Multi-Center Validation of Lipidomic Biomarkers: From Discovery to Clinical Application
The integration of lipidomics with machine learning is revolutionizing non-invasive diagnostic biomarker discovery for a wide range of diseases, including cancer, rheumatoid arthritis, and congenital conditions.
Multi-Center Validation of Lipidomic Biomarkers: From Discovery to Clinical Application
Abstract
The integration of lipidomics with machine learning is revolutionizing non-invasive diagnostic biomarker discovery for a wide range of diseases, including cancer, rheumatoid arthritis, and congenital conditions. This article provides a comprehensive framework for the development and rigorous multi-center validation of lipidomic biomarkers, a critical step for clinical translation. We explore foundational concepts in lipid metabolism and its dysregulation in disease, detail methodological workflows combining untargeted and targeted lipidomics with advanced data analytics, address key challenges in model optimization and reproducibility, and finally, present robust validation strategies across diverse, independent cohorts. Designed for researchers, scientists, and drug development professionals, this review synthesizes current best practices and emerging trends, highlighting how validated lipidomic signatures are paving the way for more accurate, early-stage disease detection and personalized medicine approaches.
The Basis of Lipidomic Biomarkers: Linking Lipid Metabolism to Disease Pathogenesis
Lipids represent a vast and diverse group of hydrophobic or amphiphilic small molecules that are fundamental to life, serving as critical structural components of cellular membranes, energy storage molecules, and potent signaling mediators [1] [2]. The study of lipids, known as lipidomics, has evolved into a major research field that complements genomics and proteomics, providing a systems-level understanding of cellular physiology and pathology [2]. The structural diversity of lipids arises from complex biosynthetic pathways, leading to hundreds of thousands of distinct lipid species [3]. This complexity is organized by the Lipid Metabolites and Pathways Strategy (LIPID MAPS) consortium, which classifies lipids into eight core categories based on their biochemical subunits: fatty acyls (FA), glycerolipids (GL), glycerophospholipids (GP), sphingolipids (SP), sterol lipids (ST), prenol lipids (PR), saccharolipids (SL), and polyketides (PK) [1] [4] [2].
In the context of multi-center validation lipidomic biomarkers research, understanding this lipid diversity is paramount. Lipidomics has emerged as a powerful tool for identifying novel biomarkers in various diseases, from cardiovascular conditions to cancer [4] [5] [6]. The technological advances in mass spectrometry and chromatography have enabled researchers to detect and quantify subtle alterations in lipid profiles that correspond to specific pathological states [4] [3]. This article provides a comprehensive comparison of key lipid classes, their biological functions in cellular structure and signaling, and the experimental frameworks essential for advancing lipid biomarker discovery and validation.
Key Lipid Classes: Structures and Core Biological Functions
Table 1: Major Lipid Classes: Composition, Structure, and Primary Biological Functions
| Lipid Category | Core Components / Structure | Primary Biological Functions | Cellular Localization |
|---|---|---|---|
| Fatty Acyls (FA) [1] | Fatty acids, Eicosanoids, Fatty alcohols | Energy sources, precursors for signaling molecules (e.g., prostaglandins), inflammation [1] [7] | Cytosol, associated with carrier proteins |
| Glycerolipids (GL) [1] | Mono-, Di-, and Triacylglycerols (Triglycerides) | Energy storage, metabolic intermediates [1] [7] | Lipid droplets, adipose tissue |
| Glycerophospholipids (GP) [1] [8] | Phosphatidylcholine (PC), Phosphatidylethanolamine (PE), Phosphatidylserine (PS), Phosphatidylinositol (PI) | Primary structural components of the plasma membrane, formation of permeability barrier, cell signaling, precursors for second messengers [1] [8] [7] | Plasma membrane (both leaflets), organelle membranes |
| Sphingolipids (SP) [1] [8] | Sphingomyelin, Ceramide, Glycosphingolipids, Sphingosine-1-phosphate | Membrane structural integrity and microdomains, cell recognition, potent signaling molecules in apoptosis, senescence, and proliferation [1] [8] [9] | Plasma membrane (primarily outer leaflet), intracellular membranes |
| Sterol Lipids (ST) [1] [8] | Cholesterol, Bile acids, Steroid hormones | Modulates membrane fluidity and rigidity, precursor for signaling molecules (hormones, bile acids) [1] [8] [7] | Plasma membrane, endoplasmic reticulum |
| Prenol Lipids (PR) [1] | Terpenes, Quinones, Carotenoids | Antioxidants (e.g., Vitamin E), electron carriers (ubiquinone), pigmentation [1] | Various intracellular membranes |
The plasma membrane exemplifies the functional collaboration between different lipid classes. Its bilayer is primarily composed of glycerophospholipids, which form the fundamental permeability barrier [8]. Sphingolipids, particularly sphingomyelin, and sterols (like cholesterol) are integrated within this bilayer, where cholesterol interacts with both phospholipids and sphingolipids to fine-tune membrane properties [8]. It can have a rigidifying effect among phospholipids but increases fluidity among sphingolipids, preventing them from forming a gel-like phase [8]. Furthermore, the membrane is asymmetrical: the inner and outer leaflets have distinct lipid compositions. For instance, charged phospholipids like phosphatidylserine (PS) and phosphatidylethanolamine (PE) are predominantly maintained in the inner leaflet, while glycosphingolipids are exclusively in the outer leaflet [8]. This organization is crucial for cellular functions, including signaling and the maintenance of membrane potential.
Lipid Signaling Pathways: From Membrane to Second Messenger
Lipids are not merely passive structural components; they are dynamic signaling molecules. Lipid signaling involves lipid messengers that bind protein targets like receptors and kinases to mediate specific cellular responses, including apoptosis, proliferation, and inflammation [9] [7]. A key feature of many lipid messengers is that they are not stored but are biosynthesized "on demand" at their site of action [9].
The Ceramide/Sphingosine-1-Phosphate Rheostat
The sphingolipid pathway is a critically important signaling axis where the balance between ceramide, sphingosine, and sphingosine-1-phosphate (S1P) determines cell fate, often described as a "rheostat" where ceramide promotes apoptosis and S1P promotes survival and proliferation [9].
Figure 1: Sphingolipid Signaling Pathway and Cell Fate Determination
Ceramide is a central molecule in sphingolipid metabolism. It can be generated by the hydrolysis of sphingomyelin by enzymes called sphingomyelinases (SMases) or synthesized de novo from serine and a fatty acyl-CoA [9]. Ceramide mediates numerous cell-stress responses, including apoptosis and senescence [9]. It can activate specific protein phosphatases (PP1, PP2A) and protein kinases (PKCζ), leading to the dephosphorylation/inactivation of pro-survival proteins like AKT [9]. This is particularly relevant in metabolic diseases; palmitate-induced ceramide accumulation can desensitize cells to insulin, linking lipid signaling to insulin resistance and diabetes [9].
Sphingosine-1-phosphate (S1P) is the product of sphingosine phosphorylation by sphingosine kinase (SK) [9]. In contrast to ceramide, S1P is a potent promoter of cell survival, migration, and inflammation [9]. Its primary mode of action is through a family of G protein-coupled receptors (S1PRs) on the cell surface [9]. The enzymes that produce S1P (sphingosine kinases) are often upregulated by growth factors and cytokines, driving a pro-survival and inflammatory program [9]. The dynamic balance between ceramide and S1P levels is thus a critical determinant of cellular fate.
Glycerophospholipid-Derived Second Messengers
Glycerophospholipids in the plasma membrane are a major source of rapid second messenger generation. When cleaved by phospholipases, they produce a variety of signaling lipids [7].
- Phosphatidylinositol (PI) and its phosphorylated derivatives (PIPs): These lipids are involved in signal transduction from the cell surface to the interior. The hydrolysis of Phosphatidylinositol (4,5)-bisphosphate (PIP2) by phospholipase C produces inositol trisphosphate (IP3) and diacylglycerol (DAG). DAG remains in the membrane and activates protein kinase C (PKC), while IP3 triggers calcium release from intracellular stores [1] [7].
- Arachidonic acid (AA): This fatty acid, often esterified in membrane phospholipids, is released by phospholipase A2. It serves as the precursor for a vast family of signaling molecules collectively known as eicosanoids, which include prostaglandins, thromboxanes, and leukotrienes [1] [7]. These molecules are potent mediators of pain, fever, inflammation, and blood clotting [7].
Experimental Lipidomics: Methodologies for Biomarker Discovery
The translational potential of lipid biology into clinical biomarkers relies on robust and precise lipidomic methodologies. The workflow is a complex, multi-step process that requires careful planning and execution [4] [3].
Figure 2: Lipidomics Biomarker Discovery Workflow
Core Analytical Strategies in Lipidomics
Lipidomics strategies can be broadly classified into three main approaches, each with distinct applications in biomarker research [3].
Table 2: Comparison of Lipidomics Analytical Approaches
| Approach | Objective | Key Technology | Applications in Biomarker Research | Advantages | Limitations |
|---|---|---|---|---|---|
| Untargeted Lipidomics [4] [3] | Comprehensive, unbiased analysis of all detectable lipids | High-Resolution MS (Q-TOF, Orbitrap) | Discovery phase: Screening for novel lipid biomarkers and pathways [4] [3] | Broad coverage, hypothesis-generating | Semi-quantitative, requires complex data analysis |
| Targeted Lipidomics [4] [3] | Precise identification and absolute quantification of a predefined set of lipids | Tandem MS (e.g., UPLC-QQQ MS) with MRM | Validation phase: Quantifying candidate biomarkers in large cohorts [4] [5] | High sensitivity, accuracy, and reproducibility | Limited to known lipids |
| Pseudo-Targeted Lipidomics [4] [3] | Combines broad coverage with improved quantification | LC-MS/MS | Bridging discovery and validation; increasing coverage of quantified lipids [4] | Improved coverage and quantitative accuracy | Method development is more complex |
A Case Study in Pancreatic Cancer Biomarker Discovery
A 2025 study on Pancreatic Ductal Adenocarcinoma (PDAC) exemplifies the application of these methodologies. The researchers used a non-targeted lipidomics approach on plasma from patients and mouse models to screen for common fatty acid alterations [5]. This discovery phase identified several lipid platforms of interest. They then moved to a targeted analysis, validating 20 specific lipids (including 18 phospholipids) that could distinguish healthy individuals from PDAC patients with high accuracy (AUC of 0.9207) [5]. This study highlights how a multi-step, cross-species lipidomics workflow can identify a panel of lipid biomarkers with performance superior to the current clinical standard, CA19-9 [5].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Lipidomics Studies
| Reagent / Material | Function and Application | Considerations |
|---|---|---|
| Chloroform-Methanol Mixtures [3] | Standard solvent system for lipid extraction from biological samples (e.g., Folch or Bligh & Dyer methods). | Allows phase separation; chloroform enriches lipids, methanol enriches polar metabolites. |
| Internal Standards (IS) [4] [5] | Stable isotope-labeled lipid analogs for mass spectrometry. | Critical for accurate quantification; corrects for sample loss and ion suppression; added at the beginning of extraction. |
| C18 and Silica Chromatography Columns [4] [3] | Solid-phase for lipid separation by liquid chromatography (LC) prior to MS analysis. | C18 for reverse-phase separation by fatty acyl chain; silica for normal-phase separation by lipid class polarity. |
| Mass Spectrometry Quality Control (QC) Pools [5] | A pooled sample created from all individual samples in a study. | Injected repeatedly throughout the analytical sequence to monitor instrument stability and data reproducibility. |
| Lipid Standards (Unlabeled) | Pure chemical standards for lipid identification and calibration curves. | Essential for defining retention time and generating fragmentation spectra for library matching. |
| Lipid Extraction Kits | Commercial kits for standardized, high-throughput lipid extraction. | Improve reproducibility across labs, a key challenge in multi-center studies [4]. |
The diversity of lipid classes, from the structural glycerophospholipids to the signaling-active sphingolipids and sterols, underpins their wide-ranging biological functions. Lipidomics provides the technological framework to decode this complexity and discover novel biomarkers for diseases like cancer, metabolic syndrome, and osteoporosis [4] [6] [3]. The path from discovery to clinical application requires a rigorous, multi-stage process. It begins with untargeted discovery, progresses to targeted validation in larger cohorts, and must culminate in multi-center validation studies to ensure reproducibility and clinical reliability [4] [5]. Despite the challenges, such as biological variability and a lack of standardized protocols, the strategic integration of lipidomics into multi-omics research holds immense promise for advancing personalized medicine, enabling earlier disease diagnosis, and informing the development of new therapeutic strategies.
Lipid metabolic reprogramming is an established hallmark of cancer, enabling tumor cells to sustain uncontrolled proliferation, survive in harsh microenvironments, and resist therapeutic interventions. This review synthesizes evidence from pancreatic, liver, and gynecological cancers to elucidate the shared and unique alterations in lipid uptake, synthesis, storage, and oxidation that drive oncogenesis. We present a comprehensive analysis of lipidomic biomarkers with diagnostic and therapeutic potential, detailing experimental protocols for their identification and validation. The integrated multi-omics approach reveals distinct lipid signatures across cancer types, while highlighting emerging targets for therapeutic intervention. Within the framework of multi-center validation lipidomic biomarkers research, we compare quantitative data from recent studies and provide standardized methodologies for reproducing key findings, offering researchers a validated toolkit for advancing diagnostic and therapeutic strategies targeting cancer lipid metabolism.
Metabolic reprogramming constitutes a core hallmark of cancer, with lipid metabolism emerging as a critical facilitator of tumor progression, metastasis, and treatment resistance across diverse malignancies [10] [11]. Cancer cells extensively rewire their lipid metabolic pathways to fulfill bioenergetic demands, generate membrane components, and produce signaling molecules that sustain proliferative programs [10]. This reprogramming encompasses enhanced lipid uptake, de novo lipogenesis, lipid storage, and fatty acid oxidation (FAO), creating metabolic dependencies that can be therapeutically exploited [10].
The tumor microenvironment (TME) further shapes lipid metabolic adaptations through hypoxia, nutrient scarcity, and metabolic crosstalk between cancer cells and stromal components [10]. Understanding the commonalities and distinctions in lipid metabolic rewiring across different cancers provides crucial insights for developing targeted interventions. This review systematically examines lipid metabolic reprogramming in three cancer types with significant metabolic dependencies: pancreatic ductal adenocarcinoma (PDAC), hepatocellular carcinoma (HCC), and gynecological cancers, with specific emphasis on endometrial cancer (EC).
Recent advances in lipidomics technologies have enabled comprehensive profiling of lipid species alterations in tumors, revealing potential diagnostic biomarkers and therapeutic targets [4] [12]. The integration of lipidomics with other omics approaches (transcriptomics, proteomics) provides unprecedented insights into the regulatory networks governing lipid metabolic reprogramming [12]. This review synthesizes findings from such integrated studies and presents standardized experimental workflows to guide future research into lipid-based biomarkers and therapies.
Lipid Metabolic Pathways in Cancer: A Comparative Analysis
Key Alterations in Lipid Metabolism Across Cancers
Table 1: Lipid Metabolic Reprogramming Across Cancer Types
| Metabolic Process | Pancreatic Cancer | Hepatocellular Carcinoma | Gynecological Cancers |
|---|---|---|---|
| Lipid Uptake | CD36 overexpression; FABP-mediated uptake [10] | CD36 upregulation; LDLR enhancement [13] | CD36 overexpression; FABP involvement [10] |
| De novo Lipogenesis | FASN, ACC, ACLY overexpression [14] [11] | FASN, ACC, ACLY upregulation [13] | FASN, ACC, ACLY overexpression [11] |
| Fatty Acid Oxidation | CPT1A/C upregulation [11] | CPT1A/C enhancement [13] | CPT1C upregulation via PPARα [15] |
| Desaturation | SCD1 overexpression [10] | SCD1 upregulation [13] | SCD1 enhancement [10] |
| Cholesterol Metabolism | LDLR upregulation; enhanced synthesis [11] | HMGCR upregulation; LDLR enhancement [13] | HMGCR overexpression; LDLR upregulation [11] |
| Key Enzymes/Transporters | ACSL1, ACSL4 [12] | ACSL1, ACSL4, GPD1 [12] | ACSL4 [15] |
| Signaling Pathways | KRAS, HIF-1α, PI3K/Akt/mTOR [14] | Wnt/β-catenin, PI3K/Akt/mTOR [13] | PPARα, E2F2 [15] |
Molecular Mechanisms and Signaling Pathways
The rewiring of lipid metabolism in cancer cells is orchestrated by oncogenic signaling pathways and environmental cues. The PI3K/Akt/mTOR pathway emerges as a central regulator across cancer types, enhancing lipid synthesis through activation of sterol regulatory element-binding proteins (SREBPs) that transcriptionally upregulate lipogenic enzymes like FASN, ACC, and ACLY [14] [13]. In pancreatic cancer, KRAS mutations drive metabolic reprogramming by enhancing lipid uptake and synthesis, while HIF-1α stabilizes under hypoxia to promote lipid storage and utilization [14].
In hepatocellular carcinoma, the Wnt/β-catenin pathway directly influences lipid metabolism by regulating glutamine synthetase expression, thereby connecting amino acid and lipid metabolic networks [13]. c-MYC amplification, common in HCC, transcriptionally upregulates enzymes involved in fatty acid synthesis and glutamine metabolism, providing precursors for lipid synthesis [13].
For gynecological cancers, particularly endometrial cancer, PPARα serves as a master regulator of lipid metabolic genes, including ACSL4 and CPT1C, creating a feedforward loop that sustains FAO [15]. Concurrently, E2F2 drives cell cycle progression while intersecting with metabolic circuits, forming a positive feedback loop with ACSL4 that coordinately enhances both proliferation and metabolic adaptation [15].
Figure 1: Regulatory Networks in Cancer Lipid Metabolic Reprogramming. This diagram illustrates how extracellular factors (hypoxia, oncogenes, obesity) activate key signaling pathways that coordinately regulate lipid metabolic processes, ultimately driving tumor progression, metastasis, and therapy resistance. Dashed lines indicate feedback mechanisms.
Cancer-Specific Lipid Metabolic Reprogramming
Pancreatic Ductal Adenocarcinoma (PDAC)
Pancreatic cancer demonstrates extensive lipid metabolic reprogramming to support its aggressive growth in a nutrient-poor, hypoxic microenvironment [14]. PDAC cells upregulate both de novo lipogenesis and exogenous lipid uptake to satisfy their substantial membrane biosynthesis and energy requirements [5]. FASN overexpression is nearly universal in PDAC and correlates with poor prognosis, while SCD1 upregulation enhances the production of monounsaturated fatty acids that maintain membrane fluidity and support signaling pathways [10] [11].
The lipid-rich, fibrotic TME of pancreatic cancer further shapes metabolic adaptations. Cancer-associated fibroblasts (CAFs) and pancreatic stellate cells (PSCs) provide lipids to cancer cells through metabolic coupling, creating a feedforward loop that sustains tumor growth [10]. PDAC cells also demonstrate enhanced fatty acid oxidation (FAO) under nutrient deprivation, with CPT1A upregulation enabling mitochondrial import and oxidation of fatty acids for ATP production [11]. This metabolic flexibility contributes to chemotherapy resistance, as FAO inhibition sensitizes PDAC cells to gemcitabine [14].
Notably, lipid droplet accumulation serves as a marker of PDAC aggressiveness and chemoresistance. Cancer stem cells within PDAC tumors exhibit higher lipid content than their differentiated counterparts, and LD-rich cells demonstrate increased resistance to cytotoxic therapies [11]. The dependence of PDAC on lipid metabolic pathways offers promising therapeutic targets, with inhibitors of FASN, SCD1, and CPT1 showing efficacy in preclinical models [14] [11].
Hepatocellular Carcinoma (HCC)
As the metabolic hub of the body, the liver normally maintains lipid homeostasis, but HCC development involves profound dysregulation of lipid metabolic pathways [13]. The connection between HCC and underlying conditions such as nonalcoholic steatohepatitis (NASH) and obesity highlights the importance of lipid metabolism in hepatocarcinogenesis [16]. HCC cells exhibit enhanced lipid uptake via CD36 and LDLR, increased de novo lipogenesis through FASN and ACLY, and upregulated cholesterol synthesis via HMGCR [13].
Integrated multi-omics analyses reveal distinctive lipid signatures in HCC tissues compared to adjacent non-tumor tissues [12]. Transcriptomic and proteomic profiling identifies key deregulated genes including ACSL1, ACSL4, GPD1, LCAT, PEMT, and LPCAT1, which coordinately enhance fatty acid activation, phospholipid remodeling, and lipid storage [12]. Metabolomic studies further demonstrate alterations in phosphatidylcholines, sphingolipids, and carnitine esters that distinguish HCC from normal liver tissue [12].
The PPARα pathway plays a central role in regulating lipid catabolism in HCC, driving the expression of genes involved in fatty acid oxidation [13]. This pathway enables HCC cells to utilize fatty acids as an energy source, particularly under conditions of metabolic stress. Additionally, the Wnt/β-catenin pathway influences lipid metabolism by regulating glutamine synthetase expression, thereby connecting amino acid and lipid metabolic networks [13]. Targeting lipid metabolic enzymes such as ACSL4 has shown promise in preclinical HCC models, impairing tumor growth and enhancing sensitivity to therapy [15].
Gynecological Cancers (Endometrial Cancer)
Endometrial cancer exhibits strong associations with obesity and metabolic syndrome, highlighting the importance of lipid metabolic reprogramming in its pathogenesis [15]. EC cells demonstrate enhanced lipogenesis mediated by FASN and ACLY upregulation, providing lipids for membrane synthesis and signaling molecules that drive proliferation [15]. The ACSL4 enzyme emerges as a critical node in EC lipid metabolism, activating polyunsaturated fatty acids for incorporation into complex lipids or oxidation [15].
Recent research has identified a novel ACSL4-E2F2 positive feedback loop that coordinately regulates lipid metabolism and cell cycle progression in endometrial cancer [15]. ACSL4 upregulates E2F2 through activation of the PPARα-CPT1C-FAO axis, while E2F2 transcriptionally enhances ACSL4 expression, creating an amplification circuit that drives both metabolic reprogramming and proliferation [15]. This mechanism links the obesity-driven lipid-rich environment directly to tumor growth, explaining the epidemiological connection between obesity and endometrial cancer incidence.
Therapeutic targeting of this axis through ACSL4 inhibition suppresses EC progression in preclinical models, validating the clinical potential of targeting lipid metabolic pathways [15]. Additionally, the dependence of EC cells on CPT1C-mediated FAO provides an alternative targeting strategy, particularly for tumors with ACSL4 overexpression [15].
Diagnostic Biomarker Discovery through Lipidomics
Lipidomic Signatures as Diagnostic Tools
Lipidomics has emerged as a powerful approach for identifying cancer biomarkers, with specific lipid signatures demonstrating diagnostic potential across multiple cancer types [4] [12] [5]. Advances in mass spectrometry-based lipid profiling enable comprehensive characterization of lipid alterations in tumors, revealing distinct patterns that can distinguish malignant from normal tissues with high accuracy [4].
Table 2: Lipidomic Biomarkers in Cancer Diagnosis
| Cancer Type | Lipid Classes Altered | Specific Lipid Biomarkers | Diagnostic Performance | Reference |
|---|---|---|---|---|
| Pancreatic Cancer | Phospholipids, Acylcarnitines, Sphingolipids, Fatty Acid Amides | 18 phospholipids, 1 acylcarnitine, 1 sphingolipid | AUC 0.9207 (0.9427 with CA19-9) | [5] |
| Hepatocellular Carcinoma | Glycerophospholipids, Sphingolipids, Fatty Acids | LCAT, PEMT, ACSL1, GPD1, ACSL4, LPCAT1 | 6 metabolites with AUC >0.8 | [12] |
| Cervical Cancer | Ceramides, Sphingosine, Phospholipids | 2 ceramides, 1 sphingosine metabolite | Discriminate HSIL from normal | [17] |
In pancreatic cancer, a recent multi-platform lipidomics study identified 20 lipid species that consistently differentiated PDAC patients from healthy controls across multiple validation sets [5]. A model incorporating 11 phospholipids achieved an AUC of 0.9207, significantly outperforming the conventional biomarker CA19-9 (AUC 0.7354) [5]. When combined with CA19-9, the diagnostic performance further improved to AUC 0.9427, demonstrating the complementary value of lipid biomarkers [5].
Hepatocellular carcinoma exhibits distinct lipidomic alterations identifiable through integrated multi-omics approaches [12]. Transcriptomic and proteomic analyses of HCC tissues reveal significant enrichment of lipid metabolism-related pathways, including fatty acid degradation and steroid hormone biosynthesis [12]. Six key genes (LCAT, PEMT, ACSL1, GPD1, ACSL4, and LPCAT1) show consistent changes at both mRNA and protein levels, correlating strongly with lipid metabolite alterations and offering diagnostic potential [12].
Cervical cancer lipidomics has identified specific ceramides and sphingosine metabolites that distinguish high-grade squamous intraepithelial lesions (HSIL) from normal tissue, independent of HPV status [17]. Plasma metabolomic profiling further reveals alterations in prostaglandins, phospholipids, and sphingolipids that differentiate cervical intraepithelial neoplasia (CIN) and invasive cancer from healthy controls [17].
Experimental Protocols for Lipidomic Biomarker Discovery
Protocol 1: Untargeted Lipidomics for Biomarker Discovery
Sample Collection and Preparation: Collect plasma/serum samples after overnight fasting or tissue samples snap-frozen in liquid nitrogen. For plasma, add antioxidant preservatives and store at -80°C until analysis [5].
Lipid Extraction: Use modified Folch or Bligh-Dyer methods with chloroform:methanol (2:1 v/v). Add internal standards for quantification [4] [5].
LC-MS/MS Analysis:
- Platform: Ultimate 3000-LTQ-Orbitrap XL or similar high-resolution mass spectrometer
- Chromatography: Reversed-phase C18 column (2.1 × 100 mm, 1.7 μm)
- Mobile Phase: (A) acetonitrile:water (60:40) with 10mM ammonium formate; (B) isopropanol:acetonitrile (90:10) with 10mM ammonium formate
- Gradient: 0-2 min 30% B, 2-25 min 30-100% B, 25-30 min 100% B
- MS Parameters: ESI positive/negative mode, mass range 150-2000 m/z, data-dependent MS/MS [5]
Data Processing: Use software (Compound Discoverer, MS-DIAL, Lipostar) for peak picking, alignment, and identification against lipid databases (LIPID MAPS, HMDB) [4].
Statistical Analysis: Apply multivariate statistics (PCA, PLS-DA) and machine learning algorithms to identify discriminatory lipid features. Validate with univariate tests and ROC analysis [5].
Protocol 2: Multi-omics Integration for Lipid Pathway Analysis
Transcriptomic Profiling: Extract total RNA, prepare cDNA libraries, and sequence on Illumina platform. Map reads to reference genome, quantify gene expression, identify differentially expressed genes (DEGs) with DESeq2 [12].
Proteomic Analysis: Homogenize tissues in lysis buffer, digest proteins with trypsin, fractionate peptides by high-pH reverse phase chromatography. Analyze on Orbitrap Fusion Lumos with LC-MS/MS. Identify differentially expressed proteins (DEPs) [12].
Integration Analysis: Map DEGs and DEPs to KEGG lipid metabolic pathways. Correlate expression changes with lipid metabolite alterations. Identify key regulatory nodes through pathway enrichment and network analysis [12].
Figure 2: Integrated Multi-omics Workflow for Lipid Biomarker Discovery. This diagram outlines the comprehensive approach for identifying and validating lipid metabolic biomarkers, encompassing sample collection, multi-omics profiling, data integration, and clinical application.
Therapeutic Targeting of Lipid Metabolism
Preclinical and Clinical Development
Therapeutic targeting of lipid metabolic pathways represents a promising strategy for cancer treatment, with several approaches in various stages of development:
FASN Inhibitors: TVB-2640 (denifanstat) has shown efficacy in preclinical models of multiple cancers, including breast, ovarian, and pancreatic cancer [11]. Phase I/II trials demonstrate acceptable safety and preliminary activity, particularly in KRAS-mutant cancers [11].
ACSL4 Inhibitors: PRGL493 and other small-molecule inhibitors suppress endometrial cancer progression in vivo by disrupting the ACSL4-PPARα-CPT1C axis and reducing cancer stemness [15]. ACSL4 inhibition also shows promise in hepatocellular carcinoma models [15].
CPT1 Inhibitors: Etomoxir, perhexiline, and other CPT1 inhibitors block fatty acid oxidation, sensitizing cancer cells to chemotherapy and targeted therapies [14] [11]. These are particularly effective in tumors reliant on FAO for energy production.
SCD1 Inhibitors: MF-438 and other desaturase inhibitors disrupt membrane fluidity and lipid signaling, demonstrating antitumor effects in pancreatic and liver cancer models [10] [11].
CD36 Antibodies: Blocking this fatty acid transporter impairs lipid uptake and suppresses metastasis in multiple cancer models, including ovarian, breast, and oral cancers [10].
Combination Therapies
Targeting lipid metabolism enhances the efficacy of conventional and targeted therapies:
Chemotherapy Sensitization: FAO inhibition reverses chemoresistance in pancreatic cancer by reducing ATP production and increasing oxidative stress [14]. FASN inhibition enhances gemcitabine efficacy in PDAC models through induction of endoplasmic reticulum stress [14].
