies Cas9 cleavage sites using adapter-ligated genomic DNA incubated with Cas9-sgRNA complexes, followed by enrichment of cleaved fragments and high-throughput sequencing, providing a quantitative measure of cleavage efficiency [13].
Digenome-seq (In vitro Digestion of Genomic DNA): A cell-free method where purified genomic DNA is digested with CRISPR/Cas9, followed by whole-genome sequencing. Cleavage sites are identified by bioinformatic analysis of sequencing reads with misaligned ends, offering a sensitive approach to map off-target sites without amplification bias [3].
Computational Prediction Tools
Computational algorithms predict potential off-target sites by searching for genomic sequences with similarity to the sgRNA, allowing for mismatches, bulges, and variations in the protospacer adjacent motif (PAM). While essential for initial risk assessment, these tools should be combined with empirical validation due to variable predictive accuracy across platforms and cell types [13].
Quantitative Assessment of Off-Target Profiles
Rigorous quantification of editing efficiency and specificity is essential for therapeutic development. The following table synthesizes quantitative data from recent studies comparing different editing platforms targeting CCR5:
Table 3: Quantitative Off-Target Profiles of Gene Editing Platforms in CCR5-Targeted Therapies
| Editing Platform | On-Target Efficiency (%) | Off-Target Sites Identified | Validation Method | Key Safety Findings |
|---|---|---|---|---|
| CRISPR/Cas9 (SpCas9) | 85-95% (in CD4+ T-cells) | 3-15 sites per sgRNA | GUIDE-seq, NGS | Off-target activity correlates with sgRNA specificity scores; minimal clinical adverse events in early trials [3] [13] |
| CRISPR/Cas12a (Cpf1) | 70-80% (in HSPCs) | 1-5 sites per crRNA | CIRCLE-seq | Different PAM requirement (TTTN) reduces overlap with Cas9 off-target sites [13] |
| ZFNs (SB-728-T) | 30-50% (in clinical T-cells) | 5-20 sites per ZFN pair | Digenome-seq | Moderate efficiency with acceptable safety profile in clinical trials [3] |
| TALENs | 60-75% (in preclinical) | 0-8 sites per TALEN pair | SITE-seq | High specificity due to longer recognition sequence [3] |
| Base Editors (ABE8e) | 40-60% (without DSBs) | Rare (primarily RNA off-targets) | RNA-seq | Minimal DNA off-targets; occasional RNA editing observed [3] |
Experimental Protocols for Safety Assessment
Comprehensive Off-Target Screening Workflow
This integrated protocol combines multiple assessment methods for thorough safety profiling:
Sample Preparation: Transfer 1×10⁶ edited cells (e.g., CD34+ hematopoietic stem cells or primary T-cells) to sterile microcentrifuge tubes. Include unedited controls from the same donor.
Genomic DNA Extraction: Use high-molecular-weight DNA extraction kits (e.g., Qiagen Blood & Cell Culture DNA Kit) according to manufacturer specifications. Determine DNA concentration using fluorometric methods (e.g., Qubit dsDNA HS Assay).
GUIDE-seq Library Preparation:
- Prepare 100 µM GUIDE-seq oligoduplex by annealing phosphorylated and HPLC-purified oligos.
- Electroporate 2µg of oligoduplex with edited cells using optimized parameters (e.g., 1400V, 10ms pulse width).
- Culture cells for 72 hours post-electroporation to allow oligo integration.
- Extract genomic DNA and sonicate to 300-500bp fragments.
- Prepare sequencing libraries with adapters containing unique molecular identifiers (UMIs) to reduce PCR bias.
High-Throughput Sequencing: Perform paired-end sequencing (2×150bp) on Illumina platforms to achieve minimum 50× coverage across the genome.
Bioinformatic Analysis:
- Align sequencing reads to the reference genome (hg38) using BWA-MEM or Bowtie2.
- Identify GUIDE-seq integration sites using specialized software (e.g., GUIDE-seq software).
- Perform off-target prediction using Cas-OFFinder and compare with empirical results.
- Annotate off-target sites with genomic features (exonic, intronic, intergenic, promoter regions).
Orthogonal Validation: Validate top candidate off-target sites (prioritizing coding regions) using targeted amplicon sequencing with minimum 1000× coverage.
Long-Term Safety and Stability Assessment
Karyotypic Analysis:
- Culture 5×10⁵ edited cells in complete medium with colcemid (0.1 µg/mL) for 4 hours.
- Harvest cells using hypotonic solution (0.075M KCl) and fix with 3:1 methanol:acetic acid.
- Prepare metaphase spreads on glass slides and stain with Giemsa for G-banding.
- Analyze 20-50 metaphase spreads per sample for chromosomal abnormalities.
Oncogenic Transformation Assay:
- Plate edited cells in soft agar at density of 1×10⁴ cells/well in 6-well plates.
- Culture for 3-4 weeks with regular medium replenishment.
- Score colony formation (>50 cells) as potential transformation indicator.
Immunogenicity Assessment:
- Co-culture edited cells with allogeneic peripheral blood mononuclear cells (PBMCs) at 1:10 ratio.
- Measure T-cell activation markers (CD69, CD25) by flow cytometry at 24, 48, and 72 hours.
- Quantify cytokine secretion (IFN-γ, IL-2, IL-6) in supernatant using multiplex ELISA.
The Scientist's Toolkit: Essential Reagents and Materials
Table 4: Essential Research Reagents for CCR5 Gene Editing Safety Assessment
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Gene Editing Platforms | CRISPR/Cas9 (SpCas9), Cas12a, ZFNs, TALENs, Base Editors | Core editing machinery for targeted CCR5 disruption |
| Delivery Systems | Lentiviral vectors, AAV6, Electroporation systems (Neon, Amaxa) | Introduction of editing components into target cells |
| Off-Target Detection Kits | GUIDE-seq oligonucleotides, CIRCLE-seq library prep kits | Empirical identification of unintended editing events |
| Cell Culture Materials | CD34+ HSPC expansion media, T-cell activation beads (CD3/CD28) | Maintenance and expansion of primary cell populations |
| Sequencing & Analysis | Illumina sequencing platforms, UMI adapters, GUIDE-seq analysis software | Detection and quantification of editing outcomes |
| Validation Assays | Karyotyping G-banding kits, Soft agar colony formation assays | Assessment of long-term genomic stability and safety |
The strategic targeting of CCR5 represents a promising avenue for achieving HIV remission or cure, inspired by the natural resistance conferred by the CCR5Δ32 mutation. As gene editing technologies advance toward clinical application, comprehensive safety assessment encompassing rigorous off-target profiling and long-term follow-up remains non-negotiable. The integrated methodologies outlined in this guide provide a framework for characterizing and mitigating risks associated with CCR5-targeted therapies. Continued refinement of both editing platforms and safety assessment protocols will be essential to balance the remarkable therapeutic potential of these approaches with the imperative of patient safety, ultimately enabling the development of effective and secure genetic interventions for HIV.
Optimizing Delivery Systems for Gene Editing Tools In Vivo
The mechanism by which the CCR5Δ32 mutation confers resistance to HIV-1 infection is well-established: this 32-base-pair deletion in the C-C chemokine receptor 5 (CCR5) gene results in a truncated, non-functional protein that is not expressed on the cell surface, thereby preventing R5-tropic HIV-1 strains from entering host CD4+ T-cells [48] [77]. This natural resistance, famously demonstrated by the "Berlin" and "London" patients cured of HIV after CCR5Δ32/Δ32 hematopoietic stem cell transplantation, provides a powerful therapeutic blueprint [48] [3]. The goal of contemporary gene editing is to recapitulate this protective phenotype in patients' own cells. The clustered regularly interspaced short palindromic repeats (CRISPR)/Cas system has emerged as a leading technology for this purpose, enabling precise genome editing to disrupt the CCR5 locus [48] [78].
However, the therapeutic potential of CRISPR-based gene editing is entirely contingent on the safe and efficient in vivo delivery of its molecular components—typically the Cas nuclease and a guide RNA (gRNA)—to target cells [79]. The ideal delivery vector must protect the nucleic acid payload from degradation, facilitate cell-specific targeting, ensure robust and precise editing, and exhibit minimal toxicity and immunogenicity. The translation of mechanistic research on CCR5 into viable in vivo therapies therefore hinges on parallel advancements in delivery vector technology. This guide provides an in-depth technical analysis of the current landscape of in vivo delivery systems for gene editing tools, framing this discussion within the context of achieving a functional cure for HIV through CCR5 disruption.
Delivery System Modalities: A Comparative Technical Analysis
The delivery systems for in vivo gene editing can be broadly categorized into viral and non-viral vectors. Each modality presents a unique profile of advantages, limitations, and optimal use cases, which are critical to consider for therapeutic application.
Viral Vector Systems
Adeno-Associated Virus (AAV) is one of the most widely deployed viral vectors for in vivo gene editing to date [79]. Its popularity stems from several key characteristics, which are summarized in Table 1 alongside other delivery modalities.
Table 1: Quantitative Comparison of Key In Vivo Delivery Systems for Gene Editing Tools
| Delivery System | Payload Capacity | Editing Duration | Key Advantages | Major Limitations & Toxicity Concerns | Clinical Trial Status for Gene Editing |
|---|---|---|---|---|---|
| Adeno-Associated Virus (AAV) | ~4.7 kb | Long-term/ Persistent | - High transduction efficiency- Multiple serotypes for tissue tropism- Established clinical manufacturing | - Limited payload capacity- Pre-existing and treatment-induced immunogenicity- Liver toxicity at high doses- Risk of genomic integration & genotoxicity | Most widely used in clinical trials to date |
| Virus-Like Particle (VLP) | Varies; can deliver pre-assembled ribonucleoprotein (RNP) | Transient | - Enables transient RNP delivery- Reduces off-target risks- Potential for cell-specific targeting | - Lower delivery efficiency vs. AAV- Challenges in large-scale production- Potential residual immunogenicity | Emerging technology; pre-clinical and early clinical development |
| Lipid Nanoparticle (LNP) | Varies; can deliver RNP or mRNA/gRNA | Transient | - Well-suited for mRNA/gRNA delivery- Scalable manufacturing- Reduced risk of genomic integration | - Primarily accumulates in liver (passive targeting)- Challenges targeting extra-hepatic tissues- Potential inflammatory responses | Used in clinical trials (e.g., NTLA-2001 for ATTR); growing application for gene editing |
Despite its advantages, AAV faces significant challenges. Immunogenicity against the capsid can neutralize the vector and prevent re-dosing, while high doses have been linked to liver toxicity [79]. Furthermore, the prolonged expression of Cas9 from AAV vectors can increase the risk of off-target editing [79].
Other Viral Vectors, such as adenoviruses, were used in early proof-of-concept studies. For instance, one study demonstrated that chimeric Ad5F35 adenoviruses could successfully deliver CCR5-targeting CRISPR/Cas9 to primary CD4+ T-cells, conferring resistance to HIV-1 infection [78]. However, due to high immunogenicity, adenoviruses are less favored for in vivo gene editing therapies compared to AAV.
Non-Viral Vector Systems
Non-viral systems are emerging as promising alternatives to mitigate the risks associated with viral vectors.
Lipid Nanoparticles (LNP) are particularly well-suited for delivering mRNA encoding the Cas9 protein, along with the gRNA. This approach leads to transient expression of the editor, significantly reducing the window for potential off-target effects and minimizing immune recognition [79]. A primary limitation of current LNP technology is the predominant accumulation in the liver after systemic administration, making passive targeting of other tissues challenging [79].
Virus-Like Particles (VLP) represent another innovative non-viral platform. VLPs are engineered to package and deliver the pre-assembled Cas9-gRNA ribonucleoprotein (RNP) complex directly into target cells [79]. This method facilitates very transient editor activity, akin to mRNA delivery, and further reduces the risk of off-target mutations and genomic integration. Recent advances include the use of "Gag-only" packaging strategies and VLPs based on the SpRY Cas9 variant to broaden targeting scope [3].
The following diagram illustrates the core mechanisms of these three leading delivery systems.
Experimental Workflows for Delivery System Evaluation
Developing and optimizing a delivery system requires a structured pipeline from vector design to functional validation. The following workflow is generalized for engineering a cell-specific LNP for delivering base editors to hematopoietic cells, a key target for CCR5 editing.
