Nested PCR vs. LAMP for SARS-CoV-2: A Comprehensive Analysis of Diagnostic Accuracy for Researchers
This article provides a systematic comparison of nested PCR and Loop-Mediated Isothermal Amplification (LAMP) for detecting SARS-CoV-2, tailored for researchers, scientists, and drug development professionals.
Nested PCR vs. LAMP for SARS-CoV-2: A Comprehensive Analysis of Diagnostic Accuracy for Researchers
Abstract
This article provides a systematic comparison of nested PCR and Loop-Mediated Isothermal Amplification (LAMP) for detecting SARS-CoV-2, tailored for researchers, scientists, and drug development professionals. It explores the foundational principles of both techniques, details methodological protocols and applications, addresses troubleshooting and optimization strategies, and presents a rigorous validation and comparative analysis based on current scientific literature. The synthesis offers evidence-based guidance for selecting appropriate molecular diagnostics in both clinical and research settings, with implications for future assay development and pandemic preparedness.
Core Principles and Diagnostic Landscape of SARS-CoV-2 Molecular Assays
Fundamental Principles of Nested PCR and LAMP Amplification
Nucleic acid amplification technologies represent cornerstone methodologies in modern molecular diagnostics, particularly for infectious disease detection. Among these techniques, polymerase chain reaction (PCR) and its advanced variants have long served as the gold standard in laboratory settings. However, the evolving demands of diagnostic testing, emphatically highlighted during the SARS-CoV-2 pandemic, have accelerated the development and adoption of alternative methods that offer rapid, sensitive, and field-deployable pathogen detection. This guide provides a comprehensive comparative analysis of two such powerful techniques: Nested PCR and Loop-Mediated Isothermal Amplification (LAMP).
The diagnostic challenges presented by COVID-19 underscored the critical need for diverse testing strategies. While reverse transcription-quantitative PCR (RT-qPCR) remains the primary reference method, its limitations—including requirements for sophisticated equipment, lengthy processing times, and highly trained personnel—have stimulated the evaluation of more accessible alternatives [1]. Within this context, both nested PCR and LAMP have emerged as viable methodologies, each with distinct operational principles and performance characteristics that suit them for particular diagnostic scenarios. This article examines their fundamental principles, relative performance metrics based on experimental data, and practical applications within SARS-CoV-2 research and beyond.
Fundamental Principles and Methodologies
Nested PCR: Enhanced Specificity Through Sequential Amplification
Nested PCR is a two-stage amplification technique designed to significantly enhance the specificity and sensitivity of nucleic acid detection compared to conventional PCR. This method utilizes two distinct sets of primers that target the same genetic region in successive amplification reactions. The initial PCR round employs an outer primer pair that amplifies a larger target sequence. The product from this first reaction then serves as the template for a second amplification round using inner primers that bind internal to the first amplicon. This sequential priming strategy minimizes non-specific amplification and dramatically increases detection sensitivity by effectively re-amplifying the target from the first reaction [2] [3].
The fundamental advantage of this nested approach lies in its robustness and reliability. By requiring four specific priming events (two in each round) for successful target detection, the method effectively reduces false-positive results caused by primer-dimer formations or non-specific binding [4]. However, this enhanced fidelity comes with operational complexities, primarily the risk of carryover contamination between reaction tubes when transferring first-round products, which necessitates meticulous laboratory technique and dedicated workspace to prevent amplicon contamination [3]. Additionally, the requirement for two sequential thermal cycling procedures extends the total assay time to typically several hours.
LAMP: Isothermal Amplification for Rapid Detection
Loop-Mediated Isothermal Amplification (LAMP) represents a fundamentally different approach to nucleic acid amplification. Unlike PCR-based methods that require precise thermal cycling between denaturation, annealing, and extension temperatures, LAMP operates at a constant temperature between 60-65°C, utilizing a DNA polymerase with strand displacement activity [5]. This reaction employs four to six specifically designed primers that recognize six to eight distinct regions on the target DNA, forming stem-loop structures that facilitate auto-cycling amplification without the need for thermal denaturation [4] [6].
The LAMP reaction proceeds with remarkable speed and efficiency, often yielding detectable amplification within 15-60 minutes [5] [6]. This kinetic advantage stems from the continuous amplification process without time loss for thermal transitions. Furthermore, result detection can be achieved through multiple simplified methods, including colorimetric changes using pH-sensitive dyes, turbidity measurements to detect magnesium pyrophosphate precipitate, or fluorescence detection with intercalating dyes [7] [5]. These detection formats eliminate the need for gel electrophoresis and enable visual interpretation, making LAMP particularly suitable for point-of-care testing and resource-limited settings where sophisticated instrumentation is unavailable [1].
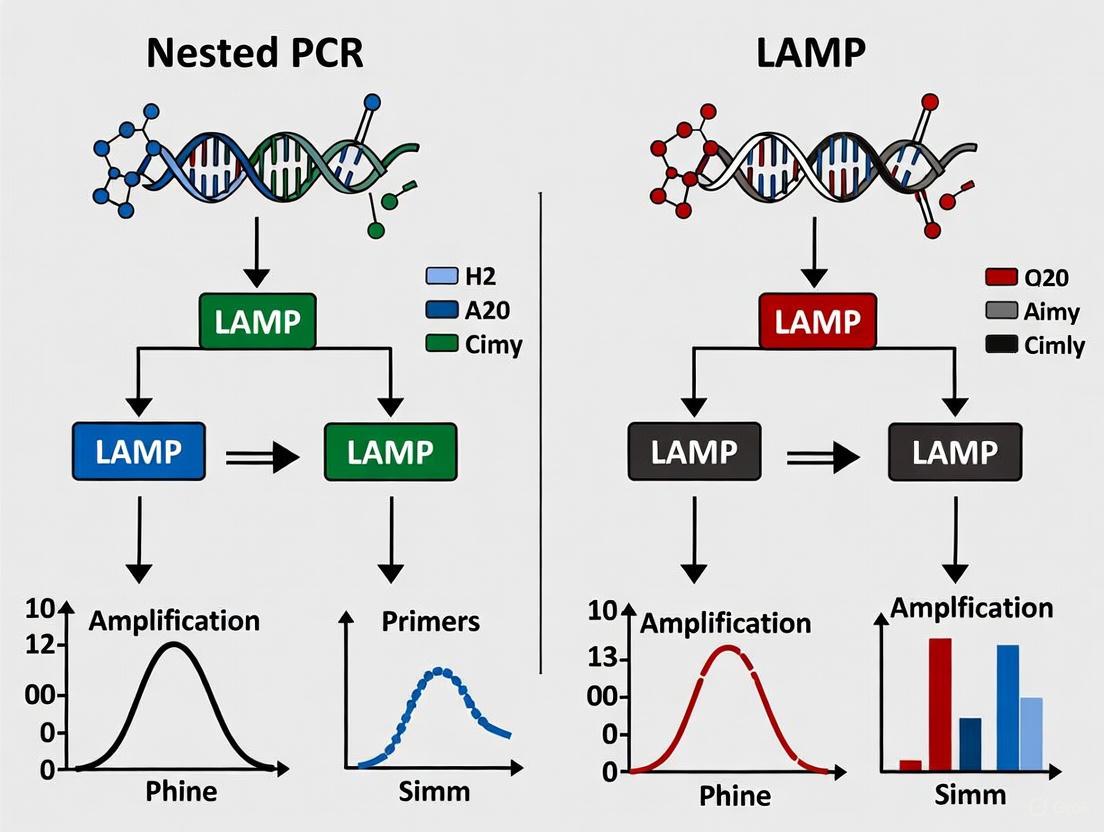
Figure 1: Comparative Workflow Principles of LAMP and Nested PCR. LAMP utilizes isothermal amplification with multiple primers enabling rapid, visual detection. Nested PCR requires thermal cycling with sequential primer sets followed typically by gel electrophoresis.
