Multiplex Droplet Digital PCR Assay Design for CCR5 Genotyping: From Foundational Principles to Clinical Validation
This comprehensive guide details the development and implementation of multiplex droplet digital PCR (ddPCR) assays for precise discrimination between CCR5 wild-type and Δ32 alleles.
Multiplex Droplet Digital PCR Assay Design for CCR5 Genotyping: From Foundational Principles to Clinical Validation
Abstract
This comprehensive guide details the development and implementation of multiplex droplet digital PCR (ddPCR) assays for precise discrimination between CCR5 wild-type and Δ32 alleles. Targeting researchers, scientists, and drug development professionals, it covers foundational principles of CCR5 biology and ddPCR technology, step-by-step methodological design for robust multiplexing, systematic troubleshooting and optimization strategies, and rigorous validation frameworks against established platforms. The content synthesizes current research demonstrating ddPCR's capability to detect mutant alleles in heterogeneous cell mixtures with sensitivity down to 0.8%, providing an essential resource for applications in HIV cure research, clinical diagnostics, and therapeutic monitoring of CCR5-targeted interventions.
CCR5 Biology and Digital PCR Fundamentals: Building Your Assay Knowledge Base
The Critical Role of CCR5 in HIV Pathogenesis and Therapeutic Targeting
The C-C chemokine receptor type 5 (CCR5) is a seven-transmembrane G-protein-coupled receptor (GPCR) expressed on various immune cells including macrophages, dendritic cells, and memory T-cells [1] [2]. Beyond its physiological role in inflammatory responses and immune cell migration, CCR5 serves as a critical co-receptor for human immunodeficiency virus type 1 (HIV-1) entry into host cells [3] [4]. The discovery that a 32-base pair deletion (Δ32) in the CCR5 gene provides strong resistance to HIV-1 infection in homozygous individuals sparked extensive research into CCR5 as a therapeutic target [4] [5]. This application note examines the role of CCR5 in HIV pathogenesis and details protocols for CCR5 genotyping using multiplex droplet digital PCR (ddPCR), providing researchers with methodologies to advance therapeutic development.
The HIV-1 entry process begins with viral envelope glycoprotein gp120 binding to the CD4 receptor on target cells, followed by interaction with CCR5, which facilitates viral fusion and entry [1] [2]. The critical nature of this interaction is evidenced by the fact that individuals homozygous for the CCR5-Δ32 mutation are highly resistant to HIV-1 infection, while heterozygotes often experience slower disease progression [4] [5]. This application note provides detailed methodologies for CCR5 allele detection, enabling research into CCR5-targeted therapies and genetic screening.
CCR5 in HIV-1 Pathogenesis: Mechanisms and Epidemiology
Molecular Mechanisms of HIV-1 Entry
CCR5 serves as the principal co-receptor for macrophage-tropic (R5) HIV-1 strains, which are responsible for the majority of primary infections [1]. The HIV-1 entry mechanism involves a multi-step process:
- Step 1: Viral gp120 binding to CD4 receptor, inducing conformational changes [1]
- Step 2: Exposure of CCR5 binding sites on gp120, facilitating coreceptor engagement [1]
- Step 3: Structural rearrangements in gp41 that promote membrane fusion and viral entry [2]
The N-terminal domain and extracellular loops of CCR5 contain critical residues for HIV-1 envelope interaction, particularly tyrosine sulfation sites at positions 3, 10, 14, and 15 that enhance gp120 binding affinity [1]. Post-translational modifications including sulfation, palmitoylation, and glycosylation further contribute to the structural diversity of CCR5, influencing viral tropism and entry efficiency [1].
Global Distribution of CCR5-Δ32 Allele
The protective CCR5-Δ32 allele demonstrates significant geographic variation in frequency, with highest prevalence in Northern European populations and near absence in African, Asian, and indigenous American populations [5]. This distribution has important implications for HIV susceptibility and therapeutic development across different ethnic groups.
Table 1: Global Frequency Distribution of CCR5-Δ32 Allele
| Population | Δ32 Allele Frequency (%) | Homozygous Frequency (%) | Source |
|---|---|---|---|
| Norwegian | 16.4 | ~2.7 | [5] |
| Faroe Islands | - | 2.3 | [5] |
| Peruvian | 1.35 | 0 | [6] |
| Ethiopian | 0 | 0 | [5] |
Recent studies of Peruvian populations with high-risk sexual behavior revealed a low CCR5-Δ32 prevalence (2.7% heterozygous, 0% homozygous), highlighting the potential influence of other genetic factors in HIV resistance in certain populations [6]. This geographical variation necessitates population-specific screening approaches when evaluating CCR5-targeted therapies.
CCR5-Targeted Therapeutic Approaches
CCR5 Knockout Strategies
The proof-of-concept for CCR5-targeted therapies was established by the cases of the "Berlin Patient" and "London Patient," both cured of HIV-1 infection following hematopoietic stem cell transplantation from CCR5-Δ32 homozygous donors [3]. This breakthrough prompted development of multiple CCR5 knockout strategies:
- Zinc Finger Nucleases (ZFNs): First genome editing technology used in clinical trials for CCR5 disruption [3] [7]
- TAL Effector Nucleases (TALENs): Offer high specificity with customizable DNA-binding domains [7]
- CRISPR/Cas9: Provides flexible and efficient gene editing capabilities [3] [8]
Recent advances include GMP-compatible, automated production of CCR5-negative CD4+ T-cells using TALEN mRNA electroporation, enabling clinical-scale generation of HIV-resistant cells (>1.5 × 10⁹ cells with >60% editing efficiency) [7]. This approach demonstrates the translational potential of CCR5 gene editing for HIV therapy.
CCR5 Blockade Strategies
Alternative therapeutic approaches focus on CCR5 receptor blockade:
- Small molecule antagonists (e.g., maraviroc) bind CCR5 transmembrane domains, preventing conformational changes required for HIV-1 entry [3]
- Monoclonal antibodies target extracellular CCR5 domains, inhibiting gp120 binding while potentially modulating receptor signaling [3]
These strategies face challenges including viral resistance development and potential interference with CCR5 physiological functions in immune response [3]. Combination approaches utilizing both gene editing and pharmacological blockade may provide synergistic benefits for long-term HIV control.
Multiplex ddPCR for CCR5 Genotyping: Application Notes
Principles of CCR5 Allele Discrimination
Droplet digital PCR (ddPCR) enables absolute quantification of CCR5 wild-type and Δ32 alleles without standard curves, providing superior precision for detecting copy number variations in heterogeneous samples [8] [9]. The fundamental principle involves partitioning nucleic acid samples into thousands of nanoliter-sized droplets, with endpoint PCR amplification and fluorescence detection allowing binary determination of target presence in each droplet [8] [9].
For CCR5 genotyping, the 32-bp deletion creates a natural sequence polymorphism that can be distinguished through probe-based detection systems. The Δ32 mutation results in a frameshift and premature stop codon, producing a truncated, non-functional receptor [8]. Multiplex ddPCR assays can simultaneously quantify wild-type and Δ32 alleles in a single reaction, significantly reducing processing time and reagent costs compared to separate duplex reactions [9].
Comprehensive Experimental Workflow
The following diagram illustrates the complete workflow for CCR5 genotyping using multiplex ddPCR:
Research Reagent Solutions
Table 2: Essential Reagents for CCR5 ddPCR Genotyping
| Reagent/Category | Specific Examples | Function & Application Notes |
|---|---|---|
| DNA Extraction | QIAamp DNA Blood Mini Kit [8] [9] | High-quality genomic DNA isolation; critical for PCR efficiency |
| ddPCR Master Mix | ddPCR Supermix for Probes (No dUTP) [9] | Optimized buffer system for probe-based digital PCR |
| CCR5 Primers/Probes | Custom FAM/HEX-labeled probes [8] [9] | Allele-specific discrimination; dual-labeling enables multiplexing |
| Reference Assay | AP3B1 Copy Number Reference Assay [9] | Internal control for DNA quality and quantity normalization |
| Droplet Generation | DG8 Cartridges, Droplet Generation Oil [8] [9] | Nanodroplet formation for partition-based digital PCR |
| Restriction Enzyme | BamHI [9] | DNA fragmentation to improve amplification efficiency |
| Positive Controls | CCR5-Δ32 plasmid standards [8] | Assay validation and quantification reference |
Detailed Protocol for Multiplex ddPCR
Reaction Setup and Optimization
The multiplex ddPCR assay allows simultaneous quantification of CCR5 wild-type and Δ32 alleles in a single reaction, reducing costs and processing time by approximately threefold compared to separate duplex reactions [9].
Reaction Master Mix Preparation:
- Combine 12.5 µL of 2× ddPCR Supermix for Probes (No dUTP)
- Add 1.25 µL of primer/probe mix (final concentrations: 1350-2700 nM primers, 250-750 nM probes)
- Include 5 µL of BamHI-treated genomic DNA (10-55 ng total)
- Adjust final volume to 25 µL with nuclease-free water [9]
Primer/Probe Design Considerations:
- CCR5 wild-type probe: FAM-labeled, targets intact CCR5 sequence
- CCR5-Δ32 probe: HEX-labeled, spans deletion junction
- Reference gene probe: Different fluorophore for normalization
- All probes should have similar Tm values for balanced amplification [8] [9]
Droplet Generation and Thermal Cycling
Droplet Generation:
- Transfer 20 µL reaction mix to DG8 cartridge wells
- Add 70 µL Droplet Generation Oil for Probes
- Process in QX200 Droplet Generator
- Collect 40 µL emulsion for PCR amplification [9]
Thermal Cycling Conditions:
- Initial denaturation: 95°C for 10 min
- 40 cycles of:
- Denaturation: 94°C for 30 s
- Annealing/Extension: 60°C for 60 s
- Final enzyme deactivation: 98°C for 10 min
- Hold at 4°C until analysis [9]
Optimal Annealing Temperature Determination:
- Test temperature gradient from 58°C to 64°C
- Select temperature with best separation between positive and negative droplets for both targets
- 60°C typically provides optimal results for CCR5 genotyping [9]
Data Acquisition and Analysis
Droplet Reading and Analysis:
- Transfer amplified droplets to QX200 Droplet Reader
- Measure fluorescence amplitude for each fluorophore
- Analyze data using QuantaSoft software (v1.7.4.0917 or higher)
- Set threshold between positive and negative droplets based on controls [8] [9]
Genotype Determination:
- Calculate target copies/µL based on Poisson distribution
- Normalize CCR5 signals to reference gene
- Determine Δ32 allele frequency:
- Homozygous wild-type: Only FAM-positive droplets
- Heterozygous: Both FAM and HEX-positive droplets
- Homozygous Δ32: Only HEX-positive droplets [8]
Quality Control Measures:
- Include no-template control (NTC) for contamination assessment
- Use known genotype controls for assay validation
- Accept samples with >10,000 total droplets for statistical reliability
- Ensure reference gene signals are consistent across samples [8] [9]
Troubleshooting and Technical Considerations
Common Optimization Challenges
Poor Droplet Resolution:
- Cause: Improper oil-to-sample ratio or viscous samples
- Solution: Verify DNA purity (A260/A280 ~1.8-2.0), ensure complete restriction digest
- Adjustment: Dilute samples if concentrated, vortex mixture before loading [9]
Low Amplitude Separation:
- Cause: Suboptimal primer/probe concentrations or thermal cycling conditions
- Solution: Titrate primer/probe ratios (typically 1:1 to 3:1 primer:probe)
- Adjustment: Optimize annealing temperature using thermal gradient [9]
Assay Sensitivity Limitations:
- The developed ddPCR system can accurately detect mutant CCR5-Δ32 alleles in heterogeneous cell mixtures down to 0.8% variant frequency [8]
- For lower frequency detection, increase DNA input and droplet count
- Ensure restriction digestion to eliminate amplification bias from DNA secondary structures [8]
Applications in HIV-1 Therapeutic Monitoring
Multiplex ddPCR for CCR5 genotyping enables several critical research applications:
- Stem cell donor screening for HIV-1 positive patients requiring transplantation [5]
- Monitoring gene editing efficiency in CCR5 knockout therapies [7]
- Quantifying Δ32 allele frequency in mixed cell populations after transplantation [8]
- Population studies of CCR5-Δ32 distribution and HIV-1 susceptibility [6] [5]
The methodology provides absolute quantification without external standards, making it ideal for clinical translation and therapeutic development workflows [8] [9].
CCR5 remains a critical therapeutic target for HIV-1 treatment, with gene editing approaches showing particular promise for long-term viral control. The multiplex ddPCR protocols detailed herein provide researchers with robust, reproducible methods for CCR5 genotyping with applications in basic research, therapeutic development, and clinical monitoring. As CCR5-targeted therapies advance, precise genotyping and editing efficiency quantification will be essential for evaluating therapeutic efficacy and optimizing treatment strategies.
Future directions include developing even more highly multiplexed assays incorporating additional HIV-related genetic markers (e.g., HLA-B*57:01 for abacavir hypersensitivity [6] [10]) and creating integrated workflows for comprehensive HIV patient stratification. The continued refinement of ddPCR technologies will further enhance sensitivity and throughput, accelerating the development of CCR5-targeted curative strategies for HIV-1 infection.
The C-C chemokine receptor type 5 (CCR5) is a seven-transmembrane G-protein coupled receptor that serves as a major co-receptor for human immunodeficiency virus (HIV) entry into host cells [11]. HIV infection requires binding to both the CD4 receptor and a coreceptor, predominantly CCR5 or CXCR4, with CCR5-tropic (R5) strains being primarily responsible for initial transmission and establishing new infections [11] [12]. The CCR5Δ32 mutation refers to a 32-base pair deletion in the CCR5 gene that results in a frameshift and premature stop codon, producing a truncated protein that is not expressed on the cell surface [8] [13]. This mutation obliterates the functional CCR5 receptor, thereby preventing R5-tropic HIV strains from entering and infecting host cells [13].
Individuals homozygous for the CCR5Δ32 mutation exhibit substantial resistance to HIV-1 infection, while heterozygous carriers show reduced susceptibility to infection and delayed progression to AIDS if infection occurs [11] [14]. This natural resistance mechanism has inspired numerous therapeutic strategies aimed at mimicking this protective effect, including gene editing approaches and pharmacological blockade of the CCR5 receptor [12] [15].
Biological Mechanism of CCR5Δ32-Mediated Resistance
CCR5 Structure and Function in HIV Entry
CCR5 is normally expressed on immature (Th0) and memory Th1 cells, monocytes, macrophages, dendritic cells, and certain neural and vascular tissue cells [11]. The receptor's natural ligands include pro-inflammatory chemokines CCL3 (MIP-1α), CCL4 (MIP-1β), and CCL5 (RANTES) [11]. For HIV entry, the viral envelope glycoprotein gp120 initially binds to CD4, inducing conformational changes that enable subsequent binding to CCR5. This binding exposes the gp41 fusion peptide, facilitating viral envelope fusion with the host cell membrane and entry of the viral core [15].
The CCR5Δ32 mutation results in a truncated protein that fails to traffic to the cell surface due to improper folding and retention in the endoplasmic reticulum [13]. Without the CCR5 coreceptor exposed on the cell surface, HIV cannot complete the entry process, thereby conferring resistance to infection for homozygous individuals [11] [13].
Signaling Pathway and Mutation Impact
The following diagram illustrates the normal HIV entry process via CCR5 and how the Δ32 mutation confers resistance:
Geographic Distribution and Population Genetics
The CCR5Δ32 mutation exhibits a distinctive geographic distribution pattern, with frequencies highest in Northern European populations and decreasing along a north-to-south cline [14] [16]. The table below summarizes the allele frequency distribution across different populations:
Table 1: CCR5Δ32 Allele Frequency Distribution by Geographic Region
| Geographic Region | Average Allele Frequency | Homozygous Frequency | Notes |
|---|---|---|---|
| Northern Europe | ~16% | ~1% | Highest frequencies in Scandinavia, Baltic regions |
| Central Europe | ~10% | ~0.5% | Moderate frequencies |
| Southern Europe | ~4-6% | <0.2% | Lower frequencies in Italy, Greece |
| Western Asia | ~2-4% | Rare | Frequencies decrease eastward |
| North Africa | <2% | Very rare | Limited distribution |
| African, East Asian, & Native American | ~0% | ~0% | Virtually absent |
This distribution pattern has inspired several hypotheses about the historical selective pressures that drove the allele to its current frequencies. Research indicates the mutation is evolutionarily young (estimated between 700-3,500 years old) yet reached relatively high frequencies, suggesting intense positive selection [14] [17] [16]. Proposed historical selective agents include:
- Bubonic plague (Black Death): Initial hypothesis suggesting plague resistance drove selection
- Smallpox: Currently favored hypothesis due to smallpox's longer historical presence and utilization of CCR5 receptor
- Other historical epidemics: Various pathogens that may have utilized CCR5
Spatial modeling studies suggest that with uniform selection across Europe, a Northern European origin with Viking-mediated dispersal best explains the distribution [14] [16]. However, when allowing for selection gradients, an origin outside Northern Europe with stronger selection intensity in northwestern regions may better fit observed patterns [16].
Clinical Significance and Therapeutic Applications
Natural HIV Resistance and Disease Modification
The clinical significance of CCR5Δ32 varies by genotype:
- Homozygous individuals: Exhibit substantial resistance to HIV-1 infection, though rare cases of infection with dual-tropic or X4-tropic strains have been reported [11]
- Heterozygous individuals: Show delayed progression to AIDS, reduced viral load set points, and increased likelihood of long-term non-progression [11] [17]
The landmark cases of the "Berlin" and "London" patients demonstrated that transplantation with hematopoietic stem cells (HSCs) from CCR5Δ32 homozygous donors could effectively cure HIV infection in patients with hematological malignancies [12] [18]. More recently, a case of sustained HIV remission after allogeneic HSCT with wild-type CCR5 donor cells was reported, suggesting that immune mechanisms beyond CCR5 ablation may contribute to viral control [18].
Therapeutic Strategies Inspired by CCR5Δ32
The protective effect of CCR5Δ32 has inspired multiple therapeutic approaches:
Table 2: CCR5-Targeted Therapeutic Strategies
| Strategy | Mechanism | Development Status |
|---|---|---|
| CCR5 Antagonists | Small molecule blockade of CCR5 receptor | Licensed (Maraviroc) |
| Gene Editing | CRISPR/Cas9, ZFNs, TALENs to disrupt CCR5 gene | Clinical trials (NCT03164135) |
| Stem Cell Transplantation | Allo-HSCT from CCR5Δ32 homozygous donors | Proven efficacy (5 reported cures) |
| Antibody-Based Approaches | CCR5-targeting antibodies to block HIV entry | Preclinical and clinical development |
| Multi-target Editing | Simultaneous targeting of CCR5, CXCR4, and HIV LTR | Experimental |
Gene editing technologies represent a particularly promising approach, with CRISPR/Cas9 enabling precise CCR5 disruption in hematopoietic stem cells or T cells to generate HIV-resistant immune populations [12] [15]. Multiplexed strategies targeting both CCR5 and CXCR4 coreceptors are being developed to prevent viral escape via tropism switching [12].
Multiplex ddPCR Assay for CCR5 Genotyping
Assay Principle and Applications
Droplet digital PCR (ddPCR) provides an accurate method for quantifying wild-type and Δ32 mutant alleles in heterogeneous cell mixtures [8]. This technology is particularly valuable for monitoring engraftment of CCR5-disrupted cells after gene therapy interventions and for precise genotyping in research and clinical settings.
The ddPCR assay works by partitioning a PCR reaction into thousands of nanoliter-sized droplets, with each droplet functioning as an individual PCR reaction. This partitioning allows absolute quantification of target DNA sequences without need for standard curves and enables detection of rare variants in mixed samples [8].
Experimental Protocol
Sample Preparation and DNA Extraction
- Cell Source: Peripheral blood mononuclear cells (PBMCs), hematopoietic stem cells, or tissue samples
- DNA Extraction: Use phenol-chloroform method or commercial kits (e.g., ExtractDNA Blood and Cells Kit)
- Quality Control: Measure DNA concentration and purity (A260/A280 ratio ~1.8-2.0)
- Storage: Store extracted DNA at -20°C until use
ddPCR Reaction Setup
- Reaction Volume: 20-22μL total volume
- DNA Input: 50-100ng genomic DNA
- Probe Design:
- FAM-labeled probe: Targets wild-type CCR5 sequence
- HEX/VIC-labeled probe: Targets CCR5Δ32 deletion junction
- Reference gene probe (optional): For normalization
- Master Mix: ddPCR Supermix for Probes (Bio-Rad)
- Droplet Generation: Use automated droplet generator (e.g., QX200 Droplet Generator)
- Thermal Cycling Conditions:
- 95°C for 10 minutes (enzyme activation)
- 40 cycles of: 94°C for 30 seconds, 58-60°C for 60 seconds
- 98°C for 10 minutes (enzyme deactivation)
- 4°C hold
Data Acquisition and Analysis
- Droplet Reading: Use droplet reader (e.g., QX200 Droplet Reader)
- Threshold Setting: Establish fluorescence thresholds based on negative controls
- Quantification: Calculate copies/μL of wild-type and Δ32 alleles using Poisson statistics
- Quality Metrics: Ensure >10,000 droplets per sample; exclude samples with poor droplet generation
The following workflow diagram illustrates the complete ddPCR process:
Data Analysis and Interpretation
The ddPCR platform generates four distinct populations of droplets:
- Double-negative droplets: No target DNA (background)
- FAM-positive droplets: Wild-type CCR5 alleles only
- HEX/VIC-positive droplets: CCR5Δ32 alleles only
- Double-positive droplets: Heterozygous or mixed samples
Table 3: Expected Genotyping Results with Multiplex ddPCR
| Genotype | FAM Signal (Wild-type) | HEX/VIC Signal (Δ32) | Interpretation |
|---|---|---|---|
| Wild-type homozygous | Positive | Negative | Two functional CCR5 alleles |
| Heterozygous | Positive | Positive | One wild-type, one Δ32 allele |
| Δ32 homozygous | Negative | Positive | Two Δ32 alleles |
| Mixed chimerism | Both populations present | Both populations present | Multiple cell populations |
The assay sensitivity allows detection of CCR5Δ32 alleles down to 0.8% in heterogeneous cell mixtures, making it suitable for monitoring engraftment efficiency in transplantation and gene therapy contexts [8].
Research Reagent Solutions
Table 4: Essential Research Reagents for CCR5Δ32 Studies
| Reagent/Category | Specific Examples | Application/Function |
|---|---|---|
| Cell Lines | MT-4 human T-cell line, Primary CD4+ T cells, HEK293T | In vitro models for HIV infection and gene editing studies |
| Genome Editing Tools | CRISPR/Cas9 (pCas9-IRES2-EGFP), ZFNs, TALENs | Introduction of CCR5Δ32 mutation or CCR5 knockout |
| ddPCR Reagents | ddPCR Supermix for Probes, FAM/HEX-labeled probes, droplet generation oil | Absolute quantification of wild-type and Δ32 alleles |
| Antibodies | Anti-CCR5 monoclonal antibodies, Flow cytometry antibodies | Detection of CCR5 surface expression |
| DNA Extraction Kits | ExtractDNA Blood and Cells Kit, Phenol-chloroform methods | High-quality genomic DNA isolation |
| Viral Assay Systems | HIV-1 reporter viruses, p24 ELISA kits, Viral culture systems | Assessment of HIV infectivity and replication |
The CCR5Δ32 mutation represents a powerful example of natural selection in humans and has provided profound insights into HIV pathogenesis and therapeutic development. The multiplex ddPCR assay for CCR5 genotyping offers researchers a robust tool for quantifying wild-type and Δ32 alleles with high precision and sensitivity. This methodology supports diverse applications from basic population genetics to clinical monitoring of cell-based therapies. As gene editing technologies advance toward clinical application, precise quantification of CCR5 disruption will remain essential for evaluating therapeutic efficacy and optimizing HIV cure strategies.
Core Principles of Digital PCR
Digital PCR (dPCR) represents a transformative approach to nucleic acid quantification by enabling absolute quantification without the need for standard curves [19]. The core principle involves partitioning a PCR reaction into thousands of individual reactions, effectively diluting the sample to a concentration where some partitions contain zero, one, or multiple target molecules [20]. After end-point PCR amplification, each partition is analyzed as positive or negative for the target sequence based on fluorescence detection [19]. The absolute quantity of the target nucleic acid is then calculated using Poisson statistics to account for the random distribution of molecules across partitions [19].
This partitioning and binary detection system fundamentally distinguishes dPCR from quantitative real-time PCR (qPCR), which relies on comparing amplification curves to standards of known concentration [21]. The "digital" nature of the readout provides direct absolute quantification by counting positive partitions rather than extrapolating from amplification kinetics [20].
Table 1: Key Advantages of Digital PCR for Absolute Quantification
| Feature | Description | Application Benefit |
|---|---|---|
| Absolute Quantification | Does not require standard curves or reference materials [19] | Eliminates reference preparation errors and inter-lab variability |
| High Precision | Partitioning creates thousands of data points for statistical robustness [19] | Enables detection of small fold-change differences with high confidence |
| Superior Sensitivity | Effective target enrichment through partitioning improves limit of detection [19] | Ideal for rare allele detection in complex backgrounds |
| Tolerance to Inhibitors | Sample partitioning reduces effective concentration of PCR inhibitors [19] | Maintains accuracy in complex sample matrices like blood, soil, and tissue |
Application to CCR5 Genotyping
In CCR5 wild-type and Δ32 allele research, dPCR provides exceptional capability for precise genotyping and quantification of allele frequencies in heterogeneous cell mixtures [8]. The CCR5Δ32 mutation, a 32-base pair deletion that confers resistance to HIV-1 infection, represents an ideal target for dPCR quantification due to the need for precise measurement of editing efficiency in therapeutic applications [8] [22].
Droplet digital PCR (ddPCR) enables researchers to accurately quantify the proportion of CCR5Δ32 alleles even at frequencies as low as 0.8% in mixed cell populations [8]. This sensitivity is critical for evaluating CRISPR/Cas9 genome editing efficiency in hematopoietic stem and progenitor cells (HSPCs) being developed for HIV-1 functional cure strategies [22]. The absolute quantification capability of dPCR allows direct measurement of mutant allele copy numbers without reference to endogenous controls, providing unambiguous data on gene editing outcomes [21].
Multiplex ddPCR Assay Design for CCR5
Multiplex dPCR enables simultaneous detection of wild-type and Δ32 alleles in a single reaction, conserving precious samples while providing internal validation [23]. Advanced dPCR systems support multiplexing through multiple detection channels and amplitude-based discrimination strategies [23].
Diagram 1: Digital PCR workflow for CCR5 genotyping. The process involves sample partitioning, endpoint amplification, fluorescence detection in multiple channels, and statistical analysis for absolute quantification.
Probe Design Strategy
For CCR5 wild-type and Δ32 discrimination, a dual-probe system employing different fluorophores enables clear allele distinction:
- Wild-type CCR5 detection: Use probes targeting the intact sequence with FAM fluorophore
- Δ32 allele detection: Use probes spanning the deletion junction with HEX/VIC fluorophore
- Reference gene: Incorporate a third channel for reference gene quantification when needed [23]
This multiplex approach allows researchers to directly calculate the ratio of wild-type to mutant alleles and determine the editing efficiency in heterogeneous samples [8].
Detailed Experimental Protocol
Multiplex ddPCR for CCR5 Genotyping
Materials and Equipment:
- QIAcuity Digital PCR System (Qiagen) or QX200 Droplet Digital PCR System (Bio-Rad)
- dPCR master mix suitable for probe-based detection
- FAM-labeled probe for CCR5 wild-type sequence
- HEX-labeled probe for CCR5 Δ32 deletion sequence
- Primers flanking the CCR5 Δ32 mutation region
- DNA template (50-100 ng genomic DNA per reaction)
- Nuclease-free water
- Partitioning cartridges or droplets generation oil as appropriate for system
Table 2: Research Reagent Solutions for CCR5 ddPCR
| Reagent | Function | Specifications | Optimization Tips |
|---|---|---|---|
| Probe-based dPCR Master Mix | Provides enzymes, nucleotides, and buffer for amplification | Contains dUTP for carryover prevention; optimized for partition stability | Use master mixes specifically formulated for dPCR to ensure stable partitions |
| FAM-labeled Wild-type Probe | Detects intact CCR5 sequence | Typically 20-30 nucleotides; Tm 65-70°C | Position probe to span wild-type specific sequence avoiding Δ32 junction |
| HEX-labeled Δ32 Probe | Detects 32-bp deletion allele | Designed to span deletion junction; Tm matched to wild-type probe | Verify specificity with both wild-type and Δ32 control templates |
| CCR5 Amplification Primers | Amplify region containing Δ32 mutation | Amplicon size: 60-150 bp; Tm 55-65°C | Place primers to generate short amplicons for optimal dPCR efficiency |
Procedure:
Reaction Setup:
- Prepare 20 μL reaction mix containing:
- 1× dPCR master mix
- 900 nM forward primer
- 900 nM reverse primer
- 250 nM FAM-labeled wild-type probe
- 250 nM HEX-labeled Δ32 probe
- 50-100 ng genomic DNA
- Nuclease-free water to volume
- Mix thoroughly by pipetting, avoid vortexing after partitioning reagents added
- Prepare 20 μL reaction mix containing:
Partitioning:
- For droplet systems: Generate droplets according to manufacturer's instructions using droplet generation oil
- For nanoplate systems: Load reaction mix into nanoplate wells and seal with appropriate foil
- Ensure proper partition formation by verifying uniformity under manufacturer-specified QC parameters
PCR Amplification:
- Perform amplification using the following cycling conditions:
- Initial denaturation: 95°C for 10 minutes
- 40 cycles of:
- Denaturation: 95°C for 30 seconds
- Annealing/Extension: 60°C for 60 seconds
- Final extension: 72°C for 5 minutes
- Signal stabilization: 4°C hold (optional, system-dependent)
- Use a ramp rate of 2°C/second for all steps
- Perform amplification using the following cycling conditions:
Signal Detection and Analysis:
- For droplet systems: Transfer plate to droplet reader for individual droplet analysis
- For nanoplate systems: Perform in-plate imaging with appropriate fluorescence filters
- Set fluorescence thresholds to distinguish positive from negative partitions
- Apply Poisson correction to calculate absolute copy numbers of wild-type and Δ32 alleles
Diagram 2: Multiplex ddPCR workflow for simultaneous CCR5 wild-type and Δ32 allele detection, showing droplet classification based on fluorescence patterns.