Diagram Title: LNP Delivery Workflow for Hematopoietic Cells
Detailed Methodologies for Key Experiments
Protocol 1: Electroporation of CRISPR Editor mRNA into Primary T-cells (Ex Vivo)
This protocol, adapted from several studies [80] [81] [82], is a cornerstone for ex vivo cell therapy production, such as for generating CCR5-edited T-cells or CAR-T cells.
- T-cell Activation and Expansion: Isolate PBMCs from a leukapheresis product. Activate and expand primary human T-cells using anti-CD3/CD28 antibody-coated beads in a culture medium supplemented with IL-2 (e.g., 100 IU/mL) for 2-3 days.
- mRNA Production: In vitro transcribe the mRNA encoding the CRISPR editor (e.g., Cas9, base editor) from a linearized plasmid DNA template. The plasmid must contain a 5' cap analog (or use a capping enzyme) and a 3' poly(A) tail (optimally 120 nucleotides) to enhance mRNA stability and translation [81]. Include modified nucleotides (e.g., N1-Methylpseudouridine) to reduce innate immune recognition.
- Ribonucleoprotein (RNP) Complex Formation (Alternative): As an alternative to mRNA, complex purified Cas9 protein with a synthetic, chemically modified gRNA targeting CCR5. Incubate at room temperature for 10-20 minutes to allow RNP assembly.
- Electroporation: Wash the activated T-cells to remove residual culture media. Resuspend the cell pellet in an electroporation buffer. Combine the cells with the mRNA (or pre-formed RNP) and electroporate using a specialized system for primary immune cells (e.g., Lonza 4D-Nucleofector). Use a pre-optimized program such as "EO-115" for human T-cells.
- Post-Transfection Culture: Immediately after electroporation, transfer the cells to pre-warmed culture medium. Analyze editing efficiency approximately 48-72 hours post-electroporation. For T-cells intended for therapy, continue expansion with IL-2 for a further 7-10 days before infusion.
Protocol 2: Assessing CCR5 Knockout Efficiency and Functional HIV Resistance
This protocol validates the success of the gene editing procedure [78] [81].
- Genomic DNA Extraction and Amplicon Sequencing (On-Target): Harvest edited cells and extract genomic DNA. Amplify the region surrounding the CCR5 target site via PCR. Use next-generation sequencing (NGS) of the PCR amplicons to quantify the percentage of insertions and deletions (indels). Editing efficiency is calculated as (number of reads with indels / total number of reads) × 100%.
- Flow Cytometry for Surface CCR5 Expression: Stain the edited and control cells with a fluorescently labeled anti-CCR5 antibody. Analyze using flow cytometry. The percentage of CCR5-negative cells in the edited population, compared to the control, provides a functional measure of knockout efficiency.
- HIV Challenge Assay: Challenge the edited T-cells (and control T-cells) with a CCR5-tropic HIV-1 strain (e.g., HIV-1BaL) or HIVenv-pseudotyped lentiviral vectors. After several days, measure the level of infection by quantifying:
- Intracellular p24 Antigen: via flow cytometry.
- Viral RNA in Supernatant: via RT-qPCR. A significant reduction in p24+ cells and viral RNA in the culture supernatant of edited cells indicates successful conferral of HIV-1 resistance.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for In Vivo Gene Editing Research
| Reagent / Tool | Function / Purpose | Example & Notes |
|---|---|---|
| CRISPR Editor mRNAs | Enables transient expression of editors (Cas9, BE, ABE) without viral vectors. | TriLink Cas9 mRNA; Custom-made mRNA for cost-effectiveness (up to 10x cheaper) [82]. |
| Ionizable Lipids | The key functional component of LNPs for encapsulating and delivering mRNA/RNP. | Proprietary lipids (e.g., from Acuitas, Moderna); ALC-0315 (in approved COVID-19 vaccines). |
| CROP-seq-CAR Vector | Allows co-delivery of a CAR transgene and a gRNA library for pooled in vivo screens. | Enables in vivo CROP-seq to simultaneously track gRNA identity and transcriptome in single cells [82]. |
| High-Resolution Melting Curve Analysis (HRMCA) | A rapid, cost-effective method for initial screening of editing efficiency in cell populations. | Used post-editing on PCR amplicons from the target locus to identify samples with indels [81]. |
| Next-Generation Amplicon Sequencing (NGS) | Gold standard for quantifying on-target editing efficiency and analyzing mutation spectra. | Illumina MiSeq/NextSeq platforms. CRISPResso2 is a standard tool for bioinformatic analysis [81]. |
| PROGNOS & TALEN Targeter | In silico bioinformatics tools for predicting potential off-target sites for TALENs and CRISPR gRNAs. | Critical for pre-design gRNA risk assessment [81]. |
The future of in vivo delivery for gene editing tools lies in overcoming the current limitations of specificity, immunogenicity, and capacity. Key areas of development include the engineering of novel cell-specific targeting ligands for LNPs and VLPs to move beyond passive liver accumulation, and the creation of advanced capsid variants for AAV with improved tropism and reduced immunogenicity. Furthermore, the adaptation of delivery systems for larger or more complex editors, such as dual base editors or prime editors, is an active area of research. The successful clinical translation of CCR5 editing strategies will depend on a synergistic approach, where a deep understanding of the therapeutic target is matched by sophisticated vector engineering to deliver the editing machinery safely and effectively.
Overcoming Incomplete Protection in Heterozygous Individuals
The CCR5Δ32 mutation represents a well-characterized genetic determinant of resistance to HIV-1 infection, with homozygous individuals demonstrating near-complete protection against CCR5-tropic virus strains. However, heterozygous carriers, while exhibiting reduced susceptibility to infection and slowed disease progression, remain vulnerable to HIV-1 acquisition and transmission. This whitepaper comprehensively examines the mechanistic basis for incomplete protection in CCR5Δ32 heterozygotes and synthesizes current research strategies aimed at overcoming this limitation. We analyze quantitative data on heterozygote susceptibility, detail experimental methodologies for investigating CCR5 expression and function, visualize key signaling pathways, and outline therapeutic approaches to augment natural protection. The findings presented herein aim to inform drug development initiatives seeking to translate heterozygous resistance phenotypes into complete protection through targeted interventions.
The C-C chemokine receptor type 5 (CCR5) serves as a principal co-receptor for HIV-1 entry into CD4+ T-cells [10] [2]. The Δ32 mutation, a 32-base-pair deletion in the CCR5 gene coding region, results in a truncated protein that fails to localize to the cell surface [4]. While individuals homozygous for CCR5Δ32 demonstrate virtually complete resistance to CCR5-tropic HIV-1 infection, heterozygotes exhibit an intermediate phenotype characterized by reduced cell surface CCR5 expression and delayed disease progression, yet ultimately remain susceptible to infection [10] [47].
This incomplete protection presents a significant challenge and opportunity for therapeutic development. Understanding the quantitative relationship between CCR5 expression, viral entry, and disease susceptibility in heterozygotes is fundamental to designing strategies that mimic or enhance this natural resistance. This review frames the heterozygous phenotype within the broader context of CCR5Δ32 research, focusing on mechanistic insights and translational applications aimed at achieving complete protection through targeted intervention.
Quantitative Assessment of Heterozygote Susceptibility
Epidemiological studies and meta-analyses provide crucial data on the relative susceptibility of heterozygous individuals compared to wild-type and homozygous groups. The table below summarizes key quantitative findings from population studies.
Table 1: HIV-1 Susceptibility and Progression by CCR5 Genotype
| Genotype | Cell Surface CCR5 Expression | Relative Susceptibility to HIV-1 Infection | Impact on Disease Progression |
|---|---|---|---|
| Wild-type (+/+) | Normal | Reference (OR=1.0) | Standard progression rate |
| Heterozygous (+/Δ32) | Reduced (~40-50% lower) | Slightly increased (OR=1.16, 95% CI=1.02-1.32) [71] | Delayed progression [47] |
| Homozygous (Δ32/Δ32) | Absent or non-functional | Significantly reduced (OR=0.25, 95% CI=0.09-0.68) [71] | Near-complete resistance [10] |
A comprehensive meta-analysis of 24 case-control studies revealed that CCR5Δ32 heterozygotes actually had a marginally increased susceptibility to HIV-1 (OR=1.16) compared to wild-type individuals when analyzing general population controls [71]. However, when using exposed uninfected (EU) individuals as controls, delta32 allele carriers showed significantly reduced susceptibility (OR=0.71) [71], suggesting that the protective effect is more evident in high-risk populations. The same analysis confirmed that homozygous individuals enjoy substantial protection (OR=0.25) [71].
Geographic and ethnic variations significantly influence heterozygous distribution and impact. For example, a study of U.S. women found heterozygosity frequencies of 11.8% in white women, 3.7% in African Americans, and 3.3% in Hispanic/Latinas [83]. These regional and ethnic variations in heterozygous frequency must be considered when studying host genetic factors and HIV-1 susceptibility across different populations [83].
Mechanistic Basis of Incomplete Protection
Molecular Consequences of Heterozygous Genotype
In heterozygous individuals, the CCR5Δ32 allele produces a truncated protein that retains the first three transmembrane domains but lacks the final four domains and the C-terminal tail [4]. This mutant protein fails to reach the cell surface and is degraded intracellularly [10]. However, the wild-type allele continues to produce functional CCR5 receptors, resulting in approximately 40-50% reduction in cell surface receptor density compared to wild-type homozygous individuals [10].
This partial receptor reduction creates a threshold effect for HIV-1 infection. Viral entry requires a critical density of CCR5 co-receptors to facilitate fusion with the host cell membrane [2]. While reduced receptor expression diminishes infection efficiency, it does not eliminate it entirely, particularly under high viral inoculum or repeated exposure scenarios. The diagram below illustrates the comparative expression and HIV-1 interaction across different CCR5 genotypes.
CCR5-Mediated Signaling in Heterozygotes
Beyond its role as an HIV-1 co-receptor, CCR5 functions as a key modulator of immune responses through its interactions with chemokines including CCL3 (MIP-1α), CCL4 (MIP-1β), and CCL5 (RANTES) [10]. These interactions mediate leukocyte trafficking to inflammatory sites and regulate T-cell activation and differentiation [10] [2]. In heterozygous individuals, while CCR5 expression is reduced, the remaining functional receptors maintain sufficient signaling capacity to support near-normal immune function, explaining the absence of severe immunodeficiency in these individuals.
The diagram below illustrates the CCR5 signaling pathway and its disruption in HIV-1 infection.
Experimental Approaches for Investigating Heterozygous Protection
Genotyping and Expression Analysis
Protocol 1: CCR5Δ32 Genotyping by PCR
- Primer Design: Forward: 5'-GTCTTCATTACACCTGCAGCTCT-3', Reverse: 5'-CACAGCCCTGTGCCTCTTCTTCTC-3'
- Reaction Setup: Prepare 25μL reactions containing 100ng genomic DNA, 1X PCR buffer, 1.5mM MgCl₂, 0.2mM dNTPs, 0.4μM each primer, and 1.25U Taq polymerase
- Thermal Cycling: Initial denaturation at 95°C for 5min; 35 cycles of 95°C for 30s, 58°C for 30s, 72°C for 45s; final extension at 72°C for 7min
- Product Analysis: Resolve products on 2% agarose gel. Wild-type allele: 735bp, Δ32 allele: 703bp [84]
Protocol 2: Cell Surface CCR5 Quantification by Flow Cytometry
- Cell Preparation: Isolate PBMCs from heterozygous and control donors by density gradient centrifugation
- Staining: Incubate 1×10^6 cells with anti-CCR5 antibody (e.g., clone 2D7/CCR5) and anti-CD4 antibody for 30min at 4°C
- Analysis: Acquire data on flow cytometer, gate on CD4+ T-cells, and compare CCR5 mean fluorescence intensity between genotypes [10] [72]
Functional Viral Entry Assays
Protocol 3: Pseudovirus Entry Assay
- Pseudovirus Production: Generate HIV-1 pseudotyped viruses expressing CCR5-tropic envelope proteins (e.g., JR-FL, Bal) and luciferase reporter gene in HEK293T cells
- Infection: Transduce target cells (primary CD4+ T-cells or cell lines) from heterozygous and control donors with normalized pseudovirus stocks
- Quantification: Measure luciferase activity 48-72 hours post-infection to quantify viral entry efficiency [2]
Table 2: Research Reagent Solutions for CCR5 Heterozygosity Studies
| Reagent/Category | Specific Examples | Research Application | Technical Notes |
|---|---|---|---|
| Genotyping Reagents | CCR5Δ32-specific primers, PCR master mixes | Genotype determination | Validate with known controls; check HWE [71] |
| Flow Cytometry Antibodies | Anti-CCR5 (clone 2D7), anti-CD4, anti-CD3 | Surface receptor quantification | Include isotype controls; use fresh PBMCs [72] |
| Viral Constructs | CCR5-tropic pseudoviruses (JR-FL, Bal) | Viral entry assays | Normalize by p24 antigen or reverse transcriptase activity [2] |
| CCR5 Modulators | Maraviroc, CCL5/RANTES, CCR5 antagonists | Functional studies | Titrate concentrations; monitor cytotoxicity [10] [2] |
| Cell Culture Models | Primary CD4+ T-cells, PM-1 cell line, HEK293T | In vitro infection models | Primary cells best reflect physiological state [72] [2] |
Therapeutic Strategies to Enhance Heterozygous Protection
CCR5 Inhibition Approaches
Several therapeutic strategies aim to augment the natural protection observed in heterozygotes by further reducing available CCR5 receptors or blocking their function:
1. Small Molecule Antagonists: Maraviroc, an FDA-approved CCR5 antagonist, binds allosterically to CCR5 and induces conformational changes that prevent HIV-1 gp120 interaction while preserving chemokine binding [2]. This approach mimics the heterozygous phenotype by functionally reducing CCR5 availability without altering receptor expression.
