Harnessing DNA Repair: How CRISPR-Cas9 Double-Strand Break Mechanisms Are Revolutionizing Genetic Disease Therapy
This article provides a comprehensive analysis of the cellular DNA repair mechanisms that determine the success of CRISPR-Cas9 gene editing for genetic diseases.
Harnessing DNA Repair: How CRISPR-Cas9 Double-Strand Break Mechanisms Are Revolutionizing Genetic Disease Therapy
Abstract
This article provides a comprehensive analysis of the cellular DNA repair mechanisms that determine the success of CRISPR-Cas9 gene editing for genetic diseases. Tailored for researchers, scientists, and drug development professionals, it explores the foundational biology of double-strand break repair pathways, examines current methodological applications in clinical trials, addresses critical troubleshooting and optimization challenges, and validates approaches through comparative analysis of editing outcomes. The content synthesizes the latest 2025 clinical updates with fundamental research to offer a practical framework for advancing therapeutic genome editing programs, highlighting both emerging opportunities and persistent hurdles in the field.
The Cellular Machinery: Understanding CRISPR-Cas9 Induced DNA Break Repair Pathways
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) system represents one of the most transformative breakthroughs in modern molecular biology. Originally discovered as an adaptive immune system in prokaryotes, CRISPR-Cas9 has been repurposed into a highly versatile, precise, and programmable genome editing tool [1] [2]. This technology has fundamentally revolutionized genetic research and therapeutic development by enabling researchers to make targeted modifications to DNA sequences in living cells with unprecedented ease and accuracy. The system's core function stems from its ability to create targeted double-strand breaks (DSBs) in DNA, which harnesses the cell's endogenous repair machinery to introduce specific genetic changes [1] [3]. For researchers and drug development professionals, understanding the precise components, mechanisms, and applications of CRISPR-Cas9 is crucial for developing novel therapeutic strategies for genetic diseases. This technical guide examines the journey of CRISPR-Cas9 from its bacterial origins to its current status as the premier "genetic scissors" driving innovation in genetic disease research.
Biological Origins: The CRISPR-Cas System as an Adaptive Immune System in Prokaryotes
The CRISPR-Cas system functions as a sophisticated adaptive immune system in bacteria and archaea, providing sequence-specific protection against invading viruses and plasmids [4] [2]. This defense system maintains a genetic memory of previous infections by incorporating short fragments of viral DNA (known as spacers) into the host's CRISPR array, which is composed of short, partially palindromic repeats separated by spacer sequences [1] [4]. The biological mechanism unfolds in three distinct stages:
- Adaptation (Spacer Acquisition): During initial viral infection, the Cas protein complex recognizes and processes protospacer sequences from the invading DNA, inserting them as new spacers into the CRISPR genomic locus [1] [4]. This creates a heritable genetic record of the infection.
- crRNA Synthesis (Expression): Following transcription of the CRISPR array into a long precursor CRISPR RNA (pre-crRNA), Cas proteins process this molecule into mature CRISPR RNAs (crRNAs), each containing a single spacer sequence [1] [3].
- Target Interference: The mature crRNAs guide Cas nucleases to complementary sequences in invading nucleic acids, leading to their specific cleavage and degradation [1] [4].
This sophisticated biological defense mechanism laid the foundation for repurposing CRISPR-Cas systems, particularly the type II system from Streptococcus pyogenes, into a powerful genome engineering platform.
Core Components of the CRISPR-Cas9 System
The repurposed CRISPR-Cas9 genome editing system requires two fundamental components: the Cas9 nuclease and a guide RNA (gRNA) [1] [5] [6]. The minimalistic nature of these components—requiring only reprogramming of the gRNA to target different genomic loci—has democratized genome editing across biological disciplines.
The Cas9 Nuclease: Architecture and Functional Domains
The Cas9 protein is a large (1368 amino acids in S. pyogenes) multidomain DNA endonuclease that functions as the catalytic engine of the system [1] [3]. Its structure is organized into two primary lobes:
- Recognition Lobe (REC Lobe): Comprising REC1, REC2, and REC3 domains, this lobe is primarily responsible for binding the guide RNA [1] [3].
- Nuclease Lobe (NUC Lobe): This lobe contains three critical domains:
The Cas9 protein remains in an inactive conformation until it forms a complex with the guide RNA and encounters a target sequence with the appropriate PAM, at which point it undergoes structural rearrangement to activate its nuclease domains [3].
Guide RNA: Design and Function
The guide RNA is a synthetic hybrid molecule that combines the functions of two natural RNA components:
- crRNA (CRISPR RNA): A 18-20 nucleotide sequence that specifies DNA target recognition through Watson-Crick base pairing with the complementary DNA strand [1] [3].
- tracrRNA (trans-activating CRISPR RNA): A structural scaffold that facilitates Cas9 nuclease binding and complex formation [1] [3].
For experimental applications, these are typically combined into a single guide RNA (sgRNA) to simplify delivery and processing [1] [3] [6]. The sgRNA can be produced either in situ (via transcription from plasmid or viral vectors) or ex situ (through in vitro transcription or chemical synthesis) [3]. Chemically synthesized sgRNAs can incorporate modifications to enhance nuclease resistance, improve target recognition efficiency, and reduce potential immune responses [3].
The Protospacer Adjacent Motif (PAM): A Critical Recognition Element
The PAM is a short (typically 2-6 base pairs) conserved DNA sequence immediately adjacent to the target site that is essential for self versus non-self discrimination in the native bacterial immune system [1] [3] [7]. For the most commonly used Cas9 from S. pyogenes, the PAM sequence is 5'-NGG-3' (where "N" can be any nucleotide) [1] [7]. The PAM-interacting domain of Cas9 recognizes this sequence, triggering local DNA melting and enabling the guide RNA to initiate strand invasion and R-loop formation [3].
Table 1: Core Components of the CRISPR-Cas9 System
| Component | Structure/Composition | Function | Key Features |
|---|---|---|---|
| Cas9 Nuclease | 1368 amino acids; REC lobe (RNA binding) and NUC lobe (DNA cleavage) | DNA endonuclease that creates double-strand breaks | Contains HNH (target strand cleavage) and RuvC (non-target strand cleavage) domains |
| Guide RNA (gRNA) | Single guide RNA (sgRNA) combining crRNA and tracrRNA | Targets Cas9 to specific DNA sequences | 20-nucleotide guide sequence complementary to target DNA; scaffold region binds Cas9 |
| PAM | Short DNA sequence (5'-NGG-3' for SpCas9) | Recognition motif for Cas9 binding | Essential for self vs. non-self discrimination; varies between Cas9 orthologs |
Molecular Mechanism: From Target Recognition to DNA Cleavage
The CRISPR-Cas9 mechanism involves a precisely coordinated sequence of molecular events that culminate in site-specific DNA cleavage. This process can be divided into three distinct stages: recognition, cleavage, and repair [1].
Target Recognition and R-loop Formation
The Cas9-gRNA complex scans the genome through 3D and 1D diffusion, searching for PAM sequences [3]. Upon PAM recognition, the Cas9 protein triggers local DNA melting, allowing the seed region (8-12 nucleotides adjacent to the PAM) of the gRNA to initiate base pairing with the target DNA [3]. If complementarity is sufficient, complete R-loop formation occurs, whereby the RNA-DNA hybrid displaces the non-complementary DNA strand and positions the target DNA within the Cas9 nuclease active sites [3]. This process induces a conformational change in Cas9 from an inactive to an active state, exposing the DNA strands to the nuclease domains [3].
DNA Cleavage and Double-Strand Break Formation
Once the stable R-loop is established, the Cas9 nuclease domains are activated to create a DSB:
- The HNH domain cleaves the target DNA strand (complementary to the gRNA) 3 base pairs upstream of the PAM [1] [3].
- The RuvC domain cleaves the non-target DNA strand 3-5 base pairs upstream of the PAM [1] [3].
This coordinated cleavage typically results in blunt-ended or slightly staggered double-strand breaks [3]. The resulting DSB then activates the cellular DNA damage response machinery, initiating repair processes that can be harnessed for genome editing.
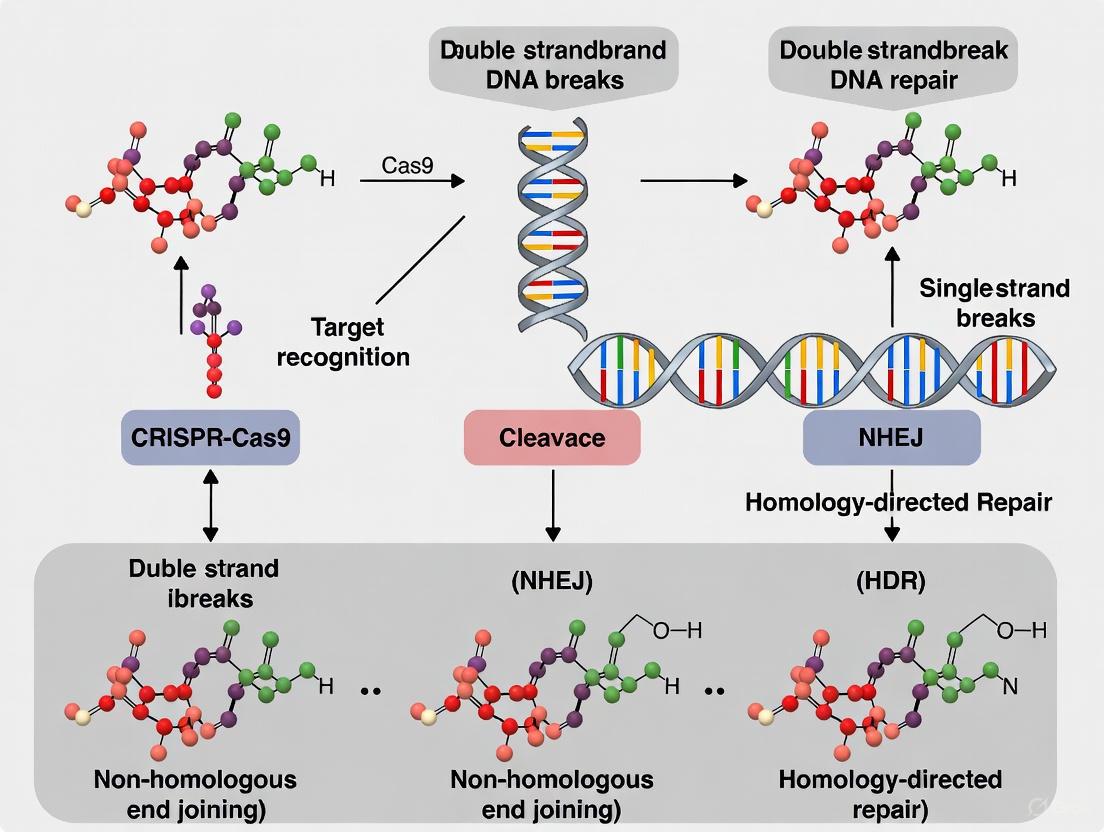
Double-Strand Break Repair Pathways and Genome Editing Outcomes
The strategic induction of DSBs represents the foundational step in CRISPR-Cas9 genome editing, as the subsequent repair by endogenous cellular machinery generates the desired genetic modifications [1] [3]. Eukaryotic cells employ multiple distinct pathways to repair DSBs, each with characteristic fidelity and resulting mutational profiles.
Non-Homologous End Joining (NHEJ)
NHEJ is the predominant DSB repair pathway in mammalian cells, active throughout the cell cycle and functioning without requiring a homologous DNA template [1] [3]. This pathway involves direct re-ligation of the broken DNA ends, but is inherently error-prone due to potential nucleotide insertions or deletions (indels) during processing [1] [3]. In genome editing applications, NHEJ is primarily harnessed for gene knockout strategies, as indels within coding sequences frequently result in frameshift mutations and premature stop codons that disrupt gene function [1] [7].
Homology-Directed Repair (HDR)
HDR is a high-fidelity repair mechanism that utilizes a homologous DNA template (typically the sister chromatid or an exogenously supplied donor template) to precisely repair the DSB [1] [3]. This pathway is most active during the late S and G2 phases of the cell cycle and enables precise gene modifications, including specific point mutations, gene insertions, or gene replacements [1] [3]. For therapeutic applications, HDR facilitates the correction of disease-causing mutations by providing a corrected donor DNA template alongside the CRISPR-Cas9 components [1] [5].
Alternative Repair Pathways
In addition to classical NHEJ and HDR, cells employ several alternative repair mechanisms:
- Microhomology-Mediated End Joining (MMEJ): An error-prone pathway that utilizes microhomology regions (5-25 bp) flanking the break site for repair, often resulting in deletions [3].
- Single-Strand Annealing (SSA): Functions when homologous repeats flank the DSB, leading to deletion of the intervening sequence [3].
Recent research using UMI-DSBseq to quantify repair dynamics at endogenous loci in tomato protoplasts revealed that precise repair accounts for a substantial proportion (up to 70%) of all repair events, highlighting the high fidelity of endogenous repair systems even in the context of CRISPR-Cas9-induced breaks [8].
Table 2: Double-Strand Break Repair Pathways in CRISPR-Cas9 Genome Editing
| Repair Pathway | Template Requirement | Fidelity | Primary Applications | Key Characteristics |
|---|---|---|---|---|
| Non-Homologous End Joining (NHEJ) | None | Error-prone | Gene knockouts, gene disruption | Active throughout cell cycle; generates indels |
| Homology-Directed Repair (HDR) | Homologous DNA template | High-fidelity | Gene correction, precise insertions | Restricted to late S/G2 phases; requires donor template |
| Microhomology-Mediated End Joining (MMEJ) | Microhomology regions | Error-prone | Targeted deletions | Utilizes 5-25 bp microhomology regions; generates predictable deletions |
Experimental Protocols and Methodologies
RNP Delivery in Protoplasts for Kinetic Studies
The delivery of preassembled ribonucleoproteins (RNPs) into plant protoplasts represents a powerful approach for studying the kinetics of CRISPR-Cas9-induced DSB repair [8]. This methodology enables synchronized DSB induction and high-resolution analysis of repair dynamics:
- RNP Complex Assembly: Purified SpCas9 protein is complexed with synthetic sgRNA in vitro to form active RNPs [8].
- Protoplast Transformation: RNPs are delivered into tomato protoplasts using PEG-mediated transformation, bypassing transcriptional and translational delays [8].
- Time-Course Sampling: Cells are harvested at multiple time points (e.g., 0, 6, 12, 24, 36, 48, 72 hours) post-transformation to capture DSB induction and repair progression [8].
- Molecular Analysis: The UMI-DSBseq protocol is employed to simultaneously quantify DSB intermediates and repair products at single-molecule resolution [8].
This approach revealed rapid DSB induction (detectable within 6 hours) with indel accumulation peaking between 48-72 hours, demonstrating the utility of this system for characterizing repair dynamics [8].
UMI-DSBseq for Single-Molecule Resolution of Repair Outcomes
UMI-DSBseq is a ligation-mediated PCR-based assay that enables multiplexed quantification of DSB intermediates and repair products through single-molecule sequencing [8]. The protocol involves:
- Adapter Ligation: Unique Molecular Identifier (UMI)-containing adapters are ligated directly to unrepaired DSBs and to intact molecules (via in vitro restriction enzyme digestion) [8].
- End Repair: Potential resected ends are repaired by fill-in of 3' overhangs prior to adapter ligation to ensure comprehensive molecule capture [8].
- Library Preparation and Sequencing: Direct preparation of Illumina sequencing-ready libraries enables simultaneous sequencing of unrepaired DSBs and repair products [8].
- Bioinformatic Analysis: Computational categorization of sequenced molecules into unrepaired DSBs, WT intact molecules, or indel-containing products [8].
This methodology revealed that 64-88% of target molecules were cleaved across three endogenous targets, with indels ranging between 15-41%, indicating that precise repair accounts for most DSBs [8].
The Scientist's Toolkit: Essential Reagents and Research Solutions
Successful implementation of CRISPR-Cas9 experiments requires carefully selected reagents and methodologies. The following table summarizes key research solutions for conducting CRISPR-Cas9 studies:
Table 3: Essential Research Reagents and Solutions for CRISPR-Cas9 Experiments
| Reagent/Solution | Function | Application Notes |
|---|---|---|
| SpCas9 Nuclease | DNA endonuclease that creates DSBs | Most widely used Cas9 ortholog; requires 5'-NGG-3' PAM |
| Synthetic sgRNA | Guides Cas9 to target sequence | Can be chemically modified to enhance stability and reduce immune responses |
| Donor DNA Template | Provides homology for HDR | Single-stranded or double-stranded DNA with homology arms |
| Lipid Nanoparticles (LNPs) | In vivo delivery vehicle | Particularly efficient for liver-targeted delivery [9] |
| Adeno-Associated Viruses (AAVs) | Viral delivery vector | Limited packaging capacity; immunogenicity concerns with redosing [9] |
| UMI-DSBseq Reagents | Quantitative DSB and repair product detection | Enables single-molecule resolution of repair dynamics [8] |
Advancements and Future Perspectives in CRISPR-Cas9 Technology
The CRISPR-Cas9 field continues to evolve rapidly, with several recent advancements addressing initial limitations and expanding therapeutic applications:
Enhanced Specificity and Delivery Systems
Substantial progress has been made in mitigating off-target effects through:
- High-fidelity Cas9 variants with reduced off-target activity
- Improved sgRNA design algorithms incorporating epigenetic and structural considerations
- Dual nickase systems requiring two adjacent sgRNAs for DSB formation [6]
Delivery technologies have also advanced significantly, with lipid nanoparticles (LNPs) emerging as a promising vehicle for in vivo therapeutic applications [9]. Unlike viral vectors, LNPs enable redosing potential due to reduced immunogenicity, as demonstrated in recent clinical trials where participants safely received multiple doses of LNP-encapsulated CRISPR therapies [9].
Clinical Applications and Therapeutic Translation
CRISPR-Cas9 has demonstrated remarkable success in clinical applications, most notably with the approval of Casgevy for sickle cell disease and transfusion-dependent beta thalassemia [9]. Ongoing clinical trials are exploring CRISPR-based therapies for diverse conditions including hereditary transthyretin amyloidosis (hATTR), hereditary angioedema (HAE), and various cancers [9]. Recent breakthroughs include the development of personalized in vivo CRISPR therapies, exemplified by a bespoke treatment for an infant with CPS1 deficiency that was developed and delivered in just six months [9].
The technology is also being applied to infectious diseases, with companies developing CRISPR-enhanced phage therapies to treat dangerous bacterial infections, and to diagnostic applications through CRISPR-based detection systems for respiratory viruses including SARS-CoV-2 and influenza [9] [10].
Emerging Challenges and Research Directions
Despite substantial progress, several challenges remain in the widespread clinical implementation of CRISPR-Cas9:
- Delivery Efficiency: Ensuring efficient targeting of therapeutic cells and tissues beyond the liver [9]
- Safety Optimization: Further reducing off-target effects and potential immune responses [5] [9]
- Manufacturing Scale-up: Developing cost-effective manufacturing processes for widespread therapeutic access [9]
- Ethical Considerations: Establishing appropriate regulatory frameworks for germline editing and other controversial applications [5]
Ongoing research focuses on developing novel Cas orthologs with distinct properties, enhancing precision editing through base and prime editing systems, and combining CRISPR with other modalities such as epigenetic engineering to expand the therapeutic potential of this transformative technology [10].
The CRISPR-Cas9 system has revolutionized biological research and therapeutic development by enabling precise genome editing through the induction of double-strand breaks (DSBs) at targeted genomic locations [3]. The Cas9 nuclease, guided by a specifically designed RNA sequence, creates a DSB in the DNA, which activates the cell's endogenous repair machinery [11]. The outcome of genome editing is not determined by the initial cut, but rather by how the cell repairs this break, making the understanding of DSB repair pathways fundamental to controlling editing outcomes [12]. In mammalian cells, three primary pathways compete to repair DSBs: non-homologous end joining (NHEJ), homology-directed repair (HDR), and microhomology-mediated end joining (MMEJ) [3]. The choice between these pathways has profound implications for the precision and safety of genome editing applications, particularly in the context of genetic disease research [11].
The balance between these repair pathways varies significantly across different cell types and states. Recent research has revealed that postmitotic cells such as neurons exhibit markedly different repair kinetics and pathway preferences compared to dividing cells, favoring NHEJ over MMEJ and displaying extended repair timelines that can continue for up to two weeks [12] [13]. Understanding these cell-type-specific differences is crucial for developing effective therapies for genetic diseases, particularly those affecting non-dividing cells in contexts like neurological disorders [12].
DNA Double-Strand Break Repair Pathways
Non-Homologous End Joining (NHEJ)
Mechanism and Key Players: NHEJ is the predominant and most active DSB repair pathway in mammalian cells, operating throughout the cell cycle but particularly dominant in G1 phase [14]. This pathway begins with the rapid recognition of DSBs by the Ku70/Ku80 heterodimer, which forms a ring-like structure that encircles the DNA ends [11]. Ku recruitment triggers a cascade of events: it attracts DNA-dependent protein kinase catalytic subunit (DNA-PKcs), which in turn activates Artemis nuclease [11]. Artemis processes the DNA ends by trimming overhangs, creating ligatable termini [11]. The final ligation step is catalyzed by a complex consisting of XRCC4, XLF, and DNA ligase IV [11] [14].
Editing Outcomes and Clinical Relevance: NHEJ is typically error-prone, often resulting in small insertions or deletions (indels) at the repair junction [11]. These indels can be harnessed to disrupt gene function by introducing frameshifts or premature stop codons, making NHEJ particularly valuable for gene knockout strategies [14]. In therapeutic contexts, NHEJ has been successfully employed in exa-cel (Casgevy), the first approved CRISPR therapy, where it disrupts the GATA1 motif in the BCL11A gene to induce fetal hemoglobin expression for treating sickle cell disease and β-thalassemia [15]. However, the error-prone nature of NHEJ also presents risks, as it can introduce unintended mutations that may have pathogenic consequences [15].
Homology-Directed Repair (HDR)
Mechanism and Key Players: HDR is a high-fidelity repair pathway that utilizes homologous DNA sequences as templates to accurately repair DSBs [11]. Unlike NHEJ, HDR is restricted to the S and G2 phases of the cell cycle when sister chromatids are available as templates [14]. The pathway initiates with DSB recognition by the MRE11-Rad50-NBS1 (MRN) complex, which coordinates DNA end resection to generate 3' single-stranded DNA overhangs [14]. These overhangs are rapidly coated by replication protein A (RPA), which is subsequently replaced by Rad51 filaments with the assistance of BRCA2, PALB2, and other mediator proteins [14]. The Rad51-nucleoprotein filament then invades the homologous donor template—either the sister chromatid or an exogenously supplied DNA donor—forming a displacement loop (D-loop) that allows DNA polymerase to extend the invading strand using the donor sequence as a template [14].
Editing Outcomes and Clinical Relevance: HDR enables precise genome editing outcomes, including specific nucleotide substitutions, gene insertions, and gene corrections [11]. This precision makes HDR the preferred pathway for therapeutic applications aimed at correcting disease-causing mutations [16]. However, HDR efficiency is generally low compared to NHEJ, especially in non-dividing cells such as neurons and cardiomyocytes [12] [11]. This limitation represents a significant challenge for therapeutic genome editing in postmitotic cells, which are relevant for many genetic diseases [12].
Microhomology-Mediated End Joining (MMEJ)
Mechanism and Key Players: MMEJ, also known as alternative end joining (alt-EJ), represents a distinct repair pathway that relies on short microhomology regions (2-20 base pairs) flanking the break site [3] [17]. The key regulator of MMEJ is DNA polymerase theta (POLQ), which is specifically inhibited by compounds such as ART558 [17]. MMEJ begins with PARP1 binding to DNA ends, competing with Ku70/Ku80 [14]. This binding stabilizes γH2AX and promotes end resection that reveals microhomology regions [14]. The exposed microhomology sequences then anneal, resulting in the deletion of the intervening sequence and generating flaps that are removed by nucleases before ligation by DNA ligase 1 or 3 [14].
Editing Outcomes and Clinical Relevance: MMEJ typically produces larger deletions than NHEJ, as the intervening sequence between microhomology regions is lost during repair [17]. The activity of MMEJ is cell cycle-dependent, with higher activity in S and G2 phases, and is generally more prominent in dividing cells compared to non-dividing cells [12]. In CRISPR genome editing, MMEJ can be exploited to create predictable deletion patterns based on the microhomology sequences flanking the target site [18]. However, MMEJ also contributes to imprecise integration in knock-in experiments and can generate larger genomic rearrangements, posing safety concerns for therapeutic applications [17] [15].
Table 1: Comparison of Key DSB Repair Pathways
| Feature | NHEJ | HDR | MMEJ |
|---|---|---|---|
| Template Dependency | Template-independent | Requires homologous template | Uses microhomology (2-20 bp) |
| Cell Cycle Phase | Active throughout, dominant in G1 | Restricted to S/G2 phases | Higher in S/G2 phases |
| Key Initiating Factors | Ku70/Ku80, DNA-PKcs | MRN complex, CtIP | PARP1, POLQ |
| Critical Effectors | DNA-PKcs, Artemis, XRCC4/LigIV | RPA, Rad51, BRCA1/2 | POLQ, MRN complex |
| Repair Fidelity | Error-prone (small indels) | High-fidelity (precise) | Error-prone (larger deletions) |
| Preference in Postmitotic Cells | Highly preferred [12] | Very low efficiency [11] | Reduced compared to dividing cells [12] |
Pathway Competition and Influencing Factors
The choice between DSB repair pathways is not random but is influenced by a complex interplay of cellular factors, experimental conditions, and cell-type-specific characteristics. Understanding these influencing factors is crucial for predicting and controlling genome editing outcomes.
Molecular Determinants of Pathway Choice
Several key molecular players act as gatekeepers directing DSBs toward specific repair pathways. The initial step of end resection represents a critical branch point, with limited resection favoring NHEJ and extensive resection promoting HDR or MMEJ [14]. The balance between 53BP1 and BRCA1 is particularly important, with 53BP1 favoring NHEJ by protecting DNA ends from resection, while BRCA1 promotes end resection and HDR [15]. Cell cycle stage exerts perhaps the most fundamental influence, with NHEJ dominating in G1 phase when sister chromatids are unavailable, while HDR and MMEJ activity increases in S/G2 phases when homologous templates are accessible [14].
The recently discovered single-strand annealing (SSA) pathway represents another relevant repair mechanism that contributes to imprecise knock-in outcomes, particularly asymmetric HDR events where only one side of the donor DNA integrates precisely [17]. SSA depends on Rad52-mediated annealing of longer homologous sequences and can be inhibited by specific Rad52 inhibitors such as D-I03 [17].
Cell-Type-Specific Repair Characteristics
Recent research has revealed striking differences in DSB repair between dividing and non-dividing cells. A 2025 study demonstrated that human neurons exhibit markedly different repair kinetics compared to genetically identical induced pluripotent stem cells (iPSCs) [12]. While iPSCs typically resolve DSBs within hours to a few days, neurons continue to accumulate indels for up to two weeks post-Cas9 delivery [12] [13]. This extended repair timeline was also observed in iPSC-derived cardiomyocytes and resting T cells, suggesting it may be a general feature of non-dividing cells [12].
Pathway preferences also differ significantly between cell types. Neurons predominantly utilize NHEJ, producing a narrower distribution of smaller indels, while dividing cells show greater MMEJ activity with larger deletions [12]. Transcriptomic profiling revealed that neurons mount a unique gene expression response to CRISPR-induced damage, upregulating non-canonical DNA repair factors including RRM2, a ribonucleotide reductase subunit [13]. Pharmacological or genetic inhibition of these neuron-specific repair factors can shift editing outcomes, providing strategies to enhance editing efficiency in these challenging cell types [13].
Table 2: Experimental Reagents for Manipulating DSB Repair Pathways
| Target Pathway | Reagent/Approach | Mechanism of Action | Effect on Editing |
|---|---|---|---|
| NHEJ Inhibition | Alt-R HDR Enhancer V2 | Inhibits key NHEJ factors | Increases HDR efficiency [17] |
| DNA-PKcs Inhibition | AZD7648 | Suppresses NHEJ initiation | Enhances HDR but increases large deletions [15] |
| MMEJ Inhibition | ART558 | Inhibits POLQ polymerase | Reduces large deletions [17] |
| SSA Inhibition | D-I03 | Inhibits Rad52 annealing | Reduces asymmetric HDR [17] |
| HDR Activation | Cell cycle synchronization | Arrests cells in S/G2 phase | Enhances HDR efficiency [14] |
| p53 Inhibition | Pifithrin-α | Transient p53 suppression | Reduces large chromosomal aberrations [15] |
Experimental Strategies for Pathway Control
Modulating Repair Pathways with Chemical Inhibitors
Strategic use of small molecule inhibitors has emerged as a powerful approach to steer DSB repair toward desired outcomes. NHEJ inhibition using compounds like Alt-R HDR Enhancer V2 has been widely adopted to enhance HDR efficiency in knock-in experiments [17]. However, recent studies have revealed that some NHEJ inhibitors, particularly DNA-PKcs inhibitors like AZD7648, can inadvertently promote large-scale genomic aberrations including kilobase- to megabase-scale deletions and chromosomal translocations [15]. These findings highlight the importance of carefully evaluating the safety implications of repair-modulating compounds.
Combination approaches targeting multiple pathways simultaneously have shown promise for enhancing precision. Co-inhibition of DNA-PKcs (NHEJ) and POLQ (MMEJ) has been demonstrated to protect against kilobase-scale deletions, though not megabase-scale events [15]. Similarly, SSA inhibition via Rad52 suppression reduces asymmetric HDR and other imprecise integration events, further enhancing knock-in accuracy [17].
Timing and Delivery Considerations
The extended repair timeline in non-dividing cells necessitates revised experimental approaches. Since neurons continue to process Cas9-induced breaks for up to two weeks, standard 2-4 day analysis windows significantly underestimate editing efficiency in these cells [12]. The remarkable longevity of Cas9 protein in neurons—remaining active for over 30 days—enables multiple cycles of cutting and repair, further extending the editing window [13].
Advanced delivery systems have been developed to overcome the challenges of editing non-dividing cells. Virus-like particles (VLPs) pseudotyped with VSVG and/or BaEVRless (BRL) glycoproteins can achieve up to 97% delivery efficiency in human neurons [12]. All-in-one lipid nanoparticles that concurrently deliver Cas9, sgRNA, and siRNAs enable coordinated manipulation of DNA repair pathways in challenging cell types [13]. For dividing cells, electroporation of Cas9 ribonucleoprotein (RNP) complexes provides efficient delivery with minimal off-target effects [12].
Advanced Visualization of DSB Repair Pathways
The following diagram illustrates the key molecular mechanisms and decision points in the competing DSB repair pathways:
DSB Repair Pathway Decision Mechanism
Research Reagent Solutions
Table 3: Essential Research Reagents for DSB Repair Studies
| Reagent Category | Specific Examples | Research Applications | Key Considerations |
|---|---|---|---|
| Pathway Inhibitors | Alt-R HDR Enhancer V2 (NHEJi), ART558 (POLQi), D-I03 (Rad52i) | Directing repair toward specific pathways | Combination treatments may have synergistic effects [17] |
| Cas9 Delivery Systems | Virus-like particles (VLPs), Lipid nanoparticles (LNPs), Electroporation of RNP | Efficient delivery to challenging cell types | VSVG/BRL-pseudotyped VLPs achieve >97% neuronal transduction [12] |
| Donor Template Designs | dsDNA with homology arms, ssODN templates, AAV-delivered donors | Enhancing HDR efficiency | Homology arm length and modification affect integration precision [11] [17] |
| Repair Reporters | Fluorescent-based pathway reporters, LAM-HTGTS, CAST-Seq | Quantifying pathway choice and detecting SVs | Long-read sequencing essential for detecting large deletions [19] [15] |
Safety Considerations and Clinical Implications
As CRISPR-based therapies advance toward clinical application, understanding and mitigating the risks associated with DSB repair has become increasingly important. Beyond well-characterized small indels, CRISPR editing can induce large structural variations (SVs) including kilobase- to megabase-scale deletions, chromosomal translocations, and chromothripsis [15]. These potentially harmful alterations have traditionally been underestimated because standard short-read amplicon sequencing fails to detect large deletions that eliminate primer binding sites, leading to overestimation of HDR efficiency and underestimation of indels [15].
The safety implications of repair pathway manipulation are particularly relevant for HDR-enhancing strategies. DNA-PKcs inhibitors used to boost HDR efficiency can increase the frequency of large deletions by a thousand-fold and promote chromosomal translocations [15]. Interestingly, not all HDR-enhancing approaches carry equivalent risks; transient 53BP1 inhibition does not increase translocation frequencies, and POLQ co-inhibition provides partial protection against kilobase-scale deletions [15]. Transient p53 suppression can reduce chromosomal aberrations but raises oncogenic concerns due to p53's tumor suppressor function [15].
These findings highlight the critical need for comprehensive genotoxicity assessment in therapeutic genome editing programs. Advanced detection methods like CAST-Seq and LAM-HTGTS are essential for identifying large SVs that would be missed by conventional sequencing [15]. Furthermore, researchers should carefully consider whether enhancing HDR efficiency is necessary for specific applications, as naturally corrected cells may gain selective advantages in some disease contexts, and even moderate editing levels may provide therapeutic benefit [15].
The competing DSB repair pathways—HDR, NHEJ, and MMEJ—represent fundamental determinants of CRISPR-Cas9 genome editing outcomes. The complex interplay between these pathways is influenced by cellular state, experimental conditions, and molecular regulators. Recent research has revealed striking cell-type-specific differences, particularly between dividing and non-dividing cells, with neurons exhibiting extended repair timelines and distinct pathway preferences. While chemical and genetic approaches to manipulate repair pathways offer powerful strategies to enhance desired editing outcomes, they must be applied with careful consideration of potential safety implications, including the risk of large structural variations. As CRISPR-based therapies continue to advance, a comprehensive understanding of DSB repair mechanisms will be essential for developing safe and effective treatments for genetic diseases.