Immunotherapy Combinations: Modulating lipid metabolism in the TME enhances antitumor immunity. CD36 blockade improves CD8+ T cell function by reducing lipid accumulation-induced dysfunction [10]. COX-2 inhibitors, which target prostaglandin synthesis, enhance checkpoint inhibitor efficacy in preclinical models [10].
Targeted Therapy Synergy: SCD1 inhibition enhances the efficacy of EGFR inhibitors in lung cancer and HER2-targeted therapies in breast cancer by disrupting membrane lipid composition and signaling [11].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for Lipid Metabolism Research
| Category | Reagent/Solution | Application | Key Features |
|---|---|---|---|
| Lipid Extraction | Modified Folch Reagent (CHCl₃:MeOH 2:1) | Lipid extraction from tissues/body fluids | Preserves lipid integrity; compatible with MS |
| Bligh & Dyer Solution | Alternative extraction method | Effective for polar lipids | |
| Internal Standards | SPLASH LIPIDOMIX Mass Spec Standard | Lipid quantification | Covers multiple lipid classes; stable isotope-labeled |
| Avanti Polar Lipids Internal Standards | Targeted lipid analysis | Individual lipid class standards | |
| LC-MS Reagents | Ammonium Formate/Ammonium Acetate | Mobile phase additives | Enhances ionization; reduces adduct formation |
| HPLC-grade Solvents (ACN, MeOH, IPA) | LC-MS mobile phases | Low UV absorbance; high purity | |
| Enzyme Inhibitors | TVB-2640 (FASN inhibitor) | Target validation; therapeutic studies | Clinical-stage inhibitor; good bioavailability |
| Etomoxir (CPT1 inhibitor) | FAO inhibition studies | Well-characterized; widely used | |
| PRGL493 (ACSL4 inhibitor) | ACSL4 pathway studies | Specific ACSL4 inhibition [15] | |
| Antibodies | Anti-ACSL4 (for WB/IHC) | Protein expression analysis | Validated for multiple applications [15] |
| Anti-CD36 (for flow/IF) | Lipid uptake studies | Cell surface staining | |
| Anti-FASN (for WB/IHC) | Lipogenesis assessment | Widely used in cancer research | |
| Cell Culture Supplements | Fatty acid-free BSA | Fatty acid delivery to cells | Controls fatty acid concentrations |
| Lipid-rich media (e.g., with oleate) | Lipid loading studies | Mimics obese/tumor microenvironment |
Lipid metabolic reprogramming represents a convergent hallmark across pancreatic, liver, and gynecological cancers, driven by both oncogenic signaling and microenvironmental pressures. The consistent alterations in lipid uptake, synthesis, storage, and oxidation pathways across these malignancies highlight the fundamental role of lipid metabolism in supporting tumor progression and therapy resistance.
Integrated multi-omics approaches have unveiled distinct lipid signatures with diagnostic potential, offering improved sensitivity and specificity over conventional biomarkers. The identification of specific lipid species and enzymes as therapeutic targets, including ACSL4, FASN, and CPT1, provides promising avenues for intervention. However, translating these findings into clinical practice requires standardized methodologies, robust validation across diverse patient populations, and careful consideration of the metabolic complexities within the tumor microenvironment.
Future research should focus on expanding multi-center validation studies for lipidomic biomarkers, developing more specific inhibitors targeting key lipid metabolic enzymes, and exploring combination strategies that exploit metabolic dependencies while minimizing systemic toxicity. The Scientist's Toolkit provided herein offers a foundation for standardizing methodologies across research laboratories, facilitating reproducibility and comparison of findings. As our understanding of cancer lipid metabolism deepens, targeting these pathways holds significant promise for improving early detection, monitoring treatment response, and developing more effective therapeutic strategies for these challenging malignancies.
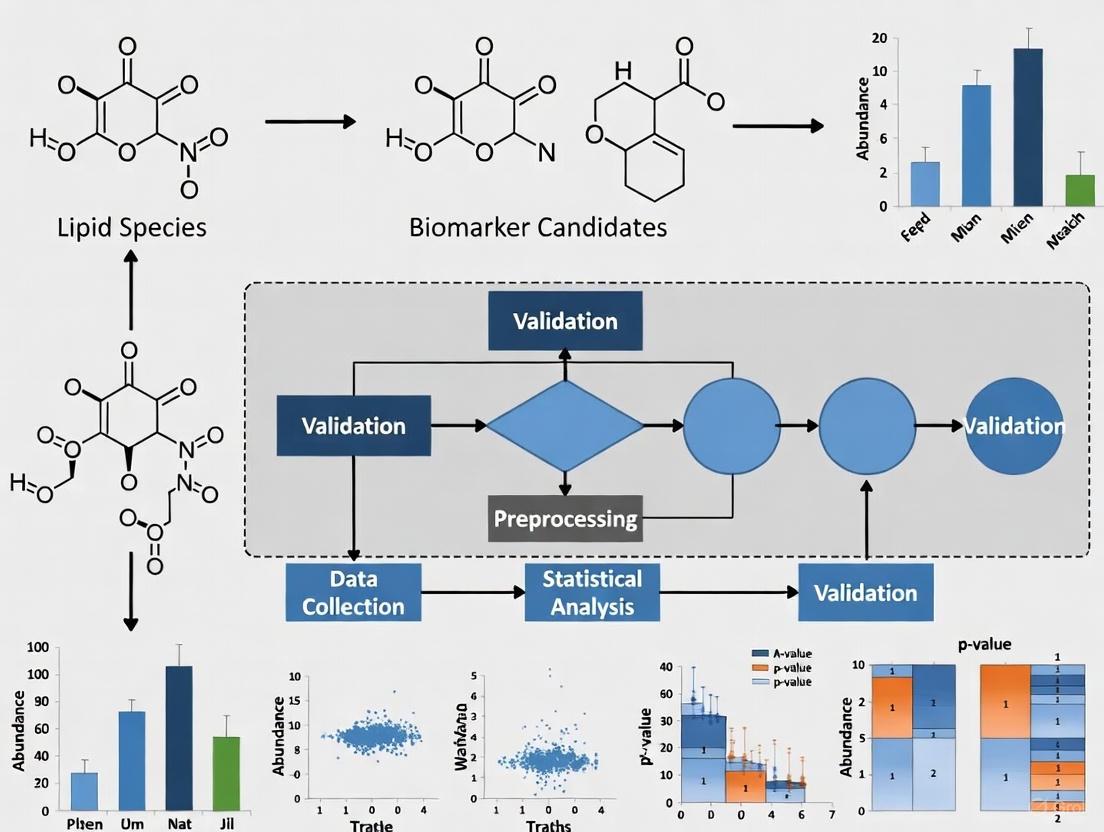
Nonsyndromic cleft lip with palate (nsCLP) is among the most common congenital craniofacial anomalies, affecting approximately 1 in 700 live births globally [18]. Current diagnostic methods primarily rely on fetal ultrasound imaging, which remains limited by factors such as fetal position and technician skill [19]. The emergence of lipidomics—the comprehensive study of lipid molecules and their biological functions—offers promising avenues for identifying molecular biomarkers that could enable earlier and more reliable detection [4]. Lipids constitute thousands of chemically distinct molecules that play vital roles in cellular processes including signaling, energy storage, and structural membrane integrity [4]. Their molecular structures largely determine their functions, and disruptions in lipid homeostasis can provide crucial information about disease mechanisms [4]. This case study examines how integrated lipidomics and machine learning approaches have identified specific altered lipid pathways in nsCLP, potentially paving the way for novel diagnostic strategies.
Experimental Design and Methodological Framework
Study Cohorts and Sampling Strategy
The investigation employed a multi-stage cohort design to ensure robust biomarker discovery and validation [19]. In the initial discovery phase, researchers conducted untargeted lipidomics profiling on maternal serum samples from a cohort of nsCLP-affected pregnancies and matched controls. This approach aimed to capture a comprehensive view of the lipid landscape without pre-selecting specific lipid classes. Promising candidate biomarkers identified in this phase were subsequently evaluated using targeted lipidomics in a separate validation cohort, ensuring that findings were reproducible across different sample sets [19]. To further strengthen the validity of results, an additional validation cohort incorporating early serum samples from the nsCLP group was also analyzed, providing temporal insights into lipid alterations [19].
Analytical Platform: Liquid Chromatography-Mass Spectrometry (LC-MS)
Table 1: Core Lipidomics Analytical Platform Specifications
| Component | Specification | Application in nsCLP Study |
|---|---|---|
| Separation Technique | Liquid Chromatography | Separation of complex lipid mixtures from serum samples |
| Detection Instrument | Mass Spectrometry (MS) | Accurate mass measurement and structural characterization |
| Analytical Approach | Untargeted & Targeted | Comprehensive discovery followed by quantitative validation |
| Data Processing | Compound Discoverer software | Feature extraction, alignment, and identification |
Lipidomic profiling was performed using liquid chromatography-mass spectrometry (LC-MS), a powerful analytical platform that combines separation capabilities with sensitive detection [19]. The untargeted approach provided a comprehensive assessment of lipid species present in the samples, regardless of whether they were previously known or unknown, thereby offering a complete picture of the lipid profile [4]. For the targeted validation phase, the methodology shifted to precise quantification of specific candidate lipids, enhancing reproducibility and reliability of the measurements [4]. This dual approach balanced discovery power with analytical rigor, addressing a common challenge in biomarker research.
Data Analysis and Machine Learning Integration
Table 2: Machine Learning and Statistical Approaches for Biomarker Discovery
| Analytical Method | Implementation Purpose | Outcome in nsCLP Study |
|---|---|---|
| Feature Selection Methods | Eight different algorithms to assess dysregulated lipids | Identified most consistently altered lipid species |
| Robust Rank Aggregation | Integrated results from multiple feature selection methods | Prioritized candidate biomarkers with consensus importance |
| Classification Models | Seven different models to retrieve biomarker panels | Identified optimal combination of diagnostic lipids |
| Multivariate Analyses | Constructed diagnostic models with selected lipids | Finalized minimal biomarker panel with high diagnostic performance |
The data analysis framework employed multiple computational techniques to ensure reliable biomarker identification [19]. Eight distinct feature selection methods were initially applied to assess dysregulated lipids from the untargeted lipidomics data. The robust rank aggregation algorithm then integrated these selections to prioritize the most consistently significant lipid species [19]. Subsequently, seven classification models were applied to retrieve a panel of candidate lipid biomarkers. This multi-algorithm approach mitigated biases inherent in any single method and increased confidence in the final biomarker selection [19].
Figure 1: Experimental Workflow for Lipid Biomarker Discovery in nsCLP
Key Findings: Altered Lipid Pathways and Diagnostic Biomarkers
Identified Lipid Biomarkers and Their Diagnostic Performance
The integrated analysis revealed a panel of three lipid biomarkers with strong diagnostic potential for nsCLP [19]. The specific lipids identified included FA (20:4), likely arachidonic acid, and LPC (18:0), a lysophosphatidylcholine species [19]. Both were significantly downregulated in early serum samples from the nsCLP group in the additional validation cohort, suggesting their potential role in early detection [19]. The diagnostic model incorporating these three lipids achieved high performance in determining nsCLP status, demonstrating the power of minimal biomarker panels when properly selected and validated [19].
Table 3: Validated Lipid Biomarkers in nsCLP
| Lipid Biomarker | Class | Regulation in nsCLP | Biological Significance |
|---|---|---|---|
| FA (20:4) | Fatty Acyl (Arachidonic Acid) | Downregulated | Precursor for signaling molecules; membrane fluidity |
| LPC (18:0) | Glycerophospholipid (Lysophosphatidylcholine) | Downregulated | Signaling lipid; involved in cell membrane structure |
| Third Lipid | Not Specified | Not Specified | Not Specified |
Biological Implications of Lipid Alterations
The observed alterations in specific lipid species provide insights into potential mechanistic pathways involved in nsCLP pathogenesis. Arachidonic acid (FA 20:4) serves as a precursor for eicosanoids, signaling molecules that play crucial roles in inflammation and embryonic development [4]. Similarly, lysophosphatidylcholines like LPC (18:0) are involved in cell signaling and membrane structure [4]. The significant downregulation of these lipids in nsCLP maternal serum suggests potential disruptions in lipid-mediated signaling pathways during critical stages of craniofacial development. These findings align with emerging understanding that lipids are not merely structural components but active participants in developmental processes, with dysregulation potentially contributing to congenital anomalies [4].
The Research Toolkit: Essential Reagents and Platforms
Table 4: Essential Research Reagents and Platforms for Lipidomics Biomarker Discovery
| Reagent/Platform Category | Specific Examples | Function in nsCLP Study |
|---|---|---|
| Chromatography Systems | Liquid Chromatography (LC) | Separation of complex lipid mixtures prior to detection |
| Mass Spectrometry Instruments | LTQ-Orbitrap XL MS | Accurate mass measurement and structural characterization |
| Data Processing Software | Compound Discoverer; MS DIAL; Lipostar | Lipid feature extraction, alignment, and identification |
| Bioinformatics Algorithms | Robust Rank Aggregation; Multiple Classifiers | Prioritization of biomarker candidates from large datasets |
| Lipid Reference Databases | LIPID MAPS Classification | Structural annotation of identified lipid species |
The implementation of lipidomics biomarker discovery requires specialized reagents and platforms [19] [4]. Liquid chromatography systems enable separation of complex lipid mixtures, while mass spectrometry instruments provide sensitive detection and structural information [5]. Bioinformatics tools are equally crucial, as they transform raw instrumental data into biologically meaningful information [19] [4]. The nsCLP study utilized specialized software for lipid feature extraction and alignment, followed by multiple statistical and machine learning algorithms for biomarker selection [19]. Reference databases such as LIPID MAPS provide standardized classification and nomenclature systems essential for consistent lipid identification across studies [4].
Multi-Center Validation Challenges and Solutions
Reproducibility and Technical Variability
A significant challenge in lipidomic biomarker research involves reproducibility across different analytical platforms and laboratories [4]. Recent studies indicate that prominent software platforms like MS DIAL and Lipostar demonstrate only approximately 14-36% agreement in lipid identifications when using default settings, even when analyzing identical LC-MS data [4]. This technical variability complicates cross-study comparisons and multi-center validation efforts. To address these challenges, researchers have emphasized standardized protocols for sample collection, processing, and data analysis [4]. Additionally, the use of targeted validation following untargeted discovery, as implemented in the nsCLP study, improves the reliability of findings [19].
Integration with Multi-Omics Data
The complexity of nsCLP pathogenesis suggests that lipid biomarkers alone may not capture the full pathological picture. Integration with other data types, including genomic, proteomic, and clinical information, provides a more comprehensive understanding [4]. Family-based trio studies examining genetic polymorphisms in genes such as ABCA4, which is involved in lipid transport, have revealed potential genetic contributors to NSCL/P that may interact with the lipid alterations observed in the lipidomics study [18]. Such integrated approaches align with systems biology perspectives that recognize complex traits like nsCLP as emerging from interactions across multiple biological layers [4].
Figure 2: Multi-Omics Data Integration for Biomarker Validation
Comparative Performance: Lipid Biomarkers Versus Conventional Methods
Table 5: Performance Comparison of Diagnostic Approaches for nsCLP
| Diagnostic Method | Strengths | Limitations | Target Population |
|---|---|---|---|
| Fetal Ultrasound (Current Standard) | Non-invasive; visualizes anatomy | Limited by fetal position, technician skill | General obstetric population |
| Genetic Risk Markers | Potential for early risk assessment | Incomplete penetrance; population-specific variants | High-risk families |
| Lipid Biomarker Panel | Objective biochemical measure; early detection | Requires validation in diverse populations | Maternal serum screening |
When compared to conventional diagnostic methods for nsCLP, lipid biomarkers offer distinct advantages and limitations. Current ultrasound-based diagnosis, while non-invasive and providing direct structural information, faces limitations including dependence on fetal position and technician expertise [19]. Genetic markers have shown promise but often exhibit population-specific variations and inconsistent replicability across ethnic groups [18]. In contrast, lipid biomarkers present an objective biochemical measure that could potentially enable earlier detection, though they require extensive validation across diverse populations before clinical implementation [19] [4]. The diagnostic model developed in the nsCLP study demonstrated high performance with just three lipids, suggesting potential clinical utility if validated in broader cohorts [19].
Future Directions and Translational Potential
The translational pathway for lipid biomarkers in nsCLP requires addressing several key challenges. While the FDA has approved very few lipid-based biomarkers to date, examples such as the Tina-quant Lipoprotein(a) assay demonstrate the feasibility of clinical implementation [4]. Future directions include leveraging artificial intelligence to enhance lipid annotation accuracy, with models like MS2Lipid demonstrating up to 97.4% accuracy in predicting lipid subclasses [4]. Additionally, interdisciplinary collaboration among lipid biologists, physicians, bioinformaticians, and regulatory scientists is essential to fully realize the potential of lipidomics in personalized medicine approaches to congenital anomaly detection and prevention [4]. The established workflow from this nsCLP case study—combining untargeted discovery, machine-learning-driven feature selection, and targeted validation—provides a template that can be adapted to other congenital disorders potentially linked to lipid metabolic disruptions.
In the evolving landscape of precision medicine, the quest for reliable, non-invasive biomarkers has become a paramount objective across therapeutic areas. Among various candidates, lipids have emerged as particularly promising targets for blood-based biomarker development. Lipids represent a fundamental component of human metabolism, constituting approximately 70% of the metabolites in plasma and playing crucial roles in cellular structure, energy storage, and signaling pathways [3]. The field of lipidomics, which enables comprehensive analysis of lipid species and their dynamic alterations, has opened new avenues for understanding disease mechanisms and identifying clinically useful biomarkers [4] [3].
The translational potential of lipid biomarkers stems from their direct reflection of pathophysiological processes occurring at the cellular level. Unlike genetic or proteomic biomarkers, which indicate potential or expressed activity, lipid profiles represent functional metabolic endpoints that capture real-time biochemical alterations in disease states [3]. This review examines the strategic advantages of lipids as non-invasive blood-based biomarkers, supported by experimental data and methodological considerations, within the critical context of multi-center validation research required for clinical adoption.
Strategic Advantages of Lipids as Blood-Based Biomarkers
Biological and Technical Rationale
Lipids offer distinctive advantages as biomarker candidates due to their structural diversity, metabolic responsiveness, and detectability in accessible biofluids. The biological system contains thousands of chemically distinct lipids classified into eight major categories: fatty acyls (FA), glycerolipids (GL), glycerophospholipids (GP), sphingolipids (SP), sterol lipids (ST), prenol lipids (PR), saccharolipids (SL), and polyketides (PK) [4]. This molecular diversity enables precise mapping of disease-specific signatures, as different pathological processes affect distinct lipid pathways.
From a technical perspective, lipids demonstrate remarkable stability in blood samples compared to more labile molecules such as proteins or RNA. This characteristic reduces pre-analytical variability and facilitates standardized clinical sampling protocols. Additionally, advances in analytical platforms, particularly high-resolution mass spectrometry (HRMS) coupled with liquid chromatography (LC), have dramatically improved the sensitivity, resolution, and throughput of lipid detection [20] [3]. The technological progress in lipidomics has greatly advanced our comprehension of lipid metabolism and biochemical mechanisms in diseases, while also offering new technical pathways for identifying potential biomarkers [3].
Physiological Reflection in Accessible Compartments
Blood-based lipid biomarkers provide a window into systemic physiology while simultaneously reflecting tissue-specific alterations. For instance, red blood cell membrane (RCM) lipids have been shown to accumulate over time and reflect chronic physiological alterations rather than fleeting changes, providing profound insight into long-term disease trajectories such as Alzheimer's disease [21]. This temporal stability enhances their diagnostic utility for chronic conditions where single timepoint plasma measurements might miss relevant pathophysiology.
The non-invasive nature of blood sampling facilitates repeated measurements, enabling dynamic monitoring of disease progression and treatment response. This addresses a critical limitation of tissue-based biomarkers that require invasive procedures such as biopsies, which carry risks of bleeding, pain, and infection [22]. The practical advantage of blood-based lipid biomarkers is particularly valuable for longitudinal studies and chronic disease management where frequent sampling is necessary.
Table 1: Comparative Advantages of Lipid Biomarkers Over Other Molecular Classes
| Feature | Lipid Biomarkers | Genetic Biomarkers | Protein Biomarkers |
|---|---|---|---|
| Stability in blood | High stability in plasma and RBC membranes | High stability | Moderate to low stability |
| Reflects current physiology | Real-time metabolic status | Predisposition only | Expressed activity |
| Technical detection | Advanced LC-HRMS methods | PCR, sequencing | Immunoassays, MS |
| Dynamic range | Extensive molecular diversity | Limited to gene variants | Moderate diversity |
| Pathway coverage | Broad metabolic pathway representation | Limited pathway insight | Signaling pathways |
Lipidomics Methodologies: Experimental Workflows for Biomarker Discovery
Analytical Platforms and Strategies
Lipidomics employs three primary analytical strategies, each with distinct advantages for biomarker discovery. Untargeted lipidomics provides comprehensive, unbiased analysis of all detectable lipid species in a sample, making it ideal for discovery-phase research [3]. This approach typically utilizes high-resolution mass spectrometry (HRMS) with data-dependent acquisition (DDA) or data-independent acquisition (DIA) modes to achieve extensive lipid coverage [3]. The exceptional mass resolution and accuracy of HRMS platforms, including Quadrupole Time-of-Flight (Q-TOF) and Orbitrap instruments, enable precise structural elucidation of lipid molecules [3].
Targeted lipidomics focuses on precise identification and quantification of predefined lipid panels with enhanced accuracy and sensitivity [3]. This approach typically employs triple quadrupole mass spectrometers operating in multiple reaction monitoring (MRM) mode, offering superior quantification capabilities for validation studies [3]. The pseudo-targeted approach represents a hybrid strategy that combines the comprehensive coverage of untargeted methods with the quantitative rigor of targeted analysis, making it suitable for complex disease characterization [3].
Standardized Workflow for Lipid Biomarker Discovery
A robust lipidomics workflow encompasses multiple critical stages from sample collection to data interpretation. The process begins with standardized sample collection using appropriate anticoagulants and strict fasting conditions to minimize pre-analytical variability [20]. For plasma preparation, samples are typically centrifuged within 2-6 hours of collection and stored at -80°C until analysis [20]. The lipid extraction step often employs modified Folch or Bligh-Dyer methods using chloroform-methanol or isopropanol-based solvents to achieve efficient recovery of diverse lipid classes [20] [21].
For LC-MS analysis, reverse-phase chromatography with C18 columns provides excellent separation of most lipid classes using binary mobile phase gradients [20] [21]. Mobile phase A typically consists of acetonitrile-water (60:40) with 10 mM ammonium formate and 0.1% formic acid, while mobile phase B contains isopropanol-acetonitrile (90:10) with the same additives [21]. Mass spectrometry detection in both positive and negative ionization modes ensures comprehensive coverage of ionizable lipid species, with capillary voltages typically set at +3.0 kV for positive mode and -2.5 kV for negative mode [21].
Table 2: Essential Research Reagents and Platforms for Lipidomics
| Category | Specific Items | Function in Workflow |
|---|---|---|
| Sample Collection | EDTA or heparin tubes, PBS, Tris-HCl buffer | Blood collection, RBC washing, hemolysis |
| Lipid Extraction | Isopropanol, methanol, acetonitrile, chloroform | Protein precipitation, lipid solubilization |
| Chromatography | UPLC systems, C18 columns (e.g., Waters ACQUITY) | Lipid separation prior to MS detection |
| Mass Spectrometry | Q-TOF, Orbitrap, Triple Quadrupole instruments | Lipid detection, identification, quantification |
| Internal Standards | PC 14:0, PC 18:0-18:1, LPC 17:0 | Quantification normalization, quality control |
| Data Processing | Progenesis QI, MS-DIAL, Lipostar | Peak alignment, identification, statistical analysis |
Diagram 1: Comprehensive lipidomics workflow for biomarker discovery and validation.
Experimental Evidence: Lipid Biomarkers in Clinical Applications
Oncology Applications
Substantial evidence demonstrates the clinical potential of lipid biomarkers in cancer detection and stratification. A 2025 study on non-muscle invasive bladder cancer (NMIBC) employed LC-HRMS to analyze plasma samples from 106 patients and 108 healthy controls [20]. The research identified a three-lipid panel comprising PE(14:1/20:0), PE(18:2/16:0), and 19-methyl-heneicosanoic acid that achieved exceptional diagnostic performance with an AUC of 0.88 in the training cohort and 0.82 in the validation cohort [20]. Notably, for distinguishing between low-grade and high-grade NMIBC, a four-lipid panel demonstrated an AUC of 0.815, with consistent performance in 10-fold cross-validation (AUC: 0.77) and leave-one-out validation (AUC: 0.77) [20].
In gynecological cancers, lipidomics has revealed disease-specific signatures with diagnostic potential. Altered lipid metabolism supports the energy demands of rapidly proliferating cancer cells, and specific lipid classes including glycerophospholipids, sphingolipids, and fatty acyls show consistent alterations in ovarian, cervical, and endometrial cancers [3]. These findings highlight the fundamental role of lipid metabolic reprogramming in cancer pathogenesis and the opportunity to leverage these changes for early detection.
Neurodegenerative Disorders
Lipid biomarkers show particular promise for neurodegenerative conditions where early diagnosis remains challenging. A 2025 study on Alzheimer's disease (AD) incorporated both plasma and red blood cell membrane (RCM) lipids to identify diagnostic signatures [21]. The investigation revealed that RCM lipids provided superior separation between normal subjects, those with amnestic mild cognitive impairment, and AD patients compared to plasma lipids alone [21]. This advantage stems from the ability of RCM lipids to reflect chronic physiological alterations rather than acute fluctuations, providing a more stable biomarker platform for progressive conditions [21].
The study identified 138 differentially expressed lipids enriched in AD-related pathways, with six lipids selected as a potential biomarker panel based on multi-dimensional criteria [21]. The incorporation of RCM lipids enhanced diagnostic performance and highlighted the value of exploring alternative blood compartments beyond plasma for biomarker discovery. This approach addresses the critical need for non-invasive alternatives to cerebrospinal fluid analysis and PET imaging, which are invasive, costly, and limited in clinical utility, especially in early disease stages [21].
Metabolic and Cardiovascular Diseases
Lipidomics has naturally found extensive application in metabolic and cardiovascular disorders where lipid metabolism plays a central pathophysiological role. Specific ceramides and phosphatidylcholines have been associated with cardiovascular risk, enabling improved risk stratification beyond conventional lipid panels [4]. Similarly, in metabolic syndrome and diabetes, distinct lipid signatures reflect underlying insulin resistance and metabolic dysregulation, offering potential for early detection and monitoring of intervention responses [23].
The FDA-approved Tina-quant immunoassay for apolipoprotein A-I and B represents a successful example of lipid-related biomarker translation, demonstrating the clinical acceptance of lipid-based assessments when supported by robust validation [4]. This precedent establishes a pathway for more comprehensive lipid panels to eventually enter clinical practice as evidence accumulates.
Table 3: Performance of Lipid Biomarkers in Various Disease Applications
| Disease Area | Biomarker Panel | Performance (AUC) | Sample Type | Study Cohort |
|---|---|---|---|---|
| Bladder Cancer (NMIBC) | PE(14:1/20:0), PE(18:2/16:0), 19-methyl-heneicosanoic acid | 0.88 (training), 0.82 (validation) | Plasma | 106 patients, 108 controls [20] |
| Bladder Cancer (Grading) | Four-lipid panel | 0.815 (10-fold CV: 0.77) | Plasma | 106 NMIBC patients [20] |
| Alzheimer's Disease | Six-lipid panel (from 138 differential lipids) | Superior separation with RCM vs plasma | RBC Membrane & Plasma | 156 individuals [21] |
| Cardiovascular Risk | Ceramides, phosphatidylcholines | Improved risk stratification | Plasma | Multiple cohorts [4] |
Multi-Center Validation: Critical Considerations and Challenges
Standardization and Reproducibility
The transition of lipid biomarkers from research discoveries to clinically useful tools faces several substantial challenges. Reproducibility across platforms and laboratories remains a significant hurdle, with studies reporting alarmingly low agreement rates (14-36%) between different lipidomics platforms when analyzing identical samples [4]. This variability stems from differences in sample preparation protocols, chromatographic separation conditions, mass spectrometry instrumentation, and data processing algorithms [4].
Addressing these challenges requires rigorous standardization of pre-analytical factors including fasting status, time of collection, processing delays, and storage conditions [20] [21]. The implementation of standard reference materials and internal standardization strategies using stable isotope-labeled lipids is essential for quantitative accuracy and cross-laboratory comparability [4]. Additionally, the field would benefit from harmonized data reporting standards that encompass lipid nomenclature, quantification units, and quality control metrics.
Biological and Analytical Complexities
The extraordinary structural diversity of lipids presents both an opportunity and a challenge for biomarker development. While this diversity enables precise disease mapping, it complicates comprehensive analysis and interpretation. The dynamic range of lipid concentrations in biological samples exceeds the detection capabilities of any single analytical platform, necessitating strategic trade-offs between coverage and sensitivity [4].