2. Gene Editing Strategies: Zinc finger nucleases (ZFN), CRISPR/Cas9, and other gene editing platforms can permanently disrupt CCR5 expression in hematopoietic stem cells or primary T-cells [2]. Clinical trials demonstrate that CCR5-edited CD4+ T-cells can persist long-term and potentially confer resistance when transplanted into HIV-1 patients [2].
3. Antibody-Mediated Blockade: Monoclonal antibodies targeting CCR5 extracellular domains can sterically hinder HIV-1 interaction while potentially modulating receptor signaling [2]. These biologics offer longer half-lives than small molecules but face delivery challenges.
Combination Strategies
The most promising approaches combine CCR5-targeting modalities with other antiviral strategies:
- CCR5 inhibition + ART: Enhanced viral suppression and potential reservoir reduction
- Gene editing + stem cell transplantation: Mimicking the "Berlin patient" approach [2]
- CCR5 blockade + latency reversal agents: Targeting both viral entry and persistent reservoirs
The experimental workflow below outlines a comprehensive approach to developing and testing these therapeutic strategies.
The incomplete protection against HIV-1 infection in CCR5Δ32 heterozygotes represents a critical intermediate phenotype that offers unique insights for therapeutic development. While natural heterozygosity reduces CCR5 expression by approximately 40-50%, this proves insufficient to reliably block HIV-1 acquisition, particularly under high exposure conditions. Successful strategies to enhance this protection will need to achieve more complete CCR5 blockade while preserving essential immune functions.
Future research should focus on several key areas: (1) defining the precise threshold of CCR5 expression required for reliable HIV-1 protection; (2) developing tissue-specific delivery mechanisms for CCR5-targeting therapeutics; (3) understanding potential compensatory mechanisms by other viral co-receptors; and (4) investigating the long-term immunological consequences of therapeutic CCR5 reduction. The continued study of CCR5Δ32 heterozygotes provides not only fundamental insights into HIV-1 pathogenesis but also a roadmap for developing novel prevention strategies that build upon natural resistance mechanisms.
Epidemiological and Clinical Evidence: Validating the Protective Phenotype
Global Distribution and Population Genetics of the CCR5Δ32 Allele
The CCR5-Δ32 mutation (a 32-base pair deletion in the C-C chemokine receptor type 5 gene) represents a critical focus in human genetics and infectious disease research due to its well-established role in conferring resistance to HIV-1 infection [12] [71]. This mutation results in a truncated protein that fails to localize to the cell surface, thereby preventing entry of R5-tropic HIV-1 strains—the viruses predominantly responsible for initial infections [12]. The global distribution of this allele is remarkably heterogeneous, with pronounced frequencies in European populations and near absence in other geographical regions [85]. This uneven distribution pattern has fueled extensive scientific investigation into both the evolutionary origins of the mutation and its contemporary implications for HIV therapeutics and public health strategies. Understanding the population genetics of CCR5-Δ32 provides not only insights into human evolutionary history but also a foundation for developing novel genetic-based interventions against HIV/AIDS.
Global Distribution and Population Frequencies
The CCR5-Δ32 allele demonstrates a distinctive geographical gradient across human populations, with the highest frequencies observed in Northern Europe and decreasing prevalence toward the south and east [76]. This pattern suggests a possible selective sweep in European populations, though the nature of the selecting agent remains debated [76]. A foundational study screening 3,342 individuals from globally distributed populations found the allele at frequencies of 2–5% throughout Europe, the Middle East, and the Indian subcontinent [85]. Isolated occurrences elsewhere in the world most likely represent recent European gene flow into indigenous populations [85].
Table 1: Global Frequency Distribution of the CCR5-Δ32 Allele
| Geographical Region | Population Group | Allele Frequency (%) | Homozygous Frequency (%) | Key Studies |
|---|---|---|---|---|
| Northern Europe | European descent | ~10% | ~1% | [32] [85] |
| Angola (Luanda) | African | 0% | 0% | [86] |
| Colombia | Mixed ancestry | Varies with European ancestry | Low (precise frequency not specified) | [87] |
| Asian Populations | Multiple | Nearly absent | Nearly absent | [85] |
Notably, a 2025 study in Luanda, Angola, a sub-Saharan African country, identified a complete absence of the CCR5-Δ32 allele (0/272 alleles screened) [86]. This finding aligns with the established pattern of the allele's near absence in African, Native American, and Asian populations [32]. Research in Colombian populations with mixed ancestry further corroborates the relationship between genetic ancestry and allele frequency, demonstrating a significant positive association between European ancestry and CCR5-Δ32 frequency, with negative (though not statistically significant) associations with African and American ancestries [87]. The low number of allele carriers in these populations indicates that finding homozygous donors for therapeutic purposes remains challenging outside of European-descended groups [87].
Mechanism of HIV Resistance
The CCR5 receptor plays a fundamental role in HIV pathogenesis. It is the primary co-receptor used by R5-tropic HIV-1 strains to gain entry into CD4+ T-cells and other target cells of the immune system [12] [13]. The binding of the viral envelope glycoprotein gp120 to both CD4 and CCR5 triggers a conformational change that facilitates viral fusion with the host cell membrane [12].
The CCR5-Δ32 mutation confers resistance through a simple yet effective mechanism: the 32-base pair deletion creates a frameshift mutation that introduces a premature stop codon, resulting in a severely truncated and non-functional receptor that is retained intracellularly and degraded [12]. Consequently, in individuals homozygous for the Δ32 allele (genotype Δ32/Δ32), CCR5 is absent from the cell surface, effectively blocking the entry pathway for R5-tropic HIV-1 viruses [12] [71]. This biological mechanism provides near-complete resistance to HIV-1 infection in homozygous individuals, as evidenced by their significant underrepresentation among HIV-positive cohorts [71].
Heterozygous carriers (genotype WT/Δ32) exhibit reduced CCR5 expression on their cell surfaces [72]. While this does not prevent infection, it is associated with slower disease progression in HIV-infected individuals, suggesting a gene-dosage effect where lower receptor availability impedes viral replication and spread [12] [71]. The pivotal "Berlin patient" (and later the "London patient"), who were cured of HIV after receiving allogeneic hematopoietic stem cell transplants from CCR5-Δ32 homozygous donors, provides the most compelling clinical validation of this protective mechanism [88] [13].
Diagram 1: Mechanism of HIV-1 resistance in CCR5-Δ32 homozygous cells. The absence of a functional CCR5 receptor on the cell surface prevents viral fusion and entry.
Evolutionary Genetics and Selective Pressure
The population genetics of the CCR5-Δ32 allele present an intriguing evolutionary puzzle. The allele is considered evolutionarily young, yet it has reached unexpectedly high frequencies in certain populations, strongly suggesting a history of intense positive selection [76]. The fundamental debate centers on identifying the specific selective pressure responsible for this rapid rise.
The timing of the selection event is a primary point of contention. HIV-1 itself has not exerted selection pressure on human populations for a sufficient duration to account for the current allele frequencies [76]. This has led researchers to propose that another, historically significant pathogen—possibly Yersinia pestis (the bacterium causing bubonic plague) or Variola virus (smallpox)—that similarly used the CCR5 receptor for entry may have driven the selection [76]. These pathogens would have created a persistent selective advantage for Δ32 carriers over many generations, allowing the allele to reach its present-day frequency in Europe.
The observed North-South cline within Europe, with higher frequencies in the north, provides additional clues for reconstructing the allele's evolutionary history [76]. This pattern could reflect the historical geographic focus of the selecting agent or, alternatively, may represent a founder effect followed by population expansion [76]. Parallel evolution observed at the CCR5 locus in other primate species further underscores the receptor's significance in pathogen defense and supports the hypothesis of recurrent selective pressure across species [76].
Research Methodologies and Experimental Protocols
Genotyping the CCR5-Δ32 Polymorphism
Conventional Polymerase Chain Reaction (PCR) followed by gel electrophoresis remains a standard, reliable method for detecting the CCR5-Δ32 genotype [86].
Table 2: Key Research Reagent Solutions for CCR5-Δ32 Research
| Reagent/Kit | Function/Application | Specific Example/Protocol |
|---|---|---|
| DNA Extraction Kit | Isolation of genomic DNA from various samples | QIAamp DNA Mini Kit (Qiagen) for extraction from DBS (Dried Blood Spots) [86]. |
| PCR Master Mix | Amplification of target DNA sequences | GoTaq Green Master Mix (Promega) or NZYTaq II 2x Green Master Mix (NZYTECH) [86]. |
| Specific Primers | Targeted amplification of the CCR5 gene region containing the Δ32 locus | CCR5-Δ32 Forward: 5′-CTTCATCATCCTCCTGACAATCG-3′CCR5-Δ32 Reverse: 5′-GACCAGCCCCAAGTTGACTATC-3′ [86]. |
| Agarose Gel | Visualization and size separation of PCR products | 2% agarose gel electrophoresis; wild-type band: 262 bp, Δ32 band: 230 bp [86]. |
| T7 Endonuclease I (T7E1) | Detection of CRISPR-induced mutations and editing efficiency by recognizing and cleaving mismatched DNA heteroduplexes [88]. |
Detailed Protocol:
- DNA Extraction: Purify genomic DNA from patient samples (e.g., whole blood, DBS) using a commercial kit [86].
- PCR Amplification: Set up a 20µL reaction mixture containing:
- 5µL of extracted DNA (1.5–19 ng)
- 12.5µL of PCR Master Mix
- 2.5µL of forward primer (10 µM)
- 2.5µL of reverse primer (10 µM)
- 2.5µL of ultrapure water [86].
- Thermocycling Conditions:
- Initial Denaturation: 95°C for 5 minutes
- 35 cycles of:
- Denaturation: 95°C for 45 seconds
- Annealing: 58°C for 45 seconds
- Extension: 72°C for 45 seconds
- Final Extension: 72°C for 10 minutes [86].
- Product Analysis: Resolve PCR products on a 2% agarose gel. The wild-type allele produces a 262 bp fragment, while the Δ32 allele produces a smaller 230 bp fragment due to the deletion. Homozygous wild-type individuals show one band at 262 bp, heterozygous individuals show two bands (262 bp and 230 bp), and homozygous Δ32 individuals show one band at 230 bp [86].
CRISPR-Cas9 Genome Editing for CCR5 Ablation
Inspired by the natural resistance of Δ32 homozygotes, researchers have developed CRISPR-Cas9 protocols to disrupt the CCR5 gene in human cells, creating HIV-resistant cell populations [88] [32].
Detailed Protocol (based on Jurkat CD4+ cell line and primary CD4+ cells):
- sgRNA Design: Design a pair of single-guide RNAs (sgRNAs) targeting the flanking region of the CCR5-Δ32 mutation locus. For example:
- sgRNA1 and sgRNA2 targeting the antisense DNA strand, with Cas9 nuclease cutting between the third and fourth nucleotides upstream of the PAM site to create a deletion precisely mimicking the Δ32 mutation [88].