The Critical Role of PAM Recognition and R-loop Formation in Target Specificity
The CRISPR-Cas9 system has revolutionized genetic engineering by providing an unprecedented tool for precise genome manipulation. Its application in researching genetic diseases hinges on the reliable formation of double-strand breaks (DSBs) at predetermined genomic loci. This fidelity is governed by two fundamental and interdependent mechanical processes: the initial recognition of a short protospacer-adjacent motif (PAM) and the subsequent directional formation of an R-loop structure. This whitepaper delves into the molecular mechanics of these steps, synthesizing recent structural and kinetic findings to outline their collective critical role in ensuring target specificity. Furthermore, it provides a detailed experimental framework for profiling Cas9 kinetics and discusses how an nuanced understanding of this mechanism is pivotal for developing safer, more effective CRISPR-based therapies for genetic disorders.
In the context of genetic disease research, the CRISPR-Cas9 system functions as a programmable DNA-endonuclease complex. Its core components are the Cas9 protein, which creates the DSB, and a single-guide RNA (sgRNA), which confers sequence specificity [20]. The repair of these induced DSBs via cellular mechanisms like non-homologous end joining (NHEJ) or homology-directed repair (HDR) enables gene knockout or correction, respectively [21] [22]. The system's utility and safety are entirely dependent on its ability to discriminate the intended target from the vast expanse of the genome. This discrimination is not a single event but a multi-stage interrogation process, beginning with PAM recognition and proceeding through R-loop formation, which together act as a two-factor authentication system for DNA cleavage [23].
Molecular Mechanism of Target Recognition
The journey to a site-specific DSB is a coordinated sequence of events that ensures only the correct DNA site is cleaved.
Step 1: Initial DNA Binding and PAM Interrogation
The Cas9-sgRNA complex first interacts with DNA through non-specific, transient contacts. The critical initial specific interaction is the recognition of a short PAM sequence by the PAM-interacting domain of Cas9. For the most commonly used Streptococcus pyogenes Cas9 (SpyCas9), this PAM is the 3'-NGG-5' sequence, where "N" is any nucleotide [21] [20].
- Function: The PAM acts as a self vs. non-self discriminator, preventing the Cas9 complex from targeting the bacterial CRISPR locus itself. In applied settings, it is the primary gatekeeper for target site selection.
- Mechanism: Recent biophysical studies reveal that Cas9 engages DNA in a two-step capture process [23]. The first step involves selective but low-affinity binding to the PAM.
- Specificity Trade-off: Engineering Cas9 variants with relaxed PAM specificity (recognizing, for example, NG instead of NGG) often comes at a cost. These variants can suffer from persistent non-selective DNA binding and kinetic traps that ultimately reduce genome-editing efficiency by hindering the transition to the next step: DNA unwinding [23].
Step 2: DNA Melting and R-loop Propagation
Following PAM binding, Cas9 triggers local DNA melting, unwinding the double helix upstream of the PAM. This creates a "R-loop," a three-stranded nucleic acid structure where:
- The target strand (complementary to the sgRNA) hybridizes with the 20-nucleotide spacer sequence of the sgRNA.
- The non-target strand is displaced.
This R-loop formation is highly directional, proceeding from the PAM-proximal end towards the PAM-distal end [24]. The stability of the R-loop is paramount; once it extends beyond approximately 14 base pairs, a conformational change in Cas9's REC3 domain "locks" the structure, activating the HNH and RuvC nuclease domains to cleave the target and non-target strands, respectively [24].
Table 1: Key Steps in the CRISPR-Cas9 Target Recognition Pathway
| Step | Molecular Event | Key Determinants | Outcome |
|---|---|---|---|
| 1. PAM Interrogation | Cas9 PAM-interacting domain scans for and binds to the correct PAM sequence (e.g., NGG). | PAM sequence identity, PAM-binding domain affinity. | Grants permission to initiate DNA unwinding; primary specificity filter. |
| 2. DNA Melting | PAM binding induces structural distortion and local unwinding of the DNA duplex. | Energy from PAM binding, protein-DNA interactions. | Creates a DNA "bubble" to allow sgRNA-DNA hybridization. |
| 3. R-loop Propagation | The sgRNA spacer sequentially base-pairs with the target DNA strand, displacing the non-target strand. | Complementarity between sgRNA spacer and DNA target strand, directionality (PAM-proximal to distal). | Forms a stable R-loop structure; the secondary specificity filter. |
| 4. Conformational Activation | Stable R-loop formation triggers REC3 docking and activates HNH and RuvC nuclease domains. | R-loop stability and length (>14 bp). | Cas9 transitions from a DNA-binding complex to an active endonuclease. |
| 5. DNA Cleavage | HNH cleaves the target strand, RuvC cleaves the non-target strand, creating a DSB. | Proper positioning of catalytic residues. | Generation of a double-strand break 3-4 nucleotides upstream of the PAM. |
The following diagram illustrates this sequential mechanism and the critical checkpoint where conformational activation occurs.
Experimental Profiling of Cas9 Kinetics and Specificity
Understanding the kinetics of the above process is essential for assessing the efficiency and fidelity of a given Cas9-sgRNA system. The following protocol outlines a robust method for quantifying Cas9 cleavage activity and turnover, adapted from recent single-molecule and biochemical studies [24].
Protocol: In Vitro Cas9 Cleavage and Turnover Assay
Objective: To determine the cleavage efficiency and multi-turnover capability of wild-type (WT) Cas9 compared to engineered sgRNAs designed for faster product release.
Materials:
- Purified Cas9 protein (WT or variant of interest)
- In vitro transcribed or synthesized sgRNAs:
- WT sgRNA (20-nt spacer, fully matched)
- Truncated sgRNA (e.g., 15-nt spacer, fully matched)
- Mismatch-engineered sgRNA (e.g., 17-nt spacer with 2 mismatches at the 5' end)
- Target DNA substrate: Plasmid DNA (e.g., pUC19 with lacZ target site) or a short, double-stranded, fluorescently-labeled DNA oligonucleotide.
- Reaction Buffer: 20 mM HEPES-KOH (pH 7.5), 100 mM KCl, 5 mM MgCl₂, 1 mM DTT, 5% glycerol.
- Equipment: Thermocycler or water bath, agarose gel electrophoresis system, fluorimeter (if using fluorescent substrates).
Methodology:
- Complex Formation: Pre-incubate Cas9 protein with each sgRNA (1:1.2 molar ratio) in reaction buffer for 10 minutes at 25°C to form the ribonucleoprotein (RNP) complex.
- Reaction Initiation: Start the cleavage reaction by adding the target DNA substrate to the RNP complex. Use a 5-fold molar excess of DNA substrate over the RNP complex to challenge the system and observe turnover. For example:
- Final RNP concentration: 100 nM
- Final plasmid DNA concentration: 500 nM (in molecules)
- Time-course Sampling: Aliquot the reaction mixture at specific time points (e.g., 0, 5, 15, 30, 60, 120, 240 minutes) and immediately stop the reaction by adding a chelating agent (e.g., 10 mM EDTA) or denaturing loading dye.
- Product Analysis:
- For plasmid substrates: Analyze samples by agarose gel electrophoresis. Resolve supercoiled (uncut), nicked (intermediate), and linear (cleaved) DNA forms. Quantify the band intensities to calculate the percentage of cleaved DNA over time.
- For fluorescent oligonucleotides: Use denaturing polyacrylamide gel electrophoresis or capillary electrophoresis to separate and quantify the cleaved product.
- Data Fitting: Plot the fraction of cleaved product versus time. Fit the data to a kinetic model to derive the observed rate constants for cleavage (k₂) and product release (k₃). WT Cas9 with a 20-nt sgRNA typically shows a rapid k₂ (~60 min⁻¹) but an extremely slow k₃ (~0.00085 min⁻¹), characteristic of single-turnover behavior [24].
Expected Outcomes:
- WT sgRNA will show rapid initial cleavage that plateaus at a stoichiometric level, indicating single-turnover kinetics.
- Engineered sgRNAs (e.g., the 2-mm variant) will show slower initial cleavage but will continue to process multiple DNA substrates, demonstrating multi-turnover kinetics and a higher final cleavage yield.
Table 2: Key Research Reagents for Profiling Cas9 Mechanism
| Research Reagent | Function / Rationale | Example Application |
|---|---|---|
| PAM-relaxed Cas9 Variants | Engineered Cas9 proteins that recognize a broader range of PAM sequences (e.g., NG). | Study the trade-off between target range and editing efficiency/kinetics [23]. |
| Truncated sgRNAs (tru-gRNAs) | sgRNAs with shortened spacer regions (e.g., 15-17 nt). Used to destabilize the PAM-distal R-loop. | Promote faster product release and multi-turnover kinetics; study R-loop stability [24]. |
| Mismatch-engineered sgRNAs | sgRNAs with intentional mismatches at the 5' end of the spacer sequence. | Enhance DNA rehybridization post-cleavage, facilitating R-loop collapse and enzyme turnover without sacrificing full activation [24]. |
| Catalytically Inactive Cas9 (dCas9) | A "dead" Cas9 with mutated nuclease domains (H840A, D10A). Binds DNA without cutting. | Serves as a control for DNA binding; base for CRISPRi/a studies; used in imaging genomic loci [20]. |
| High-Fidelity Cas9 Variants | Engineered Cas9 proteins (e.g., eSpCas9, SpCas9-HF1) with enhanced specificity. Contain mutations that destabilize non-specific RNA-DNA interactions. | Reduce off-target effects in therapeutic applications and functional genomics screens [24]. |
Implications for Specificity and Off-Target Effects
The two-step mechanism has direct consequences for off-target editing. While stable R-loop formation ensures efficient on-target cleavage, it also means that near-complementary off-target sites with correct PAMs can be cleaved, especially if the mismatches are tolerated in the PAM-distal region [20] [24]. The kinetic proofreading inherent in the sequential process—where PAM binding must be followed by successful R-loop propagation—provides natural protection, but it is not foolproof.
Recent research indicates that Cas9's single-turnover nature (prolonged binding to the cleaved product) also impacts specificity. This product inhibition sequesters Cas9, but when displaced by cellular machinery, it can inadvertently bias repair outcomes or become available for new targeting cycles [24]. Strategies that promote multi-turnover, such as using mismatch-engineered sgRNAs, may therefore influence both the efficiency and the specificity profile of the editing reaction by altering the enzyme's cellular residence time and availability.
The critical role of PAM recognition and R-loop formation in defining CRISPR-Cas9's target specificity is a cornerstone of its application in genetic disease research. The PAM serves as the initial, essential gatekeeper, while the directional and stable formation of the R-loop acts as the definitive verification step. The interplay between these two stages—optimized for both speed and fidelity—ensures precise DSB formation. Continued biochemical and structural insights into this mechanism, such as the engineering of multi-turnover Cas9 systems, are driving the development of next-generation editors with enhanced safety and efficacy profiles. For researchers and drug developers, a deep understanding of this process is not merely academic; it is fundamental to the rational design of sgRNAs, the interpretation of editing outcomes, and the ultimate translation of CRISPR-based therapies into clinical reality.
How Cellular Context and Cell Cycle Stage Influence Repair Pathway Choice
The CRISPR-Cas9 system has revolutionized genetic engineering by enabling precise induction of double-strand breaks (DSBs) at targeted genomic loci. However, the ultimate outcome of genome editing is not determined by the cutting event itself, but by the cellular machinery that repairs these breaks. The choice of DNA repair pathway—a process profoundly influenced by cellular context and cell cycle stage—represents a critical determinant of editing success. In dividing cells, the full repertoire of repair pathways is available, while in nondividing cells, options are more constrained. Understanding and controlling these variables is essential for advancing therapeutic genome editing, particularly for genetic diseases affecting non-proliferative tissues like neurons and cardiomyocytes.
DNA Repair Pathways: Mechanisms and Regulators
Major Pathways for DSB Repair
Cellular repair of CRISPR-Cas9-induced DSBs occurs primarily through three competing pathways: non-homologous end joining (NHEJ), microhomology-mediated end joining (MMEJ), and homology-directed repair (HDR). Each pathway possesses distinct mechanistic features and leaves characteristic molecular signatures at the repair site [25] [26].
- Non-Homologous End Joining (NHEJ) operates throughout the cell cycle but dominates in G1 phase. This pathway directly ligates broken DNA ends without a template, often resulting in small insertions or deletions (indels). While efficient, NHEJ is error-prone and typically produces gene knockouts [26] [27].
- Microhomology-Mediated End Joining (MMEJ) utilizes short homologous sequences (5-25 bp) flanking the break site for repair. This pathway, active in S and G2 phases, typically generates larger deletions than NHEJ and contributes to complex structural variations when dysregulated [25] [28].
- Homology-Directed Repair (HDR) requires a DNA template with homologous sequences and is restricted to late S and G2 phases when sister chromatids are available. HDR enables precise gene editing but generally occurs at lower frequencies than error-prone pathways [29] [16].
Table 1: Characteristics of Major DNA Repair Pathways
| Pathway | Cell Cycle Phase | Template Required | Fidelity | Primary Outcome |
|---|---|---|---|---|
| NHEJ | All phases (predominant in G1) | No | Error-prone | Small insertions/deletions (indels) |
| MMEJ | S/G2 phases | Microhomology sequences | Error-prone | Larger deletions with microhomology |
| HDR | Late S/G2 phases | Homologous DNA template | High-fidelity | Precise sequence modifications |
Cell Cycle Regulation of Repair Pathways
The cell cycle exerts stringent control over DNA repair pathway choice through both regulatory proteins and substrate availability. Key regulatory mechanisms include:
- Cyclin-Dependent Kinase (CDK) Activity: CDK-mediated phosphorylation activates resection enzymes like CtIP and EXO1, promoting end resection that channels DSBs toward HDR and MMEJ rather than NHEJ [27].
- Template Availability: HDR predominantly utilizes the sister chromatid as a repair template, restricting this pathway to S and G2 phases after DNA replication [16].
- Pathway-Specific Factor Expression: The expression and activation of repair factors like BRCA1, RAD51, and DNA-PKcs fluctuate across the cell cycle, creating permissive environments for specific pathways [28].
Diagram 1: Cell cycle phase determines repair pathway choice. NHEJ dominates in G1, MMEJ in S phase, and HDR in G2 phase.
Impact of Cellular Context on Repair Outcomes
Dividing vs. Nondividing Cells
Recent research has revealed striking differences in how dividing and nondividing cells respond to CRISPR-Cas9-induced DNA damage. A 2025 study comparing induced pluripotent stem cells (iPSCs) to isogenic iPSC-derived neurons demonstrated that postmitotic cells resolve DSBs over dramatically extended timescales—with indel accumulation continuing for up to two weeks post-transduction compared to days in dividing cells [12].
The repair pathway preferences also differ substantially between cellular states. Dividing cells predominantly utilize MMEJ, generating larger deletions, while neurons favor NHEJ, producing smaller indels and exhibiting more precise repair outcomes. This divergence stems from differential expression of DNA repair factors, with neurons upregulating non-canonical DNA repair machinery in response to Cas9 exposure [12].
Table 2: CRISPR Repair Outcomes in Dividing vs. Nondividing Cells
| Parameter | Dividing Cells (iPSCs) | Nondividing Cells (Neurons) |
|---|---|---|
| Time to peak indel accumulation | 1-3 days | Up to 16 days |
| Dominant repair pathway | MMEJ | NHEJ |
| Characteristic indels | Larger deletions | Smaller insertions/deletions |
| Ratio of insertions to deletions | Lower | Significantly higher |
| HDR efficiency | Moderate | Very low |
Repair in Specialized Cell Types
Differentiated cells exhibit specialized DNA repair responses reflecting their physiological contexts:
- Neurons: As long-lived postmitotic cells, neurons prioritize error-free repair to maintain genomic integrity over decades. They engage specialized repair factors and resolve DSBs gradually, avoiding cell death triggers that would eliminate damaged dividing cells [12].
- Cardiomyocytes: Similar to neurons, iPSC-derived cardiomyocytes show prolonged indel accumulation timelines, suggesting common repair mechanisms in postmitotic cells [12].
- Immune Cells: Resting versus activated T cells display different repair capacities, with activated T cells supporting more efficient editing—a consideration crucial for CAR-T cell therapies [12] [27].
- Plant Cells: Studies in tomato protoplasts reveal that precise repair accounts for most DSB resolution, with 64-88% of molecules cleaved but only 15-41% containing indels, highlighting species- and cell-type-specific repair characteristics [8].
Experimental Approaches and Methodologies
Controlling for Cell Cycle Effects
Researchers have developed multiple strategies to bias repair outcomes by manipulating cell cycle status or exploiting cycle-dependent repair preferences:
- Cell Cycle Synchronization: Chemical inhibitors like thymidine, nocodazole, or RO-3306 can arrest cells at specific cell cycle stages, enriching for populations competent for HDR (S/G2) [27].
- Engineered Cas9 Variants: The vCas9 variant (S55R-R976A-K1003A-T1314R) creates staggered DNA ends that suppress NHEJ while promoting MMEJ and HDR, effectively bypassing cell cycle restrictions [25].
- Small Molecule Inhibitors: Compounds targeting NHEJ factors (e.g., DNA-PKcs inhibitors) or enhancing HDR (e.g., RS-1) can shift repair balance toward precise editing in a cell cycle-dependent manner [29].
Delivery Methods for Different Cellular Contexts
Effective CRISPR editing requires delivery strategies adapted to specific cellular contexts:
- Virus-Like Particles (VLPs): Engineered VLPs pseudotyped with VSVG/BRL envelopes achieve up to 97% delivery efficiency in human iPSC-derived neurons, enabling controlled Cas9-RNP delivery to postmitotic cells [12].
- Electroporation of RNP Complexes: Preassembled Cas9 ribonucleoprotein (RNP) complexes electroporated into primary cells minimize toxicity and enable efficient editing, particularly in immune cells [27].
- Lipid Nanoparticles (LNPs): LNPs efficiently deliver CRISPR components to hepatocytes in vivo, showing natural liver tropism and enabling therapeutic editing for liver-expressed diseases [9].
Diagram 2: Experimental workflow for studying cellular influences on CRISPR repair. Approaches combine tailored delivery methods with cell cycle manipulation, followed by sophisticated outcome analysis.
Quantitative Assessment of Repair Dynamics
Advanced molecular tools enable precise quantification of repair kinetics and outcomes:
- UMI-DSBseq: This method uses unique molecular identifiers (UMIs) in ligation-mediated PCR to simultaneously quantify DSB intermediates and repair products at single-molecule resolution, revealing repair dynamics across time courses [8].
- Kinetic Modeling: Mathematical modeling of time-course data quantifies rates of DSB induction, end processing, and repair pathway choice, revealing that indel accumulation depends on the balance between these processes [8].
- Long-Term Tracking: In neurons, editing outcomes must be assessed over extended periods (up to 16 days) to account for slow repair kinetics, unlike the 24-72 hour timelines sufficient for most dividing cells [12].
The Scientist's Toolkit: Essential Reagents and Methods
Table 3: Key Research Reagents and Methods for Studying Repair Pathways
| Reagent/Method | Function | Application Context |
|---|---|---|
| vCas9 variant | Engineered nuclease producing staggered cuts that bias repair toward HDR/MMEJ | Dividing and nondividing cells [25] |
| VSVG/BRL-pseudotyped VLPs | High-efficiency delivery of Cas9-RNP to neurons | Postmitotic cells including iPSC-derived neurons [12] |
| Chemical synchronizers | Arrest cells at specific cell cycle stages | Enhancing HDR efficiency in dividing cells [27] |
| NHEJ inhibitors | Suppress dominant error-prone pathway | Increasing HDR:NHEJ ratio [16] |
| Preassembled RNPs | Complex of Cas9 protein and sgRNA for transient editing | Primary cells, including T cells and stem cells [27] |
| UMI-DSBseq | High-resolution quantification of DSBs and repair products | Kinetic studies of repair pathway engagement [8] |
| Modified sgRNAs | Chemical modifications improve stability and efficiency | Challenging primary cells (e.g., resting T cells) [27] |
Implications for Therapeutic Genome Editing
Challenges in Genetic Disease Therapy
The cellular context dependence of DNA repair presents both challenges and opportunities for therapeutic genome editing:
- Neurological Diseases: Dominant neurological disorders requiring allele-specific inactivation represent promising CRISPR targets, but the slow, precise repair in neurons necessitates extended timelines and specialized delivery methods [12].
- Cardiovascular Diseases: The prolonged indel accumulation observed in cardiomyocytes suggests similar considerations for treating inherited heart conditions [12].
- Ex Vivo Therapies: For CAR-T cell and stem cell therapies, optimizing editing during proliferative stages followed by differentiation may maximize outcomes [9] [27].
Strategic Considerations
Successful therapeutic editing strategies must account for cellular context:
- Timeline Expectations: Editing outcomes in postmitotic cells should be evaluated over weeks rather than days to account for extended repair kinetics [12].
- Delivery Optimization: Cell-specific delivery systems (VLPs for neurons, LNPs for hepatocytes, electroporation for ex vivo therapies) are essential for efficient editing [12] [9].
- Pathway Bias: Engineered editors like vCas9 or base editors can circumvent repair pathway limitations in nondividing cells [25].
- Patient-Specific Factors: Age, disease state, and tissue homeostasis may further influence repair pathway choices beyond fundamental cell biology principles.
Future Directions
Emerging research directions promise to enhance control over DNA repair outcomes in diverse cellular contexts:
- Cell-Type Specific Repair Mapping: Comprehensive characterization of repair preferences across human cell types would enable predictive editing outcome models.
- Temporal Control: Inducible editing systems that activate Cas9 during specific cell cycle phases could maximize desired repair pathways.
- Epigenetic Engineering: CRISPR-based epigenetic editors that reversibly modulate gene expression without DSBs avoid repair pathway constraints entirely [10].
- Machine Learning: AI-driven analysis of editing outcomes across cellular contexts may reveal previously unrecognized predictors of repair pathway choice [10].
- Novel Editor Development: Continued engineering of Cas variants with customized break geometries and repair biases will expand the toolkit for specific therapeutic applications [25].
The CRISPR-Cas9 system has revolutionized genetic engineering by enabling precise induction of double-strand breaks (DSBs) at targeted genomic locations. The ultimate editing outcome is determined not by the initial cut, but by the complex cellular repair processes that follow. This technical guide examines the molecular journey from DSB induction to the formation of insertions/deletions (indels) or precise edits, with particular emphasis on the competing non-homologous end joining (NHEJ) and homology-directed repair (HDR) pathways. Within the context of genetic disease research, we explore how understanding and manipulating these repair pathways is critical for developing effective therapeutic applications, from sickle cell disease to rare metabolic disorders.
The CRISPR-Cas9 system functions as a programmable DNA-cutting machine composed of two core components: the Cas9 nuclease and a guide RNA (gRNA) [1]. The gRNA, through its 20-nucleotide spacer sequence, directs Cas9 to a specific genomic locus by complementary base pairing [30]. Successful binding and DNA cleavage require the presence of a protospacer adjacent motif (PAM) immediately downstream of the target site—typically a 5'-NGG-3' sequence for the most commonly used Streptococcus pyogenes Cas9 [3] [1].
Upon recognition of the target sequence, the Cas9 enzyme undergoes conformational changes that activate its two nuclease domains: the HNH domain cleaves the DNA strand complementary to the gRNA, while the RuvC domain cleaves the non-complementary strand [3] [1]. This coordinated action results in a blunt-ended DSB approximately 3-4 nucleotides upstream of the PAM site [1] [30]. The cell perceives this break as DNA damage and rapidly initiates repair processes, setting the stage for either mutagenic or precise editing outcomes.
DNA Repair Pathways for Double-Strand Breaks
Non-Homologous End Joining (NHEJ)
The NHEJ pathway represents the dominant and most efficient DSB repair mechanism in mammalian cells, operating throughout the cell cycle without requiring a repair template [1] [16]. This pathway involves the direct ligation of broken DNA ends through a series of coordinated steps: (1) recognition of the DSB by repair proteins, (2) processing of the broken ends, and (3) ligation to reseal the DNA backbone [3].
While efficient, NHEJ is inherently error-prone due to nucleolytic processing of DNA ends before ligation [30]. The Ku70/Ku80 heterodimer initiates NHEJ by binding to DNA ends and recruiting additional factors, including DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and the XRCC4-DNA ligase IV complex [3]. End processing often results in small insertions or deletions (indels) at the junction site, which typically range from 1 to 20 nucleotides [31]. When these indels occur within coding sequences, they frequently cause frameshift mutations that introduce premature stop codons, effectively knocking out the target gene [30].
Homology-Directed Repair (HDR)
HDR provides a template-dependent, high-fidelity repair mechanism that is most active during the S and G2 phases of the cell cycle when sister chromatids are available [1] [16]. This pathway requires a homologous DNA template—either the sister chromatid, homologous chromosome, or an exogenously supplied donor template—to accurately repair the break [16].
The HDR process involves extensive end resection to create 3' single-stranded DNA overhangs, which then invade the homologous template sequence [3]. Key mediators include the MRN complex (Mre11-Rad50-Nbs1), CtIP, and BRCA1/2 proteins [3]. The invading strand primes DNA synthesis using the homologous template, ultimately resulting in accurate restoration of the original sequence or precise incorporation of desired edits when an exogenous donor is provided [16].
Alternative End Joining Pathways
Beyond classical NHEJ, cells possess additional error-prone repair mechanisms, including microhomology-mediated end joining (MMEJ) and single-strand annealing (SSA) [3]. These pathways typically operate when canonical NHEJ is compromised or unavailable and often result in larger deletions [3]. MMEJ specifically utilizes short homologous sequences (5-25 bp) flanking the break site for alignment and repair, frequently generating predictable deletion patterns based on the location of microhomology regions [3] [31].
Table 1: Characteristics of Major DNA Double-Strand Break Repair Pathways
| Repair Pathway | Template Requirement | Fidelity | Cell Cycle Phase | Primary Proteins Involved | Typical Outcomes |
|---|---|---|---|---|---|
| Non-Homologous End Joining (NHEJ) | None | Error-prone | All phases | Ku70/80, DNA-PKcs, XRCC4-LigIV | Small indels (1-20 bp) |
| Homology-Directed Repair (HDR) | Homologous DNA template | High-fidelity | S/G2 phases | MRN complex, BRCA1/2, Rad51 | Precise edits, gene corrections |
| Microhomology-Mediated End Joining (MMEJ) | Microhomology regions | Error-prone | All phases | PARP1, Polθ, FEN1 | Larger deletions flanked by microhomology |
| Single-Strand Annealing (SSA) | Direct repeats | Error-prone | All phases | Rad52, ERCC1, XPF | Large deletions between repeats |
Pathway Competition and Dynamics
The choice between competing repair pathways is influenced by multiple factors, including cell cycle stage, chromatin context, and the nature of the break ends [3]. Recent quantitative studies in tomato protoplasts revealed that precise repair (restoration of the original sequence) accounts for a substantial proportion of repair events—up to 70% at some targets—highlighting the remarkable fidelity of endogenous repair systems [8]. Kinetic modeling of DSB repair dynamics along a 72-hour time-course demonstrated that indel accumulation is determined by the combined effect of DSB induction rates, processing of broken ends, and the balance between precise versus error-prone repair [8].
The following diagram illustrates the competitive dynamics between these major repair pathways following a CRISPR-Cas9-induced double-strand break:
Factors Determining Editing Outcomes
Target Sequence Determinants
Large-scale analyses of indel profiles at over 1,000 genomic sites in human cells have revealed that CRISPR editing outcomes are not random but follow predictable patterns based primarily on local sequence context [31]. The precision of DNA editing—defined as the recurrence of a specific indel—varies considerably among target sites, with some loci showing one highly preferred indel and others displaying numerous infrequent indels [31].
A critical determinant of editing precision is the fourth nucleotide upstream of the PAM site [31]. Targets with a thymine at this position typically exhibit more predictable outcomes with a single dominant indel, while other sequences often result in heterogeneous mutation patterns [31]. Additionally, the presence of microhomology regions flanking the cut site strongly predisposes toward MMEJ-mediated larger deletions [31].
Chromatin and Epigenetic Influences
Chromatin accessibility and epigenetic modifications significantly influence both CRISPR cutting efficiency and repair pathway choice [31]. Euchromatic regions with open chromatin configurations generally show higher editing efficiency compared to heterochromatic regions [31]. However, the effect of chromatin states appears more pronounced for imprecise targets, while precise targets with highly dominant indels remain relatively unaffected by epigenetic context [31].
Cellular Environment and Experimental Conditions
The cellular environment plays a decisive role in determining repair outcomes. Key factors include:
- Cell cycle stage: HDR is preferentially active in S/G2 phases, while NHEJ operates throughout the cell cycle [3] [16]
- Cell type: Primary cells and differentiated tissues often show different repair preferences compared to immortalized cell lines [8]
- DNA repair protein expression: Relative levels of NHEJ, HDR, and MMEJ pathway components vary between cell types and influence pathway choice [3]
Experimental parameters such as delivery method (RNP vs. plasmid), Cas9 expression levels, and timing of analysis also significantly impact observed outcomes [8] [30]. For instance, direct delivery of preassembled ribonucleoproteins (RNPs) enables more synchronized DSB induction, facilitating kinetic studies of repair dynamics [8].
Table 2: Quantitative Analysis of CRISPR-Cas9 Repair Outcomes at Endogenous Loci
| Target Locus | Cleavage Efficiency | Indel Frequency | Precise Repair Rate | Dominant Repair Pathway | Time to Peak Indel Accumulation |
|---|---|---|---|---|---|
| PhyB2 (Tomato) | 88% | 41% | ~47% | NHEJ/MMEJ | 48-72 hours |
| Psy1 (Tomato) | 64% | 15% | ~49% | Precise repair | 48-72 hours |
| CRTISO (Tomato) | 68% | 22% | ~46% | Balanced | 48-72 hours |
| Human sites (average) | 50-90%* | 20-60%* | 15-30%* | Context-dependent | 5 days (plateau) |
*Ranges based on large-scale studies in human cells [31]; precise repair rates in human cells estimated from comparative studies [8]
Methodologies for Studying Repair Outcomes
UMI-DSBseq for Single-Molecule Resolution
The UMI-DSBseq (Unique Molecular Identifier-Double-Strand Break sequencing) method enables multiplexed quantification of DSB intermediates and repair products at single-molecule resolution [8]. This approach combines a target-specific primer with a DSB-flanking restriction enzyme site to capture both DSBs and intact molecules simultaneously [8].
Key Protocol Steps:
- DSB capture: Following RNP delivery and incubation, genomic DNA is extracted at multiple time points
- End repair: DSB ends are repaired by fill-in of 3' overhangs to enable adaptor ligation
- UMI adaptor ligation: Adaptors containing unique molecular identifiers are ligated directly to unrepaired DSBs and to intact molecules at flanking restriction sites
- Library preparation and sequencing: Illumina sequencing-ready libraries are prepared in a single-tube reaction
- Bioinformatic analysis: Molecules are categorized as unrepaired DSBs, WT intact molecules, or indel products using specialized computational pipelines (github.com/daniebt/UMI-DSBseq) [8]
This method allows simultaneous tracking of DSB induction, processing intermediates, and final repair products along time-courses, providing unprecedented resolution of repair dynamics [8].
Single-Cell DNA Sequencing
Recent advances in single-cell DNA sequencing technologies, such as the Tapestri platform, enable comprehensive analysis of editing outcomes at single-cell resolution [32]. This approach characterizes the genotype of edited cells simultaneously at more than 100 loci, including editing zygosity, structural variations, and cell clonality [32]. Studies using this technology have revealed nearly unique editing patterns in every edited cell, highlighting the importance of single-cell resolution for safety assessment in therapeutic applications [32].
The following workflow diagram illustrates the key methodological approaches for analyzing CRISPR editing outcomes:
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Studying CRISPR Repair Outcomes
| Reagent/Tool | Function | Application Examples |
|---|---|---|
| SpCas9 Nuclease | RNA-guided DNA endonuclease that induces DSBs at target sites | Genome editing across diverse cell types and organisms [1] [30] |
| High-Fidelity Cas9 Variants | Engineered Cas9 with reduced off-target effects (eSpCas9, SpCas9-HF1, HypaCas9) | Applications requiring high specificity [30] |
| Preassembled RNPs | Cas9 protein complexed with synthetic sgRNA; enables rapid, synchronized editing | Kinetic studies of DSB repair dynamics [8] |
| Nickase Cas9 (Cas9n) | D10A mutant that creates single-strand breaks; improves specificity when used in pairs | Reduced off-target effects while maintaining on-target activity [30] |
| dCas9 (catalytically dead) | Binds DNA without cleavage; base for fusion proteins | Gene regulation, imaging, and epigenetic modifications [30] |
| UMI-DSBseq Reagents | Molecular toolkit for capturing DSB intermediates and repair products | Quantitative analysis of repair dynamics at single-molecule resolution [8] |
| HDR Donor Templates | Single-stranded or double-stranded DNA with homology arms | Precise gene correction or insertion via HDR [16] |
| NHEJ Inhibitors | Small molecules that suppress NHEJ pathway (e.g., SCR7) | Enhancing HDR efficiency in replicating cells [16] |
Implications for Genetic Disease Research and Therapeutics
The balance between indel formation and precise editing has profound implications for developing CRISPR-based therapies for genetic diseases. Two fundamental therapeutic strategies have emerged:
Disruption Strategies for Loss-of-Function Therapies
For diseases caused by toxic gain-of-function mutations or where gene disruption provides therapeutic benefit, NHEJ-mediated indel formation is harnessed to disrupt problematic genes [9]. Approved therapies like Casgevy for sickle cell disease and beta-thalassemia utilize this approach by disrupting the BCL11A gene to reactivate fetal hemoglobin production [9]. Similarly, ongoing clinical trials for hereditary transthyretin amyloidosis (hATTR) and hereditary angioedema (HAE) employ CRISPR to disrupt disease-causing genes in the liver [9].
Precision Correction Strategies
For diseases caused by loss-of-function mutations, HDR-mediated precise correction is required. The recent landmark case of an infant with CPS1 deficiency demonstrates the therapeutic potential of customized base editing, where the patient's specific mutation was corrected using a bespoke CRISPR therapy delivered via lipid nanoparticles [33]. This approach successfully corrected the faulty enzyme while allowing for multiple doses to increase editing efficiency—an advantage of LNP delivery over viral vectors [9] [33].