Biological variability introduced by factors such as diet, circadian rhythms, medications, and gut microbiota further complicates biomarker validation [4]. These confounding influences must be carefully controlled through study design and statistical adjustment to distinguish disease-specific signatures from background noise. The implementation of fasting blood collection in the morning hours, as practiced in multiple cited studies, helps mitigate some sources of variability [20] [21].
Diagram 2: Key challenges in the validation pathway for lipid biomarkers.
Emerging Technologies and Approaches
The future of lipid biomarker development is intrinsically linked to technological advancements and integrated analytical frameworks. Artificial intelligence and machine learning are playing an increasingly important role in deciphering complex lipidomic data, with models such as MS2Lipid demonstrating up to 97.4% accuracy in predicting lipid subclasses [4]. These computational approaches enable more efficient pattern recognition and feature selection from high-dimensional datasets.
The integration of lipidomics with other omics technologies (genomics, transcriptomics, proteomics) provides a systems biology perspective that enhances biomarker specificity and mechanistic understanding [3] [6]. This multi-omics approach is particularly valuable for addressing the context-dependent nature of lipid alterations and establishing causal relationships between lipid changes and disease processes [4]. Additionally, the exploration of alternative blood compartments such as red blood cell membranes extends the diagnostic potential beyond conventional plasma analysis [21].
Pathway to Clinical Translation
The successful translation of lipid biomarkers into clinical practice requires a coordinated multidisciplinary effort addressing technical, clinical, and regulatory considerations. Large-scale multi-center studies with standardized protocols are essential to establish robust reference ranges and validate performance across diverse populations [4] [24]. The prioritization of biomarkers with clear pathophysiological relevance over those with merely statistical associations will enhance clinical adoption and utility [22].
The evolving regulatory framework for biomarker qualification necessitates early engagement with regulatory agencies to align validation strategies with clinical requirements [22]. The demonstrated success of selected lipid-based tests, such as the FDA-approved apolipoprotein assays, provides a template for this translation process [4]. As evidence accumulates, lipid biomarkers are anticipated to emerge as central elements in personalized medicine, enabling early detection, risk stratification, and targeted therapeutic interventions across a spectrum of human diseases [23].
In conclusion, lipids represent ideal candidates for non-invasive blood-based biomarkers due to their physiological relevance, metabolic responsiveness, and detectable alterations in accessible biofluids. While significant challenges remain in standardization and validation, the strategic integration of advanced analytical platforms, computational methods, and multi-center collaborative research provides a clear pathway for clinical translation. As the field matures, lipid biomarkers are poised to make substantial contributions to precision medicine, fundamentally enhancing our approach to disease detection, monitoring, and management.
Methodological Workflow: Integrating Lipidomics and Machine Learning for Biomarker Discovery
Strategic Application of Untargeted vs. Targeted Lipidomics in Discovery and Validation Phases
Lipidomics, the large-scale study of pathways and networks of cellular lipids, has emerged as a crucial discipline for understanding cellular processes, disease mechanisms, and identifying potential therapeutic targets [25] [26]. In the context of multi-center validation studies for lipidomic biomarkers, the strategic selection of analytical approaches is paramount to generating reliable, reproducible, and clinically relevant data. The lipidome comprises thousands of molecular species with diverse chemical structures and functions, broadly classified by the LIPID MAPS consortium into eight categories: fatty acyls, glycerolipids, glycerophospholipids, sphingolipids, sterol lipids, prenol lipids, saccharolipids, and polyketides [25] [27]. These molecules regulate critical biological processes including cellular structure, energy storage, signaling, inflammation, and metabolic homeostasis [23] [27].
Two principal methodologies—untargeted and targeted lipidomics—have evolved with complementary strengths and applications in biomarker research. Untargeted lipidomics provides a comprehensive, unbiased analysis of the entire lipid profile, while targeted lipidomics focuses on precise quantification of predefined lipid species [26] [27]. This guide objectively compares these approaches within the framework of discovery and validation research phases, providing experimental data, methodological protocols, and practical considerations for their strategic application in multi-center studies aimed at clinical translation.
Fundamental Methodological Comparisons
Core Analytical Philosophies and Workflows
The fundamental distinction between untargeted and targeted lipidomics lies in their analytical philosophies. Untargeted lipidomics is a discovery-oriented approach that aims to detect and relatively quantify as many lipid species as possible without prior selection, enabling hypothesis generation and novel biomarker discovery [26]. In contrast, targeted lipidomics is a hypothesis-driven approach that focuses on precise identification and absolute quantification of specific, predefined lipids, typically employing internal standards for accurate measurement [26] [28]. This methodological divergence creates complementary applications: untargeted methods excel in comprehensive profiling and novel discoveries, while targeted approaches provide rigorous validation and precise quantification essential for clinical application [27].
The workflow for both approaches begins with careful sample preparation but diverges significantly in data acquisition and analysis. Untargeted workflows typically involve liquid chromatography (LC) separation followed by high-resolution mass spectrometry (HRMS) detection, generating complex datasets requiring sophisticated bioinformatics processing [29]. Targeted methods often employ differential mobility spectrometry (DMS) or LC separation coupled with multiple reaction monitoring (MRM) on triple quadrupole instruments, producing more focused datasets with streamlined analysis [30] [27].
Technical Specifications and Performance Metrics
Table 1: Technical Comparison of Untargeted and Targeted Lipidomics Approaches
| Parameter | Untargeted Lipidomics | Targeted Lipidomics |
|---|---|---|
| Analytical Philosophy | Discovery-oriented, unbiased | Hypothesis-driven, focused |
| Primary Instrumentation | LC-HRMS (Q-TOF, Orbitrap) | LC-MS/MS (QQQ), Lipidyzer platform |
| Quantification Approach | Relative quantification (peak areas) | Absolute quantification with internal standards |
| Lipid Identification | Based on m/z, retention time, fragmentation spectra | Predefined transitions with authentic standards |
| Typical Coverage | Hundreds to thousands of features | Dozens to hundreds of predefined lipids |
| Data Complexity | High, requiring advanced bioinformatics | Moderate, with more straightforward processing |
| Ideal Application Phase | Discovery, hypothesis generation | Validation, clinical application |
| Throughput | Moderate (longer chromatographic separations) | High (streamlined methods) |
Table 2: Experimental Performance Metrics from Comparative Studies
| Performance Metric | Untargeted LC-MS | Targeted Lipidyzer Platform |
|---|---|---|
| Median Intra-day Precision (CV%) | 3.1% | 4.7% |
| Median Inter-day Precision (CV%) | 10.6% | 5.0% |
| Technical Repeatability (Median CV%) | 6.9% | 4.7% |
| Median Accuracy | 6.9% | 13.0% |
| Correlation Between Platforms (Median r) | 0.71 (for commonly detected lipids) | 0.71 (for commonly detected lipids) |
| Lipid Coverage in Mouse Plasma | 337 lipids across 11 classes | 342 lipids across 11 classes |
| Overlap Between Platforms | 196 lipid species (35% of untargeted detections) | 196 lipid species (57% of targeted detections) |
Data derived from a cross-platform comparison study analyzing aging mouse plasma [30]. The correlation was calculated for lipids detected by both platforms using endogenous plasma lipids in the context of aging.
Experimental Designs and Protocol Specifications
Untargeted Lipidomics Workflow
Untargeted lipidomics employs a comprehensive analytical workflow designed to capture the broadest possible lipid profile:
Sample Preparation Protocol:
- Extraction: Lipids are extracted from biological matrices (plasma, tissue, cells) using organic solvents such as chloroform-methanol or methyl tert-butyl ether (MTBE) mixtures [26] [29]. The classic Folch or Bligh-Dyer methods are commonly employed.
- Internal Standards: Addition of isotope-labeled internal standards early in the process enables quality control and normalization. Standards should cover multiple lipid classes to account for extraction efficiency variations [29].
- Quality Controls: Preparation of pooled quality control (QC) samples from all study samples is essential for monitoring instrument performance and data quality throughout the analysis [29].
Chromatographic Separation:
- Reversed-Phase LC: Most commonly used for separating lipid species by hydrophobicity, effectively resolving molecular species within the same lipid class [30] [29].
- HILIC Chromatography: Useful for separating lipids by class based on polar head groups.
- Optimized Conditions: Typical UHPLC methods use C8 or C18 columns with acetonitrile-water mobile phases containing ammonium formate/acetate modifiers, with run times of 15-30 minutes per sample [29].
Mass Spectrometry Analysis:
- High-Resolution MS: Quadrupole time-of-flight (Q-TOF) or Orbitrap mass analyzers provide accurate mass measurements (<5 ppm mass error) for confident lipid identification [27] [29].
- Data Acquisition: Both data-dependent acquisition (DDA) and data-independent acquisition (DIA) modes are employed, with DDA providing MS/MS spectra for structural elucidation of the most abundant ions, while DIA fragments all ions simultaneously [27].
- Ionization Modes: Both positive and negative electrospray ionization modes are typically required to cover the diverse lipidome [29].
Data Processing and Lipid Identification:
- Peak Detection and Alignment: Software tools (e.g., XCMS, MS-DIAL) detect chromatographic peaks and align them across samples [29].
- Lipid Identification: Matching of accurate mass, isotopic patterns, retention time, and MS/MS fragmentation spectra against lipid databases (LIPID MAPS, HMDB) [26].
- Statistical Analysis: Multivariate statistics (PCA, PLS-DA) and univariate analyses identify lipids differentially abundant between experimental groups [29].
Targeted Lipidomics Workflow
Targeted lipidomics employs a focused approach designed for precise quantification:
Sample Preparation Protocol:
- Extraction: Similar extraction protocols as untargeted approaches, but with emphasis on reproducibility and recovery of specific lipid classes of interest [30].
- Comprehensive Internal Standardization: Addition of multiple stable isotope-labeled internal standards (typically 50+ IS covering all targeted lipid classes) to correct for extraction efficiency, matrix effects, and instrument variability [30] [28].
- Calibration Curves: For absolute quantification, calibration curves with authentic standards are prepared in matching matrix [28].
Lipid Separation and Detection:
- DMS and LC Separation: Some targeted platforms (e.g., Lipidyzer) incorporate differential mobility spectrometry (DMS) for lipid class separation, while others use conventional LC separation [30].
- Mass Spectrometry Analysis: Triple quadrupole instruments operating in multiple reaction monitoring (MRM) mode provide high sensitivity and specificity for predefined lipid transitions [30] [27].
- Optimized Transitions: Pre-optimized precursor-product ion transitions for each targeted lipid ensure selective detection and accurate quantification [30].
Data Processing and Quantification:
- Peak Integration: Automated integration of chromatographic peaks for each targeted transition.
- Concentration Calculation: Lipid concentrations calculated based on internal standard response and calibration curves, reported in absolute quantities (nmol/g or μM) [30] [28].
- Quality Assessment: Monitoring of internal standard response and QC sample performance throughout the analysis.
Comparative Performance in Biomarker Research
Lipid Coverage and Complementarity
Direct comparative studies reveal both overlapping and complementary coverage between untargeted and targeted approaches. In a cross-platform comparison using aging mouse plasma, untargeted LC-MS detected 337 lipids across 11 classes, while the targeted Lipidyzer platform detected 342 lipids across similar classes [30]. However, the overlap was only 196 lipid species, representing just 35% of untargeted detections and 57% of targeted detections, highlighting their complementary nature [30].
Each approach offers distinct advantages for specific lipid classes. Untargeted methods better capture ether-linked phospholipids (plasmalogens) and phosphatidylinositols, while targeted approaches excel at detecting free fatty acids and cholesterol esters [30]. For triacylglycerol (TAG) speciation, untargeted LC-MS provides identification of all three fatty acyl chains (e.g., TAG(16:0/18:1/18:2)), while targeted platforms typically report total carbon number and unsaturation (e.g., TAG52:3-FA16:0) [30].
The combined application of both approaches significantly expands lipid coverage, with one study demonstrating 700 unique lipid molecular species detected in mouse plasma when integrating data from both platforms [30]. This complementarity is particularly valuable in discovery phases where comprehensive lipidome assessment is critical.
Quantitative Performance and Reproducibility
Quantitative performance metrics demonstrate specific strengths for each approach. Targeted lipidomics generally shows superior precision, with median inter-day CV of 5.0% compared to 10.6% for untargeted methods, and technical repeatability of 4.7% versus 6.9% [30]. This enhanced precision makes targeted approaches particularly valuable for longitudinal studies and clinical applications where detecting subtle changes is essential.
Untargeted methods demonstrated slightly better accuracy (6.9% vs 13.0%) in spiked recovery experiments, though targeted accuracy improved to comparable levels when excluding the highest concentration samples where signal plateau was observed [30]. This highlights the importance of maintaining calibration within linear dynamic ranges for targeted quantification.
Correlation between platforms is strong for quantitative measurements, with a median correlation coefficient of 0.71 for endogenous plasma lipids in aging studies [30]. This supports the practice of using untargeted discovery followed by targeted validation, as quantitative patterns are generally conserved between platforms.
Strategic Implementation in Multi-Center Studies
Integrated Workflow for Biomarker Discovery and Validation
The most effective application of lipidomics in multi-center biomarker research involves a phased approach that strategically employs both untargeted and targeted methodologies. This integrated workflow leverages the complementary strengths of each platform while mitigating their respective limitations.
Phase 1: Discovery Using Untargeted Lipidomics Initial discovery phases should employ untargeted lipidomics to generate comprehensive lipid profiles across appropriately powered sample sets. This enables identification of potentially novel lipid biomarkers without pre-existing biases [26]. The output is a set of candidate biomarkers showing significant association with the disease state or intervention.
Phase 2: Verification Using Targeted Methods Promising candidates from discovery are transitioned to targeted quantification methods for verification in expanded sample sets. This phase confirms the analytical robustness of the measurements and refines effect size estimates [27].
Phase 3: Multi-Center Validation Fully validated targeted assays are deployed across multiple centers for large-scale validation studies. The standardized nature of targeted methods ensures consistency across sites and enables pooling of data for definitive evaluation of clinical utility [25].
Addressing Multi-Center Reproducibility Challenges
Reproducibility across different laboratories and platforms remains a significant challenge in lipidomic biomarker research. Studies have reported agreement rates as low as 14-36% between different lipidomics platforms analyzing identical samples [25]. This variability stems from differences in sample preparation, chromatographic separation, mass spectrometry instrumentation, and data processing algorithms.
Standardization Strategies for Multi-Center Studies:
- Reference Materials: Implementation of standardized reference materials and quality control samples across all participating centers.
- Harmonized Protocols: Development and adherence to detailed standard operating procedures for sample collection, storage, preparation, and analysis.
- Cross-Platform Calibration: Use of standardized lipid mixtures to calibrate responses across different instrument platforms.
- Data Harmonization: Statistical adjustment for center-specific effects and batch-to-batch variability.
Targeted lipidomics generally demonstrates better inter-laboratory reproducibility due to standardized assays and internal standardization, making it preferable for the final validation phases of multi-center studies [25]. However, the discovery phase benefits from the broader coverage of untargeted approaches, even with their greater variability between platforms.
Essential Research Tools and Reagents
Table 3: Essential Research Reagent Solutions for Lipidomics Studies
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Internal Standards | Deuterated lipid standards (e.g., d7-Cholesterol, d5-TAG, d9-PC) | Quantification normalization, recovery correction, and quality control |
| Extraction Solvents | Chloroform, methanol, MTBE, isopropanol | Lipid extraction from biological matrices with protein precipitation |
| LC Mobile Phases | Acetonitrile, water, isopropanol with ammonium formate/acetate | Chromatographic separation of lipid classes and molecular species |
| Quality Control Materials | Pooled plasma, NIST SRM 1950, in-house QC pools | Monitoring instrument performance and inter-batch reproducibility |
| Calibration Standards | Authentic lipid standards with certified concentrations | Construction of calibration curves for absolute quantification |
| Sample Preparation Kits | Commercial lipid extraction kits (e.g., Matyash method) | Standardized extraction protocols for improved reproducibility |
The strategic selection between untargeted and targeted lipidomics approaches depends on the specific research objectives, study phase, and required data quality. Untargeted lipidomics provides the discovery power and comprehensive coverage essential for initial biomarker identification, while targeted approaches deliver the precision, accuracy, and reproducibility required for clinical validation and translation.
In the context of multi-center biomarker studies, an integrated approach that leverages both methodologies in sequential phases offers the most robust pathway from initial discovery to clinical application. This strategy maximizes both biomarker coverage and analytical reliability, addressing the critical need for reproducible, clinically relevant lipid biomarkers across diverse populations and settings.
As lipidomics technologies continue to evolve, emerging approaches such as pseudo-targeted lipidomics [27] and enhanced computational methods [25] promise to further bridge the gap between comprehensive coverage and precise quantification, potentially offering improved solutions for the unique challenges of multi-center biomarker research.
The following table provides a high-level comparison of the core technical and performance characteristics of UHPSFC/MS, LC-MS/MS, and direct infusion (shotgun) mass spectrometry platforms for lipidomic profiling.
Table 1: Core Platform Comparison for Lipidomic Profiling
| Feature | UHPSFC/MS | LC-MS/MS (Reversed-Phase) | Shotgun MS (Direct Infusion) |
|---|---|---|---|
| Primary Separation Mechanism | Lipid class (by polarity of headgroup) [31] | Lipid species (by fatty acyl chain properties) [31] | No chromatographic separation |
| Analysis Speed | Very High (~8 min for 33 lipid classes) [32] | Moderate to High (method-dependent) | Highest |
| Chromatographic Resolution | High for lipid classes; capable of resolving isomers [33] [31] | High for lipid species within a class; resolves chain-length isomers [31] | Not Applicable |
| Ionization Efficiency | Enhanced sensitivity for some sterols; APCI common [33] [32] | Robust ESI for most lipids; well-characterized [34] | Prone to severe ion suppression from complex matrices |
| Key Strength | High-throughput, orthogonal selectivity for classes, green technology [32] | Gold standard for species-level quantification, robust workflows | Ultimate throughput for high-sample number screening |
| Major Limitation | Mobile phase compatibility in 2D setups, newer technique [31] | Longer run times for comprehensive analysis, higher solvent consumption | Inability to separate isomers, high matrix effects, less confident IDs |
| Ideal for Multi-Center Studies | Rapid screening of large cohorts, validating class-level biomarkers [32] | Targeted, absolute quantification of predefined biomarker panels | Discovery-phase fingerprinting across very large population cohorts |
Ultra-High Performance Supercritical Fluid Chromatography/Mass Spectrometry (UHPSFC/MS)
UHPSFC/MS utilizes supercritical carbon dioxide (CO₂) as the primary mobile phase component, mixed with organic modifiers like methanol or ethanol. The supercritical fluid possesses low viscosity and high diffusivity, enabling faster separations and higher efficiency compared to traditional liquid chromatography [32]. This technique provides orthogonal separation selectivity, primarily based on the polarity of lipid headgroups, effectively separating lipid classes from each other [31]. This makes it a powerful complementary technique to reversed-phase LC-MS.
Performance and Experimental Data
Recent methodological advances demonstrate the robust performance of UHPSFC/MS for comprehensive lipidomics.
Table 2: Representative UHPSFC/MS Method Performance
| Metric | Performance Data | Experimental Context |
|---|---|---|
| Analysis Throughput | 8 minutes per sample (including equilibration) [32] | Analysis of 33 lipid classes from human plasma |
| Lipid Coverage | 298 lipid species from 16 subclasses [31] | Four-dimensional analysis of human plasma lipid extract |
| Separation Power | Resolution of 15 stereoisomers and 17 positional steroids [33] | Targeted steroid analysis in a complex panel of 36 steroids |
| Peak Capacity | 10x and 18x higher than 1D RP-UHPLC and 1D UHPSFC, respectively [31] | Online comprehensive RP-UHPLC × UHPSFC/MS/MS system |
Detailed Experimental Protocol
Protocol: Fast Lipidomic Profiling of Human Plasma using UHPSFC/Q-TOF MS [32]
Sample Preparation:
- Perform protein precipitation by mixing 25 μL of plasma with 2 mL of chloroform and 1 mL of methanol.
- Ultrasonicate the mixture for 15 minutes at ambient temperature.
- Add 600 μL of 250 mM ammonium carbonate buffer, followed by another 15-minute ultrasonication and centrifugation for 3 minutes at 886×g.
- Collect the organic phase, evaporate it under a gentle stream of nitrogen, and reconstitute the residue in 50 μL of chloroform/methanol (1:1, v/v) [31].
Chromatography Conditions:
- Column: Diol or HILIC column (e.g., 150 x 3.0 mm, 1.7 μm).
- Mobile Phase A: CO₂ (4.5 grade, 99.995% purity).
- Mobile Phase B: Methanol or ethanol with a modifier such as 10 mM ammonium acetate.
- Gradient: Linear gradient from 1% to 30-40% B over 5-6 minutes.
- Flow Rate: 1.0 - 1.5 mL/min.
- Column Temperature: 35-55 °C.
- Back Pressure Regulator (BPR): 120-150 bar.
Mass Spectrometry Detection:
- Platform: Q-TOF or Triple Quadrupole (QQQ) mass spectrometer.
- Ionization: ESI or APCI in positive and negative polarity switching mode.
- Acquisition Mode: Full-scan MS (for untargeted) or Multiple Reaction Monitoring (MRM) for targeted quantification.
The workflow for this protocol is standardized and efficient.
Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS)
LC-MS/MS is the cornerstone technique for lipidomic analysis, leveraging the high sensitivity and specificity of tandem mass spectrometry coupled with robust chromatographic separations. Reversed-phase (RP) LC, typically using C18 columns, separates lipids based on their hydrophobicity, which is determined by the length and unsaturation of their fatty acyl chains. This provides excellent resolution of individual lipid species within a class [31] [35]. The technique is highly versatile, supporting both untargeted discovery and highly precise targeted quantification.
Performance and Experimental Data
LC-MS/MS is renowned for its quantitative rigor and deep coverage of the lipidome.
Table 3: Representative LC-MS/MS Method Performance
| Metric | Performance Data | Experimental Context |
|---|---|---|
| Quantification Mode | High sensitivity and stability for low-abundance analytes [27] | Targeted lipidomics using UPLC-QQQ MS |
| Identification Confidence | MS³ capability improves identification of toxic natural products in serum [36] | LC-HR-MS³ with a spectral library of 85 natural products |
| Application Versatility | Widespread use in pharmaceutical analysis, food safety, and clinical diagnostics [34] | Broad applicability across scientific disciplines |
Detailed Experimental Protocol
Protocol: Targeted Lipid Quantification using UPLC-QQQ MS [27]
Sample Preparation:
- Select an appropriate extraction method (e.g., Folch, Bligh & Dyer, or MTBE) based on the sample matrix and lipid classes of interest [35].
- Add internal standards (ideally stable isotope-labeled) for each lipid class to ensure accurate quantification.
Chromatography Conditions (Reversed-Phase):
- Column: C18 column (e.g., 150 mm long, sub-2 μm particles).
- Mobile Phase A: Water with 10 mM ammonium acetate or formate.
- Mobile Phase B: Acetonitrile/2-propanol or methanol.
- Gradient: Steep gradient from high aqueous to high organic phase over 10-20 minutes.
- Flow Rate: 0.2 - 0.4 mL/min.
- Column Temperature: 45-60 °C.
Mass Spectrometry Detection:
- Platform: Triple Quadrupole (QQQ) mass spectrometer.
- Ionization: ESI in positive or negative mode.
- Acquisition Mode: Multiple Reaction Monitoring (MRM). Precursor and characteristic product ions are defined for each target lipid.
- Data Analysis: Quantify lipids based on the peak area ratio of the analyte to its corresponding internal standard.
Shotgun Lipidomics (Direct Infusion MS)
Shotgun lipidomics involves the direct infusion of a crude lipid extract into the mass spectrometer without prior chromatographic separation. This approach relies entirely on the mass spectrometer to resolve lipids based on their mass-to-charge ratio (m/z) and fragmentation patterns. Its primary advantage is extremely high throughput, making it suitable for screening large sample cohorts in population-scale studies [32]. However, the lack of separation makes it highly susceptible to ion suppression from co-eluting matrix components and limits its ability to resolve isomeric lipids [35].
Performance and Experimental Data
While less chromatographically sophisticated, shotgun approaches are powerful for specific applications.
Table 4: Considerations for Shotgun Lipidomics
| Aspect | Consideration & Impact |
|---|---|
| Throughput | Maximum speed, enabling analysis of hundreds to thousands of samples [32]. |
| Ion Suppression | High, due to simultaneous introduction of all lipids and matrix components, affecting quantification accuracy [35]. |
| Isomer Separation | Not possible without prior separation, leading to potential misidentification [35]. |
| Data Complexity | High, requires high-resolution mass spectrometers and advanced software for deconvolution. |
Detailed Experimental Protocol
Protocol: High-Throughput Screening via Direct Infusion-Orbitrap MS
Sample Preparation:
- Use a simple one-phase extraction (e.g., BuOH:MeOH) or protein precipitation with a solvent compatible with direct infusion (e.g., isopropanol) [35].
- Include a cocktail of internal standards.
Mass Spectrometry Analysis:
- Platform: High-resolution mass spectrometer (e.g., Orbitrap, FT-ICR, or Q-TOF).
- Ionization: ESI in positive and negative polarity modes (separate runs).
- Infusion: Directly infuse the sample extract at a low, constant flow rate (e.g., 5-10 μL/min) using a syringe pump.
- Acquisition: Full-scan MS at high resolution (e.g., R > 140,000 @ m/z 200) and data-dependent MS/MS (dd-MS²) or data-independent acquisition (DIA) to obtain structural information.
The choice of platform strategy should be guided by the specific goals of the research phase.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 5: Key Reagents and Materials for Lipidomics Workflows
| Item | Function & Application | Critical Notes |
|---|---|---|
| CO₂ (4.5 Grade) | Primary mobile phase for UHPSFC. | Requires high purity (99.995%) to ensure baseline stability and avoid ion source contamination [32]. |
| Stable Isotope-Labeled Internal Standards (SIL-IS) | Normalization for extraction efficiency and MS ionization variability; essential for absolute quantification. | Should be added as early as possible in sample preparation; ideally one per lipid class analyzed [33]. |
| Chloroform, Methanol, Methyl-tert-butyl ether (MTBE) | Solvents for liquid-liquid extraction (e.g., Folch, Bligh & Dyer, MTBE methods). | Chloroform is highly effective but toxic; MTBE offers a less toxic alternative [35]. |
| Ammonium Acetate/Formate | Mobile phase additive to promote [M+NH₄]⁺ or [M+HCOO]⁻ adduct formation in ESI, improving ionization efficiency and consistency. | Commonly used in concentrations of 5-20 mM [32]. |
| Butanol:MeOH Mixtures | For one-phase extraction (OPE) protocols like BUME; efficient for less polar lipids and high-throughput workflows [35]. | Simplifies sample preparation by eliminating phase separation steps. |
| Antioxidants (e.g., BHT) | Added to solvents to prevent oxidation of unsaturated lipids during extraction and storage, preserving sample integrity [35]. | Crucial for samples rich in polyunsaturated fatty acids (PUFAs). |
In the rapidly evolving field of biomedical research, lipidomics has emerged as a powerful tool for discovering biomarkers associated with various diseases. Lipids, comprising thousands of chemically distinct molecules involved in vital cellular processes, offer a rich source of potential diagnostic and prognostic indicators [25]. The transition of lipid research from bench to bedside relies heavily on the discovery of clinically reliable biomarkers that can be validated across diverse populations [25]. However, the high-dimensional nature of lipidomic data, characterized by numerous lipid species with complex interactions, presents significant analytical challenges. This is where machine learning (ML) approaches become indispensable, enabling researchers to extract meaningful patterns from complex lipidomic datasets and identify robust biomarkers with clinical utility.
The integration of machine learning in lipidomics is particularly crucial within the context of multi-center validation studies, which are essential for establishing biomarker reliability but introduce additional variability. Ensemble methods for feature selection and classification algorithms including Naïve Bayes (NB), Random Forest (RF), and Support Vector Machines (SVM) have demonstrated significant potential in addressing these challenges. This guide provides a comprehensive comparison of these ML techniques, supported by experimental data and detailed protocols from recent lipidomics research, to inform their application in robust, multi-center biomarker discovery pipelines.
Lipidomics in Biomarker Research: A Primer
Lipidomics, a subfield of metabolomics, involves the comprehensive study of lipid molecular species and their biological functions within biological systems [25]. The lipidome encompasses tremendous structural diversity, with lipids classified into eight major categories: fatty acyls (FA), glycerolipids (GL), glycerophospholipids (GP), sphingolipids (SP), sterol lipids (ST), prenol lipids (PR), saccharolipids (SL), and polyketides (PK) [25] [27]. These molecules play crucial roles in cellular structure, energy storage, and signaling processes, making them prime candidates for biomarker discovery.
The application of lipidomics in clinical biomarker research has gained substantial traction across various disease domains, including cancer, cardiovascular disorders, diabetes, and neurodegenerative conditions [25]. Specific ceramides and phosphatidylcholines have been associated with cardiovascular risk, while alterations in sphingolipids and glycerophospholipids are being investigated in contexts of multiple sclerosis and cancer [25]. In gynecological cancers, for instance, lipid metabolism is reprogrammed to support the energy demands of rapidly proliferating cancer cells [27].