Comparative Performance Analysis
Sensitivity and Detection Limits
Sensitivity, defined as the lowest detectable concentration of target nucleic acid, represents a critical performance parameter for diagnostic assays. Direct comparative studies reveal distinct sensitivity profiles for nested PCR and LAMP, though their relative performance can vary depending on the specific target pathogen and assay optimization.
In SARS-CoV-2 detection, a highly sensitive one-step nested RT-PCR (OSN-qRT-PCR) demonstrated exceptional performance, achieving a limit of detection (LoD) of 189.1 copies/mL for the N gene, surpassing both conventional qRT-PCR (LoD: 528.1 copies/mL) and droplet digital PCR (LoD: 336.8 copies/mL) in direct comparisons [2]. This enhanced sensitivity translated to superior clinical performance, with the nested format detecting SARS-CoV-2 in 82.35% (28/34) of patient samples compared to 58.82% (20/34) for conventional qRT-PCR [2].
LAMP assays also demonstrate robust sensitivity, though typically slightly lower than nested formats. For SARS-CoV-2 detection, RT-LAMP assays reliably detected viral RNA at concentrations of 100-1000 copies per reaction [5], with one study reporting an LoD of 6.7 copies/reaction [1]. In non-COVID applications, a comparative study on Entamoeba histolytica detection reported that LAMP outperformed all PCR formats, detecting DNA equivalent to a single trophozoite, while both qPCR and nested PCR required 100 trophozoites for detection [3]. Similarly, for Alternaria solani detection, nested PCR was 100-fold more sensitive than LAMP, which was itself 10-fold more sensitive than conventional PCR [6].
Table 1: Comparative Sensitivity of Molecular Detection Methods
| Pathogen | LAMP LoD | Nested PCR LoD | qPCR LoD | Reference |
|---|---|---|---|---|
| SARS-CoV-2 | 6.7 copies/reaction [1] | 189.1 copies/mL [2] | 528.1 copies/mL [2] | |
| Entamoeba histolytica | 1 trophozoite [3] | 100 trophozoites [3] | 100 trophozoites [3] | |
| Alternaria solani | 100 fg DNA [6] | 10 fg DNA [6] | 1 fg DNA [6] | |
| Mycobacterium marinum | Equivalent to nested PCR in clinical samples [8] | 10-fold more sensitive than LAMP with pure DNA [8] | Not reported | |
| Fusarium tricinctum | 31 fg/μL [7] | 31 fg/μL [7] | 3.1 fg/μL [7] |
Diagnostic Accuracy for SARS-CoV-2 Detection
The diagnostic performance of LAMP for SARS-CoV-2 detection has been extensively evaluated against the reference standard of RT-qPCR across multiple clinical studies. These investigations consistently demonstrate that LAMP maintains high diagnostic accuracy, particularly during the acute phase of infection when viral loads are highest.
A comprehensive evaluation of RT-LAMP using 124 nasopharyngeal swab samples from 24 COVID-19 patients revealed time-dependent performance characteristics [1]. During the first 9 days after symptom onset—when viral loads typically peak—RT-LAMP demonstrated 92.8% positivity with 100% sensitivity and specificity compared to RT-qPCR [1]. However, this performance declined in later disease stages, with positivity rates dropping below 25% after the 10th day post-onset as viral loads diminished [1]. This pattern underscores that LAMP performs with diagnostic accuracy equivalent to RT-qPCR during the acute symptomatic phase when patients are most infectious and in need of rapid diagnosis.
Nested PCR formats have demonstrated even enhanced detection capabilities for SARS-CoV-2. The one-step nested qRT-PCR approach detected 82.35% of clinical samples compared to 58.82% for conventional qRT-PCR and 67.65% for droplet digital PCR, indicating superior identification of patients with low viral loads [2]. This enhanced sensitivity positions nested PCR as a valuable tool for detecting SARS-CoV-2 in challenging samples where target concentration is limited.
Speed, Throughput, and Operational Considerations
The temporal requirements for diagnostic results represent a crucial practical consideration, particularly during outbreak responses where rapid case identification informs public health interventions.
LAMP exhibits significant advantages in terms of assay speed and simplicity. Typical LAMP reactions are completed within 30-60 minutes at a constant temperature, with some assays generating detectable signals in as little as 20 minutes [5] [6]. This rapid turnaround stems from the isothermal nature of the reaction, which eliminates time-consuming thermal cycling steps. Furthermore, the ability to visualize results through color changes or turbidity without electrophoresis streamlines the detection process [7] [5].
In contrast, nested PCR protocols inherently require more time due to their sequential amplification design. The initial PCR round typically takes 1-2 hours, followed by product transfer and a second amplification round of similar duration, resulting in total processing times of 2-4 hours [3]. Additionally, the requirement for post-amplification analysis by gel electrophoresis further extends the time to result and increases hands-on technical requirements.
Table 2: Operational Comparison of LAMP and Nested PCR
| Parameter | LAMP | Nested PCR |
|---|---|---|
| Amplification Time | 15-60 minutes [5] [6] | 2-4 hours (including two rounds) [3] |
| Temperature Requirement | Single isothermal temperature (60-65°C) [4] | Thermal cycling (2-3 temperatures) [3] |
| Result Detection | Visual (color change), turbidity, or fluorescence [7] [5] | Typically requires gel electrophoresis [3] |
| Equipment Needs | Simple heat block or water bath [1] | Thermal cycler, electrophoresis equipment [3] |
| Risk of Contamination | Lower (single closed-tube reaction) [4] | Higher (tube transfer between rounds) [3] |
| Throughput Potential | High (simple procedure) [5] | Moderate (technically demanding) [3] |
| Ease of Use | Suitable for point-of-care and field use [1] | Requires laboratory setting and technical expertise [3] |
Experimental Protocols and Reagent Requirements
Standardized Nested PCR Protocol for SARS-CoV-2 Detection
The following protocol outlines a optimized one-step nested RT-PCR approach for SARS-CoV-2 detection as validated in comparative studies [2]:
RNA Extraction: Extract viral RNA from clinical samples (nasopharyngeal swabs, saliva) using commercial nucleic acid extraction kits (e.g., QIAamp Viral RNA Mini Kit) with automated extraction devices recommended for consistency.
First Amplification Round:
- Prepare 25 μL reaction mixture containing:
- 12.5 μL of 2× PCR buffer
- 1 μL of outer forward primer (10 μM)
- 1 μL of outer reverse primer (10 μM)
- 2 μL of template RNA
- Nuclease-free water to volume
- Thermal cycling conditions:
- Reverse transcription: 50°C for 15 minutes
- Initial denaturation: 95°C for 2 minutes
- 35 cycles of: 95°C for 15 seconds, 58-62°C for 30 seconds, 72°C for 30 seconds
- Final extension: 72°C for 5 minutes
- Prepare 25 μL reaction mixture containing:
Second Amplification Round:
- Prepare 50 μL reaction mixture containing:
- 25 μL of 2× PCR master mix
- 1 μL of inner forward primer (10 μM)
- 1 μL of inner reverse primer (10 μM)
- 1 μL of first-round product (diluted 1:10)
- Nuclease-free water to volume
- Thermal cycling conditions identical to first round
- Prepare 50 μL reaction mixture containing:
Product Analysis: Analyze amplified products by agarose gel electrophoresis (2%) with ethidium bromide staining and visualize under UV light.
This protocol specifically targets the ORF1ab and N genes of SARS-CoV-2, with primer sequences designed to ensure specificity for the internal regions [2].