Enhancing Therapeutic Precision
Current research focuses on improving the precision and safety of CRISPR therapeutics through several approaches:
- Optimized gRNA design: Selecting targets with predictable editing outcomes based on sequence context [31]
- Delivery optimization: Using LNPs that enable redosing and tissue-specific targeting [9]
- Temporal control: Regulating Cas9 expression or activity to align with HDR-favorable cell cycle stages [16]
- Pathway modulation: Co-delivering HDR-enhancing factors or NHEJ inhibitors to bias repair toward precise outcomes [16]
The journey from CRISPR-induced double-strand break to final mutation outcome represents a complex interplay between targeted DNA damage and cellular repair machinery. The balance between error-prone NHEJ/MMEJ and high-fidelity HDR pathways determines whether indels or precise edits result, with target sequence context, chromatin environment, and cellular state all contributing to this decision. Understanding these dynamics is crucial for advancing both basic research and therapeutic applications of CRISPR technology. As methodologies for quantifying repair outcomes continue to improve in resolution and scale, and as clinical applications demonstrate the very real impact of controlling these processes, the field moves closer to realizing the full potential of precise genome editing for treating genetic diseases.
From Bench to Bedside: Therapeutic Applications and Clinical Trial Strategies
The therapeutic application of CRISPR-Cas9 technology for correcting genetic diseases hinges on the efficient delivery of editing components to target cells in vivo. The mechanism of CRISPR-Cas9 revolves around creating precise double-strand breaks (DSBs) in DNA, which are subsequently repaired by the cell's endogenous repair machinery [3]. The success of this editing process is profoundly influenced by the delivery system, which determines the kinetics, distribution, and persistence of the CRISPR components within the organism [34] [35]. Adeno-associated virus (AAV) vectors and lipid nanoparticles (LNPs) have emerged as the two leading platforms for in vivo delivery, each with distinct biological properties that influence DSB repair outcomes and therapeutic efficacy. This technical guide provides a comprehensive comparison of these systems for researchers and drug development professionals, focusing on their implications for CRISPR-Cas9-mediated DSB repair in genetic disease research.
Core Biology and Mechanisms
CRISPR-Cas9 and DNA Repair Pathways
CRISPR-Cas9 functions as a programmable nuclease that induces DSBs at specific genomic locations guided by RNA molecules. The Cas9 enzyme, derived from bacterial immune systems, complexes with a guide RNA (gRNA) to recognize and cleave DNA sequences adjacent to a protospacer adjacent motif (PAM) [3]. The resulting DSBs trigger cellular repair mechanisms, primarily non-homologous end joining (NHEJ) or homology-directed repair (HDR). NHEJ is an error-prone pathway that often results in small insertions or deletions (indels) that can disrupt gene function, while HDR enables precise gene correction using a donor DNA template [3] [36]. The balance between these pathways significantly influences editing outcomes in therapeutic contexts.
The repair pathway choice for Cas9-induced DSBs is influenced by multiple factors, including cell cycle stage, chromatin accessibility, and the nature of the DSB itself. While HDR is active primarily in the S and G2 phases and requires a template, NHEJ operates throughout the cell cycle without a template [3]. Additional repair pathways such as microhomology-mediated end joining (MMEJ) and single-strand annealing (SSA) represent alternative error-prone mechanisms [3]. Understanding these pathways is essential for designing effective gene therapies, as the delivery system can influence which repair mechanisms predominate.
Adeno-Associated Virus (AAV) Vectors
AAV is a non-pathogenic, single-stranded DNA virus that has been engineered as a gene delivery vector. Recombinant AAV (rAAV) retains the viral inverted terminal repeats (ITRs) but replaces viral genes with therapeutic expression cassettes, achieving a packaging capacity of approximately 4.7 kb [37] [38]. AAV vectors are characterized by their broad tissue tropism, which varies by serotype, and their ability to establish long-term transgene expression through episomal persistence in post-mitotic cells [37] [38]. The primary viral components include VP1-3 capsid proteins that determine tropism and mediate cell entry through receptor binding and endocytosis [37].
The AAV lifecycle involves several intracellular steps post-entry, including endosomal trafficking, nuclear import, capsid uncoating, and second-strand DNA synthesis—a rate-limiting step for transgene expression [38]. AAV vectors typically trigger minimal immune responses compared to other viral vectors, though pre-existing neutralizing antibodies remain a significant challenge for clinical application [37]. For CRISPR delivery, AAV's limited packaging capacity often requires creative solutions such as splitting Cas9 and gRNA expression components across multiple vectors or using smaller orthologs like SaCas9 [39].
Lipid Nanoparticles (LNPs)
LNPs are sophisticated nanocarriers composed of ionizable lipids, phospholipids, cholesterol, and PEGylated lipids that self-assemble into particles typically measuring 50-120 nm in diameter [34] [40]. These components function synergistically to encapsulate nucleic acid payloads, facilitate cellular uptake, and enable endosomal escape—a critical step for delivering CRISPR components to the cytoplasm [34] [36]. The ionizable lipids are particularly crucial, as their pH-dependent charge characteristics enable efficient RNA encapsulation during formulation and facilitate endosomal membrane disruption following cellular uptake [34] [40].
Following systemic administration, LNPs demonstrate a natural tropism for hepatocytes, making them particularly suitable for liver-directed gene editing therapies [34] [36]. However, recent advances in surface functionalization with targeting ligands such as designed ankyrin repeat proteins (DARPins) have demonstrated promising results for redirecting LNP distribution to extrahepatic tissues, including T cells [34]. Unlike AAV, LNPs provide transient expression of CRISPR components, which can reduce off-target editing risks but may require repeated administration for certain therapeutic applications [34].
Comparative Analysis of Delivery Systems
Table 1: Comparative Analysis of AAV versus LNP Delivery Systems for CRISPR-Cas9
| Parameter | Adeno-Associated Virus (AAV) | Lipid Nanoparticles (LNPs) |
|---|---|---|
| Packaging Capacity | Limited (~4.7 kb); requires miniaturized editors or dual-vector systems [37] [39] | Higher capacity; can deliver multiple CRISPR components simultaneously [34] [36] |
| Expression Kinetics | Slow onset (days to weeks); long-term persistence (months to years) [37] [38] | Rapid onset (hours to days); transient expression (days to weeks) [34] [36] |
| Immunogenicity | Pre-existing neutralizing antibodies in population; T-cell responses against capsid/transgene possible [37] | Lower immunogenicity; suitable for repeated administration [34] |
| Manufacturing | Complex biological process; weeks for production; difficult to scale [34] [37] | Synthetic process; 1-2 days for production; easily scalable [34] |
| Biodistribution | Broad tissue tropism (serotype-dependent); natural targeting of liver, muscle, CNS [37] | Primarily hepatotropic; requires engineering for extrahepatic targeting [34] [36] |
| Editing Profile | Sustained editor expression increases potential for off-target effects [34] [39] | Transient expression reduces off-target risks [34] [36] |
| Clinical Translation | Multiple approved gene therapies (e.g., Luxturna); established regulatory path [37] | Approved for COVID-19 vaccines; emerging for CRISPR (e.g., personalized therapy in 2025) [34] |
Table 2: DNA Repair Outcomes by Delivery System
| Repair Aspect | AAV-Mediated Delivery | LNP-Mediated Delivery |
|---|---|---|
| Primary Repair Pathway | HDR favored in dividing cells; NHEJ predominant in post-mitotic tissues [3] | NHEJ predominates due to transient expression; HDR possible with optimized timing [36] |
| Editing Efficiency | High in permissive tissues; varies by serotype and promoter [37] | High in hepatocytes; improving for other cell types [34] |
| Therapeutic Window | Single administration sufficient; editing persistence months to years [37] [38] | May require repeated dosing for sustained effect; enables "dose to effect" approach [34] |
| Genomic Safety | Potential for AAV genome integration; extended editor expression increases off-target risks [37] [39] | Minimal genomic integration; transient expression limits off-target exposure [34] [36] |
Experimental Protocols
AAV Vector Production and Validation
Protocol: Production of rAAV via Triple Transfection in HEK293 Cells
Materials:
Methodology:
- Cell Culture: Expand HEK293 cells in appropriate medium to 70-80% confluency for adherent cultures or optimal density for suspension cultures.
- Triple Transfection: Co-transfect cells with the three plasmid system at a predetermined optimal ratio (typically 1:1:1) using transfection reagent.
- Harvesting: Collect cells and media 48-72 hours post-transfection.
- Purification: Lyse cells to release viral particles, then purify via cesium chloride gradient ultracentrifugation or affinity chromatography.
- Titration: Determine viral genome titer using quantitative PCR and assess purity via SDS-PAGE and analytical ultracentrifugation.
- Quality Control: Test for endotoxin, sterility, and replication-competent AAV [37] [38].
LNP Formulation and Characterization
Protocol: Microfluidic Formulation of CRISPR-LNPs
Materials:
Methodology:
- Lipid Preparation: Dissolve lipid components in ethanol at specific molar ratios (e.g., 50:10:38.5:1.5 for ionizable lipid:DSPC:cholesterol:PEG-lipid).
- Aqueous Phase Preparation: Dilute CRISPR payload in citrate buffer (pH 4.0) to promote electrostatic interactions with ionizable lipids.
- Microfluidic Mixing: Use a staggered herringbone micromixer with precise flow rates (typically 1:3 aqueous:ethanol ratio) to facilitate rapid mixing and LNP self-assembly.
- Dialyzation: Dialyze formulated LNPs against PBS (pH 7.4) to remove ethanol and establish neutral pH.
- Concentration: Concentrate LNPs using tangential flow filtration to desired concentration.
- Characterization: Measure particle size (dynamic light scattering), polydispersity index, encapsulation efficiency (RiboGreen assay), and in vitro potency [34] [36] [40].
Visualization of Mechanisms and Workflows
Diagram 1: LNP-mediated CRISPR Delivery Mechanism. This workflow illustrates the complete pathway from nanoparticle formulation to intracellular gene editing.
Diagram 2: AAV-mediated CRISPR Delivery Pathway. This visualization details the intracellular journey of AAV vectors from initial receptor binding to eventual genome editing.
Diagram 3: DNA Repair Pathways for CRISPR-Cas9-Induced Double-Strand Breaks. This diagram illustrates the competing cellular mechanisms for repairing DSBs, highlighting the different outcomes that influence therapeutic strategy selection.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Delivery System Development
| Reagent Category | Specific Examples | Research Function |
|---|---|---|
| Ionizable Lipids | ALC-0315, SM-102, ALC-0315 [34] [40] | Enable RNA encapsulation and endosomal escape through pH-dependent charge reversal |
| Helper Lipids | DSPC, DOPE, Cholesterol [36] [40] | Enhance LNP stability, membrane fusion, and structural integrity |
| PEGylated Lipids | ALC-0159, PEG-DMG [34] [40] | Improve nanoparticle stability, reduce aggregation, and modulate pharmacokinetics |
| AAV Serotypes | AAV2, AAV8, AAV9, AAVrh74 [37] | Provide distinct tissue tropism profiles for targeting different organs |
| Capsid Engineering | DARPins, Peptide Ligands [34] [39] | Enhance tissue specificity and transduction efficiency through targeted modifications |
| Promoter Systems | CAG, CBh, SYN, MiniPromoters [37] [39] | Control transgene expression levels and cell-type specificity |
| CRISPR Payloads | saCas9, mRNA, RNP, Base Editors [36] [39] | Provide genome editing functionality with varying sizes and persistence |
| Analytical Tools | ddPCR, NSEM, AUC, RiboGreen [37] [40] | Characterize vector properties, titer determination, and encapsulation efficiency |
The selection between AAV and LNP delivery systems for CRISPR-Cas9-mediated in vivo editing represents a critical strategic decision that directly influences DSB repair outcomes and therapeutic efficacy. AAV vectors offer the advantage of durable transgene expression and established clinical applications but face challenges related to packaging capacity, immunogenicity, and manufacturing scalability [37] [39]. In contrast, LNPs provide flexible payload capacity, transient expression that may enhance safety, and streamlined manufacturing, though they currently exhibit limited tropism beyond hepatocytes and relatively short persistence [34] [36].
The optimal delivery system varies based on therapeutic objectives, target tissue, and desired editing profile. For long-term correction of genetic diseases in post-mitotic tissues, AAV's persistent expression remains advantageous. For applications requiring precise temporal control or multiple administrations, LNP's transient nature provides superior flexibility. Future directions include engineering hybrid approaches that combine favorable attributes of both systems, developing novel capsids and lipid formulations with enhanced tissue specificity, and optimizing delivery strategies to favor precise HDR over error-prone repair pathways. As both platforms continue to evolve, they will undoubtedly expand the therapeutic landscape for CRISPR-based interventions against genetic diseases.
The approval of Casgevy (exagamglogene autotemcel) by the U.S. Food and Drug Administration (FDA) in December 2023 marked a historic milestone in medicine, representing the first-ever authorized therapeutic application of CRISPR-Cas9 genome editing technology [41]. This autologous cell-based gene therapy is indicated for patients aged 12 years and older with sickle cell disease (SCD) experiencing recurrent vaso-occlusive crises and for those with transfusion-dependent β-thalassemia (TDT) [42] [43]. The regulatory endorsement of Casgevy validates nearly a decade of clinical development and establishes a precedent for the approval of genome editing therapies, signaling a transformative shift from conventional disease management toward potentially curative genetic interventions.
The scientific and regulatory significance of Casgevy extends beyond its therapeutic applications for hemoglobinopathies. As the inaugural CRISPR-based therapy to navigate the rigorous FDA review process, it has established a regulatory framework for assessing the safety, efficacy, and manufacturing requirements of precision gene editing products [44]. This case study examines the technical mechanism, clinical evidence, and regulatory considerations of Casgevy within the broader context of CRISPR-Cas9-mediated double-strand break repair mechanisms in genetic disease research.
CRISPR-Cas9 and DNA Repair Mechanisms: Technical Foundations
The CRISPR-Cas9 Molecular Machinery
The CRISPR-Cas9 system functions as a precise DNA-targeting platform derived from bacterial adaptive immunity. The therapeutic application in Casgevy utilizes two fundamental components: the Cas9 enzyme, which acts as a programmable molecular scissor to create double-strand breaks (DSBs) in DNA, and a single-guide RNA (sgRNA), which directs Cas9 to a specific genomic locus through complementary base-pairing [42] [43]. This complex induces a site-specific DSB at the target sequence, activating the cell's innate DNA repair mechanisms.
The precision of this system enables targeting of the erythroid-specific enhancer region of the BCL11A gene, a key transcriptional regulator of the developmental switch from fetal to adult hemoglobin [42]. This targeted approach distinguishes Casgevy from viral vector-based gene therapies by directly modifying endogenous genetic regulation rather than adding supplemental genetic material.
DNA Repair Pathways in Genome Editing
Following DSB induction, mammalian cells primarily activate two distinct repair pathways with different therapeutic outcomes:
- Non-Homologous End Joining (NHEJ): An error-prone repair pathway that directly ligates broken DNA ends, often resulting in small insertions or deletions (indels) at the cleavage site [45]. In Casgevy, NHEJ repair following cleavage of the BCL11A enhancer region introduces disruptive mutations that reduce BCL11A expression specifically in erythroid lineage cells [42].
- Homology-Directed Repair (HDR): A precise repair mechanism that uses a DNA template to faithfully restore sequence at the breakpoint [45]. While HDR offers theoretical advantages for certain applications, its lower efficiency in hematopoietic stem cells compared to NHEJ makes it less suitable for the knockout strategy employed in Casgevy.
Emerging research reveals that CRISPR editing can occasionally generate more complex genomic alterations beyond simple indels, including large structural variations (SVs), chromosomal translocations, and megabase-scale deletions [45]. These findings highlight the importance of comprehensive genomic safety assessments in therapeutic genome editing.
The following diagram illustrates the molecular mechanism of CRISPR-Cas9 and the subsequent cellular repair pathways:
Casgevy Therapeutic Mechanism: From Genetic Target to Physiological Outcome
BCL11A as a Genetic Master Switch
Casgevy's therapeutic approach leverages fundamental insights into hemoglobin biology and regulation. The BCL11A gene encodes a transcription factor that functions as a master regulator of the fetal-to-adult hemoglobin switch [42]. Shortly after birth, increased BCL11A expression represses γ-globin gene expression, thereby reducing fetal hemoglobin (HbF) production and activating adult β-globin synthesis [46]. In both SCD and TDT, this developmental transition coincides with the onset of disease pathology:
- In SCD, a single nucleotide substitution in the β-globin gene leads to production of sickle hemoglobin (HbS), which polymerizes under deoxygenated conditions, causing erythrocyte sickling, vaso-occlusion, and tissue ischemia [42].
- In TDT, mutations in the β-globin gene cause reduced or absent β-globin synthesis, creating an imbalance in α- and non-α-globin chains that leads to ineffective erythropoiesis, hemolytic anemia, and compensatory transfusion dependence [42].
Mimicking a Natural Protective Variant
Casgevy's mechanism mimics the hereditary persistence of fetal hemoglobin (HPFH), a natural condition where individuals continue to express elevated HbF levels into adulthood due to genetic variations [42]. HPFH attenuates the clinical severity of SCD and TDT, providing a validated physiological model for therapeutic intervention.
The therapeutic workflow involves several key stages:
- HSC Collection: CD34+ hematopoietic stem cells are collected from the patient via apheresis after mobilization from bone marrow [46].
- Ex Vivo Editing: Cells are electroporated with CRISPR-Cas9 components targeting the BCL11A enhancer region [42].
- Myeloablative Conditioning: Patients receive busulfan conditioning to create niche space for edited cells [46].
- Reinfusion: Edited CD34+ cells are infused back into the patient [46].
- Engraftment and Differentiation: Edited HSCs engraft in bone marrow and reconstitute hematopoiesis with reduced BCL11A expression in erythroid lineage [42].
The edited erythroid precursor cells exhibit reduced BCL11A expression, leading to derepressed γ-globin transcription and significantly increased HbF production [42]. In SCD, elevated HbF concentrations prevent HbS polymerization, reducing sickling and its associated complications. In TDT, increased γ-globin chains pair with excess α-globin chains, improving hemoglobinization and red blood cell survival while reducing transfusion requirements.
The following workflow diagrams the therapeutic process from cell collection to physiological effect:
Clinical Trial Data and Efficacy Outcomes
Clinical Trial Design and Patient Populations
The regulatory approval of Casgevy was supported by evidence from ongoing single-arm, multi-center trials evaluating safety and efficacy in defined patient populations [41]:
- For SCD, the trial enrolled patients aged 12-35 with history of at least two severe vaso-occlusive crises (VOCs) annually in the two years preceding screening [41].
- For TDT, the trial included patients with documented β-thalassemia who required regular red blood cell transfusions (≥6 transfusions annually in the two years before screening) [42].
The primary efficacy endpoints were disease-specific:
- SCD: Freedom from severe VOCs for at least 12 consecutive months within the 24-month follow-up period [41].
- TDT: Maintenance of transfusion-free status for at least 12 consecutive months with maintained weighted average hemoglobin ≥9 g/dL [42].
Quantitative Efficacy Results
Clinical trials demonstrated substantial treatment effects across both disease populations, with outcomes sustained through follow-up periods.
Table 1: Casgevy Efficacy Outcomes in Sickle Cell Disease Clinical Trials
| Parameter | SCD Trial Results | Follow-up Duration |
|---|---|---|
| Patients with sufficient follow-up | 31 of 44 treated patients | 24 months |
| Achieving primary endpoint (freedom from severe VOCs) | 29 patients (93.5%) | ≥12 consecutive months |
| Successful engraftment rate | 44 patients (100%) | 24 months |
| Graft failure or rejection | 0 patients | 24 months |
Table 2: Casgevy Efficacy Outcomes in Transfusion-Dependent β-Thalassemia Clinical Trials
| Parameter | TDT Trial Results | Follow-up Duration |
|---|---|---|
| Patients achieving transfusion independence | >90% of patients | ≥12 consecutive months |
| Weighted average hemoglobin level | Maintained ≥9 g/dL | During transfusion-free period |
| Successful engraftment rate | >90% of patients | Through follow-up period |
The high response rates observed in clinical trials demonstrate the potentially transformative nature of Casgevy for eligible patients. The durable elimination of VOCs in SCD and achievement of transfusion independence in TDT represent unprecedented therapeutic outcomes for these previously incurable genetic disorders [42] [41].
Regulatory Pathway and Safety Assessment
Regulatory Designations and Review Process
Casgevy received multiple regulatory designations that facilitated its development and review:
- Breakthrough Therapy Designation: Granted based on preliminary clinical evidence indicating substantial improvement over available therapies [41].
- Orphan Drug Designation: Assigned for treatment of SCD, reflecting the rare disease status of the indication [41].
- Regenerative Medicine Advanced Therapy (RMAT): Provided to expedite development and review of promising regenerative medicine products [41].
- Priority Review: Designated for applications that would provide significant improvements in safety or effectiveness [41].
The FDA's comprehensive evaluation included assessment of on-target editing efficiency, potential off-target editing risks, cellular product purity and potency, and long-term safety monitoring [44]. The agency required a Risk Evaluation and Mitigation Strategy (REMS) program and long-term follow-up studies spanning 15 years to monitor delayed adverse events.
Safety Profile and Risk Management
Clinical trials identified a predictable safety profile consistent with myeloablative conditioning and autologous hematopoietic stem cell transplantation:
Table 3: Casgevy Safety Profile and Adverse Events
| Safety Parameter | Observed Events | Management Strategy |
|---|---|---|
| Most common adverse events | Low platelet/white blood cell levels, mouth sores, nausea, musculoskeletal pain, abdominal pain, vomiting, febrile neutropenia, headache, itching [41] | Supportive care, growth factors, antimicrobial prophylaxis |
| Neutrophil engraftment | Achieved in all patients (median time ~29 days) [42] | Monitor absolute neutrophil counts; manage infections per guidelines |
| Platelet engraftment | Delayed in some patients [42] | Frequent platelet counts; monitor for bleeding |
| Hypersensitivity reactions | Potential risk to cryopreservatives (DMSO, dextran 40) [42] | Monitor during and after infusion |
| Off-target editing risk | Not observed in clinical trials but cannot be ruled out [42] | Comprehensive genomic assessment in product testing |
The FDA approval recognized the favorable benefit-risk profile of Casgevy, particularly for patients with severe disease manifestations despite available treatments [41]. The absence of graft rejection or failure in clinical trials represents a significant advantage over allogeneic transplantation [41].
Manufacturing and Quality Control
Production Workflow and Timeline
Casgevy manufacturing involves a complex, patient-specific process requiring stringent quality controls:
- Cell Collection (~1 week): CD34+ hematopoietic stem cells are collected via apheresis after mobilization with granulocyte colony-stimulating factor (G-CSF) and plerixafor [46].
- Cell Editing and Expansion (~3-6 months): Cells are electroporated with CRISPR-Cas9 ribonucleoprotein complex targeting the BCL11A enhancer region, then expanded ex vivo [42].
- Quality Control Testing: Comprehensive assessments include viability, potency, sterility, and genomic integrity testing [42].
- Cryopreservation and Shipping: The final product is cryopreserved and shipped to the treatment center [46].
- Product Administration: After myeloablative conditioning, cells are thawed and infused into the patient [46].
The extended manufacturing timeline (up to 6 months from cell collection to infusion) presents unique challenges for patient management during the interim period [46].
Critical Reagents and Research Solutions
The development and production of Casgevy requires specialized reagents and platform technologies that enable precise genome editing:
Table 4: Essential Research Reagents and Platform Technologies for CRISPR Therapeutics
| Reagent/Technology | Function in Casgevy Production | Technical Specifications |
|---|---|---|
| CRISPR-Cas9 RNP Complex | Site-specific DNA cleavage at BCL11A enhancer | High-purity Cas9 nuclease complexed with synthetic sgRNA |
| Electroporation System | Non-viral delivery of editing components | Optimized parameters for HSC transfection |
| CD34+ Cell Selection Reagents | Isolation of hematopoietic stem cells | Immunomagnetic bead-based separation (e.g., CliniMACS) |
| Stem Cell Mobilization Agents | Enhance HSC release from bone marrow | G-CSF and plerixafor (CXCR4 antagonist) |
| Myeloablative Conditioning Agent | Create marrow niche for engraftment | Busulfan chemotherapy |
| Cryopreservation Medium | Long-term storage of cell product | DMSO-based cryoprotectant formulation |
Emerging Challenges and Future Directions
Safety Considerations in Therapeutic Genome Editing
While Casgevy has demonstrated a favorable safety profile in clinical trials, emerging research highlights potential challenges in therapeutic genome editing that require continued investigation:
- Structural Variations: Recent studies identified that CRISPR editing can induce large structural variations (SVs), including chromosomal translocations and megabase-scale deletions, particularly when using DNA-PKcs inhibitors to enhance editing efficiency [45].
- On-Target Genomic Aberrations: Beyond intended edits, CRISPR can cause kilobase- to megabase-scale deletions at the on-target site that may delete critical regulatory elements or adjacent genes [45].
- p53-Mediated Stress Responses: DNA damage from editing may activate p53 pathways, potentially selecting for p53-deficient cell clones with oncogenic potential [45].
For Casgevy specifically, studies have reported large kilobase-scale deletions upon BCL11A editing in hematopoietic stem cells, though the clinical significance of these findings requires further investigation [45]. These observations underscore the importance of advanced analytical methods like CAST-Seq and LAM-HTGTS that can detect complex genomic rearrangements beyond simple indels [45].
Clinical Implementation and Access Considerations
The translation of Casgevy from regulatory approval to widespread clinical application faces several practical challenges:
- Treatment Capacity: As of mid-2025, over 75 authorized treatment centers (ATCs) have been activated globally, with approximately 115 patients having completed cell collection and 29 patients having received Casgevy infusions [47].
- Economic Considerations: With anticipated costs exceeding $2 million per treatment, securing reimbursement from payers represents a significant barrier to patient access [9].
- Manufacturing Scalability: The patient-specific, multi-month manufacturing process creates inherent limitations on treatment capacity compared to conventional pharmaceuticals [47].
Next-generation approaches under development aim to address these limitations, including targeted conditioning agents (anti-CD117 antibody-drug conjugates) and in vivo editing platforms that could eliminate the need for ex vivo manufacturing and myeloablative conditioning [47].
The approval of Casgevy represents a watershed moment for genetic medicine, establishing CRISPR-Cas9 genome editing as a viable therapeutic modality. Its success validates a sophisticated mechanism-based approach that mimics protective natural genetic variants rather than simply compensating for defective genes. The rigorous regulatory assessment of Casgevy has created a pathway for future genome editing therapies, with implications extending far beyond hemoglobinopathies to encompass numerous genetic disorders.
Ongoing monitoring of long-term safety outcomes and continued technological innovations to enhance precision, efficiency, and accessibility will shape the future evolution of CRISPR-based therapeutics. As the inaugural FDA-approved CRISPR therapy, Casgevy has transformed the therapeutic landscape for severe hemoglobinopathies while establishing foundational regulatory and manufacturing precedents that will guide the next generation of genetic medicines.
The advent of in vivo CRISPR/Cas9-based genome editing represents a paradigm shift in the treatment of genetic disorders. This whitepaper examines the clinical progress of Intellia Therapeutics' two leading in vivo gene editing programs: nexiguran ziclumeran (nex-z) for hereditary transthyretin (ATTR) amyloidosis and lonvoguran ziclumeran (lonvo-z) for hereditary angioedema (HAE). Both programs utilize lipid nanoparticle (LNP) delivery to target disease-causing genes in the liver, demonstrating durable protein reduction and promising clinical efficacy across multiple Phase 1/2 trials. Recent Phase 3 developments include a clinical hold for nex-z following a serious adverse event of liver toxicity, highlighting the critical importance of ongoing safety monitoring as these pioneering therapies advance through clinical development. The data summarized herein provide compelling evidence for the potential of in vivo liver editing to achieve systemic therapeutic effects with a single administration.
The CRISPR/Cas9 system functions as a precise DNA-targeting platform derived from bacterial adaptive immunity. For in vivo therapeutic applications, the system requires two key components: (1) the Cas9 nuclease enzyme that creates double-strand breaks in DNA, and (2) a guide RNA (gRNA) that directs Cas9 to specific genomic sequences via Watson-Crick base pairing. Upon binding to target DNA, Cas9 induces a double-strand break (DSB) that is subsequently repaired by endogenous cellular mechanisms, primarily the non-homologous end joining (NHEJ) pathway. NHEJ is an error-prone repair process that often results in insertions or deletions (indels) that disrupt the targeted gene's function, making it ideal for therapeutic gene inactivation [15] [48].
The liver has emerged as a prime target for in vivo gene editing due to its accessibility to systemically administered vectors, its role in producing numerous plasma proteins, and its tolerogenic immune environment. Intellia's platform employs LNPs to deliver CRISPR components specifically to hepatocytes, leveraging the natural tropism of LNPs for hepatic tissue after intravenous administration. This approach enables transient expression of the editing machinery, potentially reducing off-target risks while achieving permanent genomic modifications [48] [49].
Therapeutic Applications and Clinical Outcomes
Nexiguran Ziclumeran (nex-z) for Hereditary ATTR Amyloidosis
Nex-z is designed as a one-time therapy for ATTR amyloidosis by inactivating the TTR gene in the liver, thereby reducing production of both wild-type and mutant transthyretin protein. The misfolding and aggregation of TTR protein forms amyloid deposits in tissues including the heart and nerves, leading to progressive organ dysfunction [50].
Table 1: Clinical Outcomes for Nex-z in ATTR Amyloidosis
| Parameter | Results | Follow-up Period | Clinical Implications |
|---|---|---|---|
| Serum TTR Reduction | Mean reduction of 87% | 36 months (n=9) | Potential to halt amyloid deposition |
| Cardiac Biomarkers | 70% patients stable/improved NT-proBNP; 85% stable/improved hs-Troponin T | 24 months | Suggests disease stabilization |
| Functional Status | 69% patients stable/improved in 6-minute walk test | 24 months | Preservation of physical capacity |
| NYHA Class | 81% patients stable/improved; 83% improvement in NYHA Class III | 24 months | Improvement in heart failure classification |
| Quality of Life | Benefit regardless of NYHA class baseline (KCCQ) | 24 months | Enhanced patient-reported outcomes |
The Phase 1 trial has demonstrated rapid, deep, and durable reductions in serum TTR, with all patients maintaining sustained response through the latest follow-up. Across multiple measures of cardiomyopathy, patients showed evidence of disease stabilization or improvement at 24 months compared to baseline [50]. Notably, these editing effects have persisted through three years of follow-up, supporting the potential for a one-time treatment to provide lifelong benefit [50].
Safety Update for Nex-z
In October 2025, Intellia announced a temporary pause in dosing and screening for the Phase 3 MAGNITUDE and MAGNITUDE-2 trials following a serious adverse event involving grade 4 liver toxicity and hyperbilirubinemia in a dosed ATTR-CM patient. The affected individual was hospitalized with life-threatening liver injury, meeting Hy's Law criteria for drug-induced liver damage. This event marks the first reported instance of severe hepatotoxicity in the nex-z clinical program, occurring in less than 1% of all patients enrolled in MAGNITUDE. Prior safety data from over 450 trial participants had shown nex-z to be generally well-tolerated, with most adverse events being mild or moderate infusion reactions. The company is investigating potential risk factors and implementing additional hepatic safety measures before resuming trials [51] [52].
Lonvoguran Ziclumeran (lonvo-z) for Hereditary Angioedema
Lonvo-z aims to prevent HAE attacks by inactivating the KLKB1 gene encoding prekallikrein, a key precursor protein in the kallikrein-kinin pathway that is dysregulated in HAE. By reducing kallikrein production, lonvo-z addresses the underlying pathophysiology of HAE, characterized by unpredictable and potentially life-threatening swelling attacks [53] [54].
Table 2: Clinical Outcomes for Lonvo-z in Hereditary Angioedema
| Parameter | Results (50 mg dose) | Patient Population | Significance |
|---|---|---|---|
| Kallikrein Reduction | Mean reduction of 89% | 24 months (n=32) | Sustained target engagement |
| Attack-Free Status | 31 of 32 patients (97%) attack-free and LTP-free | As of data cutoff | Near-complete clinical response |
| Durability of Response | 24 of 32 patients (75%) attack-free for ≥7 months (up to 32 months) | Patients with longest follow-up | Long-term efficacy demonstration |
| Phase 2 Cohort | 10 of 11 patients attack-free and LTP-free | Original 50 mg recipients | Consistent treatment effect |
Pooled analysis of 32 patients who received the 50 mg dose of lonvo-z demonstrated profound and durable reductions in plasma kallikrein, with corresponding dramatic decreases in HAE attack frequency. The vast majority of patients became completely free from both attacks and long-term prophylaxis medications, with effects maintained for up to 32 months of follow-up [53] [54]. The Phase 3 HAELO trial completed enrollment in September 2025, with topline data expected by mid-2026 and potential U.S. commercial launch in the first half of 2027 [51].
Safety Profile of Lonvo-z
Lonvo-z has demonstrated a well-tolerated safety profile with up to three years of patient follow-up and no new long-term risks identified. The most frequent treatment-emergent adverse events within 28 days of infusion were infusion-related reactions, fatigue, and headache. Beyond 28 days, the most common events were nasopharyngitis, upper respiratory tract infection, back pain, arthralgia, and COVID-19. A single Grade 2 AST elevation was reported, which spontaneously resolved within days. One serious adverse event of pulmonary embolism occurred in a patient with multiple risk factors one year post-infusion and resolved without sequelae [53] [54].
Molecular Mechanisms and Editing Outcomes
The therapeutic effects of nex-z and lonvo-z stem from their precise targeting of disease-causing genes in hepatocytes, leveraging the cell's endogenous DNA repair machinery to achieve permanent gene inactivation.