Despite its promise, lipidomic biomarker discovery faces several challenges, including biological variability, lipid structural diversity, inconsistent sample processing, and a lack of standardized procedures [25]. These factors complicate reproducibility and clinical validation, necessitating sophisticated computational approaches like machine learning to identify robust signatures amidst complex data.
Machine Learning Methodologies for Lipidomics
Experimental Workflows in Lipidomics
The typical workflow for ML-driven lipidomic biomarker discovery integrates laboratory processes with computational analysis, progressing from sample collection through to model validation. Key stages include sample preparation, lipid extraction, chromatographic separation, mass spectrometric analysis, data preprocessing, feature selection, model building, and validation [27] [37].
Feature Selection with Ensemble Methods
Feature selection is a critical step in lipidomic analysis, addressing the "curse of dimensionality" by identifying the most informative lipid species while reducing noise and computational complexity. Ensemble feature selection methods combine multiple selection techniques to produce more robust and stable feature sets, which is particularly valuable for multi-center studies where technical and biological variability can impact results.
Several ensemble-compatible feature selection methods have been successfully applied in lipidomics research:
- Entropy-based methods: These approaches evaluate the information gain provided by each feature, effectively capturing non-linear relationships in lipidomic data [37].
- Boruta algorithm: A wrapper method that compares the importance of original features with shadow features created by random permutation, selecting all features that perform significantly better than their randomized counterparts [6] [37].
- Recursive Feature Elimination (RFE): Iteratively constructs models and eliminates the least important features, often used with SVM (SVM-RFE) [6].
- LASSO Regression: Applies L1 regularization to shrink coefficients of less important features to zero, performing simultaneous feature selection and regularization [6].
Table 1: Performance Comparison of Feature Selection Methods in Lipidomic Studies
| Method | Application Context | Key Strengths | Performance Metrics |
|---|---|---|---|
| Entropy-based | Breast cancer tissue classification [37] | Captures non-linear relationships | Accuracy: 0.9843, AUC: 0.97 (cancerous vs normal) |
| Boruta | Osteonecrosis biomarker discovery [6] | Robust against overfitting | Identified 3 stable biomarkers (CREBBP, GLB1, PSAP) |
| SVM-RFE | Nonsyndromic cleft lip with palate [38] | Effective for high-dimensional data | Selected 35 candidate lipid biomarkers |
| LASSO | Osteonecrosis of femoral head [6] | Performs feature selection and regularization | Identified key genes in lipid metabolism processes |
Model Building with NB, RF, and SVM
Algorithm Comparison and Performance
Once informative lipid features are selected, classification algorithms build predictive models for disease diagnosis or stratification. Naïve Bayes, Random Forest, and Support Vector Machines each have distinct characteristics that make them suitable for different aspects of lipidomic analysis.
Support Vector Machines (SVM) seek to find an optimal hyperplane that separates classes with maximum margin in high-dimensional space. They are particularly effective for lipidomic data due to their strength with high-dimensional datasets and ability to model complex boundaries using kernel functions [39] [40] [37].
Random Forest is an ensemble method that constructs multiple decision trees and aggregates their predictions, reducing overfitting and providing native feature importance measures [41] [37]. This makes RF valuable for both prediction and biomarker interpretation.
Naïve Bayes operates on the principle of conditional probability with the "naïve" assumption of feature independence. While this assumption is often violated in lipidomic data due to metabolic correlations, NB remains competitive in many applications, particularly with appropriate feature preprocessing [39] [40].
Table 2: Comparative Performance of Classification Algorithms in Lipidomic Studies
| Algorithm | Application | Advantages | Limitations | Reported Performance |
|---|---|---|---|---|
| SVM | Breast cancer tissue classification [37] | Effective in high dimensions; Robust to overfitting | Memory intensive; Requires careful parameter tuning | SVM-Polynomial: Superior performance in LC-MS positive/negative modes |
| Random Forest | Fatigue life prediction [41] | Native feature importance; Handles non-linear relationships | Can be biased toward features with more categories | Ensemble neural networks outperformed RF in predictive accuracy |
| Naïve Bayes | Sentiment analysis [39] [40] | Computational efficiency; Works well with small samples | Strong feature independence assumption | Accuracy: 80% (vs. SVM: 87%) in sentiment classification |
Integrated ML Approaches in Practice
Recent studies demonstrate the power of integrating multiple ML approaches in lipidomic biomarker discovery. A study on nonsyndromic cleft lip with palate (nsCLP) employed eight feature selection methods combined with seven classification models, identifying a panel of 35 candidate lipid biomarkers that was subsequently refined to a 3-lipid diagnostic signature [38]. This multi-algorithm approach enhances the robustness of discovered biomarkers, which is crucial for multi-center validation.
In breast cancer research, a comprehensive methodology combining biological feature selection (median log2 Fold Change) with algorithmic approaches (Boruta, MLP, Entropy-based, VIP) identified distinctive lipid signatures, including elevated saturated and monounsaturated phospholipids in cancerous tissues [37]. The integration of univariate and multivariate analyses with machine learning classification created a more reliable biomarker discovery pipeline.
Experimental Protocols for Multi-Center Lipidomic Studies
Standardized Lipidomics Workflow
To ensure reproducibility across multiple centers, standardized experimental protocols are essential:
Sample Preparation Protocol:
- Collect biological samples (plasma, serum, tissue) using standardized collection tubes and procedures
- Implement immediate freezing at -80°C after collection
- Use identical protein precipitation or lipid extraction methods across centers (e.g., Folch, Bligh-Dyer, or MTBE-based extraction)
- Add internal standards at the beginning of extraction to account for technical variability
LC-MS Analysis:
- Employ uniform liquid chromatography conditions (column chemistry, gradient profiles, flow rates)
- Standardize mass spectrometry parameters (ionization modes, resolution, mass accuracy)
- Implement quality control pools from representative sample mixtures for instrument monitoring
- Use reference standards for lipid identification and quantification
Data Preprocessing:
- Apply consistent peak picking, alignment, and normalization algorithms
- Implement blank subtraction to remove background interference
- Use quality assurance metrics to identify and address batch effects
- Apply missing value imputation methods appropriate for lipidomic data
Machine Learning Implementation Protocol
Feature Selection Phase:
- Apply multiple filter methods (variance-based, correlation-based) for initial feature reduction
- Implement ensemble wrapper methods (Boruta, RFE) to identify stable feature subsets
- Use embedded methods (LASSO, Elastic Net) for regularized selection
- Aggregate results from multiple methods to create consensus feature sets
Model Building Phase:
- Split data into training (70%), validation (15%), and test (15%) sets, maintaining class proportions
- Implement stratified k-fold cross-validation (k=5 or 10) to optimize hyperparameters
- Train multiple classifier types (SVM, RF, NB) on identical feature sets
- Apply appropriate class balancing techniques (SMOTE, class weighting) for imbalanced datasets
Validation Phase:
- Evaluate model performance on held-out test sets using multiple metrics (accuracy, precision, recall, F1-score, AUC-ROC)
- Assess generalizability using external validation cohorts from different centers
- Perform permutation testing to establish statistical significance of model performance
- Implement calibration checks to ensure predicted probabilities reflect true likelihoods
Essential Research Reagents and Computational Tools
Successful implementation of ML-driven lipidomic biomarker discovery requires both wet-lab reagents and computational resources.
Table 3: Essential Research Resources for ML-Driven Lipidomic Studies
| Category | Item | Specification/Function | Application Context |
|---|---|---|---|
| Analytical Standards | Internal standards | Stable isotope-labeled lipids for quantification | All lipidomic workflows [25] |
| LC-MS Columns | C18 reverse phase | 1.7-1.8μm particle size, 100mm length | Lipid separation [27] |
| Data Processing | MS-DIAL, Lipostar | Peak detection, alignment, identification | Untargeted lipidomics [25] |
| Programming Languages | R, Python | Statistical analysis, machine learning | All computational phases [38] [6] [37] |
| ML Libraries | scikit-learn, caret | Implementation of NB, RF, SVM algorithms | Model building [38] [37] |
| Feature Selection | Boruta, Glmnet | Dimensionality reduction, feature importance | Preprocessing [6] [37] |
Multi-Center Validation Considerations
The transition from single-center discovery to multi-center validation introduces additional complexity that must be addressed through methodological rigor:
Technical Variability Mitigation:
- Implement standardized protocols across all participating centers
- Use randomized block designs for sample processing and analysis
- Include common reference samples in each batch to monitor and correct for technical variation
- Apply batch correction algorithms (ComBat, EigenMS) before model building
Model Generalizability Assessment:
- Train models on data from some centers and validate on entirely separate centers
- Assess performance consistency across demographic and clinical subgroups
- Evaluate feature stability using metrics like selection frequency in bootstrap samples
- Monitor model drift over time and across populations
Statistical Robustness:
- Ensure adequate sample sizes through power calculations that account for center effects
- Use mixed-effects models to account for center-specific variability
- Report confidence intervals alongside performance metrics
- Conduct sensitivity analyses to assess model robustness to protocol variations
The integration of machine learning, particularly ensemble feature selection methods and classification algorithms like NB, RF, and SVM, has significantly advanced lipidomic biomarker discovery. Each approach offers distinct advantages: SVM excels in high-dimensional spaces, RF provides native feature importance measures, and NB offers computational efficiency. Ensemble methods enhance robustness by aggregating across multiple selection techniques, which is particularly valuable for multi-center validation studies where technical and biological variability must be addressed.
The future of lipidomic biomarker research lies in the continued refinement of integrated ML workflows, with artificial intelligence models already demonstrating up to 97.4% accuracy in predicting lipid subclasses [25]. As these methodologies mature and standardization improves, ML-driven lipidomics will play an increasingly pivotal role in delivering clinically validated biomarkers for personalized medicine, ultimately fulfilling the promise of precise disease diagnosis, stratification, and treatment monitoring.
The discovery of lipidomic biomarkers for diseases like liver cancer represents a frontier in precision medicine. However, the transition from research findings to clinically validated tools is significantly hampered by the "black-box" nature of traditional machine learning (ML) models, which often provide high accuracy at the expense of interpretability [42] [43]. This lack of transparency creates a barrier to clinical adoption, as practitioners require understanding the reasoning behind a model's predictions for trust and ethical decision-making [44]. Explainable AI (XAI) directly addresses this challenge by providing mechanisms to elucidate model predictions, thereby building accountability and reducing bias [44]. This guide objectively compares the performance of different XAI-integrated approaches for lipidomic biomarker discovery, framing the analysis within the critical context of multi-center validation, a necessary step for clinical translation [45] [25].
Comparative Performance of XAI-Integrated Machine Learning Models
Research demonstrates that integrating XAI with ML models not only provides interpretability but can also enhance diagnostic performance for lipidomic biomarker discovery. The following table summarizes the performance of various models reported in recent studies focused on liver cancer detection.
Table 1: Performance Comparison of ML Models for Lipidomic-Based Liver Cancer Detection
| Model / Study | Accuracy (%) | Sensitivity (%) | Specificity (%) | AUC | Key XAI Technique |
|---|---|---|---|---|---|
| Explainable Boosting Machine (EBM) [44] | 87.0 | 87.7 | 86.3 | 0.918 | Model-specific self-interpretation |
| AdaBoost [46] | ~87.5* | N/R | N/R | 0.875 | SHAP |
| Random Forest [44] | N/R | N/R | N/R | <0.918 | SHAP |
| XGBoost [44] | N/R | N/R | N/R | <0.918 | SHAP |
Note: AUC = Area Under the Receiver Operating Characteristic Curve; N/R = Not Explicitly Reported in the context of model comparison; *Estimated from the AUC performance and other metrics.
The Explainable Boosting Machine (EBM) demonstrated superior performance in a head-to-head comparison, achieving the highest accuracy and AUC [44]. EBMs are a type of glass-box model that combines the power of boosting with inherent interpretability, allowing researchers to see the contribution of each feature to individual predictions without post-hoc methods.
In a separate study, AdaBoost coupled with SHAP (SHapley Additive exPlanations) analysis also showed strong classification performance for liver cancer, achieving an AUC of 0.875 [46]. SHAP is a model-agnostic XAI technique that quantifies the marginal contribution of each feature to a model's prediction, providing both global and local interpretability [46] [42]. While often high-performing, traditional ensemble models like Random Forest and XGBoost are inherently black-box and rely on post-hoc XAI methods like SHAP for interpretability, which can add complexity [44].
Experimental Protocols and Workflow for XAI-Based Biomarker Discovery
A typical pipeline for discovering interpretable lipidomic biomarkers integrates robust laboratory protocols, data processing, and XAI-driven analysis. The following workflow diagram and detailed methodology outline this process.
Diagram 1: Experimental workflow for XAI-based lipidomic biomarker discovery.
Sample Collection and Laboratory Analysis
- Study Cohort: The referenced studies analyzed serum samples from 219 patients diagnosed with liver cancer and 219 matched healthy controls. Controls were matched based on age, date of blood collection, and sample handling procedures to minimize pre-analytical variability [46] [44].
- Sample Preparation: Blood samples were collected from participants following an overnight fast. Serum was separated via centrifugation, aliquoted, and stored at -70°C until analysis to preserve lipid integrity [46].
- Lipidomic Profiling: Untargeted lipidomic analysis was performed using Liquid Chromatography Quadrupole Time-of-Flight Mass Spectrometry (LC-QTOF-MS). This high-resolution platform allows for the semi-quantitative measurement of a wide range of lipid species. A dual-polarity acquisition approach (positive and negative electrospray ionization mode) is often used to broaden lipid coverage [46] [44].
- Lipid Identification: Lipids are identified by aligning experimental data (accurate mass and retention time) against reference spectral libraries, such as LipidBlast. This typically results in putative annotations (MSI Level 2-3) [44].
Data Processing and Machine Learning
- Data Pre-processing: Raw data from MS instruments undergoes peak picking, alignment, and normalization. The use of the SERRF (Systematic Error Reduction using Random Forest) tool is common to correct for undesirable systematic variance and batch effects [46].
- Feature Selection: Lipid species are filtered and selected using univariate and multivariate statistical methods. Common techniques include fold change (FC) analysis, t-tests with false discovery rate (FDR) adjustment, and multivariate methods like Partial Least Squares Discriminant Analysis (PLS-DA) with Variable Importance in Projection (VIP) scores [46].
- Model Training and Validation: The dataset is split into training and test sets (e.g., 4:1 ratio) using stratified random sampling to maintain class distribution. Models are trained and their performance is evaluated on the held-out test set. To ensure robustness, this process is often repeated multiple times (e.g., 100 iterations) with different random seeds [46].
Explainable AI (XAI) Interpretation
- SHAP (SHapley Additive exPlanations): Applied to black-box models like AdaBoost or Random Forest, SHAP calculates the contribution of each lipid feature to a specific prediction. This allows researchers to generate global feature importance plots (showing which lipids matter most overall) and local explanations (for a single patient's diagnosis) [46] [42].
- Explainable Boosting Machine (EBM): As a glass-box model, the EBM inherently produces feature importance scores and visualizes the relationship between the concentration of a specific lipid and its contribution to the model's output, making the decision-making process fully transparent [44].
Multi-Center Validation and Standardization of Lipidomic Biomarkers
For lipidomic biomarkers to achieve clinical utility, they must be validated across multiple independent laboratories and populations. This process faces significant challenges but is essential for demonstrating reliability.
Table 2: Key Reagents and Kits for Standardized Lipidomics Analysis
| Research Reagent / Solution | Function in the Workflow | Role in Multi-Center Validation |
|---|---|---|
| MxP Quant 500 Kit [45] | Targeted quantification of 630+ metabolites and lipids from 26 compound classes using UHPLC and FIA-MS/MS. | Provides a standardized SOP, reagents, and software to minimize inter-laboratory variability. |
| Internal Standards (Isotope-Labeled) [45] [25] | Added to samples to correct for analytical variability during sample preparation and MS analysis. | Essential for accurate quantification and ensuring data comparability across different instrument setups. |
| NIST SRM 1950 Reference Plasma [45] | A standardized reference material of human plasma with characterized analyte concentrations. | Serves as a quality control to monitor and correct for analytical drift and performance between runs and labs. |
| SERRF Normalization Tool [46] | A normalization algorithm that uses quality control samples to reduce systematic error. | A computational tool to post-process data and enhance comparability in multi-batch studies. |
A recent large-scale interlaboratory study evaluated the MxP Quant 500 kit across 14 laboratories. The kit demonstrated a median coefficient of variation (CV) of 14.3%, with CVs for 494 metabolites in the NIST SRM 1950 reference plasma being below 25% [45]. This high level of reproducibility is a promising indicator that standardized kits can enable robust multi-center studies.
However, challenges remain. The lack of reproducibility across different analytical platforms is a major obstacle. Prominent software platforms can agree on as low as 14–36% of lipid identifications from identical data, highlighting the need for standardized workflows and data processing methods [25]. Furthermore, transitioning from a research finding to an approved diagnostic tool requires navigating incomplete regulatory frameworks specifically for lipidomic biomarkers [25].
Identified Lipid Biomarkers and Biological Pathways in Liver Cancer
XAI models have successfully identified specific lipid species that are consistently dysregulated in liver cancer, providing both diagnostic value and biological insight. The following diagram summarizes the key altered lipid pathways.
Diagram 2: Key lipid alterations and pathways in liver cancer identified by XAI.
The identified biomarkers highlight crucial disruptions in lipid metabolism. The consistent decrease in specific sphingomyelins (SMs) and the increase in certain phosphatidylcholines (PCs) and fatty acids (FAs) point to a metabolic rewiring where cancer cells alter their membrane architecture and signaling pathways to support rapid proliferation and evade cell death [46] [44]. For instance, SHAP analysis in one study identified PC 40:4 as the most impactful lipid for model predictions, while decreased levels of SM d41:2 and SM d36:3 were associated with an increased cancer risk [46]. Another study using EBM highlighted PC 38:2 and SM d40:2 as key discriminators [44]. This convergence on specific lipid classes from independent studies strengthens the case for their biological relevance and potential as robust biomarkers.
Lipids are crucial biomolecules involved in various biological processes, and changes in lipid profiles are closely linked to the development of multiple disorders [47]. The field of lipidomics, which entails the large-scale measurement of molecular lipids in biological specimens, is moving from a basic research tool to a cornerstone of precision health [48]. This guide objectively compares the methodologies and analytical techniques used to transform raw lipidomic data into robust, clinically applicable diagnostic algorithms. The process typically integrates advanced mass spectrometry with machine learning to identify lipid signatures that can distinguish between health and disease states with high accuracy, a approach firmly set within the critical context of multi-center biomarker validation [49] [50].
Experimental Workflows for Lipidomic Profiling
Building a diagnostic model begins with rigorous experimental design to generate high-quality lipidomic data. The two primary mass spectrometry approaches offer distinct advantages and are often used in tandem.
Table: Comparison of Lipidomic Profiling Approaches
| Feature | Untargeted Lipidomics | Targeted Lipidomics |
|---|---|---|
| Primary Objective | Hypothesis-generating, global discovery of lipid signatures [49] | Hypothesis-testing, validation of specific candidate biomarkers [49] |
| Metabolite Coverage | High; potential to identify 1,000+ lipid species across many classes [49] | Focused on a pre-defined panel of lipids |
| Data Output | Relative abundance of a wide range of lipids | Absolute quantification of specific lipids |
| Typical Application | Discovery phase to identify differential lipids [49] | Validation phase to confirm biomarkers in independent cohorts [49] [50] |
| Key Strength | Unbiased, comprehensive screening [49] | High sensitivity, specificity, and reliability for validated lipids [49] |
Integrated Discovery-to-Validation Pipeline
A robust diagnostic model employs a sequential pipeline. An initial discovery cohort is analyzed using untargeted lipidomics to identify a wide spectrum of dysregulated lipids [49]. Promising candidate biomarkers from this stage are then evaluated using targeted lipidomics (e.g., Multiple Reaction Monitoring) in an independent validation cohort to confirm their diagnostic performance [49] [50]. This two-step approach was successfully implemented in a study for nonsyndromic cleft lip with palate (nsCLP), which used untargeted lipidomics on a discovery set and then targeted lipidomics for validation [49].
Data Processing and Statistical Analysis
Following data acquisition, raw lipidomic data undergoes preprocessing and statistical analysis to identify lipids with significant diagnostic value.
Data Preprocessing and Quality Control
Data preprocessing is critical for ensuring data quality. Steps include peak picking, alignment, and normalization. A key quality control measure involves analyzing Quality Control (QC) samples. A common standard is that over 98% of lipid features in QC samples should have a relative standard deviation (RSD) of ≤ 30%, indicating satisfactory stability and repeatability of the analysis [49].
Statistical and Multivariate Analysis
Both univariate and multivariate statistical techniques are used to find diagnostic signals.
- Univariable Analysis: Initial univariable comparisons (e.g., t-tests) identify individual molecular lipids that are significantly altered between disease and control groups. For example, a pediatric IBD study found 45 altered molecular lipids in IBD compared to symptomatic controls [50].
- Multivariate Analysis: These methods analyze the entire lipid profile simultaneously.
- OPLS-DA: Orthogonal Partial Least Squares-Discriminant Analysis is a supervised method that maximizes the separation between pre-defined groups (e.g., control vs. treatment) [51]. It produces a score plot to visualize group separation and a loadings plot to identify the specific driver lipids responsible for the segregation [51]. The model's reliability should be checked with a permutation test to rule out overfitting [51].
- PCA: Principal Component Analysis is an unsupervised method that visualizes the major variance in a dataset without using group labels, helping to identify outliers and overall data structure [51].
Machine Learning for Diagnostic Model Building
Machine learning (ML) is used to create a high-performance classification algorithm from the lipidomic features selected in earlier stages.
Feature Selection and Model Training
To build a robust model, identifying the most informative lipids is crucial. An ensemble feature selection strategy can be employed, where multiple feature selection methods are used to rank lipids, and a robust rank aggregation (RRA) algorithm integrates these rankings into a unified list [49]. This list is then evaluated using multiple classification models to find the optimal panel of lipid biomarkers [49].
Table: Performance Comparison of Machine Learning Classifiers
| Machine Learning Model | Application Context | Diagnostic Performance (AUC) | Key Findings / Advantage |
|---|---|---|---|
| Naive Bayes (NB) | nsCLP Diagnosis [49] | 0.95 (Discovery, top 35 features) [49] | Achieved high classification accuracy without overfitting [49] |
| SCAD (Logistic Regression) | Pediatric IBD Diagnosis [50] | 0.87 (Discovery), 0.85 (Validation) [50] | Selected a parsimonious model with 30 lipids; outperformed hsCRP (AUC=0.73) [50] |
| Random Forest (RF), Decision Tree (DT), Adaboost (ADA) | nsCLP Diagnosis [49] | Training accuracy ~1.0, Testing accuracy <0.8 [49] | Prone to overfitting; poor generalization to testing sets [49] |
| Stacking (Ensemble) | Pediatric IBD Diagnosis [50] | High performance in validation [50] | Leveraged strengths of multiple algorithms for improved classification [50] |
Multi-Center Validation and Benchmarking
A diagnostic model's true value is confirmed through rigorous validation. This involves:
- Independent Validation: Testing the model's performance in a completely separate cohort of patients [49] [50].
- Comparison to Standard Biomarkers: Benchmarking the lipidomic signature against current clinical standards. The pediatric IBD lipidomic signature (AUC=0.85) significantly outperformed high-sensitivity C-reactive protein (hsCRP, AUC=0.73) [50].
- Multi-Center Confirmation: Further validating the findings in a third cohort to confirm the directionality and significance of the lipid changes (e.g., increased LacCer(d18:1/16:0) and decreased PC(18:0p/22:6) in pediatric IBD) [50].
Data Visualization and Interpretation
Effective visualization is key to interpreting complex lipidomic data and communicating findings.
- PCA and OPLS-DA Plots: These plots graphically visualize lipidomic differences between groups. PCA shows the major variance in the dataset, while OPLS-DA is superior for highlighting structured variation between two specific groups [51].
- Volcano Plots: Used to visualize univariate results, plotting the negative log of the p-value against the log fold change to highlight lipids that are both statistically significant and substantially altered [52].
- Lipidome Projector: This web-based software uses a neural network to embed lipid structures in a 2D or 3D space so that structurally similar species are positioned near each other. It allows for interactive exploration of lipidomes, displaying abundances and structures for intuitive interpretation [53].
The Scientist's Toolkit: Essential Reagents and Materials
Table: Key Research Reagents and Solutions for Lipidomics
| Item | Function in the Experimental Protocol |
|---|---|
| Quality Control (QC) Samples | A pooled sample from all samples used to monitor the stability and repeatability of the mass spectrometry run [49]. |
| Discovery Cohort Samples | The initial set of biological samples (e.g., serum/plasma) used for untargeted lipidomics to discover a wide range of candidate biomarkers [49]. |
| Validation Cohort Samples | An independent set of samples from a different patient cohort, used to confirm the diagnostic performance of the candidate biomarkers via targeted lipidomics [49] [50]. |
| Internal Standards | Stable isotope-labeled lipid standards spiked into samples for accurate quantification, particularly in targeted lipidomics. |
| Constraints List (for Lipidome Projector) | A file specifying allowed fatty acyls and long-chain bases for an organism, used to filter plausible lipid isomers when precise structural data is unavailable from MS [53]. |
Overcoming Challenges: Ensuring Robustness and Reproducibility in Lipidomic Studies
Technical variance arising from inconsistent sample handling, analytical procedures, and data processing presents a significant challenge in multi-center lipidomics research, particularly for biomarker discovery and validation. The reproducibility of lipidomic data across different laboratories and platforms is crucial for translating research findings into clinically applicable tools. This guide objectively compares quality control (QC) protocols and assesses sample stability parameters, providing researchers with experimental data and methodologies to standardize lipidomic workflows. As lipidomics advances toward clinical application, establishing robust QC frameworks becomes imperative for ensuring data reliability across multiple research sites. This comparison examines current QC approaches, their performance metrics, and practical implementation strategies to mitigate technical variability in large-scale lipidomic studies.
Comparative Analysis of QC Approaches
Table 1: Comparison of Quality Control Strategies in Lipidomics
| QC Approach | Methodology | Key Performance Metrics | Advantages | Limitations |
|---|---|---|---|---|
| Pooled QC (PQC) Samples | Repeated analysis of pooled representative samples throughout batch sequence [54] | Retention time stability (<2% RSD), peak intensity variance (<15-25% RSD) [55] [54] | Monitors instrumental performance; corrects for batch effects | May not capture all biological variability |
| Surrogate QC (sQC) | Commercial reference materials as long-term references [54] | Inter-laboratory comparability; long-term reproducibility | Provides standardization across multiple sites | Potential matrix differences from study samples |
| Multiplexed Targeted Assays | NPLC-HILIC MRM with internal standards [55] | Inter-assay variability (<25% for 700+ lipids); quantitative accuracy [55] | High reproducibility; absolute quantification | Requires predefined lipid panels; instrument-specific optimization |
| Cross-Platform Validation | Multiple software platforms (MS DIAL, Lipostar) on identical data [56] | Identification agreement (14-36% between platforms) [56] | Reveals platform-specific biases | Highlights need for manual curation |
Table 2: Sample Stability Considerations in Lipidomics
| Pre-Analytical Factor | Stability Impact | Recommended Protocols | Supporting Evidence |
|---|---|---|---|
| Sample Collection | Plasma vs. serum differences; anticoagulant effects | Consistent blood collection tubes; processing within 2 hours [57] | Standardized in multi-center studies [57] [50] |
| Storage Conditions | Lipid degradation over time; temperature effects | -80°C storage; freeze-thaw cycle minimization [55] | Validated in long-term biomarker studies [58] [50] |
| Extraction Methodology | Lipid recovery variance; oxidation prevention | Modified Folch or BUME extraction; antioxidant addition (BHT) [56] | Reproducible across cell lines and biofluids [56] |
| Analytical Batch Effects | Signal drift over sequences; column degradation | Randomized sample analysis; PQC every 6-10 samples [54] | <25% RSD achieved in validated assays [55] |
Experimental Protocols for QC Implementation
Multiplexed Targeted Lipidomics Validation
A rigorously validated multiplexed normal phase liquid chromatography-hydrophilic interaction chromatography (NPLC-HILIC) multiple reaction monitoring (MRM) method exemplifies a comprehensive QC protocol [55]. This approach quantifies over 900 lipid molecular species across 20 lipid classes within a single 20-minute analysis, addressing challenges such as in-source fragmentation, isomer separation, and concentration dynamics [55]. The methodology employs lipid class-based calibration curves with predefined acceptance criteria for quality control samples according to FDA Bioanalytical Method Validation Guidance [55].