Optimized RT-LAMP Protocol for SARS-CoV-2 Detection
The following protocol describes a standardized RT-LAMP procedure for SARS-CoV-2 detection as implemented in clinical evaluations [5] [1]:
RNA Extraction: Extract viral RNA using standardized methods (consistent with nested PCR requirements).
Reaction Setup:
- Prepare 25 μL reaction mixture containing:
- 15 μL of LAMP reaction mix (commercial kits available)
- 1.6 μM each of FIP and BIP primers
- 0.2 μM each of F3 and B3 primers
- 0.4 μM each of Loop F and Loop B primers (if using 6-primer system)
- 10 μL of template RNA
- Primer sets typically target SARS-CoV-2 ORF1ab (RdRP, nsp3) or N genes [5]
- Prepare 25 μL reaction mixture containing:
Amplification:
- Incubate reaction at 62-65°C for 30-45 minutes in a heat block or water bath
- No thermal cycling required
Result Detection:
- Colorimetric: Positive reactions change from pink to yellow via pH-sensitive dyes
- Fluorescent: Add SYBR Green dye and visualize under UV light - green fluorescence indicates amplification
- Turbidity: Measure precipitation of magnesium pyrophosphate in real-time turbidimeter
Figure 2: Experimental Workflows for LAMP and Nested PCR Detection. LAMP utilizes a single-tube isothermal amplification with direct detection, while nested PCR requires sequential amplification with product transfer between rounds followed by gel analysis.
Essential Research Reagent Solutions
Table 3: Essential Reagents for Molecular Detection Assays
| Reagent/Category | Specific Examples | Function in Assay |
|---|---|---|
| Nucleic Acid Extraction | QIAamp Viral RNA Mini Kit [1], Column Fungal DNAout Kit [7] | Isolation of high-quality template DNA/RNA from clinical samples |
| Polymerase Enzymes | Bst DNA polymerase (LAMP) [5], Taq DNA polymerase (nested PCR) [3] | DNA amplification with strand displacement (Bst) or thermal stability (Taq) |
| Primer Sets | SARS-CoV-2 ORF1ab/N gene primers [2] [5], CYP51C gene primers [7] | Target-specific sequence recognition and amplification initiation |
| Amplification Buffers | WarmStart Colorimetric LAMP Master Mix [5], Taq PCR Master Mix [3] | Optimal reaction environment with Mg²⁺, dNTPs, and stabilizers |
| Detection Systems | Hydroxy naphthol blue dye [7], SYBR Green fluorescence [5], Ethidium bromide gels [3] | Visual or instrumental detection of amplification products |
| Positive Controls | Synthetic SARS-CoV-2 RNA controls [5] [9], Cloned target sequences [6] | Assay validation and quality control |
Application Contexts and Selection Guidelines
Scenario-Based Method Selection
The choice between nested PCR and LAMP amplification methodologies should be guided by specific application requirements, available infrastructure, and performance priorities.
LAMP is distinctly superior in settings requiring:
- Rapid point-of-care testing: The isothermal nature, visual detection, and short turnaround time make LAMP ideal for clinical settings, field testing, and resource-limited environments [1].
- High-throughput screening: Simplified procedures enable processing of large sample volumes without sophisticated equipment [5].
- Resource-limited settings: Minimal equipment requirements (simple heat blocks) and reduced technical expertise lower implementation barriers [8].
Nested PCR demonstrates advantages for:
- Maximum sensitivity requirements: When detecting low-abundance targets is critical, particularly in late-stage infections or environmental samples with minimal pathogen load [2].
- Research applications: Where definitive target identification is prioritized over speed, and specialized equipment is available [3].
- Challenging samples: The two-step amplification process dilutes inhibitors, improving performance with complex matrices like soil or stool samples [3].
Technical Implementation Considerations
Successful implementation of either methodology requires careful attention to technical considerations specific to each platform:
For LAMP assays, key considerations include:
- Primer design complexity: Six primer regions must be carefully designed for optimal reaction kinetics and specificity [5].
- False-positive management: Robust contamination control measures are essential due to high amplification efficiency and product accumulation [5].
- Inhibitor susceptibility: While generally robust, some sample matrices may require purification or dilution to overcome inhibition [6].
For nested PCR protocols, critical factors include:
- Contamination prevention: Physical separation of pre- and post-amplification areas and dedicated equipment are mandatory to prevent false positives [3].
- Primer compatibility: Inner and outer primer sets must be designed to avoid dimerization and ensure efficient second-round amplification [2].
- Protocol optimization: Reaction conditions for both rounds require independent optimization and validation [7].
Nested PCR and LAMP represent two powerful yet fundamentally distinct approaches to nucleic acid amplification with complementary strengths and applications. Nested PCR offers exceptional sensitivity and specificity through its sequential amplification process, making it invaluable for detecting low-abundance targets in research and specialized diagnostic applications. LAMP technology provides unmatched speed and operational simplicity with its isothermal amplification and visual detection capabilities, positioning it as an ideal platform for rapid point-of-care testing and resource-limited settings.
Within the context of SARS-CoV-2 diagnostics, both methods have demonstrated clinical utility, with LAMP performing equivalently to RT-qPCR during acute infection phases [1], and nested PCR formats achieving superior sensitivity for low viral load detection [2]. The selection between these methodologies should be guided by specific application requirements, with nested PCR preferred for maximum sensitivity in laboratory settings, and LAMP recommended for rapid deployment, field use, and point-of-care testing scenarios. As molecular diagnostics continue to evolve, both techniques will undoubtedly play significant roles in the diagnostic landscape, offering powerful options for pathogen detection across diverse settings and applications.
The Role of Molecular Diagnostics in SARS-CoV-2 Surveillance and Research
Molecular diagnostics have been pivotal in managing the COVID-19 pandemic, enabling detection of SARS-CoV-2, tracking its spread, and informing public health interventions. This guide provides a comparative analysis of two key molecular techniques—Nested PCR and Loop-Mediated Isothermal Amplification (LAMP)—focusing on their diagnostic accuracy and application in research and surveillance.
The accurate detection of the SARS-CoV-2 virus is a cornerstone of the public health response to the COVID-19 pandemic. Molecular diagnostics target the genetic material of the virus and have served as the primary tool for confirming active infections, conducting surveillance, and supporting research into viral dynamics. Reverse transcription real-time quantitative polymerase chain reaction (RT-qPCR) is widely regarded as the gold standard for clinical detection [10]. However, techniques like nested PCR and LAMP have been developed and optimized to address some of RT-qPCR's limitations, such as potential false-negative results in samples with low viral loads and the need for sophisticated laboratory equipment [11] [10]. The ongoing evolution of the virus and the integration of SARS-CoV-2 surveillance into routine respiratory virus monitoring underscore the continuous need for reliable, sensitive, and accessible testing methods [12].
This section details the fundamental principles and procedures of nested PCR and LAMP assays.
Nested PCR
Nested PCR is a highly sensitive technique that utilizes two sequential amplification reactions, each with a different pair of primers. The second set of primers binds internal to the first set, leading to a second round of amplification that increases both the sensitivity and specificity of the assay by reducing non-specific amplification products [11]. A one-step nested quantitative real-time PCR (OSN-qRT-PCR) format has been developed to combine both amplification reactions in a single tube, minimizing the risk of cross-contamination that can occur in traditional two-step protocols [11].
- Experimental Protocol (One-Step Nested RT-PCR): The following protocol is adapted from validation studies for SARS-CoV-2 detection [11].
- RNA Extraction: Total RNA is extracted from clinical samples (e.g., throat swabs, sputum) using membrane adsorption kits. The purified RNA is eluted in a small volume of elution buffer.
- Reaction Setup: The OSN-qRT-PCR reaction mixture is prepared with a kit containing a master mix and enzyme mixture. The template RNA is added.
- Amplification: The reaction is run on a real-time PCR system under the following conditions:
- Reverse Transcription: 50°C for 30 minutes.