DNA Repair Pathways and Editing Outcomes
The double-strand breaks induced by Cas9 are primarily repaired through non-homologous end joining (NHEJ) in post-mitotic hepatocytes. This repair pathway is particularly active in non-dividing cells and typically results in small insertions or deletions (indels) at the cleavage site. When these indels occur within protein-coding exons and disrupt the reading frame, they lead to premature stop codons and effective gene knockout [12] [48]. Recent research has revealed that DNA repair outcomes can differ significantly between dividing and non-dividing cells, with post-mitotic cells like hepatocytes and neurons exhibiting prolonged timelines for indel accumulation and distinct repair factor expression patterns compared to rapidly dividing cells [12].
Structural Variation Risks
Beyond intended indels, CRISPR/Cas9 editing can potentially lead to larger-scale genomic rearrangements including chromosomal translocations and megabase-scale deletions. These structural variations represent significant safety considerations for therapeutic editing applications. The risk of such events may be exacerbated by certain editing enhancement strategies, particularly the use of DNA-PKcs inhibitors to promote homology-directed repair. Conventional short-read sequencing methods often fail to detect these large deletions, potentially leading to overestimation of precise editing outcomes [15]. Intellia's clinical data, with extended follow-up exceeding three years, have not indicated emergence of such events, suggesting that LNP-delivered editing without repair pathway manipulation may mitigate these risks [53] [50].
Experimental Protocols and Methodologies
Clinical Trial Designs
The clinical development programs for both nex-z and lonvo-z employ methodically dose-escalation studies to establish safety and efficacy profiles:
Nex-z Phase 1 Protocol:
- Design: Open-label, dose-escalation trial in adults with ATTRv-PN or ATTR-CM
- Dosing: Single intravenous infusion of nex-z at 0.1, 0.3, 0.7, or 1.0 mg/kg
- Endpoints: Safety, tolerability, pharmacodynamics (serum TTR reduction), clinical measures (mNIS+7 for ATTRv-PN, cardiac biomarkers for ATTR-CM)
- Follow-up: Long-term monitoring for durability and late-emerging effects [50]
Lonvo-z Phase 1/2 Protocol:
- Design: Two-part global trial in adults with HAE Types I or II
- Dosing: Single IV infusion at 25 mg or 50 mg
- Endpoints: Safety, kallikrein reduction, monthly HAE attack rate, use of rescue medication
- Phase 3 HAELO Trial: Randomized, double-blind, placebo-controlled with 50 mg dose [53] [51]
Analytical Methods for Editing Assessment
Comprehensive assessment of editing outcomes requires multiple complementary techniques:
- Digital droplet PCR (ddPCR): Quantification of editing frequencies in circulating cells
- LC-MS/MS: Precise measurement of target protein reduction in serum (TTR, kallikrein)
- Next-generation sequencing (NGS): Detection of on-target editing spectra and identification of potential off-target events
- Long-read sequencing and CAST-Seq: Identification of large structural variations and chromosomal rearrangements that may be missed by short-read sequencing [15]
Safety Considerations and Risk Mitigation
The clinical development of in vivo CRISPR therapeutics requires meticulous safety assessment encompassing both anticipated and potential unforeseen risks:
On-Target and Off-Target Editing
Traditional safety concerns for CRISPR-based therapies have focused on off-target editing at genomic sites with sequence similarity to the intended target. However, emerging evidence suggests that on-target structural variations may represent a more significant risk. Intellia's approach utilizes LNP-delivered Cas9-gRNA ribonucleoprotein (RNP) complexes, which provide transient editing activity that may limit both on-target and off-target effects compared to viral vector delivery methods that enable prolonged nuclease expression [15] [49].
Immune Responses and Hepatotoxicity
As demonstrated by the recent clinical hold for nex-z, hepatotoxicity remains a critical safety consideration for LNP-delivered therapies targeting the liver. The grade 4 liver transaminase elevations and hyperbilirubinemia observed in one patient highlight the importance of careful patient selection, monitoring, and potential prophylactic management. The company has responded by implementing enhanced laboratory monitoring protocols across all clinical sites while investigating potential contributing factors [51] [52].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for In Vivo Liver Editing Studies
| Reagent/Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Delivery Systems | LNPs, AAVs, Virus-like Particles (VLPs) | In vivo delivery of editing machinery | LNP preferred for liver: transient expression, redosing capability, scalability [49] |
| Nuclease Systems | Cas9, base editors, prime editors | Inducing DNA modifications | Cas9 for gene knockout; base/prime editors for precise point mutations [48] |
| gRNA Design Tools | In silico prediction algorithms | Target selection and off-target risk assessment | Critical for maximizing on-target efficiency, minimizing off-target effects [15] |
| Analytical Methods | ddPCR, NGS, CAST-Seq, LAM-HTGTS | Assessment of editing outcomes and safety | Multi-platform approach essential for comprehensive evaluation [15] |
| Cell Models | iPSCs, iPSC-derived hepatocytes, primary hepatocytes | Preclinical testing of editing efficiency | Species-specific differences require careful translation to human applications [12] |
Intellia's nex-z and lonvo-z programs represent pioneering advancements in in vivo therapeutic genome editing, demonstrating compelling clinical proof-of-concept for CRISPR-based interventions targeting the liver. The durable TTR and kallikrein reduction observed in these trials supports the potential for one-time treatments to provide lifelong benefit for patients with ATTR amyloidosis and HAE, respectively.
The recent serious adverse event with nex-z underscores that despite promising efficacy, the safety profile of in vivo editing requires continued rigorous evaluation. As the field advances, key areas for development include optimized delivery systems with enhanced tissue specificity, novel editing platforms with improved precision, and comprehensive analytical methods to fully characterize editing outcomes. The anticipated Phase 3 data for lonvo-z in mid-2026 may mark a significant milestone in establishing in vivo CRISPR editing as a transformative therapeutic modality.
Researchers and clinicians should maintain cautious optimism regarding these technologies, recognizing their extraordinary potential while acknowledging the need for meticulous safety assessment and long-term monitoring as these innovative treatments progress through clinical development.
Novel 2025 Frontiers: Personalized CRISPR for Ultra-Rare Diseases and Epigenetic Editing
The year 2025 marks a pivotal transformation in CRISPR-based therapeutics, moving from one-size-fits-all treatments toward two revolutionary paradigms: personalized gene editing for ultra-rare diseases and epigenetic editing for sophisticated transcriptional control. These approaches are fundamentally linked through their shared reliance on the core mechanisms of DNA double-strand break (DSB) repair. This technical guide examines the cutting-edge scientific advances, experimental protocols, and clinical trial designs that are forging a new era of precision medicine. We detail how platform-based "umbrella" trials are overcoming regulatory hurdles for personalized therapies and how all-RNA epigenetic editors are achieving durable multiplexed gene silencing without genotoxic risks. For researchers and drug development professionals, this whitepaper provides a comprehensive analysis of the methodologies, reagents, and mechanistic insights driving the next generation of CRISPR therapeutics.
Personalized CRISPR Platforms for Ultra-Rare Diseases
The traditional drug development model is economically unfeasible for ultra-rare diseases, where patient populations may number only in the dozens worldwide. The landmark case of "Baby KJ" represents a paradigm shift, demonstrating a platform approach to creating patient-specific therapies within a clinically relevant timeframe [55].
Case Study: Personalized In Vivo Editing for CPS1 Deficiency
Background: Carbamoyl phosphate synthetase 1 (CPS1) deficiency is a one-in-a-million autosomal recessive urea cycle disorder that causes lethal ammonia accumulation due to mutations in the CPS1 gene [56]. Conventional management requires a strict protein-restricted diet and eventual liver transplantation.
Therapeutic Strategy: Researchers at CHOP and Penn Medicine developed a bespoke CRISPR-Cas9 therapy to correct a private mutation in the CPS1 gene specifically for Baby KJ [9]. The approach utilized:
- Delivery System: Lipid nanoparticles (LNPs) for in vivo delivery to hepatocytes
- Editing Component: CRISPR-Cas9 system designed to correct the specific point mutation via homology-directed repair (HDR)
- Dosing Regimen: Initial low dose followed by two higher doses, leveraging the re-dosability of LNP delivery [9]
Table 1: Clinical Outcomes for Baby KJ Following Personalized CRISPR Treatment
| Parameter | Pre-Treatment Status | Post-Treatment Observation | Significance |
|---|---|---|---|
| Ammonia Levels | Required medication control | Reduced medication dependence | Metabolic function improvement |
| Protein Tolerance | Severely restricted | Increased dietary protein intake | Functional enzyme restoration |
| Stress Response | High risk during illness | Successfully overcame infections | Clinical resilience achieved |
| Safety Profile | N/A | No serious adverse events | LNP delivery platform validated |
Regulatory Innovation: The "Umbrella Trial" Framework
A critical breakthrough enabling personalized CRISPR involves redefining the regulatory pathway. Rather than treating each variant-specific therapy as a new drug, researchers have reached agreement with the FDA on "umbrella" trial designs [56].
Protocol Overview:
- Platform Design: Single clinical trial protocol enrolling patients with different variants across multiple genes
- Unified Mechanism: All therapies utilize the same core editing platform with only the guide RNA swapped to address specific mutations
- Statistical Endpoints: Approval potentially achievable with 5-10 successfully treated participants per variant, dramatically reducing recruitment barriers [56]
2026 Clinical Trial Implementation: Researchers plan to launch an umbrella trial for 7 different urea cycle disorders caused by variants in any of 7 genes, all correctable by the same gene editor used in Baby KJ's therapy [56].
Advanced Epigenetic Editing Technologies
While traditional CRISPR-Cas9 induces DNA double-strand breaks, epigenetic editing represents a more nuanced approach that modifies gene expression without altering the underlying DNA sequence. The CRISPRoff platform exemplifies this next-generation technology.
CRISPRoff: Durable Multiplexed Gene Silencing
System Architecture: CRISPRoff utilizes a catalytically inactive dCas9 fused to DNA methyltransferase domains (DNMT3A, DNMT3L) and the KRAB repression domain from ZNF10 [57]. This fusion protein enables targeted DNA methylation and subsequent gene silencing without DNA cleavage.
Delivery Optimization: Researchers systematically compared seven mRNA variants differing in:
- Cap structure (Cap1 selected)
- Nucleoside base modifications (1-methylpseudouridine incorporation)
- Codon optimization algorithms [57]
The optimal design (CRISPRoff 7) achieved complete silencing in 85-99% of primary human T-cells at high mRNA doses with no observed toxicity [57].
Table 2: Performance Comparison of Gene Silencing Technologies in T-Cells
| Technology | Editing Mechanism | Durability | Multiplexing Capacity | Cytotoxicity |
|---|---|---|---|---|
| CRISPR-Cas9 Nuclease | DNA double-strand break | Permanent (indels) | High toxicity with 3-5 targets | Significant chromosomal abnormalities |
| CRISPRi (Interference) | Transcriptional repression | Progressive loss over time | Limited by sustained protein expression | Low |
| CRISPRoff (Epigenetic) | DNA methylation | Stable through ~30-80 cell divisions | 65.8% efficiency with 5 targets | Minimal toxicity |
Experimental Protocol: CRISPRoff in CAR-T Cell Engineering
Workflow for Enhanced CAR-T Cell Manufacturing:
- mRNA Delivery: Electroporation of optimized CRISPRoff mRNA into primary human T-cells
- Target Selection: Design guide RNAs against immunosuppressive checkpoint genes (e.g., RASA2, FAS, PTPN2)
- Epigenetic Silencing: Incubation for methylation establishment and stable gene repression
- CAR Integration: Orthogonal CRISPR-Cas12a RNP for targeted CAR knock-in at TRAC locus
- Functional Validation: In vitro and in vivo assessment of antitumor activity [57]
Key Findings: RASA2-silenced CAR-T cells maintained superior tumor control in NSG mice bearing Nalm6 leukemia and demonstrated persistent silencing even after adoptive transfer and tumor infiltration [57].
Molecular Mechanisms: DSB Repair in Therapeutic Context
The divergent approaches of personalized correction versus epigenetic silencing represent different strategic engagements with the DNA repair machinery. Understanding these pathways is essential for rational therapeutic design.
DSB Repair Pathways in CRISPR Applications
Non-Homologous End Joining (NHEJ):
- Therapeutic Context: Utilized in Baby KJ's therapy for precise gene correction via HDR
- Key Proteins: Ku70/Ku80 heterodimer, DNA-PKcs, XRCC4, Ligase IV [58]
- Clinical Consideration: While HDR offers precision, it is inherently less efficient than NHEJ and restricted to replicating cells [58]
RNA-Mediated DSB Repair Mechanisms: Emerging research reveals RNA's active role in DSB repair through:
- RNA-Bridging: Nascent transcripts hold broken DNA ends in proximity [59]
- RNA-Templated Repair: Transcripts serve as templates for accurate repair, with DNA polymerase η exhibiting reverse transcriptase activity [59]
- R-NHEJ/R-MMEJ: RNA-guided end joining pathways that operate independently of DNA replication [59]
Diagram 1: DSB Repair Pathways in CRISPR Applications. The flowchart illustrates how different CRISPR therapeutic strategies engage with DNA repair mechanisms. Traditional nuclease-based approaches intentionally create double-strand breaks that are resolved through either error-prone NHEJ or precise HDR pathways. In contrast, epigenetic editing platforms like CRISPRoff bypass DSB formation entirely, instead establishing stable transcriptional repression through targeted DNA methylation, thereby avoiding genotoxic risks associated with DNA cleavage [57] [58] [59].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of these advanced CRISPR methodologies requires carefully selected reagents and delivery systems. The following table details critical components referenced in the featured studies.
Table 3: Research Reagent Solutions for Advanced CRISPR Applications
| Reagent / System | Function | Application Example | Key Characteristics |
|---|---|---|---|
| Lipid Nanoparticles (LNPs) | In vivo delivery of CRISPR components | Baby KJ's personalized therapy [9] | Liver-tropic; re-dosable; avoids immune activation seen with viral vectors |
| CRISPRoff mRNA (Variant 7) | Epigenetic editing machinery | Multiplexed gene silencing in T-cells [57] | Cap1 structure; 1-methylpseudouridine; specific codon optimization |
| GalNAc-LNP | Targeted liver delivery | VERVE-102 for cardiovascular disease [60] | N-acetylgalactosamine conjugation enhances hepatocyte specificity |
| All-RNA Delivery System | Electroporation of mRNA encoding editors | CRISPRoff delivery to primary T-cells [57] | Avoids DNA integration; transient expression; high efficiency |
| Cas12a RNP | Orthogonal genome editing | CAR integration alongside CRISPRoff [57] | Compatible with epigenetic editing; precise knock-in capability |
Emerging Frontiers and Implementation Tools
AI-Powered Experimental Design
The complexity of designing personalized therapies and epigenetic editing approaches is being addressed through artificial intelligence tools. CRISPR-GPT, developed at Stanford Medicine, represents a significant advancement in this space [61].
Capabilities:
- Experimental Planning: Generates CRISPR design strategies based on 11 years of published data
- Off-Target Prediction: Identifies potential off-target sites and assesses their functional impact
- Accessibility: Functions at beginner, expert, and Q&A levels to support researchers across experience spectra [61]
Validation: In laboratory testing, researchers using CRISPR-GPT successfully executed experiments targeting multiple genes in lung cancer cells on their first attempt, dramatically reducing the traditional trial-and-error timeline [61].
Clinical Trial Landscape Expansion
The field is witnessing rapid diversification beyond early monogenic targets. As of February 2025, the CRISPR Medicine News database tracks approximately 250 clinical trials involving gene-editing therapeutic candidates [62]. Notable expansions include:
- Cardiovascular Diseases: VERVE-101/102 (base editing for PCSK9) and CTX310 (ANGPTL3 knockout) for hypercholesterolemia [60]
- Autoimmune Conditions: Multiple trials for lupus, multiple sclerosis, and refractory autoimmune diseases [62]
- Oncology: Enhanced CAR-T approaches leveraging epigenetic editing for solid tumors [57]
- Infectious Diseases: CRISPR-enhanced phage therapies targeting antibiotic-resistant bacteria [9]
The convergence of personalized CRISPR platforms with advanced epigenetic editing technologies represents a fundamental shift in therapeutic development. The Baby KJ case demonstrates that patient-specific therapies for ultra-rare diseases are now clinically feasible, while CRISPRoff technology provides a sophisticated toolkit for durable transcriptional control without genotoxic risks. Both approaches are united through their sophisticated engagement with DNA repair mechanisms, from traditional HDR pathways to RNA-mediated repair processes. For researchers and drug developers, these advances create unprecedented opportunities to address previously untreatable conditions through rational design of gene regulation strategies. As AI tools accelerate experimental planning and regulatory frameworks evolve to support platform-based approval pathways, the translation of these technologies from boutique applications to standard care appears increasingly imminent.
The CRISPR-Cas9 system has revolutionized genetic engineering by providing an adaptable platform for precise genome modification. Its fundamental mechanism relies on creating targeted DNA double-strand breaks (DSBs) that harness the cell's endogenous repair machinery to introduce specific genetic changes [3]. This process begins with the Cas9 nuclease, guided by a synthetic RNA molecule, inducing DSBs at predetermined genomic locations. The subsequent repair of these breaks through either error-prone non-homologous end joining (NHEJ) or high-fidelity homology-directed repair (HDR) pathways determines the final genetic outcome [63] [64]. The therapeutic application of this technology across diverse disease domains demonstrates its remarkable versatility.
This technical guide examines how CRISPR-Cas9-mediated DSB repair is being deployed against three major disease categories: cardiovascular disorders, neurodegenerative conditions, and infectious diseases. Each domain presents unique challenges and opportunities for genome editing interventions. Cardiovascular applications focus on correcting monogenic mutations and modulating risk factors, neurodegenerative strategies aim to disrupt pathogenic protein production or correct underlying mutations, while infectious disease approaches directly target pathogen genomes or enhance host defenses [65] [63] [66]. Understanding the interplay between Cas9-induced DSBs and the cellular repair mechanisms in each context is essential for developing effective therapies.
Core Mechanism: CRISPR-Cas9-Induced DSB Repair Pathways
The CRISPR-Cas9 system functions as a programmable DNA endonuclease derived from bacterial adaptive immune systems. The core machinery consists of two components: the Cas9 protein, which performs the DNA cleavage, and a guide RNA (gRNA) that confers sequence specificity through complementary base pairing [3] [64]. The system's operation unfolds in three critical phases: target identification, DNA cleavage, and cellular repair.
Following the formation of the Cas9-gRNA complex, the system scans DNA for both the target sequence (complementary to the gRNA) and an adjacent protospacer adjacent motif (PAM), typically a short 5'-NGG-3' sequence for Streptococcus pyogenes Cas9 [64]. PAM recognition triggers local DNA melting, allowing gRNA-target hybridization and activation of Cas9's nuclease domains. The HNH domain cleaves the DNA strand complementary to the gRNA, while the RuvC domain cleaves the opposite strand, generating a blunt-ended DSB [3] [26].
The fate of the DSB is determined by competing cellular repair pathways, each with distinct mutational outcomes and experimental applications:
- Non-Homologous End Joining (NHEJ): This dominant pathway in mammalian cells directly ligates broken DNA ends without a template, often resulting in small insertions or deletions (indels) that can disrupt gene function, making it ideal for gene knockout strategies [3] [64].
- Homology-Directed Repair (HDR): This precise, template-dependent pathway uses homologous DNA (either sister chromatids or exogenous donor templates) to accurately repair the break, enabling specific gene corrections or insertions [65] [26].
- Microhomology-Mediated End Joining (MMEJ): An error-prone alternative pathway that utilizes microhomologous sequences (5-25 bp) for end joining, typically resulting in deletions [3] [63].
Figure 1: CRISPR-Cas9-Induced DNA Repair Pathways. Following a Cas9-mediated double-strand break, cellular repair machinery proceeds through one of three primary pathways, each yielding distinct genetic outcomes.
The balance between these pathways significantly impacts editing efficiency and specificity. Recent studies utilizing single-molecule sequencing (UMI-DSBseq) to track repair dynamics in tomato protoplasts revealed that precise repair accounts for up to 70% of all repair events, highlighting the importance of high-fidelity repair in limiting mutagenic outcomes [8]. Understanding and manipulating this repair pathway choice is therefore crucial for optimizing CRISPR-based therapeutic strategies.
Cardiovascular Disease Applications
Cardiovascular diseases (CVDs) remain a leading cause of global mortality, with many conditions having underlying genetic components that make them promising targets for CRISPR-based interventions [65] [64]. The applications in cardiovascular medicine primarily focus on correcting monogenic disorders, modulating risk factors, and creating research models to better understand disease pathophysiology.
Key Therapeutic Targets and Approaches
Familial Hypercholesterolemia and Atherosclerosis: Approaches target genes involved in lipid metabolism, particularly PCSK9, which regulates LDL receptor degradation. CRISPR-mediated disruption of PCSK9 in animal models has demonstrated significant reductions in LDL cholesterol and atherosclerosis risk [65] [64].
Inherited Cardiomyopathies: Mutations in genes encoding cardiac sarcomere proteins, such as MYBPC3, cause hypertrophic cardiomyopathy. CRISPR strategies aim to correct these mutations through HDR-mediated repair or disrupt dominant-negative alleles [65].
Channelopathies and Arrhythmias: Conditions like long QT syndrome, caused by mutations in potassium channel genes (KCNQ1, KCNH2), are being targeted for correction to normalize cardiac repolarization and prevent lethal arrhythmias [65].
Duchenne Muscular Dystrophy (DMD): While primarily a neuromuscular disorder, DMD significantly impacts cardiac function. CRISPR approaches aim to restore dystrophin expression through exon skipping or mutation correction [64].
Table 1: CRISPR-Cas9 Applications in Cardiovascular Disease Models
| Disease Target | Genetic Target | Approach | Experimental Model | Key Outcome |
|---|---|---|---|---|
| Familial Hypercholesterolemia | PCSK9 | NHEJ-mediated knockout | Mouse models | >60% reduction in LDL cholesterol |
| Hereditary Transthyretin Amyloidosis (hATTR) | TTR gene | NHEJ-mediated knockout | Phase I/II Clinical Trial (Intellia) | ~90% reduction in TTR protein levels [9] |
| Hypertrophic Cardiomyopathy | MYBPC3 | HDR-mediated correction | Human iPSC-derived cardiomyocytes | Restoration of normal sarcomere structure |
| Long QT Syndrome | KCNQ1/KCNH2 | HDR-mediated correction | Mouse models | Normalization of action potential duration |
Experimental Protocol: In Vivo CRISPR Treatment for hATTR
The recent clinical success with in vivo CRISPR treatment for hereditary transthyretin amyloidosis (hATTR) demonstrates a viable pathway for cardiovascular gene therapy applications [9]:
gRNA Design and Validation: Design sgRNAs targeting the human TTR gene with high on-target efficiency and minimal off-target potential. Validate cleavage efficiency in human hepatocyte cell lines using T7E1 assay or sequencing.
LNP Formulation: Encapsulate Cas9 mRNA and sgRNA in lipid nanoparticles (LNPs) optimized for hepatocyte tropism. Characterize LNP size (80-100 nm), encapsulation efficiency (>90%), and stability.
In Vivo Delivery: Administer via systemic intravenous injection at doses ranging from 0.5-1.5 mg/kg. The LNPs preferentially accumulate in the liver through ApoE-mediated uptake.
Efficacy Assessment: Monitor serum TTR protein levels weekly using immunoassays. Successful editing typically shows >80% reduction in TTR levels within 4 weeks post-treatment.
Safety Evaluation: Assess liver enzymes (ALT, AST), inflammatory markers, and conduct off-target analysis using CIRCLE-seq or similar methods to ensure specificity.
This protocol represents the first systemic in vivo CRISPR administration in humans and establishes a framework for liver-targeted cardiovascular therapies [9]. The sustained TTR reduction (>2 years) observed in clinical trial participants demonstrates the potential long-term durability of this approach.
Neurodegenerative Disease Applications
Neurodegenerative disorders present unique challenges for therapeutic intervention due to the blood-brain barrier and the post-mitotic nature of neuronal cells. CRISPR-Cas9 applications in this field focus on disrupting pathogenic processes, correcting causal mutations, and creating more accurate disease models for drug screening [63] [67].
Key Therapeutic Strategies
Alzheimer's Disease (AD): Approaches target the amyloid precursor protein (APP) gene to reduce production of amyloid-β peptides, or presenilin genes (PSEN1/2) to modify γ-secretase activity [63].
Parkinson's Disease (PD): Strategies focus on genes involved in autosomal dominant forms (SNCA, LRRK2) using CRISPR to reduce expression of toxic protein aggregates, or recessive forms (PARK2, PINK1) through gene correction [63].
Huntington's Disease (HD): The CAG repeat expansion in the huntingtin (HTT) gene is targeted for selective disruption of the mutant allele while preserving wild-type function using allele-specific gRNAs [63].
Amyotrophic Lateral Sclerosis (ALS): Both familial forms (targeting SOD1, C9orf72, FUS, TARDBP) and sporadic cases are being investigated, with approaches ranging from direct mutation correction to knockdown of toxic gain-of-function alleles [63] [67].
Spinocerebellar Ataxia (SCA): Various polyglutamine expansion disorders are targeted using similar approaches to Huntington's disease, with particular focus on selective mutant allele disruption [63].
Table 2: CRISPR-Cas9 Applications in Neurodegenerative Disease Research
| Disease Target | Genetic Target | Approach | Experimental Model | Key Outcome |
|---|---|---|---|---|
| Huntington's Disease | mutant HTT allele | NHEJ-mediated disruption | Human iPSC-derived neurons | Selective reduction of mutant HTT protein aggregates |
| Alzheimer's Disease | APP gene | HDR-mediated correction | Mouse models | Reduced Aβ plaque formation |
| Parkinson's Disease | LRRK2 G2019S | Base editing | Human iPSC-derived dopaminergic neurons | Correction of point mutation without DSBs |
| Amyotrophic Lateral Sclerosis | SOD1 mutations | NHEJ-mediated knockout | Mouse models | Delayed disease onset and extended survival |
Experimental Protocol: Gene Editing in Patient-Derived iPSCs
The use of induced pluripotent stem cells (iPSCs) enables modeling of neurodegenerative diseases in human neuronal cultures and provides a platform for therapeutic development [63]:
iPSC Generation and Culture: Derive iPSCs from patient fibroblasts using non-integrating Sendai virus encoding Yamanaka factors. Maintain cultures on Matrigel-coated plates with mTeSR1 medium.
CRISPR Delivery: Electroporate ribonucleoprotein (RNP) complexes of purified Cas9 protein and synthetic sgRNA using the Neon Transfection System (1100V, 20ms, 2 pulses). Alternatively, use AAV vectors for hard-to-transfect cells.
Clone Isolation and Validation: Following editing, dissociate cells to single cells and plate at clonal density. Expand individual colonies and screen for desired edits using PCR followed by restriction fragment length polymorphism (RFLP) analysis or Sanger sequencing.
Neuronal Differentiation: Differentiate edited iPSCs into relevant neuronal subtypes using established protocols. For cortical neurons, use dual-SMAD inhibition with Noggin and SB431542.
Phenotypic Rescue Assessment: Evaluate functional correction using disease-specific assays: immunostaining for pathogenic protein aggregates, electrophysiology for neuronal function, and RNA-seq for transcriptional profiles.
This ex vivo approach allows for precise genetic correction while avoiding delivery challenges associated with targeting the central nervous system directly. The edited iPSCs can subsequently be differentiated into disease-relevant cell types for transplantation or further mechanistic studies [63].
Infectious Disease Applications
CRISPR-Cas9 technology provides innovative approaches for combating infectious diseases through direct pathogen targeting, enhanced host immunity, and improved diagnostic capabilities. The applications leverage both the DNA-targeting Cas9 and RNA-targeting Cas13 systems for comprehensive anti-infective strategies [66] [68].
Therapeutic and Diagnostic Platforms
Antiviral Strategies: For DNA viruses like HIV-1, CRISPR-Cas9 excises integrated proviral DNA from the host genome. The EBT-101 therapy, currently in clinical trials, uses AAV delivery to target multiple conserved regions of the HIV genome for complete eradication [68]. For RNA viruses including SARS-CoV-2, Cas13 degrades viral RNA within infected cells, with studies demonstrating reduced viral load in human airway epithelial cultures [66] [68].
Antibacterial Approaches: CRISPR-engineered bacteriophages (crPhages) selectively target antibiotic-resistant pathogens. Locus Biosciences has developed crPhages containing Cas3 that effectively eliminate Escherichia coli in urinary tract infections, showing promise in Phase 1b clinical trials [68]. Conjugative plasmids deliver CRISPR systems to pathogens like Salmonella enterica, selectively eliminating antibiotic resistance genes [68].
Antifungal Applications: CRISPR enables genetic manipulation of previously intractable fungal pathogens like Aspergillus fumigatus through MMEJ-mediated mutagenesis, identifying novel drug targets and mechanisms of resistance [68].
Diagnostic Platforms: CRISPR-based diagnostics like SHERLOCK (Cas13) and DETECTR (Cas12) provide rapid, sensitive detection of pathogens including SARS-CoV-2, Ebola, and Zika viruses with single-copy sensitivity and results in under 45 minutes [66].
Table 3: CRISPR-Based Applications in Infectious Diseases
| Application Type | Pathogen Target | CRISPR System | Delivery Method | Key Outcome |
|---|---|---|---|---|
| Antiviral Therapy | HIV-1 | Cas9 | AAV vector | Excision of integrated provirus from host genome [68] |
| Antibacterial Therapy | Escherichia coli | crPhage with Cas3 | Engineered bacteriophage | Specific killing of antibiotic-resistant strains [68] |
| SARS-CoV-2 Detection | SARS-CoV-2 | Cas13 (SHERLOCK) | Lateral flow readout | 95% PPA, 100% NPA vs. PCR, 45min turnaround [66] |
| Host-directed Therapy | Multiple viruses | Cas9 knockout | Lentiviral delivery | Identification of host dependency factors |
Experimental Protocol: CRISPR Phage Therapy for Bacterial Infections
The development of CRISPR-engineered bacteriophages represents a novel approach to targeting antibiotic-resistant bacteria [68]:
Phage Selection and Engineering: Select temperate bacteriophages with broad host range against target pathogen (e.g., E. coli). Engineer phage genome to incorporate CRISPR-Cas system with guides targeting essential bacterial genes or antibiotic resistance genes.
CRISPR Array Design: Design gRNAs targeting conserved essential genes (e.g., gyrA, recA) or specific antibiotic resistance genes (e.g., blaCTX-M, mecA) in the bacterial genome. Cloning multiple gRNAs increases efficacy and reduces escape mutant formation.
Phage Propagation and Purification: Amplify engineered phages in bacterial cultures at mid-log phase (OD600 = 0.4-0.6). Purify through polyethylene glycol precipitation and cesium chloride gradient centrifugation. Determine titer via plaque assay.
In Vitro Efficacy Testing: Evaluate bacterial killing kinetics by measuring optical density and colony-forming units over 24 hours. Compare to wild-type phage and antibiotic controls.
Resistance Monitoring: Passage bacteria with sublethal phage concentrations for 10-15 generations to assess resistance development. Sequence resistant colonies to identify escape mechanisms.
This approach leverages the natural specificity of bacteriophages while enhancing their killing potential through targeted CRISPR-mediated destruction of the bacterial genome. The specificity of CRISPR targeting minimizes damage to beneficial microbiota, addressing a significant limitation of conventional antibiotics [68].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of CRISPR-based therapeutic strategies requires carefully selected reagents and methodologies. The following toolkit summarizes critical components for developing CRISPR interventions across disease areas.
Table 4: Essential Research Reagents for CRISPR-Based Disease Modeling and Therapy
| Reagent Category | Specific Examples | Function | Considerations |
|---|---|---|---|
| Cas Nucleases | SpCas9, SaCas9, Cas12a, Cas13 | DNA/RNA cleavage with varying PAM requirements | SaCas9 smaller size beneficial for AAV packaging [26] |
| Delivery Systems | AAVs, LNPs, Electroporation | Transport editing components to target cells | LNPs enable redosing; AAVs have limited capacity [9] [26] |
| gRNA Design Tools | CRISPOR, ChopChop | Predict on-target efficiency and off-target sites | Include specificity scores and potential off-target loci |
| Validation Assays | T7E1, TIDE, NGS | Confirm editing efficiency and specificity | NGS provides most comprehensive analysis [8] |
| Repair Templates | ssODNs, dsDNA donors | Facilitate HDR for precise edits | ssODNs suitable for small edits; dsDNA for larger inserts |
| Cell Models | iPSCs, Organoids, Primary cells | Disease modeling and therapeutic testing | iPSCs enable patient-specific models [63] |
Technical Challenges and Future Directions
Despite significant progress, several technical hurdles remain in translating CRISPR therapies to clinical practice. Delivery efficiency to specific tissues, particularly across the blood-brain barrier for neurodegenerative applications, requires further optimization [26]. Immune responses to Cas proteins and pre-existing immunity to AAV vectors present additional challenges for in vivo applications [9] [26].
Off-target effects continue to be a concern, though improved gRNA design, high-fidelity Cas variants, and novel detection methods like UMI-DSBseq are addressing these limitations [8] [26]. The emergence of new technologies like base and prime editing offers more precise genetic modifications without inducing DSBs, potentially improving safety profiles [26].
The future of CRISPR-based therapeutics lies in combining technological advancements with interdisciplinary approaches. Machine learning algorithms are being deployed to predict gRNA efficiency and specificity, while novel delivery vehicles such as cell-type-specific LNPs and engineered viral vectors are expanding the range of targetable tissues [26]. As these technologies mature, CRISPR-based interventions promise to transform treatment paradigms across cardiovascular, neurodegenerative, and infectious diseases.
Figure 2: Challenges and Solutions in CRISPR Therapeutic Development. Major technical hurdles in clinical translation of CRISPR therapies and emerging approaches to address these limitations.