Key experimental steps include:
- Sample Preparation: Modified Folch extraction using chilled methanol/chloroform (1:2 v/v) supplemented with 0.01% butylated hydroxytoluene (BHT) to prevent oxidation [56]
- Internal Standardization: Addition of stable isotope-labeled internal standards (SIL IS) or commercially available quantitative internal standard mixtures (e.g., Avanti EquiSPLASH LIPIDOMIX) before extraction [55] [56]
- Chromatographic Separation: Multiplexed NPLC-HILIC separation using a binary gradient with eluent A (60:40 acetonitrile/water) and B (85:10:5 isopropanol/water/acetonitrile), both supplemented with 10 mM ammonium formate and 0.1% formic acid [55] [56]
- Mass Spectrometry Analysis: Scheduled MRM on triple quadrupole instrumentation; utilization of multiple MS/MS product ions per lipid species to improve identification confidence and determine relative abundances of positional isomers [55]
- Quality Control Samples: Analysis of pooled quality control (PQC) samples and commercial reference materials (e.g., NIST-SRM-1950 plasma) throughout analytical sequences [55] [54]
Cross-Platform Lipid Identification Protocol
The reproducibility challenge in lipid identification was systematically evaluated by processing identical LC-MS spectra through two open-access lipidomics platforms, MS DIAL (v4.9.221218) and Lipostar (v2.1.4) [56]. This protocol revealed significant discrepancies, with only 14.0% identification agreement using default settings and 36.1% agreement when utilizing MS2 spectra [56].
Methodology for platform comparison:
- Sample Analysis: LC-MS analysis of PANC-1 cell line lipid extracts using microflow separation on a C18 column with positive ionization mode [56]
- Data Processing: Identical raw data files processed through both platforms using maximally comparable settings [56]
- Identification Agreement Assessment: Formula-level comparison of putative identifications from both platforms [56]
- Manual Curation: Systematic verification of conflicting identifications through manual inspection of spectral data [56]
- Outlier Detection: Implementation of support vector machine regression with leave-one-out cross-validation to identify potential false positives [56]
Visualizing QC Workflows
QC Workflow for Multi-center Lipidomics
Essential Research Reagent Solutions
Table 3: Essential Research Reagents for Lipidomics QC
| Reagent Category | Specific Examples | Function in QC Protocols |
|---|---|---|
| Internal Standards | Avanti EquiSPLASH LIPIDOMIX; Stable isotope-labeled (SIL) lipids [55] [56] | Normalization of extraction efficiency; quantitative calibration |
| Reference Materials | NIST-SRM-1950 plasma; Commercial plasma pools [55] [54] | Inter-laboratory standardization; long-term performance monitoring |
| Antioxidants | Butylated hydroxytoluene (BHT) [56] | Prevention of lipid oxidation during extraction and storage |
| Extraction Solvents | HPLC-grade methanol, chloroform, methyl-tert-butyl ether (MTBE) [57] [55] | Efficient lipid recovery with minimal degradation |
| Mobile Phase Additives | Ammonium formate, formic acid [55] [56] | Enhancement of ionization efficiency; chromatographic separation |
The implementation of robust quality control protocols is fundamental to addressing technical variance in multi-center lipidomic biomarker research. Based on comparative analysis, integrated approaches combining pooled QC samples, surrogate reference materials, standardized extraction methodologies, and cross-platform validation demonstrate the most effective strategy for ensuring data reproducibility. The documented performance metrics provide benchmarks for researchers establishing lipidomic QC protocols, with the optimal approaches achieving less than 25% inter-assay variability for hundreds of lipid species. As lipidomics advances toward clinical applications, these standardized QC frameworks will be essential for generating reliable, reproducible data across multiple research sites, ultimately accelerating the translation of lipidomic biomarkers into clinical practice.
The pursuit of robust, multi-center validated lipidomic biomarkers is a cornerstone of modern precision medicine, yet it is fraught with two formidable computational challenges: high-dimensionality and batch effects. High-dimensional data, characterized by a vast number of lipid features (often thousands per sample) relative to a small number of biological specimens, introduces the "curse of dimensionality" [59]. This phenomenon leads to data sparsity, increased computational complexity, and a heightened risk of model overfitting, where algorithms learn noise instead of true biological signals [59] [60]. Concurrently, batch effects are non-biological technical variations introduced when samples are processed in different batches, using different reagent lots, by different personnel, or across different sequencing platforms or centers [61] [62]. In multi-center studies, these effects are inevitable and can be profound; if left uncorrected, they act as confounders that dilute genuine biological signals, reduce statistical power, and critically, can lead to irreproducible findings and misleading conclusions [61] [63]. For a lipidomic signature to be clinically translatable, it must be demonstrably robust to these technical variations, making the mastery of the following correction strategies not merely beneficial, but essential.
Understanding High-Dimensional Lipidomic Data
The Nature and Challenge of High-Dimensionality
In lipidomics, high-dimensionality arises from the technological capacity to measure a staggering number of distinct lipid molecular species from a single biological sample. A single profiling experiment can quantify hundreds to thousands of unique lipid entities, transforming each sample into a point in an extremely high-dimensional space [27]. The primary challenge, known as the curse of dimensionality, is that as the number of features grows, the volume of the feature space expands so exponentially that the data becomes sparse [59] [60]. This sparsity breaks down traditional statistical methods and distance metrics, making it difficult to find meaningful patterns. Furthermore, with so many features, the risk of overfitting is acute—models may appear to perform perfectly on the initial dataset but fail to generalize to new samples from a different batch or center [59]. This directly undermines the goal of multi-center validation.
Dimensionality Reduction Techniques: A Comparative Analysis
To combat these challenges, dimensionality reduction techniques are employed to project the data into a lower-dimensional, more manageable space while preserving the essential biological information. These methods can be broadly categorized into linear and non-linear approaches, each with distinct strengths for lipidomic data.
Table 1: Comparison of Dimensionality Reduction Techniques for Lipidomic Data
| Technique | Type | Key Principle | Strengths | Weaknesses | Suitability for Lipidomics |
|---|---|---|---|---|---|
| PCA [59] [64] | Linear | Finds orthogonal axes of maximum variance in the data. | Computationally efficient, preserves global structure, simple to interpret. | Assumes linear relationships, may miss complex non-linear structures. | High - Excellent for initial exploratory analysis and noise reduction. |
| t-SNE [59] [64] | Non-linear | Models pairwise similarities to preserve local neighborhood structure. | Excellent at revealing clusters and local data structure. | Computationally heavy, cannot transform new data, stochastic (results vary). | Medium - Ideal for final visualization and cluster validation. |
| UMAP [60] [64] | Non-linear | Uses manifold learning to preserve both local and global structure. | Faster than t-SNE, better at preserving global structure. | More complex to tune, relatively newer than other methods. | High - Great for visualization and as a pre-processing step for clustering. |
| Autoencoders [59] [63] [64] | Non-linear | Neural networks learn a compressed, efficient representation of the data. | Can learn highly complex, non-linear relationships. | "Black box" nature, requires large datasets and computational resources. | Medium-High - Powerful for large, multi-center datasets where complex effects exist. |
The following diagram illustrates the workflow for applying these techniques to high-dimensional lipidomic data:
Tackling Batch Effects in Multi-Center Studies
Batch effects are a pervasive threat to the integrity of multi-center lipidomic studies. Their sources can be traced to virtually every step of the experimental workflow [61]. During sample preparation, variations in centrifugal forces, storage temperature, duration, and freeze-thaw cycles can introduce significant technical variance. In data generation, differences between mass spectrometry platforms, calibration, reagent lots (e.g., different batches of fetal bovine serum), and even the personnel handling the samples can create batch-specific signals [61] [27]. The impact of these effects is not merely noise; they can completely obscure biological truth. For instance, a study comparing human and mouse tissues found that gene expression differences were initially attributed to species, but after batch correction, the data clustered by tissue type, demonstrating that the reported "species-specific" signatures were actually batch effects introduced by processing the species' samples three years apart [61]. In a clinical context, a change in an RNA-extraction solution batch led to an incorrect risk classification for 162 patients, 28 of whom received unnecessary chemotherapy [61]. Such examples underscore that batch effects are not a nuisance but a fundamental source of irreproducibility that can invalidate study conclusions.
Batch Effect Correction Algorithms (BECAs)
A suite of computational tools has been developed to identify and remove batch effects, a process crucial for integrating data from multiple centers. The choice of algorithm often depends on the data type and the nature of the study.
Table 2: Comparison of Batch Effect Correction Algorithms (BECAs)
| Algorithm/Method | Underlying Principle | Key Features | Reported Performance | Considerations for Multi-Center Lipidomics |
|---|---|---|---|---|
| ComBat [63] | Empirical Bayes framework. | Adjusts for known batch sources, can preserve biological variance of interest. | Established benchmark for bulk data; can be outperformed by newer methods on complex data. | Well-understood but may over-correct if study variables are confounded with batch. |
| Harmony [62] [63] | Iterative clustering and correction. | Uses a concept of "anchors" (shared cell types/States) to integrate datasets. | Excels at integrating single-cell data while preserving fine-grained cellular populations. | Highly relevant for complex lipidomic data with multiple underlying cell type-driven signatures. |
| Mutual Nearest Neighbors (MNN) [62] [63] | Identifies mutual nearest neighbors across batches to define "correction vectors." | Makes minimal assumptions about the data distribution. | Effective at aligning similar cell populations across batches in scRNA-seq data. | Its success depends on the existence of shared biological states across all batches. |
| Deep Learning (e.g., scVI) [63] | Uses deep generative models (e.g., variational autoencoders) to learn a batch-invariant latent representation. | Models complex non-linear effects, can handle uncertainty. | Shows superior performance on large, complex single-cell datasets [63]. | Requires significant computational resources and expertise; ideal for very large, multi-center cohorts. |
| Seurat Integration [62] | Identifies "anchors" between datasets and uses them to integrate data into a shared space. | Widely used in single-cell genomics community, well-documented. | Consistently performs well in independent benchmarks of data integration tasks. | A robust, widely-adopted choice that can be a good starting point for lipidomic data integration. |
The strategic process for diagnosing and correcting for batch effects is outlined below:
Case Study: Integrated Strategies in Biomarker Discovery
Experimental Protocol: A Lipidomic Signature for Pediatric IBD
A landmark study published in Nature Communications in 2024 provides a exemplary blueprint for successfully navigating high-dimensionality and batch effects in a multi-center context [50]. The research aimed to identify and validate a blood-based diagnostic lipidomic signature for pediatric inflammatory bowel disease (IBD). The experimental design was rigorously structured across three independent cohorts: a discovery cohort (n=94), an independent validation cohort (n=117), and a confirmation cohort (n=263), all comprising treatment-naïve patients and symptomatic controls [50].
Methodology Overview:
- Lipidomics Profiling: Blood samples were subjected to high-resolution mass spectrometry (HRMS)-based lipidomics to quantify a wide array of molecular lipids.
- Handling High-Dimensionality: The team employed a suite of seven different machine learning algorithms (including regularized logistic regression like SCAD) to identify a minimal, diagnostic lipid signature from hundreds of initial lipid features. This approach directly addresses overfitting by using penalized models that shrink the coefficients of non-informative lipids to zero [65] [50].
- Multi-Center Validation: The signature derived from the discovery cohort was directly applied to the independent validation and confirmation cohorts without re-fitting, a critical step for testing generalizability and robustness to batch effects inherent in different patient cohorts.
- Performance Evaluation: The diagnostic performance of the lipidomic signature was compared against established biomarkers like high-sensitivity C-reactive protein (hsCRP) and fecal calprotectin using Area Under the Curve (AUC) metrics [50].
Key Findings and Comparative Performance
The study successfully identified a compact, diagnostic signature comprising just two lipids: increased Lactosyl Ceramide (d18:1/16:0) and decreased phosphatidylcholine (18:0p/22:6). The performance of this signature highlights the efficacy of the integrated strategy.
Table 3: Performance Comparison of Lipidomic Signature vs. Traditional Biomarker
| Model / Biomarker | Cohort | Performance (AUC) & 95% CI | Key Outcome |
|---|---|---|---|
| 30-Lipid SCAD Model [50] | Discovery | AUC = 0.87 (0.79 - 0.93) | Demonstrated strong initial predictive power. |
| 2-Lipid Signature [50] | Validation | AUC = 0.85 (0.77 - 0.92) | Signature validated in an independent cohort. |
| hsCRP [50] | Validation | AUC = 0.73 (0.63 - 0.82) | Lipid signature significantly outperformed hsCRP (P < 0.001). |
| 2-Lipid Signature [50] | Confirmation | Consistent direction of change confirmed | Robustness of the two key lipids confirmed in a third cohort. |
This case study demonstrates that a rigorous analytical workflow combining machine learning for dimensionality reduction and multi-center validation for batch effect robustness can yield a clinically actionable biomarker.
The Scientist's Toolkit: Essential Research Reagent Solutions
The reliability of lipidomics data is fundamentally dependent on the quality and consistency of the reagents and materials used throughout the workflow. The following table details key research reagent solutions and their critical functions in ensuring data quality and mitigating batch variations.
Table 4: Essential Research Reagent Solutions for Robust Lipidomics
| Reagent / Material | Function in Workflow | Importance for Minimizing Batch Effects |
|---|---|---|
| Internal Standard Mixture [27] | Added to each sample prior to lipid extraction for absolute quantification. | Corrects for variations in extraction efficiency and instrument response drift; crucial for inter-batch comparability. |
| Quality Control (QC) Pool [61] [27] | A pooled sample from all study samples injected repeatedly throughout the analytical run. | Monitors instrument stability over time. Drift in QC data signals a batch effect, allowing for post-hoc correction. |
| Standardized Lipid Extraction Solvents [61] [27] | Used for liquid-liquid extraction (e.g., MTBE, chloroform:methanol) of lipids from biological matrices. | Consistent reagent purity and supplier are vital. Variations between lots or suppliers can introduce major batch artifacts. |
| Chromatography Columns [61] | Stationary phase for separating lipid molecules prior to MS detection (e.g., C18 reversed-phase). | Column performance degrades over time. Using columns from the same manufacturing lot across centers helps minimize a major source of variation. |
| Calibration Solutions [27] | Standard solutions of known concentration for mass spectrometer mass/charge (m/z) and retention time calibration. | Ensures measurement accuracy and allows alignment of lipid identities across different instruments and batches. |
In machine learning, a model's true value is determined not by its performance on familiar training data but by its ability to make accurate predictions on new, unseen data. The phenomenon where a model learns the training data too well—capturing noise and random fluctuations alongside genuine patterns—is known as overfitting. An overfitted model essentially memorizes the training set, excelling with known data but failing to generalize to novel datasets [66]. This challenge is particularly acute in high-dimensional fields like lipidomics biomarker research, where models must navigate complex biological data with limited samples, making robust validation and regularization not merely beneficial but essential for scientific credibility [67] [5].
Conversely, underfitting presents an opposite problem, occurring when a model is too simplistic to capture the underlying trend in the data, performing poorly on both training and validation sets [66]. The core objective for researchers and drug development professionals is to navigate between these extremes to achieve a well-fit model that generalizes effectively. This article provides a comparative guide to the techniques that make this balance possible—cross-validation and regularization—framed within the critical context of multi-center lipidomic biomarker validation.
Cross-Validation: The Gold Standard for Generalization Assessment
Core Concepts and Techniques
Cross-validation (CV) is a family of resampling techniques designed to provide a realistic estimate of a model's performance on unseen data. It systematically partitions the available data into subsets, iteratively using some for training and others for validation. This process stands in contrast to a simple train-test split, which can yield unreliable performance estimates dependent on a single, arbitrary data division [68] [69].
The following table compares the most common cross-validation techniques:
Table 1: Comparison of Common Cross-Validation Techniques
| Technique | Core Methodology | Advantages | Limitations | Ideal Use Cases |
|---|---|---|---|---|
| K-Fold CV [68] [69] | Data randomly split into K equal folds; each fold serves as validation once. | Balanced bias-variance trade-off; efficient data use. | Assumes IID data; unsuitable for temporal data. | General-purpose model evaluation on IID data. |
| Stratified K-Fold [68] [69] | Preserves original class distribution in each fold. | Essential for imbalanced datasets; more reliable performance estimate. | Primarily for classification tasks. | Classification problems with class imbalance. |
| Leave-One-Out (LOOCV) [68] [69] | K equals the number of samples; one sample left out for validation each time. | Low bias; uses maximum data for training. | Computationally expensive; high variance in estimate. | Very small datasets where data is precious. |
| Time Series Split [69] | Maintains temporal order; trains on past, validates on future. | Prevents data leakage; realistic for forecasting. | Earlier training sets are smaller. | Time-series data, chronological datasets. |
Experimental Protocol and Implementation
Implementing k-fold cross-validation follows a standardized workflow. The diagram below illustrates the core process for a 5-fold cross-validation.
In Python, the scikit-learn library provides a straightforward implementation. The following code demonstrates a typical k-fold CV protocol for a support vector machine classifier [68] [70]:
This protocol outputs the performance from each fold, providing both an average performance metric and insight into the model's consistency across different data subsets.
Regularization: Constraining Model Complexity
Core Techniques and Their Mechanisms
While cross-validation helps detect overfitting, regularization techniques help prevent it by discouraging over-complex models. These methods impose constraints during training, guiding models toward simpler representations that capture fundamental patterns rather than noise [71] [66].
Table 2: Comparison of Common Regularization Techniques
| Technique | Core Mechanism | Key Hyperparameter(s) | Architectural Considerations |
|---|---|---|---|
| L1 & L2 Regularization [71] [66] | Adds penalty to loss function based on weight magnitude. | λ (regularization strength). | L1 promotes sparsity; L2 discourages large weights. |
| Dropout [71] [66] | Randomly disables neurons during training. | Dropout rate (fraction of neurons to disable). | Forces network to develop redundant representations. |
| Early Stopping [71] [66] | Halts training when validation performance stops improving. | Patience (epochs to wait before stopping). | Prevents the model from continuing to memorize noise. |
| Data Augmentation [71] | Artificially expands training set via transformations. | Type and magnitude of transformations. | Domain-specific (e.g., image rotations, noise injection). |
Experimental Insights from Architectural Comparisons
A systematic study comparing regularization techniques on different convolutional neural network (CNN) architectures for image classification provides compelling experimental data. Using the Imagenette dataset, researchers evaluated how dropout and data augmentation impact generalization in baseline CNNs versus ResNet-18 models [71].
Table 3: Performance Comparison of Regularized CNN vs. ResNet-18 on Imagenette Dataset
| Model Architecture | Key Regularization Techniques | Reported Validation Accuracy | Generalization Gap Reduction |
|---|---|---|---|
| Baseline CNN [71] | Dropout, Data Augmentation | 68.74% | Significant reduction in overfitting |
| ResNet-18 [71] | Dropout, Data Augmentation, Batch Normalization | 82.37% | Superior generalization with reduced gap |
| ResNet-18 with Transfer Learning [71] | Fine-tuning, Early Stopping | >82.37% (Baseline) | Faster convergence and higher accuracy |
The results demonstrate that while both architectures benefit from regularization, ResNet-18 achieved superior validation accuracy. The study confirmed that regularization consistently reduced overfitting and improved generalization across all scenarios. Furthermore, it highlighted that the effectiveness of specific techniques varies with architecture; for instance, the residual connections in ResNet models can alter how dropout and batch normalization interact with the learning process [71].
Application in Lipidomic Biomarker Research: A Multi-Center Context
Case Studies in Disease Diagnostics
The integration of cross-validation and regularization is paramount in lipidomics, where high-dimensional data and limited sample sizes create a high risk of overfitting. The following case studies illustrate their application in real-world biomarker discovery.
Table 4: Lipidomic Biomarker Discovery Case Studies Employing Robust Validation
| Study Focus | ML & Validation Approach | Reported Performance (AUC) | Key Lipidomic Signature |
|---|---|---|---|
| Pancreatic Cancer (PDAC) Detection [5] | Logistic Regression (LR), Random Forest (RF), SVM with radial basis kernel; Multi-cohort validation. | 0.9207 (LR Model), 0.9427 (with CA19-9) | 18 phospholipids, 1 acylcarnitine, 1 sphingolipid |
| Pediatric Inflammatory Bowel Disease (IBD) [50] | Seven ML algorithms with stacking; SCAD regularization; Independent inception cohort validation. | 0.85 (Discovery), 0.85 (Validation) | Lactosyl ceramide (d18:1/16:0), Phosphatidylcholine (18:0p/22:6) |
| Osteonecrosis of Femoral Head (ONFH) [6] | LASSO, SVM-RFE, Boruta algorithms; Multi-database validation. | >0.7 for all biomarkers | CREBBP, GLB1, PSAP (Lipid metabolism related) |
These studies exemplify the rigorous methodology required for credible biomarker development. The pancreatic cancer study, for instance, utilized three distinct human plasma sets (Sets A, B, and C) for discovery, verification, and validation, ensuring the identified lipid signature was not an artifact of a particular sample group [5]. The workflow for such a multi-center study typically follows a structured path to ensure robustness.
The Scientist's Toolkit: Essential Research Reagents and Solutions
The experimental protocols cited rely on a suite of analytical technologies and bioinformatics tools. The following table details key resources essential for conducting rigorous, reproducible lipidomic research.
Table 5: Essential Research Reagent Solutions for Lipidomics Biomarker Discovery
| Tool / Reagent | Specific Example / Platform | Primary Function in Workflow |
|---|---|---|
| LC-MS/MS System | Ultimate 3000-LTQ-Orbitrap XL [5] | High-resolution lipid separation, detection, and quantification. |
| Metabolite Identification Software | Compound Discoverer 3.3 [5] | Processes raw MS data; identifies metabolites via database matching. |
| Multivariate Analysis Software | SIMCA [67] or MetaboAnalyst | Performs PCA, PLS-DA for exploratory data analysis and dimensionality reduction. |
| Machine Learning Environment | R (glmnet, Boruta, e1071) [6] or Python (scikit-learn) [70] | Implements regularized models (LASSO, SVM) and feature selection algorithms. |
| Bioinformatics Databases | Reactome, KEGG, Human Protein Atlas [6] | Provides biological context; enables functional enrichment analysis of candidate biomarkers. |
Achieving a generalizable model in lipidomics requires a synergistic application of both cross-validation and regularization. Cross-validation provides the unbiased performance estimate needed to guide model selection and hyperparameter tuning, including the strength of regularization. Regularization, in turn, produces the more robust models that yield stable results during cross-validation. This interplay is critical for developing biomarkers that can reliably transfer across multiple clinical centers and diverse patient populations.
Based on the experimental data and case studies reviewed, the following integrated protocol is recommended for lipidomic biomarker studies:
- Experimental Design: Partition data into discovery, verification, and hold-out validation sets from the outset. Employ k-fold cross-validation (typically k=5 or k=10) within the discovery set for model development [68] [69].
- Model Training with Regularization: Apply regularized models (e.g., LASSO, SVM with RBF kernel) to the high-dimensional lipidomic data. Use the cross-validation results to tune hyperparameters, including regularization strength (λ, C) and dropout rates [71] [6].
- Validation and Interpretation: Apply the final model to the independent validation set for an unbiased performance assessment. Use multivariate statistical methods like PCA and PLS-DA to visualize and interpret the model's findings in a biological context [67].
In conclusion, mitigating overfitting is not a single-step solution but a rigorous process enforced by methodological discipline. Cross-validation and regularization are not competing techniques but complementary pillars of robust machine learning. For researchers and drug development professionals working on the frontier of lipidomic biomarkers, their diligent application is the foundation upon which clinically viable and commercially successful diagnostic signatures are built.
The reproducibility and clinical translation of lipidomic biomarkers depend critically on overcoming significant methodological variations across research sites. Inconsistent pre-analytical handling, analytical protocols, and data processing methodologies introduce substantial variability that compromises data quality and hinders the validation of clinically relevant lipid biomarkers [25]. Multi-center studies face particular challenges in lipidomics, where structural diversity of lipids, pre-analytical variables, and analytical platform discrepancies collectively contribute to what has been termed the "reproducibility crisis" in biomedical research [72].
The magnitude of this challenge is evident from inter-laboratory comparisons showing alarmingly low concordance. When identical liquid chromatography-mass spectrometry (LC-MS) lipidomic data were processed through different software platforms (MS DIAL and Lipostar) using default settings, the identification agreement was merely 14.0% for MS1 data and 36.1% when including MS2 fragmentation data [73]. This reproducibility gap highlights the critical need for standardized approaches across all phases of multi-center lipidomic studies, from sample collection to data annotation.
Pre-analytical Standardization: Foundation for Reproducible Lipidomics
Critical Pre-analytical Variables and Their Impacts
Pre-analytical variables introduce significant variability in lipidomic measurements, potentially overshadowing true biological signals. Evidence from standardized protocols demonstrates that controlling these factors substantially improves inter-laboratory reproducibility [74].
Table 1: Key Pre-analytical Variables and Standardization Recommendations for Multi-Center Lipidomics
| Pre-analytical Phase | Variable | Impact on Lipidomics Data | Standardization Recommendation |
|---|---|---|---|
| Sample Collection | Blood collection tube type | Affects MP counts and lipid profiles | Use specified citrate tubes (3.2%, 3.5mL minimum); avoid CTAD, EDTA, heparin, ACD tubes, glass tubes [74] |
| Tourniquet use & venipuncture | Alters lipid concentrations | Apply light tourniquet; discard first 2-3mL blood; use ≥21-gauge needle [74] | |
| Time of collection | Diurnal lipid variation | Collect from fasting subjects in morning (08:00-11:00) [74] | |
| Sample Handling | Time to processing | Lipid degradation and modification | Process within <2 hours at room temperature (20-24°C) [74] |
| Transportation conditions | Artificial MP formation | Use steady vertical position boxes; avoid agitation [74] | |
| Sample Processing | Centrifugation protocol | Dramatically affects MP and lipid particle counts | First centrifugation: 2500×g, 15min, room temperature, no brake [74] |
| Plasma collection | Cellular contamination | Leave 1cm plasma above buffy coat; use precise pipetting (1000μL then 200μL) [74] | |
| Sample Storage | Freezing method | Lipid stability | Snap-freeze in liquid nitrogen; store at -80°C [74] |
| Freeze-thaw cycles | Lipid degradation | Limit freeze-thaw cycles; document any deviations |
The implementation of a common pre-analytical protocol across 14 laboratories demonstrated a measurable improvement in inter-laboratory reproducibility, reducing the mean coefficient of variation of platelet-derived microparticle (PMP) counts from 80% with individual laboratory protocols to 66% with standardized methods [74]. Centrifugation force was identified as particularly critical, exhibiting an inverse correlation with MP counts [74].
Pre-analytical Workflow Standardization
The following workflow diagram illustrates a standardized pre-analytical protocol for multi-center lipidomics studies:
Analytical Standardization: Harmonizing Lipid Detection and Quantification
Analytical Platform Variability and Harmonization Approaches
Significant variability in lipid identification and quantification arises from differences in analytical platforms, instrumentation, and data processing software. The CLINSPECT-M consortium conducted a systematic round-robin study comparing six proteomic laboratories analyzing identical plasma and cerebrospinal fluid samples [75]. Each laboratory applied their own "best practice" protocols for sample preparation and LC-MS measurement, followed by a second round with harmonized methods based on shared learnings.
Table 2: Lipidomics Software Comparison and Identification Consistency
| Software Platform | Identification Agreement | Key Limitations | Quality Control Requirements |
|---|---|---|---|
| MS DIAL | 14.0% (MS1)36.1% (MS2) | Default libraries may yield inconsistent identifications; co-elution issues | Manual curation; retention time validation; ECN model verification [73] [76] |
| Lipostar | 14.0% (MS1)36.1% (MS2) | Alignment methodologies affect peak identification; library dependencies | Cross-platform validation; fragmentation pattern confirmation [73] |
| Lipid Data Analyzer (LDA) | N/A | Specialized for phospholipids and glycerolipids | Platform-independent; uses ECN model for retention validation [77] |
| Rule-Based Approaches | 1-10% false positive rate (class-dependent) | Requires lipid class-specific fragmentation rules | Must detect class-specific fragments (e.g., m/z 184.07 for PC) [76] |
Following protocol harmonization and transparent exchange of methods between laboratories, the CLINSPECT-M study demonstrated improved identification rates, data completeness, and reproducibility in the second measurement round [75]. This highlights the value of expert-driven exchange of best practices for direct practical improvements in multi-center studies.
Lipid Identification Confidence Framework
Correct lipid identification requires multiple lines of evidence to minimize false positives. Analyses of published lipid datasets reveal that a significant proportion of reported identifications may be unreliable when relying solely on software annotations without manual validation [76]. The following workflow ensures comprehensive lipid identification:
Experimental Protocols for Multi-Center Harmonization
Protocol for Inter-Laboratory Method Harmonization
The CLINSPECT-M consortium established a systematic approach for analytical harmonization across multiple sites [75]:
Initial Benchmark Phase: Each participating laboratory receives identical aliquots of reference plasma or other biological samples. Laboratories apply their current best-practice protocols for sample preparation and LC-MS analysis without constraints.
Centralized Data Analysis: All generated MS data are analyzed using a common pipeline to eliminate variability introduced by different bioinformatic tools. In the CLINSPECT-M study, this involved using MaxQuant for data-dependent acquisition data with consistent parameters and false discovery rate set to 1% for both peptide spectrum matches and protein level [75].
Transparent Method Exchange: Participating laboratories share detailed protocols, including sample preparation techniques, LC gradients, MS instrument settings, and data processing parameters.
Method Optimization and Re-analysis: Laboratories refine their methods based on collective insights and re-analyze the same samples using improved, harmonized protocols.