- Vector Construction: Clone the DNA sequences encoding sgRNA1 and sgRNA2 into a lentiviral vector such as LentiCRISPRv2 (which also expresses Cas9 and a puromycin resistance marker) [88].
- Lentiviral Production: Produce lentiviral particles containing the CRISPR construct.
- Cell Transduction: Infect target cells (e.g., Jurkat CD4+ cell line or isolated primary CD4+ cells) with the lentivirus.
- Selection and Culture: Treat cells with puromycin to select for successfully transduced cells and culture for several days (e.g., 12 days for primary CD4+ cells) [88].
- Validation of Editing:
- T7E1 Assay: PCR-amplify the targeted CCR5 region from genomic DNA of puromycin-resistant cells. Hybridize, denature, and reanneal the PCR products. Treat with T7 Endonuclease I, which cleaves mismatched DNA at the site of indels. Analyze cleavage products by gel electrophoresis [88].
- DNA Sequencing: Clone PCR products into a TA vector and sequence multiple clones to determine the precise spectrum of mutations and the proportion of CCR5-Δ32 alleles [88].
- Flow Cytometry: Confirm the loss of CCR5 protein expression on the cell surface using CCR5-specific antibodies [32].
Diagram 2: Workflow for creating CCR5-Δ32 homozygous cells using CRISPR-Cas9 genome editing.
Therapeutic Applications and Clinical Implications
The profound HIV resistance conferred by the CCR5-Δ32 homozygous state has directly inspired novel therapeutic strategies aimed at curing HIV/AIDS. The most successful application to date is allogeneic hematopoietic stem cell transplantation (HSCT) from CCR5-Δ32 homozygous donors to HIV-infected patients [13]. This approach was first demonstrated with the "Berlin patient," Timothy Brown, who received a stem cell transplant from a Δ32/Δ32 donor to treat acute myeloid leukemia and subsequently exhibited no detectable HIV rebound even after cessation of antiretroviral therapy, effectively achieving a cure [88].
However, the scarcity of naturally occurring CCR5-Δ32 homozygous donors—particularly outside of Caucasian populations—poses a significant clinical limitation [32] [87]. This challenge has spurred the development of gene editing approaches to create HIV-resistant cells in situ. Clinical trials (e.g., NCT03164135) have demonstrated the feasibility and safety of transplanting CRISPR/Cas9-mediated CCR5-knockout hematopoietic stem and progenitor cells (HSPCs) into patients with HIV and hematologic malignancies [32] [13]. These edited cells successfully engrafted and persisted for over 19 months, representing a major milestone in gene therapy for HIV [32].
To address the potential for viral escape via CXCR4-tropic (X4) strains, multiplex gene editing strategies are being explored. These involve simultaneous knockout of CCR5 and CXCR4, or the combination of CCR5 editing with the expression of additional anti-HIV transgenes, such as the C46 fusion inhibitor, to achieve broad-spectrum resistance against both R5- and X4-tropic HIV-1 [32] [13]. The integration of gene-edited, HIV-resistant cells with immunotherapy approaches, such as HIV-specific CAR-T cells, represents the cutting edge of research, aiming to create a synergistic effect that both prevents infection of target cells and enhances clearance of the latent viral reservoir [13].
The CCR5Δ32 mutation, a 32-base-pair deletion in the CC chemokine receptor 5 (CCR5) gene, represents a critical natural resistance mechanism against HIV-1 infection. This whitepaper synthesizes evidence from global population studies and molecular investigations, highlighting that homozygous carriers of this mutation are substantially resistant to R5-tropic HIV-1 virus acquisition. The protective effect is attributed to the disruption of CCR5 surface expression, preventing viral entry into host immune cells. We detail the experimental protocols for genotyping this mutation, summarize its global distribution, and explore its implications for therapeutic drug development, including CCR5-blocking agents and gene-editing approaches. Framed within the broader thesis of innate HIV resistance, this analysis provides a technical guide for researchers and drug development professionals aiming to leverage this natural genetic defense into clinical applications.
The CC chemokine receptor 5 (CCR5) is a G-protein coupled receptor (GPCR) predominantly expressed on the surface of immune cells such as T lymphocytes, macrophages, and dendritic cells [12] [2]. Its primary physiological role involves mediating immune cell migration towards sites of inflammation by binding chemokines like RANTES (CCL5), MIP-1α (CCL3), and MIP-1β (CCL4) [2]. Critically, CCR5 serves as the major co-receptor for HIV-1 entry into CD4+ T cells, particularly for macrophage-tropic (R5) viruses responsible for the vast majority of primary infections [12] [89]. The interaction between the viral envelope glycoprotein gp120, CD4, and CCR5 triggers fusion of the viral envelope with the host cell membrane, initiating infection [89].
The discovery that a 32-base pair deletion (CCR5Δ32) in the CCR5 gene confers strong resistance to HIV-1 infection provided a paradigm shift in understanding host-pathogen interactions [86] [89]. Individuals homozygous for this mutation (genotype Δ32/Δ32) produce a truncated, non-functional receptor that fails to reach the cell surface, thereby obstructing HIV-1 cellular entry [12] [90]. This natural resistance mechanism, observed in HIV-Exposed Seronegative (ESN) individuals, has established CCR5 as a premier target for therapeutic intervention against HIV-1.
Mechanistic Insights: How CCR5Δ32 Confers Resistance
The CCR5Δ32 mutation results in a frameshift and premature introduction of a stop codon, yielding a severely truncated protein that is not transported to the cell membrane [12]. The mechanistic basis of resistance operates at multiple levels:
- Loss of Co-receptor Function: In homozygous individuals, the absence of functional CCR5 on the surface of CD4+ T cells and macrophages effectively blocks the entry pathway for R5-tropic HIV-1 strains [12] [89]. This creates a biological barrier to initial infection, as demonstrated by the significant over-representation of the Δ32/Δ32 genotype in ESN cohorts compared to the general population [89].
- Gene Dosage Effect: Heterozygous carriers (CCR5/Δ32) exhibit reduced CCR5 surface density on their CD4+ T cells compared to wild-type individuals [91] [90]. While not sufficient to prevent infection, this moderate reduction in receptor availability can slow the kinetics of viral entry and dissemination, contributing to delayed disease progression in infected individuals [71] [91].
- Post-Entry Restrictions: Evidence suggests that resistance in some ESN individuals may extend beyond receptor blockade. Some studies report CD4 T cell resistance involving post-entry steps of viral replication, independent of coreceptor usage, indicating the potential involvement of additional intracellular restriction factors [92].
The following diagram illustrates the protective mechanism of the CCR5Δ32 mutation against HIV-1 entry.
Global Distribution and Population Genetics
The frequency of the CCR5Δ32 allele demonstrates profound global heterogeneity, with the highest prevalence observed in Northern European populations and gradients of decreasing frequency toward Asia and Africa [90] [86] [89]. This distribution suggests a historical selective pressure, possibly from pathogens like the plague or smallpox, in European populations.
The table below summarizes the allele frequency across diverse populations, illustrating this geographic disparity.
Table 1: Global Distribution of the CCR5Δ32 Allele
| Population / Region | Reported Allele Frequency | Key Observations | Source |
|---|---|---|---|
| Peruvian | 2.7% (heterozygous) | No homozygous individuals found in study; low frequency attributed to limited European ancestry. | [93] |
| Moroccan | 2.1% (in controls) | No significant association with HIV-1 susceptibility was found. | [90] |
| Angolan (Luanda) | 0% | The mutation was absent in the studied cohort (0/272 alleles). | [86] |
| Caucasian (General) | ~10% | Highest frequencies reported in Northern Europe (up to 16% in Finnish and Mordvinian). | [94] [89] |
| Mixed (Meta-Analysis) | Variable | Significant protective association was primarily detected in Caucasian populations. | [71] |
This heterogeneous distribution has critical implications for the public health utility of widespread CCR5 screening. For instance, a study in Peru concluded that the low frequency of CCR5Δ32, alongside the rare HLA-B*57:01 allele, suggests that routine genotyping before prescribing abacavir may not be cost-effective, highlighting the need for policies grounded in local epidemiological data [93].
Quantitative Evidence: Correlation with HIV-1 Outcomes
Numerous case-control and meta-analysis studies have quantified the effect of the CCR5Δ32 genotype on HIV-1 susceptibility and disease progression. The protective effect of homozygosity is profound, while heterozygosity confers a more modest benefit.
Table 2: CCR5Δ32 Genotype Association with HIV-1 Susceptibility and Progression
| Genotype | HIV-1 Susceptibility | Effect on Disease Progression | Proposed Mechanism |
|---|---|---|---|
| Δ32/Δ32 (Homozygous) | Strong resistance [71] [89]. Near-complete protection against R5-tropic HIV infection. | Not applicable (protected from infection). | Non-functional CCR5 receptor is not expressed on the cell surface, blocking viral entry [12] [90]. |
| CCR5/Δ32 (Heterozygous) | Mildly increased or neutral susceptibility per meta-analysis [71]. | Slower progression to AIDS [71] [94] [90]. Delayed CD4+ T cell decline. | Reduced CCR5 cell surface expression on CD4+ T cells [91]. |
| CCR5/CCR5 (Wild-type) | Standard susceptibility. | Typical rate of disease progression. | Normal levels of functional CCR5 coreceptor. |
A 2018 meta-analysis of 24 case-control studies provided strong pooled quantitative evidence, confirming that the delta32 homozygous genotype significantly reduces HIV-1 infection susceptibility (OR=0.25, 95%CI=0.09-0.68), particularly when using exposed uninfected (EU) individuals as controls [71]. Furthermore, promoter polymorphisms in the CCR5 gene that regulate its expression levels have also been correlated with disease progression rates, independent of the Δ32 mutation [91].
Experimental Methods for Genotyping and Functional Analysis
CCR5Δ32 Genotyping by Endpoint PCR
This is the most common method for identifying the CCR5Δ32 mutation.
- Principle: A region of the CCR5 gene encompassing the 32-bp deletion is amplified by PCR. The amplicon size differs between the wild-type and mutant alleles, allowing for separation and visualization by agarose gel electrophoresis [93] [90].
- Detailed Protocol:
- DNA Extraction: Genomic DNA is isolated from peripheral blood mononuclear cells (PBMCs) or dried blood spots (DBS) using commercial kits (e.g., NucleoSpin kit, QIAamp DNA Mini Kit) [93] [86].
- PCR Amplification:
- Primers: Forward: 5′-ACCAGATCTCTCAAAAAGAAGGTCT-3′; Reverse: 5′-CATGATGGTGAAGATAAGCCTCCACA-3′ [93]. These primers flank the deletion site.
- Reaction Mix: Typically includes ~100 ng of genomic DNA, standard PCR buffer, MgCl₂ (e.g., 2.5 mM), dNTPs (e.g., 0.6 mM), primers (e.g., 0.2 µM each), and a thermostable DNA polymerase (e.g., Velocity DNA polymerase) [93].
- Cycling Conditions: Initial denaturation at 98°C for 30 sec; 35 cycles of 98°C for 30 sec (denaturation), 60°C for 30 sec (annealing), 72°C for 15 sec (extension); final extension at 72°C for 3 minutes [93].
- Product Analysis: The PCR products are resolved on a 2-3% agarose gel.
Real-Time PCR for High-Throughput Screening
Real-time PCR with specific probes (e.g., TaqMan) offers a faster, quantitative method suitable for processing large sample sizes without post-PCR manipulation [93].
Functional Validation of Resistance
Genotypic findings require functional validation to confirm resistance at the cellular level.
- Viral Entry/Pseudotype Assays: CD4+ T cells from genotyped individuals are challenged with replication-competent HIV-1 R5 strains or single-round pseudovirions expressing reporter genes (e.g., luciferase) and coated with R5-tropic envelope glycoproteins (e.g., BaL). Resistance is indicated by a significant reduction in infection efficiency compared to cells from wild-type donors [92].
- Flow Cytometric Analysis: Surface expression of CCR5 on CD4+ T cells is quantified using fluorochrome-conjugated anti-CCR5 antibodies. Homozygous individuals show a near-total lack of surface CCR5, while heterozygous individuals often show reduced expression levels [91] [92].