Navigating Challenges: Off-Target Effects, Efficiency, and Cell-Type Specific Repair
The CRISPR-Cas9 system has revolutionized genome editing by enabling precise modification of target genes, creating unprecedented opportunities for treating human genetic diseases. However, this therapeutic potential is tempered by a significant challenge: off-target effects, where unintended genomic modifications occur at sites other than the intended target. These off-target events raise substantial genotoxicity concerns that can delay clinical translation [69] [70]. The wild-type Streptococcus pyogenes Cas9 (SpCas9) exhibits a concerning level of promiscuity, tolerating between three and five base pair mismatches between the guide RNA (gRNA) and target DNA, potentially creating double-stranded breaks at numerous sites across the genome [71]. For researchers and drug development professionals, minimizing these effects is not merely an optimization challenge but a fundamental safety requirement, particularly as CRISPR-based therapies like Casgevy have received regulatory approval and are being administered to patients [9] [71]. This technical guide examines the integrated approaches of gRNA design, high-fidelity Cas9 variants, and artificial intelligence (AI) tools that collectively address the off-target challenge within the broader context of CRISPR-Cas9 mechanisms for double-strand break repair in genetic disease research.
Fundamentals of gRNA Design for Enhanced Specificity
The design of the guide RNA represents the most critical determinant of editing specificity, as the sequence composition directly influences where the Cas nuclease will bind and cleave. Optimal gRNA design requires careful consideration of multiple sequence-based factors to minimize off-target recognition while maintaining robust on-target activity.
Core Principles of gRNA Design:
- Sequence Specificity: Select gRNA sequences with minimal similarity to other genomic sites, particularly in the PAM-proximal "seed" region (nucleotides 1-10), which is less tolerant of mismatches [72] [73]. Computational tools like CRISPOR can identify candidate guides with high specificity by scanning the reference genome for similar sequences.
- GC Content Optimization: Maintain a balanced GC content (typically 40-60%) in the gRNA sequence. Higher GC content stabilizes the DNA:RNA duplex, enhancing on-target editing, while excessively high GC content may increase off-target binding affinity [71].
- Chemical Modifications: Incorporate synthetic modifications such as 2'-O-methyl analogs (2'-O-Me) and 3' phosphorothioate bonds (PS) to reduce off-target editing while potentially increasing on-target efficiency [71]. These modifications enhance gRNA stability and alter binding kinetics to favor specific interactions.
- Length Optimization: Consider truncated gRNAs (17-19 nucleotides instead of 20) to reduce off-target activity while potentially maintaining sufficient on-target efficiency, as shorter gRNAs have reduced tolerance for mismatches [74] [71].
Table 1: Key Factors in gRNA Design for Minimizing Off-Target Effects
| Design Factor | Optimal Range/Type | Impact on Specificity | Considerations |
|---|---|---|---|
| Seed Region Mismatches | 0 mismatches in PAM-proximal 10bp | Critical for specificity | Mismatches in seed region dramatically reduce cleavage |
| GC Content | 40-60% | Balanced stability | <30%: poor efficiency; >70%: potential increased off-target risk |
| gRNA Length | 17-20 nucleotides | Shorter gRNAs increase specificity | Truncated gRNAs may reduce on-target efficiency |
| Chemical Modifications | 2'-O-Me, PS | Reduces off-target activity | Enhances nuclease resistance and alters binding kinetics |
| Target Sequence Complexity | High Shannon index | Inverse correlation with off-target count | More complex sequences have fewer potential off-target sites [72] |
High-Fidelity Cas9 Variants: Engineering Precision
While optimal gRNA design reduces off-target potential, the inherent properties of the Cas nuclease itself play a crucial role in determining specificity. Wild-type SpCas9 possesses flexible energy dynamics that enable cleavage at mismatched sites, prompting the development of engineered high-fidelity variants with enhanced discrimination capabilities.
Mechanism and Development of SpCas9-HF1
The development of SpCas9-HF1 (High-Fidelity variant #1) exemplifies a structure-guided rational design approach to enhancing specificity. Based on the "excess energy" hypothesis that SpCas9-sgRNA complexes possess more energy than needed for target recognition, researchers systematically mutated four residues (N497A, R661A, Q695A, and Q926A) that form non-specific contacts with the DNA phosphate backbone [74]. These strategic alterations reduced non-specific interactions while preserving the energy landscape necessary for on-target cleavage. In comprehensive testing, SpCas9-HF1 demonstrated comparable on-target activities to wild-type SpCas9 for 86% (32/37) of sgRNAs tested while rendering nearly all off-target events undetectable by genome-wide break capture and targeted sequencing methods [74].
Comparative Performance of High-Fidelity Variants
Several high-fidelity Cas9 variants have been developed using different engineering strategies, each with distinct characteristics and performance profiles. When selecting a high-fidelity variant for therapeutic applications, researchers must consider the balance between on-target efficiency and off-target reduction, as some variants achieve enhanced specificity at the cost of reduced activity at certain targets.
Table 2: High-Fidelity Cas9 Variants and Their Characteristics
| Variant | Key Mutations/Features | On-Target Efficiency | Off-Target Reduction | Key Applications |
|---|---|---|---|---|
| SpCas9-HF1 | N497A, R661A, Q695A, Q926A | >70% of wild-type for 86% of guides [74] | Undetectable for most guides [74] | Standard non-repetitive sequences |
| eSpCas9(1.1) | Enhanced specificity mutations | Varies by guide | Significant reduction | Mismatch-sensitive applications |
| HypaCas9 | Enhanced fidelity mutations | Maintained for most targets | Strong reduction | High-precision editing |
| xCas9 | Altered PAM recognition | Broad PAM compatibility | Reduced off-targets | Expanded targeting range |
| SpCas9-NG | Engineered PAM recognition | NG PAM sites | Varies by site | Expanded targeting range |
Experimental Protocol: Evaluating High-Fidelity Variants
Objective: Compare the off-target profiles of wild-type SpCas9 and high-fidelity variants for a candidate therapeutic gRNA.
Materials:
- Plasmids encoding wild-type SpCas9 and high-fidelity variants (e.g., SpCas9-HF1)
- Synthetic gRNA or plasmid for expression
- Target cell line (e.g., HEK293T)
- Transfection reagent
- Genomic DNA extraction kit
- GUIDE-seq or BreakTag reagents [72] [71]
- Next-generation sequencing platform
Methodology:
- Transfection: Co-transfect cells with Cas9 expression plasmid (wild-type or variant) and gRNA expression plasmid. Include controls (untransfected cells).
- Genomic DNA Extraction: Harvest cells 72 hours post-transfection and extract genomic DNA.
- Off-Target Detection:
- GUIDE-seq: Transfect cells with double-stranded oligodeoxynucleotide (dsODN) tag alongside CRISPR components. Capture tag integration sites through PCR amplification and sequencing [74].
- BreakTag: Digest genomic DNA with Cas9-sgRNA ribonucleoproteins (RNPs) in vitro. Perform end repair/A-tailing, ligate adaptors with unique molecular identifiers (UMIs), conduct tagmentation, and PCR amplification for sequencing [72].
- Sequencing and Analysis: Sequence libraries and analyze using dedicated bioinformatics pipelines (e.g., BreakInspectoR for BreakTag data) to identify and quantify on-target and off-target cleavage events [72].
- Validation: Perform targeted amplicon sequencing of nominated off-target sites to confirm editing frequencies.
The experimental workflow for evaluating nuclease specificity encompasses both cellular and biochemical methods, each providing complementary information about the off-target landscape.
AI and Machine Learning Approaches for Prediction and Optimization
Artificial intelligence, particularly deep learning, has emerged as a transformative technology for predicting and minimizing CRISPR off-target effects. By analyzing large-scale datasets of gRNA activity and editing outcomes, AI models can identify complex patterns that elude traditional rule-based design approaches.
AI-Driven gRNA Design and Optimization
Machine learning models have demonstrated remarkable capability in predicting both on-target efficiency and off-target propensity, leveraging diverse feature sets including sequence composition, epigenetic context, and mismatch tolerance patterns. These models address the critical challenge in gRNA design: balancing high on-target activity with minimal off-target risk.
Key AI Models and Their Applications:
- CRISPRon: A deep learning framework that integrates gRNA sequence features with epigenomic information (e.g., chromatin accessibility) to predict Cas9 on-target knockout efficiency with improved accuracy compared to sequence-only predictors [75] [73].
- DeepSpCas9: A convolutional neural network (CNN) model trained on high-throughput screening of 12,832 target sequences in human cells, demonstrating better generalization across different datasets compared to existing models [75].
- CRISPR-Net: Combines CNN and bidirectional gated recurrent unit (GRU) architectures to analyze guides with up to four mismatches or indels relative to targets, outputting scores for both cleavage activity and specificity [73].
- Multitask Models: Hybrid deep learning approaches that simultaneously predict on-target efficacy and off-target cleavage, internalizing the trade-offs between sequence features that enhance one versus the other [73].
Experimental Protocol: AI-Guided gRNA Selection and Validation
Objective: Employ AI tools to design high-specificity gRNAs and experimentally validate their off-target profiles.
Materials:
- AI prediction platforms (CRISPRon, DeepSpCas9, etc.)
- Target genomic sequence
- gRNA synthesis platform
- Cell culture system
- Next-generation sequencing capability
- Data analysis software
Methodology:
- Target Identification: Define the target genomic region and PAM constraints based on the selected Cas nuclease.
- In Silico gRNA Design:
- Input target sequence into multiple AI prediction tools (e.g., CRISPRon, DeepSpCas9).
- Extract on-target efficiency scores and off-target propensity predictions for all candidate gRNAs.
- Apply filters based on GC content, presence of homopolymer repeats, and sequence complexity.
- gRNA Selection: Prioritize gRNAs with optimal balanced scores (high on-target, low off-target predictions).
- Experimental Validation:
- Synthesize top-ranked gRNAs and corresponding controls.
- Transfert cells with Cas9-gRNA complexes.
- Assess on-target efficiency using T7 Endonuclease I assay or sequencing.
- Evaluate off-target profiles using GUIDE-seq or BreakTag methods.
- Model Refinement: Feed experimental results back to improve prediction algorithms, closing the iterative design loop.
The integration of AI into the gRNA design workflow creates an iterative improvement cycle where experimental results continuously refine predictive models, progressively enhancing design accuracy.
The Scientist's Toolkit: Essential Research Reagents and Methods
Implementing a comprehensive strategy for minimizing off-target effects requires specialized reagents, tools, and methodologies. This toolkit summarizes key resources available to researchers for designing, executing, and validating high-specificity CRISPR experiments.
Table 3: Essential Research Reagents and Methods for Off-Target Assessment
| Tool/Reagent | Function | Key Features | Applications |
|---|---|---|---|
| High-Fidelity Cas9 Variants (e.g., SpCas9-HF1) | Engineered nucleases with reduced off-target activity | Alanine substitutions at non-specific DNA contact points; maintain high on-target efficiency [74] | Therapeutic genome editing; sensitive cell models |
| BreakTag Method | Profiles Cas9-induced DNA double-strand breaks (DSBs) | Maps DSB end structures; identifies ~150,000 on/off-target sites; works with ~3,500 sgRNAs [72] | Genome-wide off-target nomination; scission profile analysis |
| GUIDE-seq | Identifies off-target sites in living cells | Uses dsODN tag integration at break sites; sensitive in cellulo detection [74] [72] | Comprehensive off-target mapping in cellular context |
| CRISPOR | Computational gRNA design tool | Off-target scoring algorithms; integrates multiple prediction models | Pre-experimental gRNA selection and ranking |
| AI Prediction Platforms (e.g., CRISPRon, DeepSpCas9) | Predicts gRNA efficiency and specificity | Deep learning models trained on large screening datasets; epigenomic integration [75] [73] | Prioritizing gRNAs with optimal on/off-target ratios |
| Chemically Modified gRNAs | Synthetic gRNAs with enhanced properties | 2'-O-Me and PS modifications reduce off-target editing [71] | Therapeutic applications; sensitive editing contexts |
| Whole Genome Sequencing | Comprehensive off-target analysis | Identifies chromosomal aberrations and unexpected edits [71] | Final safety assessment for clinical candidates |
The integration of optimized gRNA design principles, high-fidelity Cas9 variants, and AI-powered prediction tools represents a comprehensive framework for minimizing off-target effects in CRISPR-based therapeutic applications. The experimental protocols and methodologies detailed in this guide provide researchers with actionable strategies for enhancing editing specificity while maintaining therapeutic efficacy. As the field advances, the convergence of these approaches—coupled with improved delivery systems such as lipid nanoparticles that enable temporal control of editing components—will be crucial for realizing the full potential of CRISPR gene therapies for genetic diseases [9] [71]. The landmark approval of Casgevy and the development of personalized CRISPR treatments for rare diseases underscore the transformative potential of these technologies, while simultaneously highlighting the critical importance of specificity and safety assessment throughout the therapeutic development pipeline [9]. Through continued refinement of these approaches and standardization of off-target assessment protocols, the research community can address the remaining challenges in clinical translation, ultimately enabling safer, more precise genetic interventions for a broad spectrum of human diseases.
The CRISPR-Cas9 system has revolutionized genetic engineering by providing a precise mechanism for creating double-strand breaks (DSBs) in DNA. For therapeutic applications in genetic diseases, the cell primarily repairs these breaks via one of two pathways: the error-prone non-homologous end joining (NHEJ), which often results in gene knockouts, or the more precise homology-directed repair (HDR), which allows for specific gene corrections or insertions using a donor DNA template [76]. A major bottleneck in both research and clinical settings is low editing efficiency, which is profoundly influenced by the strategies used to deliver the CRISPR components (Cas nuclease and guide RNA) and control their expression within the target cell. This guide synthesizes current optimization strategies to overcome this critical barrier.
Optimizing CRISPR-Cas9 Delivery Vehicles
Efficient intracellular delivery of CRISPR components is a fundamental prerequisite for successful gene editing. The cargo can be delivered as a DNA plasmid, mRNA, or pre-assembled Ribonucleoprotein (RNP) complex. RNP delivery is increasingly favored for its rapid activity, reduced off-target effects, and transient presence, which minimizes immune responses [77]. The choice of delivery vehicle is critical and must be matched to the specific experimental or therapeutic context.
Table 1: Comparison of Primary CRISPR-Cas9 Delivery Methods
| Delivery Method | Mechanism | Advantages | Disadvantages | Best Use Cases |
|---|---|---|---|---|
| Adeno-Associated Virus (AAV) | Viral transduction | Favorable safety profile, low immunogenicity [77] | Limited packaging capacity (~4.7kb) [77] | In vivo delivery of smaller Cas variants or sgRNA alone |
| Lentivirus (LV) | Viral transduction, genomic integration | Infects dividing & non-dividing cells, large cargo capacity [77] | Permanent integration raises safety concerns [77] | In vitro studies, stable cell line generation |
| Lipid Nanoparticles (LNPs) | Non-viral; encapsulation & fusion | Low immunogenicity, scalable, tunable formulations [9] [77] | Can be trapped in endosomes, requires optimization for cell type [78] | Systemic in vivo delivery (especially liver), clinical applications [9] |
| Electroporation | Physical; electrical pore formation | High efficiency in hard-to-transfect cells (e.g., primary T cells, stem cells) [76] | Can cause significant cell stress and reduced viability [76] | Ex vivo editing of immune cells, stem cells |
| Extracellular Vesicles (EVs) | Non-viral; natural intercellular carriers | Low toxicity, innate ability to cross biological barriers, serum stability [79] | Complex manufacturing, heterogeneity [77] [79] | Emerging therapeutic delivery, modular RNP loading [79] |
Advanced and Hybrid Delivery Systems
Innovation in delivery platforms continues to advance the field. Spherical Nucleic Acids (SNAs), which consist of a nanoparticle core (such as an LNP) coated with a dense shell of DNA, represent a structural breakthrough. In recent studies, these Lipid Nanoparticle-SNAs (LNP-SNAs) demonstrated a threefold increase in gene-editing efficiency and a more than 60% improvement in precise HDR repair rates compared to standard LNPs, while also reducing cellular toxicity [78]. Their unique architecture promotes superior cellular uptake and endosomal escape.
Another sophisticated approach uses engineered Extracellular Vesicles (EVs). One modular strategy involves fusing RNA-binding MS2 coat proteins to EV membrane proteins. These bind with high affinity to MS2 aptamers engineered into the sgRNA, effectively loading pre-formed Cas9 RNP complexes into EVs. This method avoids direct fusion to Cas9, maintaining the functionality of diverse Cas9 variants (e.g., base editors, activators) and enabling efficient editing upon delivery [79].
The following diagram illustrates the core decision-making workflow for selecting and optimizing a CRISPR delivery strategy.
Expression Strategies and Cas9 Selection
Beyond delivery, the choice of Cas9 variant and the design of its expression cassette are crucial for enhancing efficiency and specificity.
Cas9 Variant Selection: While the commonly used Streptococcus pyogenes Cas9 (SpCas9) is highly efficient, its large size is incompatible with AAV delivery. For such cases, smaller natural variants like Staphylococcus aureus Cas9 (SaCas9) or engineered compact Cas9s are essential [76]. Furthermore, high-fidelity Cas9 variants, base editors, and prime editors can significantly improve specificity and enable precise single-nucleotide changes without requiring a DSB or a donor template, thereby increasing the efficiency of desired edits [76] [79].
Vector Design and Control: Using cell-type-specific promoters to drive Cas9 expression can enhance precision and reduce off-target effects in non-target tissues. For advanced experimental control, inducible systems (e.g., tetracycline- or light-activated) allow temporal regulation of editing, which is valuable for studying gene function or minimizing cellular stress [76]. For HDR-based correction, the co-delivery of a donor template is critical. Single-stranded oligodeoxynucleotides (ssODNs) are suitable for small edits, while double-stranded DNA (dsDNA) or circular single-stranded DNA (cssDNA) templates can be used for larger insertions, with cssDNA showing knock-in efficiencies of up to 70% in induced pluripotent stem cells (iPSCs) [76].
Quantitative Assessment of Editing Efficiency
Accurately measuring the outcomes of a CRISPR experiment is necessary to evaluate and optimize delivery and expression strategies. Different methods offer varying levels of sensitivity, throughput, and information depth.
Table 2: Methods for Quantifying CRISPR-Cas9 Editing Efficiency
| Method | Detection Principle | Sensitivity | Throughput | Key Advantage | Key Limitation |
|---|---|---|---|---|---|
| T7 Endonuclease 1 (T7E1) | Mismatch cleavage & gel electrophoresis | Low | Medium | Low cost, simple | Semi-quantitative, low sensitivity [80] |
| Sanger Sequencing + ICE/TIDE | Sequence deconvolution by algorithms | Medium | Medium | Quantitative, accessible | Lower sensitivity for <5% edits [80] |
| Droplet Digital PCR (ddPCR) | Target-specific fluorescent probes | High | High | Absolute quantification, high sensitivity [80] | Requires specific probe design |
| PCR-Capillary Electrophoresis (IDAA) | Amplicon size separation | High | High | Accurate size calling, high sensitivity [80] | Limited to smaller indels |
| Targeted Amplicon Sequencing (AmpSeq) | Next-generation sequencing | Very High | Low (per sample) | Gold standard, detects all edits quantitatively [80] | Higher cost, longer turnaround [80] |
Protocol: Rapid Screening of Editing Outcomes via eGFP-to-BFP Conversion
This protocol provides a rapid, flow cytometry-based method to quantitatively assess NHEJ and HDR efficiencies in a cell population [81].
- Generation of eGFP-positive Cell Line: Create a stable cell line expressing enhanced Green Fluorescent Protein (eGFP) via lentiviral transduction.
- Transfection of Editing Reagents: Transfect the eGFP-positive cells with CRISPR-Cas9 components, including a gRNA targeting the eGFP sequence.
- For NHEJ Assessment: Co-transfect Cas9 and the eGFP-targeting gRNA. Successful NHEJ-mediated knockout will result in a loss of green fluorescence.
- For HDR Assessment: Co-transfect Cas9, the eGFP-targeting gRNA, and a Blue Fluorescent Protein (BFP) donor template. Successful HDR will convert eGFP fluorescence to BFP fluorescence.
- Cell Handling and Analysis: 48-72 hours post-transfection, harvest cells and analyze using Flow Cytometry (FACS).
- Data Processing and Interpretation:
- The percentage of cells that have lost eGFP signal indicates the total editing rate (largely NHEJ).
- The percentage of cells that have gained BFP signal indicates the successful HDR rate.
- The ratio of BFP-positive to total eGFP-negative cells provides a normalized measure of HDR efficiency.
The workflow for this protocol is summarized in the following diagram.
The Scientist's Toolkit: Key Reagents and Materials
Table 3: Essential Research Reagents for CRISPR Delivery Optimization
| Item | Function/Description | Example Use Case |
|---|---|---|
| Pre-assembled Cas9 RNP | Ribonucleoprotein complex of Cas9 protein and sgRNA; enables immediate editing with reduced off-target effects [82] [77]. | Direct delivery via electroporation or nanoparticles for high-efficiency, transient editing. |
| Lipid Nanoparticles (LNPs) | Synthetic, biodegradable nanoparticles for encapsulating and delivering CRISPR cargo (mRNA, RNP) [9] [78]. | Systemic in vivo delivery to the liver; basis for advanced structures like LNP-SNAs. |
| Adeno-Associated Virus (AAV) | A viral vector for efficient gene delivery with low pathogenicity. Often used with smaller Cas9 variants [77] [76]. | Delivery of CRISPR components in vivo where packaging size allows. |
| MS2-MCP EV Loading System | A modular system using MS2 aptamers (in sgRNA) and MS2 Coat Protein (MCP) fusions to load Cas9 RNP into Extracellular Vesicles [79]. | Modular loading of various Cas9 effectors (nuclease, base editors) into EVs for delivery. |
| eGFP-BFP Reporter Cell Line | A cellular biosensor where successful HDR converts eGFP to BFP, allowing quantitative FACS analysis [81]. | Rapid, high-throughput screening of HDR efficiency across different delivery methods. |
| HDR Donor Template (cssDNA) | Circular single-stranded DNA donor template; shown to improve HDR knock-in efficiency compared to linear dsDNA [76]. | Precise gene insertion or correction in primary cells like iPSCs and immune cells. |
Optimizing the delivery and expression of CRISPR-Cas9 components is a multifaceted challenge that is central to achieving high editing efficiency. As the field progresses, the convergence of advanced delivery platforms like LNPs and EVs, the use of AI-designed editors [83], and more sensitive analytical methods will continue to push the boundaries of what is possible. By systematically addressing these factors—choosing the right cargo, tailoring the delivery vehicle to the target cell, employing optimal Cas9 variants, and using accurate quantification methods—researchers can significantly enhance the efficacy and safety of CRISPR-Cas9 for genetic disease research and therapeutic development.
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) technology has revolutionized biomedical research by enabling precise genomic modifications, holding particular promise for treating genetic diseases [84] [35]. The technology operates by creating a double-strand break (DSB) at a specific genomic locus directed by a guide RNA (gRNA) [85]. However, the ultimate editing outcome is not determined by the cut itself, but by the cellular DNA repair machinery's response to that DNA perturbation [12]. DSB repair proceeds primarily via two competing pathways: the error-prone non-homologous end joining (NHEJ), which often results in small insertions or deletions (indels), and the more precise homology-directed repair (HDR), which requires a DNA template to accurately repair the break [84] [85].
A critical and often overlooked factor is that DNA repair pathway activity and efficiency are not uniform across all cell types. This is particularly true for postmitotic cells—cells that have exited the cell cycle, such as neurons and cardiomyocytes [12]. These cell types must maintain genomic integrity over a human lifetime without the benefit of replication-associated repair mechanisms. Pathways like HDR, which are active primarily in the S and G2 phases of the cell cycle, are presumed to be less accessible in non-dividing cells [12] [86]. This fundamental biological difference creates unique challenges for achieving desired CRISPR editing outcomes in these clinically relevant cells, impacting strategies for treating neurodegenerative and cardiac genetic diseases. This whitepaper details the distinct DNA repair landscapes in postmitotic neurons and cardiomyocytes and outlines experimental strategies for overcoming these challenges.
Distinct DNA Repair Kinetics and Pathway Usage in Postmitotic Cells
Repair Kinetics Are Dramatically Slower in Postmitotic Cells
Research comparing human induced pluripotent stem cells (iPSCs) with isogenic iPSC-derived neurons reveals profound differences in the timeline of CRISPR-Cas9 editing. In dividing cells like iPSCs, Cas9-induced indels typically plateau within a few days, as DSBs are rapidly resolved to prevent cell cycle arrest and apoptosis [12]. In stark contrast, indel accumulation in postmitotic neurons continues for up to two weeks after transient Cas9 RNP delivery, indicating a significantly prolonged DSB repair process [12] [13]. This extended time course is not due to delivery deficits, as base editing (which does not rely on DSB repair) occurs efficiently in neurons within three days [12]. A similar weeks-long timeline of indel accumulation has been observed in iPSC-derived cardiomyocytes, suggesting that prolonged resolution of DSBs may be a common feature of nondividing cells [12].
Pathway Choice Favors NHEJ and Suppresses MMEJ
The choice of DNA repair pathway differs markedly between dividing and postmitotic cells. Dividing iPSCs exhibit a broad range of indel outcomes, including a high proportion of larger deletions characteristic of the microhomology-mediated end joining (MMEJ) pathway, an end resection-dependent process [12] [13]. Conversely, neurons display a much narrower distribution of editing outcomes, dominated by small indels associated with classical NHEJ (cNHEJ) and a higher ratio of insertions to deletions [12]. This indicates that postmitotic neurons predominantly utilize NHEJ while suppressing MMEJ, a pathway typically restricted to the S/G2/M phases of the cell cycle [12]. This preference for NHEJ over MMEJ has been replicated across multiple sgRNAs, including targets relevant to neurodegenerative diseases [12].
Table 1: Comparative Analysis of DNA Repair in Dividing vs. Postmitotic Cells
| Characteristic | Dividing Cells (e.g., iPSCs) | Postmitotic Cells (Neurons, Cardiomyocytes) |
|---|---|---|
| Primary DSB Repair Pathways | NHEJ, MMEJ, and some HDR | Predominantly NHEJ; MMEJ is suppressed [12] |
| Kinetics of Indel Accumulation | Plateaus within a few days [12] | Continues for up to 2 weeks [12] [13] |
| Spectrum of Indel Outcomes | Broad range, including large deletions [12] | Narrow range, predominantly small indels [12] |
| HDR Efficiency | Higher, as HDR is active during cell cycle | Traditionally considered low, but new strategies show promise [86] [16] |
Experimental Models and Methodologies for Studying Repair
iPSC-Derived Models and Delivery Systems
A key methodology for studying cell-type-specific repair involves using human iPSC-derived neurons and cardiomyocytes [12]. These models provide a genetically identical background to their parent iPSCs, allowing for direct comparison of DNA repair between dividing and postmitotic states. Immunocytochemistry confirms postmitotic status (e.g., Ki67-negative) and cell-type purity (e.g., NeuN-positive for neurons) [12].
Delivering CRISPR machinery to neurons requires specialized methods. A highly efficient approach uses virus-like particles (VLPs) pseudotyped with envelopes like VSVG or BaEVRless (BRL) to deliver Cas9 ribonucleoprotein (RNP) [12]. These VLPs can achieve transduction efficiencies of up to 97% in human iPSC-derived neurons [12]. For both neurons and cardiomyocytes, all-in-one lipid nanoparticles have been developed that can co-deliver Cas9, sgRNA, and siRNAs to modulate DNA repair pathways [13]. In T cells and some other models, electroporation of Cas9 RNP is a viable alternative [12].
Protocol: Assessing Editing Outcomes and Kinetics
Workflow:
- Differentiation: Differentiate human iPSCs into postmitotic cortical-like neurons or cardiomyocytes using established protocols [12].
- Delivery: Transduce cells with VSVG/BRL-pseudotyped VLPs containing Cas9 RNP at a defined multiplicity of infection. Include controls with isogenic dividing iPSCs [12].
- Time-Course Sampling: Harvest genomic DNA at multiple time points post-transduction (e.g., days 1, 3, 7, 14, 21) [12].
- Analysis: Amplify the targeted genomic locus by PCR and perform deep sequencing (e.g., Illumina). Analyze sequences for indel spectra and frequency using tools like CRISPResso2 [12].
- Pathway Interrogation: To manipulate outcomes, repeat the experiment with VLP or lipid nanoparticles that co-deliver siRNAs or chemical inhibitors targeting specific DNA repair factors (e.g., RRM2) identified via transcriptomic profiling [12] [13].
Strategies for Enhancing Precision Editing in Postmitotic Cells
Manipulating Endogenous Repair Pathways
The unique repair environment of postmitotic cells can be leveraged and manipulated to improve editing outcomes. Transcriptomic analyses reveal that neurons upregulate non-canonical DNA repair factors, such as the ribonucleotide reductase subunit RRM2, in response to Cas9-induced damage [12] [13]. Pharmacological or siRNA-mediated inhibition of RRM2 has been shown to shift editing outcomes in neurons, increasing deletion size and overall indel efficiency [13]. This demonstrates that targeted perturbation of the postmitotic repair response provides a strategy to direct editing toward desired outcomes.
Enabling Homology-Directed Repair (HDR)
While HDR has been considered inefficient in non-dividing cells, recent advances challenge this notion. The CASAAV-HDR platform, which co-delivers Cas9, sgRNA, and an HDR template via adeno-associated virus (AAV), has achieved precise genome modification in postmitotic adult cardiomyocytes and neurons [86]. Remarkably, HDR efficiency in cardiomyocytes was comparable when AAV was delivered to fetal, neonatal, or mature mice, indicating that HDR can occur independently of cardiomyocyte proliferation [86]. Efficiency correlated strongly with target gene expression level, with robustly expressed genes (TPM >100) achieving editing in ≥20% of cardiomyocytes [86].
Table 2: Research Reagent Solutions for Postmitotic Cell Genome Editing
| Reagent / Tool | Function | Application in Postmitotic Cells |
|---|---|---|
| iPSC-Derived Neurons/Cardiomyocytes | Clinically relevant human model system | Provides a genetically defined, pure population of postmitotic cells for studying cell-type-specific repair [12]. |
| Virus-Like Particles (VLPs) | Protein cargo delivery | Efficiently delivers Cas9 RNP to hard-to-transfect neurons (up to 97% efficiency); pseudotyping (VSVG/BRL) modulates tropism [12]. |
| All-in-One Lipid Nanoparticles (LNPs) | Co-delivery platform | Simultaneously delivers Cas9 RNP and repair-modulating siRNAs (e.g., anti-RRM2) to tune editing outcomes [13]. |
| CASAAV-HDR System | HDR template delivery | Enables precise gene insertion/correction in postmitotic cardiomyocytes and neurons via AAV-borne template [86]. |
| siRNA / Chemical Inhibitors | DNA repair pathway modulation | Knocking down or inhibiting specific factors (e.g., RRM2) can shift the balance of repair outcomes toward larger deletions [12] [13]. |
The evidence is clear that postmitotic neurons and cardiomyocytes possess a unique DNA repair environment characterized by slower kinetics and a distinct preference for NHEJ over other pathways like MMEJ. These differences directly impact the efficiency and outcome of CRISPR-Cas9 genome editing, presenting both a challenge and an opportunity for therapeutic development. Ignoring these cell-type-specific nuances risks the failure of otherwise promising gene therapies.
Future research must focus on further elucidating the molecular mechanisms governing DNA repair in these nondividing cells. The integration of artificial intelligence (AI) for designing novel CRISPR systems and predicting repair outcomes holds great promise [85] [83]. Furthermore, the development of advanced delivery vehicles, such as cell-type-specific LNPs, and next-generation editors like prime editors, which avoid DSBs altogether, may bypass many of the current hurdles [85] [16]. By moving beyond the paradigms established in dividing cells and directly addressing the unique biology of postmitotic cells, researchers can unlock the full therapeutic potential of CRISPR for a host of devastating neurodegenerative and cardiac genetic diseases.
The therapeutic application of CRISPR-Cas9 technology for genetic diseases hinges on creating targeted double-strand breaks (DSBs) in DNA and harnessing cellular repair mechanisms to achieve desired genetic outcomes. However, a fundamental roadblock persists: our inability to control how those perturbations are repaired without triggering cellular toxicity or genotoxic stress [12]. The DNA damage response (DDR) activated by Cas9-induced DSBs can lead to cell cycle arrest, apoptosis, or unintended genomic alterations that compromise both efficacy and safety [15] [87]. For CRISPR-based therapies to fulfill their potential, researchers must navigate the delicate balance between achieving therapeutic levels of editing and maintaining cell viability. This technical guide examines the sources of CRISPR-related toxicity, provides methodologies for its detection and quantification, and outlines strategies to optimize the safety profile of genome editing interventions for genetic disease research.
Molecular Mechanisms of CRISPR-Induced Toxicity
DNA Damage Response to Cas9-Induced DSBs
The core mechanism of CRISPR-Cas9 involves creating DSBs at target genomic locations, which immediately triggers a complex DDR. This response includes the recruitment of repair proteins such as 53BP1, activation of the p53 pathway, and phosphorylation of histone H2AX (γH2AX) [12] [87]. While this response is essential for facilitating repair, excessive or persistent activation can lead to several cytotoxic outcomes:
- Cell cycle arrest and senescence: DSB recognition activates checkpoints that halt cell cycle progression, potentially leading to permanent growth arrest in edited cells [15].
- Apoptosis: Cells with unresolved DSBs may initiate programmed cell death, particularly in sensitive cell types like neurons or stem cells [12] [15].
- Inflammatory responses: DSBs can trigger cGAS-STING pathway activation via micronuclei formation, promoting pro-inflammatory cytokine production [87].
Spectrum of Unintended Genomic Alterations
Beyond immediate DDR activation, CRISPR editing can generate a range of unintended genetic outcomes that threaten genomic integrity and cell viability:
Table 1: Types of Unintended Genomic Alterations in CRISPR Editing
| Alteration Type | Size Range | Potential Consequences | Detection Challenges |
|---|---|---|---|
| Small indels | 1-100 bp | Frameshifts, gene disruption | Standard amplicon sequencing |
| Large deletions | kb-Mb scale | Loss of regulatory elements, gene clusters | Missed by short-read sequencing |
| Chromosomal translocations | Multi-Mb scale | Oncogenic fusions, genomic instability | Specialized methods (CAST-Seq, LAM-HTGTS) |
| Chromothripsis | Chromosome-arm | Massive genomic rearrangement, cell death | Whole genome sequencing |
The particularly concerning finding is that large structural variations (SVs) including kilobase- to megabase-scale deletions and chromosomal translocations represent a significantly underappreciated safety concern in therapeutic editing [15]. These alterations are especially problematic because they may delete critical cis-regulatory elements or tumor suppressor genes, with consequences that extend far beyond the immediate target locus.