Data Analysis and Interpretation
Calculating Absolute Quantification: The absolute concentration of targets is calculated using Poisson statistics:
- Copies/μL = -ln(1 - p) × (1/partition volume) where p = ratio of positive partitions [19]
- For a typical droplet system with 0.91 nL droplets: Copies/μL = -ln(1 - p) × (1/0.00091)
- Most dPCR instruments automatically perform these calculations with proprietary software
Key Quality Control Parameters:
- Partition number: Minimum of 10,000 valid partitions for reliable quantification
- Partition uniformity: Ensure consistent partition volume and fluorescence intensity
- Threshold setting: Set fluorescence thresholds to clearly separate positive and negative populations
- Negative controls: Include no-template controls to assess contamination and background signal
Case Study: CCR5Δ32 Quantification in HIV Cure Research
A 2022 study demonstrated the power of ddPCR for quantifying CRISPR/Cas9-generated CCR5Δ32 mutations in heterogeneous cell mixtures [8]. Researchers developed a multiplex ddPCR assay to accurately measure the content of mutant CCR5Δ32 alleles down to 0.8% in the MT-4 human T-cell line [8].
The experimental approach involved:
- Generating CCR5Δ32 mutations using CRISPR/Cas9 with two guide RNAs
- Sorting single cells to generate monoclonal cell lines
- Using multiplex ddPCR to quantify the editing efficiency in mixed cell populations
- Validating results with sequencing to confirm mutation specificity
This application highlights dPCR's critical role in advancing HIV cure strategies by providing precise quantification of gene editing outcomes essential for therapeutic development [22].
Troubleshooting and Optimization
Common Challenges and Solutions:
- Rain Effect (partitions with intermediate fluorescence): Optimize annealing temperature and probe concentration
- Low Partition Number: Ensure proper partitioning technique and check for master mix compatibility
- Poor Cluster Separation: Titrate primer and probe concentrations; verify probe specificity
- Inhibition Issues: Despite dPCR's tolerance to inhibitors, excessive inhibition may require sample dilution or cleanup
Optimization Strategies:
- Perform primer/probe concentration gradients in singleplex before multiplexing
- Validate assay specificity with known wild-type and mutant controls
- Optimize thermal cycling conditions for specific instrument platforms
- Use low-binding plastics to minimize sample loss during preparation [21]
The precision of dPCR for absolute quantification of CCR5 alleles provides researchers and drug developers with robust data for evaluating gene editing therapies, monitoring patient responses, and advancing innovative approaches for HIV-1 functional cure [8] [22].
Advantages of ddPCR Over qPCR for Rare Allele Detection and Quantification
The detection and quantification of rare genetic alleles present significant challenges for conventional quantitative PCR (qPCR). This application note details the superior performance of droplet digital PCR (ddPCR) for identifying low-frequency mutations, with a specific focus on detecting the CCR5-Δ32 allele, a co-receptor knockout mutation conferring resistance to HIV infection. We demonstrate that ddPCR provides absolute quantification without standard curves, enhances sensitivity for variants with allele frequencies below 1%, and offers greater resilience to PCR inhibitors. Detailed protocols and reagent solutions are provided to facilitate the implementation of robust multiplex ddPCR assays for CCR5 wild-type and Δ32 allele research and drug development.
Accurate detection and quantification of rare alleles are critical in numerous research and clinical diagnostics fields, from monitoring subpopulations in cancer to assessing genome editing efficiency. The C-C chemokine receptor type 5 (CCR5) represents a prime example, where a 32-base pair deletion (CCR5-Δ32) results in a non-functional receptor and confers resistance to HIV-1 infection [8]. Research into curative strategies for HIV, including hematopoietic stem cell transplantation from CCR5-Δ32 donors or the use of CRISPR/Cas9 to recreate this mutation, requires sensitive methods to accurately quantify the proportion of mutant alleles in heterogeneous cell mixtures [8].
While quantitative real-time PCR (qPCR) has been the workhorse for nucleic acid quantification, it falters in applications demanding the detection of rare sequence variants or minute fold-changes. Droplet digital PCR (ddPCR), by contrast, transforms these measurements by providing absolute quantification with a precision that is unattainable with qPCR for low-abundance targets [24] [25]. This note outlines the theoretical and practical advantages of ddPCR, providing a validated framework for its application in sensitive CCR5 genotyping.
Comparative Analysis: ddPCR vs. qPCR
The fundamental difference between the two techniques lies in their approach to quantification. qPCR is an "analog" method that relies on comparing the amplification curve of a sample to a standard curve derived from samples of known concentration, with data collected during the exponential phase of amplification. Conversely, ddPCR is a "digital" method that partitions a sample into thousands of nanoliter-sized droplets, performs end-point PCR on each droplet, and uses Poisson statistics to count the absolute number of target DNA molecules from the ratio of positive to negative droplets, without the need for a standard curve [25] [26].
Table 1: Core Technical Differences Between qPCR and ddPCR
| Feature | Quantitative PCR (qPCR) | Droplet Digital PCR (ddPCR) |
|---|---|---|
| Quantification Basis | Relative (requires standard curve) | Absolute (no standard curve) |
| Signal Measurement | Real-time, during exponential phase | End-point, after amplification is complete |
| Sample Handling | Bulk reaction | Partitioned into ~20,000 droplets |
| Tolerance to Inhibitors | Lower | Higher [24] [25] |
| Tolerance to Amplification Efficiency Variations | Sensitive | Robust [24] [25] |
| Precision | Detects mutation rates >1% [24] | Detects mutation rates ≥ 0.1% [24] [25] |
For rare allele detection, such as quantifying the fraction of CCR5-Δ32 alleles in a wild-type background, the partitioning step of ddPCR is crucial. It effectively concentrates the rare target within isolated microreactors, reducing competition from abundant wild-type sequences and thereby improving the signal-to-noise ratio [26]. Studies have shown that ddPCR can accurately measure the content of cells with the CCR5-Δ32 mutation down to 0.8% [8], a level of sensitivity that is challenging for standard qPCR assays.
The Scientist's Toolkit: Research Reagent Solutions
The following table lists key reagents and materials essential for setting up a multiplex ddPCR assay for CCR5 wild-type and Δ32 alleles, based on published protocols [8].
Table 2: Essential Reagents for CCR5 ddPCR Assay Development
| Item | Function/Description | Example (from Literature) |
|---|---|---|
| ddPCR Supermix | Optimized master mix for droplet generation and digital PCR. | Bio-Rad ddPCR Supermix for Probes |
| FAM-labeled Probe | Detects the mutant CCR5-Δ32 allele sequence. | Sequence specific to the Δ32 deletion junction |
| HEX/VIC-labeled Probe | Detects the wild-type CCR5 allele sequence. | Sequence spanning the wild-type deletion region |
| Primers | Amplify a conserved region of the CCR5 gene flanking the Δ32 site. | F: CCCAGGAATCATCTTTACCAR: GACACCGAAGCAGAGTTT [8] |
| Droplet Generator | Partitions the PCR reaction into ~20,000 nanoliter-sized droplets. | Bio-Rad QX200 Droplet Generator |
| Droplet Reader | Performs end-point fluorescence reading of each droplet. | Bio-Rad QX200 Droplet Reader |
| Thermal Cycler | Standard instrument for PCR amplification. | C1000 Touch Thermal Cycler (Bio-Rad) [8] |
| Cell Lysis Buffer | For preparing crude lysates from limited cell samples without DNA extraction. | SuperScript IV CellsDirect cDNA Synthesis Kit Lysis Buffer [27] |
Experimental Protocol: Multiplex ddPCR for CCR5 Genotyping
This protocol is adapted from a study that successfully used ddPCR to quantify an artificial CCR5-Δ32 mutation generated by CRISPR/Cas9 in heterogeneous cell mixtures [8].
Sample Preparation: Crude Lysate Method for Limited Samples
For samples with limited cell numbers (e.g., < 1000 cells), a crude lysate protocol is recommended to avoid target loss during DNA extraction [27]. This method has been validated for the absolute quantification of rare gene targets.
- Cell Lysis: Resuspend 200 - 10,000 cells in PBS and lyse using a buffer such as that from the SuperScript IV CellsDirect cDNA Synthesis Kit ("Buffer 2").
- Viscosity Breakdown (Critical Step): To reduce viscosity from intact oligonucleotides that can impede droplet formation, subject the lysate to a viscosity breakdown protocol. This step has been shown to improve reliability and accuracy by preventing anomalous droplet patterns and overestimation of target copies [27].
- Lysate Use: Use the crude lysate directly in the ddPCR reaction mix. A study quantifying T-Cell Receptor Excision Circles (TRECs) demonstrated that this method produces droplets of consistent volume (average ~0.70 nL) and shows strong linearity and accuracy compared to standard ddPCR with extracted DNA [27].
ddPCR Reaction Setup and Droplet Generation
- Prepare Reaction Mix (20 µL total volume):
- ddPCR Supermix for Probes (1X)
- FAM-labeled CCR5-Δ32 probe (e.g., 250 nM)
- HEX-labeled CCR5-WT probe (e.g., 250 nM)
- Forward and Reverse Primers (e.g., 900 nM each)
- Template: ~20 ng of genomic DNA or 2-5 µL of crude cell lysate
- Generate Droplets: Transfer the entire reaction mix to a DG8 cartridge for the droplet generator. Following the manufacturer's instructions, generate approximately 20,000 droplets per sample.
- Transfer and Seal: Carefully transfer the generated emulsion to a 96-well PCR plate. Seal the plate with a foil heat seal.
Thermal Cycling
Amplify the target sequences using the following standard cycling conditions on a standard thermal cycler:
- Enzyme activation: 95°C for 10 minutes.
- Denaturation: 94°C for 30 seconds.
- Annealing/Extension: 55-60°C (assay-specific) for 60 seconds.
- Repeat steps 2-3 for 40 cycles.
- Enzyme deactivation: 98°C for 10 minutes.
- Hold at 4°C.
Droplet Reading and Data Analysis
- Read Droplets: Place the PCR plate in the droplet reader. The instrument will aspirate each sample and read the fluorescence (FAM and HEX) of each droplet.
- Analyze Data: Use the accompanying software (e.g., QuantaSoft) to analyze the results. The software will generate a 2D plot showing four droplet populations:
- FAM-positive/HEX-negative (mutant Δ32 alleles)
- HEX-positive/FAM-negative (wild-type alleles)
- Double-positive (potential heterozygotes or artifacts)
- Double-negative (no target present)
- Quantify Absolute Copy Numbers: The software uses Poisson statistics to calculate the absolute concentration of wild-type and mutant targets in copies per microliter of the original reaction. The fraction of mutant alleles can be determined as: (Δ32 copies) / (Δ32 copies + WT copies).
Workflow and Data Analysis
The following diagram illustrates the core ddPCR workflow and its logical advantage in rare allele detection.
The statistical foundation of ddPCR is based on binomial probability and Poisson statistics. The concentration of the target (λ) is calculated from the proportion of positive droplets (k) over the total number of partitions (n) using the formula: λ = -ln(1 - k/n) [26]. This mathematical framework defines the precision of the technique, with optimal performance achieved when a significant proportion of partitions are negative, a condition easily met for rare alleles.
Droplet digital PCR represents a significant technological advancement for applications requiring the detection and absolute quantification of rare alleles. Its superior sensitivity, precision, and robustness compared to qPCR make it an indispensable tool for advanced genetic research. In the context of CCR5 research, the implementation of the multiplex ddPCR assays and protocols described herein provides researchers and drug development professionals with a powerful method to accurately monitor CCR5-Δ32 allele frequencies, thereby accelerating the development of next-generation therapies for HIV and beyond.
The C-C chemokine receptor type 5 (CCR5) is a transmembrane protein that serves as a co-receptor for the human immunodeficiency virus (HIV), particularly the R5-tropic strains which are the most common and contagious [8] [4]. A natural 32-base pair deletion in the CCR5 gene, known as the CCR5Δ32 allele, results in a non-functional receptor. Individuals homozygous for this mutation (CCR5Δ32/Δ32) exhibit high-level resistance to HIV-1 infection, as the virus cannot effectively enter their CD4+ T-cells [12] [4]. This discovery, stemming from observations of exposed seronegative individuals and the documented cures of the "Berlin" and "London" HIV patients who received CCR5Δ32/Δ32 hematopoietic stem cell transplants (HSCT), has established CCR5 as a paramount target for novel therapeutic and curative strategies against HIV [8] [12] [28].
The frequency of the CCR5Δ32 allele varies significantly across global populations, being present in approximately 10% and 1% of the Northern European population in heterozygous and homozygous states, respectively [8]. However, its prevalence is low or absent in other regions, such as Africa and South America. A 2025 study in the Peruvian population found a heterozygous prevalence of only 2.7%, with no homozygous cases detected [6]. Similarly, a 2025 study in Angola reported a 0% frequency for the CCR5Δ32 allele among the 272 individuals tested [29]. This geographical distribution influences the global applicability of therapies centered on this mutation.
Table 1: Global Distribution of the CCR5Δ32 Allele
| Population | Heterozygous Frequency | Homozygous Frequency | Citation |
|---|---|---|---|
| Northern European | ~10% | ~1% | [8] |
| Peruvian | 2.7% | 0% | [6] |
| Angolan (Luanda) | 0% | 0% | [29] |
CCR5 in HIV Cure Strategies
Allogeneic Hematopoietic Stem Cell Transplantation
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) using cells from CCR5Δ32/Δ32 donors has proven to be a curative intervention for a small number of HIV-positive individuals with hematological malignancies [12] [28]. The procedure reconstitutes the patient's immune system with donor-derived cells that inherently resist R5-tropic HIV infection. Recent advancements have challenged the assumption that a CCR5Δ32/Δ32 donor is absolutely necessary. A landmark 2024 case report described an individual who achieved sustained HIV remission for over 32 months after ART interruption following allo-HSCT with wild-type CCR5 donor cells [18]. This suggests that factors beyond CCR5 ablation, such as the conditioning regimen and graft-versus-host disease (GvHD) reactions, may contribute to reservoir reduction and long-term control [18].
Gene Editing and Autologous Transplantation
To overcome the limitations of allogeneic transplants—including donor scarcity and morbidity—researchers are developing autologous transplantation strategies using gene-edited hematopoietic stem and progenitor cells (HSPCs). The goal is to reproduce the CCR5Δ32 phenotype in a patient's own cells. Technologies like CRISPR/Cas9 allow for precise disruption of the CCR5 gene [8] [12] [28].
A critical 2025 pre-clinical study demonstrated that high-frequency editing (>90%) of CCR5 in human HSPCs is necessary to confer protective benefit upon transplantation into a mouse xenograft model. Titration experiments revealed that lower editing frequencies (e.g., between 54% and 26%) provided a negligible protective effect against HIV challenge [28]. This establishes a clear threshold effect for a successful gene-editing-based cure strategy, underscoring the need for highly efficient protocols.
Table 2: Key Gene Editing Technologies for CCR5
| Technology | Mechanism of Action | Advantages | Limitations |
|---|---|---|---|
| Zinc Finger Nucleases (ZFNs) | Custom zinc finger proteins fuse to FokI nuclease for DNA cleavage. | Early clinical trial data available (e.g., SB-728-T). | Complex design; higher risk of off-target effects [12]. |
| TALENs | Transcription activator-like effector proteins fuse to FokI nuclease. | Modular design offers improved specificity over ZFNs. | Technically demanding; large size hinders viral delivery [12]. |
| CRISPR/Cas9 | Guide RNA (gRNA) directs Cas9 nuclease to target DNA. | Easy design; high efficiency; allows for multiplexing. | Off-target effects; potential immunogenicity to Cas9 [12] [28]. |
Multiplex ddPCR for CCR5 Genotyping and Quantification
Principles and Advantages of ddPCR
Droplet digital PCR (ddPCR) is a powerful method for the absolute quantification of nucleic acid targets. In CCR5 research, it enables precise measurement of wild-type and Δ32 allele proportions in heterogeneous cell mixtures, which is crucial for monitoring the success of gene editing or transplantation protocols [8]. The technology partitions a single PCR reaction into thousands of nanoliter-sized droplets, effectively creating an endpoint PCR in each droplet. This allows for absolute quantification without the need for a standard curve and provides high sensitivity for detecting rare alleles in a background of wild-type sequences.
Application Note: Quantifying CCR5 Editing Efficiency
Objective: To accurately quantify the proportion of CCR5Δ32 mutant alleles in a heterogeneous cell population following CRISPR/Cas9 genome editing.
Experimental Protocol:
- DNA Extraction: Isolate genomic DNA from the cell mixture (e.g., edited HSPCs or peripheral blood mononuclear cells) using a commercial kit (e.g., ExtractDNA Blood and Cells Kit, Evrogen). Quantify DNA concentration and assess purity using a spectrophotometer [8].
- ddPCR Reaction Setup:
- Prepare a multiplex ddPCR reaction mixture containing:
- DNA template (approximately 50-100 ng).
- Two primer/probe sets: one specific for the wild-type CCR5 allele (e.g., labeled with FAM) and one specific for the CCR5Δ32 deletion (e.g., labeled with HEX/VIC) [8].
- ddPCR Supermix for Probes (No dUTP).
- Nuclease-free water.
- Prepare a multiplex ddPCR reaction mixture containing:
- Droplet Generation: Transfer the reaction mixture to a droplet generator cartridge. Following manufacturer guidelines, generate approximately 20,000 droplets using a droplet generator (e.g., Bio-Rad QX200 Droplet Generator) [8].
- PCR Amplification: Carefully transfer the emulsified samples to a 96-well PCR plate. Seal the plate and perform PCR amplification in a thermal cycler using optimized cycling conditions for the selected assay.
- Droplet Reading and Analysis: Place the plate in a droplet reader (e.g., Bio-Rad QX200 Droplet Reader). The instrument sequentially reads each droplet, classifying it as FAM-positive (wild-type), HEX-positive (Δ32), double-positive (heterozygous), or negative. Use associated software to analyze the data and calculate the copy number concentration (copies/μL) for each target [8].
- Calculation of Editing Efficiency:
- The percentage of CCR5Δ32 alleles is calculated as:
[Δ32 copies / (Wild-type copies + Δ32 copies)] * 100. - This system has been demonstrated to detect mutant alleles down to 0.8% in a mixture, highlighting its exceptional sensitivity [8].
- The percentage of CCR5Δ32 alleles is calculated as:
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CCR5 ddPCR and Gene Editing Studies
| Item/Category | Specific Examples | Function in Workflow |
|---|---|---|
| Cell Culture | RPMI-1640 medium, Fetal Bovine Serum (FBS) | Maintenance and expansion of cell lines (e.g., MT-4 T-cells) [8]. |
| Genomic DNA Isolation | Phenol-chloroform method, ExtractDNA Blood and Cells Kit (Evrogen) | High-quality DNA extraction for downstream PCR applications [8]. |
| CRISPR/Cas9 System | pU6-gRNA vector, pCas9-IRES2-EGFP plasmid, SpCas9 protein | Delivery of Cas9 nuclease and guide RNAs for targeted CCR5 gene disruption [8] [28]. |
| Electroporation | Gene Pulser Electroporation Buffer/Cuvettes (Bio-Rad) | Physical method for introducing CRISPR components into hard-to-transfect cells like HSPCs [8]. |
| ddPCR Core Reagents | ddPCR Supermix for Probes (No dUTP), DG8 Cartridges and Gaskets (Bio-Rad) | Master mix and consumables for partitioning and amplifying the DNA sample [8]. |
| Target-Specific Assays | Primer/Probe sets for CCR5 WT and Δ32, RNase P (reference assay) | Multiplexed detection and absolute quantification of specific alleles [8]. |
Visualizing Key Concepts and Workflows
CCR5 Signaling and HIV Entry Pathway
Experimental Workflow for CCR5 Gene Editing and Validation
The study of CCR5 wild-type and Δ32 alleles remains a cornerstone of biomedical research, directly informing the development of curative strategies for HIV. The integration of multiplex ddPCR provides a critical tool for sensitive and accurate genotyping and quantification, enabling precise monitoring of therapeutic interventions like gene editing. Furthermore, advanced gene-editing technologies, particularly CRISPR/Cas9, are being harnessed to recreate the protective CCR5Δ32 phenotype autologously, moving beyond the limitations of allogeneic transplants. As research progresses, the combination of multiplex ddPCR for robust analytics and sophisticated gene-editing techniques for therapeutic manipulation represents a powerful paradigm for advancing personalized medicine and achieving functional cures for HIV and other diseases.
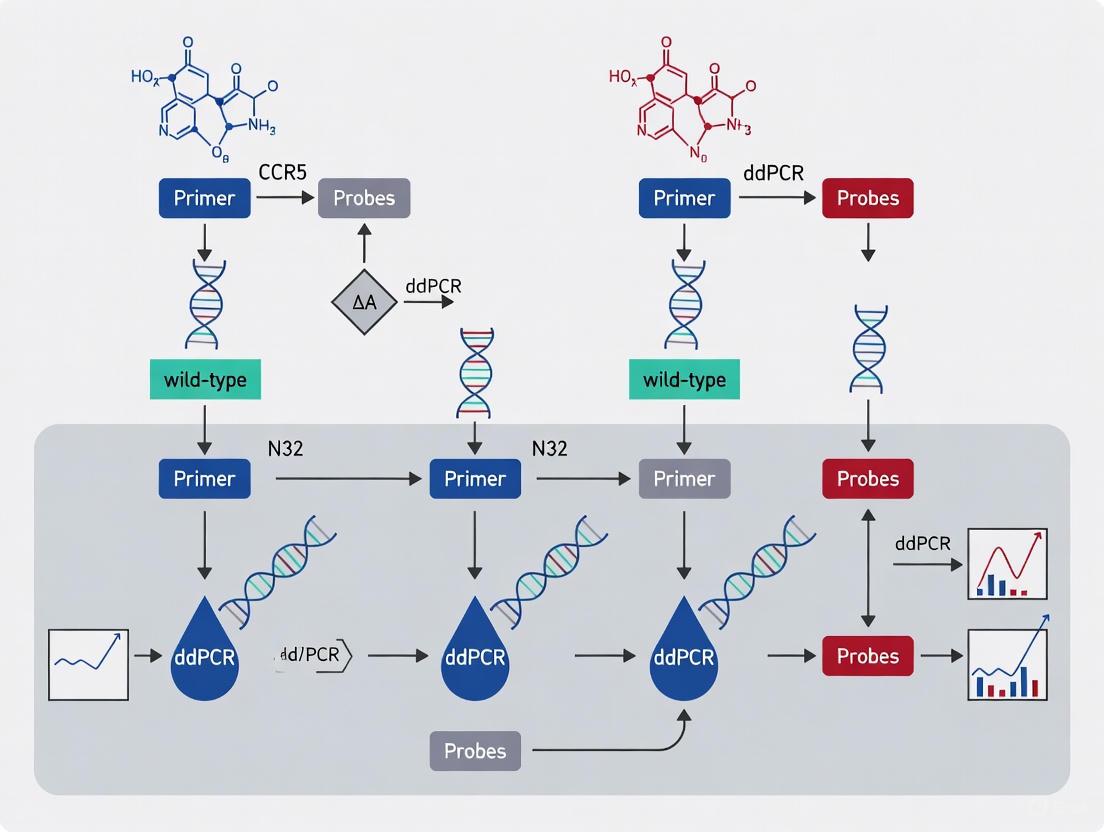
Step-by-Step Multiplex ddPCR Assay Development for CCR5 Genotyping
Primer and Probe Design Strategies for Wild-type and Δ32 Allele Discrimination
The C-C chemokine receptor type 5 (CCR5) serves as a crucial co-receptor for human immunodeficiency virus (HIV) entry into T-cells [8]. A naturally occurring 32-base pair deletion (CCR5Δ32) results in a non-functional receptor that confers resistance to HIV R5-tropism strains in homozygous individuals [8]. With the demonstration that hematopoietic stem cell transplantation from CCR5Δ32 homozygous donors can cure HIV infection, and the advent of CRISPR/Cas9 technology to create this mutation in autologous cells, accurate methods for discriminating between wild-type and Δ32 alleles have become increasingly important for both research and therapeutic applications [8].
Droplet digital PCR (ddPCR) has emerged as a powerful tool for precise quantification of mutant alleles in heterogeneous cell mixtures, offering absolute quantification without need for standard curves and detection sensitivity down to 0.8% mutant allele frequency [8]. This application note details optimized strategies for designing primers and probes to effectively distinguish between CCR5 wild-type and Δ32 alleles using multiplex ddPCR assays, providing researchers with a framework for accurate genotyping and quantification in various experimental contexts.
Core Design Principles
Primer Design Guidelines
Effective primer design forms the foundation of successful allele discrimination in ddPCR assays. The table below summarizes key parameters for optimal primer design:
Table 1: Primer Design Specifications for Allele Discrimination Assays
| Parameter | Recommended Specification | Rationale |
|---|---|---|
| Length | 18-30 bases | Balances specificity and binding efficiency [30] |
| Melting Temperature (Tm) | 60-64°C (ideal 62°C) | Ensures efficient annealing while maintaining specificity [30] |
| Tm Difference Between Primers | ≤2°C | Promotes simultaneous binding and efficient amplification [30] |
| GC Content | 35-65% (ideal 50%) | Provides sequence complexity while avoiding extreme stability [30] |
| 3' End Specificity | Perfect match to target allele | Critical for allele-specific amplification [31] |
| Secondary Structures | Free of strong hairpins and self-dimers (ΔG > -9.0 kcal/mol) | Prevents non-specific amplification and primer artifacts [30] |
Primers should be positioned to flank the 32-bp deletion region, with the 3' ends designed for optimal allele specificity. For assays targeting the Δ32 mutation, the reverse primer should be positioned downstream of the deletion site to ensure both wild-type and mutant alleles are amplified with similar efficiency [8].
Probe Design Strategies
Hydrolysis probes (TaqMan) provide the specificity required for discriminating between wild-type and Δ32 alleles in multiplex ddPCR assays. The following design principles apply:
Table 2: Hydrolysis Probe Design Guidelines
| Parameter | Wild-type Probe | Δ32 Probe | Rationale |
|---|---|---|---|
| Location | Spanning deletion junction | Spanning deletion junction | Enables differentiation based on deletion presence [32] |
| Melting Temperature (Tm) | 5-10°C higher than primers | 5-10°C higher than primers | Ensures probe binding before primer extension [30] |
| Fluorophore Labels | FAM or HEX | VIC or Cy5 | Distinct emission spectra for multiplex detection [32] [33] |
| Quencher | BHQ, TAMRA, or MGB | BHQ, TAMRA, or MGB | Efficient fluorescence quenching [34] |
| Length | 20-30 bases (single-quenched) | 20-30 bases (single-quenched) | Optimal for discrimination without background [30] |
| 5' Base | Avoid G | Avoid G | Prevents fluorophore quenching [30] |
For the Δ32 mutation, which involves a 32-bp deletion, probes should be designed to span the deletion junction, creating a distinct binding site that differentiates it from the wild-type sequence [32]. Double-quenched probes with ZEN or TAO internal quenchers are recommended for reduced background fluorescence, particularly for longer probe sequences [30].
Figure 1: Workflow for designing and implementing CCR5 wild-type and Δ32 allele discrimination assays, from initial primer design to final multiplex detection.