Performance Assessment: Key metrics including identification numbers, data completeness, retention time precision, quantitative accuracy, and inter-laboratory reproducibility are compared between phases.
Lipid Extraction and Analysis Protocol for Multi-Center Studies
A standardized lipid extraction and analysis protocol based on modified Folch extraction provides consistency across sites [73]:
Sample Preparation:
- Add 0.75mL methanol to 100μL plasma sample in glass tube with Teflon-lined cap
- Vortex thoroughly to mix
- Add 2.5mL methyl-tert-butyl ether (MTBE)
- Incubate 1 hour at room temperature with shaking
- Induce phase separation with 0.625mL MS-grade water
- Incubate 10 minutes at room temperature
- Centrifuge at 1000×g for 10 minutes
- Collect upper organic phase
- Re-extract lower phase with 1mL MTBE:methanol (2.5:0.75) solvent mixture
- Combine organic phases and dry under nitrogen
- Reconstitute in 100μL isopropanol for UHPLC-MS/MS analysis
LC-MS Analysis:
- System: UHPLC system coupled to high-resolution mass spectrometer
- Column: Reversed-phase C18 column (e.g., 50×0.3mm, 1.7μm)
- Mobile Phase: A: 60:40 acetonitrile:water; B: 85:10:5 isopropanol:water:acetonitrile
- Both supplemented with 10mM ammonium formate and 0.1% formic acid
- Gradient: 0-0.5min (40% B), 0.5-5min (99% B), 5-10min (99% B), 10-12.5min (40% B), 12.5-15min (40% B)
- Mass spectrometry: Positive and negative ion modes; data-dependent MS2 acquisition
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for Standardized Multi-Center Lipidomics
| Category | Specific Product/Type | Function in Lipidomics Workflow | Standardization Importance |
|---|---|---|---|
| Blood Collection | 3.2% citrate tubes (3.5mL minimum) | Anticoagulation without lipid interference | Consistent anticoagulant concentration critical for MP analysis [74] |
| Internal Standards | Avanti EquiSPLASH LIPIDOMIX | Deuterated lipid internal standards for quantification | Enables cross-laboratory quantification accuracy [73] |
| Extraction Solvents | HPLC-grade methanol, MTBE, chloroform | Lipid extraction with minimal degradation | Solvent purity affects extraction efficiency and MS compatibility [73] |
| LC-MS Mobile Phase | Ammonium formate, formic acid | LC buffer systems for positive/negative mode MS | Consistent additive concentration crucial for retention time reproducibility [73] |
| Quality Controls | HeLa standard digest (Pierce) | System suitability and performance monitoring | Allows inter-laboratory performance comparison [75] |
| Retention Time Calibration | iRT peptides (PROCAL, JPT) | Retention time standardization across gradients | Enables cross-laboratory retention alignment [75] |
| Reference Materials | Synthetic lipid standards | Retention time and fragmentation confirmation | Essential for confident lipid identification [76] [77] |
Quality Control and Data Standards Framework
Minimum Reporting Standards for Lipidomics
The Lipidomics Standards Initiative has developed reporting guidelines to improve reproducibility and data quality [77]:
Identification Confidence Levels:
- Level 1: Identified by comparison to authentic standard using accurate mass, retention time, and fragmentation spectrum
- Level 2: Putatively annotated by accurate mass and characteristic fragmentation pattern
- Level 3: Putatively characterized by accurate mass and retention behavior
- Level 4: Accurate mass only
Chromatographic Quality Metrics:
- Signal-to-noise ratio: ≥5:1 for quantification, ≥3:1 for detection
- Data points across peak: Minimum 6-10 points for reliable integration
- Retention time stability: ≤2% relative standard deviation for quality controls
- Peak shape: Gaussian shape without significant fronting or tailing
Machine Learning Approaches for Quality Control
Advanced computational methods can augment traditional quality control. Support vector machine (SVM) regression combined with leave-one-out cross-validation (LOOCV) can identify potential false positive identifications by detecting outliers in retention time behavior [73]. These approaches leverage the predictable relationship between lipid structure and chromatographic retention, encapsulated in the Equivalent Carbon Number (ECN) model:
ECN Model Equation: RT(x,y) = A × (1 - B × x^(-C)) + D × e^(-E × y + F × x) + G
Where x = number of carbon atoms, y = number of double bonds, RT = retention time, and A-G are fitted parameters for each lipid class and chromatographic setup [76].
Successful multi-center lipidomic studies require integrated standardization across pre-analytical, analytical, and computational phases. Evidence from collaborative consortia demonstrates that systematic harmonization approaches significantly improve inter-laboratory reproducibility and data quality [75] [74]. The convergence of standardized protocols, reference materials, quality control frameworks, and data standards creates a foundation for clinically translatable lipidomic biomarkers. Implementation of these practices across research networks will accelerate the validation of lipid biomarkers and their translation into clinical practice, ultimately supporting personalized medicine approaches in cardiometabolic diseases, cancer, and neurodegenerative disorders [25] [78].
In the rigorous field of clinical lipidomics, researchers face a fundamental dilemma: untargeted lipidomics provides extensive coverage of lipid species but often lacks the quantitative precision required for clinical validation, while targeted lipidomics offers exceptional accuracy but is restricted to predefined analytes, potentially missing novel biological insights [4] [27] [79]. This methodological gap becomes particularly problematic in multi-center biomarker research, where both comprehensive coverage and precise quantification are prerequisites for developing clinically applicable diagnostic tools [4] [80].
The pseudo-targeted metabolomics strategy has emerged as a hybrid solution to this challenge, effectively bridging the critical gap between discovery-oriented and validation-focused approaches [81]. First conceptualized in 2012 and subsequently optimized for liquid chromatography-mass spectrometry (LC-MS) platforms, pseudo-targeted methodology transforms untargeted lipidomic data into a targeted analysis framework, enabling highly sensitive monitoring of thousands of lipid species without sacrificing quantitative reliability [81]. This balanced approach has proven particularly valuable in clinical research settings, where it supports both biomarker discovery and verification phases within a unified analytical workflow.
For research aimed at clinical translation, the pseudo-targeted approach addresses several persistent challenges in lipidomics. Prominent among these are the reproducibility issues observed across different analytical platforms, with studies reporting disconcertingly low agreement rates (as low as 14-36%) when identical samples are analyzed using different lipidomic software platforms [4]. By establishing a predefined set of lipid targets derived from initial untargeted discovery, the pseudo-targeted method enhances cross-laboratory consistency while maintaining the breadth of coverage necessary for comprehensive biomarker research.
Core Methodologies: A Comparative Analysis of Lipidomics Approaches
Technical Foundations and Workflows
Lipidomics methodologies can be broadly categorized into three distinct approaches, each with characteristic strengths and limitations for biomarker research. Untargeted lipidomics provides a comprehensive, unbiased analysis of all detectable lipids in a sample, typically utilizing high-resolution mass spectrometry (HRMS) platforms such as Quadrupole Time-of-Flight (Q-TOF) or Orbitrap instruments [27]. This approach is ideally suited for hypothesis generation and discovery of novel lipid biomarkers, as it requires no prior knowledge of the lipid species present in a sample. However, its limitations include lower quantitative accuracy, greater susceptibility to matrix effects, and more complex data processing requirements [79] [80].
In contrast, targeted lipidomics employs triple quadrupole (TQ) or Q-Trap mass spectrometers operating in multiple reaction monitoring (MRM) or parallel reaction monitoring (PRM) modes to achieve precise identification and quantification of specific, predefined lipid molecules [27] [79]. This approach provides superior sensitivity, linear dynamic range, and quantitative precision—attributes essential for clinical assay validation. However, its targeted nature inherently restricts the scope of analysis to known lipids, potentially overlooking unexpected biomarkers [79].
The pseudo-targeted approach strategically integrates elements from both methodologies, creating a streamlined workflow that begins with untargeted analysis to identify candidate lipids, followed by development of a targeted method for precise quantification [81]. The standardized workflow encompasses several critical stages: initial sample collection and metabolite extraction; untargeted data acquisition using UHPLC-HRMS with data-independent (DIA) or data-dependent acquisition (DDA); identification of optimal ion pairs using specialized software; and finally, targeted analysis and quantification using triple quadrupole or Q-Trap systems [81].
Table 1: Comparative Analysis of Lipidomics Approaches for Biomarker Research
| Parameter | Untargeted Lipidomics | Pseudo-Targeted Lipidomics | Targeted Lipidomics |
|---|---|---|---|
| Metabolite Coverage | Broad (1000s of features) | High (100s-1000s of features) | Narrow (10s-100s of analytes) |
| Quantitative Accuracy | Limited (semi-quantitative) | Good to High | Excellent |
| Analytical Sensitivity | Moderate | High | Very High |
| Best Application | Discovery phase, novel biomarker identification | Biomarker verification & validation | Clinical assay validation, absolute quantification |
| Throughput | Lower (complex data processing) | Moderate | High |
| Standardization Needs | High | Moderate | Lower (already standardized) |
| Multi-center Reproducibility | Challenging | Achievable with protocols | Most achievable |
Visualizing the Pseudo-Targeted Lipidomics Workflow
The following diagram illustrates the integrated workflow of pseudo-targeted lipidomics, highlighting how it bridges untargeted and targeted approaches:
Experimental Evidence: Performance in Multi-Center Studies
Rheumatoid Arthritis Diagnostic Biomarker Development
A comprehensive multi-center investigation across seven cohorts exemplifies the rigorous application of pseudo-targeted methodology in clinical biomarker research [80]. This study, encompassing 2,863 blood samples from five medical centers across three geographically diverse regions, initially employed untargeted metabolomics to identify candidate biomarkers, then transitioned to targeted approaches for validation. The research ultimately identified six metabolites—including imidazoleacetic acid, ergothioneine, and N-acetyl-L-methionine—as promising diagnostic biomarkers for rheumatoid arthritis (RA) [80].
The classification models developed from these biomarkers demonstrated robust discriminatory power across multiple independent validation cohorts. For RA versus healthy control classification, the models achieved area under the curve (AUC) values ranging from 0.8375 to 0.9280, while RA versus osteoarthritis classifiers achieved moderate to good accuracy (AUC range: 0.7340–0.8181) [80]. Importantly, the classifier performance remained independent of serological status, effectively identifying seronegative RA cases that often present diagnostic challenges. This study highlights how pseudo-targeted methodologies can facilitate the development of clinically applicable models that perform consistently across diverse patient populations and geographic locations.
Pancreatic Cancer Biomarker Discovery
In pancreatic ductal adenocarcinoma (PDAC) research, pseudo-targeted methodologies have demonstrated exceptional utility in identifying lipid-based biomarkers with clinical potential [5]. A recent investigation analyzed plasma samples from 202 individuals (99 normal, 103 PDAC) using non-targeted approaches to identify candidate lipids, followed by targeted validation. The research identified 20 lipid biomarkers—including acylcarnitines, sphingolipids, and phospholipids—that consistently distinguished healthy individuals from cancer patients [5].
When incorporated into logistic regression models, panels containing 11 or more phospholipids achieved a separation performance of 0.9207 AUC between healthy individuals and cancer patients, significantly outperforming the conventional biomarker CA19-9, which achieved an AUC of only 0.7354 in the same validation set [5]. The combined model integrating both phospholipid biomarkers and CA19-9 further improved performance to 0.9427 AUC, demonstrating the complementary value of lipidomic biomarkers to existing clinical tools. This study exemplifies how pseudo-targeted approaches can yield biomarker panels with enhanced diagnostic performance compared to conventional single-protein biomarkers.
Table 2: Performance Metrics of Lipid Biomarkers in Multi-Center Studies
| Disease Context | Biomarker Class | Sample Size | Analytical Approach | Performance (AUC) | Reference |
|---|---|---|---|---|---|
| Rheumatoid Arthritis | 6-metabolite panel | 2,863 participants (7 centers) | Untargeted discovery → Targeted validation | 0.8375-0.9280 (vs. healthy controls) | [80] |
| Pancreatic Cancer | 18 phospholipids | 202 participants | Non-targeted screening → Targeted validation | 0.9207 (vs. CA19-9 at 0.7354) | [5] |
| Ovarian Cancer | Multi-omic (lipids, gangliosides, proteins) | ~1,000 symptomatic women | LC-MS + immunoassays + machine learning | 0.89-0.92 (early-stage detection) | [82] |
| Extranodal NK/T-cell Lymphoma | Triglycerides + ApoA1 | 1,017 patients + matched controls | LC-MS-based metabolomic profiling | HR=1.33 (PFS), 1.37 (OS) for high TG | [83] |
The Scientist's Toolkit: Essential Reagents and Platforms
Successful implementation of pseudo-targeted lipidomics requires specific analytical tools and reagents carefully selected to ensure reproducibility and quantification accuracy. The following table details essential components of the lipidomics research pipeline:
Table 3: Essential Research Reagent Solutions for Pseudo-Targeted Lipidomics
| Category | Specific Examples | Function in Workflow | Technical Considerations |
|---|---|---|---|
| Chromatography Systems | UHPLC (e.g., Vanquish, Ultimate 3000) | Lipid separation prior to MS analysis | Compatibility with reverse-phase and HILIC columns for comprehensive coverage |
| Mass Spectrometry Platforms | Q-TOF (e.g., Orbitrap Exploris), Triple Quadrupole (e.g., UPLC-QQQ) | Lipid detection & quantification | HRMS for discovery, TQ for validation; ensure mass accuracy <5 ppm |
| Chromatography Columns | Waters ACQUITY BEH Amide, C18 columns | Compound separation | Different column chemistries for various lipid classes |
| Internal Standards | Deuterated lipid standards, isotope-labeled compounds | Quantification normalization | Critical for correcting matrix effects & ionization efficiency |
| Sample Preparation Kits | Protein precipitation kits, lipid extraction kits | Sample clean-up & metabolite extraction | Methanol:acetonitrile (1:1) common for protein precipitation |
| Data Processing Software | Compound Discoverer, MS DIAL, Lipostar | Lipid identification & quantification | Automated peak picking, alignment, and database matching |
Implementation Framework for Multi-Center Studies
Standardization of protocols across participating sites represents perhaps the most critical challenge in multi-center lipidomic biomarker research. The pseudo-targeted approach offers distinct advantages in this context by establishing a predefined set of transitions for monitoring, thereby reducing inter-laboratory variability [4] [80]. Successful implementation requires careful attention to several key aspects:
Sample Collection and Processing: Standardization begins at sample collection, with consistent use of anticoagulants (EDTA for plasma), processing protocols, and storage conditions (-80°C or liquid nitrogen) across all sites [80]. Studies indicate that plasma is generally preferred over serum for biomarker discovery due to greater consistency and easier handling, though researchers should note that lipid profiles differ between these sample types [79].
Quality Control Procedures: Implementation of rigorous quality control measures is essential, including use of pooled quality control (QC) samples, internal standards, and regular instrument calibration [80]. Incorporating deuterated internal standards for quantification normalization helps correct for matrix effects and variations in ionization efficiency, significantly enhancing data quality and cross-site comparability [79].
Data Integration and Analysis: For pseudo-targeted lipidomics, the integration of untargeted and targeted data streams requires specialized bioinformatics approaches. Machine learning algorithms have demonstrated particular utility in analyzing these complex multi-omic datasets, revealing disease-specific signatures that might be overlooked using conventional statistical methods [82] [80].
The following diagram illustrates the strategic integration of pseudo-targeted lipidomics within a multi-center biomarker validation framework:
Pseudo-targeted lipidomics represents a strategically balanced approach that successfully addresses the critical tension between metabolite coverage and quantification accuracy in clinical biomarker research. By integrating the discovery power of untargeted methods with the precision of targeted approaches, this methodology provides an optimal framework for biomarker development and validation across multiple research centers [81] [80].
The future trajectory of pseudo-targeted lipidomics will likely be shaped by several technological innovations. Artificial intelligence and machine learning are already demonstrating remarkable potential in lipidomics, with models like MS2Lipid achieving up to 97.4% accuracy in predicting lipid subclasses [4]. Further development of these computational approaches, coupled with expanded lipid databases and standardized reporting frameworks, will enhance the efficiency and reliability of pseudo-targeted methods.
Additionally, the growing integration of lipidomics with other omics technologies—including genomics, transcriptomics, and proteomics—will provide more comprehensive insights into disease mechanisms and strengthen the biological validation of candidate biomarkers [4] [6]. As these multi-omic approaches mature, pseudo-targeted lipidomics will undoubtedly play an increasingly pivotal role in translating lipid biomarker discoveries into clinically applicable tools for precision medicine.
For research teams embarking on multi-center lipidomic studies, the pseudo-targeted approach offers a practical pathway to generate both comprehensive discovery data and clinically actionable validation results within a unified analytical framework. This balanced methodology effectively addresses the dual imperatives of innovation and translation that define modern biomarker research.
Multi-Center Validation and Benchmarking: The Path to Clinical Utility
Multi-center validation studies represent a critical step in translating biomarker research into clinically applicable tools. Within lipidomics biomarker research, these studies are particularly vital for assessing the generalizability and robustness of candidate biomarkers across diverse patient populations and clinical settings. Multi-center validation involves confirming that a biomarker's performance remains consistent and reliable when applied to data collected from multiple independent institutions, rather than just the single center where it was developed [84] [85]. This process helps identify biomarkers that truly reflect biological phenomena rather than site-specific artifacts.
The validation of lipidomic biomarkers presents unique methodological challenges. Lipid molecules exhibit considerable structural diversity and are influenced by numerous pre-analytical factors, including sample processing protocols, storage conditions, and analytical platforms [4]. When designing multi-center studies, researchers must carefully control for these variables to ensure that observed lipid profile differences genuinely reflect disease states rather than technical variations. Furthermore, the integration of lipidomic data with clinical, genomic, and proteomic data requires sophisticated statistical approaches and machine learning frameworks to achieve sufficient predictive power for clinical application [4] [5].
Fundamental Considerations for Cohort Selection
Defining Eligibility Criteria and Recruitment Strategy
The foundation of any robust multi-center validation study lies in careful cohort selection. Eligibility criteria must strike a balance between being sufficiently specific to target the intended population and broad enough to ensure recruitment feasibility and generalizability. Research indicates that clearly defined inclusion and exclusion criteria are essential for minimizing selection bias across participating centers [86]. For lipidomic biomarker studies, this typically involves specifying diagnostic criteria, prior treatment history, demographic factors, and comorbid conditions that might influence lipid metabolism.
The recruitment strategy must account for potential differences in patient populations across centers. As demonstrated in a multicenter lipidomic study for pancreatic cancer diagnosis, researchers successfully enrolled participants from multiple institutions, with a final cohort comprising 165 normal controls and 180 pancreatic cancer patients across two sample sets [5]. This approach ensured adequate representation of both case and control populations. When designing recruitment targets, consider that tertiary referral centers may enroll patients with more advanced disease stages compared to secondary-level hospitals, potentially influencing biomarker performance [84].
Determining Appropriate Sample Size
Sample size determination must account for both the expected effect size and the multi-center design. The table below summarizes key considerations for sample size calculation in multi-center biomarker validation studies:
Table 1: Key Factors in Sample Size Calculation for Multi-Center Studies
| Factor | Consideration | Impact on Sample Size |
|---|---|---|
| Primary Outcome | Binary (e.g., disease presence) vs. Continuous (e.g., risk score) | Larger sample needed for binary outcomes [87] |
| Effect Size | Minimum detectable difference in biomarker performance | Smaller effect sizes require larger samples [87] |
| Statistical Power | Probability of detecting true effect | Higher power (typically 80-95%) requires larger samples [87] [88] |
| Significance Level | Risk of false-positive findings (Type I error) | Lower alpha (e.g., 0.01 vs. 0.05) requires larger samples [87] |
| Center Heterogeneity | Variability between participating centers | Greater heterogeneity requires larger samples [89] |
| Attrition Rate | Expected loss to follow-up or missing data | Higher anticipated attrition requires larger initial recruitment [87] |
Statistical power, typically set at 80-95%, must be sufficient to detect clinically meaningful differences in biomarker performance [87] [88]. The sample size must also account for center-level effects and potential imbalances in recruitment across sites. Specialized statistical methods that incorporate random effects for center are recommended to properly account for between-center heterogeneity [89].
Addressing Missing Data and Attrition
Proactive planning for missing data is essential in multi-center studies. Research shows that missing data patterns can vary significantly between institutions due to differences in local protocols, data storage systems, and documentation practices [85]. A study validating a machine learning model for postoperative mortality reported missing value proportions ranging from 3.76% to 19.66% across different hospitals, with specific variables (e.g., body mass index, ASA-PS grade) particularly affected at certain sites [85]. Establishing uniform data collection standards across all participating centers and implementing systematic monitoring can help minimize missing data. Statistical techniques such as multiple imputation should be pre-specified in the analytical plan to handle inevitable missing values appropriately.
Powering Considerations for Multi-Center Studies
Statistical Framework for Sample Size Calculation
Power analysis for multi-center studies requires specialized statistical approaches that account for the clustered nature of the data. Conventional sample size calculations that assume simple random sampling may substantially overestimate power in multi-center settings. For continuous outcomes, mixed models that incorporate random effects for center provide a more appropriate framework for sample size estimation [89]. These models account for the intra-class correlation that arises when patients from the same center are more similar to each other than to patients from different centers.
The sample size calculation must also consider the allocation ratio between groups and potential imbalances across centers. Block randomization with fixed block length is commonly used to maintain balance within centers, but the choice of block size involves trade-offs [89]. Smaller blocks minimize the risk of substantial imbalance but may increase the risk of unmasking the allocation sequence. The anticipated distribution of patients across treatment or exposure groups should be incorporated into power calculations, as imbalance between groups can result in substantial power loss, particularly when center heterogeneity is present [89].
Accounting for Center-Level Variability
Center-level variability can substantially impact the statistical power of multi-center studies. This variability may arise from differences in patient populations, local treatment practices, or technical aspects of biomarker measurement. The degree of center-level variability is often quantified by the intra-class correlation coefficient (ICC) or the variance component (τ²) in mixed models [89]. Larger values of these parameters indicate greater between-center heterogeneity and necessitate larger sample sizes to maintain statistical power.
The number and size of participating centers also influence power. Studies with many small centers may face challenges related to center-level imbalance, while studies with few large centers may be more vulnerable to the influence of center-specific practices. A simulation-based approach to power calculation can help optimize the trade-offs between the number of centers and the number of participants per center [89]. Research suggests that including centers with differing levels of care (secondary-level general hospitals and tertiary-level academic referral hospitals) enhances the generalizability of validation findings [84].
Experimental Protocols for Lipidomic Biomarker Validation
Standardized Sample Processing and Analysis
The experimental protocol for multi-center lipidomic biomarker validation must prioritize standardization across participating sites. Based on successful lipidomic studies, the following workflow represents best practices:
Table 2: Essential Research Reagents and Platforms for Lipidomic Biomarker Studies
| Category | Specific Examples | Function in Lipidomic Analysis |
|---|---|---|
| Sample Collection | EDTA plasma tubes, serum separator tubes | Standardized biological sample collection |
| Internal Standards | Stable isotope-labeled lipid standards | Quantification normalization & quality control |
| Extraction Solvents | Methyl tert-butyl ether (MTBE), chloroform-methanol | Lipid extraction from biological samples |
| LC-MS Platforms | Ultra-performance liquid chromatography systems | Lipid separation prior to mass analysis |
| Mass Spectrometers | Q-TOF, Orbitrap instruments | High-resolution lipid identification and quantification |
| Quality Control | Pooled quality control samples, NIST SRM 1950 | Monitoring analytical performance across batches |
| Bioinformatics | MS-DIAL, Lipostar, Compound Discoverer | Lipid identification, alignment, and statistical analysis |
The workflow should begin with standardized sample collection procedures, specifying details such as fasting status, time of day for collection, processing timelines, and storage conditions (-80°C) [4] [5]. For lipidomic analyses, the use of internal standards is particularly important for normalizing quantification across batches and sites. The National Institute of Standards and Technology Standard Reference Material (NIST SRM) 1950 represents a commonly used quality control material for lipidomic analyses [4].
Lipid extraction should follow validated protocols, such as methyl tert-butyl ether (MTBE) or chloroform-methanol methods, with detailed specifications for solvent ratios, mixing procedures, and phase separation [5]. Either targeted or untargeted lipidomics approaches may be employed, depending on the study objectives. Untargeted approaches provide comprehensive lipid profiling but may face challenges in structural identification and quantification, while targeted approaches offer better sensitivity and quantification for pre-specified lipid classes [4]. As demonstrated in a pancreatic cancer biomarker study, a combination of non-targeted discovery followed by targeted validation represents an effective strategy [5].
Quality Control Procedures
Rigorous quality control procedures are essential for generating reliable lipidomic data across multiple centers. These should include: (1) pooled quality control samples analyzed regularly throughout analytical batches to monitor instrument stability; (2) technical replicates to assess precision; (3) standard reference materials to evaluate accuracy; and (4) blank samples to identify contamination [4]. For multi-center studies, a central laboratory coordinating quality assurance is highly recommended. This center can distribute standardized protocols, reference materials, and acceptance criteria for analytical performance.
Data quality metrics should be pre-specified, including thresholds for retention time stability, mass accuracy, peak intensity variability, and extraction efficiency [4]. Recent studies highlight concerning variability in lipid identification across different software platforms, with agreement rates as low as 14-36% when using default settings [4]. To address this, multi-center studies should implement harmonized data processing protocols, including consistent parameter settings for peak picking, alignment, and identification.
Data Analysis and Interpretation
Statistical Analysis Framework
The statistical analysis plan for multi-center lipidomic biomarker studies should address both the multi-center design and the high-dimensional nature of lipidomic data. For biomarker classification performance, receiver operating characteristic (ROC) analysis is commonly used, with area under the curve (AUC) values reported along with confidence intervals [5] [85]. A recent multi-center study of postoperative complications reported AUC values ranging from 0.789 to 0.925 for different complications across validation cohorts, demonstrating robust performance [84].
Machine learning algorithms have shown particular promise for handling the complexity of lipidomic data. Successful multi-center validation studies have employed various algorithms, including logistic regression, random forests, support vector machines, and gradient boosting methods [5] [85] [86]. For example, a multicenter study on pancreatic cancer diagnosis utilized logistic regression, random forest, and support vector machine models, achieving AUC values up to 0.9427 when combining lipid biomarkers with CA19-9 [5]. Similarly, a study predicting postoperative mortality found that XGBoost and logistic regression models maintained strong performance across external validation sites, with AUC values up to 0.941 [85].
The analysis plan must account for the multi-center design through appropriate statistical methods. Mixed effects models that include center as a random effect can help account for between-center variability [89]. Alternatively, stratified analyses or meta-analytic approaches that combine results across centers may be employed. Covariate adjustment for patient characteristics that vary across centers (e.g., age, disease severity, comorbidities) is essential to minimize confounding.
Validation and Generalizability Assessment
External validation performance should be compared against derivation cohort performance to assess generalizability. A significant decrease in performance upon external validation suggests potential overfitting or limited transportability of the biomarker. Successful multi-center validations demonstrate consistent performance across sites with different patient populations and practice patterns [84] [85] [86].
The following diagram illustrates the complete workflow for a multi-center lipidomic biomarker validation study:
Multi-Center Lipidomic Biomarker Validation Workflow
When interpreting results, researchers should consider both statistical significance and clinical utility. Decision curve analysis can help evaluate the clinical net benefit of incorporating the biomarker into decision-making [86]. Additionally, comparing the novel biomarker against established standards is essential. In the pancreatic cancer lipidomic study, researchers demonstrated that their biomarker panel significantly outperformed the FDA-approved CA19-9 biomarker, with AUC values of 0.9207 versus 0.7354 in the validation set [5].
Designing a robust multi-center validation study for lipidomic biomarkers requires meticulous attention to cohort selection, power calculations, and experimental standardization. By implementing rigorous protocols for sample processing, data generation, and statistical analysis across participating centers, researchers can generate compelling evidence regarding biomarker performance and generalizability. The framework presented in this review provides a roadmap for conducting methodologically sound multi-center studies that will accelerate the translation of lipidomic biomarkers from research discoveries to clinically useful tools.
The Three-Phase (Discovery, Qualification, Verification) Validation Model
The discovery of lipidomic biomarkers has been accelerated by advancements in high-resolution mass spectrometry, enabling the identification of thousands of lipid species across biological systems [4]. However, the transition of these findings from research settings to clinically applicable tools faces significant challenges, with biological variability, lipid structural diversity, inconsistent sample processing, and a lack of defined procedures exacerbating reproducibility and validation issues [4]. The three-phase validation model (discovery, qualification, verification) provides a systematic framework to address these challenges, offering a structured pathway for biomarker development that enhances reliability and clinical translatability.
This framework is particularly crucial in the context of multi-center validation studies, where standardized protocols ensure consistency across different analytical platforms and laboratories [4]. Recent studies have highlighted alarming discrepancies in lipidomic analyses, with prominent software platforms agreeing on as little as 14-36% of lipid identifications when using identical LC-MS data [4]. The three-phase model directly addresses these concerns through its graduated approach to validation, progressively increasing analytical stringency while expanding cohort sizes to establish both analytical validity and clinical utility.