Cell-Type Specific Vulnerability Factors
DNA repair is especially understudied in nondividing cells like neurons, limiting the efficiency and precision of genome editing in many clinically relevant tissues [12]. Recent research reveals that postmitotic human neurons repair Cas9-induced DNA damage fundamentally differently than dividing cells, taking longer to fully resolve damage and upregulating non-canonical DNA repair factors in the process [12]. This extended repair timeline (up to 2 weeks versus days in dividing cells) creates a prolonged window of vulnerability to genotoxic stress. Similar prolonged indel accumulation has been observed in iPSC-derived cardiomyocytes and primary T cells, suggesting this may be a general property of nondividing cells [12].
Diagram 1: CRISPR toxicity mechanisms from DSB to cell fate. This pathway illustrates how Cas9-induced double-strand breaks trigger different repair pathways that can lead to either therapeutic outcomes or cytotoxic consequences through genomic instability.
Advanced Detection Methods for CRISPR Genotoxicity
Comprehensive Off-Target and Structural Variation Analysis
Traditional amplicon sequencing approaches frequently miss large-scale genomic alterations because these events delete primer binding sites, rendering them "invisible" to standard analysis [15]. This limitation can lead to overestimation of HDR rates and concurrent underestimation of indels, creating a false impression of editing precision and safety. To address this, researchers should implement orthogonal detection methods:
- GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing): Captures DSBs with a double-stranded oligonucleotide, which is then used as a priming site for sequencing [88].
- BLESS (Direct in Situ Breaks Labeling, Enrichment on Streptavidin, and NGS): Biochemical ligation of NGS sequencing adapters to exposed gDNA ends, applicable to tissue samples from in vivo models [88].
- Digenome-seq: In vitro nuclease-digested whole genome sequencing that identifies cleavage sites in cell-free genomic DNA [88].
- CAST-Seq and LAM-HTGTS: Specialized methods for detecting chromosomal translocations and large structural variations [15].
Chromatin Context Assessment
Recent research has revealed that chromatin context dramatically influences DNA repair pathway balance [89] [90]. The local chromatin environment can determine whether a DSB is repaired via non-homologous end joining (NHEJ), microhomology-mediated end joining (MMEJ), or homology-directed repair (HDR). One study found that NHEJ is broadly biased toward euchromatin, while the contribution of MMEJ is higher in specific heterochromatin contexts [89]. A separate screen of 519 DNA repair proteins found that 89 (17.1%) showed significant chromatin context-dependency in how they modulate the MMEJ:NHEJ balance [90].
Table 2: Chromatin Context-Dependent Repair Protein Synergies
| Synergy Type | Chromatin Features | Representative Proteins | Functional Impact |
|---|---|---|---|
| N-synergy (Favors NHEJ) | H3K4me3, H3K27ac, TT-seq, POL2, H3K36me3 | BRCA2, POLL, LIG4, XLF | Enhanced precise repair in active regions |
| M-synergy (Favors MMEJ) | H3K9me2/3, late replication, LMNB1 | RAD50, FANC complex, ATM, POLθ | Increased microhomology use in heterochromatin |
| M-synergy (Favors MMEJ) | H3K27me3-marked heterochromatin | Multiple resection factors | Context-specific repair pathway choice |
Experimental Strategies to Minimize Toxicity
Delivery System Optimization
The choice of delivery method significantly impacts both editing efficiency and cellular toxicity. Recent advances have demonstrated that virus-like particles (VLPs) can efficiently deliver Cas9 ribonucleoprotein to challenging cell types like neurons with up to 97% efficiency while potentially reducing off-target effects compared to plasmid-based delivery [12]. For in vivo applications, lipid nanoparticles (LNPs) have emerged as a promising delivery vehicle due to their reduced immunogenicity and potential for redosing, as demonstrated in recent clinical trials for hereditary transthyretin amyloidosis (hATTR) and hereditary angioedema (HAE) [9].
DNA Repair Pathway Modulation
Directing DNA repair toward desired outcomes represents a powerful approach for reducing toxicity. However, strategies that manipulate repair pathways must be carefully evaluated:
- DNA-PKcs inhibition: While initially promising for enhancing HDR, recent findings reveal that DNA-PKcs inhibitors can lead to exacerbated genomic aberrations, including thousand-fold increases in chromosomal translocation frequencies [15].
- Alternative HDR enhancement: Transient inhibition of 53BP1 may enhance HDR without increasing translocation frequencies, offering a potentially safer alternative [15].
- p53 modulation: Transient p53 suppression can reduce large chromosomal aberrations but raises oncogenic concerns given p53's critical tumor suppressor role [15].
High-Specificity CRISPR Systems
Engineering improved CRISPR systems with enhanced specificity represents a cornerstone of toxicity reduction:
- High-fidelity Cas9 variants: Engineered Cas9 proteins with reduced off-target activity while maintaining on-target efficiency [88].
- Cas12a systems: Alternative Cas proteins with different PAM requirements and potentially lower off-target rates [91].
- Prime editing and base editing: Systems that avoid DSBs altogether, significantly reducing genotoxic risk [12] [15].
Diagram 2: Comprehensive toxicity assessment workflow. This experimental pipeline outlines orthogonal methods for detecting diverse genotoxic outcomes of CRISPR editing, from large on-target deletions to genome-wide off-target effects and cellular viability measures.
The Scientist's Toolkit: Essential Reagents and Methodologies
Table 3: Research Reagent Solutions for Toxicity Mitigation
| Reagent Category | Specific Examples | Function & Application | Toxicity Consideration |
|---|---|---|---|
| High-fidelity Nucleases | HiFi Cas9, eSpCas9 | Reduced off-target cleavage while maintaining on-target activity | Lower unintended genomic alterations |
| Specificity-enhanced Cas variants | Cas12a, xCas9 | Alternative PAM requirements, different fidelity profiles | Context-dependent specificity improvements |
| DNA Repair Modulators | AZD7648 (DNA-PKcs inhibitor), pifithrin-α (p53 inhibitor) | Shift repair pathway balance toward HDR | Risk of increased structural variations; requires validation |
| Delivery Systems | VLPs, LNPs, AAVs | Efficient RNP delivery with reduced immunogenicity | Cell-type specific toxicity profiles |
| Detection Tools | GUIDE-seq oligos, CAST-Seq reagents | Comprehensive identification of off-target effects and structural variations | Critical for safety assessment |
| Cell Health Assays | Caspase-3/7 assays, γH2AX staining, cell viability dyes | Quantification of apoptosis, DNA damage response, and overall cell health | Early indicators of editing-associated toxicity |
Protocol: Comprehensive Assessment of Editing Outcomes and Viability in Non-Dividing Cells
Background and Principles
This protocol adapts methodologies from recent studies on DNA repair in nondividing cells [12], enabling researchers to simultaneously quantify editing efficiency, genotoxicity, and cellular viability in postmitotic cell types such as neurons, cardiomyocytes, and primary T cells.
Specialized Materials
- Human iPSC-derived neurons, cardiomyocytes, or primary T cells
- Virus-like particles (VLPs) pseudotyped with VSVG and/or BaEVRless (BRL) for efficient transduction
- Cas9 RNP complexes with target-specific sgRNAs
- Immunocytochemistry antibodies: γH2AX, 53BP1, NeuN, Ki67
- Next-generation sequencing library preparation kit
- Cell viability assay (e.g., Calcein AM/propidium iodide staining)
- Long-range PCR amplification reagents
- ddPCR or qPCR reagents for relative viral copy number quantification
Step-by-Step Procedure
Day 1: Cell Preparation and VLP Transduction
- Culture human iPSC-derived neurons (≥95% NeuN-positive, ≥99% Ki67-negative) in appropriate maintenance medium.
- Prepare VLP particles containing Cas9 RNP at optimized concentrations (see [12] for pseudotype optimization).
- Transduce cells with VLP particles at appropriate multiplicity of infection (MOI), determined through pilot titration experiments.
- Include control groups: untreated cells, VLP-only (no RNP), and RNP-only (no VLP).
Days 2-16: Time-Course Monitoring
- Monitor editing kinetics over an extended time course (up to 16 days for neurons) as indel accumulation in nondividing cells follows prolonged kinetics.
- Collect time-point samples at days 1, 2, 4, 7, 11, and 16 post-transduction for comprehensive analysis.
- At each time point, assess:
- Cell viability via Calcein AM/propidium iodide staining
- DNA damage markers (γH2AX, 53BP1) via immunocytochemistry
- Editing efficiency via targeted NGS of genomic DNA
Day 16: Comprehensive Endpoint Analysis
- Harvest cells for genomic DNA extraction using silica column-based methods.
- Perform long-range PCR (≥5kb) around the target site to detect large deletions missed by standard amplicon sequencing.
- Conduct targeted deep sequencing of on-target region with 2x300bp paired-end reads to capture spectrum of indels.
- Implement orthogonal off-target detection method (GUIDE-seq or Digenome-seq) for comprehensive risk assessment.
- Fix remaining cells for immunocytochemical analysis of DNA damage foci and cell type-specific markers.
Data Analysis and Interpretation
- Editing Efficiency Calculation: Process NGS data with automated indel detection pipelines (e.g., CRISPResso2). Calculate percentage of reads with indels at each time point.
- Genotoxicity Assessment: Quantify γH2AX/53BP1 foci per nucleus across experimental conditions. Compare to untreated controls to establish baseline.
- Viability Correlation: Normalize editing efficiency to viability measurements at each time point. Calculate "therapeutic window" as the ratio between desired editing outcomes and cell death.
- Structural Variation Analysis: Assess long-range PCR products by agarose gel electrophoresis for anomalous band sizes indicating large deletions.
- Kinetic Modeling: Plot indel accumulation over time. Note that nondividing cells typically show continued accumulation for up to 2 weeks versus plateau within days in dividing cells.
Troubleshooting Notes
- If VLP transduction efficiency is suboptimal, optimize pseudotype combination or modulate nuclear localization signals [12].
- If cytotoxicity is observed predominantly in edited cells, consider alternative sgRNAs with different repair outcome distributions.
- If large deletions are detected, implement additional specificity measures such as high-fidelity Cas9 variants or paired nickases.
- For cell types with particularly low viability, consider Cas9 base editors or prime editors that avoid DSB formation altogether.
Balancing editing efficacy with cell viability requires a multifaceted approach that addresses the complexity of DNA repair biology across different cell types and chromatin contexts. The strategies outlined here—including careful delivery system selection, comprehensive genotoxicity assessment, and context-aware repair modulation—provide a roadmap for developing safer CRISPR-based therapies. As the field progresses, standardized assessment of both on-target and off-target genomic alterations, combined with improved understanding of cell-type specific repair mechanisms, will be essential for advancing therapeutic genome editing while minimizing toxicities. The recent clinical successes in lipid nanoparticle-mediated in vivo editing demonstrate that these challenges are not insurmountable, offering promise for a new generation of precisely targeted genetic medicines [9] [92].
The CRISPR-Cas9 system has revolutionized genetic engineering by enabling precise induction of double-strand breaks (DSBs) at targeted genomic loci. However, the ultimate editing outcome is determined not by the cutting event itself, but by the cellular DNA repair machinery that responds to the damage. While DSB repair has been extensively studied in dividing cells, understanding these processes in non-dividing cells is particularly crucial for therapeutic applications in genetic diseases affecting postmitotic tissues such as neurons and cardiomyocytes.
Recent research has revealed a fundamental biological difference: non-dividing cells exhibit dramatically prolonged indel accumulation kinetics compared to their dividing counterparts [12] [13]. This discovery has significant implications for the development of CRISPR-based therapies for neurological and cardiovascular diseases, as it challenges conventional expectations about editing timelines and efficiency that were largely derived from studies in proliferative cell types.
Fundamental Kinetic Differences Between Cell Types
Distinct Repair Timelines in Dividing vs. Non-Dividing Cells
In dividing cells such induced pluripotent stem cells (iPSCs), Cas9-induced DSBs are typically resolved relatively quickly, with indel frequencies plateauing within a few days. Quantitative studies in human cell lines have estimated the repair half-life of Cas9-induced DSBs to be between 1 and 10 hours, with most editing outcomes stabilizing within approximately 24-48 hours [93] [94].
In striking contrast, postmitotic cells such as neurons display markedly prolonged repair kinetics. Research using iPSC-derived neurons has demonstrated that indel accumulation continues to increase for up to 16 days following Cas9 delivery, with no evidence of plateauing before this timepoint [12] [95]. This weeks-long timeline of editing activity represents a fundamental shift in our understanding of DNA repair dynamics in non-dividing cells and suggests that postmitotic cells may employ different regulatory mechanisms for DNA damage response.
Table 1: Comparative Kinetics of INDEL Accumulation in Different Cell Types
| Cell Type | Cell Cycle Status | Time to INDEL Plateau | Predominant Repair Pathways | Key Regulatory Features |
|---|---|---|---|---|
| iPSCs | Dividing | 2-3 days | MMEJ, NHEJ | Cell cycle-dependent repair pathway choice |
| iPSC-derived Neurons | Postmitotic | >16 days | NHEJ | Limited MMEJ, unique repair factor expression |
| iPSC-derived Cardiomyocytes | Postmitotic | >14 days | NHEJ | Similar to neuronal repair patterns |
| Primary T Cells (Resting) | Non-dividing | Prolonged | NHEJ | Cell cycle-independent repair |
| K562 Cells | Dividing | ~60 hours | C-NHEJ, MMEJ | Standard dividing cell kinetics |
Preferential DNA Repair Pathway Utilization
The prolonged indel accumulation in non-dividing cells coincides with fundamental differences in the deployment of specific DNA repair pathways. Dividing cells utilize a broad repertoire of repair mechanisms, including microhomology-mediated end joining (MMEJ) which typically generates larger deletions and is active in specific cell cycle phases (S/G2/M) [12].
In contrast, postmitotic neurons predominantly rely on non-homologous end joining (NHEJ), resulting in a narrower distribution of smaller indels [12] [95]. This pathway preference was consistent across multiple sgRNAs targeting different genomic loci, suggesting it represents a fundamental characteristic of neuronal DNA repair rather than a target-specific phenomenon.
The differential pathway utilization has practical implications for therapeutic editing outcomes. While dividing cells produce a broad spectrum of indel sizes, the predominance of NHEJ in neurons yields predominantly smaller indels, which may be advantageous for gene disruption strategies but presents challenges for approaches requiring larger sequence modifications.
Mechanisms Underlying Prolonged Kinetics
Cell Cycle-Independent Repair Regulation
The prolonged kinetics of indel accumulation in non-dividing cells can be partially explained by freedom from replication-associated pressure. Dividing cells face stringent DNA damage checkpoints that can trigger cell cycle arrest or apoptosis if DSBs remain unresolved [12]. This pressure encourages rapid, albeit potentially error-prone, repair to permit cell cycle progression.
Postmitotic cells, lacking replication checkpoints, can tolerate unresolved DSBs for extended periods without triggering cell death pathways [12]. This permits a more measured repair process that may prioritize accuracy over speed, potentially explaining the extended timeline for complete resolution of Cas9-induced damage.
Unique Molecular Responses in Non-Dividing Cells
Transcriptomic profiling of Cas9-treated neurons has revealed that they mount a unique gene expression response to DNA damage compared to dividing cells [13]. Notably, neurons upregulate unexpected DNA repair factors, including replication-associated genes such as RRM2 (a ribonucleotide reductase subunit) that are not typically associated with postmitotic DNA repair [13].
This atypical response suggests that neurons may employ non-canonical mechanisms to process DSBs, potentially contributing to the extended repair timeline. Pharmacological or genetic inhibition of these upregulated factors, including RRM2, has been shown to influence editing outcomes by increasing deletion sizes and overall indel efficiency [13], providing both experimental evidence for their functional role and potential strategies for modulating repair outcomes.
Experimental Models and Methodologies
Advanced Delivery Systems for Non-Dividing Cells
Studying DNA repair in non-dividing cells requires specialized delivery approaches, as standard transfection methods optimized for dividing cells often prove inefficient for postmitotic cells. Recent research has leveraged virus-like particles (VLPs) to deliver Cas9 ribonucleoprotein (RNP) complexes to human iPSC-derived neurons with high efficiency (up to 97%) [12] [95].
These VLP systems, pseudotyped with VSVG and/or BaEVRless (BRL) envelope proteins, enable efficient transduction of human neurons while providing transient delivery of active Cas9 complexes [12]. This delivery method has been crucial for enabling kinetic studies in postmitotic cells, as it bypasses the need for nuclear import mechanisms that may be inefficient in non-dividing cells.
Table 2: Essential Research Reagents for Studying DNA Repair in Non-Dividing Cells
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Delivery Systems | VSVG/BRL-pseudotyped VLPs | Efficient Cas9 RNP delivery to neurons | Optimize pseudotype for specific cell type |
| Cell Models | iPSC-derived neurons, iPSC-derived cardiomyocytes | Physiologically relevant postmitotic models | Ensure purity (>95% NeuN+ for neurons) |
| Kinetic Assays | Time-course sequencing, γH2AX/53BP1 immunofluorescence | Monitoring repair progression and DSB resolution | Extended timelines (2+ weeks) for non-dividing cells |
| Pathway Modulators | RRM2 inhibitors, siRNA against repair factors | Directing repair outcomes | Cell-type specific optimization required |
| Analysis Tools | UMI-DSBseq, single-molecule sequencing | High-resolution repair mapping | Enables precise kinetic measurements |
Quantitative Kinetic Measurement Approaches
Accurately measuring repair kinetics requires methodologies that can distinguish between intact, broken, and repaired molecules across extended timecourses. UMI-DSBseq (unique molecular identifier DSB sequencing) represents a recent advancement that enables simultaneous quantification of DSB intermediates and repair products at single-molecule resolution [8].
This approach involves ligating adaptors containing UMIs directly to unrepaired DSBs as well as to intact molecules, allowing precise tracking of repair progression over time [8]. When applied to time-course experiments in Cas9-treated cells, this method can capture the gradual accumulation of indels and the resolution of DSB intermediates, providing quantitative data for modeling repair kinetics.
Protocol: Measuring INDEL Accumulation Kinetics in iPSC-Derived Neurons
Experimental Workflow for Kinetic Analysis:
Cell Differentiation: Differentiate human iPSCs into cortical-like excitatory neurons using established protocols [12] [95]. Validate postmitotic status through immunocytochemistry for Ki67 (negative) and NeuN (positive) to ensure >95% purity by day 7 of differentiation.
Cas9 Delivery: Prepare VSVG/BRL-pseudotyped Friend Murine Leukemia Virus (FMLV) VLPs containing Cas9 ribonucleoprotein complexes. Transduce neurons at optimized MOI to achieve high efficiency delivery while maintaining cell viability.
Time-Course Sampling: Collect cells at multiple timepoints post-transduction (e.g., 6h, 24h, 72h, 7d, 14d, 21d). Include early timepoints to capture initial repair dynamics and extended timepoints to track the prolonged accumulation phase characteristic of neurons.
Genomic Analysis: Extract genomic DNA and amplify target loci by PCR. Utilize high-throughput sequencing with minimum 10,000x coverage per timepoint. Employ UMI-based approaches where possible to enable single-molecule resolution of repair outcomes.
Quantitative Modeling: Analyze sequencing data to quantify intact, broken, and indel-containing molecules at each timepoint. Fit mathematical models to determine rate constants for cutting (kc), perfect repair (kp), and mutagenic repair (km) based on established kinetic frameworks [94].
Implications for Therapeutic Genome Editing
Challenges for Gene Therapy Applications
The prolonged kinetics of indel accumulation in non-dividing cells presents both challenges and opportunities for therapeutic genome editing. The extended timeline means that therapeutic outcomes in neuronal tissues may require weeks to fully manifest, complicating the assessment of editing efficiency in clinical trials and potentially delaying therapeutic effects [12] [95].
Additionally, the persistent presence of active Cas9 nuclease in non-dividing cells (reportedly remaining active for over 30 days in neurons) [13] increases the potential for off-target effects, as the editing window remains open for extended periods. This necessitates careful consideration of delivery modalities and Cas9 persistence when designing therapeutic approaches for non-dividing tissues.
Strategies for Modulating Repair Outcomes
The unique repair environment of non-dividing cells also presents opportunities for therapeutic intervention. Research has demonstrated that manipulating the DNA damage response through chemical or genetic perturbations can influence editing outcomes in neurons [12] [13]. For example, inhibition of upregulated repair factors like RRM2 can shift the distribution of indel outcomes, providing a potential strategy for favoring therapeutic editing results.
Advanced delivery systems such as all-in-one lipid nanoparticles that concurrently deliver Cas9, sgRNA, and siRNAs against specific repair factors enable coordinated editing and pathway modulation [13]. This approach allows researchers to tailor the cellular repair environment to favor desired outcomes, potentially overcoming the natural biases of neuronal repair pathways.
The prolonged indel accumulation kinetics in non-dividing cells represents a fundamental biological phenomenon with significant implications for therapeutic genome editing. The extended repair timeline, predominant use of NHEJ over MMEJ, and unique molecular responses to DNA damage in postmitotic cells collectively shape distinct editing outcomes that differ dramatically from those observed in dividing cells.
Understanding these cell-type-specific repair mechanisms is crucial for developing effective CRISPR-based therapies for neurological and cardiovascular diseases. Future research directions should focus on elucidating the molecular regulators of these kinetic differences and developing strategies to harness these unique properties for improved therapeutic outcomes. As the field advances, consideration of these cell-type-specific repair kinetics will be essential for translating CRISPR technologies into effective treatments for genetic disorders affecting non-dividing tissues.
Ensuring Safety and Efficacy: Analytical Methods and Outcome Assessment
The CRISPR-Cas9 system has revolutionized genetic engineering by providing an efficient and programmable method for making targeted double-strand breaks (DSBs) in the genome. This system consists of two key components: the Cas9 nuclease and a single-guide RNA (sgRNA) that directs Cas9 to a specific DNA sequence adjacent to a protospacer adjacent motif (PAM) [1]. Once bound, the Cas9 enzyme cleaves both DNA strands, creating a DSB that is subsequently repaired by the cell's endogenous repair mechanisms, primarily non-homologous end joining (NHEJ) or homology-directed repair (HDR) [1]. While NHEJ is efficient, it is error-prone and often results in small insertions or deletions (indels) that can disrupt gene function, making it particularly useful for gene knockout strategies [96] [1].
A significant challenge in CRISPR-Cas9 applications, especially for therapeutic purposes, is the potential for off-target effects—unintended edits at genomic locations other than the intended target site. These occur primarily because the Cas9 nuclease can tolerate mismatches between the sgRNA and genomic DNA, particularly when these mismatches are located distal to the PAM sequence [96]. As CRISPR-based therapies advance toward clinical applications, with the first FDA approval granted for Casgevy (exa-cel) for sickle cell disease and transfusion-dependent beta thalassemia in 2023 [97] [9], comprehensive off-target profiling has become an essential safety requirement. Regulatory agencies like the FDA now recommend using multiple methods, including genome-wide analysis, to measure off-target editing events [97].
Traditional in silico prediction tools (e.g., Cas-OFFinder, CCTop, CFD) that rely solely on sequence homology have significant limitations as they insufficiently consider the complex intranuclear microenvironment, including epigenetic and chromatin organization states [96]. This has driven the development of unbiased experimental methods for genome-wide off-target detection, including GUIDE-seq, BLESS, and Digenome-seq, which provide comprehensive profiling without a priori knowledge of potential off-target sites [96] [97]. This technical guide examines these three key methods, providing detailed methodologies and comparative analysis to inform their application in genetic disease research and therapeutic development.
The landscape of off-target detection methods can be broadly categorized into biochemical approaches (using purified genomic DNA) and cellular approaches (using living or fixed cells) [97]. Each approach offers distinct advantages and limitations in sensitivity, biological relevance, and technical complexity.
Table 1: Classification of Featured Off-Target Detection Methods
| Method | Approach Category | Input Material | Detection Context | Key Advantage |
|---|---|---|---|---|
| GUIDE-seq [96] [97] | Cellular | Living cells (edited) | Native chromatin + cellular repair | Reflects true cellular activity; identifies biologically relevant edits |
| BLESS [96] [97] | In situ | Fixed/permeabilized cells or nuclei | Chromatinized DNA in native location | Preserves genome architecture; captures breaks in situ |
| Digenome-seq [96] [97] [98] | Biochemical | Purified genomic DNA | Naked DNA (no chromatin) | Ultra-sensitive; comprehensive; standardized |
Table 2: Comprehensive Comparison of Technical Specifications
| Specification | GUIDE-seq | BLESS | Digenome-seq |
|---|---|---|---|
| Sensitivity | High sensitivity for off-target DSB detection [97] | Moderate; detects DSBs but limited by labeling efficiency [97] | Moderate; requires deep sequencing to detect off-targets [97] |
| Detects Indels | Yes [97] | No [97] | Yes (via downstream validation) [98] |
| Detects Translocations | No [97] | No [97] | No |
| Workflow Complexity | Requires efficient delivery of dsODN; limited by transfection efficiency [96] [97] | Technically complex; involves in situ labeling in fixed cells [97] | Simplified in vitro workflow; no cell culture needed [97] [98] |
| Primary Limitation | Limited by transfection efficiency [96] [97] | Only identifies off-target sites at the time of detection [96] | Lacks biological context; may overestimate cleavage [97] |
Detailed Experimental Protocols
GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing)
Principle: GUIDE-seq relies on the incorporation of a double-stranded oligodeoxynucleotide (dsODN) tag into DSBs generated by CRISPR-Cas9 in living cells through the NHEJ repair pathway. These tagged genomic locations are then enriched and identified via next-generation sequencing [96] [97].
Step-by-Step Protocol:
- Transfection: Co-deliver the Cas9/sgRNA complex (as plasmid, RNA, or ribonucleoprotein) along with the synthetic, blunt-ended, 5'-phosphorylated dsODN tag into the target cells. Typical dsODN concentration is 50-100 nM [97].
- Incubation: Allow cells to grow for 48-72 hours to enable CRISPR cutting, dsODN integration via NHEJ, and repair.
- Genomic DNA Extraction: Harvest cells and extract genomic DNA using standard methods.
- Library Preparation:
- Fragment the genomic DNA by sonication or enzymatic digestion.
- Repair the ends of the DNA fragments and ligate to sequencing adapters.
- Perform PCR amplification using one primer specific to the ligated adapter and another primer specific to the integrated dsODN tag. This selectively enriches for genomic regions containing the dsODN.
- Sequencing and Analysis: Sequence the amplified library using next-generation sequencing. Map the reads to the reference genome to identify genomic locations that contain the integrated dsODN tag, which correspond to both on-target and off-target DSB sites [97].
GUIDE-seq tags DSBs in living cells via NHEJ repair.
BLESS (Breaks Labeling, Enrichment on Streptavidin, and Sequencing)
Principle: BLESS directly captures and labels DSBs in situ within fixed cells, preserving the native genomic architecture and providing a snapshot of cleavage events at a specific time point [96] [97].
Step-by-Step Protocol:
- Cell Fixation and Permeabilization: Treat cells with CRISPR-Cas9, then fix with formaldehyde and permeabilize to preserve nuclear architecture while allowing reagent access.
- In Situ Ligation: In fixed nuclei, ligate biotinylated adapters or linkers directly to the ends of the DSBs using T4 DNA ligase.
- Genome Extraction and Capture: Extract and purify genomic DNA. Shear the DNA by sonication and capture the biotinylated fragments (containing the DSB sites) using streptavidin-coated magnetic beads.
- Library Preparation: On-bead, ligate sequencing adapters to the captured DNA fragments and amplify via PCR.
- Sequencing and Analysis: Perform next-generation sequencing. Map the reads to the reference genome, identifying the genomic locations of the DSB sites based on the junction between the genome and the known adapter sequence [96] [97].
BLESS captures DSBs in fixed cells to preserve genomic context.
Digenome-seq (Digested Genome Sequencing)
Principle: Digenome-seq involves digesting purified, cell-free genomic DNA with the Cas9 ribonucleoprotein (RNP) complex in vitro. The cleaved DNA is then subjected to whole-genome sequencing, and the resulting reads are computationally analyzed to identify sites of cleavage based on characteristic alignment patterns [96] [97] [98].
Step-by-Step Protocol:
- Genomic DNA Preparation: Isolve high-quality, high-molecular-weight genomic DNA from the cell type of interest.
- In Vitro Digestion: Incubate the purified genomic DNA with preassembled Cas9/sgRNA RNP complexes in an appropriate reaction buffer.
- Whole-Genome Sequencing: Directly subject the digested DNA to whole-genome sequencing without an enrichment step, generating high-coverage data.
- Computational Analysis:
- Map the sequencing reads to the reference genome.
- Use a specialized scoring algorithm to identify genomic positions where many reads start and end precisely, forming a bimodal distribution. This pattern indicates a Cas9 cleavage site.
- The computational program searches the entire genome for these systematically aligned reads to call off-target sites [99] [98].
- Validation: Candidate off-target sites typically require validation in cellular systems using targeted amplicon sequencing to confirm indel induction [98].
Digenome-seq detects cleavage patterns in purified DNA.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of these off-target profiling methods requires specific reagents and tools. The table below details essential components for establishing these assays.
Table 3: Research Reagent Solutions for Off-Target Profiling
| Reagent / Tool | Function | Considerations |
|---|---|---|
| Recombinant Cas9 Nuclease | Catalyzes the DNA double-strand break at target sites. | Source and purity (e.g., S. pyogenes) are critical for consistent in vitro (Digenome-seq) and cellular activity [1] [30]. |
| Synthetic sgRNA or Plasmid | Guides Cas9 to the specific genomic target. | Quality is crucial; sgRNAs transcribed from plasmid templates reduce false-positive bulge-type off-targets compared to those from oligonucleotide duplexes [99]. |
| Double-Stranded Oligodeoxynucleotide (dsODN) | Serves as the tag incorporated into DSBs in GUIDE-seq. | Must be blunt-ended, 5'-phosphorylated, and HPLC-purified for efficient NHEJ integration [97]. |
| Biotinylated Adapters | Labels DSB ends for capture in BLESS and SITE-seq. | Linker design and ligation efficiency are key factors determining sensitivity [96] [97]. |
| Streptavidin Magnetic Beads | Enriches biotinylated DNA fragments (DSB sites) in BLESS. | Bead capacity and purity affect the background noise and specificity of the assay [97]. |
| Next-Generation Sequencer | Enables genome-wide sequencing of libraries. | Required for all methods; Digenome-seq needs deeper WGS coverage, while others require less depth due to enrichment steps [97] [100]. |
| Analysis Software/Pipeline | Maps sequencing reads and calls off-target sites. | Method-specific algorithms are needed (e.g., bimodal peak calling for Digenome-seq, tag alignment for GUIDE-seq) [99] [97]. |
GUIDE-seq, BLESS, and Digenome-seq each provide powerful, complementary strategies for unbiased genome-wide profiling of CRISPR-Cas9 off-target effects. The choice of method depends on the specific research goals: GUIDE-seq offers high sensitivity within a cellular context, BLESS provides spatial information about breaks in situ, and Digenome-seq delivers ultra-sensitive discovery in a controlled biochemical environment [96] [97].
For robust off-target assessment in preclinical therapeutic development, a combination of these methods is increasingly recommended. Initial broad discovery using a sensitive in vitro method like Digenome-seq can be followed by validation in physiologically relevant cell models using a cellular method like GUIDE-seq to identify biologically relevant off-target sites [97]. As the field advances, emerging improvements such as multiplex Digenome-seq—which allows profiling of up to 11 sgRNAs simultaneously—are enhancing throughput and reducing costs [99]. Furthermore, the application of these methods is revealing insights into the fundamental mechanisms of DSB repair [101], informing the development of next-generation CRISPR systems with enhanced fidelity [96] [30] and paving the way for safer genetic therapies for a wide range of human diseases.
The CRISPR-Cas9 system has revolutionized genetic research and therapeutic development by enabling precise double-strand breaks (DSBs) at specific genomic loci [102]. This RNA-guided nuclease system functions as programmable molecular scissors, where the Cas9 enzyme complexes with a single-guide RNA (sgRNA) to identify and cleave target DNA sequences adjacent to a protospacer adjacent motif (PAM) [3] [103]. However, the CRISPR-Cas9 machinery itself does not perform genetic modifications; it merely creates the DSB, and the subsequent editing outcome is entirely determined by the cell's endogenous DNA repair mechanisms [102] [104]. The competition between these repair pathways—primarily non-homologous end joining (NHEJ) and homology-directed repair (HDR)—represents a critical juncture in therapeutic genome editing, with each pathway offering distinct advantages and limitations for different disease contexts [11] [105].
The therapeutic application of CRISPR-Cas9 hinges on manipulating this DNA repair competition. While HDR enables precise, template-driven corrections ideal for monogenic disorders, its efficiency remains a significant challenge, particularly in non-dividing cells [11] [16]. Conversely, NHEJ offers higher efficiency across cell types but introduces unpredictable insertions or deletions (indels) [102] [104]. This technical analysis examines the efficiency-precision dichotomy between NHEJ and HDR pathways across therapeutic contexts, evaluates current enhancement strategies, and explores their implications for clinical translation.
Non-Homologous End Joining (NHEJ): The Efficient First Responder
NHEJ serves as the cell's primary DSB repair mechanism, characterized by rapid ligation of broken DNA ends without requiring a homologous template [11]. This pathway operates throughout all cell cycle phases, making it highly active in both dividing and postmitotic cells [105]. The repair process initiates with the Ku70-Ku80 heterodimer recognizing and binding to broken DNA ends, forming a ring-like structure that encircles the DNA [11] [106]. This Ku-DNA complex then recruits DNA-dependent protein kinase catalytic subunit (DNA-PKcs), which activates Artemis nuclease to process damaged ends [11] [106]. Finally, the XRCC4-DNA ligase IV complex catalyzes ligation, completing the repair [11] [106].