Multiplex ddPCR Assay Protocol
Reaction Setup
The ddPCR reaction should be assembled with careful attention to component concentrations and partitioning efficiency. The following protocol has been optimized for CCR5 allele discrimination:
Table 3: ddPCR Reaction Setup for CCR5 Genotyping
| Component | Final Concentration | Volume per 20 μL Reaction | Notes |
|---|---|---|---|
| ddPCR Supermix | 1X | 10 μL | Use probe-based supermix for hydrolysis assays [33] |
| Forward Primer | 500 nM | 0.4 μL (from 10 μM stock) | Optimize concentration (100-500 nM) [35] |
| Reverse Primer | 500 nM | 0.4 μL (from 10 μM stock) | Optimize concentration (100-500 nM) [35] |
| WT Probe (FAM-labeled) | 250 nM | 0.2 μL (from 10 μM stock) | Titrate for optimal signal (50-800 nM) [32] |
| Δ32 Probe (VIC-labeled) | 250 nM | 0.2 μL (from 10 μM stock) | Titrate for optimal signal (50-800 nM) [32] |
| Template DNA | 10-100 ng | 1-5 μL | Adjust based on DNA concentration [8] |
| Nuclease-free Water | - | To 20 μL | - |
For the QX200 Droplet Digital PCR System, droplets should be generated according to manufacturer instructions, typically yielding approximately 20,000 droplets per sample [32]. Proper negative controls (no-template) and positive controls (known wild-type, heterozygous, and Δ32 homozygous samples) should be included in each run.
Thermal Cycling Conditions
The thermal cycling protocol must be optimized for efficient amplification while maintaining probe specificity:
Table 4: Thermal Cycling Protocol for CCR5 ddPCR
| Step | Temperature | Time | Cycles | Purpose |
|---|---|---|---|---|
| Enzyme Activation | 95°C | 10 minutes | 1 | Hot-start enzyme activation [32] |
| Denaturation | 95°C | 30 seconds | 40 | Template denaturation |
| Annealing/Extension | 62°C | 60 seconds | 40 | Primer annealing and probe hydrolysis |
| Enzyme Deactivation | 98°C | 10 minutes | 1 | Enzyme inactivation |
| Hold | 4-12°C | ∞ | - | Short-term storage |
The annealing temperature should be optimized for each primer set, typically 5°C below the primer Tm [30]. For CCR5 assays, 62°C has been demonstrated as an effective annealing/extension temperature [32].
Essential Reagents and Equipment
Successful implementation of CCR5 genotyping assays requires specific reagents and instrumentation:
Table 5: Research Reagent Solutions for CCR5 Allele Discrimination
| Category | Specific Product/Type | Application Notes |
|---|---|---|
| Digital PCR System | QX200 Droplet Digital PCR System (Bio-Rad) or equivalent | Enables absolute quantification by partitioning samples into nanoliter droplets [8] [33] |
| PCR Mastermix | ddPCR Supermix for Probes (Bio-Rad) | Optimized for droplet stability and PCR efficiency in partitioned reactions [32] |
| DNA Polymerase | Hot-start Taq Polymerase | Reduces non-specific amplification and primer-dimer formation [35] |
| Fluorophores | FAM, VIC, HEX, Cy5 | Selection depends on instrument channel availability and multiplexing capacity [32] [36] |
| Quenchers | BHQ, TAMRA, MGB | MGB probes offer improved specificity for SNP discrimination [34] |
| DNA Extraction Kit | QIAamp DNA Mini Kit (Qiagen) or equivalent | High-quality DNA extraction is critical for accurate quantification [33] |
| Droplet Generator | QX200 Droplet Generator (Bio-Rad) | Creates uniform droplets for partitioning [8] |
| Droplet Reader | QX200 Droplet Reader (Bio-Rad) | Measures fluorescence in individual droplets [8] |
Data Analysis and Interpretation
Quantification Methods
In ddPCR, target concentration is calculated using Poisson statistics based on the fraction of positive droplets [32]. The concentration of wild-type and Δ32 alleles is expressed as copies/μL, allowing for direct calculation of mutant allele frequency without standard curves [8].
For CCR5Δ32 detection, the mutant allele frequency in heterogeneous samples can be calculated as:
[ \text{Mutant Allele Frequency (\%)} = \frac{[\Delta32]}{[WT] + [\Delta32]} \times 100 ]
where [WT] and [Δ32] represent the concentration of wild-type and mutant alleles in copies/μL, respectively.
Sensitivity and Specificity Considerations
The developed assay should demonstrate a limit of detection (LOD) of at least 0.8% for mutant alleles in wild-type backgrounds, as previously established for CCR5Δ32 detection [8]. Specificity should be validated using control samples with known genotypes, with clear discrimination between wild-type, heterozygous, and homozygous mutant clusters in 2D amplitude plots [32].
Figure 2: Expected fluorescence patterns for different CCR5 genotypes in a multiplex ddPCR assay, showing distinct clusters for wild-type, heterozygous, and homozygous Δ32 samples.
Troubleshooting and Optimization
Common challenges in CCR5 allele discrimination assays include:
- Poor cluster separation: Optimize probe concentrations and annealing temperature
- Low dynamic range: Check DNA quality and quantity; ensure proper droplet generation
- Non-specific amplification: Verify primer specificity using BLAST analysis; implement hot-start polymerase [30]
- High background fluorescence: Switch to double-quenched probes; optimize probe design to avoid G at 5' end [30]
Assay validation should include testing on samples with known genotypes and determination of intra- and inter-assay precision. For quantitative applications, a minimum of 10,000 droplets per reaction is recommended to ensure accurate quantification of low-frequency mutants [32].
The strategies outlined in this application note provide a robust framework for developing and implementing multiplex ddPCR assays to discriminate CCR5 wild-type and Δ32 alleles, supporting research in HIV therapeutics, genetic studies, and clinical diagnostics.
The precise quantification of the CCR5 wild-type and Δ32 alleles is a critical component of research into potential cures for HIV, ranging from allogeneic hematopoietic stem cell transplantation to novel CRISPR/Cas9-based gene editing approaches [8] [37]. The droplet digital PCR (ddPCR) platform is uniquely suited for this task, as it allows for the absolute quantification of mutant allele fractions in heterogeneous cell mixtures with a demonstrated sensitivity down to 0.8% [8]. Achieving this level of performance requires meticulous optimization of the reaction parameters, specifically the master mix composition and thermal cycling conditions, which this application note will detail for researchers and drug development professionals.
Master Mix Composition Optimization
The foundation of a robust multiplex ddPCR assay lies in the careful formulation of the master mix. The following components require specific attention.
Core Reaction Components
DNA Polymerase and Master Mix: The use of a ddPCR supermix for probes is standard. For the CCR5 Δ32 assay, reactions can be set up using 1X ddPCR Supermix for Probes in a final volume of 20 μL [8] [38]. This supermix provides the necessary buffer, dNTPs, and a hot-start DNA polymerase optimized for droplet-based reactions.
Oligonucleotide Concentration: Optimal primer and probe concentrations are paramount for strong signal separation and minimal rain. While "normal" concentrations (e.g., 900 nM for primers and 250 nM for probes) can be a starting point, empirical optimization is recommended [39]. The "experience matrix" approach, which evaluates assay performance based on fluorescence signal separation, has proven effective for identifying optimal concentrations [39] [40].
Magnesium and Additives: The MgCl₂ concentration in commercial supermixes is often sufficient. However, for difficult targets, the addition of cosolvents such as dimethyl sulfoxide (DMSO), glycerol, or betaine can be beneficial. These additives help prevent the stalling of DNA polymerization by destabilizing secondary structures within the template DNA, which is particularly relevant for GC-rich regions [41].
Probe and Primer Design for Multiplexing
A successful duplex assay for CCR5 wild-type and Δ32 alleles requires primers and probes that function efficiently together.
- Primer Design: All primer pairs in the multiplex should have similar optimum annealing temperatures. Primers should be 18–30 bp long with a GC content of 35–60% and should not display significant homology to each other to prevent the formation of primer-dimers [41].
- Probe Selection: Use hydrolysis probes (e.g., TaqMan) labelled with distinct fluorophores such as FAM and HEX/VIC [38] [39]. This allows for independent detection of the wild-type and Δ32 alleles in a single reaction.
Table 1: Recommended Master Mix Components for CCR5 ddPCR Assay
| Component | Final Concentration | Function & Notes |
|---|---|---|
| ddPCR Supermix | 1X | Provides buffer, dNTPs, and hot-start DNA polymerase. |
| Forward/Reverse Primers | 500–900 nM each | Concentration must be optimized to balance efficiency and specificity [39]. |
| FAM/HEX Probes | 100–250 nM each | Hydrolysis probes for specific detection of wild-type and Δ32 alleles. |
| DNA Template | 5–30 ng/reaction | Amount depends on sample type (gDNA, cfDNA). |
| Nuclease-free Water | To volume | Adjust to the final reaction volume (e.g., 20 μL). |
Thermal Cycling Condition Optimization
After assembling the master mix, the thermal cycling profile must be fine-tuned to maximize amplification efficiency and specificity.
Denaturation
- Initial Denaturation: A single cycle of 95°C for 1–3 minutes is standard. This step fully denatures the complex template DNA, inactivates contaminants, and activates the hot-start polymerase [42].
- Cyclic Denaturation: Subsequent cycles typically use a shorter denaturation step of 94°C for 30 seconds. For GC-rich templates or long amplicons, a higher temperature (98°C) or longer duration (up to 2 minutes) may be required [42].
Annealing and Extension
- Annealing Temperature: The annealing temperature is the most critical parameter to optimize. It should be calculated based on the primer Tm and validated experimentally. A general rule is to start 3–5°C below the lowest Tm of the primer set [42]. Using a thermal cycler with a gradient function is highly recommended for this optimization. For the CCR5 Δ32 assay, an annealing temperature of 56–60°C is an effective starting point [8] [38].
- Annealing and Extension Time: An annealing/extension time of 1 minute is usually sufficient for amplicons under 150 bp [42]. The CCR5 Δ32 assay uses a combined annealing/extension step at 56°C for 1 minute [38].
- Cycle Number: 40–50 cycles are standard for ddPCR to ensure endpoint amplification for accurate digital quantification [8] [38].
Denaturation-Enhanced ddPCR (dddPCR)
For maximum sensitivity, particularly with limited input DNA such as in liquid biopsies, a denaturation-enhanced ddPCR (dddPCR) protocol can be employed. This involves a complete denaturation of the double-stranded DNA (95°C for 1 min) immediately prior to droplet generation [38]. This separates the sense and antisense strands, effectively doubling the number of available template molecules. For fragmented DNA (e.g., cfDNA), an end-repair step prior to denaturation is recommended to ensure both strands are amplifiable, restoring the full 2-fold increase in positive droplets [38].
Table 2: Optimized Thermal Cycling Profile for CCR5 ddPCR Assay
| Step | Temperature | Time | Cycles | Purpose |
|---|---|---|---|---|
| Initial Denaturation | 95 °C | 5–10 min | 1 | DNA denaturation, enzyme activation. |
| Cyclic Denaturation | 94 °C | 30 s | 40–50 | Denature DNA for each cycle. |
| Annealing/Extension | 56–60 °C | 60 s | 40–50 | Primer binding and amplification. |
| Enzyme Deactivation | 98 °C | 10 min | 1 | Deactivate the polymerase. |
| Hold | 4–10 °C | ∞ | 1 | Short-term storage of plates. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Kits for CCR5 ddPCR
| Reagent/Kits | Function/Application | Example (Supplier) |
|---|---|---|
| ddPCR Supermix for Probes | Core reaction mix for probe-based digital PCR. | ddPCR Supermix for Probes (Bio-Rad) [38] [39] |
| FAM/HEX Labelled Probes | Target-specific hydrolysis probes for multiplex detection. | Custom TaqMan Probes (IDT Technologies) [38] |
| Droplet Generation Oil & Cartridges | Physical partitioning of the reaction into nanoliter droplets. | DG Cartridges & Droplet Generation Oil (Bio-Rad) [39] |
| Genomic DNA Extraction Kit | High-quality DNA isolation from cells or tissues. | NucleoSpin Kit (Macherey-Nagel) [43] / Maxwell 16 (Promega) [39] |
| DNA End-Repair Enzyme Mix | For dddPCR; creates blunt-ended fragments for optimal denaturation. | NEBNext Ultra II End Prep Enzyme Mix (NEB) [38] |
| Restriction Enzymes | May be used to digest gDNA to reduce complexity and viscosity. | HindIII-HF (NEB) [38] |
Experimental Workflow and Data Analysis
The following diagram outlines the complete optimized workflow for the CCR5 ddPCR assay, from sample preparation to data analysis.
Workflow for CCR5 Genotyping by ddPCR
Key Data Analysis Considerations
- Threshold Setting: Accurate quantification requires clear separation between positive and negative droplet clusters. Software like QuantaSoft is used to set fluorescence thresholds for each channel (FAM and HEX) [38] [39].
- Managing "Rain": Droplets with intermediate fluorescence ("rain") can complicate analysis. This is minimized by optimizing annealing temperature and oligonucleotide concentrations, which improves cluster separation [39] [40].
- Calculating Mutant Fraction: The fraction of CCR5 Δ32 alleles is calculated as: (Concentration of Δ32 alleles) / (Concentration of WT alleles + Concentration of Δ32 alleles). The absolute concentrations (in copies/μL) are provided directly by the ddPCR software [8].
The development of effective treatments and potential cures for HIV-1 has been significantly advanced through research on the C-C chemokine receptor type 5 (CCR5). A 32-base pair deletion in the CCR5 gene (CCR5Δ32) results in a non-functional receptor that confers resistance to R5-tropic HIV-1 infection [8] [22]. The successful application of CCR5Δ32/Δ32 hematopoietic stem cell transplantation in achieving HIV-1 remission in multiple patients underscores the critical importance of this genetic target [22] [44]. Accurate detection and quantification of both wild-type and Δ32 CCR5 alleles is therefore essential for advancing therapeutic strategies, including CRISPR/Cas9 genome editing approaches and transplantation monitoring [8] [22].
Droplet digital PCR (ddPCR) technology provides the precision and sensitivity required for CCR5 genotyping applications, particularly when quantifying low-frequency mutations or assessing editing efficiency in heterogeneous cell mixtures [8]. This application note details optimized multiplexing methodologies for fluorescence channel selection and panel configuration specifically for simultaneous detection of CCR5 wild-type and Δ32 alleles, enabling researchers to accurately quantify mutant allelic fractions down to 0.8% [8].
Principles of Fluorescence Multiplexing in ddPCR
Multiplex ddPCR enables simultaneous detection of multiple targets in a single reaction by leveraging different fluorescence channels and amplitude-based discrimination. The fundamental principle involves partitioning a PCR reaction into thousands of nanodroplets, effectively creating individual reaction chambers where target amplification occurs independently. Each target is detected using sequence-specific probes labeled with different fluorophores, and the fluorescence signature of each droplet is read upon completion of amplification [36].
Advanced multiplexing approaches combine spectral imaging and combinatorics to expand beyond traditional 4-6 color limits. By creating covalent combinations of fluorophores and measuring emission spectra across multiple excitation channels, the effective multiplexing capacity can be increased approximately 4-5 fold compared to conventional approaches [45]. This principle forms the basis for techniques like Multiplexing using Spectral Imaging and Combinatorics (MuSIC), which enables robust demultiplexing of multiple probes within a limited spectral window [45].
For CCR5 genotyping applications, a drop-off assay design is particularly advantageous as it can detect multiple proximal genetic alterations within a short genomic interval using a single assay [46]. This approach uses two probes: a wild-type probe that spans the mutation hotspot and is complementary only to the wild-type sequence, and a reference probe that binds to a stable region adjacent to the mutation site [46].
Table 1: Comparison of Fluorescence Multiplexing Approaches for CCR5 Genotyping
| Method | Multiplexing Capacity | Key Advantages | Limitations | Suitable Applications |
|---|---|---|---|---|
| 2-Color Channel ddPCR | 2-3 targets | Widely available instrumentation, simple data analysis | Limited multiplexing capacity | Basic WT/Δ32 discrimination |
| Amplitude-Based Multiplexing | 4-6 targets | Increases multiplexing without additional fluorophores | Requires extensive optimization, signal crowding at high concentrations | Complex mutation profiling |
| Sral Imaging & Combinatorics | 9+ targets | Significant multiplexing expansion, preserves sample | Specialized instrumentation required, complex data analysis | High-plex biomarker panels |
| Drop-Off Assay | Multiple mutations in one region | Detects various mutation types in hotspot regions | Limited to defined genomic intervals | KRAS, BRAF, CCR5 mutation profiling |
Fluorescence Channel Configuration for CCR5 Genotyping
Probe Design Strategy
The selection of appropriate fluorophores and their configuration across available channels is critical for successful multiplexing. For CCR5 wild-type and Δ32 allele discrimination, a drop-off assay design provides optimal performance [46]. This approach utilizes:
- A wild-type (drop-off) probe spanning the 32bp deletion hotspot, labeled with a high-energy fluorophore (e.g., Cy5)
- A reference probe binding adjacent to the deletion region, labeled with a different fluorophore (e.g., FAM)
- Target-specific primers amplifying a region encompassing the deletion
This configuration generates three distinct populations in a 2D scatter plot: double-positive droplets (wild-type alleles), single-positive reference droplets (Δ32 mutant alleles), and double-negative droplets (non-template) [46].
Fluorophore Selection and Concentration Optimization
Effective multiplexing requires careful balancing of fluorophore concentrations to ensure clear cluster separation. Based on the 9-plex viral detection assay validated in wastewater samples, targets can be categorized as "high" or "low" based on their fluorescence signal intensity [36]:
- High targets: Primer/probe sets at 900nM/300nM final concentration
- Low targets: Primer/probe sets at 400-450nM/100-150nM final concentration
This tiered concentration approach enables formation of upper and lower clusters in the same fluorescence channel, effectively doubling the multiplexing capacity of each channel [36].
Table 2: Recommended Fluorophore Combinations for CCR5 ddPCR
| Target | Assay Type | Recommended Fluorophore | Alternative Fluorophores | Optimal Concentration | Cluster Position |
|---|---|---|---|---|---|
| CCR5 Reference | Reference | FAM | HEX, VIC | 900nM primer/300nM probe | High |
| CCR5 Wild-Type | Drop-off | Cy5 | ROX, Texas Red | 900nM primer/300nM probe | High |
| CCR5 Δ32 Mutant | Drop-off | - | - | - | Low |
| Endogenous Control | Internal control | HEX | ATTO590, TET | 450nM primer/150nM probe | Low |
| Exogenous Control | Process control | ATTO590 | Cy5.5, Quasar 705 | 400nM primer/100nM probe | Low |
Experimental Protocol: CCR5 Wild-Type/Δ32 Genotyping Assay
Sample Preparation and DNA Extraction
Cell Culture and Harvesting: Culture MT-4 human T-cells or other relevant cell lines in RPMI-1640 medium with 10% FBS at 37°C with 5% CO2. Harvest approximately 6×10^6 cells for genomic DNA extraction [8].
Genomic DNA Extraction: Extract genomic DNA using phenol-chloroform method or commercial kits (e.g., ExtractDNA Blood and Cells Kit). Measure DNA concentration and purity using spectrophotometry (A260/A280 ratio of 1.8-2.0 indicates acceptable purity) [8].
DNA Fragmentation (if needed): For high molecular weight DNA, fragment to ~200bp using restriction enzymes (e.g., Tru1I) or sonication. Verify that the restriction enzyme does not target the amplicon region [46].
ddPCR Reaction Setup
Prepare Master Mix (calculate for n+1 samples to account for pipetting loss):
- 5.0 μL One-step RT-ddPCR Advanced Supermix (for RNA targets) or ddPCR Supermix (for DNA)
- 2.0 μL Reverse Transcriptase (omit for DNA targets)
- 1.0 μL 300mM DTT (for RNA targets)
- CCR5 reference probe (FAM-labeled): 900nM primer/300nM probe final concentration
- CCR5 wild-type probe (Cy5-labeled): 900nM primer/300nM probe final concentration
- Endogenous control probe (HEX-labeled): 450nM primer/150nM probe final concentration
- 5 μL DNA template (optimize concentration based on extraction)
- Nuclease-free water to final volume of 20 μL [36] [8]
Droplet Generation: Transfer 20μL reaction mix to droplet generator cartridges according to manufacturer instructions. Generate droplets using appropriate oil for your system.
PCR Amplification:
- Transfer droplets to 96-well PCR plate
- Seal plate properly
- Run with following thermal cycling conditions:
Droplet Reading and Data Analysis
Plate Reading: Load plate into droplet reader (e.g., QX600 Droplet Reader). Ensure adequate droplet count (>10,000 valid droplets per well) [36].
Threshold Setting: Set thresholds for each channel to clearly distinguish positive and negative populations. For drop-off assays, three populations should be visible [46]:
- Quadrant 1 (FAM+/Cy5+): Wild-type alleles
- Quadrant 4 (FAM+/Cy5-): Δ32 mutant alleles
- Quadrant 3 (FAM-/Cy5-): Empty droplets
Mutant Allelic Fraction (MAF) Calculation: Calculate using the formula:
MAF = Cₘᵤₜ / (CWT + Cₘᵤₜ)
Where:
- Cₘᵤₜ = -ln(1 - (P₁₀ / (P₁₀ + P₀₀))) / v
- CWT = -ln(1 - (P₁₁ / (P₁₁ + P₀₀ + P₁₀))) / v
- P₁₁ = double-positive droplets (wild-type)
- P₁₀ = single-positive FAM droplets (mutant)
- P₀₀ = double-negative droplets
- v = partition volume (μL) [46]
Research Reagent Solutions
Table 3: Essential Reagents for CCR5 ddPCR Genotyping
| Reagent/Category | Specific Examples | Function | Considerations |
|---|---|---|---|
| ddPCR Systems | QX600 (Bio-Rad), Naica (Stilla), QIAcuity (Qiagen) | Partitioning, thermal cycling, droplet reading | Channel availability (2-6 color), throughput needs |
| PCR Master Mixes | One-step RT-ddPCR Advanced Kit, ddPCR Supermix | Enzymes, nucleotides, buffer for amplification | RNA vs. DNA targets, inhibitor resistance |
| DNA Extraction Kits | ExtractDNA Blood and Cells Kit, Enviro Wastewater TNA Kit | Nucleic acid purification, inhibitor removal | Sample type (cells, wastewater, blood), yield |
| Fluorophore-Labeled Probes | FAM-, HEX-, Cy5-, ATTO590-labeled hydrolysis probes | Target-specific detection | Spectral overlap, quenching efficiency (ZEN/Iowa Black) |
| Primer/Probe Design Tools | In silico design tools | Assay development for conserved regions | Specificity, efficiency, multiplex compatibility |
| Reference Materials | Synthetic gBlocks, cell lines with known genotypes | Assay validation, quantification standards | Sequence verification, stability |
Workflow Visualization
Diagram 1: CCR5 Genotyping Workflow. The complete process from sample preparation to result interpretation for CCR5 wild-type and Δ32 allele detection using ddPCR.
Diagram 2: Drop-off Assay Design. Schematic representation of the probe binding and droplet classification strategy for discriminating wild-type and Δ32 CCR5 alleles.
Optimized fluorescence channel selection and panel configuration are critical for robust multiplex ddPCR assays targeting CCR5 genotypes. The drop-off assay design provides an efficient approach for detecting the 32bp deletion characteristic of the CCR5Δ32 allele, with sensitivity sufficient for quantifying mutant allelic fractions as low as 0.8% [8]. By implementing the tiered probe concentration strategy and appropriate fluorophore combinations described in this protocol, researchers can reliably assess CCR5 editing efficiency in heterogeneous cell mixtures [8], monitor transplant engraftment [22] [44], and advance therapeutic strategies for HIV-1 treatment. The standardized methodologies presented here ensure accurate, reproducible results across research and potential clinical applications.
The accuracy of any multiplex droplet digital PCR (ddPCR) assay is fundamentally dependent on the quality and integrity of the input nucleic acid material. This document provides detailed application notes and protocols for the preparation of genomic DNA (gDNA) and processing of heterogeneous cell mixtures, framed within the context of a broader thesis on multiplex ddPCR assay design for CCR5 wild-type and Δ32 alleles research. The CCR5-Δ32 mutation, a 32-base pair deletion in the CCR5 gene, confers resistance to HIV-1 infection, and its accurate quantification in cell mixtures is crucial for developing curative autologous stem cell therapies [8] [22]. This guide is intended for researchers, scientists, and drug development professionals requiring robust, reproducible methods for sensitive genotyping and quantification applications.
Background and Significance
The C-C chemokine receptor type 5 (CCR5) serves as a co-receptor for human immunodeficiency virus (HIV). A mutant form of the gene, CCR5-Δ32, results from a 32-nucleotide deletion that causes a frameshift and a knockout of gene function [8]. Individuals homozygous for this mutation are largely resistant to infection by the most common strains of HIV-1 [8] [22]. Consequently, transplantation of hematopoietic stem cells with the CCR5-Δ32 knockout mutation represents a promising path toward a complete cure for HIV, as demonstrated by the "Berlin" and "London" patients [8] [22].
With the advent of CRISPR/Cas9 genome editing, it is now possible to reproducibly create the CCR5-Δ32 mutation in wild-type cells, paving the way for autologous transplantation strategies [8] [22]. A critical requirement for monitoring the success of such therapies is the ability to accurately quantify the fraction of cells carrying the mutant allele in a background of wild-type cells. Multiplex ddPCR is uniquely suited for this task, as it allows for absolute quantification of target DNA sequences with a high degree of precision and sensitivity, down to fractional abundances of 0.8% [8]. The reliability of this powerful analytical technique is entirely contingent upon proper sample preparation, from cell culture to genomic DNA isolation.
Cell Culture and Sample Collection
The starting biological material for gDNA extraction can vary, ranging from established cell lines to primary cells. The protocol below outlines a general method using a human T-cell line, which is relevant for CCR5 research.
Materials
- Cell Line: A relevant human T-cell line (e.g., MT-4) [8].
- Culture Medium: Roswell Park Memorial Institute medium (RPMI-1640) supplemented with 10% Fetal Bovine Serum (FBS) [8].
- Culture Conditions: Humidified incubator at 37°C with 5% CO2 [8].
Protocol: Cell Culture and Harvesting
- Culture Maintenance: Maintain cells in recommended growth medium, passaging them regularly to maintain logarithmic growth.
- Cell Counting: Determine cell concentration and viability using a hemocytometer or an automated cell counter.
- Cell Harvesting: For suspension cells, collect the cell suspension in a centrifuge tube.
- Centrifugation: Pellet cells by centrifugation at approximately 300 × g for 5 minutes.
- Washing: Carefully aspirate the supernatant and wash the cell pellet with 1X Phosphate-Buffered Saline (PBS).
- Final Pellet: Re-pellet the cells and aspirate the PBS supernatant. The cell pellet can now be processed immediately for DNA extraction or frozen at -20°C or -80°C for future use.
Genomic DNA Extraction
High-quality, high-molecular-weight gDNA is essential for ddPCR. The following protocol describes a standard phenol-chloroform extraction method.
Materials
- Lysis Buffer: A buffer containing Tris-HCl, EDTA, and SDS.
- Enzymes: Proteinase K and RNase A.
- Extraction Reagents: Phenol, chloroform, and isoamyl alcohol mixture.
- Precipitation Reagents: Isopropanol and 70% ethanol.
- Elution Buffer: TE buffer (10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0) or nuclease-free water.
- Commercial Kits: Alternatively, use a commercial kit such as the "ExtractDNA Blood and Cells Kit" [8].
Protocol: Phenol-Chloroform gDNA Extraction
- Cell Lysis: Resuspend the cell pellet in lysis buffer. Add Proteinase K and RNase A, and mix by inversion.
- Incubation: Incubate the mixture at an appropriate temperature (e.g., 56°C) for several hours or overnight until the solution appears clear.
- Phenol-Chloroform Extraction:
- Add an equal volume of phenol:chloroform:isoamyl alcohol (25:24:1) to the lysate.
- Mix thoroughly by inversion for 2-5 minutes.
- Centrifuge at ≥12,000 × g for 5 minutes to separate the phases.
- Aqueous Phase Recovery: Carefully transfer the upper, aqueous phase (which contains the DNA) to a fresh tube.
- DNA Precipitation:
- Add an equal volume of room-temperature isopropanol to the aqueous phase. Mix gently by inversion until the DNA precipitates as a stringy white mass.
- Centrifuge at ≥12,000 × g for 5-10 minutes to pellet the DNA.
- Wash: Carefully decant the supernatant. Wash the DNA pellet with 1 mL of 70% ethanol. Centrifuge again and carefully remove the ethanol.
- Air-Dry: Allow the pellet to air-dry for 5-15 minutes to evaporate residual ethanol. Do not over-dry.
- Resuspend: Dissolve the purified DNA pellet in TE buffer or nuclease-free water.
- Quantification and Quality Control: Measure the DNA concentration and purity using a spectrophotometer (e.g., NanoPhotometer). High-quality DNA should have an A260/A280 ratio of ~1.8 and an A260/A230 ratio of ~2.0-2.2 [8].