- Initial Denaturation & Enzyme Activation: 95°C for 1 minute.
- First Stage Amplification: 20 cycles of:
- 95°C for 30 seconds (denaturation)
- 70°C for 40 seconds (annealing/extension)
- Second Stage Amplification: 40 cycles of:
- 95°C for 15 seconds (denaturation)
- 60°C for 30 seconds (annealing/extension)
A significant increase in fluorescence during the second stage amplification indicates a positive result.
Loop-Mediated Isothermal Amplification (LAMP)
LAMP is an isothermal nucleic acid amplification technique that uses 4 to 6 primers targeting 6 to 8 distinct regions of the genome. This provides high specificity. Amplification occurs at a constant temperature (usually 60–65°C) using a strand-displacing DNA polymerase, eliminating the need for a thermal cycler [10]. Results can be visualized through methods such as colorimetric change (e.g., a pH indicator) or fluorescence [13].
- Experimental Protocol (One-Step Colorimetric RT-LAMP): This protocol is adapted for the detection of SARS-CoV-2 in saliva, utilizing a colorimetric readout [13].
- Sample Preparation: Saliva samples are minimally processed, often involving a simple heating step (e.g., 95°C for 2-5 minutes) to inactivate the virus and release RNA, bypassing the need for RNA extraction.
- Reaction Setup: The LAMP reaction mixture is prepared containing:
- A primer set (e.g., targeting the N or E gene of SARS-CoV-2).
- Bst DNA/RNA Polymerase with reverse transcriptase activity.
- dNTPs.
- A colorimetric pH indicator (e.g., phenol red).
- The prepared sample.
- Amplification & Detection: The reaction is incubated at 60–65°C for 20–40 minutes. A color change from pink (alkaline) to yellow (acidic) indicates a positive test due to a drop in pH from nucleotide hydrolysis. Results should be read dynamically using a handheld instrument to improve accuracy over endpoint observation [13].
Visual Comparison of Workflows
The diagram below illustrates the key procedural differences between the two methods.
Performance Data and Comparative Analysis
Direct comparisons of nested PCR and LAMP reveal distinct performance characteristics, particularly regarding sensitivity and suitability for different settings.
Quantitative Performance Comparison
The table below summarizes key performance metrics from validation studies.
Table 1: Diagnostic Performance Comparison of Nested PCR and LAMP vs. RT-qPCR
| Method | Sensitivity | Specificity | Limit of Detection (LoD) | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Nested PCR | 82.35% [11] | High (kit-dependent) [11] | 189.1 copies/mL (N gene) [11] | Superior sensitivity for low viral loads [11] | Higher risk of amplicon contamination; longer process [14] |
| LAMP | 79% (overall) [15] | 97% (overall) [15] | ~50 genomes/reaction (saliva) [13] | Speed, simplicity, minimal equipment [16] [13] | Moderate overall sensitivity; can be affected by sample inhibitors [15] |
| LAMP (with RNA Extraction) | 88% [15] | High (comparable to overall) [15] | Improved vs. without extraction [15] | Improved sensitivity | Adds time and complexity |
| LAMP (without RNA Extraction) | 50% [15] | High (comparable to overall) [15] | Reduced vs. with extraction [15] | Maximum speed and simplicity | Significantly reduced sensitivity |
Analysis of Key Findings
Sensitivity in Low Viral Load Cases: Nested PCR demonstrates a clear advantage in detecting low viral loads. One study directly comparing OSN-qRT-PCR to ddPCR and standard RT-qPCR found that nested PCR had a higher positive detection rate (82.35%) than both ddPCR (67.65%) and RT-qPCR (58.82%) in clinical samples from confirmed patients [11]. This makes it particularly valuable for confirming infection in convalescent patients or those with low viral shedding.
Impact of RNA Extraction on LAMP: A meta-analysis of LAMP studies concluded that RNA extraction is a critical factor influencing test sensitivity. The sensitivity of LAMP with RNA extraction was 88%, but it dropped to just 50% without RNA extraction [15]. This highlights a trade-off between the simplicity of direct assays and diagnostic accuracy.
Operational and Practical Considerations: LAMP is notably faster (results in under 2 hours) and can be performed with minimal equipment (a heating block or water bath), making it suitable for point-of-care or resource-limited settings [13] [10]. In contrast, nested PCR, while highly sensitive, is a longer process and carries an inherent risk of laboratory contamination from amplicon exposure, which requires stringent controls [14].
Essential Research Reagents and Materials
Successful implementation of these molecular assays requires specific reagent solutions. The following table lists key materials and their functions.
Table 2: Research Reagent Solutions for SARS-CoV-2 Molecular Assays
| Reagent / Material | Function | Example Application |
|---|---|---|
| Primer Sets (Nested PCR) | Target-specific amplification in two rounds (external and internal primers) for high sensitivity/specificity [14] [11]. | Amplification of SARS-CoV-2 N gene [14]. |
| LAMP Primer Sets | Set of 4-6 primers recognizing 6-8 distinct genome regions for specific isothermal amplification [16] [13]. | Targeting SARS-CoV-2 E1 and N2 genes [13]. |
| Bst DNA/RNA Polymerase | Strand-displacing DNA polymerase with reverse transcriptase activity for one-step RT-LAMP [16]. | Isothermal amplification in LAMP assays [16]. |
| Colorimetric pH Indicator | Visual detection of amplification by color change (e.g., pink to yellow) [13]. | Phenol red for endpoint or real-time LAMP readout [13]. |
| Viral RNA Extraction Kit | Purification of high-quality RNA from clinical samples, critical for assay sensitivity [11] [15]. | Membrane adsorption-based kits for nested PCR and LAMP [11]. |
| One-Step RT-PCR Master Mix | Contains reverse transcriptase and DNA polymerase for combined cDNA synthesis and PCR [14] [11]. | One-Step Nested RT-PCR kits [11]. |
Application in Surveillance and Research
Molecular diagnostics are crucial for public health surveillance and research. Techniques like LAMP are well-suited for rapid screening and early outbreak detection in community settings due to their speed and simplicity [15]. The CDC monitors key indicators such as test positivity and emergency department visits as early signals of potential increases in COVID-19 activity [17]. Genomic surveillance, often relying on PCR-based methods, is used to track the emergence and prevalence of variants like XFG and NB.1.8.1 [12].
In research, highly sensitive methods like nested PCR are invaluable for studies requiring detection of low viral loads, such as investigating long COVID pathogenesis or assessing viral clearance [11] [18]. The ability to accurately detect pre-symptomatic or asymptomatic infection using host-response transcriptomic signatures like IFI27 further expands the utility of molecular tools in understanding infection dynamics [19].
Both nested PCR and LAMP are powerful molecular techniques with distinct roles in SARS-CoV-2 research and surveillance. The choice between them depends on the specific application requirements.
- Nested PCR is the more sensitive technique, making it the preferred choice for diagnostic confirmation in research settings where detecting low viral loads is critical, and where laboratory infrastructure can mitigate contamination risks.
- LAMP offers a rapid, simple, and equipment-minimal alternative, ideal for rapid screening, point-of-care testing, and surveillance in resource-limited environments, especially when performed with RNA extraction to maintain adequate sensitivity.
The continued development and refinement of both technologies will enhance our preparedness for future outbreaks and deepen our understanding of viral diseases.