Therapeutically, NHEJ is particularly valuable for gene knockout strategies where disrupting gene function is the objective [102] [104]. The error-prone nature of NHEJ often generates frameshift mutations that effectively knockout gene function, making it ideal for treating conditions like sickle cell disease and beta-thalassemia where disrupting a harmful gene provides therapeutic benefit [9]. The high efficiency of NHEJ (typically 20-60% across cell types) compared to HDR (often <10% in many primary cells) further enhances its therapeutic utility [11] [16].
Homology-Directed Repair (HDR): The Precision Engineer
HDR represents the cell's precise DSB repair mechanism, utilizing homologous DNA sequences as templates for error-free repair [102] [105]. This pathway is restricted to the S and G2 phases of the cell cycle when sister chromatids are available, limiting its activity in non-dividing cells [105]. The HDR mechanism begins with the MRN complex (MRE11-RAD50-NBS1) recognizing the DSB and initiating 5' to 3' end resection in cooperation with CtIP, creating 3' single-stranded DNA overhangs [105] [106]. Replication protein A (RPA) then stabilizes these ssDNA tails, after which RAD51 displaces RPA to form nucleoprotein filaments that mediate strand invasion into the homologous donor template [105]. DNA polymerase extends the invading strand using the donor sequence as a template, followed by resolution of the resulting DNA structures to complete precise repair [105].
HDR's precision makes it indispensable for therapeutic applications requiring specific genetic corrections, such as point mutation repairs, targeted insertions, or gene additions [11] [16]. However, HDR faces significant therapeutic challenges due to its inherently low efficiency and competition with the more active NHEJ pathway [11] [105]. Additionally, the requirement for co-delivery of donor DNA templates and its cell cycle dependence creates technical and biological barriers that must be overcome for successful clinical application [16] [106].
Table 1: Fundamental Characteristics of NHEJ and HDR Pathways
| Feature | Non-Homologous End Joining (NHEJ) | Homology-Directed Repair (HDR) |
|---|---|---|
| Template Requirement | No template required; ligates broken ends directly [102] | Requires homologous donor template (endogenous or exogenous) [11] |
| Primary Proteins | Ku70/Ku80, DNA-PKcs, Artemis, XRCC4, DNA Ligase IV [11] [106] | MRN complex, CtIP, RPA, RAD51, BRCA1/2 [105] [106] |
| Cell Cycle Activity | Active throughout all phases (G1, S, G2) [105] | Primarily restricted to S and G2 phases [105] |
| Mutation Profile | Error-prone; generates small insertions/deletions (indels) [102] [104] | High-fidelity; enables precise base changes or insertions [11] |
| Therapeutic Application | Gene knockouts (e.g., BCL11A for sickle cell disease) [9] | Gene correction (e.g., point mutations), targeted knockins [11] [16] |
Diagram 1: DNA Repair Pathways After CRISPR-Cas9 Cleavage
Quantitative Efficiency and Precision Analysis
Pathway Efficiency Metrics Across Cell Types
The efficiency disparity between NHEJ and HDR pathways varies significantly across different cell types and experimental conditions. In standard in vitro models using immortalized cell lines, NHEJ typically demonstrates 20-80% efficiency in generating indels, while HDR efficiency generally ranges from 1-20% depending on the specific modification and delivery method [11]. This efficiency gap widens considerably in therapeutically relevant primary cells; in hematopoietic stem cells (HSCs), NHEJ efficiencies of 40-60% are commonly reported, whereas HDR rates typically remain below 10% without enhancement strategies [11] [16]. The challenge is most pronounced in postmitotic cells like neurons and cardiomyocytes, where HDR efficiency falls to negligible levels (<1%) due to cell cycle restrictions, while NHEJ maintains substantial activity (10-30%) [11] [105].
Recent clinical data further illustrates this efficiency-precision tradeoff. In the landmark Casgevy (exa-cel) trial for sickle cell disease, NHEJ-mediated knockout of the BCL11A gene achieved therapeutic levels of fetal hemoglobin in 91% of patients with high efficiency [9]. Conversely, HDR-based approaches in clinical trials for monogenic disorders like hereditary transthyretin amyloidosis (hATTR) demonstrate lower but still therapeutic editing efficiencies, with Intellia Therapeutics reporting approximately 90% reduction in disease-related protein levels through HDR-mediated gene disruption [9].
Table 2: Efficiency and Precision Comparison in Therapeutic Contexts
| Therapeutic Context | Typical NHEJ Efficiency | Typical HDR Efficiency | Primary Applications |
|---|---|---|---|
| Immortalized Cell Lines | 20-80% indel formation [11] | 1-20% precise editing [11] [16] | Target validation, screening assays |
| Hematopoietic Stem Cells | 40-60% indel formation [11] | 5-10% precise editing (with enhancement) [16] | Sickle cell disease, beta-thalassemia, immunodeficiencies |
| Primary Lymphocytes | 50-80% indel formation [11] | 2-10% precise editing [16] | CAR-T engineering, adoptive cell therapies |
| Postmitotic Cells (e.g., neurons) | 10-30% indel formation [11] [105] | <1% precise editing [11] [105] | Neurodegenerative disorders (limited application) |
| In Vivo Liver Editing | 30-60% indel formation [9] | 5-15% precise editing (reported as protein reduction) [9] | hATTR, hypercholesterolemia, metabolic disorders |
Molecular and Technical Factors Influencing Pathway Choice
Multiple technical parameters significantly influence the balance between NHEJ and HDR outcomes. Delivery method substantially impacts this balance; viral vectors (especially AAV) can enhance HDR efficiency by providing persistent donor template expression, while electroporation of ribonucleoprotein (RNP) complexes favors NHEJ due to rapid nuclease activity and clearance [11] [16]. The format of the donor template also critically affects HDR efficiency; single-stranded oligodeoxynucleotides (ssODNs) typically yield higher HDR rates for small edits (<100 bp), while double-stranded DNA templates with extended homology arms (800-1000 bp) are superior for larger insertions [16] [106].
Cell cycle status represents perhaps the most fundamental biological constraint on pathway choice. HDR is strongly favored in S/G2 phases due to cyclin-dependent kinase (CDK) activity that promotes DNA end resection, while NHEJ dominates in G1 and early S phases [105]. This dependency explains the particularly low HDR efficiency observed in non-dividing cell populations. Additionally, CRISPR-Cas9 kinetics influence editing outcomes; prolonged Cas9 expression increases NHEJ-dominated re-cutting of successfully edited alleles, whereas transient RNP delivery can improve HDR yields by limiting nuclease activity duration [3] [105].
Strategic Enhancement of Repair Pathways
HDR Enhancement Methodologies
Multiple strategic approaches have been developed to enhance HDR efficiency for therapeutic applications. These methodologies generally fall into four categories: biochemical inhibition of competing pathways, cell cycle synchronization, donor template optimization, and engineered nuclease systems.
NHEJ Inhibition represents a direct approach to favor HDR. Small molecule inhibitors targeting key NHEJ components, such as DNA-PKcs inhibitors (NU7441, AZD7648), Scr7 (DNA Ligase IV inhibitor), and 53BP1 knockdown strategies have demonstrated 2-5 fold increases in HDR efficiency across multiple cell types [11] [106]. However, recent studies reveal significant safety concerns with this approach, particularly that DNA-PKcs inhibition can exacerbate genomic aberrations including kilobase- to megabase-scale deletions and chromosomal translocations [45] [105].
Cell Cycle Synchronization leverages the natural restriction of HDR to S/G2 phases. Chemical treatments including nocodazole, thymidine, and lovastatin can synchronize cells in HDR-permissive phases, typically increasing HDR efficiency by 2-3 fold [11] [16]. The non-specific cytotoxicity of these compounds presents challenges for therapeutic applications, particularly in sensitive primary cells like HSCs.
Donor Template Engineering offers a more refined approach to enhance HDR. Strategies including chemically modified donor oligonucleotides (e.g., phosphorothioate backbones), asymmetric donors with longer homology arms on the PAM-distal side, and viral vector delivery systems have demonstrated consistent improvements in HDR efficiency without increasing genotoxic risk [16] [106].
Engineered Nuclease Systems represent the most technologically advanced approach. Fusion of Cas9 to HDR-promoting domains like RAD51, BRCA2, or CtIP can locally enhance HDR factors at the target site [106]. Additionally, "base editing" and "prime editing" systems capable of precise changes without DSBs circumvent the NHEJ-HDR competition entirely, though they are currently limited in their scope of editable sequences and insert size capacity [103] [105].
Diagram 2: HDR Enhancement Methods and Considerations
Experimental Protocols for Pathway Modulation
Protocol 1: HDR Enhancement via Small Molecule Inhibition
- Reagent Preparation: Prepare stock solutions of DNA-PKcs inhibitor (e.g., AZD7648 at 10 mM in DMSO) and store at -20°C [11] [106].
- Cell Treatment: Apply inhibitor at 1-10 μM concentration 2 hours prior to CRISPR delivery. Maintain inhibitor throughout the first 24-48 hours post-editing [106].
- Control Setup: Include DMSO-only vehicle control and untreated control to assess baseline editing efficiency and toxicity.
- Efficiency Assessment: Harvest cells 72-96 hours post-editing. Analyze HDR and NHEJ frequencies via next-generation sequencing of target locus. Validate potential structural variations using CAST-Seq or LAM-HTGTS if therapeutic application intended [45].
Protocol 2: Cell Cycle Synchronization for HDR Enhancement
- Synchronization: Treat dividing cells with 100 ng/mL nocodazole or 2 mM thymidine for 16-24 hours to arrest cells in M or G1/S phase, respectively [11] [16].
- Release and Edit: Release synchronization by washing and transferring to fresh media. Deliver CRISPR components immediately following release to target the subsequent S/G2 phases.
- Validation: Confirm synchronization efficiency by flow cytometry analysis of DNA content (propidium iodide staining) parallel to editing experiment.
- Timing Consideration: Optimal editing window typically occurs 4-8 hours post-release for most mammalian cell types [16].
Therapeutic Applications and Clinical Translation
NHEJ-Dominated Therapeutic Strategies
NHEJ-based therapies have demonstrated remarkable clinical success in conditions where gene disruption provides therapeutic benefit. The pioneering example is Casgevy (exa-cel) for sickle cell disease and transfusion-dependent beta thalassemia, where NHEJ-mediated knockout of the BCL11A gene disrupts its repressive activity on fetal hemoglobin expression [9]. This approach achieved 91% freedom from severe vaso-occlusive crises in sickle cell patients, demonstrating the therapeutic potential of efficient NHEJ utilization [9]. Similar NHEJ-dominated strategies are advancing for HIV treatment through CCR5 receptor knockout, lipid disorders via ANGPTL3 disruption, and various monogenic diseases where eliminating a harmful gene product provides clinical benefit [9].
The primary advantage of NHEJ-based therapies lies in their high efficiency and applicability to both dividing and non-dividing cells, enabling robust editing in therapeutically relevant stem cell populations. However, concerns regarding potential genotoxic effects have emerged, with recent studies identifying unexpected large-scale structural variations, including megabase-scale deletions and chromosomal rearrangements at on-target sites [45]. These findings highlight the critical need for comprehensive genomic safety assessment in NHEJ-dominated therapeutic applications.
HDR-Based Therapeutic Approaches
HDR-based strategies offer the potential for precise genetic correction, making them ideally suited for monogenic disorders caused by specific point mutations or small deletions. Clinical progress, however, has been slower due to efficiency limitations. Notable advances include the first personalized in vivo CRISPR therapy for CPS1 deficiency, where an infant received a bespoke HDR-based treatment developed and delivered in just six months [9]. The therapy, delivered via lipid nanoparticles (LNPs), enabled multiple doses to increase editing percentages, with the patient showing improvement in symptoms and decreased medication dependence [9].
Intellia Therapeutics' HDR-based approach for hereditary transthyretin amyloidosis (hATTR) represents another promising application, demonstrating sustained approximately 90% reduction in disease-related protein levels in clinical trial participants [9]. Similarly, their hereditary angioedema (HAE) program reported 86% reduction in kallikrein protein and significant reduction in inflammatory attacks following HDR-mediated editing [9]. These successes highlight the potential of HDR-based strategies despite efficiency challenges, particularly when edited cells gain selective advantage or when delivery systems enable repeated dosing.
Table 3: Clinical Applications of NHEJ vs. HDR in Gene Therapy
| Therapeutic Approach | Disease Target | Editing Strategy | Clinical Status | Key Outcomes |
|---|---|---|---|---|
| NHEJ-Mediated Knockout | Sickle Cell Disease, Beta Thalassemia | BCL11A disruption to induce fetal hemoglobin [9] | FDA/EMA Approved (Casgevy) | 91% freedom from severe vaso-occlusive crises [9] |
| NHEJ-Mediated Knockout | hATTR, HAE | TTR/Kallikrein reduction [9] | Phase III Trials | ~90% protein reduction, symptom improvement [9] |
| HDR-Mediated Correction | CPS1 Deficiency | Precise mutation correction [9] | Personalized Therapy (N=1) | Symptom improvement, reduced medications [9] |
| HDR-Mediated Knockin | CAR-T Therapies | TCR knockout + CAR insertion [16] | Multiple Phase I/II Trials | Improved persistence, reduced exhaustion |
| Hybrid Approach | Various Monogenic Disorders | HDR correction + NHEJ selection marker [16] | Preclinical Development | Enriched HDR population |
The Scientist's Toolkit: Essential Reagents for DNA Repair Studies
Table 4: Key Research Reagents for NHEJ and HDR Studies
| Reagent Category | Specific Examples | Primary Function | Considerations |
|---|---|---|---|
| NHEJ Inhibitors | NU7441, AZD7648 (DNA-PKcs inhibitors); Scr7 (Ligase IV inhibitor) [11] [106] | Enhance HDR efficiency by suppressing competing NHEJ pathway | Risk of increased structural variations; optimal timing critical [45] |
| HDR Enhancers | RS-1 (RAD51 stimulator); L755507 (β-3 adrenergic receptor agonist) [106] | Stimulate key HDR factors to improve precise editing efficiency | Cell type-specific responses; potential off-target effects |
| Cell Cycle Synchronizers | Nocodazole (M phase arrest); Thymidine (G1/S arrest); Abemaciclib (CDK4/6 inhibitor) [11] [16] | Enrich for HDR-permissive S/G2 cell populations | Cytotoxicity concerns; requires careful timing of editing delivery |
| Donor Templates | ssODNs (50-200 nt); dsDNA with homology arms (800-1000 bp); AAV donors [16] [106] | Provide homologous sequence for HDR-mediated repair | ssODNs ideal for small edits; dsDNA for larger insertions; AAV for persistent expression |
| Analysis Tools | T7E1 assay; Sanger sequencing; NGS (amplicon); CAST-Seq [45] | Quantify editing efficiency and detect structural variations | Short-read NGS may miss large deletions; CAST-Seq specialized for SV detection |
The therapeutic application of CRISPR-Cas9 continues to navigate the fundamental tradeoff between the high efficiency of NHEJ and the precision of HDR. While NHEJ-dominated strategies have achieved landmark clinical approvals, emerging safety concerns regarding structural variations necessitate more sophisticated genomic safety assessments [45]. Conversely, HDR-based approaches, though currently limited by efficiency barriers, offer the potential for truly curative precise corrections for monogenic disorders.
Future developments will likely focus on three key areas: First, the refinement of HDR enhancement strategies that minimize genotoxic risks, particularly through engineered nuclease systems that bypass the NHEJ-HDR competition entirely [103] [105]. Second, improved delivery systems, especially LNPs that enable redosing as demonstrated in recent clinical cases [9]. Third, more sophisticated predictive models that account for cell-type specific repair preferences and chromatin context to better anticipate editing outcomes.
The ongoing clinical successes demonstrate that both pathways have significant roles in the future of genetic medicine. NHEJ will continue to dominate applications where gene disruption provides therapeutic benefit, while HDR-based approaches will advance toward disorders requiring precise correction. As the field matures, the strategic selection between these pathways will increasingly be guided by comprehensive risk-benefit analyses specific to each therapeutic context, ultimately expanding the scope of treatable genetic diseases.
The therapeutic application of CRISPR-Cas9 for correcting double-strand breaks (DSBs) in genetic diseases represents a paradigm shift in medicine. However, its clinical translation hinges on comprehensive safety validation addressing two fundamental concerns: unintended immunological consequences and the long-term stability of editing outcomes. The bacterial origin of CRISPR components triggers both innate and adaptive immune responses that can compromise therapeutic efficacy and patient safety [107]. Simultaneously, the persistence and behavior of edited cells over time remain poorly characterized, necessitating robust monitoring frameworks. This technical guide examines current methodologies for validating clinical safety, with emphasis on immune response characterization and long-term outcome assessment within the context of CRISPR-mediated DSB repair mechanisms.
Immune Response Mechanisms and Mitigation Strategies
Immunogenicity of CRISPR Components
CRISPR therapeutics comprise multiple components, each capable of eliciting distinct immune responses. The Cas9 nuclease, guide RNA (gRNA), and delivery vectors all present unique immunogenic profiles that must be characterized during safety validation.
Pre-existing immunity presents a substantial barrier to CRISPR therapies. Adaptive immune responses against commonly used Cas orthologs are prevalent in the general population due to previous exposure to source bacteria. Research demonstrates that pre-existing anti-Cas9 antibodies are detectable in up to 58-95% of healthy individuals, while T-cell responses occur in 67-100% [107]. Table 1 summarizes the prevalence of pre-existing immunity against various CRISPR effectors.
Table 1: Prevalence of Pre-existing Immunity to CRISPR Effectors in Healthy Populations
| CRISPR Effector | Source Organism | Antibody Prevalence (%) | T-cell Response Prevalence (%) | Study Size (n) |
|---|---|---|---|---|
| SpCas9 | Streptococcus pyogenes | 58-95% | 67-95% | 125-143 |
| SaCas9 | Staphylococcus aureus | 78-95% | 78-100% | 6-123 |
| Cas12a | Acidaminococcus sp. | N/A | 100% | 6 |
| RfxCas13d | Ruminococcus flavefaciens | 89% | 96-100% | 19-24 |
Beyond pre-existing immunity, de novo immune responses can develop following therapeutic administration. In murine models, both humoral and cellular immune responses occur after exposure to SpCas9 and SaCas9 [107]. Additionally, gRNAs can trigger innate immune responses through interaction with pattern recognition receptors, particularly when in vitro transcribed 5'-triphosphate gRNAs are used instead of chemically synthesized 5'-hydroxylated versions [107].
Immunogenicity Mitigation Approaches
Several strategies have emerged to mitigate CRISPR immunogenicity:
Protein Engineering: Structure-based computational tools have enabled redesign of Cas9 and Cas12 proteins to eliminate immunodominant epitopes while retaining editing function. Researchers have used mass spectrometry to identify immune-triggering sequences in Cas9 and Cas12, then engineered variants with significantly reduced immune responses in humanized mouse models [108].
Delivery Method Optimization: The choice of delivery system significantly influences immunogenicity. Lipid nanoparticles (LNPs) demonstrate favorable immunogenic profiles compared to viral vectors, enabling safe redosing as evidenced in clinical trials for hereditary transthyretin amyloidosis (hATTR) and a personalized treatment for CPS1 deficiency [9].
Ex Vivo Editing: For cell-based therapies, editing cells ex vivo followed by transplantation avoids direct exposure to CRISPR components. The successful implementation of this approach requires rigorous quality control to ensure minimal residual Cas9 protein in the final product [107].
Methodologies for Monitoring Long-Term Outcomes
DNA Repair Outcome Characterization
Comprehensive long-term monitoring requires understanding how different cell types resolve CRISPR-induced DSBs. Recent research reveals significant differences in repair kinetics and pathway utilization between dividing and non-dividing cells relevant to therapeutic applications.
In postmitotic cells such as neurons and cardiomyocytes, CRISPR-induced indels accumulate over an extended period of up to two weeks, significantly longer than the repair timeline in dividing cells [12] [13]. Neurons predominantly employ non-homologous end joining (NHEJ) over microhomology-mediated end joining (MMEJ), resulting in a narrower distribution of smaller indels compared to the broader range of outcomes in dividing cells [12]. This prolonged editing window correlates with persistent Cas9 activity for over 30 days in these cell types [13].
Table 2 compares DNA repair characteristics between dividing and non-dividing cells.
Table 2: DNA Repair Characteristics in Dividing vs. Non-Dividing Cells
| Characteristic | Dividing Cells (iPSCs) | Non-Dividing Cells (Neurons, Cardiomyocytes) |
|---|---|---|
| Primary Repair Pathways | MMEJ-predominant | NHEJ-predominant |
| Indel Distribution | Broad range, larger deletions | Narrow range, smaller indels |
| Repair Kinetics | Plateau within days | Continues for up to 2 weeks |
| Insertion:Deletion Ratio | Lower | Significantly higher |
| Cas9 Persistence | Short-lived | Active for over 30 days |
Experimental models for characterizing these differences utilize induced pluripotent stem cells (iPSCs) and iPSC-derived neurons or cardiomyocytes, enabling comparison of editing outcomes in genetically identical dividing and non-dividing systems [12]. Delivery methods include virus-like particles (VLPs) pseudotyped with VSVG and/or BaEVRless (BRL) glycoproteins, achieving up to 97% transduction efficiency in human neurons [12].
Analytical Methods for Safety Monitoring
Robust safety validation requires multiple complementary analytical approaches:
Next-Generation Sequencing (NGS): Comprehensive sequencing of target loci identifies intended edits, off-target effects, and complex rearrangements. Unique molecular identifiers and targeted capture approaches enhance sensitivity for detecting low-frequency events.
Immunological Assays: Enzyme-linked immunospot (ELISpot) and intracellular cytokine staining detect Cas-specific T-cells, while multiplex immunoassays characterize humoral responses against CRISPR components [107].
Longitudinal Molecular Tracking: For in vivo therapies, droplet digital PCR of circulating DNA can monitor editing persistence, while RNA sequencing characterizes transcriptomic changes associated with editing.
The following diagram illustrates the key differences in DNA repair pathways and outcomes between dividing and non-dividing cells:
Experimental Protocols for Safety Validation
Immune Monitoring Protocol
Objective: Comprehensive characterization of adaptive immune responses to CRISPR components.
Materials:
- Peripheral blood mononuclear cells (PBMCs) from recipients
- Cas protein antigens (SpCas9, SaCas9, etc.)
- ELISpot kits for IFN-γ, IL-2, and IL-6
- Peptide libraries spanning Cas protein sequences
- HLA-typing reagents
Procedure:
- Baseline Assessment: Collect pre-treatment PBMCs and serum for baseline immunoreactivity.
- T-cell Response Monitoring:
- Culture PBMCs with Cas-derived peptide pools (15-mers with 11-amino acid overlap)
- Perform IFN-γ ELISpot after 24-hour incubation
- For positive responses, conduct intracellular cytokine staining for Th1/Th2/Th17 cytokines
- Identify immunodominant epitopes through truncated peptide libraries
- Humoral Response Assessment:
- Screen serial serum samples by ELISA for anti-Cas IgG/IgM/IgA
- Confirm specificity by western blot against recombinant Cas proteins
- Perform neutralization assays measuring Cas9 function inhibition
- Data Analysis: Correlate immune responses with clinical outcomes, editing efficiency, and adverse events.
Long-Term Editing Stability Assessment
Objective: Longitudinal tracking of editing outcomes and persistence in target tissues.
Materials:
- Tissue biopsies (longitudinal when feasible)
- Circulating cell-free DNA from plasma
- Single-cell RNA sequencing reagents
- Digital PCR systems
- Long-range PCR reagents
Procedure:
- Baseline Genotyping: Comprehensive sequencing of target locus pre-editing.
- Short-Term Assessment (1-4 weeks):
- Bulk sequencing of target region to characterize initial editing spectrum
- RNA sequencing to assess transcriptomic changes
- Digital PCR for quantitative editing efficiency
- Long-Term Monitoring (3-24 months):
- Track editing persistence through circulating biomarkers (cell-free DNA)
- Single-cell RNA sequencing of accessible cells to assess heterogeneity
- Immunofluorescence for tissue-specific markers and DNA damage response (γH2AX, 53BP1)
- Off-Target Analysis:
- In silico prediction of potential off-target sites
- CIRCLE-seq or GUIDE-seq for unbiased off-target identification
- Amplicon sequencing of predicted off-target loci
Clinical Translation and Regulatory Considerations
Clinical Trial Design for Safety Validation
Robust clinical validation of CRISPR therapies requires specialized trial designs incorporating comprehensive safety monitoring:
Extended Follow-Up Periods: Traditional trial timelines are insufficient for CRISPR therapies. Monitoring should extend for multiple years to detect delayed immune responses or late-onset consequences of editing.
Biomarker Development: Validated biomarkers should include:
- Cas-specific T-cell and antibody assays
- Circulating editing efficiency markers
- Tissue-specific damage markers when accessible
Risk Mitigation Strategies: Protocols should incorporate pre-screening for pre-existing immunity, step-up dosing to manage immune responses, and detailed management plans for immune-related adverse events.
Recent clinical advances demonstrate the feasibility of these approaches. In the landmark case of a personalized CRISPR treatment for CPS1 deficiency, the patient safely received three LNP-delivered doses with careful immune monitoring, establishing a precedent for redosing regimens [9]. Similarly, Intellia Therapeutics' hATTR trial demonstrated sustained TTR reduction for over two years with acceptable safety profiles [9].
Table 3: Key Research Reagents for CRISPR Safety Validation
| Reagent Category | Specific Examples | Function/Application |
|---|---|---|
| CRISPR Delivery Systems | VSVG/BRL-pseudotyped VLPs, Lipid Nanoparticles (LNPs), AAV vectors | Efficient delivery to target cells including non-dividing cells |
| Immune Monitoring Tools | Cas9 peptide libraries, HLA tetramers, IFN-γ ELISpot kits | Detection and characterization of cellular immune responses |
| DNA Repair Assays | γH2AX/53BP1 antibodies, NHEJ/MMEJ reporter constructs, Long-range PCR kits | Analysis of repair pathway utilization and DNA damage response |
| Sequencing Platforms | Next-generation sequencers, Digital PCR systems, Single-cell RNAseq | Comprehensive characterization of editing outcomes and heterogeneity |
| Cell Culture Models | iPSCs, iPSC-derived neurons/cardiomyocytes, Primary T cells | Physiologically relevant systems for safety assessment |
The following workflow diagram outlines the key stages in clinical safety validation for CRISPR therapies:
Comprehensive clinical safety validation for CRISPR-based therapies requires integrated assessment of immune responses and long-term editing outcomes. The methodologies outlined in this guide provide a framework for characterizing these critical safety parameters throughout therapeutic development. As CRISPR technologies advance, continued refinement of safety monitoring approaches will be essential for realizing the full therapeutic potential of precision genome editing while ensuring patient safety. The emerging clinical experience with approved CRISPR therapies demonstrates that with appropriate safety validation, these transformative treatments can be safely administered, paving the way for broader application across genetic diseases.
The CRISPR-Cas9 system has revolutionized genetic engineering by providing a programmable means to manipulate genomes. However, its fundamental reliance on double-strand break (DSB) repair introduces significant limitations for therapeutic applications, including unpredictable editing outcomes and reliance on specific cellular repair pathways that are inefficient in non-dividing cells [109] [110]. The imperative to overcome these constraints has driven the development of two innovative classes of "alternative editors": base editors and prime editors. These technologies represent a paradigm shift from cutting to directly rewriting genetic information, offering unprecedented precision while avoiding the pitfalls of DSB generation.
This technical guide provides a comprehensive comparison of traditional CRISPR-Cas9 with base editing and prime editing strategies, with particular emphasis on their mechanisms, capabilities, and therapeutic potential within the context of genetic disease research. As the field advances toward clinical applications, understanding the distinct advantages and limitations of each editing platform becomes paramount for selecting the optimal strategy for specific research or therapeutic objectives.
Fundamental Mechanisms and Components
CRISPR-Cas9: The Foundation
The CRISPR-Cas9 system functions as a programmable DNA endonuclease. The core components include the Cas9 protein and a single guide RNA (sgRNA) that directs Cas9 to a specific genomic locus complementary to a 20-nucleotide spacer sequence [109]. Upon recognition of a protospacer adjacent motif (PAM) adjacent to the target site, Cas9 undergoes conformational changes that activate its two nuclease domains, HNH and RuvC, which cleave opposing DNA strands to generate a DSB [109].
Cellular repair of this DSB occurs primarily through two pathways: error-prone non-homologous end joining (NHEJ), which often results in insertions or deletions (indels) that disrupt gene function, or the more precise homology-directed repair (HDR), which requires a donor DNA template to facilitate accurate sequence restoration or modification [109] [110]. A significant limitation is that HDR is restricted primarily to dividing cells and is generally inefficient compared to NHEJ, making precise editing challenging in many therapeutically relevant cell types [109].
Base Editing: Chemical Conversion without DSBs
Base editors represent the first major innovation beyond DSB-dependent editing. These systems fuse a catalytically impaired Cas protein (nickase Cas9, or nCas9, that cuts only one DNA strand) to a single-stranded DNA deaminase enzyme, enabling direct chemical conversion of one base pair to another without generating DSBs [109] [111].
Cytosine Base Editors (CBEs) combine nCas9 with a cytidine deaminase enzyme, which converts cytosine (C) to uracil (U) within a narrow editing window. The cellular DNA repair machinery then interprets the U as a thymine (T), resulting in a C•G to T•A conversion during subsequent replication. Key innovations like the incorporation of uracil glycosylase inhibitor (UGI) prevent undesired repair pathways and enhance editing efficiency [111].
Adenine Base Editors (ABEs) utilize an engineered tRNA adenosine deaminase (TadA) to deaminate adenine (A) to inosine (I), which is read as guanine (G) by polymerases, effecting an A•T to G•C conversion [111]. More recently, C-to-G Base Editors (CGBEs) have been developed to expand transversion capabilities by leveraging uracil N-glycosylase to initiate base excision repair [111].
Prime Editing: A Search-and-Replace Platform
Prime editing further expands editing capabilities through a novel "search-and-replace" mechanism that requires neither DSBs nor donor DNA templates. The system employs a prime editor protein consisting of nCas9 (H840A) fused to an engineered reverse transcriptase (RT), programmed by a specialized prime editing guide RNA (pegRNA) [112] [113].
The pegRNA serves dual functions: it directs the complex to the target genomic locus and also encodes the desired edit within an extended RNA template. The editing mechanism involves: (1) nicking of the target DNA strand by nCas9, (2) hybridization of the 3' end of the nicked DNA to the primer binding site (PBS) on the pegRNA, (3) reverse transcription of the edit-containing template from the pegRNA, and (4) resolution and integration of the edited DNA strand into the genome [112] [113]. An additional sgRNA is often used in PE3 and PE3b systems to nick the non-edited strand, further enhancing editing efficiency [112].
Table 1: Core Components of CRISPR Editing Systems
| Component | CRISPR-Cas9 | Base Editors | Prime Editors |
|---|---|---|---|
| Catalytic Protein | Wild-type Cas9 (DSB creation) | Nickase Cas9 (nCas9) fused to deaminase | Nickase Cas9 (nCas9, H840A) fused to reverse transcriptase |
| Guide Molecule | sgRNA (targeting only) | sgRNA (targeting only) | pegRNA (targeting + edit template) |
| Additional Components | Donor DNA (for HDR) | UGI (for CBEs) | Second nicking sgRNA (PE3/PE3b systems) |
| Key Mechanism | DSB → NHEJ/HDR repair | Chemical deamination of ssDNA | Reverse transcription from pegRNA template |
Comparative Technical Specifications
Editing Scope and Precision
Each editing platform offers distinct capabilities regarding the types of genetic modifications possible, with direct implications for therapeutic application selection.
Table 2: Editing Capabilities of Different CRISPR Systems
| Editing Type | CRISPR-Cas9 | Base Editors | Prime Editors |
|---|---|---|---|
| DSB Formation | Yes | No | No |
| Point Mutations | Limited (via HDR, inefficient) | All four transitions (C→T, G→A, A→G, T→C); some transversions with newer editors | All 12 possible point mutations |
| Insertions | Possible with donor template | No | Yes, up to dozens of base pairs |
| Deletions | Yes (via NHEJ, unpredictable) | No | Yes, targeted small deletions |
| Theoretical Correction Scope | Limited by HDR efficiency | ~30% of known pathogenic SNPs [109] | Up to 89% of known pathogenic variants [109] |
| Product Purity | Low (mixed indels common) | High (few indels) | High (few indels) |
Efficiency and Practical Performance
Editing efficiency varies significantly across platforms and is highly dependent on target sequence, cell type, and delivery method. The following table summarizes key performance characteristics.
Table 3: Performance Characteristics of Editing Systems
| Characteristic | CRISPR-Cas9 | Base Editors | Prime Editors |
|---|---|---|---|
| Editing Efficiency | High for gene disruption (NHEJ); Low for precise edits (HDR) | Generally high (20-50% in many cell types) [111] | Variable (10-50% with optimized systems) [112] |
| Indel Formation | High with NHEJ (often >10%) | Low (<1% with optimized editors) | Very low (<0.1% with PE2) [112] |
| Bystander Edits | Not applicable | Can occur within editing window | Minimal with optimized pegRNA design |
| Cell Cycle Dependence | HDR requires S/G2 phase; NHEJ active throughout | Active in both dividing and non-dividing cells | Active in both dividing and non-dividing cells |
| Delivery Challenge | Moderate (Cas9 + sgRNA) | Moderate (BE protein + sgRNA) | High (large PE protein + long pegRNA) |
DNA Repair Pathway Considerations in Therapeutic Editing
The cellular response to genome editing varies dramatically between dividing and non-dividing cells, with critical implications for therapeutic strategy selection. Recent research has revealed that postmitotic cells like neurons and cardiomyocytes repair CRISPR-induced DNA damage with fundamentally different kinetics and pathway utilization compared to dividing cells [12] [13].