Table 1: Troubleshooting Guide for gDNA Extraction
| Problem | Potential Cause | Solution |
|---|---|---|
| Low DNA Yield | Insufficient cell input, incomplete lysis | Increase starting material; ensure complete lysis; extend Proteinase K digestion. |
| Low A260/A280 Ratio (<1.8) | Phenol or protein contamination | Repeat the phenol-chloroform extraction step; ensure careful phase separation. |
| Low A260/A230 Ratio (<2.0) | Salt or solvent carryover | Perform an additional 70% ethanol wash; ensure the pellet is adequately dried. |
| DNA Degradation | Improper storage or nuclease contamination | Use fresh, sterile reagents and tubes; store DNA at -20°C. |
Preparation of Heterogeneous Cell Mixtures for Assay Validation
A key application of multiplex ddPCR in CCR5 research is quantifying the proportion of CCR5-Δ32 alleles in a mixture of wild-type and mutated cells, mimicking the heterogeneous environment post-therapy [8].
Protocol: Creating Artificial Cell Mixtures
- Source Cells: Obtain two distinct cell populations: one homozygous for the CCR5 wild-type allele and another homozygous for the CCR5-Δ32 allele. The mutant line can be generated via CRISPR/Cas9 editing of the wild-type line [8].
- Cell Counting: Precisely count both cell populations using a hemocytometer or automated counter.
- Mixture Formulation: Combine the two cell populations in pre-determined ratios to create a series of mixtures with known mutant allele fractions (e.g., 0%, 1%, 5%, 10%, 50%, 100%).
- gDNA Extraction: Extract gDNA from each of these mixture samples using the protocol detailed in Section 4.2.
- Assay Validation: Use these calibrated gDNA samples to validate the precision, accuracy, and linearity of the multiplex ddPCR assay for CCR5 genotyping [8].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagent Solutions for CCR5 ddPCR
| Item | Function/Application | Example Product/Composition |
|---|---|---|
| Cell Culture Medium | Supports the growth and maintenance of cell lines used in research. | RPMI-1640 supplemented with 10% FBS [8] |
| gDNA Extraction Kit | Isulates high-purity genomic DNA from cell pellets. | "ExtractDNA Blood and Cells Kit" or phenol-chloroform protocol [8] |
| ddPCR Supermix | Optimized buffer for probe-based ddPCR reactions. | ddPCR Supermix for Probes (No dUTP) [8] [47] |
| Primers & Probes | Target-specific oligonucleotides for amplifying and detecting CCR5 WT and Δ32 alleles. | FAM-labeled Δ32 probe; HEX-labeled WT probe [8] |
| Droplet Generation Oil | Creates the water-in-oil emulsion necessary for partitioning the PCR reaction. | Droplet Generation Oil for Probes [48] |
| Nuclease-Free Water | Serves as a diluent in reaction mixes to prevent enzymatic degradation of nucleic acids. | Molecular Biology Grade Water [48] |
Experimental Workflow Diagram
The following diagram summarizes the complete workflow from cell sample to data analysis in the context of CCR5 allelic quantification.
Diagram 1: Sample to Data Workflow
Data Presentation and Analysis
Upon completion of a ddPCR run, the data is analyzed using proprietary software (e.g., Bio-Rad's QuantaSoft). The software automatically calculates the concentration of target DNA molecules (copies/μL) for each target (wild-type and Δ32) based on the Poisson distribution [49] [50].
Key Calculations
- Mutant Allele Frequency: The fraction of mutant alleles in the sample is calculated as: [ \text{Mutant Allele Frequency} = \frac{[\text{CCR5-Δ32}]}{[\text{CCR5-Δ32}] + [\text{CCR5-WT}]} \times 100\% ]
- Total DNA Content: The sum of wild-type and mutant concentrations provides the total DNA content at the CCR5 locus.
Table 3: Example Data from a Heterogeneous Cell Mixture Experiment
| Sample Description | Theoretical Δ32 Fraction | Measured [WT] (copies/μL) | Measured [Δ32] (copies/μL) | Calculated Δ32 Fraction |
|---|---|---|---|---|
| Pure Wild-Type | 0% | 152.4 | 0.8 | 0.5% |
| 1:99 Mixture | 1% | 981.2 | 11.1 | 1.1% |
| 1:19 Mixture | 5% | 452.5 | 25.3 | 5.3% |
| 1:9 Mixture | 10% | 321.0 | 37.2 | 10.4% |
| 1:1 Mixture | 50% | 105.6 | 98.9 | 48.4% |
| Pure Δ32 | 100% | 1.2 | 165.7 | 99.3% |
This data demonstrates the high accuracy and sensitivity of a well-optimized multiplex ddPCR assay, capable of reliably distinguishing mutant allele fractions down to 1% and below, which is critical for monitoring engraftment of CCR5-modified cells [8].
Threshold Determination and Copy Number Calculation in Multiplex ddPCR for CCR5 Genotyping
The C-C chemokine receptor type 5 (CCR5) serves as a principal co-receptor for human immunodeficiency virus (HIV) entry into T-cells [8]. A natural 32-base pair deletion in the CCR5 gene (CCR5Δ32) confers resistance to HIV-1 infection in homozygous individuals and delays disease progression in heterozygotes [8]. Accurate quantification of wild-type and Δ32 alleles is crucial for advancing therapeutic strategies, including hematopoietic stem cell transplantation (HSCT) from CCR5Δ32 donors and CRISPR/Cas9 gene editing approaches [8]. This application note details experimental and analytical frameworks for implementing multiplex droplet digital PCR (ddPCR) to determine analytical thresholds and calculate copy number variations for CCR5 alleles in heterogeneous cell populations.
Theoretical Foundations and Assay Design
Biological Significance of CCR5 Genotyping
The CCR5Δ32 mutation results in a frameshift and premature stop codons, producing a non-functional receptor absent from the cell surface [8]. This knockout provides natural immunity against R5-tropic HIV strains, with approximately 10% and 1% of Northern European populations carrying heterozygous and homozygous mutations, respectively [8]. Recent clinical evidence demonstrates that allogeneic hematopoietic stem cell transplantation with CCR5Δ32/Δ32 donor cells can achieve sustained HIV remission, establishing this genomic target as critical for curative interventions [18]. Furthermore, research explores artificial CCR5Δ32 introduction via CRISPR/Cas9 editing, necessitating precise quantification methods to assess editing efficiency [8].
Principles of Multiplex ddPCR
Multiplex ddPCR enables simultaneous quantification of multiple DNA targets within a single reaction by partitioning samples into thousands of nanoliter-sized droplets [51]. Each droplet functions as an individual PCR microreactor, allowing absolute quantification without standard curves. This technology provides superior precision for detecting low-frequency alleles and copy number variations compared to traditional real-time PCR [8] [51]. For CCR5 genotyping, multiplexing permits co-amplification of wild-type and Δ32 alleles with distinct fluorescent probes, enabling direct calculation of allele ratios and zygosity determination [8].
Table 1: Comparative Analysis of Nucleic Acid Amplification Technologies
| Technology | Quantification Method | Multiplexing Capacity | Detection Sensitivity | Throughput |
|---|---|---|---|---|
| Endpoint PCR | Semi-quantitative (gel electrophoresis) | Moderate (size-based separation) | Low | Moderate |
| Real-time qPCR | Relative quantification (Cq values) | High (fluorophore-based) | Moderate | High |
| Capillary Electrophoresis | Fragment analysis | High (size/fluorescence) | Moderate | High |
| Digital Droplet PCR | Absolute quantification (counting) | High (fluorophore-based) | Very High (<1% variant detection) | Moderate |
| Recombinase Polymerase Amplification | Endpoint detection | Low to Moderate | High | Rapid (isothermal) |
Experimental Protocols
Primer and Probe Design for CCR5 Alleles
Effective multiplex ddPCR requires careful design of target-specific primers and differentially labeled probes:
- Target Regions: Amplify sequences spanning the 32-bp deletion junction in CCR5 [8]
- Probe Labeling: Use FAM-labeled probes for wild-type CCR5 and HEX/VIC-labeled probes for Δ32 allele detection
- Amplicon Size: Design compact amplicons (≤150 bp) to enhance amplification efficiency in partitioned droplets
- Specificity Validation: Verify minimal cross-reactivity between probe sets using control templates
Sample Preparation and DNA Extraction
- Cell Culture: Maintain MT-4 human T-cell lines in RPMI-1640 medium with 10% fetal bovine serum at 37°C with 5% CO₂ [8]
- DNA Extraction: Isolate genomic DNA using phenol-chloroform method or commercial kits (e.g., ExtractDNA Blood and Cells Kit) [8]
- Quality Assessment: Measure DNA concentration and purity using spectrophotometry (A260/A280 ratio of 1.8-2.0 recommended) [8]
- DNA Normalization: Dilute samples to working concentrations (10-100 ng/μL) in low-EDTA TE buffer for optimal partitioning
Multiplex ddPCR Reaction Setup
Establish robust reaction conditions through systematic optimization:
Table 2: Multiplex ddPCR Master Mix Configuration
| Component | Final Concentration | Function |
|---|---|---|
| ddPCR Supermix | 1X | Provides optimized buffer, dNTPs, and polymerase |
| FAM-labeled CCR5 WT Probe | 250 nM | Detects wild-type allele sequence |
| HEX-labeled CCR5 Δ32 Probe | 250 nM | Detects deletion allele sequence |
| Forward Primer | 900 nM | Targets both CCR5 alleles |
| Reverse Primer | 900 nM | Targets both CCR5 alleles |
| Genomic DNA Template | 10-100 ng | Sample nucleic acids |
| Nuclease-free Water | To volume | Reaction balance |
- Thermal Cycling Conditions: Initial denaturation at 95°C for 10 min; 40 cycles of 94°C for 30 s and 60°C for 60 s; final enzyme deactivation at 98°C for 10 min [8]
- Droplet Generation: Use automated droplet generator with appropriate oil phase according to manufacturer specifications
- Post-amplification Analysis: Transfer droplets to droplet reader for simultaneous fluorescence measurement in FAM and HEX/VIC channels
Data Analysis Framework
Threshold Determination Strategy
Accurate allele quantification depends on establishing appropriate fluorescence thresholds to distinguish positive and negative droplet populations:
- Two-dimensional Analysis: Plot FAM (wild-type) versus HEX (Δ32) fluorescence intensity for each droplet
- Cluster Identification: Define four distinct populations using density-based clustering: (1) double-negative (empty droplets), (2) FAM-positive (wild-type only), (3) HEX-positive (Δ32 only), and (4) double-positive (heterozygous samples) [8]
- Threshold Optimization: Manually adjust thresholds to maximize separation between clusters while minimizing intermediate populations
- Quality Metrics: Require minimum of 10,000 accepted droplets per reaction and clear cluster separation with minimal rain (intermediate droplets)
Copy Number Calculation
Absolute quantification of CCR5 alleles follows Poisson distribution statistics applied to partitioned reactions:
Concentration Calculation: Apply Poisson correction to raw counts: Copies/μL = -ln(1 - p) × (Total droplets / Volume per droplet) where p = positive droplets / total droplets [8]
Allele Frequency Determination: Δ32 Allele Frequency = (Δ32-positive droplets) / (Total CCR5-positive droplets)
Limit of Detection: Established at 0.8% for minor alleles in heterogeneous mixtures [8]
Zygosity Assessment:
- Homozygous wild-type: >95% FAM-positive, <5% HEX-positive
- Homozygous Δ32: <5% FAM-positive, >95% HEX-positive
- Heterozygous: ∼50% FAM-positive, ∼50% HEX-positive, high double-positive
Figure 1: Multiplex ddPCR Workflow for CCR5 Genotyping
The Scientist's Toolkit
Table 3: Essential Research Reagents and Materials
| Reagent/Material | Specification | Application | Supplier Examples |
|---|---|---|---|
| ddPCR Supermix | No dUTP, optimized for probe-based assays | PCR amplification in droplets | Bio-Rad |
| FAM-labeled Probes | CCR5 wild-type specific, BHQ-1 quencher | Wild-type allele detection | Integrated DNA Technologies |
| HEX-labeled Probes | CCR5 Δ32 specific, BHQ-1 quencher | Δ32 allele detection | Integrated DNA Technologies |
| Droplet Generation Oil | surfactant-enhanced emulsion stability | Partitioning aqueous phase | Bio-Rad |
| DNA Extraction Kits | silica-membrane technology | high-quality gDNA isolation | Evrogen, Qiagen |
| 96-well Plates | semi-skirted, clear wells | ddPCR reaction setup | Eppendorf, Bio-Rad |
| Thermal Sealers | foil-based seals | preventing well cross-contamination | Bio-Rad |
Troubleshooting and Quality Control
Common Technical Challenges
- Poor Cluster Separation: Optimize probe concentrations (50-300 nM range) and annealing temperature (55-65°C range)
- Low Droplet Count: Verify proper droplet generator function and sample viscosity
- High Background Signal: Include no-template controls to identify contamination
- Rain Between Clusters: Increase input DNA quality and optimize thermal cycling conditions
Validation Approaches
- Control Samples: Include known wild-type, heterozygous, and homozygous Δ32 controls in each run
- Reproducibility Assessment: Perform technical replicates to determine intra-assay variability (<5% CV expected)
- Limit of Detection Verification: Validate with serial dilutions of Δ32 DNA in wild-type background
- Correlation with Alternative Methods: Compare results with Sanger sequencing or quantitative PCR for method validation
Applications in HIV Research
The multiplex ddPCR platform enables critical applications in HIV cure research and therapeutic development:
- Transplantation Monitoring: Track donor cell engraftment and CCR5Δ32 expansion in HIV patients receiving HSCT [8] [18]
- Gene Editing Verification: Quantify CRISPR/Cas9 editing efficiency in autologous T-cell and stem cell therapies [8]
- Reservoir Studies: Assess proviral DNA dynamics in patients undergoing treatment interruption protocols [18]
- Clinical Trial Stratification: Identify eligible patients based on CCR5 genotype for targeted therapeutic interventions
Figure 2: Data Analysis Pipeline for Thresholding and Quantification
The accurate detection of specific genetic alleles within mixed cell populations is a critical challenge in molecular biology, particularly in the development of cell and gene therapies. For research on the CCR5 wild-type and Δ32 alleles, this capability is paramount. The CCR5 co-receptor is a primary binding site for human immunodeficiency virus (HIV), and a 32-base pair deletion (CCR5Δ32) confers natural resistance to HIV infection in homozygous individuals [8]. Consequently, quantifying the proportion of cells carrying the CCR5Δ32 mutation in a heterogeneous mixture—such as following stem cell transplantation or CRISPR-Cas9 genome editing—is essential for evaluating therapeutic efficacy [8] [22]. Droplet Digital PCR (ddPCR) has emerged as a powerful tool for this purpose, enabling absolute quantification of target sequences with high precision and sensitivity, even in complex backgrounds [8] [52]. This application note details a validated protocol for using multiplex ddPCR to assess the CCR5 genotype in heterogeneous cell mixtures, providing key data on the assay's sensitivity and specificity to support research and drug development.
Key Performance Data
The following tables summarize the core quantitative performance metrics of the multiplex ddPCR assay for CCR5 genotyping, as established during validation.
Table 1: Assay Sensitivity and Dynamic Range
| Parameter | Value | Experimental Condition |
|---|---|---|
| Limit of Detection (LOD) | 0.8% | Content of CCR5Δ32 mutant alleles in wild-type background [8]. |
| Dynamic Range | 0.8% - 100% | mutant allele frequency [8]. |
| Accuracy (Precision) | High | Enabled by absolute quantification via Poisson statistics; no calibration curve required [52]. |
Table 2: Assay Specificity and Multiplexing Capacity
| Parameter | Description | Function |
|---|---|---|
| Specificity | Distinguishes wild-type CCR5 from the Δ32 deletion based on probe binding to the 32bp deleted region [8]. | Prevents false positives from wild-type sequence. |
| Multiplexing | Simultaneously quantifies wild-type and Δ32 alleles in a single reaction [8] [53]. | Provides direct ratio calculation, saves sample and reagents. |
| Discrimination Power | High | Based on clear separation of fluorescent clusters in 2D ddPCR plots [8]. |
Experimental Protocol
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions
| Reagent/Material | Function/Description | Example/Catalog Number |
|---|---|---|
| Cell Line | Source of genomic DNA (gDNA) for assay development and control. | MT-4 human T-cell line [8]. |
| gDNA Extraction Kit | Isolation of high-quality, pure genomic DNA from cell mixtures. | ExtractDNA Blood and Cells Kit (Evrogen) [8]. |
| ddPCR Supermix | Optimized reaction mix for probe-based ddPCR. | ddPCR Supermix for Probes (Bio-Rad) |
| FAM-labeled Probe | Detects the mutant CCR5Δ32 allele. | Custom TaqMan probe [8]. |
| HEX/VIC-labeled Probe | Detects the CCR5 wild-type allele. | Custom TaqMan probe [8]. |
| Droplet Generator | Partitions the PCR reaction into thousands of nanodroplets. | QX200 Droplet Generator (Bio-Rad) [52]. |
| Droplet Reader | Performs end-point fluorescence reading of each droplet. | QX200 Droplet Reader (Bio-Rad) [52]. |
Step-by-Step Workflow
The following diagram illustrates the complete experimental workflow for the ddPCR assay, from sample preparation to data analysis.
Detailed Protocol
Step 1: Genomic DNA (gDNA) Extraction and Quality Control
- Extract gDNA from the heterogeneous cell mixture (e.g., edited and unedited cells) using a commercial kit, such as the ExtractDNA Blood and Cells Kit [8].
- Accurately quantify the DNA concentration using a spectrophotometer (e.g., NanoPhotometer P-Class P360). Ensure the A260/A280 ratio is between 1.8 and 2.0, indicating pure DNA [8].
Step 2: Prepare Multiplex ddPCR Reaction
- On ice, prepare the reaction mix according to the table below. The total reaction volume is typically 20-22 µL.
- Note: The primer and probe sequences targeting the CCR5 wild-type and Δ32 loci are as described in [8].
Table 4: ddPCR Reaction Master Mix
| Component | Final Concentration | Volume per Reaction (µL) |
|---|---|---|
| ddPCR Supermix for Probes (2X) | 1X | 10 |
| FAM-labeled CCR5Δ32 Probe (20X) | 1X | 1 |
| HEX-labeled CCR5 WT Probe (20X) | 1X | 1 |
| Forward Primer (18µM) | 900 nM | 1 |
| Reverse Primer (18µM) | 900 nM | 1 |
- Add 50-100 ng of template gDNA.
- Adjust the final volume to 20 µL with nuclease-free water.
Step 3: Droplet Generation
- Transfer the 20 µL reaction mix to the sample well of a DG8 cartridge.
- Add 70 µL of Droplet Generation Oil to the oil well.
- Place the cartridge and a rubber gasket into the QX200 Droplet Generator. Following the manufacturer's protocol, this device will partition each sample into ~20,000 nanoliter-sized droplets [52].
Step 4: PCR Amplification
- Carefully transfer 40 µL of the generated droplets to a 96-well PCR plate. Seal the plate with a foil heat seal.
- Perform PCR amplification in a thermal cycler using the following protocol:
Table 5: Thermal Cycling Conditions
| Step | Temperature | Time | Cycles |
|---|---|---|---|
| Enzyme Activation | 95°C | 10 minutes | 1 |
| Denaturation | 94°C | 30 seconds | 40 |
| Annealing/Extension | 60°C | 60 seconds | |
| Enzyme Deactivation | 98°C | 10 minutes | 1 |
| Hold | 4°C | ∞ |
Step 5: Droplet Reading and Analysis
- Place the PCR plate into the QX200 Droplet Reader.
- The reader will aspirate each sample and flow droplets single-file past a two-color (FAM/HEX) optical detection system [52].
- Analyze the data using the associated software (e.g., QuantaSoft). The software applies Poisson statistics to the count of positive and negative droplets to provide an absolute concentration of both wild-type and Δ32 targets in copies/µL [8] [52].
Data Analysis and Interpretation
The output of the ddPCR assay is analyzed by plotting the fluorescence amplitude of each droplet, which allows for clear discrimination between different allele populations.
- Cluster Identification: The analysis software will generate a 2D plot showing four distinct droplet clusters:
- Concentration Calculation: The software calculates the concentration of each target (in copies/µL) using the formula derived from Poisson statistics: ( \text{Concentration} = -\ln(1 - p) / V ) where ( p ) is the ratio of positive droplets to total droplets, and ( V ) is the droplet volume [52].
- Determining Mutant Allele Frequency: The proportion of cells with the CCR5Δ32 mutation is calculated as: ( \text{Mutant Allele Frequency} = [\text{Δ32 concentration}] / ([\text{Δ32 concentration}] + [\text{WT concentration}]) )
This method provides a highly sensitive and specific approach for validating gene editing outcomes or tracking donor cell engraftment in heterogeneous samples, crucial for advancing CCR5-targeted therapies [8] [22].
Troubleshooting CCR5 ddPCR Assays: Solving Common Challenges
Optimization of Primer and Probe Concentrations for Multiplex Reactions
Within the framework of multiplex droplet digital PCR (ddPCR) assay design for CCR5 genotyping, the precise optimization of primer and probe concentrations is a critical determinant for achieving specific and accurate quantification of both wild-type and Δ32 mutant alleles. The CCR5 coreceptor serves as a principal entry point for human immunodeficiency virus (HIV), and a naturally occurring 32-base pair deletion (CCR5Δ32) confers resistance to HIV infection [8]. Research into curative strategies, including allogeneic hematopoietic stem cell transplantation from CCR5Δ32 homozygous donors and CRISPR/Cas9-mediated gene editing to recreate this mutation, necessitates reliable methods for quantifying the mutant allele fraction in heterogeneous cell populations [8] [22]. Multiplex ddPCR enables the simultaneous, absolute quantification of multiple targets, making it an ideal platform for this application. However, its success hinges on the careful balancing of assay components to ensure high sensitivity, specificity, and low background signal. These Application Notes provide a detailed protocol for optimizing primer and probe concentrations to develop a robust multiplex ddPCR assay for concurrent detection of CCR5 wild-type and Δ32 alleles.
Core Optimization Strategies
Optimizing a multiplex ddPCR assay requires a systematic approach to overcome challenges such as preferential amplification, formation of primer-dimers, and suboptimal fluorescence separation between target populations. The following strategies are foundational.
Empirical Testing and the "Experience Matrix"
A systematic, empirical approach is recommended over theoretical prediction. The use of an "experience matrix" is a validated strategy to condense ddPCR performance parameters into a graphical format for straightforward evaluation [39]. This matrix should catalog key variables and their impact on a droplet separation value, an objective metric that combines the absolute fluorescence signal distance between positive and negative droplet populations and their internal variation.
- Parameters to Test: The matrix should include data on:
- Singleplex vs. duplex ddPCR configuration.
- Primer and probe concentrations (e.g., "normal" vs. "high").
- Annealing/extension temperature.
- Thermal cycler used.
- Probe manufacturer and dye label (FAM, HEX/VIC).
- Performance Classification: The combination of separation value and the experience matrix allows for the rating of different assay parameter sets, guiding the selection of the most robust conditions for reliable quantification [39].
Oligonucleotide Design and Concentration Balancing
The design and relative concentrations of primers and probes are paramount for a successful multiplex reaction.
- Primer Design: Primers for all targets in a multiplex reaction should have similar annealing temperatures and minimal complementarity to prevent the formation of primer-dimers [41]. Primer length is typically 18–30 bp with a GC content of 35–60%.
- Concentration Titration: While singleplex assays may perform well with standard concentrations, multiplex formats often require re-optimization. A common approach is to test a range of concentrations, such as 900 nM for each primer and 250 nM for each probe, compared to lower, standard concentrations [39]. Balanced concentrations help prevent preferential amplification of one target over another, a phenomenon known as PCR bias [41].
Table 1: Example Oligonucleotide Concentration Ranges for Optimization
| Component | Low Concentration Range | High Concentration Range | Function in Assay |
|---|---|---|---|
| Primers | 100 - 500 nM | 600 - 900 nM | Target sequence amplification |
| Hydrolysis Probes | 50 - 150 nM | 200 - 250 nM | Specific detection of amplified target |
| Template DNA | 1 - 100 ng/reaction | N/A | Source of target alleles |
Thermal Cycling Optimization and "Rain" Reduction
The annealing and extension temperature is a critical parameter that can significantly impact assay performance by influencing the specificity of amplification and the magnitude of "rain"—droplets with intermediate fluorescence that complicate threshold setting [39].
- Temperature Gradient: A thermal gradient experiment should be performed to identify the temperature that maximizes the separation between positive and negative droplet clusters while minimizing rain.
- Hot-Start PCR: The use of a hot-start DNA polymerase is essential to minimize non-specific amplification and primer-dimer formation that can occur during reaction setup at lower temperatures [41].
Detailed Experimental Protocol: Multiplex ddPCR for CCR5Δ32 Allele Quantification
This protocol is adapted from published methodologies for detecting CCR5Δ32 mutant alleles [8] and optimizing multiplex ddPCR assays [47].
Materials and Equipment
- DNA Samples: Genomic DNA extracted from cell lines (e.g., MT-4) or patient samples. Include control samples with known genotypes (wild-type, heterozygous, and homozygous for CCR5Δ32) [8].
- Oligonucleotides:
- Primers and probes for CCR5 wild-type allele.
- Primers and probes for CCR5 Δ32 allele.
- Probe Labels: Use distinct fluorescent dyes for each allele (e.g., FAM for Δ32, HEX/VIC for wild-type).
- ddPCR Master Mix: ddPCR Supermix for Probes (no dUTP) (Bio-Rad).
- Equipment: QX200 Droplet Digital PCR System (Bio-Rad), including droplet generator, thermal cycler, and droplet reader [47].
Step-by-Step Procedure
Step 1: Preliminary Singleplex Assay Setup For each target (wild-type and Δ32), set up singleplex ddPCR reactions to confirm individual assay performance. Use a standard concentration (e.g., 500 nM primers, 100 nM probe) and a standardized thermal cycling protocol.
Step 2: Multiplex Assay Optimization Matrix Set up a series of duplex reactions with varying concentrations of primers and probes. A suggested matrix is below.
Table 2: Example Optimization Matrix for Duplex CCR5 ddPCR
| Reaction | Wild-Type Primer Conc. (nM) | Δ32 Primer Conc. (nM) | Wild-Type Probe Conc. (nM) | Δ32 Probe Conc. (nM) | Annealing Temp. (°C) |
|---|---|---|---|---|---|
| 1 | 500 | 500 | 100 | 100 | 55 |
| 2 | 500 | 500 | 100 | 100 | 60 |
| 3 | 900 | 900 | 250 | 250 | 55 |
| 4 | 900 | 900 | 250 | 250 | 60 |
| 5 | 500 | 900 | 100 | 250 | 58 |
| 6 | 900 | 500 | 250 | 100 | 58 |
Step 3: Reaction Assembly and Droplet Generation
- Prepare a 22 µL reaction mix containing:
- 11 µL of 2x ddPCR Supermix for Probes.
- Primers and probes at the concentrations defined in your matrix.
- 1–100 ng of genomic DNA template.
- Nuclease-free water to volume.
- Generate droplets using the QX200 Droplet Generator according to the manufacturer's instructions.
- Transfer the emulsified samples to a 96-well PCR plate and seal.
Step 4: Thermal Cycling Amplify the target sequences using a thermal cycler with the following profile:
- Enzyme activation: 95°C for 10 minutes.
- 40 cycles of:
- Denaturation: 95°C for 30 seconds.
- Annealing/Extension: [55–60°C] for 1 minute (optimize temperature based on matrix).
- Enzyme deactivation: 98°C for 10 minutes.
- Hold at 12°C (optional, but recommended before reading).
Step 5: Droplet Reading and Analysis
- Load the plate into the QX200 Droplet Reader.
- Analyze the data using the associated software (QuantaSoft).
- For each sample, set fluorescence thresholds to distinguish positive and negative droplets for each channel (FAM and HEX/VIC).
Data Analysis and Interpretation
- Calculate Copy Numbers: The software will provide absolute copy numbers (copies/µL) for each target based on Poisson statistics.
- Determine Mutant Allele Fraction: The fraction of CCR5Δ32 alleles can be calculated as: (Δ32 copies) / (Δ32 copies + Wild-type copies).