The global response to the COVID-19 pandemic has fundamentally relied on molecular diagnostics for detecting the presence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The virus possesses a ~30 kb positive-sense single-stranded RNA genome that encodes several structural and non-structural proteins [20]. Among these, three key genetic targets have emerged as critical for diagnostic assays: the nucleocapsid (N) gene, open reading frame 1ab (ORF1ab), and spike (S) gene [20]. These targets are prioritized due to their conservation, abundance, and unique genomic characteristics that facilitate reliable detection. The N gene encodes the RNA-binding nucleocapsid protein that packages the viral genome, the ORF1ab region encodes proteins essential for viral replication including RNA-dependent RNA polymerase, and the S gene encodes the spike protein responsible for host cell entry [20]. This guide provides a comprehensive comparison of two prominent amplification techniques—nested PCR and loop-mediated isothermal amplification (LAMP)—for detecting these key genetic targets, with supporting experimental data from recent studies to inform researchers, scientists, and drug development professionals.
Comparative Performance of Genetic Targets
Analytical Characteristics of Key Genetic Targets
Different genetic targets offer varying advantages for SARS-CoV-2 detection based on their abundance, conservation, and susceptibility to mutation. The following table summarizes the key characteristics of the primary genetic targets used in diagnostic assays.
Table 1: Key genetic targets for SARS-CoV-2 detection
| Genetic Target | Function | Abundance | Conservation | Mutation Concerns |
|---|---|---|---|---|
| N Gene | Encodes nucleocapsid protein that packages viral RNA | High | High | Lower mutation rate compared to S gene |
| ORF1ab | Encodes replicase polyproteins essential for viral replication | Moderate | High | Partial target failure reported in some variants [21] |
| S Gene | Encodes spike protein responsible for host cell entry | Moderate | Lower due to selective pressure | Higher mutation rate, potential for target failure [22] |
Performance Comparison of Detection Methods
Multiple studies have directly compared the performance of nested PCR and LAMP methods for detecting SARS-CoV-2. The following table synthesizes key performance metrics from experimental studies.
Table 2: Performance comparison of nested PCR versus LAMP for SARS-CoV-2 detection
| Parameter | Nested PCR | LAMP | Experimental Context |
|---|---|---|---|
| Sensitivity | 100% [23] | 98.33% [24] | Compared to RT-qPCR as reference |
| Specificity | 100% [23] | 98.73% [24] | Compared to RT-qPCR as reference |
| Limit of Detection | 0.015 ng/μL RNA [23] | 6.7 copies/reaction [1] | Using serial dilutions of reference material |
| Time to Result | ~3-4 hours (including reverse transcription) [23] | ~30-60 minutes [16] [1] | From extracted RNA to result |
| Optimal Detection Window | Not specifically defined | Up to 9 days post-symptom onset [1] | Based on clinical sample evaluation |
| Equipment Requirements | Thermal cyclers, electrophoresis equipment | Isothermal equipment (water bath or block) | Laboratory infrastructure |
Experimental Protocols and Methodologies
Nested PCR Protocol for N Gene Detection
A validated nested PCR protocol targeting the N gene of SARS-CoV-2 demonstrated high sensitivity and specificity in clinical samples [23]. The methodology involves the following steps:
RNA Extraction: Viral RNA is extracted using commercial kits such as the ISOLATE II RNA Mini Kit. Extracted RNA should be evaluated for purity using spectrophotometry (260/280 ratio ~2.0) [23].
Reverse Transcription: Convert RNA to cDNA using reverse transcriptase enzyme. A typical 20 μL reaction contains: 7 μL extracted RNA, 8 μL DEPC-treated water, 4 μL TransAmp buffer, and 1 μL reverse transcriptase enzyme. Reaction conditions: 25°C for 10 minutes, 42°C for 15 minutes, and 80°C for 5 minutes [23].
First Round PCR: Amplify the target using external primers. Reaction mixture (25 μL): 12.5 μL My Taq HS red mix, 4 μL cDNA, 1 μL each external primer (10 pmol/μL), and 6.5 μL PCR grade water. Primers target the N gene: Ext2019nCorVF (5′-GGCAGTAACCAGAATGGAGA-3′) and Ext2019nCorVR (5′-CTCAGTTGCAACCCATATGAT-3′) positioned at 28346-28365 and 28681-28661, respectively [23]. Thermal cycling: Initial denaturation at 95°C for 1 minute, followed by 35 cycles of 95°C for 15 seconds, 55°C for 15 seconds, and 72°C for 20 seconds.
Second Round (Nested) PCR: Use the first PCR product as template with internal primers. Reaction mixture (25 μL): 12.5 μL My Taq HS red mix, 0.5 μL first PCR product, 1 μL each internal primer (10 pmol/μL), and 10 μL PCR grade water. Internal primers: intF (5′-CACCGCTCTCACTCAACAT-3′) and intR (5′-CATAGGGAAGTCCAGCTTCT-3′) positioned at 28432-28450 and 28643-28624, respectively [23]. Use the same thermal cycling conditions as the first round.
Product Analysis: Analyze amplified products by gel electrophoresis (2% agarose, 120-150V for 30 minutes) with expected band sizes of 335 bp (first round) and 212 bp (nested) [23].
LAMP Protocol for SARS-CoV-2 Detection
A optimized one-step RT-LAMP protocol for detecting SARS-CoV-2 targeting the N gene demonstrates rapid detection with high sensitivity [16]:
RNA Extraction: Extract RNA using commercial viral RNA extraction kits. Evaluate RNA purity and concentration using spectrophotometry (260/280 ratio ~2.0) [16].
LAMP Reaction Preparation: Prepare a 25 μL reaction mixture containing: 5-10 μL of extracted RNA template, 40 pmol each of FIP and BIP primers, 5 pmol each of F3 and B3 primers, 20 pmol each of LF and LB loop primers, 1 μL of Bst DNA/RNA Polymerase (8 U/μL), and appropriate reaction buffer [16].
Primer Design: LAMP primers should recognize eight distinct regions of the target sequence. For the N gene, design primers using software such as Primer Explorer V5 and verify specificity using BLAST against the SARS-CoV-2 reference database [16].
Amplification: Incubate the reaction mixture at 62-65°C for 30-60 minutes. No initial denaturation or reverse transcription step is required in optimized one-step protocols [16] [1].
Result Detection: Monitor amplification in real-time using turbidimetry or detect endpoint results using colorimetric change (visible color change from positive samples) or fluorescence under UV light [1].
Research Reagent Solutions
The following table outlines essential research reagents and their functions for implementing SARS-CoV-2 detection assays.
Table 3: Essential research reagents for SARS-CoV-2 detection assays
| Reagent/Category | Specific Examples | Function/Purpose |
|---|---|---|
| RNA Extraction Kits | ISOLATE II RNA Mini Kit [23], QIAamp Viral RNA Mini Kit [1], MiniBEST Viral RNA/DNA Extraction Kit [25] | Isolation of high-quality viral RNA from clinical specimens |
| Enzymes | Bst DNA/RNA Polymerase [16], My Taq HS Polymerase [23], SuperScript IV VILO [22] | Nucleic acid amplification through polymerase activity |
| Primer Sets | ARTIC primer pools [22], N gene-specific primers [23], ORF1ab-specific primers [25] | Target-specific binding for amplification |
| Probe Systems | Ru(bpy)32+ labeled probes [25], TaqMan probes [20] | Signal generation for detection |
| Master Mixes | SensiFAST cDNA synthesis kit [23], TaqMan Fast Virus 1-Step Master Mix [1] | Optimized reaction components for efficient amplification |
| Controls | Twist Synthetic SARS-CoV-2 RNA Control [22], inactivated SARS-CoV-2 isolate [23] | Assay validation and quality assurance |
Methodological Workflows
The following diagram illustrates the key procedural steps and comparative features of nested PCR and LAMP methodologies:
Discussion and Research Implications
The comparative analysis of nested PCR and LAMP methods for detecting key SARS-CoV-2 genetic targets reveals distinct advantages and limitations for each approach. Nested PCR demonstrates exceptional sensitivity (100%) and specificity (100%) when targeting the N gene, making it particularly valuable for confirmatory testing and situations requiring high confidence in results [23]. However, this method requires more sophisticated equipment, longer processing times (3-4 hours), and specialized personnel, limiting its application in point-of-care or resource-limited settings.