In dividing cells, Cas9-induced DSBs are rapidly resolved, with indel accumulation typically plateauing within days. In contrast, neurons exhibit prolonged editing timelines, with indel accumulation continuing for up to two weeks post-transduction. This extended repair window is attributed to both persistent Cas9 activity and distinct repair pathway preferences, with neurons favoring classical NHEJ over microhomology-mediated end joining (MMEJ) pathways that are more prevalent in dividing cells [12]. These findings highlight the critical importance of considering cell-type-specific repair mechanisms when designing therapeutic editing approaches, particularly for neurological disorders.
DNA Repair Pathways in Different Cell Types
The diagram above illustrates the divergent DNA repair responses to CRISPR-Cas9-induced DSBs in dividing versus non-dividing cells, and how base editing and prime editing circumvent these pathway dependencies altogether.
Experimental Workflows and Methodologies
Implementing Base Editing
Key Reagents:
- Base editor plasmid (e.g., BE4max for C→T editing or ABE8e for A→G editing)
- Target-specific sgRNA plasmid or synthetic sgRNA
- Delivery system (viral vectors, LNPs, electroporation)
Protocol Overview:
- Target Selection: Identify target site with the desired base within the editing window (typically positions 4-8 for SpCas9-based editors, counting the PAM as positions 21-23).
- Editor Selection: Choose appropriate base editor based on desired conversion (CBE, ABE, or CGBE).
- Vector Assembly: Clone sgRNA expression cassette into appropriate backbone.
- Delivery: Co-deliver base editor and sgRNA to target cells using optimized method.
- Validation: Assess editing efficiency by Sanger sequencing or next-generation sequencing; evaluate potential off-target effects.
Implementing Prime Editing
Key Reagents:
- Prime editor plasmid (e.g., PE2, PE3, or PE5 systems)
- Designed pegRNA plasmid or synthetic pegRNA
- Optional: nicking sgRNA for PE3 systems
Protocol Overview:
- pegRNA Design: Design pegRNA with (a) spacer sequence complementary to target site, (b) primer binding site (PBS, typically 10-15 nt), and (c) reverse transcription template (RTT) containing desired edit.
- Editor Selection: Choose appropriate prime editor version based on application (PE2 for basic editing, PE3 with additional nicking sgRNA for enhanced efficiency, PE5 with MMR inhibition for improved yield).
- Vector Assembly: Clone pegRNA expression cassette; for PE3 systems, clone additional nicking sgRNA.
- Delivery: Co-deliver prime editor and pegRNA to target cells; consider optimized delivery methods for large constructs.
- Validation: Sequence target locus to confirm edit incorporation; assess fidelity and potential byproducts.
Table 4: Research Reagent Solutions for Genome Editing
| Reagent Type | Specific Examples | Function | Considerations |
|---|---|---|---|
| Cas9 Variants | SpCas9, SaCas9, CjCas9 | DNA recognition and cleavage | Size, PAM requirements, specificity |
| Base Editors | BE4max (CBE), ABE8e (ABE), CGBE | Targeted base conversion without DSBs | Editing window, bystander edits, sequence context |
| Prime Editors | PE2, PE3, PE5, PE6 variants | Precise insertions, deletions, all point mutations | pegRNA design complexity, efficiency optimization |
| Delivery Systems | AAV, Lentivirus, LNPs, Electroporation | Intracellular delivery of editing components | Packaging capacity, cell type tropism, toxicity |
| Repair Modulators | NHEJi, ART558 (POLQi), D-I03 (Rad52i) | Manipulate DNA repair pathways to favor desired outcomes | Cell-type specific effects, timing, toxicity |
Clinical Applications and Therapeutic Advancements
The therapeutic potential of these editing platforms is rapidly being realized in clinical settings. Base editing has demonstrated remarkable success in ex vivo applications, with clinical trials underway for sickle cell disease, beta-thalassemia, and various oncological indications [9]. Prime editing, though newer, has shown promising preclinical results for diverse genetic disorders including Hurler syndrome, Batten disease, and cystic fibrosis [114].
Notably, the first personalized in vivo prime editing treatment was successfully administered to an infant with CPS1 deficiency in 2025. This landmark case demonstrated the development, FDA approval, and delivery of a bespoke therapy within just six months, establishing a regulatory precedent for rapid approval of platform therapies [9]. The patient safely received multiple doses delivered via lipid nanoparticles (LNPs), with each subsequent dose increasing editing efficiency and clinical improvement [9].
For monogenic disorders caused by premature termination codons (PTCs), a novel prime editing approach called PERT (prime editing-mediated readthrough of premature termination codons) has been developed. This strategy uses prime editing to convert a dispensable endogenous tRNA into an optimized suppressor tRNA, enabling readthrough of PTCs in a disease-agnostic manner [114]. This approach has demonstrated successful protein rescue in models of Batten disease, Tay-Sachs disease, and cystic fibrosis, with extensive pathology rescue in a Hurler syndrome mouse model [114].
The landscape of genome editing has evolved dramatically from the initial CRISPR-Cas9 system to increasingly sophisticated base editing and prime editing platforms. Each technology offers distinct advantages: CRISPR-Cas9 remains optimal for gene disruption applications; base editing provides efficient, precise single-nucleotide changes without DSBs; and prime editing offers unparalleled versatility for diverse genetic modifications.
The choice of editing platform for therapeutic applications must consider multiple factors including the specific genetic alteration required, target cell type, delivery constraints, and potential off-target effects. As these technologies continue to advance, ongoing optimization of editing efficiency, specificity, and delivery will further expand their therapeutic potential. The recent success of in vivo prime editing in clinical settings heralds a new era of precision genetic medicine, where previously untreatable genetic disorders may be addressed through tailored editing strategies.
The field of CRISPR-Cas9 gene editing stands at a pivotal crossroads in 2025, characterized by both remarkable therapeutic breakthroughs and significant safety revelations. This technology, which leverages a bacterial adaptive immune system to create programmable double-strand breaks (DSBs) in DNA, has transitioned from theoretical concept to clinical reality with the first regulatory approvals of CRISPR-based medicines [58]. The recent approval of Casgevy for sickle cell disease (SCD) and transfusion-dependent beta thalassemia (TBT) marked a historic milestone, demonstrating that precise genetic modifications can permanently correct deleterious mutations underlying genetic diseases [9]. However, the clinical landscape has simultaneously been reshaped by emerging challenges, including unexpected safety events and structural variations that demand renewed focus on the fundamental mechanisms of DNA repair.
The clinical trajectory of CRISPR therapeutics reflects a complex interplay between therapeutic promise and biological complexity. While the first six months of 2025 witnessed a landmark achievement with the first personalized in vivo CRISPR treatment for an infant with CPS1 deficiency—developed and delivered in just six months—this period also saw concerning safety developments, including the pausing of Phase 3 trials for Intellia's transthyretin amyloidosis therapy following a serious adverse event [9] [115]. These concurrent advances and setbacks provide a critical opportunity to benchmark success not merely by editing efficiency, but through a more comprehensive understanding of how CRISPR-induced DSBs are processed through cellular repair pathways, and how these mechanisms influence both therapeutic outcomes and potential risks.
Quantitative Analysis of Recent Clinical Trial Outcomes
Table 1: Efficacy and Safety Outcomes from Select Recent CRISPR Clinical Trials
| Therapeutic Area | Drug/Target | Phase | Key Efficacy Results | Safety Findings | Status/Notes |
|---|---|---|---|---|---|
| Hemoglobinopathies | Casgevy (BCL11A) | Approved | Sustained fetal hemoglobin induction; transfusion independence in TDT; resolution of vaso-occlusive crises in SCD [9] | Generally manageable safety profile relative to underlying disease | First approved CRISPR therapy; >65 treatment centers activated globally [116] |
| Transthyretin Amyloidosis | Nex-z (TTR) | Phase 3 (Paused) | ~90% reduction in TTR protein sustained over 2 years; functional stabilization/improvement [9] | Grade 4 liver transaminases and increased bilirubin in one patient (2025); trial dosing paused [115] | Paused Oct 2025 following serious adverse event; prior Orphan Drug and RMAT designations [115] |
| Hereditary Angioedema | NTLA-2002 (KLKB1) | Phase 1/2 | 86% reduction in kallikrein; 8 of 11 high-dose participants attack-free over 16 weeks [9] | No serious adverse events reported in initial results | In vivo LNP delivery; further trials ongoing |
| Cardiovascular Disease | CTX310 (ANGPTL3) | Phase 1 | Dose-dependent reductions: up to 82% in triglycerides, 81% in LDL [116] | Well-tolerated; no treatment-related SAEs; no clinically significant liver enzyme changes | LNP-mediated liver editing; data cutoff April 2025 [116] |
| Personalized Therapy | CPS1 deficiency | N/A (Compassionate) | Symptom improvement; decreased medication dependence after three LNP doses [9] | No serious side effects; successful redosing | First personalized in vivo CRISPR; developed in 6 months [9] |
Table 2: Delivery Approaches and Their Clinical Implications
| Delivery Method | Therapeutic Examples | Advantages | Limitations/Challenges |
|---|---|---|---|
| Ex Vivo HSC Editing | Casgevy (SCD, TBT) | Controlled editing environment; patient-specific product; no vector clearance issues [9] | Requires myeloablative conditioning; complex manufacturing; high cost [9] |
| LNP (Liver-Tropic) | NTLA-2002 (hATTR), CTX310 (ANGPTL3) | In vivo administration; potential for redosing (unlike viral vectors); natural liver tropism [9] [116] | Primarily targets liver; limited tissue specificity; immune reactions possible [9] |
| AAV Vectors | (Preclinical) | Potential for diverse tissue targeting; long-term expression | Limited packaging capacity; pre-existing immunity; persistent expression concerns [35] |
| Lipid Nanoparticles (Non-Liver) | (In development) | Potential for tissue-specific targeting | Early research stage; not yet validated in clinical trials [9] |
DNA Repair Mechanisms: The Foundation of CRISPR Editing Outcomes
The therapeutic efficacy and safety risks of CRISPR-Cas9 genome editing are fundamentally governed by the cellular response to double-strand breaks, primarily through two competing DNA repair pathways: non-homologous end joining (NHEJ) and homology-directed repair (HDR) [58]. The Cas9 nuclease, guided by RNA molecules, induces DSBs at specific genomic locations, activating a complex DNA damage response that determines the ultimate editing outcome [35].
Non-Homologous End Joining (NHEJ)
NHEJ represents the dominant repair pathway in mammalian cells, operating throughout the cell cycle by directly ligating broken DNA ends without requiring a template [58]. This pathway is highly efficient but error-prone, often resulting in small insertions or deletions (indels) at the cleavage site [58]. Therapeutically, NHEJ is exploited for gene knockouts, such as in Casgevy where disruption of the BCL11A enhancer restores fetal hemoglobin expression [9] [45], or in NTLA-2002 where TTR gene inactivation reduces pathogenic protein production [9]. While invaluable for therapeutic gene disruption, NHEJ's error-prone nature also underlies significant safety concerns, including the potential for large-scale structural variations [45].
Homology-Directed Repair (HDR)
HDR provides a template-dependent repair mechanism that enables precise genetic modifications, including nucleotide substitutions or gene insertions [58]. However, HDR is inherently less efficient than NHEJ and is restricted to late S and G2 phases of the cell cycle, presenting particular challenges for editing non-dividing or slowly dividing cells such as neurons or cardiomyocytes [58]. While HDR offers the potential for precise gene correction, its clinical application has been limited by efficiency barriers and the risks associated with strategies to enhance HDR, particularly through perturbation of competing repair pathways [45].
Diagram 1: CRISPR-Cas9 DNA Repair Pathways. DSBs are primarily repaired via NHEJ or HDR, each with distinct outcomes and limitations.
Emerging Safety Concerns: Beyond Off-Target Effects
While early safety assessments focused predominantly on off-target effects at sites with sequence similarity to the intended target, recent evidence reveals a more complex landscape of genomic alterations with profound implications for clinical safety [45].
On-Target Structural Variations
Advanced detection methods have uncovered that CRISPR editing can generate large-scale structural variations at on-target sites, including kilobase- to megabase-scale deletions, chromosomal truncations, and rearrangements [45]. These alterations were substantially underestimated in earlier studies due to technical limitations of short-read sequencing, which fails to detect large deletions that eliminate primer-binding sites [45]. In the context of Casgevy therapy, while effective for hemoglobinopathies, studies have identified frequent kilobase-scale deletions at the BCL11A target site in hematopoietic stem cells, warranting continued scrutiny given BCL11A's role in lymphoid development and cellular senescence [45].
Chromosomal Translocations
Simultaneous cleavage at target and off-target sites can promote chromosomal translocations between heterologous chromosomes, with particularly concerning implications when tumor suppressor genes or proto-oncogenes are involved [45]. The risk of such events appears exacerbated by strategies to enhance HDR efficiency, especially through inhibition of DNA-PKcs, which has been shown to increase translocation frequencies by up to a thousand-fold in some studies [45]. These findings underscore the critical balance between editing efficiency and genomic integrity, highlighting the need for more sophisticated safety assessment approaches in clinical development.
Intervention-Related Risks
Recent clinical experience has revealed unexpected safety challenges, including the October 2025 pausing of Intellia's Phase 3 trials for nex-z following a Grade 4 hepatotoxicity event characterized by elevated liver transaminases and bilirubin [115]. This event occurred despite earlier phase trials showing favorable safety profiles, emphasizing the limitations of extrapolating from limited early-stage data to broader patient populations. The case highlights the complex interplay between the therapeutic mechanism—liver-targeted TTR gene inactivation—and potential organ-specific toxicity, even with LNP delivery systems generally considered less immunogenic than viral vectors [9] [115].
Advanced Methodologies for Assessing Editing Outcomes
BreakTag: A Novel Approach for DSB Profiling
The recently developed BreakTag method represents a significant advancement in mapping CRISPR-Cas9-induced double-strand breaks genome-wide [117]. This next-generation sequencing-based methodology enables comprehensive profiling of DSB frequency, location, and end structure at nucleotide resolution through a streamlined protocol.
Table 3: BreakTag Experimental Protocol
| Step | Procedure | Key Details | Purpose |
|---|---|---|---|
| 1. End Preparation | End repair/A-tailing of genomic DNA | Prepares ends for adaptor ligation | Standardizes end structures for sequencing |
| 2. Adaptor Ligation | Ligation with UMI-barcoded adaptor | Unique Molecular Identifiers for DSB counting; sample barcodes for multiplexing | Enables precise quantification and sample pooling |
| 3. Tagmentation | Tn5 transposase treatment | Fragments DNA for sequencing library preparation | Creates sequencing-ready fragments |
| 4. Amplification | PCR amplification | Targets ligated fragments only | Enriches for DSB-containing fragments |
| 5. Analysis | BreakInspectoR bioinformatics pipeline | Identifies and quantifies DSBs from sequencing data | Provides comprehensive DSB mapping |
BreakTag has demonstrated exceptional reproducibility and sensitivity in profiling Cas9 activity, identifying approximately 150,000 endogenous on-target and off-target sites across 3,500 sgRNAs in one study [117]. The method revealed that approximately 35% of SpCas9 DSBs produce staggered ends rather than blunt cuts, with cleavage configuration influenced by DNA:gRNA complementarity and specific Cas9 variants [117].
Diagram 2: BreakTag DSB Profiling Workflow. This method enables comprehensive mapping of CRISPR-induced breaks.
Detection of Structural Variations
Conventional amplicon sequencing approaches frequently miss large-scale structural variations due to their dependence on intact primer-binding sites, which may be eliminated in deletion events [45]. Advanced methods including CAST-Seq and LAM-HTGTS have been developed specifically to detect these larger aberrations, revealing a previously underappreciated spectrum of CRISPR-induced genomic alterations [45]. These approaches are increasingly recognized as essential complements to traditional off-target assessment, particularly for clinical applications.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 4: Key Research Reagents for CRISPR Safety and Efficacy Assessment
| Reagent Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Cas9 Variants | HiFi Cas9, eSpCas9 | Enhanced specificity reduces off-target effects [45] | May still generate on-target structural variations; potential trade-off with efficiency |
| DSB Detection Tools | BreakTag, GUIDE-seq, CIRCLE-seq | Genome-wide mapping of on-target and off-target cleavage [117] | Varying sensitivity/specificity; BreakTag offers streamlined workflow and end-structure analysis |
| Structural Variation Assays | CAST-Seq, LAM-HTGTS | Detect large deletions, translocations, chromosomal rearrangements [45] | Essential complement to amplicon sequencing; reveal previously undetected aberrations |
| Delivery Systems | LNPs, AAV, Electroporation | Enable CRISPR component delivery to target cells [9] [35] | Tissue tropism, immunogenicity, payload capacity vary significantly between approaches |
| DNA Repair Modulators | DNA-PKcs inhibitors, 53BP1 inhibitors | Enhance HDR efficiency in research settings [45] | Clinical concerns about increased structural variations; require careful risk-benefit assessment |
| Cell Models | Immortalized lines, primary cells, iPSCs | Model physiological responses to editing [118] | Primary cells show greater editing difficulty but better biological relevance |
The current landscape of CRISPR clinical trials presents both unprecedented opportunities and complex challenges. Successful clinical translation requires balancing therapeutic efficacy with comprehensive safety assessment, particularly as emerging evidence reveals previously underappreciated risks of structural variations. The field is evolving from a singular focus on editing efficiency toward a more nuanced understanding of how DNA repair pathway dynamics influence both therapeutic outcomes and patient safety. Future success will depend on integrating advanced detection methods like BreakTag and structural variation assays throughout therapeutic development, carefully evaluating the risk-benefit profile of DNA repair modulation strategies, and maintaining rigorous safety monitoring even as promising early-stage results accelerate clinical advancement. As CRISPR medicine continues to mature, this comprehensive approach to benchmarking success will be essential for realizing the full potential of gene editing while ensuring patient safety.
Conclusion
The therapeutic application of CRISPR-Cas9 fundamentally depends on harnessing and directing the cell's intrinsic DNA repair machinery. While foundational understanding of HDR and NHEJ pathways has enabled groundbreaking therapies like Casgevy, emerging 2025 research reveals critical nuances in cell-type-specific repair, particularly in non-dividing cells. The field must now address the dual challenges of maximizing on-target efficiency while minimizing off-target effects through improved delivery systems, advanced Cas variants, and sophisticated predictive algorithms. Future directions will likely focus on personalized in vivo treatments for rare diseases, enhanced epigenetic editing, and combining CRISPR with other modalities. As clinical experience grows, developing standardized validation frameworks and understanding long-term outcomes will be crucial for realizing the full potential of CRISPR-based genetic disease therapeutics. The convergence of basic DNA repair biology with innovative clinical applications continues to push the boundaries of what's possible in medicine.
References
- 1. Mechanism and Applications of CRISPR/Cas-9-Mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8388126/]
- 2. CRISPR-Cas9: A fascinating journey from bacterial ... [https://pubmed.ncbi.nlm.nih.gov/33685600/]
- 3. DNA repair pathway choices in CRISPR-Cas9 mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8187289/]
- 4. The CRISPR-Cas immune system: Biology, mechanisms ... [https://www.sciencedirect.com/science/article/pii/S0300908415001042]
- 5. What is CRISPR/Cas9? - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4975809/]
- 6. What is CRISPR-Cas9? [https://www.yourgenome.org/theme/what-is-crispr-cas9/]
- 7. CRISPR/Cas9 gene editing [https://www.genoway.com/technologies/crispr-cas9-gene-editing]
- 8. Uncovering the dynamics of precise repair at CRISPR ... [https://www.nature.com/articles/s41467-024-49410-x]
- 9. CRISPR Clinical Trials: A 2025 Update [https://innovativegenomics.org/news/crispr-clinical-trials-2025/]
- 10. CMN Weekly (31 October 2025) [https://crisprmedicinenews.com/news/cmn-weekly-31-october-2025-your-weekly-crispr-medicine-news/]
- 11. CRISPR-Cas9-mediated homology-directed repair for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11531618/]
- 12. Characterizing and controlling CRISPR repair outcomes in ... [https://www.nature.com/articles/s41467-025-66058-3]
- 13. Neurons repair CRISPR damage with unique timing [https://crisprmedicinenews.com/news/neurons-repair-crispr-damage-with-unique-timing/]
- 14. strategies to enhance the frequency of homology-directed ... [https://www.bmbreports.org/journal/view.html?uid=1289]
- 15. The hidden risks of CRISPR/Cas: structural variations and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12325979/]
- 16. CRISPR-Cas9-mediated homology-directed repair for ... [https://www.sciencedirect.com/science/article/pii/S2162253124002312]
- 17. Comparative analysis of multiple DNA double-strand break ... [https://www.nature.com/articles/s42003-025-08187-5]
- 18. Rational Selection of CRISPR-Cas9 Guide RNAs for ... [https://www.sciencedirect.com/science/article/pii/S1525001620305402]
- 19. Influence of PARP1 on CRISPR/Cas9 induced double ... [https://pubmed.ncbi.nlm.nih.gov/41127819/]
- 20. Target specificity of the CRISPR-Cas9 system - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4338555/]
- 21. Exploring CRISPR-Cas: The transformative impact of gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12506487/]
- 22. CRISPR/Cas9 technology in tumor research and drug ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1552741/full]
- 23. Rapid two-step target capture ensures efficient CRISPR- ... [https://www.sciencedirect.com/science/article/pii/S1097276525003016]
- 24. Visualization of a multi-turnover Cas9 after product release [https://www.nature.com/articles/s41467-025-60668-7]
- 25. Altered DNA repair pathway engagement by engineered ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10242711/]
- 26. Challenges and Opportunities in the Application of CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12503735/]
- 27. CRISPR Editing in Primary Cells [https://www.synthego.com/crispr-primary-cells/]
- 28. Cell-cycle dependent DNA repair and replication unifies ... [https://www.nature.com/articles/s41467-025-58245-z]
- 29. CRISPR/Cas9-mediated homology-directed repair by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6215429/]
- 30. CRISPR Guide [https://www.addgene.org/guides/crispr/]
- 31. Target-Specific Precision of CRISPR-Mediated Genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6395888/]
- 32. Precise measurement of CRISPR genome editing ... [https://www.sciencedirect.com/science/article/pii/S2329050125000440]
- 33. World's First Patient Treated with Personalized CRISPR ... [https://www.chop.edu/news/worlds-first-patient-treated-personalized-crispr-gene-editing-therapy-childrens-hospital]
- 34. Advancing gene editing: the role of lipid nanoparticles in ... [https://www.drugtargetreview.com/article/188056/advancing-gene-editing-the-role-of-lipid-nanoparticles-in-crispr-delivery/]
- 35. CRISPR/Cas9 therapeutics: progress and prospects [https://www.nature.com/articles/s41392-023-01309-7]
- 36. Lipid nanoparticles: The game-changer in CRISPR-Cas9 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10823087/]
- 37. Adeno-associated virus as a delivery vector for gene ... [https://www.nature.com/articles/s41392-024-01780-w]
- 38. Adeno-associated virus vector as a platform for gene therapy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6927556/]
- 39. AAV-based CRISPR-Cas9 genome editing: Challenges ... [https://www.sciencedirect.com/science/article/pii/S2468451123000739]
- 40. Lipid nanoparticles for delivery of RNA therapeutics [https://pmc.ncbi.nlm.nih.gov/articles/PMC9691360/]
- 41. FDA Approves First Gene Therapies to Treat Patients with ... [https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapies-treat-patients-sickle-cell-disease]
- 42. How Does CASGEVY® Work? [https://www.casgevyhcp.com/mechanism-of-action]
- 43. Revolutionary breakthrough: FDA approves CASGEVY, the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11305803/]
- 44. The Approval of Casgevy, the First CRISPR Therapy in ... [https://www.synthego.com/blog/the-groundbreaking-approval-of-casgevy-the-first-crispr-therapy-in-the-us/]
- 45. The hidden risks of CRISPR/Cas: structural variations and ... [https://www.nature.com/articles/s41467-025-62606-z]
- 46. How Does CASGEVY® Work? [https://www.casgevy.com/sickle-cell-disease/how-casgevy-works]
- 47. Press Release [https://ir.crisprtx.com/news-releases/news-release-details/crispr-therapeutics-provides-business-update-and-reports-13]
- 48. In vivo liver targeted genome editing as therapeutic approach [https://pmc.ncbi.nlm.nih.gov/articles/PMC11378722/]
- 49. On Course: Intellia CSO Offers Clinical Update of In Vivo ... [https://www.genengnews.com/topics/genome-editing/on-course-intellia-cso-offers-clinical-update-of-in-vivo-gene-editing-therapies-at-esgct/]
- 50. Release Details [https://ir.intelliatx.com/news-releases/news-release-details/intellia-therapeutics-presents-positive-longer-term-phase-1-data]
- 51. Intellia Therapeutics Announces Third Quarter 2025 ... [https://ir.intelliatx.com/news-releases/news-release-details/intellia-therapeutics-announces-third-quarter-2025-financial]
- 52. Intellia Pauses Phase 3 CRISPR Trials after Life- ... [https://pharmacally.com/intellia-pauses-phase-3-crispr-trials-after-life-threatening-liver-injury-in-transthyretin-amyloidosis-attr-patient/]
- 53. Release Details [https://ir.intelliatx.com/news-releases/news-release-details/intellia-therapeutics-presents-positive-pooled-phase-12-data]
- 54. Intellia Therapeutics Presents Positive Pooled Phase 1/2 [https://www.globenewswire.com/news-release/2025/11/08/3184092/0/en/Intellia-Therapeutics-Presents-Positive-Pooled-Phase-1-2-Data-of-Lonvoguran-Ziclumeran-lonvo-z-in-Patients-with-Hereditary-Angioedema.html]
- 55. Infant with rare, incurable disease is first to successfully ... [https://www.nih.gov/news-events/news-releases/infant-rare-incurable-disease-first-successfully-receive-personalized-gene-therapy-treatment]
- 56. Researchers Behind Personalized CRISPR Therapy Plan ... [https://www.chop.edu/news/researchers-behind-personalized-crispr-therapy-plan-launch-new-type-clinical-trial]
- 57. CRISPR Epigenetic Editing Delivers Durable Multiplexed ... [https://crisprmedicinenews.com/news/crispr-epigenetic-editing-delivers-durable-multiplexed-gene-silencing-in-t-cells/]
- 58. Advancing CRISPR genome editing into gene therapy clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12094669/]
- 59. The multiple layers of RNA response in double-strand ... [https://www.nature.com/articles/s12276-025-01572-4]
- 60. CRISPR Clinical Trials to Follow [https://www.synthego.com/blog/crispr-clinical-trials/]
- 61. AI-powered CRISPR could lead to faster gene therapies ... [https://med.stanford.edu/news/all-news/2025/09/ai-crispr-gene-therapy.html]
- 62. Overview CRISPR Clinical Trials 2025 - Learn | Innovate [https://crisprmedicinenews.com/clinical-trials/]
- 63. CRISPR/Cas9 genome editing for neurodegenerative diseases [https://pmc.ncbi.nlm.nih.gov/articles/PMC10450213/]
- 64. CRISPR-Cas9 in Cardiovascular Medicine: Unlocking New ... [https://www.mdpi.com/2073-4409/14/2/131]
- 65. The Use of CRISPR-Cas9 Genetic Technology in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11078688/]
- 66. CRISPR-based strategies in infectious disease diagnosis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7779109/]
- 67. CRISPR/Cas9 system and its applications in nervous ... [https://www.sciencedirect.com/science/article/pii/S2352304223001459]
- 68. Fighting Infectious Disease with CRISPR [https://www.synthego.com/blog/crispr-infectious-disease/]
- 69. Off-target effects in CRISPR-Cas genome editing for ... [https://www.sciencedirect.com/science/article/pii/S2162253125001908]
- 70. Off-target effects in CRISPR-Cas genome editing for ... [https://pubmed.ncbi.nlm.nih.gov/40777742/]
- 71. CRISPR Off-Target Editing: Prediction, Analysis, and More [https://www.synthego.com/blog/crispr-off-target-editing/]
- 72. Linking CRISPR–Cas9 double-strand break profiles to ... [https://www.nature.com/articles/s41587-024-02238-8]
- 73. Artificial Intelligence for CRISPR Guide RNA Design [https://arxiv.org/html/2508.20130v1]
- 74. High-fidelity CRISPR-Cas9 variants with undetectable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4851738/]
- 75. Revolutionizing CRISPR technology with artificial intelligence [https://www.nature.com/articles/s12276-025-01462-9]
- 76. Optimizing CRISPR delivery for drug discovery [https://www.drugdiscoverynews.com/optimizing-crispr-delivery-for-drug-discovery-16487]
- 77. CRISPR Delivery Methods: Cargo, Vehicles, and Challenges [https://www.synthego.com/blog/delivery-crispr-cas9/]
- 78. Scientists just made CRISPR three times more effective [https://www.sciencedaily.com/releases/2025/09/250907024543.htm]
- 79. A modular strategy for extracellular vesicle-mediated ... [https://www.nature.com/articles/s41467-025-65995-3]
- 80. Comprehensive benchmarking of genome editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12144422/]
- 81. Protocol to rapidly screen CRISPR-Cas9 gene editing ... [https://www.sciencedirect.com/science/article/pii/S2666166725003569]
- 82. Optimization of the Genome Editing CRISPR-Cas9 ... [https://pubmed.ncbi.nlm.nih.gov/41023259/]
- 83. Design of highly functional genome editors by modelling ... [https://www.nature.com/articles/s41586-025-09298-z]
- 84. CRISPR–Cas9 Gene Editing: Curing Genetic Diseases by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10980556/]
- 85. Revolution of Biotechnology with CRISPR - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12322092/]
- 86. Efficient in vivo homology-directed repair within ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9157414/]
- 87. Weaponizing CRISPR/Cas9 for selective elimination of ... [https://www.sciencedirect.com/science/article/pii/S1568786425000369]
- 88. Methods for Optimizing CRISPR-Cas9 Genome Editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4976696/]
- 89. Impact of chromatin context on Cas9-induced DNA double ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8153251/]
- 90. Widespread chromatin context-dependencies of DNA ... [https://www.nature.com/articles/s41467-024-49232-x]
- 91. Leveraging CRISPR gene editing technology to optimize ... [https://www.nature.com/articles/s41375-024-02444-y]
- 92. CRISPR Therapeutics Reports Positive Phase 1 Data for ... [https://www.quiverquant.com/news/CRISPR+Therapeutics+Reports+Positive+Phase+1+Data+for+CTX310%C2%AE+Demonstrating+Significant+Lipid+Reductions+at+AHA+Scientific+Sessions+2025]
- 93. Kinetics and Fidelity of the Repair of Cas9-Induced Double ... [https://pubmed.ncbi.nlm.nih.gov/29804829/]
- 94. Kinetics and Fidelity of the Repair of Cas9-Induced Double ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5993873/]
- 95. Neuronal DNA repair reveals strategies to influence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11230251/]
- 96. Off-target effects in CRISPR/Cas9 gene editing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10034092/]
- 97. BLOG: Selecting the Right Gene Editing Off-Target Assay [https://seqwell.com/guide-to-selecting-right-gene-editing-off-target-assay/]
- 98. Digenome-seq for Analysis of Genome-Wide CRISPR ... [https://www.cd-genomics.com/biomedical-ngs/resource/digenome-seq-analysis-genome-wide-crispr-cas9-off-target.html]
- 99. Genome-wide target specificities of CRISPR-Cas9 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4772022/]
- 100. CRISPR Genome Editing & NGS | Off-target analysis and ... [https://www.illumina.com/techniques/popular-applications/genome-editing.html]
- 101. Comparative analysis of multiple DNA double-strand break ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12075812/]
- 102. HDR vs. NHEJ: CRISPR Gene Editing Techniques [https://www.zeclinics.com/blog/hdr-vs-nhej-in-crispr-gene]
- 103. Precision gene editing: The power of CRISPR-Cas in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12590234/]
- 104. HDR vs NHEJ: DNA Repair Pathway Comparison [https://invivobiosystems.com/crispr/hdr-vs-nhej/]
- 105. Fine-Tuning Homology-Directed Repair (HDR) for ... [https://www.mdpi.com/1422-0067/26/9/4067]
- 106. Advance trends in targeting homology-directed repair for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9376165/]
- 107. Immunogenicity of CRISPR therapeutics—Critical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9978118/]
- 108. Scientists engineer CRISPR enzymes that evade ... [https://www.broadinstitute.org/news/scientists-engineer-crispr-enzymes-evade-immune-system]
- 109. CRISPR-Cas9 DNA Base-Editing and Prime-Editing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7503568/]
- 110. From bench to bedside: cutting-edge applications of base editing and prime editing in precision medicine | Journal of Translational Medicine | Full Text [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-024-05957-3]
- 111. Base Editing and Prime Editing | SpringerLink [https://link.springer.com/chapter/10.1007/978-3-031-46150-7_2]
- 112. Emerging trends in prime editing for precision genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12322275/]
- 113. Prime Editing as a Precision Gene Editing Tool [https://www.synthego.com/guide/crispr-methods/prime-editing/]
- 114. Prime editing-installed suppressor tRNAs for disease ... [https://www.nature.com/articles/s41586-025-09732-2]
- 115. Intellia Pauses Phase 3 Clinical Trials of CRISPR-Based ... [https://globalgenes.org/raredaily/intellia-pauses-phase-3-clinical-trials-of-crispr-based-therapy/]
- 116. CRISPR Therapeutics Provides First Quarter 2025… [https://crisprtx.com/about-us/press-releases-and-presentations/crispr-therapeutics-provides-first-quarter-2025-financial-results-and-announces-positive-top-line-data-from-phase-1-clinical-trial-of-ctx310-targeting-angptl3]
- 117. Linking CRISPR – Cas - 9 ... | Nature Biotechnology double strand break [https://www.nature.com/articles/s41587-024-02238-8?error=cookies_not_supported&code=95eff2ec-0a2c-468d-a223-1af80611c8b8]
- 118. State of CRISPR Gene Editing in the Drug Discovery Sector [https://www.synthego.com/blog/survey-report-crispr-therapeutics/]