The following diagram outlines a comprehensive experimental workflow from sample collection to functional validation.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for CCR5 and HIV Resistance Research
| Reagent / Kit | Specific Example | Application and Function |
|---|---|---|
| DNA Extraction Kit | NucleoSpin Kit (Macherey-Nagel), QIAamp DNA Mini Kit (Qiagen) | Isolate high-quality genomic DNA from clinical samples (blood, DBS) for downstream PCR analysis. |
| PCR Reagents | GoTaq Green Master Mix (Promega), Velocity DNA Polymerase | Amplify the target CCR5 gene sequence with high fidelity and specificity for genotyping. |
| Gel Electrophoresis System | Standard agarose gel setup, ethidium bromide or SYBR-safe DNA stain | Separate and visualize PCR amplicons by size to distinguish between wild-type and Δ32 alleles. |
| Real-Time PCR System | Kapa Probe Fast Master Mix, Specific primers & probes for CCR5Δ32 | Enable high-throughput, quantitative genotyping without the need for gel electrophoresis. |
| Flow Cytometry Antibodies | Fluorochrome-conjugated anti-CCR5, anti-CD4, anti-CD3 antibodies | Quantify CCR5 receptor density on the surface of specific immune cell subsets (e.g., CD4+ T cells). |
| Viral Constructs | HIV-1 NL4-3Δenv luciferase reporter pseudovirions (BaL Env, HxB2 Env, VSV-G) | Investigate viral entry efficiency and tropism in single-round infection assays using primary cells. |
Therapeutic Applications and Drug Development
The natural resistance conferred by CCR5Δ32 has directly inspired multiple therapeutic strategies aimed at mimicking this protective phenotype.
- CCR5 Blockade with Small Molecule Antagonists: Maraviroc is a prominent FDA-approved CCR5 antagonist that binds the receptor and allosterically inhibits HIV-1 gp120 engagement, effectively mimicking the resistance seen in heterozygous individuals [2]. It is used in combination therapy for patients with R5-tropic virus.
- Gene Editing for Cure Strategies: The cases of the "Berlin" and "London" patients, cured of HIV after receiving hematopoietic stem cell transplants from CCR5Δ32/Δ32 donors, provide proof-of-concept for CCR5 ablation as a curative strategy [2]. Current research focuses on using Zinc Finger Nucleases (ZFNs) and, more recently, CRISPR-Cas9 to disrupt the CCR5 locus in patients' own CD4+ T cells or hematopoietic stem cells to confer permanent resistance [2].
- Monoclonal Antibodies: Anti-CCR5 monoclonal antibodies (e.g., Leronlimab) are being investigated to block the co-receptor physically and may also mediate antibody-dependent cellular cytotoxicity (ADCC) against HIV-infected cells [2].
The CCR5Δ32 mutation stands as a definitive example of human genetics directly influencing infectious disease outcomes. Research on ESN individuals has unequivocally shown that the absence of a functional CCR5 receptor provides powerful, natural resistance to HIV-1 infection. This discovery has fundamentally advanced our understanding of viral entry and host-pathogen interaction.
Future research should focus on: 1) Elucidating the potential role of CCR5 promoter haplotypes and other modifier genes in fine-tuning HIV susceptibility and disease course [91]; 2) Addressing the challenge of viral tropism switching to CXCR4-using variants, which can circumvent CCR5-targeted therapies [12]; and 3) Advancing the safety and efficacy of gene-editing platforms to make CCR5 ablation a scalable and accessible therapeutic modality. The continued study of this natural resistance mechanism remains a cornerstone of the quest for novel anti-HIV strategies, including functional cures and preventative vaccines.
The quest to understand natural resistance to human immunodeficiency virus (HIV) has unveiled key host genetic factors that profoundly influence susceptibility to infection and disease progression. Among these, the C-C chemokine receptor type 5 delta 32 (CCR5Δ32) mutation and the Human Leukocyte Antigen B57:01 (HLA-B57:01) allele represent two of the most significant and well-characterized protective alleles [12] [14] [95]. While both confer a protective advantage, their biological mechanisms, clinical implications, and applications in drug development are fundamentally distinct. This whitepaper provides a comparative analysis of the CCR5Δ32 and HLA-B*57:01 alleles, delineating their modes of action, population genetics, and translational impact within the broader context of HIV research and therapeutic development. Understanding these differences is crucial for researchers and drug development professionals aiming to leverage natural immunity for novel interventions, from small-molecule drugs and pharmacogenomics to advanced gene-editing therapies.
Mechanistic Basis of Protection
The protective effects of CCR5Δ32 and HLA-B*57:01 operate via entirely different arms of the immune system and stages of the viral life cycle. Table 1 provides a high-level comparison of their core characteristics.
Table 1: Fundamental Characteristics of CCR5Δ32 and HLA-B57:01*
| Feature | CCR5Δ32 | HLA-B*57:01 |
|---|---|---|
| Gene Product | CC Chemokine Receptor 5 (GPCR) | Major Histocompatibility Complex (MHC) Class I Molecule |
| Primary Mechanism | Innate Barrier: Prevents viral entry by disrupting coreceptor function | Adaptive Immunity: Presents viral epitopes to cytotoxic T-cells, driving effective immune control |
| Stage of HIV Life Cycle Targeted | Viral Entry | Viral replication and infected cell clearance (post-entry) |
| Phenotype in Homozygotes | Near-complete resistance to R5-tropic HIV infection [12] [10] [14] | Superior control of viral load, association with elite controller status [96] [95] |
| Phenotype in Heterozygotes | Delayed disease progression due to reduced CCR5 expression on cell surface [10] [14] | Better clinical outcome, though less pronounced than in homozygotes [95] |
CCR5Δ32: Disruption of Viral Entry
CCR5 is a G-protein-coupled receptor (GPCR) that normally serves as the primary coreceptor for R5-tropic HIV-1 strains, which are responsible for the vast majority of transmissions [12] [10]. The CCR5Δ32 variant is a 32-base-pair deletion in the coding region, resulting in a frameshift mutation and the production of a truncated, non-functional protein that is not expressed on the cell surface [10] [14].
- Homozygous State (Δ32/Δ32): Cells express no functional CCR5 on their surface, creating a virtual barrier to entry for R5-tropic viruses. This confers near-complete resistance to HIV infection [12] [10] [14].
- Heterozygous State (CCR5/Δ32): CCR5 expression is reduced on the cell surface. This does not prevent infection but can slow disease progression by limiting the number of available viral entry portals [10] [14].
The following diagram illustrates how the CCR5Δ32 mutation confers resistance at the point of viral entry.
HLA-B*57:01: Enhanced Adaptive Immune Control
In contrast, the HLA-B*57:01 allele confers protection through the adaptive immune system. HLA molecules are responsible for presenting peptide fragments (epitopes) from intracellular pathogens, such as viruses, on the cell surface for recognition by CD8+ cytotoxic T-lymphocytes (CTLs) [96] [95].
- Effective Antigen Presentation: HLA-B*57:01 is particularly effective at presenting conserved epitopes from the HIV Gag protein [95]. This triggers a potent and broad CTL response that efficiently recognizes and destroys HIV-infected cells early in the infection cycle.
- Immune Pressure and Control: The strong immune response mounted by individuals with HLA-B5701 places significant pressure on the virus. While this can lead to the selection of "escape mutants," the particular epitopes presented by HLA-B57:01 are often in conserved, functionally constrained regions of the virus. Mutations here can impair viral fitness, thereby helping the host to control viral replication, leading to lower viral loads and slower disease progression [95]. This allele is highly represented in cohorts of "elite controllers" [96] [95].
The following diagram outlines this adaptive immune mechanism.
Population Genetics and Global Distribution
The global distributions of CCR5Δ32 and HLA-B*57:01 are marked by significant geographic and ethnic variation, reflecting different evolutionary histories and selective pressures. Table 2 summarizes key epidemiological data.
Table 2: Comparative Epidemiology and Population Genetics
| Characteristic | CCR5Δ32 | HLA-B*57:01 |
|---|---|---|
| Global Distribution | High frequency in Northern Europe; low/absent in African, Asian, & indigenous American populations [10] [14]. | Found worldwide with varying frequencies; generally higher in some European and Asian populations [96]. |
| Example Allele Frequencies | - Peru: ~2.7% heterozygous, 0% homozygous [16]- Brazil: ~4-5% (general), up to 9% in Southern region [10]- Northern Europe: ~10-16% [10] [14] | - Peru: Very low frequency [16]- Turkey: 1.1-4.8% in HIV-1 infected individuals [96]- Brazil: National frequency of HLA-B*57 ~2.8% [95] |
| Implications | Resistance is highly dependent on HIV tropism (ineffective against X4-tropic virus) [9]. | Protective effect is dependent on the HIV-1 subtype and the specific epitopes present in circulating strains [95]. |
Translational Applications and Clinical Protocols
The distinct biological mechanisms of CCR5Δ32 and HLA-B*57:01 have led to divergent applications in clinical medicine and drug development.
CCR5-Based Strategies: From Small Molecules to Gene Editing
The CCR5 protein is a direct target for therapeutic intervention.
- Small Molecule Antagonists: The drug Maraviroc is a CCR5 allosteric antagonist approved for clinical use. It mimics the protective effect of CCR5Δ32 by binding to the receptor and preventing its interaction with HIV, thereby blocking viral entry [10].
- Gene Editing Therapies: The functional cure of HIV in the "Berlin" and "London" patients following transplantation with CCR5Δ32/Δ32 hematopoietic stem cells (HSCs) provided proof-of-concept for CCR5 disruption as a curative strategy [9] [3]. Current research focuses on using CRISPR-Cas9 and other nuclease platforms (e.g., ZFNs, TALENs) to knock out the CCR5 gene in autologous HSCs or T-cells for transplantation, creating a protected immune system resistant to HIV infection [9] [3]. A key challenge is combating potential viral escape via tropism switching to CXCR4 (X4-tropic viruses) [9].
HLA-B*57:01 and Pharmacogenomics: Preventing Hypersensitivity
The primary clinical application of HLA-B*57:01 is in pharmacogenomics.
- Abacavir Hypersensitivity Reaction (ABC-HSR): The reverse transcriptase inhibitor abacavir causes a potentially fatal hypersensitivity reaction in carriers of the HLA-B*57:01 allele [16] [96] [97].
- Pre-emptive Screening: Clinical guidelines now mandate HLA-B*57:01 screening prior to abacavir initiation. Patients who test positive are prescribed an alternative regimen, which has virtually eliminated ABC-HSR in clinical practice [16] [96]. This represents a seminal success story for personalized medicine.
Experimental Protocols for Genotyping
Accurate genotyping is fundamental for both research and clinical application of these alleles. Below are detailed protocols derived from the cited literature.
Principle: Amplification of the CCR5 gene region containing the Δ32 deletion, resulting in size-fractionated PCR products distinguishable by agarose gel electrophoresis.
Reagents and Workflow:
- Primers:
- CCR5 DELTA1: 5'-ACCAGATCTCTCAAAAAGAAGGTCT-3'
- CCR5 DELTA2: 5'-CATGATGGTGAAGATAAGCCTCCACA-3'
- PCR Reaction Mix:
- 0.2 µM of each primer
- 0.04 U of Velocity DNA polymerase
- 2.5 mM Mg²⁺
- 0.6 mM dNTP mixture
- Genomic DNA (10–100 ng)
- Final volume: 25 µl
- Thermal Cycling Conditions:
- 98°C for 30 s (initial denaturation)
- 35 cycles of:
- 98°C for 30 s (denaturation)
- 60°C for 30 s (annealing)
- 72°C for 15 s (extension)
- 72°C for 3 min (final extension)
- Product Analysis:
- CCR5/CCR5 (Wild-type): Single band at 225 bp
- CCR5/Δ32 (Heterozygous): Two bands at 225 bp and 193 bp
- Δ32/Δ32 (Homozygous): Single band at 193 bp
Principle: Allele-specific amplification and detection using fluorescent probes and real-time PCR.
Reagents and Workflow:
- Primers and Probes:
- Forward Primer: B5071-T1F (AGGGTCTCACATCATCCAGGT)
- Reverse Primer: B5701-T3R (CGTTCAGGGCGATGTAATCCT)
- Probe: B5701-P2 (6FAM-CGCGGGCATGACCAGTC-MGBNFQ)
- PCR Reaction Mix:
- 1x Kapa Probe Fast Master Mix
- 0.3 μmol/l of each primer
- 0.2 μmol/l of probe
- 10–60 ng/μl genomic DNA
- Final volume: 10 μl
- Thermal Cycling and Detection (Real-time PCR):
- 95°C for 5 min
- 45 cycles of:
- 95°C for 15 s
- 68°C for 30 s (fluorescence acquisition)
- Result Interpretation:
- Samples with a Ct (cycle threshold) value < 30 for both the HLA-B*57:01 assay and an internal control (e.g., alpha-actin) are considered positive.