LAMP technology offers significant advantages in terms of speed (30-60 minutes), operational simplicity, and minimal equipment requirements, needing only a constant temperature water bath or heating block for amplification [16] [1]. The method maintains high sensitivity (98.33%) and specificity (98.73%) compared to RT-qPCR, particularly during the acute phase of infection (up to 9 days post-symptom onset) [1] [24]. These characteristics make LAMP ideal for rapid screening programs, field deployment, and settings where technical expertise may be limited.
The selection of genetic targets presents another critical consideration for assay design. The N gene remains a preferred target due to its high abundance and relatively lower mutation rate compared to the S gene [22]. The ORF1ab region, while highly conserved, has demonstrated instances of partial target failure in emerging variants such as BA.2.12.1, highlighting the importance of monitoring assay performance against circulating strains [21]. For robust surveillance, multiplex approaches targeting multiple genetic regions provide insurance against diagnostic escape due to viral evolution.
Future research directions should focus on developing improved primer sets that account for viral evolution, optimizing multiplex detection systems, and integrating these amplification methods with novel detection platforms such as electrochemiluminescent biosensors [25]. Additionally, standardized validation protocols across laboratories would enhance the comparability of results and facilitate more rapid implementation of improved detection methodologies.
This guide provides an objective comparison of Nested PCR and Loop-Mediated Isothermal Amplification (LAMP) for SARS-CoV-2 detection, focusing on the technical requirements and analytical performance crucial for researchers and drug development professionals.
Performance Comparison: Nested PCR vs. LAMP
The following table summarizes the key performance characteristics of Nested PCR and LAMP assays based on experimental data from SARS-CoV-2 and other pathogen detection studies.
Table 1: Comparative Analytical Performance of Nested PCR and LAMP
| Performance Characteristic | Nested PCR | LAMP |
|---|---|---|
| Limit of Detection (LoD) | 0.015 ng/μL (SARS-CoV-2 RNA) [26]; 10-100x more sensitive than conventional PCR [6] | ~23 RNA copies/mL (SARS-CoV-2) [27]; ~10x more sensitive than conventional PCR [6] |
| Sensitivity (Clinical) | 100% (in a SARS-CoV-2 validation study) [26] | 89% (nasopharyngeal RNA), 80% (crude saliva) [28] |
| Specificity (Clinical) | 100% (in a SARS-CoV-2 validation study) [26] | 95% (nasopharyngeal RNA), 99% (crude saliva) [28] |
| Assay Speed | Slower; involves two sequential amplification rounds [29] | Faster; results in <60 minutes [28] [6] |
| Throughput | Lower; requires post-first-round manipulation, increasing hands-on time [29] | Higher; single-tube isothermal reaction is amenable to streamlining [7] |
| Risk of Contamination | Higher; due to transfer of first-round amplicon to a second tube [29] | Lower; single-tube closed-tube system [7] |
| Result Visualization | Typically requires gel electrophoresis or real-time analysis [26] | Multiple options: gel electrophoresis, turbidity, or colorimetric (visible to naked eye) [7] [28] |
Experimental Protocols and Workflows
The core difference between the two techniques lies in their amplification process, as illustrated in the following workflows.
Detailed Nested PCR Protocol for SARS-CoV-2
A validated protocol for detecting SARS-CoV-2, targeting the N gene, is as follows [26]:
- Primer Design: Two primer sets (external and internal) are designed to bind to the target gene. For example:
- External Primers: Ext2019nCorVF (5'-GGCAGTAACCAGAATGGAGA-3') and Ext2019nCorVR (5'-CTCAGTTGCAACCCATATGAT-3'), producing a 335 bp product.
- Internal Primers: intF (5'-CACCGCTCTCACTCAACAT-3') and intR (5'-CATAGGGAAGTCCAGCTTCT-3'), producing a 212 bp product [26].
- First Round PCR:
- Reaction Mix: 12.5 μL My Taq HS red mix, 4 μL cDNA template, 1 μL each external primer (10 pmol/μL), and PCR grade water to 25 μL.
- Cycling Conditions: Initial denaturation at 95°C for 1 min; 35-40 cycles of denaturation (95°C, 15 s), annealing (54-58°C, 15 s), and extension (72°C, 15 s); final extension at 72°C for 1 min [26].
- Second Round PCR:
- Reaction Mix: 12.5 μL My Taq HS red mix, 0.5-1 μL of the first PCR product as template, 1 μL each internal primer (10 pmol/μL), and PCR grade water to 25 μL.
- Cycling Conditions: Same as the first round.
- Detection: Products are analyzed via 2% agarose gel electrophoresis. A clear band at the expected size (e.g., 212 bp) confirms a positive result [26].
Detailed LAMP Protocol for SARS-CoV-2
A standardized colorimetric RT-LAMP protocol used in a multicountry study is outlined below [28]:
- Primer Design: Four to six primers (F3, B3, FIP, BIP, LF, LB) are designed to recognize six distinct regions of the target sequence. Primer design software like Primer Explorer V5 is typically used [7].
- Reaction Setup:
- The reaction mixture includes a buffer with betaine, MgSO₄ (4-8 mM), dNTPs (1.0-1.6 mM), Bst DNA polymerase, and the primer set.
- Template (extracted RNA or crude saliva) is added to the mix. The use of crude saliva requires an initial heating step (e.g., 95°C for 5 min) to inactivate the virus and release RNA [28].
- Amplification:
- The reaction is incubated at a constant temperature of 60-65°C for 30-60 minutes in a heating block, water bath, or portable incubator [28].
- Detection:
Equipment, Infrastructure, and Expertise
The fundamental difference in amplification principles dictates distinct infrastructure needs for each technique.
Table 2: Technical Requirements for Nested PCR and LAMP
| Technical Aspect | Nested PCR | LAMP |
|---|---|---|
| Core Instrument | Thermal Cycler (capable of precise temperature cycling) | Heating Block / Water Bath (maintaining a single isothermal temperature) [7] |
| Infrastructure Needs | Stable electrical supply; dedicated pre- and post-PCR areas to prevent contamination [29] | Minimal infrastructure; suitable for field deployment and resource-limited settings [28] |
| Technical Expertise | High; requires skilled pipetting for tube transfer and knowledge of PCR optimization | Moderate to Low; simpler procedure with minimal hands-on steps [7] |
| Cost & Accessibility | Higher instrument cost; requires specific consumables | Lower instrument cost; more accessible for peripheral laboratories [28] [10] |
Essential Research Reagent Solutions
The table below lists key reagents and their critical functions in establishing Nested PCR and LAMP assays.