- Assess Assay Performance: The optimized assay should demonstrate a clear separation between positive and negative droplet clusters, minimal rain, and a low false-positive rate in negative controls. The developed system should accurately quantify the mutant allele content down to 0.8% in cell mixtures [8].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Kits for Multiplex ddPCR Assay Development
| Item | Function/Application | Example Product (Supplier) |
|---|---|---|
| ddPCR System | Partitioning, amplification, and absolute quantification of nucleic acids. | QX200 Droplet Digital PCR System (Bio-Rad) [47] |
| ddPCR Master Mix | Optimized buffer, dNTPs, and polymerase for probe-based ddPCR. | ddPCR Supermix for Probes (no dUTP) (Bio-Rad) [47] |
| gBlock Gene Fragments | Synthetic double-stranded DNA for use as positive controls and extraction spike-ins. | gBlock Gene Fragments (Integrated DNA Technologies) [47] |
| Genomic DNA Isolation Kit | High-quality DNA extraction from cells or tissues. | ExtractDNA Blood and Cells Kit (Evrogen) [8] |
| CRISPR/Cas9 System | For generating isogenic cell lines with specific mutations (e.g., CCR5Δ32) for assay validation. | pCas9-IRES2-EGFP & pU6-gRNA plasmids [8] |
| Reference DNA | Genomic DNA with known genotype for assay calibration and controls. | Wild-type Human Genomic DNA (Promega) [47] |
| Cell Line | Model system for method development and optimization. | MT-4 human T-cell line [8] |
Workflow and Logical Relationships
The following diagram illustrates the logical workflow for optimizing a multiplex ddPCR assay, from initial setup to data analysis, integrating the key concepts and strategies discussed in this protocol.
Temperature Gradient Assessment for Specific Amplification
The accurate discrimination between CCR5 wild-type and Δ32 alleles is a critical component in advanced HIV cure research, particularly in the development of autologous hematopoietic stem cell transplants with engineered HIV resistance. A key challenge in multiplex droplet digital PCR (ddPCR) assay design for this application is the precise determination of optimal annealing temperatures to ensure specific amplification of both alleles with high efficiency and without cross-reactivity. This protocol details a systematic approach for temperature gradient assessment to establish robust thermal cycling conditions for simultaneous CCR5 wild-type and Δ32 allele quantification. The ability to accurately quantify the content of mutant CCR5Δ32 alleles in heterogeneous cell mixtures is essential for monitoring engineered cell populations, with recent studies demonstrating detection sensitivity down to 0.8% [8]. The methodology described herein provides researchers with a standardized framework for optimizing the critical parameter of annealing temperature, which directly impacts assay precision, sensitivity, and reliability for both research and potential clinical applications.
Theoretical Background
CCR5 Genotyping and HIV Research Context
The CCR5 co-receptor serves as a principal binding site for human immunodeficiency virus (HIV), making it a critical target for therapeutic intervention. A naturally occurring 32-base pair deletion (CCR5Δ32) results in a non-functional receptor that confers resistance to HIV infection in homozygous individuals [8]. This genetic insight has paved the way for innovative cure strategies, including allogeneic hematopoietic stem cell transplantation from CCR5Δ32/Δ32 donors and CRISPR/Cas9-mediated gene editing of autologous cells [28]. Multiplex ddPCR assays capable of precisely quantifying both wild-type and Δ32 alleles are essential for evaluating the editing efficiency and population dynamics of engineered cell products. Recent clinical evidence suggests that high-frequency CCR5 editing (>90%) is likely necessary to achieve clinical protection from HIV, highlighting the critical importance of accurate quantification methods [28].
Fundamentals of ddPCR and Temperature Optimization
Droplet digital PCR represents a refinement of conventional PCR methods, where the reaction mixture is partitioned into thousands of nanoliter-sized droplets prior to amplification. Each droplet functions as an individual micro-reactor, allowing for absolute quantification of target nucleic acids without the need for standard curves [54]. In multiplex ddPCR applications, temperature optimization is particularly crucial as it must accommodate multiple primer-probe sets with potentially different ideal annealing temperatures. Suboptimal temperatures can lead to preferential amplification of one target, reduced fluorescence amplitude separation between targets, or increased non-specific amplification, all of which compromise data quality and quantification accuracy. The temperature gradient assessment protocol described below systematically addresses these challenges to establish conditions that ensure balanced amplification efficiency and specific target discrimination.
Materials and Equipment
Research Reagent Solutions
Table 1: Essential reagents and materials for temperature gradient optimization
| Item | Function/Description | Examples/Specifications |
|---|---|---|
| ddPCR Supermix | Reaction buffer for droplet generation and amplification | Bio-Rad ddPCR Supermix for Probes (no dUTP) [55] |
| Primer/Probe Sets | Target-specific amplification and detection | FAM-labeled probe for CCR5 Δ32; HEX/VIC-labeled probe for CCR5 wild-type [8] |
| Template DNA | Target nucleic acids for assay validation | Genomic DNA from heterozygous and homozygous control samples [56] |
| Droplet Generation Oil | Immiscible fluid for creating reaction partitions | Droplet Generation Oil for Probes [55] |
| Restriction Enzymes | DNA fragmentation to improve amplification efficiency | HindIII (optional, for complex genomic regions) [57] |
| Thermal Sealer | Secure sealing of reaction plates | Pierceable foil heat seal [55] |
| DG8 Cartridges | Microfluidic droplet generation | Bio-Rad DG8 Cartridges [55] |
Specialized Equipment
- QX200 Droplet Generator: Creates uniform droplets from the reaction mixture [8] [55]
- C1000 Touch Thermal Cycler: Precisely controlled thermal cycling with gradient functionality [8]
- QX200 Droplet Reader: Quantifies fluorescence in each droplet post-amplification [55]
- S3e Cell Sorter: Optional equipment for cell sorting if analyzing specific cell populations [8]
Experimental Protocol
Preliminary Assay Design
- Primer and Probe Selection: Design or select validated primer pairs and hydrolysis probes for CCR5 wild-type and Δ32 targets. The Δ32 assay should span the deletion junction to ensure specificity.
- Control Preparation: Establish genomic DNA controls with known genotypes (wild-type homozygous, Δ32 heterozygous, and Δ32 homozygous). Cell lines with artificial CCR5Δ32 mutations created via CRISPR/Cas9 can serve as valuable reference materials [8].
- Reaction Assembly: Prepare master mix containing ddPCR supermix, primers, probes, and nuclease-free water. Aliquot equal volumes to each reaction well before adding template DNA.
Temperature Gradient Setup
- Gradient Configuration: Program the thermal cycler with a temperature gradient spanning across the columns of a 96-well plate. A recommended starting range is 55°C to 65°C, covering typical annealing temperatures for TaqMan-based assays.
- Reaction Partitioning: Transfer the reaction mixture to the droplet generator to create stabilized water-in-oil emulsions. According to standard protocols, each 20μL reaction typically generates approximately 20,000 droplets [55].
- Thermal Cycling: Place the droplet emulsion plate in the thermal cycler and run the following protocol:
- Initial denaturation: 95°C for 10 minutes
- 40-50 cycles of:
- Denaturation: 94°C for 30 seconds
- Annealing/Extension: Gradient temperature for 60 seconds
- Enzyme deactivation: 98°C for 10 minutes
- Final hold: 4°C [55]
Post-Amplification Analysis
- Droplet Reading: Transfer the plate to the droplet reader, which measures the fluorescence in each droplet (FAM and HEX/VIC channels).
- Data Acquisition: Use analysis software to count positive and negative droplets for each target at each temperature condition.
- Threshold Determination: Apply appropriate fluorescence thresholds to distinguish positive from negative droplets, ensuring consistent application across all temperature conditions.
Data Analysis and Interpretation
Quantitative Assessment Metrics
Table 2: Key performance indicators for temperature optimization
| Parameter | Optimal Range | Interpretation |
|---|---|---|
| Total Droplet Count | 10,000-20,000 per reaction | Indicates successful droplet generation and integrity throughout thermal cycling [54] |
| Mutant Allele Frequency | Matches expected ratio in control samples | Validates accurate Δ32 detection, crucial for monitoring editing efficiency [8] |
| Fluorescence Amplitude | Clear separation between positive and negative populations | Reflects specific amplification; minimal intermediate droplets indicate robust assay performance [57] |
| Channel Crosstalk | <1% in non-target channels | Confirms probe specificity, particularly important in multiplex applications [57] |
| Ratio Accuracy | 1:1 for reference genes in control samples | Validates balanced amplification efficiency between targets [57] |
Temperature Optimization Criteria
- Droplet Count Stability: Identify temperatures that maintain consistent droplet counts across replicates, indicating stable emulsion integrity throughout thermal cycling.
- Amplification Efficiency: Select temperatures yielding the highest fluorescence amplitude separation between positive and negative droplets for both targets.
- Specificity Assessment: Choose conditions that minimize rain (intermediate fluorescence droplets) and channel crosstalk.
- Balanced Multiplexing: Identify the temperature that provides the most balanced amplification efficiency between wild-type and Δ32 targets, as evidenced by expected copy number ratios in control samples.
- Precision Evaluation: Calculate coefficient of variation for replicate measurements at each temperature, prioritizing conditions with CV < 10%.
Application to CCR5 Genotyping
The optimized temperature conditions established through this protocol enable precise quantification of CCR5Δ32 mutant alleles in heterogeneous cell mixtures, a capability essential for monitoring CRISPR/Cas9 editing efficiency in hematopoietic stem cells [8] [28]. The ddPCR system described has demonstrated sensitivity down to 0.8% for detecting CCR5Δ32 mutations in mixed cell populations, making it suitable for tracking engineered cell expansion in preclinical and clinical settings [8]. Furthermore, the multiplex nature of the assay allows for simultaneous quantification of both wild-type and mutant alleles, providing a comprehensive view of the genetic landscape in edited cell products. This precise quantification is particularly important given recent findings that demonstrate a potential threshold effect, with higher CCR5 editing frequencies (>90%) conferring significantly greater protective benefit against HIV infection [28].
Troubleshooting Guide
Table 3: Common issues and solutions in temperature optimization
| Problem | Potential Causes | Solutions |
|---|---|---|
| Low droplet count | Improper droplet generation, reagent issues | Verify oil:sample ratio, ensure fresh reagents, check droplet generator gaskets [55] |
| High background signal | Non-specific amplification, probe degradation | Optimize annealing temperature, use fresh probes, consider touchdown PCR [56] |
| Unbalanced amplification | Different optimal annealing temperatures for targets | Adjust primer concentrations, modify probe sequences, or implement modified thermal profile [57] |
| Excessive rain | Suboptimal annealing temperature, inhibitor presence | Fine-tune temperature gradient, purify template DNA, add enhancers [56] |
| Poor channel separation | Spectral overlap, high probe concentrations | Adjust probe concentrations, verify filter settings, ensure proper quencher selection [57] |
Figure 1: Experimental workflow for temperature gradient assessment in multiplex ddPCR assay development.
Figure 2: Decision pathway for identifying optimal annealing temperature based on droplet data quality assessment.
Systematic temperature gradient assessment is a fundamental step in developing robust multiplex ddPCR assays for CCR5 genotyping. The protocol outlined herein provides a standardized approach for identifying optimal annealing temperatures that ensure specific, efficient, and balanced amplification of both wild-type and Δ32 alleles. The resulting optimized conditions enable precise quantification of CCR5 editing efficiency, which is essential for advancing HIV cure strategies based on hematopoietic stem cell engineering. As CRISPR-based therapies continue to evolve toward clinical application, reliable ddPCR assays with thoroughly optimized parameters will play an increasingly important role in quantifying editing outcomes and correlating them with functional efficacy.
Addressing Fluorescence Spillover and Spectral Overlap
In the field of molecular diagnostics, droplet digital PCR (ddPCR) has emerged as a powerful technology for the absolute quantification of nucleic acids, enabling highly sensitive detection of rare genetic variants [52] [58]. This application note addresses a critical technical challenge in multiplex ddPCR assay design: fluorescence spillover and spectral overlap. Specifically framed within CCR5 wild-type and Δ32 alleles research, this document provides detailed protocols and solutions for researchers developing assays to quantify the HIV-associated CCR5Δ32 mutation.
The CCR5 (C-C chemokine receptor type 5) protein serves as a co-receptor for human immunodeficiency virus (HIV) entry into T-cells [8]. A naturally occurring 32-base pair deletion (CCR5Δ32) confers resistance to HIV infection when homozygous, making it a critical target for therapeutic development and clinical monitoring [8] [7]. Accurate quantification of mutant CCR5Δ32 alleles in heterogeneous cell mixtures is essential for advancing HIV cure strategies, including hematopoietic stem cell transplantation and CRISPR/Cas9 genome editing approaches [8].
Understanding Fluorescence Spillover in ddPCR
Fundamentals of ddPCR Detection
Droplet digital PCR operates by partitioning a PCR reaction into thousands of nanoliter-sized droplets, each functioning as an individual microreactor [58]. Following amplification, each droplet is analyzed for fluorescence using optical detection systems. In multiplex assays, different targets are detected using fluorophores with distinct emission spectra. Fluorescence spillover occurs when the emission spectrum of one fluorophore is detected in the channel designated for another fluorophore, leading to compromised data quality and inaccurate quantification [59].
The underlying principle of ddPCR quantification relies on Poisson statistics applied to the distribution of target molecules across partitions [58]. The concentration of the target nucleic acid is calculated from the fraction of positive droplets using the formula: λ = -ln(1-p), where λ represents the average number of target molecules per partition and p is the fraction of positive partitions [58]. Spectral overlap can distort the accurate determination of positive partitions, directly impacting the precision of absolute quantification.
Impact on CCR5 Genotyping Assays
For CCR5 wild-type and Δ32 allele discrimination, researchers typically employ a dual-probe system where each allele is labeled with a different fluorophore [8] [7]. Accurate genotyping requires clear discrimination between four droplet populations: wild-type-only, mutant-only, double-positive (heterozygous), and double-negative. Fluorescence spillover blurs the boundaries between these populations, potentially leading to:
- Misclassification of heterozygous samples
- Underestimation of rare mutant alleles in wild-type backgrounds
- Reduced sensitivity for detecting low-frequency mutations
- Compromised accuracy in quantifying gene editing efficiency
Experimental Strategies for Minimization and Compensation
Probe Selection and Assay Design
Careful fluorophore selection forms the foundation for addressing spectral overlap. The table below summarizes characteristics of commonly used fluorophores in ddPCR applications:
Table 1: Fluorophore Properties for Multiplex ddPCR
| Fluorophore | Excitation Max (nm) | Emission Max (nm) | Recommended Quencher | Compatible Dyes for Multiplexing |
|---|---|---|---|---|
| FAM | 495 | 520 | BHQ-1 or TAMRA | HEX, VIC, TET |
| HEX | 535 | 556 | BHQ-1 | FAM, TEXAS RED |
| VIC | 538 | 554 | BHQ-1 | FAM, NED |
| CY5 | 649 | 670 | BHQ-2 or BHQ-3 | FAM, HEX |
| TEXAS RED | 589 | 615 | BHQ-2 | FAM, CY5 |
Design considerations for CCR5 assays:
- Probe positioning: For CCR5Δ32 detection, design wild-type probes to span the deletion junction, ensuring they cannot bind to mutant sequences [8]
- Probe length: Optimize probe length to balance specificity and signal intensity (typically 20-30 nucleotides)
- Quencher selection: Incorporate efficient dark quenchers (BHQ series) to minimize background fluorescence
- Dye separation: Select dye combinations with maximal spectral separation to reduce cross-talk
Experimental Optimization Protocol
Protocol: Fluorescence Compensation Matrix Setup
This protocol establishes a fluorescence compensation matrix to correct for spectral overlap in multiplex ddPCR assays.
Materials Required:
- Individual probe assays for each target (CCR5 wild-type and CCR5Δ32)
- Single-dye control samples for each fluorophore
- QX200 Droplet Generator (Bio-Rad) or equivalent system [8] [55]
- ddPCR Supermix for Probes (no dUTP) [55]
- DG8 Cartridges and Gaskets (Bio-Rad)
- Droplet Generation Oil
- 96-well PCR plate
- Thermal sealer
- C1000 Touch Thermal Cycler (Bio-Rad) or equivalent
- QX200 Droplet Reader (Bio-Rad)
Procedure:
- Prepare single-dye controls:
- Create three separate reaction mixtures:
- FAM control: Contains only CCR5 wild-type probe with FAM label
- HEX/VIC control: Contains only CCR5Δ32 probe with HEX/VIC label
- No-template control: Contains no nucleic acid template
- Use genomic DNA known to be homozygous for each allele as template for respective single-dye controls
- Create three separate reaction mixtures:
Partitioning and amplification:
- Follow manufacturer's instructions for droplet generation [55]
- Perform PCR amplification with the following cycling conditions:
- 95°C for 10 minutes (enzyme activation)
- 40 cycles of:
- 94°C for 30 seconds (denaturation)
- 56-60°C for 60 seconds (annealing/extension) [8]
- 98°C for 10 minutes (enzyme deactivation)
- 4°C hold
Droplet reading:
- Load plates into the droplet reader
- Ensure proper droplet discrimination settings before proceeding with experimental samples
Compensation matrix calculation:
- Using the data from single-dye controls, calculate the spillover coefficients using the following formula:
- FAM→HEX = Median HEX signal of FAM-only control / Median FAM signal of FAM-only control
- HEX→FAM = Median FAM signal of HEX-only control / Median HEX signal of HEX-only control
- Apply these coefficients to generate a compensation matrix in your analysis software
- Using the data from single-dye controls, calculate the spillover coefficients using the following formula:
Troubleshooting tips:
- If spillover exceeds 10%, consider switching to more spectrally distinct fluorophores
- Ensure single-dye controls have strong positive signals without saturation
- Verify that no-template controls show minimal background in both channels
CCR5Δ32 Detection Workflow and Data Analysis
The following workflow diagrams illustrate the complete process for CCR5 genotyping using multiplex ddPCR, incorporating strategies to address spectral overlap.
Figure 1: Comprehensive workflow for CCR5 genotyping using ddPCR, highlighting key steps where spectral overlap considerations are incorporated.
Data Analysis with Spectral Compensation
Following droplet reading and application of the compensation matrix, data analysis proceeds with droplet classification:
Figure 2: Data analysis workflow showing droplet classification after spectral compensation, leading to accurate allele frequency calculation.
Quantification formula: After droplet classification, apply Poisson statistics to calculate the target concentration:
[ \text{Target Concentration (copies/μL)} = \frac{-\ln(1 - \frac{Np}{Nt}) \times D}{V} ]
Where:
- (N_p) = number of positive partitions for the target
- (N_t) = total number of partitions analyzed
- (D) = dilution factor
- (V) = volume of partition (nL)
For CCR5Δ32 mutation detection, the mutant allele frequency is calculated as:
[ \text{Mutant Allele Frequency} = \frac{[\text{CCR5Δ32}]}{[\text{CCR5 WT}] + [\text{CCR5Δ32}]} \times 100\% ]
The developed system has demonstrated sensitivity for detecting CCR5Δ32 mutations down to 0.8% in heterogeneous cell mixtures [8].
Research Reagent Solutions
The table below outlines essential reagents and materials required for implementing robust CCR5 genotyping assays with minimal spectral overlap.
Table 2: Essential Research Reagents for CCR5 ddPCR Assays
| Reagent/Material | Function/Application | Example Product | Key Considerations |
|---|---|---|---|
| ddPCR Supermix | Provides optimized buffer, enzymes, and dNTPs for probe-based ddPCR | ddPCR Supermix for Probes (no dUTP) [55] | Select "no dUTP" format for assays not requiring contamination control with UDG |
| FAM-labeled Probe | Detection of CCR5 wild-type allele | Custom TaqMan probe targeting CCR5 WT sequence [8] | Position probe to span deletion site for specific discrimination of WT vs Δ32 |
| HEX/VIC-labeled Probe | Detection of CCR5Δ32 mutant allele | Custom TaqMan probe targeting CCR5Δ32 junction [8] | Validate specificity with homozygous WT and Δ32 control samples |
| Droplet Generation Oil | Creates stable water-in-oil emulsion for partitioning | Droplet Generation Oil for Probes [55] | Ensure proper storage and avoid moisture contamination |
| DG8 Cartridges & Gaskets | Microfluidic chips for droplet generation | DG8 Cartridges (Bio-Rad) | Single-use components; ensure proper sealing |
| Quantification Standards | Assay validation and quality control | Genomic DNA with known CCR5 genotypes | Include homozygous WT, homozygous Δ32, and heterozygous controls |
Advanced Applications and Future Directions
The principles outlined in this application note extend beyond basic CCR5 genotyping to advanced research applications:
Monitoring Gene Editing Outcomes
With the development of CRISPR/Cas9 genome editing to introduce CCR5Δ32 mutations, ddPCR provides a sensitive method for quantifying editing efficiency [8]. Multiplex assays can simultaneously detect intended mutations and potential off-target effects, with spectral optimization ensuring accurate quantification of each event.
Single-Cell Analysis
Emerging microfluidic technologies enable single-cell ddPCR analysis, allowing researchers to investigate cellular heterogeneity in CCR5 expression and editing [60]. These applications place even greater demands on fluorescence detection sensitivity, making optimal dye selection and spillover compensation critical.
Liquid Biopsy Applications
For translational studies, ddPCR assays can detect CCR5 mutations in cell-free DNA from liquid biopsies, enabling non-invasive monitoring of therapeutic interventions [61] [62]. The exceptional sensitivity of ddPCR for rare allele detection (down to 0.001% variant allele frequency) makes it ideal for these applications [61].
Addressing fluorescence spillover and spectral overlap is essential for developing robust multiplex ddPCR assays for CCR5 genotyping. Through careful fluorophore selection, experimental optimization, and computational compensation, researchers can achieve accurate quantification of CCR5 wild-type and Δ32 alleles across diverse research applications. The protocols and strategies outlined herein provide a foundation for advancing HIV cure research, gene editing validation, and precision medicine approaches targeting the CCR5 co-receptor.
Minimizing Non-Specific Amplification and Primer-Dimer Formation
The accurate quantification of CCR5 wild-type and Δ32 alleles using multiplex droplet digital PCR (ddPCR) is a cornerstone of advanced HIV cure research, informing studies on natural resistance and the outcomes of stem cell transplantation [8] [18]. A primary technical obstacle in this sensitive genotyping workflow is non-specific amplification and primer-dimer formation. These artifacts compete for reaction reagents, reduce the dynamic range and sensitivity of the assay for the intended target, and can lead to inaccurate copy number quantification [63] [64] [65]. This application note details targeted strategies and a optimized protocol to suppress these undesired amplification events, ensuring data integrity for critical therapeutic development decisions.
Root Causes and Strategic Optimization
Non-specific amplification occurs when primers bind to non-target sequences or to each other, instead of to the intended template. The formation of primer-dimers is often the most prevalent form, typically resulting from complementary regions at the 3' ends of primers [63] [64]. In ddPCR, these artifacts are particularly detrimental as they can lead to false-positive droplets, skewing the Poisson statistics used for absolute quantification.
The table below summarizes the primary causes and corresponding strategic solutions to minimize these effects.
Table 1: Root Causes of Non-Specific Amplification and Strategic Countermeasures
| Root Cause | Impact on Assay | Primary Optimization Strategy |
|---|---|---|
| 3' Primer Complementarity [63] | Primer-dimer formation; consumption of dNTPs and enzyme. | Meticulous in silico primer design to avoid self- and cross-dimers. |
| Excessive Primer Concentration [64] | Increased chance of primer-primer interactions. | Titration of primer and probe concentrations to optimal levels [66]. |
| Sub-Optimal Annealing Temperature [63] | Promotes off-target binding and dimer stabilization. | Empirical determination of the highest possible annealing temperature. |
| Polymerase Activity at Low Temperatures [63] | Enzymatic extension of dimers formed during reaction setup. | Use of hot-start DNA polymerase. |
| PCR Inhibitors in Sample [67] [65] | Reduced amplification efficiency; can exacerbate non-specific background. | Use of high-purity DNA and inhibitor-tolerant ddPCR chemistry. |
A Workflow for Robust ddPCR Assay Development
The following diagram illustrates a systematic workflow for developing and optimizing a multiplex ddPCR assay to minimize non-specific amplification.
Figure 1: A systematic workflow for developing a robust ddPCR assay, from in-silico design to experimental validation.
Detailed Experimental Protocol for CCR5 Δ32/wt Multiplex ddPCR
This protocol is adapted from methodologies successfully used to quantify CCR5Δ32 mutant alleles in heterogeneous cell mixtures, a key requirement for monitoring engraftment in HIV stem cell therapy research [8] [68].
Reagent Preparation and Primer/Probe Design
Research Reagent Solutions:
Table 2: Essential Reagents for CCR5 ddPCR Genotyping Assay
| Item | Function/Description | Example/Note |
|---|---|---|
| ddPCR Supermix for Probes | Provides optimized buffer, dNTPs, and hot-start Taq polymerase for probe-based assays. | Bio-Rad ddPCR Supermix for Probes (No dUTP) is standard. |
| Sequence-Specific Primers | Amplifies the target CCR5 wild-type and Δ32 regions. | Design for high specificity; final concentration ~0.5-0.9 µM each [65]. |
| FAM- and HEX-labeled Probes | Enables multiplex detection by binding specifically to wild-type or Δ32 sequences. | Final concentration ~0.25 µM; ensure no overlap between quencher and dye emission [65]. |
| Nuclease-Free Water | Solvent for reagents; ensures no RNase/DNase contamination. | Critical for preventing nucleic acid degradation. |
| Restriction Enzyme | Digests high-molecular-weight DNA to ensure uniform partitioning and accurate quantification. | Use an enzyme that does not cut within the amplicon sequence [65]. |
| QX200 Droplet Generator Oil | Creates the water-in-oil emulsion for droplet formation. | For use with Bio-Rad QX200 systems. |
Primer and Probe Design:
- Target: Exon 4 of the CCR5 gene, spanning the 32-bp deletion region [8] [69].
- Principles: Design primers with a length of 18-22 bases, a melting temperature (Tm) of ~60°C, and a GC content of 40-60%. Crucially, analyze sequences to avoid 3' self-complementarity and cross-dimers, especially between the two primer sets in the multiplex reaction. Tools like NCBI BLAST and specialized primer design software should be used.
- Probe Design: Probes should have a Tm 5-10°C higher than the primers. For the CCR5 Δ32 assay, design one probe to bind the wild-type sequence and a second, labeled with a different fluorophore (e.g., HEX), to bind the sequence spanning the Δ32 deletion junction [8].
Primer and Probe Storage: Resuspend lyophilized oligonucleotides in TE buffer (pH 8.0) to create stock solutions. Avoid repeated freeze-thaw cycles by preparing small aliquots; store at -20°C. Fluorescently labeled probes are stable for 6-9 months under these conditions [65].
Sample Preparation and Restriction Digestion
- DNA Extraction: Use high-quality DNA extraction kits (e.g., phenol-chloroform or commercial column-based kits) from cell lines (e.g., MT-4) or patient samples. Assess DNA purity via spectrophotometry (A260/A280 ratio ~1.8) [8] [65].
- Restriction Digestion (Recommended): To ensure accurate quantification, particularly for high-molecular-weight genomic DNA, perform a restriction digest prior to ddPCR. This step reduces viscosity, prevents uneven partitioning, and physically separates linked gene copies, ensuring each is counted independently.
- Procedure: Digest 1 µg of genomic DNA with a suitable restriction enzyme that does not cut within the CCR5 amplicon. Incubate for 1 hour at the enzyme's optimal temperature, followed by enzyme inactivation [65].
ddPCR Reaction Setup and Thermal Cycling
Reaction Mix Composition (20 µL total volume):
- 10 µL of 2x ddPCR Supermix for Probes
- Primers: 1 µL each (final concentration 0.9 µM)
- Probes: 0.5 µL each (final concentration 0.25 µM)
- 2 µL of template DNA (optimized amount, e.g., 50-100 ng of human gDNA)
- Nuclease-free water to 20 µL
Note: Primer and probe concentrations are higher than typical qPCR to increase fluorescence amplitude and improve cluster separation [65]. The optimal concentration should be determined empirically.
Droplet Generation: Transfer the 20 µL reaction mix to a DG8 cartridge. Add 70 µL of droplet generation oil and generate droplets using the QX200 Droplet Generator according to the manufacturer's instructions. This will create ~20,000 nanoliter-sized droplets per sample.
Thermal Cycling:
- Enzyme Activation: 95°C for 10 minutes.
- Amplification (45-50 cycles):
- Enzyme Deactivation: 98°C for 10 minutes.
- Hold: 4°C ∞.
- Ramp Rate: Use a slow ramp rate (e.g., 2°C/second) between steps to ensure precise temperature control.
Data Acquisition and Analysis
- Droplet Reading: After amplification, place the plate in the QX200 Droplet Reader. The reader will measure the fluorescence (FAM and HEX) of each droplet.
- Analysis with QuantaSoft: The software applies Poisson statistics to the count of positive and negative droplets to provide an absolute copy number per microliter of reaction.
- Critical Controls:
- No-Template Control (NTC): Essential for identifying primer-dimer formation and reagent contamination. Positive signals in the NTC indicate a need for further optimization [63] [65].
- Positive Controls: Include known wild-type, heterozygous, and homozygous Δ32 DNA samples to validate assay specificity and sensitivity.
Troubleshooting and Quality Control
The following flowchart guides the systematic investigation and resolution of persistent non-specific amplification or primer-dimer issues.
Figure 2: A troubleshooting flowchart for resolving non-specific amplification, starting with the most common and easily adjustable parameters.