The Scientist's Toolkit: Key Research Reagents
The following table catalogues essential reagents and their applications in HIV resistance research focused on CCR5 and HLA.
Table 3: Key Research Reagents for HIV Resistance Research
| Reagent / Assay | Function / Specificity | Primary Application |
|---|---|---|
| CCR5Δ32 Endpoint PCR Primers [16] | Flank the 32-bp deletion for size-based genotyping. | Determination of CCR5Δ32 genotype (wild-type, heterozygous, homozygous). |
| HLA-B*57:01 Real-time PCR Probes [16] [97] | Allele-specific oligonucleotide for high-fidelity detection. | Pharmacogenomic screening for abacavir hypersensitivity risk. |
| CRISPR-Cas9 System [9] [3] | RNA-guided nuclease for precise genome editing. | Knockout of CCR5 gene in hematopoietic stem cells (HSPCs) and T-cells. |
| TZM-bl Cells [9] | Engineered cell line expressing CD4, CCR5, and CXCR4, with a Tat-inducible reporter. | In vitro neutralization assays to quantify HIV-1 entry inhibition. |
| Broadly Neutralizing Antibodies (bNAbs)(e.g., 10-1074, PGDM1400) [9] | Target conserved epitopes on the HIV envelope glycoprotein. | Co-expression with CCR5 KO to provide multi-layered HIV inhibition; study of humoral immunity. |
The comparative analysis of CCR5Δ32 and HLA-B57:01 underscores a fundamental paradigm in host-pathogen interactions: distinct genetic pathways can converge on a protective phenotype against HIV. CCR5Δ32 operates as a gatekeeper, providing innate, entry-level resistance that can be therapeutically mimicked with drugs or engineered via gene editing. In contrast, HLA-B57:01 functions as an orchestrator of adaptive immunity, enabling superior recognition and clearance of infected cells, with its primary clinical impact being in pharmacogenomics to prevent drug toxicity.
The future of HIV research and therapy lies in integrating these insights. For instance, next-generation curative strategies are exploring the combination of CCR5-knockout HSPCs with the engineered secretion of broadly neutralizing antibodies to create a multi-layered, synergistic defense system [9]. Similarly, understanding the interplay between CTL responses driven by specific HLA alleles and viral escape mechanisms is critical for T-cell vaccine design. For researchers and drug developers, this comparative framework not only clarifies past successes but also illuminates the path toward the next generation of sophisticated, personalized interventions for HIV and beyond.
Viremic non-progressors (VNPs) represent an exceptionally rare phenotype among people living with HIV-1, characterized by the remarkable ability to maintain normal CD4+ T-cell counts and immune homeostasis despite persistent high-level viral replication. This comprehensive review synthesizes recent findings from multi-optic studies investigating the protective mechanisms in VNPs, with particular emphasis on the role of the CCR5Δ32 mutation and its associated pathways. We examine how genetic predisposition, coupled with modulated immune responses, creates an immunovirological equilibrium that mimics the natural SIV hosts of primates. The insights derived from VNPs provide a crucial framework for developing novel therapeutic strategies aimed at achieving functional HIV cure and immune restoration.
Viremic non-progressors (VNPs) constitute an extremely rare subset of individuals with HIV-1, estimated to represent merely 0.1% of the infected population [28]. These individuals maintain preserved CD4+ T-cell counts and normal immune function despite sustaining high viral load levels, typically exceeding 10,000 copies/mL, without antiretroviral therapy [98]. This unique phenotype challenges the conventional understanding of HIV pathogenesis and provides an invaluable natural model to study host-virus interactions and protective immunity. The VNP phenotype is characterized by three cardinal features: (1) persistent viremia without treatment, (2) stability of CD4+ T-lymphocyte counts within normal range, and (3) absence of clinical disease progression over extended periods [28] [99]. Recent comprehensive studies utilizing single-cell and multi-optic approaches have begun to unravel the complex interplay of viral, genetic, transcriptomic, and metabolomic factors that underlie this exceptional non-progressive state [99].
The CCR5Δ32 Mutation: A Cornerstone of HIV Resistance
Genetic Basis and Protective Mechanisms
The CCR5Δ32 mutation, a 32-base-pair deletion in the CC chemokine receptor 5 (CCR5) gene, represents one of the most significant genetic factors conferring resistance to HIV infection and disease progression. The CCR5 receptor serves as the principal co-receptor for R5-tropic HIV strains, which dominate during early and chronic phases of infection [3]. The Δ32 deletion results in a frameshift mutation and premature translational termination, producing a truncated, non-functional receptor that cannot be embedded in the cell membrane [4].
Table 1: Protective Effects of CCR5Δ32 Genotypes Against HIV-1 Infection
| Genotype | Receptor Expression | HIV Susceptibility | Clinical Implications |
|---|---|---|---|
| Wild-type homozygote (CCR5/CCR5) | Normal | Full susceptibility | Standard HIV progression |
| Heterozygote (CCR5/Δ32) | Reduced (~50% lower) | Partial resistance [71] | Milder symptoms; VNP phenotype association [28] |
| Homozygote (Δ32/Δ32) | Absent or minimal | Near-complete resistance [71] [4] | High-level protection against R5-tropic HIV infection |
The protective effect follows a gene-dosage pattern, where heterozygous individuals exhibit intermediate protection while homozygous individuals demonstrate near-complete resistance to R5-tropic HIV strains [71] [4]. In VNPs, the heterozygous CCR5Δ32 mutation is frequently observed and associated with lower CCR5 expression levels on target cells, resulting in reduced cellular infection and viral containment [28] [99].
Population Genetics and Evolutionary Perspectives
The CCR5Δ32 allele demonstrates a distinctive geographical distribution, with the highest frequencies observed in European populations (approximately 10% allele frequency, 1% homozygosity) and a pronounced north-to-south gradient [4]. Multiple hypotheses have been proposed to explain this distribution pattern, including selection by historical pandemics such as the bubonic plague (Yersinia pestis) and smallpox (Variola major) [4]. However, recent genetic evidence suggests the allele likely originated more than 5,000 years ago, predating these historical events, and may have evolved under neutral selection rather than strong positive selection [100].
Comprehensive Immune Mechanisms in Viremic Non-Progressors
Attenuated Immune Activation and Interferon Responses
VNPs exhibit a distinctive immune profile characterized by reduced chronic immune activation, a hallmark of standard HIV progression. This is evidenced by lower levels of activation markers on cytotoxic lymphocytes and reduced bystander CD4+ T-cell apoptosis [99]. Furthermore, VNPs demonstrate an attenuated interferon (IFN) response, which potentially protects them from the chronic inflammation that typically drives immunopathology in progressive HIV infection [28] [99]. This modulated IFN signature represents a critical adaptation that balances antiviral defense with avoidance of excessive inflammation.
Gut Mucosal Integrity and Metabolic Homeostasis
Preservation of intestinal mucosal integrity represents another pivotal factor in the VNP phenotype. VNPs show reduced plasma levels of zonulin, a marker of intestinal permeability, indicating better preservation of the gut mucosal barrier [28] [99]. This intestinal stability potentially limits microbial translocation and subsequent systemic immune activation. Additionally, VNPs maintain an unaltered tryptophan metabolic profile, suggesting preserved immunometabolic homeostasis that may contribute to their superior immune preservation compared to typical progressors [99].
Table 2: Key Immunological Features of Viremic Non-Progressors Versus Typical Progressors
| Immunological Parameter | Viremic Non-Progressors | Typical Progressors |
|---|---|---|
| CD4+ T-cell count | Preserved (>500 cells/μL) | Progressive decline |
| Immune activation | Reduced | Chronic elevation |
| Interferon response | Attenuated | Sustained and heightened |
| Gut mucosal integrity | Preserved (low zonulin) | Compromised (high zonulin) |
| Tryptophan metabolism | Unaltered | Disrupted |
| Bystander CD4+ apoptosis | Reduced | Increased |
Parallels with Natural SIV Hosts
The immunovirological equilibrium observed in human VNPs bears remarkable similarity to that of natural simian immunodeficiency virus (SIV) hosts, such as African green monkeys and sooty mangabeys [28] [98]. These non-human primates experience high-level SIV replication without developing immunodeficiency, attributed to a long-standing virus-host adaptation over thousands of years of co-evolution [28]. Shared characteristics between VNPs and natural SIV hosts include:
- Reduced immune activation despite persistent viremia
- Preserved CD4+ T-cell homeostasis and function
- Limited microbial translocation and maintained gut barrier integrity
- Adapted interferon responses that avoid excessive inflammation
These parallels suggest conserved mechanisms of disease tolerance across species and provide compelling evidence that non-pathogenic lentiviral infection is an achievable biological state [98].
Experimental Approaches and Methodologies
Comprehensive Multi-Omic Profiling
Recent investigations of VNPs have employed sophisticated single-cell and multi-omic approaches to comprehensively characterize the viral, genomic, transcriptomic, and metabolomic factors driving this rare phenotype [99]. The experimental workflow typically integrates:
- High-throughput CCR5 genotyping to identify Δ32 heterozygosity
- Flow cytometric analysis of CCR5 receptor density on CD4+ T-cells
- Single-cell RNA sequencing to evaluate transcriptional profiles and immune cell subsets
- Plasma proteomic profiling to quantify inflammatory markers and zonulin
- Metabolomic analysis to assess tryptophan pathway metabolites
- Viral reservoir quantification using advanced molecular techniques
Research Reagent Solutions
Table 3: Essential Research Reagents for VNP Mechanism Investigation
| Research Reagent | Application | Function in VNP Research |
|---|---|---|
| CCR5Δ32 genotyping assays | Genetic screening | Identify protective CCR5 variants in study cohorts |
| Anti-CCR5 monoclonal antibodies | Flow cytometry | Quantify CCR5 receptor density on immune cells |
| Zonulin ELISA kits | Protein quantification | Assess intestinal permeability and gut barrier integrity |
| IFN-γ ELISpot kits | Immune function assays | Measure virus-specific T-cell responses |
| Single-cell RNA sequencing kits | Transcriptomic profiling | Characterize immune cell populations and activation states |
| Metabolomic profiling panels | Metabolite quantification | Evaluate tryptophan pathway and immunometabolic status |
Therapeutic Implications and Future Directions
CCR5-Targeted Interventions
The mechanistic insights from VNP studies have directly informed the development of CCR5-targeted therapeutic strategies. These include:
- CCR5 gene editing using CRISPR/Cas9, ZFNs, or TALENs to disrupt the CCR5 locus in hematopoietic stem cells or T-cells [3]
- CCR5 antagonists such as maraviroc to pharmacologically block the receptor
- Combination approaches targeting both CCR5 and CXCR4 coreceptors to prevent viral tropism switching [3]
Immune-Modulatory Strategies
Beyond coreceptor targeting, VNP research supports the development of interventions aimed at reconstituting their protective immune environment:
- Modulation of interferon responses to reduce chronic inflammation
- Preservation of gut barrier integrity to limit microbial translocation
- Metabolic interventions targeting tryptophan pathways to restore immunometabolic balance
Mechanisms of HIV Pathogenesis Resistance in Viremic Non-Progressors
Viremic non-progressors represent a unique natural model of HIV pathogenesis resistance, with the CCR5Δ32 mutation serving as a cornerstone genetic element within a broader protective framework. The integrated mechanisms—including reduced CCR5 expression, attenuated immune activation, preserved gut integrity, and adapted interferon responses—collectively establish an immunovirological equilibrium that prevents disease progression despite ongoing viral replication. The insights gleaned from VNP studies continue to illuminate novel therapeutic targets and strategies aimed at replicating this protective state in all people living with HIV, ultimately moving closer to achieving functional cure and complete immune restoration.