Core Principles of the Three-Phase Validation Model
Phase Definitions and Objectives
The three-phase validation model represents a sequential, evidence-based approach to biomarker development that systematically addresses both analytical performance and biological significance. Each phase serves a distinct purpose in the biomarker development pipeline:
Discovery Phase: This initial phase focuses on untargeted lipidomic profiling to identify differentially abundant lipid species between case and control groups [27]. Using high-resolution mass spectrometry techniques such as Quadrupole Time-of-Flight Mass Spectrometry (Q-TOF MS) and Orbitrap MS, researchers comprehensively analyze lipid profiles without prior selection biases [27]. The objective is to generate hypotheses about potential lipid biomarkers by capturing global alterations in lipid metabolism associated with specific disease states.
Qualification Phase: In this intermediate stage, potential biomarker candidates from the discovery phase undergo preliminary validation using targeted or pseudo-targeted lipidomics approaches [27]. The focus shifts to verifying the directional consistency of lipid alterations and assessing technical variability using techniques such as multiple reaction monitoring (MRM) and parallel-reaction monitoring [27]. This phase typically employs larger sample sizes than the discovery phase and establishes initial performance characteristics of the biomarker signature.
Verification Phase: The final stage involves rigorous validation of the refined biomarker panel in independent, well-characterized cohorts that reflect the intended-use population [90]. This phase utilizes fully optimized targeted assays to quantify specific lipid molecules with high accuracy, precision, and sensitivity [27]. The verification phase aims to establish clinical validity and determine operational performance characteristics such as sensitivity, specificity, and area under the curve (AUC) values in realistic clinical scenarios.
Integration with Multi-Center Study Designs
The three-phase model is particularly well-suited for multi-center lipidomic biomarker research, as it incorporates geographical and technical validation directly into the development pathway. By validating biomarker candidates across different institutions and analytical platforms, researchers can address concerns about reproducibility and generalizability early in the development process [4]. This approach also facilitates the establishment of standardized operating procedures for sample collection, processing, storage, and analysis – critical elements for successful multi-center studies [4].
Table 1: Key Considerations for Multi-Center Studies in Each Validation Phase
| Phase | Primary Cohort Consideration | Analytical Standardization Needs | Data Harmonization Approach |
|---|---|---|---|
| Discovery | Homogeneous populations for clear signal detection | Consistent sample preparation protocols | Centralized data processing and lipid identification |
| Qualification | Expanded single-center cohort | Cross-platform analytical consistency | Harmonized quality control measures |
| Verification | Independent multi-center cohorts | Standardized operating procedures across sites | Batch effect correction and normalized reporting |
Application Across Disease Models: Comparative Performance Analysis
Early-Onset Lung Cancer Detection
The application of the three-phase validation model in early-onset lung cancer (EOLC) research demonstrates its utility in addressing clinically significant gaps in screening protocols. In a recent study, researchers employed targeted lipidomics analysis of plasma samples from 117 EOLC cases and 121 non-EOLC controls (all aged 18-49 years) in the discovery phase, identifying 843 lipids with 60 differentially expressed species [91]. Through cluster analysis, specific lipid metabolites were associated with risk factors including passive smoking (OR: 2.75, 95% CI: 1.08-7.29) and current smoking (OR: 15.65, 95% CI: 2.55-142.10) [91].
In the qualification phase, researchers refined the biomarker candidates using LASSO-bootstrap regression combined with the Boruta algorithm, selecting the most robust lipid markers while minimizing overfitting [91]. The verification phase employed a random forest model validated in an independent cohort, achieving an area under the curve (AUC) of 0.874 [91]. This final model incorporated only 6 lipids, demonstrating how the three-phase process efficiently distills complex lipidomic signatures into clinically applicable tools.
Pediatric Inflammatory Bowel Disease Diagnosis
In pediatric inflammatory bowel disease (IBD), the three-phase model addressed the challenge of differentiating IBD from non-IBD symptomatic controls using blood-based lipid biomarkers. The discovery phase analyzed plasma samples from a treatment-naïve pediatric cohort (58 IBD, 36 symptomatic controls), identifying 45 molecular lipids that differentiated IBD from controls [90]. Machine learning algorithms, including regularized logistic regression, were applied to identify optimal biomarker combinations.
The qualification phase refined the signature to 30 molecular lipids, which achieved an AUC of 0.87 (95% CI 0.79-0.93) in the discovery cohort [90]. The critical verification phase validated this signature in an independent inception cohort (80 IBD, 37 controls), maintaining strong performance with an AUC of 0.85 (95% CI 0.77-0.92) – significantly outperforming high-sensitivity C-reactive protein (AUC = 0.73, 95% CI 0.63-0.82, P < 0.001) [90]. The verified signature comprised just lactosyl ceramide (d18:1/16:0) and phosphatidylcholine (18:0p/22:6), demonstrating successful translation to a minimal biomarker panel.
Cross-Disease Comparison of Validation Performance
Table 2: Performance Metrics Across Disease-Specific Lipidomic Biomarker Studies
| Disease Application | Sample Size (Discovery/Validation) | Initial Lipid Candidates | Final Verified Signature | Performance (AUC) |
|---|---|---|---|---|
| Early-Onset Lung Cancer [91] | 117 cases/121 controls (discovery) + independent validation | 60 differentially expressed lipids | 6 lipids | 0.874 |
| Pediatric IBD [90] | 58 IBD/36 controls (discovery) + 80 IBD/37 controls (validation) | 45 differential lipids | 2 lipids | 0.85 (0.77-0.92) |
| Ischemic Stroke [92] | 20 cases/20 controls | 294 lipids assayed | 56 differential lipids | Significant differentiation (P<0.05) |
Methodological Frameworks and Experimental Protocols
Lipidomics Workflow and Analytical Platforms
The experimental methodology supporting the three-phase validation model relies on sophisticated lipidomics platforms that evolve in specificity and precision throughout the validation pipeline. The foundational workflow encompasses sample preparation, lipid extraction, chromatographic separation, mass spectrometric analysis, data processing, and statistical interpretation [27].
In the discovery phase, untargeted lipidomics utilizing high-resolution mass spectrometry (HRMS) platforms such as Quadrupole Time-of-Flight (Q-TOF) and Orbitrap instruments provides comprehensive lipidome coverage [27]. Data acquisition typically employs data-dependent acquisition (DDA) or data-independent acquisition (DIA) modes to capture global lipid profiles [27]. For example, in the EOLC study, targeted lipidomics combined with logistic regression was applied to plasma samples to identify differentially expressed lipid species [91].
The qualification phase typically transitions to pseudo-targeted approaches that combine the coverage advantages of untargeted methods with the quantitative rigor of targeted assays [27]. This phase often employs scheduled multiple reaction monitoring (MRM) or parallel reaction monitoring (PRM) on triple quadrupole or Q-TOF instruments to confirm the direction and magnitude of lipid alterations in expanded sample sets.
The verification phase utilizes fully optimized targeted methods on highly sensitive and specific platforms such as ultra-performance liquid chromatography-triple quadrupole mass spectrometry (UPLC-QQQ MS) [27]. These methods focus on precise quantification of the refined biomarker panel with enhanced sensitivity, linear dynamic range, and throughput suitable for larger validation cohorts.
Statistical and Bioinformatics Approaches
Each validation phase employs distinct statistical frameworks appropriate for the research objectives and data characteristics. The discovery phase emphasizes multivariate statistical methods such as principal component analysis (PCA) and orthogonal partial least squares discriminant analysis (OPLS-DA) to visualize group separations and identify lipid species contributing most to these separations [67] [92]. These are complemented by univariate analyses with appropriate multiple testing corrections (e.g., false discovery rate) [92].
The qualification phase incorporates machine learning algorithms for feature selection and model building. Common approaches include least absolute shrinkage and selection operator (LASSO) regression, random forests, and support vector machines [91] [90]. For instance, in the pediatric IBD study, researchers employed seven different machine learning algorithms with stacking to identify optimal biomarker combinations [90].
The verification phase focuses on establishing clinical performance metrics through receiver operating characteristic (ROC) analysis, calculation of area under the curve (AUC), sensitivity, specificity, and positive/negative predictive values [91] [90]. These analyses are performed on independent validation cohorts to ensure unbiased performance estimation.
Research Reagent Solutions and Essential Materials
Table 3: Essential Research Reagents and Platforms for Lipidomic Biomarker Validation
| Category | Specific Products/Platforms | Primary Application | Key Characteristics |
|---|---|---|---|
| MS Platforms | Q-TOF MS (Sciex, Waters), Orbitrap MS (Thermo), UPLC-QQQ MS (Waters, Agilent) | Untargeted (Discovery) vs. Targeted (Verification) | High resolution (>35,000), mass accuracy (<3 ppm), linear dynamic range (4-5 orders) |
| Chromatography | C18 columns (1.7-1.8 μm), HSS T3, CSH, BEH (Waters), ZORBAX (Agilent) | Lipid separation | Sub-2μm particles, 100mm length, stable at high pressures (>1000 bar) |
| Lipid Standards | SPLASH LIPIDOMIX, Avanti Polar Lipids standards | Quantitation | Deuterated internal standards covering major lipid classes |
| Sample Prep Kits | Matyash/MTBE protocol [92], Bligh & Dyer, Single-phase extractions | Lipid extraction | Protein precipitation, comprehensive recovery, minimal bias |
| Data Analysis | MS-DIAL, Lipostar, LipidSearch, XCMS, MetaboAnalyst | Lipid identification/quantitation | Spectral matching, peak alignment, false discovery control |
Challenges and Implementation Considerations
Analytical and Biological Variability
The implementation of the three-phase validation model must address several methodological challenges to ensure robust biomarker development. Pre-analytical variability represents a critical concern, as differences in sample collection, processing, and storage can significantly impact lipid profiles [4]. Standardized protocols for blood collection tubes, centrifugation conditions, storage temperature, and freeze-thaw cycles are essential, particularly in multi-center studies where consistency across sites must be maintained.
Analytical variability presents another substantial challenge, with different lipidomics platforms often yielding divergent results from identical samples [4]. The disconcertingly low agreement rates (14-36%) between specialized lipidomics software platforms highlight the need for standardized data processing pipelines and identification criteria [4]. Implementing standardized quality control measures, including quality control pools, internal standards, and system suitability tests, helps monitor and control technical variability throughout the validation pipeline.
Biological variability must be carefully considered in cohort design and data interpretation. Lipid profiles exhibit diurnal variations and are influenced by factors including age, sex, diet, medication, and comorbid conditions [4] [90]. Appropriate matching of cases and controls for these confounding factors, along with comprehensive clinical metadata collection, enables proper adjustment in statistical models.
Pathway Mapping and Biological Interpretation
Beyond technical validation, the three-phase model facilitates biological interpretation by mapping altered lipid species to relevant metabolic pathways. In the ischemic stroke study, enrichment analysis identified glycerophospholipid metabolism as significantly altered (FDR-adjusted P = 0.009, impact score = 0.216) [92]. Such pathway analyses strengthen the biological plausibility of biomarker signatures and may reveal novel mechanistic insights into disease pathophysiology.
The three-phase validation model provides a robust framework for advancing lipidomic biomarkers from initial discovery to clinical application. Future developments will likely focus on enhancing automation and standardization to improve reproducibility across laboratories [4]. Artificial intelligence and machine learning approaches show particular promise, with tools like MS2Lipid demonstrating up to 97.4% accuracy in predicting lipid subclasses [4]. These computational advances will complement analytical improvements to accelerate biomarker development.
The integration of lipidomics with other omics technologies (genomics, proteomics, transcriptomics) represents another important frontier [27]. Such multi-omics approaches can provide deeper insights into the mechanistic foundations of lipid alterations and strengthen the biological rationale for selected biomarker panels. Furthermore, the development of reference materials and standardized protocols will be crucial for establishing lipidomics as a reproducible and reliable approach for clinical biomarker development [4].
In conclusion, the three-phase validation model offers a systematic, evidence-based pathway for translating lipidomic discoveries into clinically useful tools. By progressively increasing analytical stringency and validation rigor while focusing on clinically relevant endpoints, this approach addresses the key challenges of reproducibility, specificity, and clinical utility that have hampered previous biomarker development efforts. As lipidomics technologies continue to evolve and standardize, this framework will play an increasingly important role in realizing the promise of precision medicine across diverse disease areas.
In multi-center validation research for lipidomic biomarkers, rigorous evaluation of performance metrics—including sensitivity, specificity, and the Area Under the Receiver Operating Characteristic (ROC) curve (AUC)—is paramount. These metrics provide a quantitative framework for assessing a biomarker's diagnostic ability to distinguish diseased from non-diseased individuals across independent cohorts, ensuring generalizability and clinical applicability. This guide objectively compares the performance of diagnostic tests, underpinned by experimental data from clinical and lipidomics research. It details standardized protocols for metric calculation and validation, crucial for researchers and drug development professionals aiming to translate lipidomic discoveries into clinically useful diagnostic tools.
In the context of lipidomic biomarker discovery and validation, evaluating diagnostic performance is a critical step in translating research findings into clinical applications. Lipidomics, a subfield of metabolomics focused on the comprehensive study of lipids, has shown significant promise in identifying biomarkers for various diseases, including cancer, cardiovascular, and neurodegenerative disorders [4]. However, the transition from bench to bedside relies on the discovery of biomarkers that are clinically reliable, repeatable, and validated in various populations [4]. This process requires a robust statistical framework to quantify how well a proposed biomarker, or "index test," can discriminate between two defined groups (e.g., diseased vs. non-diseased) compared to a gold standard reference test [93].
The cornerstone of this evaluation lies in understanding sensitivity, specificity, and the AUC. Sensitivity measures the probability that a test result will be positive when the disease is present (true positive rate), while specificity measures the probability that a test result will be negative when the disease is not present (true negative rate) [94]. These metrics are often visualized and summarized using the Receiver Operating Characteristic (ROC) curve, a plot of the true positive rate (sensitivity) against the false positive rate (1-specificity) for different cut-off points of a parameter [93] [94]. The Area Under the ROC Curve (AUC) is a single, summary metric that reflects the test's overall ability to distinguish between the two groups, with values ranging from 0.5 (no discriminative power, equivalent to chance) to 1.0 (perfect discrimination) [93]. For lipidomic biomarkers, which often yield continuous numerical results, ROC analysis is an indispensable tool for identifying the optimal cut-off value that maximizes both sensitivity and specificity, thereby determining the biomarker's potential clinical utility [93] [4].
Defining and Calculating Core Metrics
The evaluation of a diagnostic test begins with a 2x2 contingency table that cross-tabulates the results of the index test with those of the gold standard reference test. This table is the foundation for calculating all core performance metrics [93] [94].
Table 1: Contingency Table for Diagnostic Test Evaluation
| Disease (Reference Standard Present) | No Disease (Reference Standard Absent) | ||
|---|---|---|---|
| Test Positive | True Positive (TP) | False Positive (FP) | a + c |
| Test Negative | False Negative (FN) | True Negative (TN) | b + d |
| a + c | b + d |
Based on this table, the key metrics are calculated as follows [94]:
- Sensitivity = ( \frac {TP} {TP + FN} ) = ( \frac {a} {a + b} )
- Specificity = ( \frac {TN} {TN + FP} ) = ( \frac {d} {c + d} )
- Positive Predictive Value (PPV) = ( \frac {TP} {TP + FP} ) = ( \frac {a} {a + c} )
- Negative Predictive Value (NPV) = ( \frac {TN} {TN + FN} ) = ( \frac {d} {b + d} )
- Positive Likelihood Ratio (+LR) = ( \frac {Sensitivity} {1 - Specificity} )
- Negative Likelihood Ratio (-LR) = ( \frac {1 - Sensitivity} {Specificity} )
It is critical to note that while sensitivity and specificity are considered stable test properties, as they are independent of disease prevalence, PPV and NPV are highly dependent on the prevalence of the disease in the target population [94]. Therefore, when reporting performance metrics from a multi-center study, it is essential to consider and report the disease prevalence in each cohort.
The ROC Curve and Area Under the Curve (AUC)
When an index test produces continuous results, such as the concentration of a specific lipid species, ROC analysis is the preferred methodology [93]. The process involves dichotomizing the continuous results at all possible threshold values and calculating the resulting sensitivity and specificity pairs for each threshold. The ROC curve is generated by plotting these sensitivity (TPF) and 1-Specificity (FPF) pairs [93] [94].
The AUC provides a single, summary measure of the test's diagnostic performance across all possible thresholds. The value of the AUC can be interpreted as the probability that the test will correctly rank a randomly chosen diseased individual higher than a randomly chosen non-diseased individual [93]. The following table offers a standard interpretation guide for AUC values in diagnostic research [93]:
Table 2: Clinical Interpretation of AUC Values
| AUC Value | Interpretation |
|---|---|
| 0.9 ≤ AUC ≤ 1.0 | Excellent |
| 0.8 ≤ AUC < 0.9 | Considerable / Good |
| 0.7 ≤ AUC < 0.8 | Fair |
| 0.6 ≤ AUC < 0.7 | Poor |
| 0.5 ≤ AUC < 0.6 | Fail (No better than chance) |
A common mistake in diagnostic research is overestimating the clinical value of a statistically significant AUC. As a rule of thumb, AUC values above 0.80 are generally considered clinically useful, while values below 0.80 indicate limited clinical utility, even if they are statistically significant [93]. Furthermore, the AUC should always be reported with its 95% confidence interval (CI). A narrow CI indicates a more precise and reliable estimate, while a wide CI, often resulting from a small sample size, suggests substantial uncertainty [93].
Experimental Protocols for Metric Validation
Validating performance metrics in independent cohorts requires a rigorous, standardized experimental workflow. This is particularly critical in lipidomics, where technical variability can impact reproducibility [4]. The following protocol outlines the key stages for a multi-center validation study of a lipidomic biomarker.
Sample Collection and Lipidomics Analysis
The first phase involves the pre-analytical and analytical processing of samples across multiple sites.
- Cohort Design and Sample Collection: Independent cohorts should be recruited, each with well-characterized individuals (diseased and non-diseased controls) based on a gold standard diagnosis. Sample collection (e.g., plasma, serum, tissue) must follow a standardized protocol across all centers to minimize pre-analytical variation. Key parameters (e.g., fasting status, time of collection, processing time, storage temperature) must be identical [4].
- Lipid Extraction: Perform lipid extraction from biological samples using a consistent method, such as liquid-liquid extraction (e.g., Folch or Bligh & Dyer methods). Internal standards (stable-isotope labeled lipids) should be added at the beginning of extraction to correct for procedural losses and matrix effects [4].
- Lipidomic Profiling: Analyze lipid extracts using a high-resolution platform, typically liquid chromatography coupled to mass spectrometry (LC-MS). The analytical method (chromatography column, mobile phase gradient, mass spectrometer settings) must be harmonized across all participating laboratories to ensure data comparability [4].
- Data Pre-processing: Process raw LC-MS data using bioinformatics software (e.g., MS-DIAL, Lipostar) for peak detection, alignment, and lipid identification. The data processing parameters and lipid database used for identification must be consistent to reduce inter-laboratory variability [4].
Statistical Analysis and Performance Evaluation
The second phase focuses on the statistical validation of the biomarker's performance.
- Data Normalization and Transformation: Normalize the lipid concentration data using internal standards and correct for batch effects. Apply necessary data transformations (e.g., log-transformation) to achieve normality if required.
- Model Building (Training Cohort): In one cohort (the training set), use statistical or machine learning models (e.g., logistic regression) to identify a panel of lipid species that best discriminates between the groups. The output of this model can be a continuous "risk score."
- ROC Analysis and Cut-off Selection (Training Cohort): Subject the model's risk score to ROC analysis in the training cohort. Calculate the AUC and its 95% CI. Identify the optimal cut-off value that maximizes sensitivity and specificity, for example, by using the Youden index (J = Sensitivity + Specificity - 1) [93] [94].
- Validation in Independent Cohorts: Apply the model developed in the training cohort to the data from the independent validation cohort(s) without any retraining or re-estimation of parameters. Calculate the sensitivity, specificity, and AUC in this new cohort to assess the generalizability of the biomarker's performance.
- Comparison of Metrics: Statistically compare the AUC values between different biomarker models or between cohorts using methods such as the DeLong test [93].
The workflow for this multi-center validation is summarized in the diagram below.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful multi-center validation of lipidomic biomarkers depends on using standardized, high-quality reagents and platforms. The following table details essential materials and their functions in the validation workflow.
Table 3: Key Research Reagent Solutions for Lipidomic Biomarker Validation
| Item | Function in Validation Protocol |
|---|---|
| Stable Isotope-Labeled Internal Standards (e.g., ^13^C or ^2^H labeled lipids) | Added to samples prior to extraction to correct for variability in lipid recovery, ionization efficiency, and matrix effects during mass spectrometry analysis. Essential for quantitative accuracy [4]. |
| Standardized Lipid Extraction Kits (e.g., MTBE-based, Folch-based) | Ensure consistent and reproducible recovery of a broad range of lipid classes (e.g., glycerophospholipids, sphingolipids, sterols) across all samples and sites, minimizing pre-analytical bias [4]. |
| High-Performance Liquid Chromatography (HPLC) Systems with orthogonal separation columns (e.g., C18, HILIC) | Separate complex lipid mixtures from biological extracts to reduce ion suppression and enhance the detection and quantification of individual lipid species by mass spectrometry [4]. |
| High-Resolution Mass Spectrometers (e.g., Q-TOF, Orbitrap) | Provide accurate mass measurements for the confident identification of lipids and enable untargeted profiling to discover novel biomarker signatures alongside targeted validation [4]. |
| Bioinformatics Software Platforms (e.g., MS-DIAL, Lipostar, XCMS) | Automate the processing of raw LC-MS data, including peak picking, alignment, lipid identification against databases, and statistical analysis for biomarker discovery and validation [4]. |
| Quality Control (QC) Pools (e.g., pooled sample from all study subjects) | Analyzed intermittently with study samples to monitor instrument stability, data reproducibility, and to correct for analytical drift over the course of the data acquisition campaign [4]. |
Comparative Performance Data in Biomarker Research
Objective comparison of biomarker performance hinges on the quantitative data derived from ROC analysis. When comparing a novel lipidomic biomarker to existing alternatives, it is essential to report and contrast their AUC values, sensitivity, and specificity at defined cut-offs, along with confidence intervals to denote precision.
Table 4: Illustrative Comparison of Biomarker Performance in an Independent Validation Cohort
| Biomarker / Test | AUC (95% CI) | Optimal Cut-off (Youden Index) | Sensitivity at Cut-off | Specificity at Cut-off | +LR | -LR |
|---|---|---|---|---|---|---|
| Novel Lipidomic Panel (e.g., Ceramide Ratio) | 0.87 (0.82 - 0.91) | Score > 1.5 | 82% | 79% | 3.90 | 0.23 |
| Established Clinical Standard | 0.75 (0.69 - 0.80) | Concentration > 100 ng/mL | 70% | 72% | 2.50 | 0.42 |
| Combined Model (Lipid Panel + Clinical Standard) | 0.91 (0.88 - 0.94) | Score > 0.6 | 85% | 88% | 7.08 | 0.17 |
Note: This table uses illustrative data to demonstrate a comparative framework. Actual values will vary based on the specific biomarker and disease context.
The data in Table 4 illustrates a scenario where a novel lipidomic panel demonstrates considerable (AUC > 0.8) discriminatory power, outperforming an established clinical standard which shows only fair performance (AUC = 0.75) [93]. The combination of both markers into a single model yields an excellent AUC (0.91), suggesting a synergistic effect and potentially greater clinical value. The positive likelihood ratio (+LR) of 7.08 for the combined model indicates a substantial increase in the post-test probability of disease when the test is positive, while a negative likelihood ratio (-LR) of 0.17 indicates a moderate decrease in post-test probability when the test is negative [94].
Critical Considerations for Multi-Center Validation
Validating performance metrics across independent cohorts presents unique challenges that must be addressed to ensure the credibility of the findings.
- Reproducibility and Standardization: A significant challenge in lipidomics is the low agreement rate (as low as 14-36%) in lipid identification across different software platforms and laboratories [4]. Mitigating this requires stringent standardization of pre-analytical protocols, analytical methods, and data processing workflows across all participating centers.
- Confidence Intervals and Statistical Power: An AUC point estimate without a confidence interval provides an incomplete picture. A wide confidence interval, often due to a small sample size in a validation cohort, indicates substantial uncertainty about the true performance of the biomarker [93]. Therefore, calculating sample size a priori to ensure adequate statistical power is a vital prerequisite for diagnostic studies.
- Clinical Utility vs. Statistical Significance: A common error is conflating a statistically significant AUC (e.g., p < 0.05) with clinical usefulness. An AUC can be statistically significantly greater than 0.5 but still be clinically inadequate if its value is below 0.80 [93]. The decision to advance a biomarker should be based on its clinical utility, not solely on statistical significance.
The logical flow of these critical considerations and their impact on the final clinical application is visualized below.
The clinical management of gastrointestinal (GI) and hepatic cancers has long relied on a limited set of serum protein biomarkers, primarily carbohydrate antigen 19-9 (CA19-9) for pancreatic and biliary cancers and alpha-fetoprotein (AFP) for hepatocellular carcinoma [95]. While these markers provide valuable clinical information, they face significant limitations in sensitivity, specificity, and clinical utility for early detection and monitoring. CA19-9, for instance, demonstrates elevated concentrations in 70-90% of patients with pancreatic cancer but also shows increases in several other GI malignancies and various benign diseases [95]. Similarly, AFP is used for diagnosis, prognosis, and monitoring of hepatocellular carcinoma but lacks ideal diagnostic performance characteristics [95].
Concurrently, lipidomics—the large-scale study of pathways and networks of cellular lipids in biological systems—has emerged as a powerful tool for understanding the complexities of lipid metabolism and its implications in health and disease [96]. The field has gained significant attention due to the crucial roles lipids play in various biological processes, including membrane structure, energy storage, and signaling. Lipid metabolic reprogramming represents a hallmark of cancer, supporting tumor proliferation and immune evasion through coordinated dysregulation of lipogenesis, aberrant lipid trafficking, and lipid raft-mediated signaling [83]. Transitioning lipid research from bench to bedside relies on the discovery of biomarkers that are clinically reliable, repeatable, and validated across various populations [4].
This comparison guide objectively evaluates the performance of emerging lipidomic panels against established biomarker standards CA19-9 and AFP within the context of multi-center validation research for lipidomic biomarkers. We present comparative analytical and clinical data, detailed methodological protocols, and practical implementation frameworks to guide researchers, scientists, and drug development professionals in advancing the field of lipidomic biomarker validation.
Comparative Analytical Performance: Methodologies and Data
Traditional Immunoassays for CA19-9 and AFP
Experimental Protocols for Immunoassays The measurement of CA19-9 and AFP typically employs immunoassay techniques on automated chemistry analyzers. In a typical method comparison study, routine patient samples are analyzed for standard diagnostic work-up. Sera are obtained after centrifuging at approximately 1000 x g for 10 minutes. Each serum aliquot is divided and analyzed on different platforms, such as the Vitros ECi analyzer (using chemiluminescence assays) and the Cobas e 411 analyzer (using electrochemiluminescence assays) according to manufacturer instructions [95]. Quality control is maintained through daily testing of commercial control samples throughout the study period, with analytical inaccuracy calculated as bias (%) and day-to-day imprecision as coefficient of variation (CV %). Statistical analysis includes Spearman correlation coefficients and Passing-Bablok regression to assess method comparability [95].
Performance Characteristics of Traditional Biomarkers Despite the clinical establishment of CA19-9 and AFP, significant analytical challenges persist. Method comparison studies reveal that although correlation coefficients between different analyzers can be high (0.978 for CA19-9 and 0.999 for AFP), Passing-Bablok regression shows that concentrations of CA19-9 and CEA (a related marker) do not align nor follow the same linearity between platforms [95]. The slopes and y-axis intercepts significantly deviate from 1 and 0, respectively, indicating substantial proportional differences. These discrepancies occur even when using the same antibodies, suggesting differences stem from calibration and method design. Consequently, monitoring patients with the same reagents on the same analyzer is essential for consistent longitudinal assessment, as transitioning between platforms can yield significantly different values and potentially impact clinical interpretation [95].
Lipidomics Approaches and Technologies
Experimental Protocols for Lipidomics Lipidomics methodologies have advanced with multiple targeted, untargeted, and pseudo-targeted techniques that improve structural lipid profiling, resolution, and quantification [4]. A notable multiplexed targeted assay was developed that couples normal phase liquid chromatography-hydrophilic interaction chromatography (NPLC-HILIC) with multiple reaction monitoring (MRM) on a triple quadrupole mass spectrometer, enabling quantification of over 900 lipid species across more than 20 lipid classes in a single 20-minute run [55]. This method addresses analytical challenges such as in-source fragmentation, isomer separations, and concentration dynamics to ensure confidence in selectivity, quantification, and reproducibility. Utilizing multiple MS/MS product ions per lipid species improves confidence in lipid identification and enables determination of relative abundances of positional isomers. The method underwent analytical validation following FDA Bioanalytical Method Validation Guidance for Industry, demonstrating repeatable and robust quantitation with over 700 lipids achieving inter-assay variability below 25% in NIST-SRM-1950 plasma reference material [55].