Minimizing non-specific amplification is not merely an optimization step but a fundamental requirement for generating publication and clinically relevant data in CCR5 genotyping research. By integrating rigorous in silico primer design, empirical optimization of reaction components and thermal profiles, and stringent quality controls, researchers can develop robust and reliable multiplex ddPCR assays. The protocol outlined here provides a clear roadmap to achieve this, supporting the accurate quantification of CCR5 Δ32 and wild-type alleles essential for advancing HIV cure strategies.
Partition Quality Control and Data Interpretation Guidelines
Droplet digital PCR (ddPCR) represents a third-generation PCR technology that enables absolute quantification of nucleic acids by partitioning samples into thousands of nanoliter-sized droplets [52]. This technology provides significant advantages for detecting the CCR5 wild-type and Δ32 alleles, including high sensitivity, absolute quantification without standard curves, and exceptional reproducibility [54]. In HIV cure research, accurate quantification of the CCR5Δ32 mutation is crucial, as this 32-base-pair deletion confers natural resistance to HIV-1 infection when homozygous [8] [18]. The partitioning process is fundamental to ddPCR performance, as it directly impacts the reliability of detecting heterogeneous cell mixtures containing mutant alleles present at frequencies as low as 0.8% [8].
Effective partition quality control ensures that droplets maintain integrity throughout amplification and detection phases, preventing false positives and negatives that could compromise experimental outcomes in therapeutic development settings. This application note establishes comprehensive guidelines for quality control and data interpretation specifically tailored to multiplex ddPCR assays targeting CCR5 genotypes.
Partition Quality Control Parameters
Essential Quality Metrics and Acceptance Criteria
Partition quality control begins with establishing baseline metrics for droplet generation and performance. The following parameters must be monitored and maintained within specified limits to ensure data integrity.
Table 1: Acceptance Criteria for Partition Quality Control in ddPCR
| Quality Parameter | Acceptance Criteria | Impact on Data Quality |
|---|---|---|
| Total Partition Count | ≥10,000 valid partitions [52] | Affects statistical power and quantification accuracy |
| Partition Uniformity | >90% monodisperse droplets [54] | Ensures consistent amplification efficiency |
| Accepted Droplets | >95% of generated droplets [52] | Minimizes data loss and maintains statistical validity |
| Droplet Intensity Separation | Clear cluster separation with >10,000 RFU between positive and negative populations | Enables accurate binary calling (positive/negative) |
| Template Loading Efficiency | 0.5-1.5 copies/partition (ideal) [52] | Optimizes for Poisson distribution analysis |
The partition count directly influences measurement precision, with higher partition counts enabling more accurate quantification of rare alleles. Per the Poisson distribution, which forms the mathematical foundation of ddPCR quantification, each partition acts as an individual reaction vessel [52]. Partitions must be monodisperse (uniform in size) to ensure consistent amplification efficiency across all reactions [54]. Significant variation in droplet volume introduces quantification errors, as larger droplets have higher probability of containing target molecules.
Visual Assessment of Partition Quality
Data interpretation begins with visual inspection of the two-dimensional droplet plot, which displays fluorescence amplitudes for each probe channel. Well-optimized assays demonstrate four distinct clusters in multiplex assays:
- Double-negative partitions (lower left quadrant): Contain no target sequences
- Wild-type CCR5-positive partitions (lower right or upper left, depending on channel assignment)
- Δ32 mutant-positive partitions (opposite single-positive cluster)
- Double-positive partitions (upper right quadrant): May indicate heterozygotes or technical artifacts
The following visualization represents the droplet interpretation workflow:
Droplet Analysis Workflow
Failed quality checks at any stage should trigger assay repetition, as compromised partition integrity invalidates the Poisson statistical foundation of ddPCR quantification.
Data Interpretation and Analysis
Applying Poisson Statistics for Absolute Quantification
The core principle of ddPCR data analysis relies on Poisson statistics to determine target concentration based on the fraction of negative partitions [52]. The fundamental equation is:
λ = -ln(1 - p)
Where λ represents the average number of target molecules per partition and p is the fraction of positive partitions. This calculation is performed automatically by ddPCR analysis software but must be validated by researchers.
For CCR5Δ32 mutation detection in heterogeneous cell mixtures, this approach enables precise quantification of mutant allele frequency, with demonstrated sensitivity down to 0.8% mutant alleles in wild-type background [8]. This exceptional sensitivity makes ddPCR particularly valuable for monitoring engraftment success in hematopoietic stem cell transplantation studies using CCR5Δ32 donors [18].
Troubleshooting Common Data Quality Issues
Table 2: Troubleshooting Guide for Partition Quality Issues
| Issue | Potential Causes | Corrective Actions |
|---|---|---|
| Low Partition Count | Microfluidic obstruction, insufficient sample, improper oil:sample ratio | Check droplet generator, ensure proper sample filtration, verify reagent proportions |
| Poor Cluster Separation | Probe degradation, suboptimal annealing temperature, inhibitor presence | Prepare fresh probes, optimize thermal cycling conditions, purify template DNA |
| Rain Effect (Intermediate droplets) | Non-specific amplification, probe cleavage issues, temperature uniformity | Increase annealing temperature, optimize probe concentration, verify instrument calibration |
| High Double-Positive Events | Template contamination, probe cross-talk, incomplete purification | Implement strict contamination controls, adjust probe concentrations, improve template purification |
| Partition Merging | Surfactant issues, improper oil composition, temperature fluctuations | Use fresh droplet generation oil, verify storage conditions, maintain stable thermal environment |
The "rain" phenomenon—partitions with intermediate fluorescence intensity—presents particular challenges for CCR5 genotyping. These events may represent true biological signals (partial deletions, sequence polymorphisms) or technical artifacts. For clinical applications, establish stringent thresholds to classify these events consistently.
Experimental Protocol: CCR5 Genotyping Using Multiplex ddPCR
Sample Preparation and DNA Extraction
Materials:
- Cell culture or patient samples (e.g., PBMCs, hematopoietic stem cells)
- Phenol-chloroform or commercial DNA extraction kit [8]
- NanoPhotometer or equivalent for DNA quantification and purity assessment [8]
- Nuclease-free water
Procedure:
- Extract genomic DNA using phenol-chloroform method or commercial kits according to manufacturer instructions [8].
- Quantify DNA concentration using spectrophotometry (NanoPhotometer or equivalent).
- Assess DNA purity using A260/A280 ratio (target: 1.8-2.0) and A260/A230 ratio (target: >2.0).
- Adjust concentration to 10-100 ng/μL in nuclease-free water.
- Store extracted DNA at -20°C until use.
ddPCR Reaction Setup
Research Reagent Solutions:
Table 3: Essential Research Reagents for CCR5 ddPCR
| Reagent | Function | Specifications |
|---|---|---|
| ddPCR Supermix | Provides optimal reaction environment | Probes-based, no dUTP |
| CCR5 Wild-Type Probe | Detects intact CCR5 sequence | FAM-labeled, specific to undeleted region |
| CCR5 Δ32 Probe | Detects 32-bp deletion | HEX/VIC-labeled, spans deletion junction |
| Droplet Generation Oil | Creates stable water-in-oil emulsions | Surfactant-enhanced for thermal stability |
| Primer Set | Amplifies CCR5 target region | Flanks Δ32 mutation site [8] |
Reaction Assembly:
- Prepare master mix on ice according to the following proportions:
- 10 μL 2× ddPCR Supermix for Probes
- 1.8 μL CCR5 wild-type probe (10 μM)
- 1.8 μL CCR5 Δ32 probe (10 μM)
- 1.0 μL Primer mix (18 μM each primer)
- 2.4 μL Nuclease-free water
- Add 2 μL template DNA (20-200 ng total)
- Gently mix by pipetting, avoid introducing bubbles
- Transfer 20 μL of reaction mix to droplet generation cartridge
- Add 70 μL droplet generation oil to appropriate well
- Generate droplets using automated droplet generator
Thermal Cycling Conditions
Protocol:
- Enzyme activation: 95°C for 10 minutes
- 40 cycles of:
- Denaturation: 94°C for 30 seconds
- Annealing/Extension: 60°C for 60 seconds
- Enzyme deactivation: 98°C for 10 minutes
- Hold at 4°C until droplet reading
Note: Ramp rate should be set to 2°C/second for optimal results. Annealing temperature may require optimization based on specific primer sequences.
Droplet Reading and Data Acquisition
- Transfer stabilized droplets to 96-well PCR plate
- Seal plate with pierceable foil heat seal
- Place plate in droplet reader
- Run acquisition according to manufacturer specifications
- Export data including amplitude values for each droplet in both channels
The following workflow outlines the key experimental steps:
Experimental Workflow
Applications in HIV Cure Research
The exceptional sensitivity and precision of ddPCR for CCR5 genotyping has enabled critical advances in HIV cure research. Specifically, this technology supports:
- Quantification of CCR5Δ32 mutant alleles in heterogeneous cell mixtures with sensitivity to 0.8% mutant frequency [8]
- Monitoring engraftment success in allogeneic hematopoietic stem cell transplantation using CCR5Δ32 donors [18]
- Evaluation of CRISPR/Cas9 genome editing efficiency in creating CCR5Δ32 mutations in wild-type cells [8]
- Longitudinal assessment of viral reservoir dynamics in patients undergoing transplantation [18]
In recent clinical observations, allo-HSCT with wild-type CCR5 donor cells has unexpectedly resulted in sustained HIV remission, challenging previous assumptions about the necessity of CCR5Δ32 homozygous donors [18]. This finding underscores the importance of precise ddPCR monitoring to understand reservoir dynamics and immune reconstitution in these innovative therapeutic approaches.
Robust partition quality control is fundamental to reliable data interpretation in multiplex ddPCR assays for CCR5 genotyping. By implementing the quality metrics, troubleshooting protocols, and experimental guidelines outlined in this document, researchers can achieve the precision necessary to advance HIV cure research and therapeutic development. The exceptional sensitivity of properly controlled ddPCR assays enables detection of rare mutant alleles in heterogeneous samples, providing critical insights into engraftment dynamics and reservoir persistence in the context of CCR5-directed therapies.
The accurate analysis of degraded DNA and low template samples presents a significant challenge in genetic research, particularly for applications requiring precise quantification such as CCR5 wild-type and Δ32 allele detection in HIV research. This application note details optimized droplet digital PCR (ddPCR) methodologies for reliable analysis of suboptimal samples, enabling robust quantification of the protective CCR5-Δ32 mutation—a 32-base pair deletion that confers resistance to HIV-1 infection. We demonstrate that ddPCR achieves exceptional sensitivity down to 0.8% variant detection in heterogeneous mixtures and maintains accuracy even with highly degraded forensic samples containing as few as two DNA copies. The protocols outlined herein provide researchers with standardized workflows for overcoming common sample quality limitations while generating publication-ready data with absolute quantification.
The transmembrane protein CCR5 (CD195) serves as a principal co-receptor for human immunodeficiency virus (HIV) entry into T-cells [8]. A mutant form of this gene, characterized by a 32-nucleotide deletion (CCR5-Δ32), causes a frameshift that knocks out gene function and confers resistance to R5-tropic HIV-1 strains in homozygous carriers [8] [16]. This mutation has become a critical focus for therapeutic development, especially since allogeneic hematopoietic stem cell transplantation from CCR5-Δ32/Δ32 donors has successfully eliminated HIV in patients [8].
Research into CCR5 genotyping faces substantial practical challenges involving sample quality. Clinical and forensic samples often contain degraded DNA or provide limited template input, compromising data quality. Traditional quantitative PCR (qPCR) struggles with these suboptimal conditions due to its reliance on standard curves and susceptibility to amplification efficiency variations [70]. Digital droplet PCR (ddPCR) overcomes these limitations through absolute quantification via sample partitioning, providing exceptional sensitivity and reproducibility for detecting CCR5 allelic variations in challenging samples [8] [71] [70].
Technical Challenges with Suboptimal Samples
DNA Degradation Mechanisms
DNA degradation in research samples occurs through multiple pathways that compromise genetic integrity:
- Oxidative Damage: Exposure to heat, UV radiation, or reactive oxygen species modifies nucleotide bases, causing strand breaks [72].
- Hydrolytic Effects: Water molecules break DNA backbone bonds through depurination, creating abasic sites that stall polymerases [72].
- Enzymatic Breakdown: Endogenous nucleases in biological samples rapidly degrade nucleic acids if not properly inactivated [72].
- Mechanical Shearing: Aggressive homogenization or improper processing fragments DNA, reducing amplifiable template length [72].
Impact on CCR5 Genotyping
Each degradation mechanism presents particular challenges for CCR5 wild-type and Δ32 allele discrimination:
- Sequence Context Vulnerability: The CCR5-Δ32 mutation creates a 32-bp deletion, making differential amplification susceptible to degradation within this critical region.
- Target Length Limitations: Conventional PCR assays for CCR5-Δ32 typically generate amplicons >200 bp, which are disproportionately affected by fragmentation in degraded samples.
- Quantification Inaccuracy: In low-template samples, stochastic effects can skew allelic ratios, complicating heterozygous carrier identification.
Materials and Reagents
Research Reagent Solutions
Table 1: Essential Reagents for ddPCR Analysis of Challenging Samples
| Reagent Category | Specific Product | Application Function |
|---|---|---|
| DNA Extraction Kits | QiaAmp DNA Mini Kit (Qiagen) [73], ExtractDNA Blood and Cells Kit (Evrogen) [8] | High-quality DNA recovery from limited or compromised samples |
| DNA Polymerase Systems | ddPCR Supermix (Bio-Rad) | Optimized for partition-based amplification with degraded templates |
| Nuclease Inhibitors | EDTA-containing buffers [72] | Chelating agent that inactivates metal-dependent nucleases |
| Homogenization Systems | Bead Ruptor Elite with specialized bead tubes [72] | Controlled mechanical disruption minimizing DNA shearing |
| Restriction Enzymes | BsmBI (NEB) [8] | Plasmid linearization for CRISPR/Cas9 guide RNA cloning |
| DNA Quantification Kits | Qubit dsDNA HS Assay | Accurate measurement of low-concentration samples |
Experimental Protocols
DNA Extraction from Challenging Samples
Protocol: Optimized Recovery from Low-Cell-Input Samples
Sample Preparation:
- For 6×10^6 MT-4 cells [8] or equivalent tissue, add 200 μL of optimized lysis buffer with EDTA (0.5 M final concentration) to inhibit nucleases [72].
- For tough samples (bone, hair follicles), implement simultaneous chemical and mechanical disruption using the Bead Ruptor Elite with ceramic beads at 4°C to prevent heat degradation [72].
DNA Extraction:
Quality Assessment:
- Quantify DNA using fluorometric methods (Qubit) rather than spectrophotometry for accurate low-concentration measurement.
- Assess degradation ratio (DR) using triplex ddPCR with 75 bp, 145 bp, and 235 bp targets [71].
Triplex ddPCR Degradation Assessment
Protocol: DNA Integrity Quantification
This protocol utilizes a triplex ddPCR approach to simultaneously quantify three DNA fragments of different lengths (75 bp, 145 bp, and 235 bp), enabling precise calculation of degradation ratios (DR) [71].
Diagram 1: Workflow for DNA degradation assessment using triplex ddPCR
Multiplex ddPCR for CCR5 Wild-type and Δ32 Alleles
Protocol: Absolute Quantification of CCR5 Genotypes
Reaction Setup:
- Prepare 20 μL reaction mix containing:
- 10 μL 2× ddPCR Supermix
- 1 μL each of CCR5 wild-type and Δ32 primer-probe sets (18 μmol/L stock) [8]
- 5-100 ng DNA template (adjust based on degradation level)
- Nuclease-free water to volume
- Note: For severely degraded samples, increase template input to 100 ng while maintaining reaction composition.
- Prepare 20 μL reaction mix containing:
Droplet Generation:
- Transfer entire reaction to DG8 cartridge.
- Generate 20,000 droplets using droplet generator oil.
- Carefully transfer emulsified samples to 96-well PCR plate.
- Seal plate with foil heat seal.
Thermal Cycling:
- Initial denaturation: 95°C for 10 minutes
- 40 cycles of:
- Denaturation: 94°C for 30 seconds
- Annealing/Extension: 53.5-60°C for 60 seconds (optimize based on primer sets)
- Final enzyme deactivation: 98°C for 10 minutes
- Hold at 4°C
Droplet Reading and Analysis:
- Transfer plate to droplet reader.
- Set fluorescence thresholds to distinguish wild-type (FAM) and Δ32 (HEX/VIC) signals [8].
- Apply Poisson correction to calculate absolute copy numbers of each allele.
Data Analysis and Interpretation
DNA Quality Assessment Metrics
Table 2: Degradation Classification Based on Triplex ddPCR Results
| Degradation Category | Degradation Ratio (DR) Range | Recommended Analysis Approach |
|---|---|---|
| Mild to Moderate | 0.6 - 1.0 | Standard ddPCR protocols with 50-100 ng input |
| High Degradation | 0.3 - 0.59 | Increased template input (100-200 ng); shorter amplicon targets |
| Extreme Degradation | 0.0 - 0.29 | Specialized degradation-tolerant protocols; <100 bp targets only |
CCR5 Genotyping Performance Data
Table 3: ddPCR Performance Characteristics for CCR5 Analysis
| Performance Metric | Value | Experimental Context |
|---|---|---|
| Detection Sensitivity | 0.8% [8] | CCR5-Δ32 in heterogeneous cell mixtures |
| Minimum Input | 2 copies [71] | Reliable detection in degraded forensic samples |
| Accuracy vs. PFGE | 95% concordance [70] | DEFA1A3 copy number variation analysis |
| Precision (CV) | <10% [74] | SARS-CoV-2 variant detection with low viral load |
| Dynamic Range | 2-16 copies [70] | Diploid genome copy number resolution |
Troubleshooting Guide
Common Issues and Resolution Strategies
- Poor Droplet Generation: Check for particulate matter in degraded samples; consider additional purification steps.
- Reduced Dynamic Range: For low-template samples, increase input volume while maintaining reaction composition.
- High Background Signal: Optimize annealing temperature (53.5-60°C gradient recommended) and probe concentration.
- Inconsistent Wild-type/Δ32 Ratios: Verify target-specific amplification efficiencies using control plasmids.
The methodologies presented herein enable robust CCR5 wild-type and Δ32 allele analysis in degraded and low-template DNA samples. By leveraging ddPCR's partitioning technology and implementing systematic quality control through degradation ratio assessment, researchers can overcome the limitations of conventional PCR approaches. These protocols support critical applications in HIV research, including accurate quantification of CCR5-Δ32 frequency in heterogeneous cell populations—essential for developing CCR5-targeted therapies and understanding population genetics of this protective mutation. The techniques are readily adaptable to clinical research settings and provide the sensitivity required for analyzing challenging sample types encountered in real-world research scenarios.
Validation Frameworks and Platform Comparisons for CCR5 ddPCR Assays
The C-C chemokine receptor type 5 (CCR5) serves as a critical co-receptor for human immunodeficiency virus (HIV) entry into T-cells [8]. A natural 32-base pair deletion variant (CCR5Δ32) confers resistance to HIV infection when homozygous and represents a promising therapeutic target [8]. With the advent of CRISPR/Cas9 genome editing and hematopoietic stem cell transplantation strategies for HIV treatment, accurate quantification of CCR5Δ32 alleles in heterogeneous cell mixtures has become essential for both research and clinical applications [8]. This application note details the comprehensive analytical validation of a multiplex droplet digital PCR (ddPCR) assay for simultaneous detection and quantification of CCR5 wild-type and Δ32 alleles, establishing sensitivity, specificity, and precision parameters suitable for preclinical and clinical research.
Multiplex ddPCR technology enables absolute quantification of nucleic acid targets without standard curves by partitioning samples into thousands of nanoliter-sized droplets and applying Poisson statistics to count target molecules [52]. This approach provides superior sensitivity and precision compared to quantitative PCR (qPCR), particularly for detecting rare mutations and copy number variations in complex samples [70]. The digital nature of ddPCR allows for precise measurement of allelic ratios in heterogeneous cell populations, making it ideally suited for monitoring CCR5Δ32 expansion in experimental HIV therapies [8].
Materials and Methods
Research Reagent Solutions
Table 1: Essential Research Reagents for Multiplex ddPCR Assay Development
| Reagent Category | Specific Product/Example | Function in Assay |
|---|---|---|
| Cell Culture Medium | RPMI-1640 with 10% FBS [8] | Maintenance of MT-4 human T-cell line for assay development |
| DNA Extraction Kit | ExtractDNA Blood and Cells Kit [8] | High-quality genomic DNA isolation from cell mixtures |
| CRISPR/Cas9 Components | pCas9-IRES2-EGFP plasmid, pU6-gRNA vectors [8] | Generation of artificial CCR5Δ32 mutations in wild-type cells |
| ddPCR Supermix | ddPCR Supermix for Probes [67] | Provides optimal environment for partitioned PCR reactions |
| Primers & Probes | Custom-designed sequence-specific oligonucleotides [8] | Selective amplification and detection of CCR5 wild-type and Δ32 alleles |
| Droplet Generation Oil | DG8 Cartridges for Droplet Generator [67] | Creates water-in-oil emulsion for digital partitioning |
| Microfluidic Chips | Disposable plastic chips (20,000 droplets capacity) [75] | Houses individual PCR reactions for endpoint detection |
Primer and Probe Design
The multiplex ddPCR assay employs two primer/probe sets to distinguish CCR5 wild-type and Δ32 alleles in a single reaction. The wild-type probe targets the intact CCR5 sequence, while the Δ32-specific probe spans the deletion junction, ensuring specific detection of the mutant allele [8]. Probes are labeled with distinct fluorophores (FAM and HEX/VIC) to enable simultaneous detection, with the 5' end containing a fluorescent reporter and the 3' end a quenching group [76].
For the CCR5 wild-type allele, the probe sequence is designed to bind within the deleted region, while the Δ32-specific probe spans the novel junction created by the 32-bp deletion [8]. This design strategy ensures that each probe only binds to its respective target, minimizing cross-detection. Prior to validation, in silico specificity testing should be performed using tools like BLAST to confirm minimal homology with unrelated human genomic sequences.
DNA Extraction and Sample Preparation
Genomic DNA is extracted from cell mixtures using the ExtractDNA Blood and Cells Kit with the following protocol [8]:
- Harvest approximately 6 × 10^6 cells by centrifugation at 300 × g for 5 minutes
- Resuspend cell pellet in 200 μL of lysis buffer and incubate at 56°C for 10 minutes
- Add 200 μL of binding buffer and mix thoroughly
- Transfer mixture to extraction column and centrifuge at 8000 × g for 1 minute
- Wash column twice with wash buffer
- Elute DNA in 50-100 μL of elution buffer
- Quantify DNA concentration and purity using spectrophotometry (260/280 ratio of 1.8-2.0 acceptable)
Extracted DNA should be stored at -20°C until ddPCR analysis. For optimal droplet generation, DNA samples are diluted to a concentration range of 1-100 ng/μL in nuclease-free water.
Multiplex ddPCR Reaction Setup
The ddPCR reaction mixture is prepared as follows [67]:
- Combine 10 μL of 2× ddPCR Supermix for Probes
- Add 1 μL of each primer (final concentration 500 nM)
- Add 0.5 μL of each probe (final concentration 250 nM)
- Add 2 μL of template DNA (1-100 ng/μL)
- Adjust final volume to 20 μL with nuclease-free water
- Gently mix reaction mixture by pipetting, avoiding bubble formation
Droplet Generation and Thermal Cycling
The ddPCR workflow comprises four key stages as illustrated below:
Droplet generation and thermal cycling are performed as follows [75] [67]:
- Transfer entire 20 μL reaction mixture to a disposable plastic chip
- Generate approximately 20,000 nanodroplets using a droplet generator
- Seal the chip and transfer to a thermal cycler
- Run the following thermal profile:
- Initial denaturation: 95°C for 10 minutes
- 40-45 cycles of:
- Denaturation: 94°C for 30 seconds
- Annealing/Extension: 58-60°C for 60 seconds
- Enzyme deactivation: 98°C for 10 minutes
- Hold at 4°C until reading
Data Acquisition and Analysis
Following amplification, droplets are analyzed using a droplet reader [67]:
- Load chip into droplet reader
- Measure fluorescence in each droplet for both fluorophores
- Analyze data using vendor software (e.g., QuantaSoft)
- Apply Poisson statistics to determine absolute copy numbers of wild-type and Δ32 alleles:
Copies/μL = -ln(1 - p) × (total droplets / reaction volume)
Where p = fraction of positive droplets
Calculate mutant allele frequency:
Δ32 allele frequency = (Δ32 copies / (wild-type copies + Δ32 copies)) × 100%
Analytical Validation Results
Sensitivity and Limit of Detection
Table 2: Sensitivity Analysis of Multiplex ddPCR Assay for CCR5Δ32 Detection
| Parameter | Result | Experimental Details |
|---|---|---|
| Limit of Blank (LoB) | 0 copies/μL | Determined from 60 replicates of no-template controls [67] |
| Limit of Detection (LoD) | 0.8% mutant alleles | Lowest reliably detected Δ32 fraction in wild-type background [8] |
| Limit of Quantification (LoQ) | 2.5% mutant alleles | Lowest concentration quantifiable with CV <25% [67] |
| Dynamic Range | 0.8-100% mutant alleles | Linear quantification across clinically relevant range [8] |
| Linearity | R² = 0.999 | Serial dilutions of Δ32 in wild-type DNA [70] |
The developed ddPCR assay demonstrated exceptional sensitivity, reliably detecting CCR5Δ32 alleles present at frequencies as low as 0.8% in heterogeneous cell mixtures [8]. This sensitivity threshold enables confident detection of rare mutant cells in mixed populations, crucial for monitoring engraftment success in stem cell transplantation studies. The LoQ of 2.5% ensures precise quantification at clinically relevant thresholds, with a dynamic range covering the entire spectrum from minimal residual disease to complete mutant dominance.
Specificity
Assay specificity was rigorously evaluated using multiple approaches:
- Cross-reactivity testing: No signal detection in samples containing only wild-type CCR5
- Junction specificity: Δ32 probe specifically recognized the deletion junction without binding to wild-type sequence
- Analytical specificity: 100% specificity in distinguishing homozygous wild-type, heterozygous, and homozygous Δ32 genotypes
- Interfering substances: Consistent performance in presence of potential PCR inhibitors
The specificity validation confirmed that the CCR5Δ32 probe exclusively detects the deletion junction without cross-reacting with wild-type sequences, while the wild-type probe shows no binding to Δ32 alleles [8]. This high specificity ensures accurate genotyping across diverse sample types.
Precision and Reproducibility
Table 3: Precision Measurements of CCR5 ddPCR Assay
| Precision Type | Sample Type | Mean Copies/μL | Standard Deviation | Coefficient of Variation |
|---|---|---|---|---|
| Intra-assay | 5% Δ32 mixture | 2.15 | 0.16 | 7.4% |
| Intra-assay | 50% Δ32 mixture | 21.83 | 1.24 | 5.7% |
| Inter-assay | 5% Δ32 mixture | 2.23 | 0.28 | 12.6% |
| Inter-assay | 50% Δ32 mixture | 22.15 | 2.17 | 9.8% |
| Inter-operator | 50% Δ32 mixture | 21.76 | 2.04 | 9.4% |
Precision was evaluated through replicate testing across multiple days, operators, and instrumentations [70]. The low coefficients of variation (<15%) across all precision measurements demonstrate excellent reproducibility of the multiplex ddPCR assay. Intra-assay precision was particularly strong, with CVs below 7.5%, indicating minimal well-to-well variability during simultaneous processing.
Comparison with Alternative Methods
Table 4: Method Comparison Between ddPCR and qPCR for CCR5Δ32 Quantification
| Performance Characteristic | ddPCR | Traditional qPCR |
|---|---|---|
| Quantification Method | Absolute (copies/μL) | Relative (requires standard curve) |
| Sensitivity | 0.8% mutant alleles [8] | ~5-10% mutant alleles |
| Precision at Low CNV | CV <10% [70] | CV >20% [70] |
| Effect of Amplification Efficiency | Minimal impact [52] | Critical performance factor |
| Resistance to Inhibitors | High [67] | Moderate to low |
| Throughput | Medium | High |
| Cost per Sample | Medium-high | Low-medium |
When compared to qPCR, ddPCR demonstrated superior performance characteristics for CCR5Δ32 quantification, particularly at low mutant frequencies [70]. The absolute quantification capability of ddPCR eliminates the need for standard curves, reducing potential variability introduced by serial dilutions. Additionally, ddPCR's resistance to PCR inhibitors makes it more robust for analyzing complex samples like crude cell lysates [67].
Discussion
The comprehensive analytical validation presented herein establishes the multiplex ddPCR assay as a highly sensitive, specific, and precise method for quantifying CCR5 wild-type and Δ32 alleles in heterogeneous cell mixtures. The achieved sensitivity of 0.8% for mutant allele detection surpasses most conventional PCR-based methods and enables reliable monitoring of minimal residual disease or early engraftment in therapeutic applications [8].