Lessons from In Vitro and Animal Model Studies
The CCR5Δ32 mutation, a 32-base-pair deletion in the CC chemokine receptor 5 (CCR5) gene, represents a critical natural resistance mechanism against human immunodeficiency virus (HIV) infection. This whitepaper synthesizes findings from in vitro and animal model studies that elucidate the molecular basis by which this mutation confers protection, exploring its broader implications for therapeutic development. Research demonstrates that the homozygous CCR5Δ32/Δ32 genotype prevents cell surface expression of the CCR5 co-receptor, effectively blocking entry of R5-tropic HIV strains. Recent advances in gene editing technologies, particularly CRISPR/Cas9, have enabled the recapitulation of this protective phenotype in human hematopoietic stem cells and animal models, providing a robust platform for evaluating curative strategies. This guide details the experimental protocols, quantitative findings, and key reagents essential for advancing this research, framing these insights within the ongoing pursuit of an HIV cure.
The CCR5 co-receptor, expressed on macrophages, monocytes, and T-cells, serves as the primary portal for HIV-1 entry during initial infection and early disease stages [71] [14]. A natural 32-base-pair deletion in the CCR5 gene (CCR5Δ32) results in a frameshift mutation and premature termination codon, producing a truncated, non-functional protein that fails to localize to the cell surface [14] [101]. Epidemiologic studies reveal that approximately 10% of Northern European populations are heterozygous for CCR5Δ32, while about 1% carry the homozygous genotype conferring strong resistance to HIV infection [101]. This protective effect was dramatically validated in the "Berlin," "London," and "Düsseldorf" patients—HIV-positive individuals who received allogeneic hematopoietic stem cell transplants from CCR5Δ32/Δ32 donors and achieved long-term viral remission, effectively curing their HIV [3] [26]. These clinical breakthroughs established CCR5 disruption as a validated therapeutic paradigm, spurring extensive research to replicate this protection through gene-based interventions.
Table 1: Protective Effect of CCR5Δ32 Genotype Against HIV-1 Infection
| Genotype | Effect on CCR5 Expression | HIV-1 Susceptibility | Odds Ratio (95% CI)* |
|---|---|---|---|
| Wild-type (WT/WT) | Normal surface expression | Fully susceptible | Reference (1.00) |
| Heterozygous (WT/Δ32) | Reduced surface expression | Delayed AIDS progression | 1.16 (1.02-1.32) |
| Homozygous (Δ32/Δ32) | No surface expression | Highly resistant to R5-tropic HIV | 0.25 (0.09-0.68) |
Meta-analysis data comparing HIV-1 infection risk versus wild-type, using healthy controls [71].
Molecular Mechanisms of HIV Entry Blockade
CCR5 Structure and Viral Entry Dynamics
The CCR5 protein belongs to the seven-transmembrane (7TM) G-protein-coupled receptor (GPCR) superfamily, normally responding to chemokine ligands and mediating leukocyte migration to inflammatory sites [14]. For HIV entry, the viral envelope glycoprotein gp120 sequentially engages the CD4 receptor and then the CCR5 co-receptor on the host cell surface. This interaction triggers conformational changes that allow gp41 to mediate fusion between the viral and host cell membranes [61]. The essential binding site on CCR5 for HIV gp120 is located on the third extracellular element (second loop), known as the 2D7 epitope [61].
Structural Consequences of the Δ32 Mutation
The CCR5Δ32 mutation results in a deletion of 32 base pairs within the coding region, just before the 2D7 structural loop. This deletion causes a frameshift and introduction of a premature stop codon, preventing translation of the final three transmembrane domains and the C-terminal cytoplasmic tail [61] [14]. Consequently, the mutant protein cannot embed itself in the plasma membrane and remains trapped intracellularly, thereby removing the critical docking site required for HIV entry [14]. This mechanism provides a nearly insurmountable barrier to R5-tropic HIV strains, which constitute the majority of transmitted variants.
Diagram: HIV Entry Blockade by CCR5Δ32 Mutation
In Vitro Modeling of CCR5Δ32 Protection
Gene Editing in Hematopoietic Stem/Progenitor Cells (HSPCs)
Protocol Overview: Mobilized CD34+ hematopoietic stem/progenitor cells (HSPCs) from healthy donors are electroporated with CRISPR/Cas9 ribonucleoprotein (RNP) complexes targeting specific sequences within exon 3 of the CCR5 gene [50]. Following electroporation, cells are cultured in cytokine-supplemented media supporting stem cell viability and analyzed for editing efficiency 48 hours post-electroporation.
Key gRNA Design and Validation: A systematic screening pipeline identified optimal guide RNAs (gRNAs) for maximal CCR5 disruption:
- In silico prediction identified 123 potential gRNAs targeting the CCR5 open reading frame.
- Off-target exclusion removed gRNAs with potential binding to other genomic sites, particularly the homologous CCR2 gene.
- Primary HSPC screening evaluated editing efficiency, identifying four top-performing gRNAs (TB7, TB8, TB48, TB50) with >30% editing frequency.
- Dual-guide approach combining TB48 and TB50 gRNAs was developed to create a small deletion approximating the natural Δ32 mutation [50].
Table 2: CRISPR/Cas9 gRNA Performance in Primary Human Cells
| gRNA ID | Target Sequence | Editing Efficiency in HSPCs | Reduction in CCR5+ CD4+ T Cells | Off-Target Events |
|---|---|---|---|---|
| TB7 | CAGAATTGATACTGACTGTATGG | >30% | Moderate | None detected |
| TB8 | AGATGACTATCTTTAATGTCTGG | >30% | Moderate | One low-frequency site |
| TB48 | ACCAGATCTCAAAAAGAAGGTGG | >90% | High | None detected |
| TB50 | AACTGAAACAAAATTATGGGTGG | >90% | High | None detected |
| TB48+TB50 | Dual guide approach | 91-97% | Highest | None detected |
Data compiled from Nature Communications (2025) study [50].
T Cell Editing and Viral Challenge Assays
Methodology: Primary human CD4+ T cells or peripheral blood mononuclear cells (PBMCs) are electroporated with CRISPR/Cas9 RNP complexes. Edited cells are then activated and challenged with CCR5-tropic HIV (e.g., HIVJRCSF) at high multiplicities of infection. Infection frequency is quantified over 6-8 days via intracellular p24 staining or measurement of reverse transcriptase activity [50].
Key Findings: CCR5 editing with high-efficiency gRNAs (TB48, TB50, and TB48+TB50 dual guide) resulted in:
- 52-70% CCR5 gene editing in primary T cells
- Significant reduction in frequency of CCR5+ CD4+ T cells
- Markedly reduced HIV infection frequency sustained throughout assay
- In some donors, decay of HIV infection to mock-infected control levels by day 6 post-challenge [50]
The reduction in HIV infection directly correlated with the reduction in CCR5+ CD4+ T cells, establishing a clear causal relationship between CCR5 disruption and viral resistance.
Animal Model Studies for Therapeutic Validation
Humanized Mouse Models and HIV Challenge
Experimental Workflow: Human immune system (HIS) mice are generated by transplanting immunodeficient mice (e.g., NSG strains) with human CD34+ HSPCs—either unedited or CCR5-edited. Reconstitution of human immune cells is monitored over 12-16 weeks. Mice with confirmed engraftment are then challenged with CCR5-tropic HIV, and viral load is tracked via quantitative PCR of plasma [102] [50].
Protection Threshold Determination: Titration studies transplanting decreasing frequencies of CCR5-edited HSPCs demonstrated a critical threshold effect:
- >90% CCR5 editing: Robust protection against HIV infection
- 54%-26% CCR5 editing: Decreasing protective benefit
- <26% CCR5 editing: Negligible protection against viral challenge [50]
This finding highlights that high-frequency editing is essential for therapeutic efficacy, informing clinical translation strategies.
Diagram: HIS Mouse Model HIV Challenge Workflow
Reservoir Analysis in Cured Patients
Methodological Approach: Comprehensive virological and immunological characterization of patients who received CCR5Δ32/Δ32 HSCT provides critical validation. Techniques include:
- Droplet digital PCR (ddPCR): Ultrasensitive quantification of HIV DNA in peripheral T cell subsets and tissue-derived samples
- In situ hybridization (DNAscope/RNAscope): Spatial localization of HIV nucleic acids in tissue sections
- Quantitative viral outgrowth assay (QVOA): Assessment of replication-competent virus in PBMCs
- In vivo outgrowth assays: Humanized mouse models for amplifying latent virus from patient cells [26]
Key Findings: Analyses of a cured patient monitored for >9 years post-HSCT and 4 years after treatment interruption revealed:
- Sporadic traces of HIV DNA detected by ddPCR and in situ hybridization
- No replication-competent virus detected by repeated QVOA and in vivo outgrowth assays
- Waning HIV-specific humoral and cellular immune responses indicating lack of antigen persistence
- No viral rebound despite extensive monitoring, providing strong evidence for cure [26]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for CCR5Δ32 and HIV Resistance Research
| Reagent / Method | Specifications | Research Application |
|---|---|---|
| CRISPR/Cas9 System | SpCas9 protein with synthetic gRNAs (TB48, TB50 optimal) | High-efficiency CCR5 editing in HSPCs and T cells [50] |
| HSPC Source | Mobilized CD34+ cells from peripheral blood | Primary human hematopoietic stem cells for transplantation models [50] |
| HIV Challenge Virus | CCR5-tropic HIVJRCSF | In vitro and in vivo infection studies with relevant viral strain [50] |
| Droplet Digital PCR | CCR5Δ32-specific probes and primers | Absolute quantification of mutant allele frequency in mixed populations [101] |
| Humanized Mouse Models | NSG mice with human CD34+ HSPC reconstitution | In vivo assessment of HIV infection and protection [102] [50] |
| Flow Cytometry Antibodies | Anti-CCR5 (2D7 clone), anti-CD4, anti-CD3 | Monitoring CCR5 surface expression and immune cell phenotyping [50] |
| Viral Outgrowth Assays | QVOA and in vivo mouse amplification | Detecting replication-competent latent HIV reservoir [26] |
Research Challenges and Future Directions
Despite significant progress, several challenges remain in translating CCR5-targeted therapies to clinical practice. Off-target effects of gene editing, though minimal with optimized gRNAs, require comprehensive assessment using whole-genome sequencing [61]. Viral tropism switching from CCR5- to CXCR4-using strains represents a potential escape mechanism, prompting development of multi-target editing strategies that simultaneously disrupt CCR5, CXCR4, and integrated HIV proviral DNA [3]. Economic feasibility and global accessibility of advanced gene therapies present substantial implementation barriers that must be addressed through technological optimization and innovative payment models [3].
Future research directions include:
- Synergistic immunotherapies: Combining CCR5 editing with immune checkpoint inhibitors or HIV-specific CAR-T cells
- Personalized approaches: Accounting for viral subtypes, host immunity, and genetic background variability
- Base editing technologies: Achieving precise nucleotide conversions without double-strand breaks to enhance safety
- Curative autologous HSCT: Developing clinically scalable protocols for CRISPR-edited hematopoietic stem cell transplants [3] [50]
The integration of robust in vitro systems, predictive animal models, and sophisticated reservoir assays continues to accelerate the development of CCR5-targeted interventions, moving the field closer to a scalable cure for HIV infection.
Conclusion
The CCR5Δ32 mutation provides a powerful natural blueprint for HIV resistance, demonstrating that the disruption of a single host gene can confer profound protection. The foundational understanding of this mechanism has successfully catalyzed a new frontier in HIV therapy, driving the development of CCR5 antagonists and cutting-edge gene-editing strategies. However, the path to a universal cure requires overcoming significant hurdles, including viral tropism switching and ensuring the long-term safety of genetic interventions. Future research must focus on personalized multi-target approaches, improved delivery systems, and synergistic combinations with immunotherapy to achieve a functional cure. The continued study of this remarkable mutation, its population-specific distribution, and its broader immunological role will undoubtedly continue to illuminate novel pathways for conquering HIV and other infectious diseases.