Table 3: Key Research Reagents for Molecular Detection Assays
| Reagent / Kit | Function / Application | Technical Notes |
|---|---|---|
| RNA Extraction Kit (e.g., ISOLATE II RNA Mini Kit [26]) | Purification of viral RNA from clinical samples (swabs, saliva). | Critical for assay sensitivity; extraction-free protocols (e.g., for LAMP with saliva) exist but may reduce sensitivity [28]. |
| cDNA Synthesis Kit (e.g., SensiFAST cDNA kit [26]) | Reverse transcription of viral RNA into complementary DNA (cDNA). | Essential for standard Nested PCR. Some LAMP and one-step RT-PCR kits integrate this step [11]. |
| Bst DNA Polymerase | The core enzyme for LAMP amplification. Possesses strand-displacement activity. | Thermostable; enables isothermal amplification at 60-65°C [7]. |
| Taq DNA Polymerase | The core enzyme for PCR amplification. | Thermostable; used in both rounds of Nested PCR [26]. |
| Primer Sets | Specific oligonucleotides that bind to the target SARS-CoV-2 sequence. | Nested PCR requires two sets (external/internal). LAMP requires 4-6 primers for high specificity [7] [26]. |
| Colorimetric LAMP Dye (e.g., Hydroxy Naphthol Blue - HNB) | Visual pH indicator for result interpretation in LAMP. | Changes color upon amplification; enables naked-eye detection without opening tubes [7] [28]. |
Current Status and Adoption in Clinical and Research Laboratories
The COVID-19 pandemic necessitated rapid and accurate diagnostic testing on an unprecedented global scale. While real-time reverse transcription quantitative PCR (RT-qPCR) emerged as the gold standard, its limitations in resource-constrained settings accelerated the development and evaluation of alternative nucleic acid amplification techniques. Among these, Nested PCR and Loop-Mediated Isothermal Amplification (LAMP) have shown significant promise, each with distinct advantages and implementation challenges. This guide provides an objective comparison of these two methodologies within the context of SARS-CoV-2 diagnostic accuracy, synthesizing experimental data to inform researchers, scientists, and drug development professionals about their current status and adoption in laboratory settings.
Nested PCR: Principles and Process
Nested PCR is a two-stage amplification method that significantly enhances detection sensitivity and specificity. The process utilizes two sets of primers: an outer primer pair for the initial amplification, followed by an inner primer pair that binds within the first amplicon for a second round of amplification. This sequential approach substantially reduces non-specific amplification and improves detection limits for low viral load samples [11] [26]. For SARS-CoV-2 detection, this method has been successfully applied to target genes such as the N gene and ORF1ab [11] [26].
LAMP: Principles and Process
LAMP is an isothermal nucleic acid amplification technique that utilizes 4-6 primers recognizing 6-8 distinct regions of the target gene. The reaction proceeds at a constant temperature (typically 60-65°C) through a complex amplification mechanism involving strand displacement DNA synthesis. This method enables rapid amplification without the need for thermal cycling equipment, making it particularly suitable for point-of-care and resource-limited settings [30] [16]. Reverse transcription LAMP (RT-LAMP) has been adapted for SARS-CoV-2 RNA detection, with amplification results often detectable within 30-70 minutes [31] [16].
Figure 1: Comparative workflows for Nested PCR and RT-LAMP detection of SARS-CoV-2
Performance Comparison: Experimental Data Analysis
Diagnostic Sensitivity and Specificity
Recent studies have directly compared the performance of Nested PCR and LAMP for SARS-CoV-2 detection. The table below summarizes key performance metrics from multiple investigations:
Table 1: Comparative performance of Nested PCR and LAMP for SARS-CoV-2 detection
| Study | Method | Sensitivity | Specificity | Limit of Detection | Sample Size | Reference Standard |
|---|---|---|---|---|---|---|
| Zhao et al. (2020) [11] | One-Step Nested qRT-PCR | 82.35% | 100% | 189.1-194.74 copies/mL | 34 clinical samples | qRT-PCR |
| Spiteri et al. (2025) [30] | RT-LAMP | 26-30% | 75% | Not specified | 118 surface samples | RT-qPCR |
| Feng et al. (2023) [8] | Nested PCR | Higher than LAMP | 100% | 10-fold more sensitive than LAMP | 6 clinical samples | Culture |
| Feng et al. (2023) [8] | LAMP | Lower than nested PCR | 100% | 10-fold less sensitive than nested PCR | 6 clinical samples | Culture |
| FCV Study (2024) [31] | Nested PCR | 31.48% | Not specified | 100-1000 times more sensitive than conventional PCR | 54 clinical samples | Virus isolation |
| FCV Study (2024) [31] | RT-LAMP | 31.48% | 100% | 14.3 × 10¹ copies/μL | 54 clinical samples | Virus isolation |
Limits of Detection and Analytical Sensitivity
The analytical sensitivity of these methods varies significantly based on target genes, primer design, and reaction optimization:
One-Step Nested qRT-PCR demonstrated a limit of detection (LoD) of 189.1-194.74 copies/mL for SARS-CoV-2, significantly lower than conventional qRT-PCR (520.1-528.1 copies/mL) and ddPCR (336.8-401.8 copies/mL) [11].
RT-LAMP showed variable LoD depending on the target pathogen and primer design. For SARS-CoV-2, one study reported 100% detection at 200 RNA copies, with reduced sensitivity (70-90%) at 100-150 copies [32]. Another study reported LoD of 14.3 × 10¹ copies/μL for Feline Calicivirus detection [31].
Conventional Nested PCR demonstrated 10-fold higher sensitivity compared to LAMP for Mycobacterium marinum detection in clinical skin specimens [8].
Clinical Application and Validation
In clinical settings, both techniques have been successfully applied to various sample types:
Nested PCR enabled detection of SARS-CoV-2 in six cat samples during the first COVID-19 wave in Bulgaria, with 100% sensitivity and specificity compared to reference methods [26].
RT-LAMP showed 79% diagnostic sensitivity for SARS-CoV-2 in clinical samples compared to RT-qPCR, with 100% of samples with Ct <30 testing positive [32]. Another study reported 93-94% agreement with RT-qPCR across saliva and nasopharyngeal samples [16].
Experimental Protocols and Methodologies
Nested PCR Protocol for SARS-CoV-2 Detection
Based on established methodologies [11] [26], the following protocol can be implemented for SARS-CoV-2 detection:
RNA Extraction and cDNA Synthesis:
- Extract RNA using approved kits (e.g., ISOLATE II RNA Mini kit)
- Perform reverse transcription using SensiFAST cDNA synthesis kit
- Reaction conditions: 25°C for 10 min, 42°C for 15 min, 80°C for 5 min
First Round PCR:
- Reaction volume: 25 μL containing 12.5 μL master mix, 4 μL cDNA, 1 μL each external primer (10 pmol/μL)
- Primers targeting N gene or ORF1ab regions
- Cycling conditions: Initial denaturation at 95°C for 5 min, 35 cycles of (95°C for 30s, 54-62°C for 50s, 72°C for 1 min), final extension at 72°C for 10 min
Second Round PCR:
- Reaction volume: 25 μL containing 12.5 μL master mix, 0.5-1 μL first PCR product, 1 μL each internal primer (10 pmol/μL)
- Cycling conditions: Similar to first round with optimized annealing temperature
- Product analysis: 2% agarose gel electrophoresis with ethidium bromide staining
RT-LAMP Protocol for SARS-CoV-2 Detection
Based on optimized protocols [16] [32], the RT-LAMP reaction can be performed as follows:
Primer Design:
- Design primers targeting conserved regions of SARS-CoV-2 N gene or ORF1ab
- Use Primer Explorer V5 software for designing 6 primers: F3, B3, FIP, BIP, LF, LB
- Verify specificity using BLAST analysis against SARS-CoV-2 databases
Reaction Setup:
- Reaction volume: 20-25 μL containing 15-20 μL reaction mix, 1.6-40 pmol each of FIP/BIP primers, 0.2-5 pmol each of F3/B3 primers, 20 pmol each of LF/LB primers
- Enzyme: 8U Bst DNA/RNA Polymerase
- Indicator: Neutral red, calcein-manganese, or SYBR green for visual detection
Amplification Conditions:
- Temperature: 56.3-65°C for 30-70 minutes
- No initial denaturation or reverse transcription separation required
- Results interpretation: Color change (pink to yellow for pH indicators) or turbidity measurement
Critical Considerations:
- Optimal primer ratio typically 1:8 (F3/B3:FIP/BIP) [8]
- Reaction times exceeding 30 minutes may cause nonspecific amplification [32]
- Orange color intermediates may occur with low viral loads, requiring confirmation [32]
Research Reagent Solutions
Table 2: Essential research reagents for Nested PCR and LAMP assays
| Category | Specific Product | Application | Function |
|---|---|---|---|
| RNA Extraction | ISOLATE II RNA Mini Kit [26] | Both methods | Viral RNA purification from clinical samples |
| QIAamp Viral Mini Kit [30] | Both methods | RNA extraction, superior for surface samples | |
| Reverse Transcription | SensiFAST cDNA Synthesis Kit [26] | Nested PCR | First-strand cDNA synthesis from RNA template |
| Amplification Enzymes | Bst DNA/RNA Polymerase [16] | RT-LAMP | Isothermal amplification with reverse transcriptase activity |
| Taq DNA Polymerase [26] | Nested PCR | Thermostable DNA polymerase for PCR amplification | |
| Detection Systems | Neutral Red Indicator [31] | RT-LAMP | Colorimetric pH change detection |
| Ethidium Bromide [26] | Nested PCR | Nucleic acid staining for gel visualization | |
| Positive Controls | SARS-CoV-2 Pseudovirus [11] | Both methods | Assay validation and quality control |
Adoption Challenges and Implementation Considerations
Laboratory Infrastructure Requirements
The implementation of these techniques requires different infrastructure considerations:
Nested PCR requires conventional thermal cyclers and gel electrophoresis equipment, which are widely available in molecular diagnostics laboratories. However, the two-step amplification process increases hands-on time and contamination risk [26] [8].