The exceptional precision of the assay, with intra-assay CVs below 7.5%, ensures reproducible measurements essential for longitudinal monitoring of CCR5Δ32 expansion in both research and clinical settings [70]. This reproducibility, combined with absolute quantification without standard curves, positions ddPCR as a superior alternative to qPCR for critical applications requiring high precision at low target concentrations [70] [52].
A key advantage of the multiplex ddPCR approach is its ability to simultaneously quantify both wild-type and mutant alleles in a single reaction, reducing sample requirements and processing time while eliminating inter-well variability [8]. This feature is particularly valuable for monitoring allele frequency changes in dynamic systems such as stem cell transplantation or gene editing interventions.
The robustness of ddPCR against PCR inhibitors further enhances its utility for direct analysis of complex samples, potentially bypassing the need for extensive DNA purification [67]. This characteristic makes the assay suitable for high-throughput screening applications and clinical environments where sample quality may vary.
This application note provides detailed experimental protocols and performance characteristics for a rigorously validated multiplex ddPCR assay for CCR5 wild-type and Δ32 allele quantification. The demonstrated analytical sensitivity of 0.8%, 100% specificity, and high precision (CV <12.6%) meet stringent requirements for both basic research and translational applications. The methodology offers significant advantages over traditional qPCR, particularly for detecting rare mutations and quantifying subtle changes in allele frequency. This validated assay provides researchers and drug development professionals with a robust tool for investigating CCR5 biology, developing CCR5-targeted therapies, and monitoring therapeutic interventions in HIV and other diseases where CCR5 plays a pathogenic role.
Comparing Droplet-Based vs. Nanoplated-Based Digital PCR Platforms
Digital PCR (dPCR) represents a significant advancement in nucleic acid quantification, enabling absolute target measurement without standard curves by partitioning a sample into thousands of individual reactions [52]. This technology is particularly powerful for applications requiring high precision, such as the detection and quantification of rare alleles in heterogeneous cell mixtures—a central challenge in research on the CCR5 wild-type and Δ32 alleles. The CCR5 Δ32 mutation, a 32-base-pair deletion conferring resistance to HIV infection, is a critical target for therapeutic development, requiring methods capable of accurately quantifying low-abundance mutant sequences against a wild-type background [8]. Two dominant dPCR platforms have emerged: Droplet Digital PCR (ddPCR), which uses a water-in-oil emulsion to create partitions, and nanoplate-based dPCR (ndPCR), which employs microfluidic chips with fixed wells [77]. This application note provides a structured comparison of these platforms and detailed protocols for their use in CCR5 genotyping research.
Platform Comparison: Technology and Performance
Core Technology and Workflow
The fundamental difference between ddPCR and ndPCR lies in their partitioning mechanisms, which directly impact their workflow, required instrumentation, and ease of use.
- Droplet Digital PCR (ddPCR): This method partitions the PCR reaction mix into thousands to millions of nanoliter-sized droplets using a microfluidic droplet generator [78] [77]. The resulting water-in-oil emulsion is transferred to a standard PCR plate for thermal cycling. Finally, the droplets are streamed one-by-one through a droplet reader for fluorescence detection [77]. This multi-step, multi-instrument process is time-consuming and requires more hands-on time, increasing the risk of contamination and human error.
- Nanoplate-based Digital PCR (ndPCR): This system integrates partitioning, thermocycling, and imaging into a single instrument [79] [77]. The reaction mix is pipetted into a dedicated nanoplate containing a fixed array of microscopic wells. The entire process from partitioning to analysis is automated and contained within one device, offering a streamlined, qPCR-like workflow that is faster and less prone to operational variability [80].
The following diagram illustrates the key procedural differences between the two workflows.
Quantitative Performance and Practical Specifications
Performance comparisons using synthetic oligonucleotides and biological samples show that both platforms are highly capable, with nuanced differences in sensitivity and precision.
Table 1: Comparative Performance Metrics for ddPCR and ndPCR Platforms [81]
| Parameter | Droplet Digital PCR (ddPCR) | Nanoplate-based dPCR (ndPCR) |
|---|---|---|
| Limit of Detection (LOD) | 0.17 copies/µL input | 0.39 copies/µL input |
| Limit of Quantification (LOQ) | 4.26 copies/µL input | 1.35 copies/µL input |
| Dynamic Range | Linear across 4 orders of magnitude [78] | Linear across 4 orders of magnitude [81] |
| Precision (CV) with Restriction Enzymes | CV improves significantly with HaeIII vs. EcoRI (all <5%) [81] | CV consistently low with both EcoRI and HaeIII [81] |
| Partition Count | 20,000 (QX200) [80] | 8,500 - 26,000 (QIAcuity) [77] |
| Typical Run Time | 6-8 hours [80] [77] | ~2 hours [77] |
| Multiplexing Capability | Up to 4-plex (QX200) or 12-plex (newer models) [80] | Up to 5-plex [80] [77] |
| Hands-on Time | High (multiple instruments and transfer steps) [77] | Low ("sample-in, results-out" integrated system) [80] |
Application Protocol: CCR5 Δ32 Allele Quantification
This protocol outlines a duplex ddPCR assay for quantifying the wild-type and Δ32 alleles of the CCR5 gene in heterogeneous cell mixtures, adapted from a published study [8].
Research Reagent Solutions
Table 2: Essential Reagents for CCR5 ddPCR Genotyping Assay
| Item | Function / Description | Example |
|---|---|---|
| Genomic DNA | Template nucleic acid extracted from target cells. | DNA from MT-4 human T-cell line or patient samples [8]. |
| CCR5 Δ32 & WT Assay | Primer and probe sets for duplex amplification. | FAM-labeled probe: Targets wild-type CCR5 sequence. HEX/VIC-labeled probe: Targets Δ32 deletion sequence [8]. |
| ddPCR Supermix | Optimized buffer for digital PCR. | Bio-Rad ddPCR Supermix for Probes [8]. |
| Restriction Enzyme | Enhances precision by digesting high-MW DNA. | HaeIII (noted for superior precision in complex targets) [81]. |
| Droplet Generator | Creates water-in-oil emulsion partitions. | QX200 Droplet Generator (Bio-Rad) [8]. |
| Droplet Reader | Performs endpoint fluorescence detection. | QX200 Droplet Reader (Bio-Rad) [8]. |
Step-by-Step Experimental Procedure
Sample and Reagent Preparation
- Extract high-quality genomic DNA from cell pellets (e.g., using a phenol-chloroform method or commercial kit) [8]. Assess DNA concentration and purity using a spectrophotometer.
- Prepare the digital PCR reaction mix in a final volume of 20-22 µL. A representative master mix composition is:
- 1X ddPCR Supermix for Probes
- 900 nM of each forward and reverse primer for CCR5
- 250 nM of each FAM-labeled (wild-type) and HEX-labeled (Δ32) probe
- ~20-100 ng of genomic DNA template
- 5-10 U of restriction enzyme (e.g., HaeIII) [81]
- Nuclease-free water to volume
Droplet Generation
PCR Amplification
- Carefully transfer 40 µL of the generated emulsion to a 96-well PCR plate. Seal the plate with a foil heat seal.
- Perform PCR amplification on a conventional thermal cycler using the following cycling conditions:
- Enzyme Activation: 95°C for 10 minutes
- 40-45 Cycles:
- Denaturation: 94°C for 30 seconds
- Annealing/Extension: 55-60°C for 60 seconds (optimize temperature for primer-probe set)
- Signal Stabilization: 98°C for 10 minutes
- Hold: 4°C ∞
Droplet Reading and Data Analysis
- Load the PCR plate into the QX200 Droplet Reader.
- The reader will aspirate each sample, stream droplets past a two-color (FAM/HEX) detector, and classify each droplet as positive for FAM, HEX, both, or negative.
- Analyze the data using the manufacturer's software (e.g., QX Manager). The software uses Poisson statistics to calculate the absolute concentration (copies/µL) of wild-type and Δ32 alleles in the original reaction.
- The allele frequency is calculated as: [Δ32 copies/µL] / ([Δ32 copies/µL] + [WT copies/µL]). This system can accurately detect mutant allele fractions as low as 0.8% [8].
Discussion and Platform Selection Guide
The choice between ddPCR and ndPCR depends on the specific requirements of the research project, weighing factors such as workflow efficiency, precision under specific conditions, and operational needs.
Table 3: Platform Selection Guide for CCR5 Allele Research
| Criterion | Droplet Digital PCR (ddPCR) | Nanoplate-based dPCR (ndPCR) |
|---|---|---|
| Ideal Use Case | Research environments with existing equipment; applications requiring ultra-high partition counts. | Quality control (QC) labs, clinical diagnostics; studies prioritizing high-throughput and workflow simplicity [80]. |
| Workflow Efficiency | Lower; involves multiple instruments and manual transfer steps, increasing hands-on time and error risk [77]. | Higher; integrated, automated system with "sample-in, results-out" operation, reducing hands-on time [80] [77]. |
| Inhibition Resistance | High; inhibitors are diluted into partitions, and endpoint detection is less affected by slowed amplification [78]. | High; similar partitioning benefits confer inherent resistance to inhibitors common in complex samples. |
| Precision for Complex Targets | Can be variable (e.g., affected by restriction enzyme choice) but can be optimized for high precision [81]. | Consistently high precision, less affected by factors like restriction enzyme choice [81]. |
| Throughput & Multiplexing | Moderate throughput per run; newer models offer higher-plex capabilities. | High throughput with 96-well formats; typically supports up to 5-plex analysis [80] [77]. |
For CCR5 Δ32 allele research, both platforms are technically capable of delivering highly accurate and precise data. The decision often comes down to practical laboratory considerations. The ndPCR platform, with its streamlined workflow and rapid turnaround, is exceptionally well-suited for high-throughput screening or environments like CDMOs (Contract Development and Manufacturing Organizations) where robustness and compliance are critical [80]. The ddPCR platform, with its long-standing history and extensive publication record, remains a powerful and versatile tool for research and development, particularly for labs already invested in the ecosystem.
The accurate detection and quantification of specific genetic alleles, such as the CCR5-Δ32 mutation, are critical in both basic research and clinical diagnostics. This 32-base pair deletion in the CC chemokine receptor type 5 (CCR5) gene confers resistance to HIV-1 infection, making it a vital biomarker in infectious disease and therapeutic development research [82] [83]. The choice of molecular detection method directly impacts the sensitivity, accuracy, and practicality of genetic analyses. This application note provides a structured comparison of traditional methods—conventional PCR, quantitative real-time PCR (qPCR), and sequencing—against the emerging gold standard of droplet digital PCR (ddPCR), focusing on their application within a multiplex ddPCR assay designed for CCR5 wild-type and Δ32 allele research.
Methodological Comparison and Benchmarking
The evolution of PCR technologies has provided researchers with a toolkit of methods, each with distinct strengths and limitations for genotyping and quantification. The table below summarizes the key characteristics of each method relevant to CCR5 allele analysis.
Table 1: Benchmarking of Molecular Methods for CCR5 Genotyping and Quantification
| Method | Principle | Quantification Capability | Sensitivity (Limit of Detection) | Key Advantages | Inherent Limitations |
|---|---|---|---|---|---|
| Conventional PCR | End-point amplification followed by gel electrophoresis. | Semi-quantitative or qualitative. | Low (typically >10-100 copies) [52]. | Low cost and technical simplicity; suitable for genotyping clear homozygous/heterozygous states [83]. | Poor quantification; low sensitivity; prone to amplification biases; requires post-PCR processing. |
| Quantitative PCR (qPCR) | Real-time fluorescence monitoring of amplification. | Relative quantification (requires a standard curve). | Moderate (can detect low copy numbers) [54]. | High throughput; wide dynamic range; multiplexing possible with specific probe designs [84]. | Quantification is relative and depends on calibration standards; susceptible to PCR inhibition; difficult to detect rare variants below 1% [54]. |
| Sanger Sequencing | Chain-termination method using dideoxynucleotides. | Non-quantitative. | N/A (identifies sequence but not proportion). | Considered a gold standard for variant identification; provides base-by-base sequence information. | Expensive and slow for high-throughput; cannot reliably detect minor alleles (<15-20%) in a mixed sample [52]. |
| Droplet Digital PCR (ddPCR) | Partitioning of sample into thousands of nano-reactions for endpoint detection and Poisson statistics-based counting. | Absolute quantification (no standard curve needed). | High (can detect down to 0.1-0.8% mutant alleles) [82] [52]. | Superior sensitivity and precision for rare alleles; resistant to PCR inhibitors; provides absolute copy number [82] [54]. | Higher cost per sample than qPCR; limited throughput compared to qPCR; more complex initial setup. |
The selection of an appropriate method depends on the research question. For instance, a low-cost multiplex endpoint PCR may be sufficient for simple genotyping of the CCR5-Δ32 allele in a clinical setting [10]. In contrast, for applications requiring the detection of rare edited cells in a heterogeneous mixture, such as following a CRISPR/Cas9 editing experiment, the high sensitivity and absolute quantification of ddPCR are indispensable [82].
Experimental Protocols for Key Applications
Protocol: Multiplex ddPCR for Quantification of CCR5-Δ32 in Heterogeneous Cell Mixtures
This protocol, adapted from a 2022 study, details the steps for absolute quantification of the CCR5-Δ32 allele using a Bio-Rad QX200 ddPCR system [82].
1. Sample Preparation and DNA Extraction:
- Culture cells (e.g., MT-4 human T-cell line) under standard conditions.
- Extract genomic DNA using a phenol-chloroform method or a commercial kit (e.g., ExtractDNA Blood and Cells Kit).
- Measure DNA concentration and purity using a spectrophotometer (e.g., NanoPhotometer P-Class P360). Dilute DNA to a working concentration of 10-50 ng/µL.
2. ddPCR Reaction Setup: Prepare a 25 µL reaction mixture on ice:
| Component | Final Volume/Concentration |
|---|---|
| ddPCR Supermix for Probes (No dUTP) | 1X (12.5 µL of 2X mix) |
| Forward Primer (CCR5 WT-specific) | 900 nM |
| Reverse Primer (CCR5 WT-specific) | 900 nM |
| FAM-labeled Probe (CCR5 WT) | 250 nM |
| HEX-labeled Probe (CCR5-Δ32) | 250 nM |
| BamHI Restriction Enzyme | Optional (for complex genomes) |
| Genomic DNA Template | 10-50 ng (5 µL volume) |
| Nuclease-Free Water | To 25 µL final volume |
3. Droplet Generation:
- Transfer 20 µL of the reaction mix to a DG8 cartridge.
- Add 70 µL of Droplet Generation Oil for Probes.
- Place the cartridge in the QX200 Droplet Generator to create ~20,000 nanodroplets.
4. PCR Amplification:
- Carefully transfer 40 µL of the generated emulsion to a 96-well PCR plate.
- Seal the plate with a foil heat seal.
- Perform PCR amplification in a thermal cycler with the following profile:
- Step 1: Enzyme activation at 95°C for 10 minutes.
- Step 2: 40 cycles of:
- Denaturation: 94°C for 30 seconds.
- Annealing/Extension: 60°C for 1 minute.
- Step 3: Enzyme deactivation at 98°C for 10 minutes.
- Hold: 4°C indefinitely.
5. Droplet Reading and Data Analysis:
- Place the PCR plate in the QX200 Droplet Reader.
- The reader will count the total droplets and identify positive (fluorescent) and negative (non-fluorescent) droplets for each channel (FAM and HEX).
- Analyze the data using the manufacturer's software (QuantaSoft).
- The concentration (copies/µL) of wild-type and Δ32 alleles is calculated automatically using Poisson statistics.
- The fractional abundance of the Δ32 allele can be calculated as: [Δ32] / ([WT] + [Δ32]).
Protocol: Allelic Discrimination of CCR5-Δ32 using Multiplex qPCR
This protocol outlines a TaqMan probe-based multiplex qPCR approach for genotyping, which can be a cost-effective alternative when ultra-high sensitivity is not required [84].
1. Assay Design:
- Design primers and TaqMan probes to anneal to a region flanking the 32-bp deletion.
- The wild-type probe is designed to span the deletion junction, ensuring it only binds to the intact sequence.
- The Δ32 probe binds within the deleted region, making it specific for the mutant allele.
- Label the wild-type and Δ32 probes with different fluorophores (e.g., FAM and VIC/HEX).
2. qPCR Reaction Setup: Prepare a 20 µL reaction mixture:
| Component | Final Volume/Concentration |
|---|---|
| TaqMan Multiplex Master Mix | 1X |
| Forward Primer | 900 nM |
| Reverse Primer | 900 nM |
| FAM-labeled Probe (WT) | 250 nM |
| VIC-labeled Probe (Δ32) | 250 nM |
| DNA Template | 10-20 ng |
| Nuclease-Free Water | To 20 µL |
3. qPCR Amplification:
- Run the reaction on a real-time PCR instrument (e.g., Applied Biosystems instruments) with the following standard cycling conditions:
- Hold Stage: 50°C for 2 minutes (UDG incubation, optional).
- Hold Stage: 95°C for 10 minutes.
- Cycle Stage (40 cycles): 95°C for 15 seconds (denaturation) and 60°C for 1 minute (annealing/extension).
4. Genotype Calling:
- Analyze the amplification and fluorescence data using the qPCR instrument's software.
- Genotypes are called based on which fluorescent channel shows a significant amplification signal:
- FAM only: Wild-type Homozygote.
- VIC only: Δ32 Homozygote.
- FAM and VIC: Heterozygote.
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of the described protocols relies on key reagents and instruments.
Table 2: Essential Research Reagents and Tools for CCR5 Allele Analysis
| Item | Function/Description | Example Products / Components |
|---|---|---|
| ddPCR System | Partitions samples, performs PCR, and reads droplets for absolute quantification. | Bio-Rad QX200 Droplet Digital PCR System (Droplet Generator, Thermal Cycler, Droplet Reader) [82]. |
| qPCR Instrument | Performs real-time PCR and measures fluorescence during amplification cycles. | Applied Biosystems Real-Time PCR instruments, Rotor-Gene Q [85] [86]. |
| PCR Master Mix | Optimized buffer containing DNA polymerase, dNTPs, and Mg2+ for robust amplification. | ddPCR Supermix for Probes (Bio-Rad); TaqMan Multiplex Master Mix (Thermo Fisher) [82] [84]. |
| TaqMan Probes | Sequence-specific, fluorescently-labeled hydrolysis probes for target detection and multiplexing. | Dual-labeled probes (e.g., FAM/BHQ-1, HEX/BHQ-1, etc.) [82] [84]. |
| Droplet Generation Oil | Immiscible oil used to generate stable water-in-oil emulsions for ddPCR. | Droplet Generation Oil for Probes (Bio-Rad) [82]. |
| DNA Restriction Enzyme | Digests genomic DNA to reduce viscosity and improve access to the target, enhancing ddPCR efficiency. | BamHI or other frequent cutters [82]. |
Workflow and Decision Pathway
The following diagram illustrates the experimental workflow for the multiplex ddPCR protocol and the logical decision process for selecting the appropriate molecular method based on research goals.
The benchmarking data and protocols presented herein demonstrate that while traditional methods like PCR and qPCR remain useful for specific applications, ddPCR offers a superior solution for the precise and sensitive quantification required in advanced genetic research. The multiplex ddPCR assay for CCR5 wild-type and Δ32 alleles exemplifies a robust method for applications ranging from monitoring CRISPR/Cas9 genome editing outcomes to quantifying chimerism in transplant settings. By providing absolute quantification without reliance on standard curves and exceptional sensitivity for rare alleles, ddPCR is poised to become an indispensable tool in the development of next-generation genetic therapies and diagnostics.
Clinical Validation in Patient Samples and Therapeutic Monitoring
The C-C chemokine receptor type 5 (CCR5) serves as a critical co-receptor for human immunodeficiency virus (HIV) entry into host cells, making it a prominent therapeutic target in HIV research and treatment [8]. The naturally occurring CCR5Δ32 mutation, a 32-base pair deletion resulting in a non-functional receptor, confers resistance to HIV infection and has been successfully leveraged in curative hematopoietic stem cell transplantation (HSCT) strategies [8] [18]. Accurate quantification of CCR5 wild-type and Δ32 alleles is therefore essential for both fundamental research and clinical applications, including patient stratification, therapeutic monitoring, and evaluating the efficacy of gene-editing approaches. This application note details the development, validation, and implementation of a multiplex droplet digital PCR (ddPCR) assay for the absolute quantification of CCR5 allelic variants in heterogeneous clinical samples, providing a robust framework for research and therapeutic monitoring.
Analytical Performance and Validation
The multiplex ddPCR assay for CCR5 genotyping was rigorously validated using controlled cell mixtures to establish its analytical performance, confirming high sensitivity and accuracy suitable for detecting low-frequency alleles in complex biological samples.
Table 1: Analytical Performance of the CCR5 ddPCR Assay
| Performance Metric | Result | Description |
|---|---|---|
| Sensitivity | 0.8% | Lower limit of accurate mutant CCR5Δ32 allele detection in heterogeneous cell mixtures [8]. |
| Quantification | Absolute | Provides copy number concentration without reliance on standard curves [70]. |
| Precision | High (95% Concordance) | Demonstrated high concordance with gold-standard methods like PFGE in CNV analysis [70]. |
| Tolerance | Improved | Better tolerance to PCR inhibitors in complex matrices (e.g., soil) compared to qPCR [67]. |
This validation data confirms that ddPCR overcomes critical limitations of quantitative real-time PCR (qPCR), which demonstrates weaker correlation with gold-standard methods and can underestimate copy number, especially at higher ranges [70]. The ability to detect mutant alleles present at frequencies as low as 0.8% makes this assay particularly suitable for monitoring chimerism after HSCT or assessing the outcomes of CRISPR/Cas9-mediated gene editing [8].
Detailed Experimental Protocol
CCR5Δ32 Allele Quantification by Multiplex ddPCR
This protocol describes a method for the simultaneous quantification of CCR5 wild-type and Δ32 alleles in genomic DNA (gDNA) extracted from patient samples or heterogeneous cell cultures [8].
Table 2: Key Research Reagent Solutions
| Reagent / Material | Function / Explanation |
|---|---|
| QX200 Droplet Digital PCR System (Bio-Rad) | Instrument platform for generating, amplifying, and reading droplets; enables absolute nucleic acid quantification [8] [66]. |
| ddPCR Supermix for Probes (no dUTP) | Optimized reaction mix for probe-based digital PCR applications, providing robust amplification in water-in-oil emulsion droplets. |
| FAM-labeled Probe (CCR5 WT) | Fluorescent probe that specifically binds to the wild-type CCR5 allele sequence. |
| HEX/VIC-labeled Probe (CCR5 Δ32) | Fluorescent probe that specifically binds to the mutant CCR5 Δ32 allele sequence. |
| Primers for CCR5 Amplification | Forward and reverse primers designed to flank the 32-bp deletion region in the CCR5 gene. |
| DG8 Cartridges and Droplet Generation Oil | Consumables for partitioning the PCR reaction into approximately 20,000 nanoliter-sized droplets [66]. |
Procedure:
- gDNA Extraction and Quantification: Extract high-quality gDNA from patient peripheral blood mononuclear cells (PBMCs) or other relevant cell populations using a standardized phenol-chloroform method or commercial kit. Precisely quantify DNA concentration and purity using a spectrophotometer [8].
- Reaction Mix Preparation: Prepare a 20 μL ddPCR reaction mixture on ice containing:
- 10 μL of 2x ddPCR Supermix for Probes
- Primers and probes at optimized final concentrations (e.g., 0.9 μM for primers and 0.25 μM for probes, based on initial optimization) [66]
- 2-5 μL of gDNA template (recommended input: 10-50 ng)
- Nuclease-free water to 20 μL
- Droplet Generation: Transfer the 20 μL reaction mix to a DG8 cartridge. Add 70 μL of droplet generation oil to the appropriate well. Place the cartridge in the QX200 Droplet Generator to create water-in-oil emulsion droplets [66].
- PCR Amplification: Carefully transfer 40 μL of the generated emulsion to a 96-well PCR plate. Seal the plate with a pierceable foil heat seal. Perform PCR amplification in a thermal cycler using the following protocol:
- Enzyme activation: 95°C for 10 minutes
- 40-50 cycles of:
- Denaturation: 94°C for 30 seconds
- Annealing/Extension: 55-60°C (assay-specific) for 60 seconds
- Enzyme deactivation: 98°C for 10 minutes
- Hold at 4°C [67]
- Droplet Reading and Analysis: Place the amplified plate in the QX200 Droplet Reader. The reader will aspirate each sample and measure the fluorescence (FAM and HEX/VIC) of each droplet. Analyze the data using the instrument's associated software (e.g., QuantaSoft). The software applies Poisson statistics to the count of positive and negative droplets for each channel to provide an absolute copy number per microliter of reaction for both the wild-type and Δ32 alleles [8] [67].
Workflow for Assay Development and Clinical Validation
The following diagram illustrates the comprehensive workflow from initial assay setup to clinical application, integrating key steps from the experimental protocol and its therapeutic context.
Clinical Applications and Therapeutic Context
The multiplex ddPCR assay for CCR5 has direct and critical applications in modern clinical practice and therapeutic development, moving beyond basic research into patient management.
Monitoring Allogeneic Hematopoietic Stem Cell Transplantation (allo-HSCT): Transplantation with cells from a CCR5Δ32 homozygous donor is a validated strategy for achieving HIV cure [8] [18]. The described ddPCR assay is vital for quantifying the content of donor-derived cells with the CCR5Δ32 mutation in patient blood and tissue post-transplant, enabling precise monitoring of engraftment success and chimerism levels [8]. This is crucial for understanding the relationship between donor cell expansion and sustained HIV remission.
Evaluating Gene Editing Therapies: The development of CRISPR/Cas9 and other genome-editing technologies allows for the artificial creation of the CCR5Δ32 mutation in autologous or immunocompatible cells [8]. This ddPCR system provides a method to quickly and accurately measure the efficiency of these gene-editing approaches by quantifying the proportion of edited alleles in heterogeneous cell populations pre- and post-therapy, down to 0.8% sensitivity [8].
Understanding HIV Remission Mechanisms: Recent evidence suggests that sustained HIV remission can be achieved even with wild-type CCR5 donor cells, as reported in a case study where a patient remained aviremic for 32 months after antiretroviral treatment interruption post-allo-HSCT [18]. In such scenarios, highly sensitive ddPCR assays for HIV DNA, alongside CCR5 genotyping, are used to evaluate the size and composition of the residual HIV reservoir, providing insights into the complex mechanisms of viral control [8] [18].
The multiplex ddPCR assay for CCR5 wild-type and Δ32 alleles represents a significant advancement in molecular diagnostics for precision medicine. Its exceptional sensitivity, precision, and ability to provide absolute quantification make it an indispensable tool for clinical validation and therapeutic monitoring in the context of HIV cure strategies, including allo-HSCT and novel gene-editing therapies. The detailed protocol and analytical validation data provided herein offer researchers and clinicians a robust framework to implement this technology, ultimately supporting the development and application of CCR5-targeted treatments.
Inter-laboratory Reproducibility and Quality Assurance Protocols
The reproducibility of experimental data across different laboratories is a fundamental pillar of scientific research, particularly in the context of clinical diagnostics and therapeutic development. For multiplex droplet digital PCR (ddPCR) assays targeting the CCR5 wild-type and Δ32 alleles—a critical application in HIV cure research—establishing robust inter-laboratory reproducibility and quality assurance protocols is paramount. The CCR5 co-receptor serves as the principal entry point for human immunodeficiency virus (HIV), and a 32-base pair deletion (CCR5Δ32) confers natural resistance to HIV-1 infection in homozygous individuals [8]. Research in this field has expanded significantly since the reported cures of HIV-positive patients following hematopoietic stem cell transplantation from CCR5Δ32/Δ32 donors [8] [18].
Multiplex ddPCR technology enables simultaneous quantification of both wild-type and Δ32 alleles in a single reaction, providing a powerful tool for monitoring transplanted cell populations and edited cells in novel HIV therapeutic approaches [8] [23]. However, the transition of these assays from research tools to clinically applicable methods requires demonstration of reliability across multiple laboratories under varying conditions. This application note outlines standardized protocols and quality assurance measures to ensure inter-laboratory reproducibility of multiplex ddPCR assays for CCR5 genotyping, framed within the broader context of multiplex ddPCR assay design for CCR5 research.
Experimental Design and Technical Considerations
Multiplex ddPCR Assay Configuration for CCR5 Genotyping
The CCR5Δ32 mutation arises from a 32-bp deletion in the CCR5 gene coding sequence, causing a frameshift and premature stop codons that knockout gene function [8]. A well-designed multiplex ddPCR assay must simultaneously distinguish and quantify three potential genotypes: wild-type homozygous, Δ32 heterozygous, and Δ32 homozygous.