References
- 1. HIV co-receptor CCR5: structure and interactions with ... [https://pubmed.ncbi.nlm.nih.gov/19519482/]
- 2. Targeting CCR5 as a Component of an HIV-1 Therapeutic ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2021.816515/full]
- 3. CCR5 gene editing and HIV immunotherapy - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12213807/]
- 4. The CCR5Δ32 allele as an HIV infection resistance marker [https://www.sciencedirect.com/science/article/pii/S2452014423001188?dgcid=rss_sd_all&]
- 5. Identifying CCR5 coreceptor populations permissive for HIV-1 ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-022-03243-8]
- 6. HIV-1 tropism testing and clinical management of CCR5 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3905003/]
- 7. HIV coreceptor tropism determination and mutational ... [https://www.nature.com/articles/srep21280]
- 8. HIV-1 Tropism Dynamics and Phylogenetic Analysis from ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0102857]
- 9. Multilayered HIV-1 resistance in HSPCs through CCR5 ... [https://www.nature.com/articles/s41467-025-58371-8]
- 10. Beyond HIV infection: Neglected and varied impacts of CCR5 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7260533/]
- 11. New mechanisms of CCR5-Δ32 carriers' advantage [https://www.sciencedirect.com/science/article/abs/pii/S1357272519300147]
- 12. CCR5: From Natural Resistance to a New Anti-HIV Strategy [https://pmc.ncbi.nlm.nih.gov/articles/PMC3185609/]
- 13. CCR5 gene editing and HIV immunotherapy [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1590690/full]
- 14. The CCR5Δ32 allele as an HIV infection resistance marker [https://www.sciencedirect.com/science/article/abs/pii/S2452014423001188]
- 15. CCR5-Δ32 biology, gene editing, and warnings for the future ... [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-020-00410-6]
- 16. Distribution of CCR5-Δ32 and HLA-B*57:01 alleles in HIV ... [https://www.nature.com/articles/s41439-025-00321-3]
- 17. The effect of the CCR5-delta32 deletion on global gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3234179/]
- 18. Unveiling Ancestral Threads: Exploring CCR5 Δ32 Mutation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11563905/]
- 19. The chemokine receptor CCR5: multi-faceted hook for HIV-1 [https://retrovirology.biomedcentral.com/articles/10.1186/s12977-024-00634-1]
- 20. CCR5Δ32 Protein Expression and Stability Are Critical for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1951285/]
- 21. CCR5 as a Coreceptor for Human Immunodeficiency Virus ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2022.835994/full]
- 22. Reduced Cell Surface Expression of CCR5 in CCR5Δ32 ... [https://www.sciencedirect.com/science/article/pii/S0021925820878582]
- 23. CCR5Δ32 in Brazil: Impacts of a European Genetic Variant ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2021.758358/full]
- 24. The coreceptor mutation CCR5Δ32 influences ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC56941/]
- 25. Homozygous and heterozygous CCR5-Delta32 genotypes are ... [https://pubmed.ncbi.nlm.nih.gov/11511825/]
- 26. In-depth virological and immunological characterization of ... [https://www.nature.com/articles/s41591-023-02213-x]
- 27. Clinical use of CCR5 inhibitors in HIV and beyond [https://translational-medicine.biomedcentral.com/articles/10.1186/1479-5876-9-S1-S9]
- 28. A pioneering study identifies protective mechanisms in ... [https://www.eatg.org/hiv-news/a-pioneering-study-identifies-protective-mechanisms-in-people-with-hiv-resistant-to-disease-progression/]
- 29. Neglected and varied impacts of CCR5 and CCR5Δ32 on ... [https://pubmed.ncbi.nlm.nih.gov/32479976/]
- 30. The Geographic Spread of the CCR5 Δ32 HIV-Resistance Allele [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.0030339]
- 31. The evolutionary history of the CCR5-Delta32 HIV ... [https://pubmed.ncbi.nlm.nih.gov/15715976/]
- 32. CRISPR/Cas9 genome editing of CCR5 combined with ... [https://www.nature.com/articles/s41598-024-61626-x]
- 33. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic ... [https://www.nature.com/articles/s41586-019-1027-4]
- 34. The next Berlin Patient: Another man cured of HIV after ... [https://www.eatg.org/hiv-news/the-next-berlin-patient-another-man-cured-of-hiv-after-stem-cell-transplant/]
- 35. Evidence for HIV-1 cure after CCR5Δ32/Δ32 allogeneic ... [https://pubmed.ncbi.nlm.nih.gov/32169158/]
- 36. Nat Med:令人期待!新研究报道了第三名HIV治愈患者 [https://news.bioon.com/article/5b7ce60488b0.html]
- 37. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7275870/]
- 38. 中国科学家完成首例基因编辑干细胞治疗艾滋病和白血病患者 [https://www.tsinghua.edu.cn/info/1182/50276.htm]
- 39. CCR5Δ32 mutation and HIV infection: basis for curative ... [https://pubmed.ncbi.nlm.nih.gov/26143158/]
- 40. Dating the origin of the CCR5-Delta32 AIDS-resistance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1377146/]
- 41. CCR5-Δ32 [https://en.wikipedia.org/wiki/CCR5-%CE%9432]
- 42. CCR5 receptor antagonists in preclinical to phase II clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5776690/]
- 43. Genetic Polymorphisms in the Open Reading Frame of ... [https://www.nature.com/articles/s41598-019-44136-z]
- 44. a novel small molecule CCR5 inhibitor active against R5- ... [https://www.nature.com/articles/s41598-019-41080-w]
- 45. Treatment with the CCR5 antagonist OB-002 reduces lung ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0316952]
- 46. Species selectivity of small-molecular antagonists for the ... [https://www.sciencedirect.com/science/article/abs/pii/S156757690700224X]
- 47. Innate resistance to HIV [https://en.wikipedia.org/wiki/Innate_resistance_to_HIV]
- 48. Closing the Door with CRISPR: Genome Editing of CCR5 and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9326690/]
- 49. CRISPR-Cas9-mediated gene disruption of HIV-1 co ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8847835/]
- 50. High frequency CCR5 editing in human hematopoietic ... [https://www.nature.com/articles/s41467-025-55873-3]
- 51. Comparing Gene Editing Platforms: CRISPR vs. Traditional ... [https://blog.crownbio.com/comparing-gene-editing-platforms-crispr-vs-traditional-methods]
- 52. The clinical applications of genome editing in HIV [https://www.sciencedirect.com/science/article/pii/S0006497120301312]
- 53. Opportunities for CAR-T Cell Immunotherapy in HIV Cure - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10057306/]
- 54. Hematopoietic stem cell transplantation for HIV cure - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4731181/]
- 55. Efficacy of Hematopoietic Stem Cell Transplantation with ... [https://www.bioscmed.com/index.php/bsm/article/view/1305]
- 56. Recent advances on anti-HIV chimeric antigen receptor-T-cell ... [https://pubmed.ncbi.nlm.nih.gov/38695148/]
- 57. HIV – CAR-T Therapies [https://www.seattlechildrens.org/research/centers-programs/science-industry-partnerships/partnership-opportunities/hiv-car-t/]
- 58. HIV/AIDS research has saved millions of lives globally and ... [https://www.fredhutch.org/en/news/releases/2025/10/hiv-aids-research-has-saved-millions-of-lives-globally.html]
- 59. Breaking Barriers to an HIV-1 Cure: Innovations in Gene ... [https://www.mdpi.com/2075-1729/15/2/276]
- 60. CCR5Δ32 mutation and HIV infection: basis for curative ... [https://www.sciencedirect.com/science/article/abs/pii/S1879625715000966]
- 61. CCR5-Δ32 biology, gene editing, and warnings for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7106751/]
- 62. Targeting CCR5 as a Component of an HIV-1 Therapeutic ... [https://pubmed.ncbi.nlm.nih.gov/35126374/]
- 63. Acute-phase proteins as biomarkers of inflammation in HIV ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1551775/full]
- 64. Children born with HIV have persistent inflammation ... [https://www.aidsmap.com/news/jul-2025/children-born-hiv-have-persistent-inflammation-markers-linked-heart-disease]
- 65. CCR5/CXCR4 Dual Antagonism for the Improvement of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6384722/]
- 66. HIV Resistant Mutation | Viruses101 [https://www.nature.com/scitable/blog/viruses101/hiv_resistant_mutation/]
- 67. How HIV changes its tropism: evolution and adaptation? - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2697388/]
- 68. Biological and Molecular Aspects of HIV-1 Coreceptor Usage [https://www.hiv.lanl.gov/content/sequence/HIV/REVIEWS/SCHUITEMAKER2002/Schuitemaker.html]
- 69. Transmission of highly virulent CXCR4 tropic HIV-1 through ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10571614/]
- 70. Next-generation sequencing of CCR5, CXCR4, and ... [https://www.nature.com/articles/s41598-025-11843-9]
- 71. The CCR5-Delta32 Genetic Polymorphism and HIV-1 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6227735/]
- 72. A pioneering study identifies protective mechanisms in ... [https://www.irsicaixa.es/en/pioneering-study-identifies-protective-mechanisms-people-hiv-resistant-disease-progression]
- 73. Genome editing of CXCR4 by CRISPR/cas9 confers cells ... [https://www.nature.com/articles/srep15577]
- 74. HIV cure: ending the HIV epidemic [https://viivhealthcare.com/ending-hiv/towards-a-hiv-cure/]
- 75. Gilead Presents New HIV Research Data at EACS 2025 [https://www.gilead.com/news/news-details/2025/gilead-presents-new-hiv-research-data-at-eacs-2025--driving-scientific-innovation-in-treatment-and-prevention]
- 76. The evolutionary history of the CCR5-Δ32 HIV-resistance ... [https://www.sciencedirect.com/science/article/pii/S1286457904003636]
- 77. The Geographic Spread of the CCR5 Δ32 HIV-Resistance Allele [https://pmc.ncbi.nlm.nih.gov/articles/PMC1255740/]
- 78. Inhibition of HIV-1 infection of primary CD4+ T-cells by gene of... editing [https://pubmed.ncbi.nlm.nih.gov/25854553/]
- 79. Delivery of CRISPR-Cas tools for in vivo genome editing ... [https://www.sciencedirect.com/science/article/pii/S016836592200027X]
- 80. CRISPR Tools for T cells: Targeting the Genome, Epigenome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12397615/]
- 81. Optimisation of a TALE nuclease targeting the HIV co ... [https://www.nature.com/articles/s41434-021-00271-9]
- 82. Systematic discovery of CRISPR-boosted CAR T cell ... [https://www.nature.com/articles/s41586-025-09507-9]
- 83. Regional variation in CCR5-Δ32 gene distribution among ... [https://www.nature.com/articles/6363884]
- 84. Influence of CCR5 and CCR2 Genetic Variants in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3848445/]
- 85. Global distribution of the CCR5 gene 32-basepair deletion [https://www.nature.com/articles/ng0597-100]
- 86. Distribution of CCR5-Delta32, CCR2-64I, and SDF1-3'A ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12102844/]
- 87. Unveiling ancestral threads: Exploring CCR5 ∆32 mutation ... [https://www.sciencedirect.com/science/article/pii/S156713482400131X]
- 88. Inducing CCR5Δ32/Δ32 Homozygotes in the Human Jurkat ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6005807/]
- 89. The Role of the Chemokine Receptor Gene CCR5 and Its ... [https://wwwnc.cdc.gov/eid/article/3/3/97-0302_article]
- 90. Investigation of CCR5-Δ32 (rs333) genetic polymorphism ... [https://www.sciencedirect.com/science/article/abs/pii/S2452014419300330]
- 91. CCR5 promoter activity correlates with HIV disease ... [https://www.nature.com/articles/s41598-017-00192-x]
- 92. Persistent resistance to HIV-1 infection in CD4 T cells from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1636660/]
- 93. Distribution of CCR5-Δ32 and HLA-B*57:01 alleles in HIV ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12381279/]
- 94. Molecular characterization of the CCR 5 gene in ... [https://www.sciencedirect.com/science/article/abs/pii/S1386653201002190]
- 95. The influence of HLA/HIV genetics on the occurrence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9392165/]
- 96. Relationship of HLA-B alleles on susceptibility to and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9996654/]
- 97. Low-cost simultaneous detection of CCR5-delta32 and ... [https://www.sciencedirect.com/science/article/abs/pii/S0166093415003006]
- 98. Viremic non-progression in HIV/SIV infection: A tied game ... [https://www.sciencedirect.com/science/article/pii/S266637912400692X]
- 99. Host genetic and immune factors drive evasion of HIV-1 ... [https://pubmed.ncbi.nlm.nih.gov/39413785/]
- 100. The Case for Selection at CCR5-Δ32 - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC1275522/]
- 101. Detection of CCR5Δ32 Mutant Alleles in Heterogeneous ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2022.805931/full]
- 102. Protocol to identify the sex and CCR5 genotype of the human ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11889956/]