Workflow for Lipidomic Analysis The general workflow for lipidomic analysis involves sample collection and preparation, followed by lipid extraction using methods such as liquid-liquid extraction. The extracted lipids are then separated using chromatographic techniques, most commonly liquid chromatography, and analyzed by mass spectrometry. Data preprocessing including noise reduction, normalization, and batch effect correction is followed by statistical analysis, pathway analysis, and biological interpretation [96].
The following diagram illustrates the core workflow for lipidomic biomarker discovery and validation:
Direct Performance Comparison
Table 1: Comparative Analytical Performance of Biomarker Platforms
| Parameter | Traditional Immunoassays (CA19-9/AFP) | Lipidomic Panels |
|---|---|---|
| Analytical Technique | Chemiluminescence/Electrochemiluminescence immunoassays | Liquid chromatography-mass spectrometry (LC-MS) |
| Throughput | High (fully automated) | Moderate to high (depending on platform) |
| Multiplexing Capacity | Single analyte per test | Hundreds to thousands of lipid species simultaneously |
| Inter-assay CV | Variable between platforms; generally <15% for controls | <25% for over 700 lipid species in validated panels [55] |
| Cross-platform Comparability | Poor (significant differences between methods) [95] | Improving with standardization efforts |
| Regulatory Status | FDA-approved for clinical use | Research use only; early phase clinical validation |
Table 2: Clinical Performance Characteristics in Cancer Detection
| Characteristic | CA19-9 (Pancreatic Cancer) | AFP (Hepatocellular Carcinoma) | Lipidomic Signatures |
|---|---|---|---|
| Reported Sensitivity | 70-90% [95] | Varies by cutoff and population | Varies by cancer type and panel |
| Specificity | Reduced in benign conditions and other malignancies [95] | Variable; elevated in benign liver conditions | Promising for specific cancer types in early studies |
| Early Detection Capability | Limited | Limited | Emerging evidence for pre-diagnostic alterations |
| Dynamic Monitoring | Established for treatment response and recurrence [95] | Established for treatment monitoring [95] | Demonstrated potential for tracking treatment response [83] |
| Biological Insight | Limited | Limited | High (reflects underlying metabolic reprogramming) |
Clinical Validation and Biological Significance
Multi-center Validation Landscape
The translational research projects and pilot trials have produced encouraging outcomes across various disciplines, including oncology, highlighting the critical need for interdisciplinary cooperation to utilize lipidomics fully in personalized medicine [4]. However, the transition from research findings to approved lipid-based diagnostic tools remains in its infancy in clinical settings due to a lack of multi-center validation studies and incomplete regulatory frameworks for lipidomic biomarkers [4].
Recent bibliometric analyses reveal that investigations into metabolic biomarkers related to cancer have demonstrated consistent growth between 2015 and 2023, with a significant surge from 2023 to 2024 [24]. China leads in publication volume, followed by the United States, the United Kingdom, Japan, and Italy, with the Chinese Academy of Sciences, Shanghai Jiao Tong University, and Zhejiang University emerging as prominent collaborative centers [24]. This growing research activity underscores the field's recognition of the vast promise that metabolic biomarkers hold for cancer diagnosis and treatment.
Biological Pathways and Mechanistic Insights
Lipids comprise thousands of chemically distinct species classified into eight major categories according to the LIPID MAPS consortium: fatty acyls, glycerolipids, glycerophospholipids, sphingolipids, sterol lipids, prenol lipids, saccharolipids, and polyketides [4] [97]. The structural diversity of lipids confers a broad spectrum of functionality, with seemingly minor structural differences—such as the number, position, and geometry of double bonds in acyl chains—serving as pivotal determinants of their functions [55].
In cancer biology, lipidomic alterations reflect fundamental reprogramming of metabolic pathways that support tumor growth, proliferation, and survival. For instance, specific ceramides and phosphatidylcholines have been associated with cardiovascular risk, while changes in sphingolipid and glycerophospholipid metabolism are being investigated in the contexts of multiple sclerosis and cancer [4]. The diagram below illustrates key lipid metabolic pathways implicated in oncogenesis:
Case Study: Lipid Biomarkers in Lymphoma
A compelling example of lipid biomarkers' clinical potential comes from a multicenter retrospective and prospective validation study of extranodal NK/T-cell lymphoma (ENKTL). This research analyzed 1,017 patients newly diagnosed with ENKTL and matched controls, integrating longitudinal LC-MS-based metabolomic profiling with pretreatment tumor transcriptomic analysis [83]. The study revealed that patients with ENKTL showed significantly elevated triglycerides (TG) and reduced apolipoprotein A1 (ApoA1) compared to controls. Most notably, elevated TG and reduced ApoA1 independently predicted inferior survival, with patients achieving an objective response demonstrating metabolic profile normalization—specifically, TG reduction and ApoA1 elevation—while patients with stable or progressive disease retained baseline profiles [83].
This research demonstrates several advantages of lipid biomarkers: their dynamic nature enables monitoring of treatment response, their quantification is cost-effective compared to complex imaging or genomic analyses, and they reflect underlying tumor biology. Transcriptome analysis further suggested tumor-intrinsic lipid dysregulation in patients with high-TG/low-ApoA1 levels, providing mechanistic insights into the observed clinical correlations [83].
Implementation Framework and Research Toolkit
Essential Research Reagent Solutions
Successful implementation of lipidomic biomarker studies requires specific reagents and materials optimized for lipid extraction, separation, and analysis. The following table details key research reagent solutions and their functions in lipidomic workflows:
Table 3: Essential Research Reagent Solutions for Lipidomics
| Reagent/Material | Function/Application | Technical Considerations |
|---|---|---|
| Sample Collection Tubes with Stabilizers | Preserves lipid integrity during sample storage and transport | Critical for pre-analytical standardization; minimizes degradation |
| Chloroform-Methanol Mixtures | Liquid-liquid extraction of lipids from biological samples | Classic Folch or Bligh & Dyer methods; ratio optimization needed for different matrices |
| Internal Standards (SIL/SIS) | Isotopically-labeled lipid standards for quantification | Essential for accurate quantification; should cover multiple lipid classes |
| Chromatography Columns (C18, HILIC) | Separation of lipid species prior to MS analysis | Choice depends on lipid classes of interest; affects resolution and sensitivity |
| Quality Control Materials (NIST SRM 1950) | Reference plasma for inter-laboratory comparison and QC | Enables standardization across platforms and laboratories |
| Mass Spectrometry Calibrants | Instrument calibration for accurate mass measurement | Critical for lipid identification; should be compatible with ionization mode |
Validation Roadmap for Lipidomic Biomarkers
The path to clinical implementation of lipidomic biomarkers requires systematic validation across multiple phases. The following framework outlines a proposed roadmap:
Phase 1: Discovery and Assay Development
- Untargeted lipidomic profiling of well-characterized sample sets
- Identification of candidate lipid biomarkers
- Development of targeted MRM assays for verification
Phase 2: Analytical Validation
- Assessment of precision, accuracy, linearity, and limits of quantification
- Evaluation of extraction efficiency and matrix effects
- Stability studies under various storage conditions
- Documentation following FDA Bioanalytical Method Validation Guidance [55]
Phase 3: Clinical Validation
- Retrospective multi-center studies with predefined endpoints
- Determination of clinical sensitivity and specificity
- Establishment of reference intervals and clinical decision limits
- Comparison against existing standards (CA19-9, AFP)
Phase 4: Regulatory Approval and Implementation
- Submission to regulatory agencies (FDA, EMA)
- Development of clinical guidelines
- Implementation in diagnostic laboratories
- Continuous monitoring of clinical performance
The comparative analysis presented in this guide demonstrates that while traditional protein biomarkers like CA19-9 and AFP maintain established roles in specific clinical contexts, lipidomic panels offer substantial advantages in biological insight, multiplexing capability, and potential for early detection. The emerging evidence from multi-center studies indicates that lipid biomarkers can provide dynamic, cost-effective monitoring of treatment response and reflect underlying metabolic reprogramming in cancer [83].
However, significant challenges remain before lipidomic panels can achieve widespread clinical implementation. The lack of reproducibility across different analytical platforms and laboratories presents a substantial obstacle, with prominent software platforms agreeing on as few as 14% of lipid identifications when using identical LC-MS data [4]. Standardization of pre-analytical procedures, analytical methodologies, and data reporting is essential to advance the field.
Future developments will likely focus on integrating lipidomic data with other omics technologies (genomics, transcriptomics, proteomics) to provide more comprehensive insights into cancer biology. Artificial intelligence and machine learning approaches are already contributing significantly, with models like MS2Lipid demonstrating up to 97.4% accuracy in predicting lipid subclasses [4]. As these technologies mature and multi-center validation studies expand, lipidomic panels promise to emerge as central components in the future of cancer prevention, diagnosis, and therapeutic monitoring, potentially complementing or surpassing the capabilities of traditional biomarkers like CA19-9 and AFP.
Lipidomics, the large-scale study of lipid pathways and networks, is emerging as a powerful tool for discovering novel biomarkers in complex human diseases. By capturing the dynamic state of cellular metabolism and signaling, lipidomic profiles offer a unique window into disease pathophysiology. This guide objectively compares validated lipidomic models for three distinct disease areas—pancreatic cancer, rheumatoid arthritis, and hepatocellular carcinoma—focusing on their experimental validation, analytical performance, and translational potential. The findings are framed within the critical context of multi-center validation, a necessary step for clinical adoption of lipidomic biomarkers.
Comparative Analysis of Validated Lipidomic Models
The table below summarizes key performance metrics and validation data for lipidomic models across the three disease areas.
| Disease Area | Key Lipid Classes Identified | Model Performance (AUC/Sensitivity/Specificity) | Validation Cohort & Multicenter Data | Comparative Advantage vs Standard Biomarkers |
|---|---|---|---|---|
| Pancreatic Cancer | Sphingomyelins, Ceramides, (Lyso)Phosphatidylcholines [98] | Sensitivity and specificity >90% [98] | 830 total samples; verified across multiple laboratories [98] | Outperforms CA 19-9 (AUC 0.735), especially at early stage [99] [5] |
| Pancreatic Cancer | Phospholipids (18 species) [99] [5] | AUC 0.9207; Sensitivity 90.74%; Specificity 86.22% [99] [5] | Validation in independent human set (Set B: 96 normal, 78 PC) [99] [5] | 25% increase in discriminatory power vs CA 19-9 alone [99] [5] |
| Rheumatoid Arthritis (RA) | Lysophosphatidylcholine, Phosphatidylcholine, Ether-linked phosphatidylethanolamine, Sphingomyelin [100] [101] | Serum lipid profile distinguishes active RA from remission and preclinical phases [100] [101] | 79 patients across different disease phases; lipidome correlates with synovitis severity [100] [101] | Identifies preclinical RA where ESR/CRP levels are normal [100] [101] |
| Rheumatoid Arthritis (RA) | Panel of 10 serum lipids [102] | Accuracy 79%; Sensitivity 71%; Specificity 86% [102] | Validation cohort differentiated seropositive and seronegative RA from OA and SLE [102] | Diagnoses seronegative RA, addressing a major clinical challenge [102] |
| Hepatocellular Carcinoma (HCC) | Phosphatidylinositol lyso 18:1, O-phosphorylethanolamine [103] | Significantly associated with progression-free survival [103] | Prospective phase II clinical trial (58 patients) [103] | Predicts long-term survival outcomes for TKI-ICI combination therapy [103] |
| Hepatocellular Carcinoma (HCC) | Glycerol lipids, Cardiolipin, Phosphatidylethanolamine [104] | SVM model AUC 0.98 for distinguishing pre-diagnostic HCC from cirrhosis [104] | 28 pre-diagnostic serum samples from patients with cirrhosis who developed HCC [104] | Identifies HCC risk in patients with cirrhosis prior to clinical diagnosis [104] |
Experimental Protocols & Methodologies
Pancreatic Cancer Lipidomics Workflow
The most robust pancreatic cancer studies employed a multi-phase biomarker discovery approach:
- Phase I (Discovery): Initial proof-of-concept using 262 PDAC patients and 102 healthy controls analyzed with UHPSFC/MS and shotgun MS. Multivariate data analysis (OPLS-DA) revealed distinct lipidome differences independent of cancer stage [98].
- Phase II (Qualification): Confirmatory phase involving 554 samples analyzed across multiple experienced laboratories using different MS-based methods (UHPSFC/MS, shotgun MS, RP-UHPLC/MS). This phase confirmed dysregulation of specific sphingomyelins, ceramides, and phosphatidylcholines across platforms [98].
- Phase III (Verification): Independent validation of discriminatory performance, demonstrating sensitivity and specificity over 90%, outperforming CA 19-9 especially at early stages [98].
Rheumatoid Arthritis Lipidomics Workflow
RA lipidomics studies have focused on correlating lipid profiles with disease evolution and activity:
- Cohort Design: Prospective enrollment of RA patients at different clinical phases (preclinical, active, sustained remission) with matched osteoarthritis controls and healthy individuals [100] [101].
- Sample Collection: Serum collected after 8-hour fast, with synovial fluid obtained during aspiration procedures. All samples processed and stored at -80°C before analysis [100] [101].
- Lipid Extraction: Modified MTBE (methyl tert-butyl ether) or Folch method using internal standard mixtures for quantification [100] [102].
- Disease Activity Correlation: Lipid profiles correlated with clinical disease activity scores (DAS28), synovitis severity on ultrasonography, and treatment response to DMARDs [100] [101].
Hepatocellular Carcinoma Lipidomics Workflow
HCC lipidomics approaches have emphasized prediagnostic biomarker discovery:
- Prediagnostic Sample Analysis: Use of serum samples collected from patients with cirrhosis who subsequently developed HCC compared to matched controls who did not develop HCC [104].
- Machine Learning Integration: Application of random forest survival analysis, LASSO regression, and Cox proportional hazards modeling to identify minimal lipid panels predictive of survival [103].
- Multi-omics Integration: Linking differentially expressed lipids with transcriptomic signatures from publicly available datasets to validate biological relevance [104].
- Therapy Response Prediction: Focus on identifying lipids that predict response to combination therapies (TKI-ICI) in advanced HCC patients [103].
Visualizing the Lipidomic Biomarker Pipeline
The following diagram illustrates the generalized workflow for developing and validating lipidomic biomarkers, as demonstrated across the success stories in this guide.
Diagram Title: Lipidomic Biomarker Development Workflow
The Scientist's Toolkit: Essential Research Reagents & Materials
| Reagent/Material | Function & Application | Examples from Literature |
|---|---|---|
| Internal Standards (IS) | Quantification correction for different lipid classes; enables inter-laboratory comparison [98] | LPC (17:0), PC (10:0/10:0), PE (10:0/10:0), SM (18:1/17:0) [100] [101] |
| Lipid Extraction Solvents | Liquid-liquid extraction of lipids from biological samples | MTBE [100] [101], Chloroform:methanol (Folch method) [102] |
| LC-MS Grade Solvents | Mobile phase preparation for chromatographic separation | Acetonitrile, isopropanol, methanol with 10mM ammonium formate/0.1% formic acid [100] [101] |
| Quality Control (QC) Samples | Monitoring analytical performance and batch effects | Pooled plasma/serum from all study subjects [98] [99] |
| Reference Materials | Harmonization of different analytical platforms | NIST 1950 reference plasma for inter-laboratory comparison [98] |
The validated lipidomic models presented herein demonstrate remarkable diagnostic and prognostic potential across three distinct disease areas. The consistent success of these approaches underscores the fundamental role of lipid metabolism in disease pathogenesis. However, translation to routine clinical practice requires addressing several challenges, including standardization of analytical protocols, harmonization of data processing pipelines, and demonstration of clinical utility in large, diverse populations. The multi-center validation frameworks established by these studies provide a roadmap for future lipidomic biomarker development. As technologies advance and collaborative efforts grow, lipidomics is poised to make significant contributions to precision medicine, enabling earlier disease detection, accurate monitoring of treatment response, and improved patient outcomes.
Conclusion
The multi-center validation of lipidomic biomarkers represents a paradigm shift in diagnostic development, moving from single-center discoveries to clinically generalizable tools. The synthesis of evidence across various diseases confirms that panels of lipid species, analyzed through integrated workflows of advanced mass spectrometry and machine learning, can achieve diagnostic performance surpassing traditional biomarkers, especially in early-stage disease. Key to this success is a rigorous, phased validation approach that proactively addresses technical and biological variability across independent cohorts. Future directions must focus on the large-scale standardization of analytical protocols, the widespread adoption of explainable AI to build clinical trust, and the initiation of prospective, longitudinal studies to firmly establish the value of these biomarkers in improving patient outcomes. As the field matures, validated lipidomic signatures are poised to become indispensable components of the precision medicine toolkit, enabling earlier intervention and more personalized therapeutic strategies.
References
- 1. - Wikipedia Lipid [https://en.wikipedia.org/wiki/Lipid]
- 2. classification, structures and tools - PMC Lipid [https://pmc.ncbi.nlm.nih.gov/articles/PMC3995129/]
- 3. The strategic role of lipidomics in biomarker identification ... [https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2025.1546512/full]
- 4. Exploring lipidomics in biomarker discovery [https://www.sciencedirect.com/science/article/pii/S0009898125005777?dgcid=rss_sd_all]
- 5. Diagnostic Biomarker Candidates Proposed Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12468859/]
- 6. Integrated multi-omics analysis identifies lipid metabolism ... [https://www.nature.com/articles/s41598-025-13703-y]
- 7. Lipid Biological Functions [https://www.news-medical.net/life-sciences/Lipid-Biological-Functions.aspx]
- 8. Structural and signaling role of lipids in plasma membrane ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7182362/]
- 9. Lipid signaling [https://en.wikipedia.org/wiki/Lipid_signaling]
- 10. Lipid metabolic reprogramming in tumor microenvironment [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-023-01498-2]
- 11. Lipid metabolic reprogramming in cancer cells | Oncogenesis [https://www.nature.com/articles/oncsis201549]
- 12. an integrated omics study of lipid pathways and their ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06698-7]
- 13. Metabolic reprogramming in hepatocellular carcinoma [https://www.nature.com/articles/s12276-025-01415-2]
- 14. The role of metabolic reprogramming in pancreatic cancer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9868425/]
- 15. ACSL4 coordinates metabolic and cell cycle ... [https://www.sciencedirect.com/science/article/abs/pii/S0898656825006709]
- 16. Lipid Metabolic Reprogramming in Hepatocellular ... [https://pubmed.ncbi.nlm.nih.gov/30445800/]
- 17. Metabolic reprogramming in cervical cancer and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8525007/]
- 18. Original Article Association of ABCA4 gene polymorphisms ... [https://www.sciencedirect.com/science/article/abs/pii/S2468785525004112]
- 19. Combining lipidomics and machine learning to identify lipid ... [https://insight.jci.org/articles/view/186629]
- 20. Plasma lipidomics for biomarker identification in non- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12584469/]
- 21. Integrated plasma and red blood cell membrane lipidomics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12309142/]
- 22. Assessing the Use of Non-Invasive Biomarkers in Drug ... [https://www.fda.gov/science-research/fda-stem-outreach-education-and-engagement/assessing-use-non-invasive-biomarkers-drug-development-pre-cirrhotic-nonalcoholic-steatohepatitis]
- 23. Exploring lipidomics in biomarker discovery [https://pubmed.ncbi.nlm.nih.gov/41197707/]
- 24. Global research dynamics in metabolic biomarkers and cancer [https://pmc.ncbi.nlm.nih.gov/articles/PMC12589725/]
- 25. Exploring lipidomics in biomarker discovery [https://www.sciencedirect.com/science/article/abs/pii/S0009898125005777]
- 26. Untargeted vs. Targeted Lipidomics—Understanding the Differences - Lipidomics|Creative Proteomics [https://lipidomics.creative-proteomics.com/resource/untargeted-vs-targeted-lipidomics-understanding-the-differences.htm]
- 27. The strategic role of lipidomics in biomarker identification and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12483926/]
- 28. Quantitative analysis of targeted lipidomics in the ... [https://www.frontiersin.org/journals/aging-neuroscience/articles/10.3389/fnagi.2025.1561831/full]
- 29. The Hitchhiker’s Guide to Untargeted Lipidomics Analysis: Practical Guidelines - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8624948/]
- 30. Cross-Platform Comparison of Untargeted and Targeted Lipidomics Approaches on Aging Mouse Plasma | Scientific Reports [https://www.nature.com/articles/s41598-018-35807-4]
- 31. Four-Dimensional Lipidomic Analysis Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11635755/]
- 32. Fast and comprehensive lipidomic analysis using ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967325000913]
- 33. Ultra High-Performance Supercritical Fluid ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267025012978]
- 34. Recent Advances in Liquid Chromatography–Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12204688/]
- 35. Recent Analytical Methodologies in Lipid Analysis [https://www.mdpi.com/1422-0067/25/4/2249]
- 36. Comparison of liquid chromatography-high-resolution ... [https://www.sciencedirect.com/science/article/pii/S2667145X23000329]
- 37. Leveraging ML for profiling lipidomic alterations in breast ... [https://www.nature.com/articles/s41598-024-71439-7]
- 38. Combining lipidomics and machine learning to identify lipid ... [https://pubmed.ncbi.nlm.nih.gov/40337862/]
- 39. Comparative Study of the Performance of Naïve Bayes ... [https://dinastipub.org/DIJEMSS/article/view/5119]
- 40. Comparison of Naive Bayes and Support Vector Machine ... [https://radiant.politeknikassalaam.ac.id/index.php/radiant/article/view/316]
- 41. Comparative analysis of ensemble learning techniques for ... [https://www.nature.com/articles/s41598-024-79476-y]
- 42. integrating explainable AI (XAI) with machine learning ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12465982/]
- 43. Demystifying the black box: A survey on explainable ... [https://www.sciencedirect.com/science/article/pii/S2001037024004495]
- 44. Identification of a Novel Lipidomic Biomarker for ... [https://www.mdpi.com/2218-1989/15/11/716]
- 45. Interlaboratory comparison of standardised metabolomics and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11601468/]
- 46. Untargeted Lipidomic Biomarkers for Liver Cancer Diagnosis [https://pmc.ncbi.nlm.nih.gov/articles/PMC11943538/]
- 47. a comprehensive data-mining-based study [https://pubmed.ncbi.nlm.nih.gov/40544396/]
- 48. Up-to-Date Look at Lipids Role in Precision Health (2025) [https://www.laboratoriosrubio.com/en/lipidomics-precision-health/]
- 49. Combining lipidomics and machine learning to identify lipid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12128965/]
- 50. Identification and validation of a blood- based diagnostic ... [https://www.nature.com/articles/s41467-024-48763-7]
- 51. Lipidomics data analysis: OPLS-DA [https://www.lipotype.com/lipidomics-applications/lipidomics-data-analysis-opls-da/?srsltid=AfmBOoouSOn-8h4t8RnxH64HozbxkySY7QffXBIzPaWyPS9EgbF4Vtai]
- 52. Steps for Visualizing Lipidomics Data Analysis [https://www.mtoz-biolabs.com/steps-for-visualizing-lipidomics-data-analysis.html]
- 53. Lipidome visualisation, comparison, and analysis in a vector ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12058142/]
- 54. Analytical quality control in targeted lipidomics [https://www.sciencedirect.com/science/article/pii/S0003267025006191]
- 55. Validation of a multiplexed and targeted lipidomics assay ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9168725/]
- 56. Challenges in Lipidomics Biomarker Identification [https://www.mdpi.com/2218-1989/14/8/461]
- 57. Research on Lipidomic Profiling and Biomarker ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11673004/]
- 58. The Lipidomic Profile Discriminates Between MASLD and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11950808/]
- 59. Managing High-Dimensional Data in Machine Learning [https://www.geeksforgeeks.org/machine-learning/managing-high-dimensional-data-in-machine-learning/]
- 60. Handling Sparse and High-Dimensional Data in Machine ... [https://medium.com/@siddharthapramanik771/handling-sparse-and-high-dimensional-data-in-machine-learning-strategies-and-techniques-for-34516e88efff]
- 61. Assessing and mitigating batch effects in large-scale omics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11447944/]
- 62. Batch Effect Correction [https://www.10xgenomics.com/analysis-guides/introduction-batch-effect-correction]
- 63. Are batch effects still relevant in the age of big data? [https://www.sciencedirect.com/science/article/pii/S0167779922000361]
- 64. Top 12 Dimensionality Reduction Techniques for Machine ... [https://encord.com/blog/dimentionality-reduction-techniques-machine-learning/]
- 65. Handling high-dimensional data with missing values by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9930810/]
- 66. How to Fix Overfitting and Underfitting | by Tahir | Oct, 2025 [https://medium.com/@tahirbalarabe2/how-to-fix-overfitting-and-underfitting-4cf8f383ea96]
- 67. Multivariate Data Analysis Methods and Their Application in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12280249/]
- 68. Cross Validation in Machine Learning [https://www.geeksforgeeks.org/machine-learning/cross-validation-machine-learning/]
- 69. Cross Validation in Machine Learning: Techniques and ... [https://www.udacity.com/blog/2025/05/cross-validation-in-machine-learning-techniques-and-best-practices.html]
- 70. 3.1. Cross-validation: evaluating estimator performance [https://scikit-learn.org/stable/modules/cross_validation.html]
- 71. Comparative Deep Learning Analysis of Regularization ... [https://www.preprints.org/manuscript/202511.0501]
- 72. How standardization of the pre-analytical phase of both ... [https://www.sciencedirect.com/science/article/abs/pii/S1871678419300706]
- 73. Challenges in Lipidomics Biomarker Identification [https://pmc.ncbi.nlm.nih.gov/articles/PMC11356033/]
- 74. Standardization of pre-analytical variables in plasma ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4395506/]
- 75. Multicenter Collaborative Study to Optimize Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10775142/]
- 76. Quality control requirements for the correct annotation of ... [https://www.nature.com/articles/s41467-021-24984-y]
- 77. Steps Toward Minimal Reporting Standards for Lipidomics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8376269/]
- 78. Convergence: Multi-omics and AI are reshaping the ... [https://www.sciencedirect.com/science/article/abs/pii/S0925443925003758]
- 79. Advancements in Mass Spectrometry-Based Targeted ... [https://www.mdpi.com/1420-3049/29/24/5934]
- 80. Development and multi-center validation of machine learning ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-07265-w]
- 81. In-depth analysis of Pseudotargeted metabolomics strategy [https://www.sciencedirect.com/science/article/abs/pii/S0026265X25017680]
- 82. AOA Dx Demonstrates High Accuracy in AI-Powered Blood ... [https://aoadx.com/aacr2025/]
- 83. Serum triglyceride and apolipoprotein A1 as dynamic ... [https://link.springer.com/article/10.1007/s00277-025-06724-0]
- 84. Multicenter validation of a scalable, interpretable, multitask ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12485121/]
- 85. Multi-center validation of machine learning model for ... [https://www.nature.com/articles/s41746-022-00625-6]
- 86. Development and external validation of a machine learning ... [https://bmcmedicine.biomedcentral.com/articles/10.1186/s12916-025-03912-7]
- 87. Sample size estimation and power analysis for clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3409926/]
- 88. Sample size calculator [https://riskcalc.org/samplesize/]
- 89. Sample size calculation in multi-centre clinical trials [https://bmcmedresmethodol.biomedcentral.com/articles/10.1186/s12874-018-0602-y]
- 90. Identification and validation of a blood- based diagnostic lipidomic ... [https://www.nature.com/articles/s41467-024-48763-7?error=cookies_not_supported&code=397ad7e0-cc19-4c8b-a952-ea90d21c04d9]
- 91. signatures as predictive Lipidomic for early-onset lung... biomarkers [https://pubmed.ncbi.nlm.nih.gov/40180245/]
- 92. Serum Lipidomics Profiling to Identify Potential Biomarkers of... [https://www.besjournal.com/en/article/doi/10.3967/bes2025.095]
- 93. A guide to interpreting the area under the curve value [https://pmc.ncbi.nlm.nih.gov/articles/PMC10664195/]
- 94. ROC curve analysis (AUC, Sensitivity, Specificity, etc.) [https://www.medcalc.org/en/manual/roc-curves.php]
- 95. Comparison of two immunoassays for CA19-9, CEA and ... [https://www.biochemia-medica.com/en/journal/20/1/10.11613/BM.2010.010/fullArticle]
- 96. How to Interpret Lipidomics Data: A Step-by-Step Guide [https://lipidomics.creative-proteomics.com/resource/how-to-interpret-lipidomics-data-a-step-by-step-guide.htm]
- 97. Lipidomics in Biomarker Research - NCBI [https://www.ncbi.nlm.nih.gov/books/NBK584293/]
- 98. Lipidomic profiling of human serum enables detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8748654/]
- 99. Diagnostic Biomarker Candidates Proposed Using ... [https://www.mdpi.com/2072-6694/17/18/2988]
- 100. Lipidome profile predictive of disease evolution and activity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8894401/]
- 101. Lipidome profile predictive of disease evolution and activity ... [https://www.nature.com/articles/s12276-022-00725-z]
- 102. Serum and urine lipidomic profiles identify biomarkers ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2024.1410365/full]
- 103. Article Metabolomics and lipidomics predictor of survival in ... [https://www.sciencedirect.com/science/article/abs/pii/S0090955625095017]
- 104. Differences in Prediagnostic Serum Metabolomic and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11389719/]