For optimal performance in a standard two-color ddPCR system, the assay should implement a probe-based detection strategy utilizing two fluorescent channels. The recommended configuration uses:
- FAM-labeled probe: Targets the wild-type CCR5 sequence
- HEX-labeled probe: Targets the Δ32 deletion junction
This design enables clear discrimination of all three genotypes based on droplet clustering patterns, with heterozygous samples appearing as double-positive droplets [23] [87]. For research applications requiring higher multiplexing capacity, amplitude-based multiplexing approaches can be employed, where targets are differentiated by fluorescence intensity within the same channel through controlled probe concentration ratios [88] [87].
Key Performance Parameters
Establishing assay performance benchmarks is essential for inter-laboratory standardization. The following table summarizes critical validation parameters and their acceptable ranges based on published studies of multiplex ddPCR assays for genetic variants:
Table 1: Performance Parameters for Multiplex ddPCR Assays
| Parameter | Target Performance | Experimental Support |
|---|---|---|
| Limit of Detection (LoD) | ≤0.01% variant allele frequency [89] | D816V KIT mutation detection down to 0.01% allele burden [89] |
| Limit of Quantification (LoQ) | ≤0.1% variant allele frequency [90] | GMO quantification at 0.1% threshold [90] |
| Dynamic Range | 5-6 orders of magnitude [47] | Accurate quantification from 10% to 0.01% allele burden [89] |
| Inter-lab Precision | CV <15-25% [91] | HCMV DNA quantification with <15% expanded uncertainty [91] |
| Trueness | >95% agreement with reference methods [88] | 100% PPA and 98.5% NPA vs. sequencing for NSCLC variants [88] |
Materials and Reagents
Research Reagent Solutions
The following table outlines essential materials and their functions for establishing reproducible multiplex ddPCR assays for CCR5 genotyping:
Table 2: Essential Research Reagents for CCR5 Multiplex ddPCR
| Item | Function/Application | Specifications |
|---|---|---|
| ddPCR System | Partitioning, amplification, and droplet reading | Bio-Rad QX200 or equivalent with two fluorescence detection channels [47] [87] |
| Supermix | PCR reaction environment | ddPCR Supermix for Probes (no dUTP) [47] |
| Primers/Probes | Target-specific amplification/detection | HPLC-purified TaqMan probes with LNA modifications for enhanced specificity [47] |
| DNA Extraction Kit | Nucleic acid isolation from samples | Validated for sample type (e.g., QIAamp DNA kits for blood, ccfDNA kits for plasma) [47] [91] |
| Reference Materials | Assay controls and standardization | Wild-type and Δ32 homozygous control DNA [47] |
| Droplet Generation Oil | Partition creation | Surfactant-containing oil for stable droplet formation [47] |
| Microplates/Seals | Reaction containment | Semi-skirted 96-well plates and pierceable foil heat seals [47] |
Standardized Experimental Protocols
Sample Collection and Nucleic Acid Extraction
Proper sample handling and processing are critical first steps in ensuring reproducible results:
- Sample Collection: Collect peripheral blood mononuclear cells (PBMCs) in appropriate anticoagulant tubes (e.g., EDTA). Process within 8 hours of collection [92].
- Nucleic Acid Extraction: Use silica membrane-based extraction methods (e.g., QIAamp DNA kits) according to manufacturer protocols [91]. Extract DNA from approximately 2×10^6 PBMCs.
- DNA Quantification and Quality Assessment: Measure DNA concentration using fluorometric methods (e.g., Qubit) and assess purity via spectrophotometry (A260/280 ratio 1.8-2.0) [8].
- Extraction Efficiency Monitoring: For low-input samples, consider spiking with a synthetic DNA control (e.g., XenT gBlock) prior to extraction to calculate and correct for extraction efficiency [47].
Multiplex ddPCR Assay Procedure
The following workflow outlines the standardized protocol for CCR5 wild-type/Δ32 multiplex ddPCR:
Reaction Setup
Prepare Reaction Mix: In a dedicated pre-PCR area, prepare the master mix containing:
Include Controls: Each run should include:
- No-template controls (NTC) with nuclease-free water
- Wild-type homozygous control DNA
- Δ32 heterozygous control DNA (if available)
- Δ32 homozygous control DNA [47]
Droplet Generation and Thermal Cycling
- Generate Droplets: Transfer 20 µL of reaction mix to DG8 cartridges and generate droplets according to manufacturer instructions [47].
- Transfer and Seal: Carefully transfer 40 µL of generated droplets to a 96-well PCR plate and heat-seal with foil.
- Amplify: Perform PCR amplification using the following thermal cycling conditions:
Droplet Reading and Data Analysis
- Plate Reading: After amplification, place plate in droplet reader and analyze according to instrument specifications.
- Threshold Setting: Set fluorescence thresholds for positive/negative droplets based on control samples, ensuring clear separation between populations.
- Data Interpretation: Use Poisson statistics to calculate absolute copy numbers for wild-type and Δ32 alleles [47].
Quality Assurance and Inter-laboratory Reproducibility
Quality Control Measures
Implementing comprehensive quality control procedures is essential for maintaining assay performance:
Pre-analytical Controls:
Analytical Controls:
- Include control samples with known genotype in each run
- Monitor no-template controls for contamination
- Track amplification efficiency and droplet count for process monitoring [47]
Post-analytical Controls:
Inter-laboratory Reproducibility Assessment
The following workflow outlines a systematic approach to establishing and maintaining inter-laboratory reproducibility:
To systematically assess inter-laboratory reproducibility, implement the following approach:
Reference Material Exchange: Distribute identical reference materials (wild-type, heterozygous, and Δ32 homozygous DNA) to all participating laboratories. These should include samples with allele frequencies spanning the expected dynamic range (0.01% to 100%) [89] [91].
Parallel Testing: All laboratories should test the same panel of samples using the standardized protocol within a defined timeframe.
Data Analysis and Comparison:
- Calculate copy number concentrations for wild-type and Δ32 alleles independently
- Compute allele frequencies (Δ32 fraction) for each sample
- Determine inter-laboratory coefficients of variation (CV) for both absolute quantification and allele frequencies
- Compare results to reference values where available [90] [91]
Statistical Analysis of Reproducibility: The table below demonstrates the level of reproducibility achievable with well-controlled ddPCR assays, as demonstrated in previous inter-laboratory studies:
Table 3: Inter-laboratory Reproducibility Metrics from Digital PCR Studies
| Study Focus | Number of Labs | Reproducibility | Key Factors |
|---|---|---|---|
| HCMV DNA Quantification | 4 | Expanded measurement uncertainties <15% [91] | Standardized DNA extraction critical [91] |
| KIT D816V Mutation | 7 | CV between 0.07 and 0.8 across allele burdens [89] | Commercial ddPCR assays showed high concordance [89] |
| GMO Soybean Quantification | 11 | Meeting EU regulatory requirements for trueness and precision [90] | Multiplex ddPCR covering 15 GM events [90] |
Troubleshooting and Optimization
Even with standardized protocols, laboratories may encounter technical challenges that affect reproducibility:
Poor Droplet Resolution:
- Cause: Improper droplet generation or probe concentrations
- Solution: Verify droplet generator function and optimize probe ratios using control DNA
Elevated False Positives:
- Cause: Contamination or non-specific amplification
- Solution: Implement strict pre-PCR area controls, use uracil-DNA glycosylase (UDG) treatment, and optimize annealing temperature [47]
Inter-laboratory Discrepancies:
- Cause: Different instrumentation or reagent lots
- Solution: Implement cross-calibration exercises and centralize critical reagents [91]
Inhibition Issues:
- Cause: Carryover of inhibitors from DNA extraction
- Solution: Include internal positive controls and assess extraction efficiency [47]
Establishing inter-laboratory reproducibility for multiplex ddPCR assays targeting CCR5 wild-type and Δ32 alleles requires meticulous attention to standardized protocols, quality control measures, and regular proficiency testing. The protocols outlined in this application note provide a framework for achieving the consistency necessary for translational research and clinical application. As CCR5-directed therapies continue to evolve, including stem cell transplantation and gene editing approaches, robust and reproducible genotyping and quantification methods will play an increasingly important role in both research and clinical care. By implementing these quality assurance protocols, laboratories can generate reliable, comparable data that advances the field of HIV cure research and contributes to the development of novel therapeutic strategies.
The C-C chemokine receptor type 5 (CCR5) serves as a critical co-receptor for human immunodeficiency virus (HIV) entry into T-cells [8]. A natural 32-base pair deletion (CCR5Δ32) confers resistance to HIV infection, making its accurate detection valuable for both fundamental research and clinical applications in HIV management [8] [7]. This application note provides a detailed cost-benefit and workflow analysis for implementing a multiplex droplet digital PCR (ddPCR) assay to simultaneously genotype CCR5 wild-type and Δ32 alleles. We frame this within a broader thesis on optimized multiplex ddPCR design, highlighting how this approach balances superior analytical performance with operational efficiency for researchers and drug development professionals.
Multiplex ddPCR vs. Alternative Genotyping Methods
Table 1: Comparative Analysis of CCR5 Genotyping Methods
| Methodological Feature | Multiplex Endpoint PCR [93] | qPCR with HRM [7] | Multiplex ddPCR (This Work) |
|---|---|---|---|
| Multiplexing Capability | Yes (2-plex: CCR5Δ32 & HLA-B*5701) | Low | Yes (2-plex or higher) |
| Absolute Quantification | No | No | Yes |
| Precision & Sensitivity | Low | Moderate | High (LoQ as low as 0.01% VAF) [94] |
| Throughput | Moderate | Moderate | High |
| Cost per Sample | Low | Moderate | Higher initial cost, competitive at scale |
| Primary Application | Low-cost screening | Research, specificity validation | Quantification of allele fractions, MRD detection |
| Key Limitation | Qualitative or semi-quantitative | Limited multiplexing scope | Higher reagent cost, specialized equipment |
Droplet digital PCR provides significant advantages for precise nucleic acid quantification by partitioning a sample into thousands of nanoliter-sized droplets and applying Poisson statistics to count target molecules [95]. The fundamental principle is that the average number of target molecules per partition (λ) is calculated based on the proportion of negative partitions, using the formula λ = -ln(1 - k/n), where n is the total partitions and k is the number of positive partitions [95]. This enables absolute quantification without standard curves.
Multiplexing within ddPCR allows concurrent amplification of multiple targets in a single reaction, which reduces technical errors, reagent consumption, and hands-on time compared to running parallel uniplex reactions [95]. For CCR5 genotyping, a multiplex assay can be configured as a competing duplex reaction, where a single primer pair amplifies both alleles, and two allele-specific probes (e.g., for wild-type and Δ32) labeled with different fluorophores bind the same region to distinguish them [95].
Experimental Protocol: Multiplex ddPCR for CCR5 Genotyping
Reagent and Instrument Setup
Table 2: Research Reagent Solutions for CCR5 ddPCR
| Reagent / Material | Function / Application | Specification / Notes |
|---|---|---|
| ddPCR Supermix | Provides optimized reagents for PCR amplification in droplets | Use a probe-specific supermix for hydrolysis probe assays. |
| FAM-labeled Probe | Detects wild-type CCR5 allele | Sequence must span the Δ32 deletion region. |
| HEX/VIC-labeled Probe | Detects mutant CCR5Δ32 allele | Designed with 3' end complementary to the deletion junction. |
| Primer Pair | Amplifies a fragment surrounding the Δ32 deletion | Amplicon length should be optimized for ddPCR efficiency. |
| DG8 Cartridges & Droplet Generation Oil | Creates water-in-oil emulsion droplets | Essential for droplet generation in Bio-Rad QX200 system. |
| ddPCR Reader | Reads fluorescence endpoint of individual droplets | Requires at least two optical detection channels. |
| CRISPR/Cas9 System | For research use: Artificially generates CCR5Δ32 mutation in control cell lines [8] | Used in assay development and validation. |
Step-by-Step Workflow
- DNA Extraction: Isolate genomic DNA from target cells (e.g., peripheral blood mononuclear cells, PBMCs) using a standard phenol-chloroform method or commercial kit. Quantify DNA concentration and assess purity using spectrophotometry [8].
- Assay Preparation:
- Prepare the ddPCR reaction mix containing ddPCR supermix, primers, and FAM-labeled wild-type and HEX/VIC-labeled Δ32 allele-specific probes [8].
- Use a typical final reaction volume of 20-22 µL.
- Add 10-100 ng of template DNA.
- Droplet Generation:
- Load the reaction mixture into a DG8 cartridge alongside droplet generation oil.
- Place the cartridge in a droplet generator to create ~20,000 nanoliter-sized water-in-oil droplets per sample [8].
- PCR Amplification:
- Carefully transfer the emulsified samples to a 96-well PCR plate.
- Seal the plate and run PCR amplification on a conventional thermal cycler using optimized cycling conditions (e.g., 95°C for 10 min, 40 cycles of 94°C for 30 s and 60°C for 60 s, 98°C for 10 min, and a 4°C hold) [8].
- Droplet Reading and Analysis:
- Place the PCR plate in a droplet reader, which sequentially aspirates droplets from each well.
- The reader measures the fluorescence in two channels (FAM and HEX/VIC) for each droplet.
- Use the instrument's software (e.g., QuantaSoft) to analyze the data. The software plots droplets in 2D amplitude plots, clusters them into four populations (FAM+/HEX-, FAM-/HEX+, FAM+/HEX+, FAM-/HEX-), and applies Poisson statistics to calculate the absolute copy number of wild-type and Δ32 alleles per microliter of the original reaction [8] [95].
Workflow Visualization
The following diagram illustrates the logical workflow and decision points in the multiplex ddPCR process for CCR5 genotyping:
Operational Considerations and Cost-Benefit Analysis
Implementing a multiplex ddPCR strategy requires evaluating operational factors against the required data quality.
Table 3: Operational Cost-Benefit Analysis
| Operational Factor | Benefits | Considerations & Mitigations |
|---|---|---|
| Reagent Costs | Single-reaction cost for two targets reduces overall reagent use compared to separate assays. | Per-reaction cost is higher than traditional PCR. Mitigation: Optimize reagent concentrations; use at scale. |
| Equipment & Capital | Requires significant capital investment in droplet generator and reader. Mitigation: Utilize core facilities; consider service providers. | |
| Labor & Time | High throughput; minimal hands-on time post-setup; reduced pipetting steps via multiplexing. | Protocol is multi-step (droplet generation, transfer, reading). Mitigation: Automated droplet generators can improve reproducibility. |
| Data Complexity | Absolute quantification without standard curves; high sensitivity for rare alleles in mixtures [8]. | Data analysis requires specific software training; 2D plot interpretation needed. Mitigation: Standardized software and analysis templates. |
| Assay Development | Robust and reproducible once optimized. | Optimization requires fine-tuning of primer/probe concentrations, annealing temperature, and template amount [94]. |
The primary financial benefit of multiplexing is the reduction in sample and reagent consumption. By detecting two alleles in one well, the number of reactions, tips, plates, and overall preparation time is nearly halved. Furthermore, the data quality benefit is substantial: ddPCR provides unparalleled precision for quantifying allele fractions in heterogeneous cell mixtures, accurately measuring mutant allele content as low as 0.8% [8]. This is crucial for applications like monitoring the engraftment of CCR5-modified cells in HIV patients following stem cell transplantation [8].
Multiplex ddPCR represents a robust and efficient methodology for the precise quantification of CCR5 wild-type and Δ32 alleles. The initial higher costs and operational complexity are justified by the exceptional data quality, sensitivity, and quantitative nature of the results, which are difficult to achieve with traditional methods. This approach is perfectly aligned with the demands of modern therapeutic development, where accurate measurement of genetic biomarkers is non-negotiable.
Future developments in the field of multiplex ddPCR will further enhance its cost-benefit profile. Emerging techniques like digital melting curve analysis (MCA) promise to overcome multiplexing limitations by differentiating multiple targets in a single fluorescence channel based on their melting temperatures, drastically reducing probe costs and increasing multiplexing capacity [96]. The continued refinement of these protocols will solidify the role of multiplex ddPCR as a cornerstone technique in advanced genetic analysis and personalized medicine.
Conclusion
Multiplex ddPCR represents a transformative methodology for CCR5 genotyping, offering unparalleled sensitivity in detecting Δ32 alleles within complex biological samples. This comprehensive analysis demonstrates that properly designed and validated assays can achieve detection limits below 1% in heterogeneous cell mixtures, enabling precise quantification essential for monitoring CRISPR-edited hematopoietic stem cells and natural allele distribution. The robust performance across platforms and sample types positions this technology as critical for advancing HIV cure strategies, particularly autologous HSCT with CCR5-modified cells. Future directions should focus on standardizing assays for clinical implementation, expanding multiplexing capabilities for simultaneous detection of additional therapeutic markers, and validating these approaches in larger patient cohorts. As CCR5-targeted therapies evolve, multiplex ddPCR will play an increasingly vital role in both basic research and translational medicine, ultimately contributing to more effective personalized interventions for HIV and other diseases influenced by CCR5 biology.
References
- 1. The chemokine receptor CCR5: multi-faceted hook for HIV-1 [https://retrovirology.biomedcentral.com/articles/10.1186/s12977-024-00634-1]
- 2. C-C chemokine receptor type five (CCR5): An emerging target ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5133339/]
- 3. Targeting CCR5 as a Component of an HIV-1 Therapeutic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8811197/]
- 4. The Role of the Chemokine Receptor Gene CCR5 and Its ... [https://wwwnc.cdc.gov/eid/article/3/3/97-0302_article]
- 5. Frequencies of gene variant CCR5-Δ32 in 87 countries ... [https://www.sciencedirect.com/science/article/pii/S0198885917305104]
- 6. Distribution of CCR5-Δ32 and HLA-B*57:01 alleles in HIV ... [https://www.nature.com/articles/s41439-025-00321-3]
- 7. Automated production of CCR5-negative CD4 + -T cells in ... [https://www.nature.com/articles/s41434-021-00259-5]
- 8. Detection of CCR5Δ32 Mutant Alleles in Heterogeneous ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8898955/]
- 9. Clinically accessible amplitude-based multiplex ddPCR ... [https://www.nature.com/articles/s41598-024-52983-8]
- 10. Low-cost simultaneous detection of CCR5-delta32 and ... [https://www.sciencedirect.com/science/article/abs/pii/S0166093415003006]
- 11. CCR5: From Natural Resistance to a New Anti-HIV Strategy [https://pmc.ncbi.nlm.nih.gov/articles/PMC3185609/]
- 12. CCR5 gene editing and HIV immunotherapy - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12213807/]
- 13. HIV Resistant Mutation | Viruses101 [https://www.nature.com/scitable/blog/viruses101/hiv_resistant_mutation/]
- 14. The Geographic Spread of the CCR5 Δ32 HIV-Resistance Allele [https://pmc.ncbi.nlm.nih.gov/articles/PMC1255740/]
- 15. Closing the Door with CRISPR: Genome Editing of CCR5 and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9326690/]
- 16. The Geographic Spread of the CCR5 Δ32 HIV-Resistance Allele [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.0030339]
- 17. The evolutionary history of the CCR5-Δ32 HIV-resistance ... [https://www.sciencedirect.com/science/article/pii/S1286457904003636]
- 18. Sustained HIV remission after allogeneic hematopoietic ... [https://www.nature.com/articles/s41591-024-03277-z]
- 19. Fundamentals of digital PCR [https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/pcr/digital-pcr/what-is-digital-pcr]
- 20. Digital PCR Principle, techniques and applications [https://www.stillatechnologies.com/digital-pcr/principle/]
- 21. Absolute vs. Relative Quantification for qPCR [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/real-time-pcr-basics/absolute-vs-relative-quantification-real-time-pcr.html]
- 22. Multilayered HIV-1 resistance in HSPCs through CCR5 ... [https://www.nature.com/articles/s41467-025-58371-8]
- 23. Guide to multiplexing dPCR assays [https://www.qiagen.com/us/applications/digital-pcr/beginners/dpcr-multiplexing]
- 24. dPCR vs qPCR [https://www.qiagen.com/us/applications/digital-pcr/beginners/dpcr-vs-qpcr]
- 25. What Are the Advantages of ddPCR Over Other PCR ... [https://mogene.com/what-are-the-advantages-of-ddpcr-over-other-pcr-variants/]
- 26. dPCR: A Technology Review - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC5948698/]
- 27. Absolute quantification of rare gene targets in limited ... [https://www.nature.com/articles/s41598-025-94115-w]
- 28. High frequency CCR5 editing in human hematopoietic ... [https://www.nature.com/articles/s41467-025-55873-3]
- 29. Distribution of CCR5-Delta32, CCR2-64I, and SDF1-3'A ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12102844/]
- 30. Rules and Tips for PCR & qPCR Primer Design | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/how-to-design-primers-and-probes-for-pcr-and-qpcr/]
- 31. Allele-Specific Quantitative PCR for Accurate, Rapid, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4931339/]
- 32. Rare Mutation Detection Digital PCR Tutorial [https://www.stillatechnologies.com/digital-pcr/primer-design/rare-mutation-detection/]
- 33. Development of a multiplex droplet digital PCR method for ... [https://ann-clinmicrob.biomedcentral.com/articles/10.1186/s12941-024-00687-2]
- 34. All About Probe-Based Real-Time qPCR Assays [https://www.goldbio.com/blogs/articles/all-about-probe-based-qpcr?srsltid=AfmBOorOVa7fWnQUnGDRPXzPB-aSr_OSHtIUfUDjQ2IVPJNVoLkxMG68]
- 35. qPCR Protocol for SNP Genotyping [https://www.thermofisher.com/us/en/home/references/protocols/nucleic-acid-amplification-and-expression-profiling/pcr-protocol/qpcr-for-snp-genotyping.html]
- 36. One Assay, Nine Targets: Advancing Viral Surveillance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12529474/]
- 37. Detection of CCR Mutant 5 in Heterogeneous Cell Mixtures... Δ 32 Alleles [https://pubmed.ncbi.nlm.nih.gov/35265670/]
- 38. Denaturation-enhanced droplet digital PCR for liquid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6310690/]
- 39. Optimization of digital droplet polymerase chain reaction for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4827695/]
- 40. Optimization of digital droplet polymerase chain reaction ... [https://www.sciencedirect.com/science/article/pii/S2214753515300127]
- 41. Multiplex PCR: Optimization and Application in Diagnostic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC88949/]
- 42. PCR Cycling Parameters—Six Key Considerations for ... [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-cycling-considerations.html]
- 43. Distribution of CCR - 5 and HLA-B*57:01 Δ in HIV-seropositive... 32 alleles [https://www.nature.com/articles/s41439-025-00321-3?error=cookies_not_supported&code=d3cc88e7-b77f-493e-adbf-6bd9bf58bfd9]
- 44. HIV-1 remission and possible cure in a woman after haplo ... [https://www.sciencedirect.com/science/article/pii/S0092867423001733]
- 45. Fluorescence Multiplexing with Spectral Imaging and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9827428/]
- 46. Detection of Mutations using Drop-off Assay Design [https://www.stillatechnologies.com/digital-pcr/primer-design/drop-off-assay-design/]
- 47. Optimisation of robust singleplex and multiplex droplet ... [https://www.nature.com/articles/s41598-019-49043-x]
- 48. AAV ddPCR Titration [https://www.addgene.org/protocols/aav-ddpcr-titration/]
- 49. Generic Multiplex Digital PCR for Accurate Quantification of T ... [https://www.ncbi.nlm.nih.gov/books/NBK586964/]
- 50. Digital PCR | IDT [https://www.idtdna.com/pages/technology/qpcr-and-pcr/digital-pcr]
- 51. Evolution of Multiplex PCR: Benefits, Challenges & Key ... [https://www.eurogentec.com/en/news/111_evolution-of-multiplex-pcr-benefits-challenges-key-applications]
- 52. Digital PCR: from early developments to its future application ... [https://pubs.rsc.org/en/content/articlehtml/2025/lc/d5lc00055f]
- 53. Highly multiplexed digital PCR assay for simultaneous ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12515721/]
- 54. Droplet digital PCR of viral DNA/RNA, current progress ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8013307/]
- 55. Droplet digital PCR-based analyses for robust, rapid, and ... [https://actaneurocomms.biomedcentral.com/articles/10.1186/s40478-022-01335-6]
- 56. Detection Of CCR5 Delta-32 Mutation Using High- ... [https://pubmed.ncbi.nlm.nih.gov/39279712/]
- 57. Multiplexed Digital PCR Reference Gene Measurement for ... [https://www.mdpi.com/2073-4409/14/19/1544]
- 58. dPCR: A Technology Review [https://www.mdpi.com/1424-8220/18/4/1271]
- 59. Novel Multiplexing Strategies for Quantification of Rare ... [https://pubmed.ncbi.nlm.nih.gov/29717449/]
- 60. Digital PCR for Single-Cell Analysis - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC10886679/]
- 61. Rapid and Sensitive Digital Droplet PCR Assays for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11683180/]
- 62. ddPCR Overcomes the CRISPR-Cas13a-Based Technique ... [https://www.mdpi.com/1422-0067/25/20/10902]
- 63. Troubleshooting primer dimer in PCR [https://www.minipcr.com/primer-dimer-pcr/]
- 64. PCR Optimization: Strategies To Minimize Primer Dimer [https://genemod.net/blog/pcr-optimization]
- 65. dPCR assay development | Digital PCR troubleshooting [https://www.qiagen.com/us/applications/digital-pcr/beginners/dpcr-guide/setup-and-troubleshooting]
- 66. Droplet Digital RT-PCR (RT-ddPCR) Optimization [https://bio-protocol.org/exchange/minidetail?id=5915757&type=30]
- 67. Development and validation of a droplet digital PCR assay ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1573949/full]
- 68. DataSheet3_Detection of CCR5Δ32 Mutant Alleles ... - Frontiers [https://frontiersin.figshare.com/articles/dataset/DataSheet3_Detection_of_CCR5_32_Mutant_Alleles_in_Heterogeneous_Cell_Mixtures_Using_Droplet_Digital_PCR_PDF/19205547]
- 69. Seamless modification of wild-type induced pluripotent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4084478/]
- 70. Digital droplet PCR is an accurate and precise method to ... [https://www.nature.com/articles/s41598-025-20944-4]
- 71. A novel droplet digital PCR method for assessing ... [https://pubmed.ncbi.nlm.nih.gov/41237531/]
- 72. How to Work with Challenging or Limited Genomic ... [https://blog.omni-inc.com/blog/how-to-work-with-challenging-or-limited-genomic-samples-updated-for-2025]
- 73. Distribution of CCR -Delta32, 5 promoter 59029... CCR 5 [https://bmcresnotes.biomedcentral.com/articles/10.1186/1756-0500-6-288]
- 74. Droplet digital RT-PCR method for SARS-CoV-2 variants ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12267190/]
- 75. Analytical and clinical validation of multiplex droplet digital ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10976995/]
- 76. Multiplex PCR method for the detection of SARS-CoV-2 [https://patents.google.com/patent/US11649511B2/de]
- 77. Comparison of digital PCR methods [https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/pcr/digital-pcr/comparison-of-digital-pcr-methods]
- 78. : Droplet and Applications in Molecular... Digital PCR Advantages [https://www.labx.com/resources/droplet-digital-pcr-advantages-and-applications-in-molecular-diagnostics/5581]
- 79. Comparison of two digital PCR platforms for quantification ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12622311/]
- 80. dPCR vs. ddPCR: Why Precision Matters in Cell and Gene ... [https://www.roslinct.com/insights/dpcr-vs-ddpcr/]
- 81. Comparing the precision of two digital PCR applications for ... [https://www.nature.com/articles/s41598-025-13143-8]
- 82. Detection of CCR5Δ32 Mutant Alleles in Heterogeneous ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2022.805931/full]
- 83. The CCR5-Delta32 Genetic Polymorphism and HIV-1 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6227735/]
- 84. What is multiplex qPCR? [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/real-time-pcr-basics/what-is-multiplex-qpcr.html]
- 85. Development and validation of a multiplex qPCR method ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11889745/]
- 86. Developmental validation of a multiplex qPCR assay for ... [https://www.sciencedirect.com/science/article/abs/pii/S1872497324001601]
- 87. Development and evaluation of a multiplex droplet digital ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.970973/full]
- 88. A rapid, multiplex digital PCR assay to detect gene variants ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10620117/]
- 89. Performance and Interlaboratory Reproducibility of Droplet ... [https://www.sciencedirect.com/science/article/pii/S0006497123129909]
- 90. Development and inter-laboratory assessment of droplet ... [https://www.nature.com/articles/s41598-017-09377-w]
- 91. Inter-laboratory assessment of different digital PCR platforms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5359388/]
- 92. Chemokine Receptor CCR5 Δ32 Genetic Analysis Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC96180/]
- 93. Low-cost simultaneous detection of CCR5-delta32 and ... [https://pubmed.ncbi.nlm.nih.gov/26341061/]
- 94. Laboratory-developed Droplet Digital PCR Assay for ... [https://www.sciencedirect.com/science/article/pii/S2699940425000190]
- 95. Fundamentals of multiplexing with digital PCR - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5154634/]
- 96. Digital Melting Curve Analysis for Multiplex Quantification of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11764372/]