RT-LAMP requires only a simple water bath or dry block heater maintained at isothermal conditions, significantly reducing equipment costs and complexity. This makes it particularly suitable for resource-limited settings and point-of-care testing [16] [33].
Contamination Control and Specificity
Nested PCR poses significant contamination risks due to tube opening between amplification rounds. Laboratory spatial separation and dedicated equipment for pre- and post-amplification steps are essential to prevent false positives [26].
RT-LAMP is less prone to contamination as reactions are typically performed in single closed tubes. The use of 6-8 primers targeting distinct regions provides inherent specificity, though primer-dimer formations can occasionally cause false positives [32] [8].
Interpretation and Standardization
Figure 2: RT-LAMP result interpretation challenges with colorimetric detection
RT-LAMP with colorimetric detection presents interpretation challenges, particularly with low viral load samples that may produce intermediate colors (orange) rather than definitive positive (yellow) or negative (pink) results [32]. These indeterminate results require confirmatory testing by RT-qPCR to avoid false diagnoses.
Nested PCR results are typically interpreted through gel electrophoresis, providing clear size-based amplicon verification. This binary interpretation (presence/absence of bands) reduces ambiguity but adds post-amplification processing time [26].
Both Nested PCR and LAMP technologies offer valuable alternatives to conventional RT-qPCR for SARS-CoV-2 detection, with complementary strengths and limitations. Nested PCR provides superior sensitivity and established reliability, making it suitable for laboratory settings where maximum detection sensitivity is required. Conversely, RT-LAMP offers rapid results, minimal equipment requirements, and point-of-care applicability, albeit with generally lower sensitivity compared to nested methods.
The selection between these methodologies should be guided by specific application requirements, including available infrastructure, required throughput, sensitivity demands, and intended use settings. For clinical diagnostics requiring the highest sensitivity, particularly with low viral load samples, Nested PCR remains preferable. For rapid screening, resource-limited settings, and point-of-care applications, RT-LAMP provides a practical and efficient alternative. Future developments in primer design, reaction optimization, and detection methodologies will likely enhance both platforms' performance and expand their adoption in clinical and research laboratories.
Protocol Development and Practical Application in Research Settings
Step-by-Step Protocol for SARS-CoV-2 Nested PCR
Nested polymerase chain reaction (PCR) is a highly sensitive molecular technique that utilizes two successive amplification reactions with two sets of primers to detect SARS-CoV-2 RNA with enhanced sensitivity and specificity. This method addresses critical limitations of conventional single-step PCR, particularly for detecting low viral loads in clinical samples. The fundamental principle involves an initial amplification round using outer primers, followed by a second round using inner primers that bind within the first amplicon, significantly reducing non-specific amplification and increasing detection capability for the SARS-CoV-2 virus [11] [3].
In the context of the COVID-19 pandemic, diagnostic sensitivity has proven crucial for identifying infected individuals, particularly those with low viral loads who may still transmit the virus. Conventional reverse transcriptase quantitative PCR (qRT-PCR), while considered the gold standard, has demonstrated variable positive rates from throat swab samples ranging from 30-60%, creating an urgent need for more sensitive detection methods [11]. One-step nested (OSN) quantitative RT-PCR platforms have emerged as promising alternatives, showing significantly improved detection limits compared to both digital PCR (ddPCR) and conventional qRT-PCR methods [11]. When compared to other amplification techniques like loop-mediated isothermal amplification (LAMP), nested PCR demonstrates distinct advantages in sensitivity and reliability, though with differing requirements for instrumentation and operational complexity [8] [6] [3].
Principle and Advantages of Nested PCR
Technical Foundations
Nested PCR operates through a two-stage amplification process that substantially improves detection accuracy for SARS-CoV-2. The first amplification round employs outer primers that target conserved regions of the viral genome, generating a primary amplicon. The second round utilizes inner primers that bind specifically to sequences within this initial amplicon, serving to verify the target identity and exponentially amplify the specific product while minimizing non-specific binding [3]. This sequential priming mechanism provides a built-in verification system that dramatically reduces false-positive results from non-specific amplification.
The one-step nested (OSN) qRT-PCR variant consolidates this process into a single-tube reaction by incorporating both primer sets in a single reaction mixture with carefully optimized cycling conditions [11]. This innovation maintains the sensitivity benefits of traditional nested PCR while reducing contamination risks associated with transferring reaction products between tubes. The OSN approach employs specific primer design where the 3' ends of inner primers differ from related coronavirus sequences, ensuring species-specific detection even in the event of viral mutations affecting one primer pair [34].
Comparative Advantages
The enhanced sensitivity of nested PCR stems from its ability to perform effectively despite inhibitors present in clinical specimens and its capacity to amplify extremely low concentrations of target RNA [3]. This characteristic is particularly valuable for SARS-CoV-2 detection in samples with low viral loads, such as from asymptomatic individuals, patients in later disease stages, or certain sample types like saliva and blood [11]. The two-stage amplification process effectively dilutes out PCR inhibitors present in biological samples, overcoming a significant limitation of conventional single-step PCR [8].
When compared with LAMP technology, nested PCR demonstrates superior sensitivity in multiple study comparisons. Research on various pathogens including Alternaria solani and Mycobacterium marinum has consistently shown nested PCR to be 10-100 times more sensitive than LAMP detection [8] [6]. This sensitivity advantage makes nested PCR particularly valuable for detecting the low viral loads frequently encountered in SARS-CoV-2 screening and surveillance programs.
Step-by-Step Protocol for SARS-CoV-2 Nested PCR
Primer Design and Selection
Effective primer design is crucial for successful SARS-CoV-2 nested PCR detection. Primers should target highly conserved regions of the SARS-CoV-2 genome, with common targets being the ORF1ab and nucleocapsid (N) genes [11]. The one-step nested approach described by Liu et al. utilizes four diagnostic primers designed to target the ORF1ab gene region with the greatest sequence differences from bat coronaviruses, ensuring human SARS-CoV-2 specificity [34].
Primer Design Considerations:
- Outer Primers: Should flank a 300-500 bp region within target genes
- Inner Primers: Should be positioned within the outer primer amplicon with a 50-150 bp separation from outer primers
- Specificity: All diagnostic primers should be species-specific with 3' end sequences differing from other coronavirus species
- Mutation Resilience: Design multiple primer pairs to ensure detection even if mutations occur in key amplification nucleotides [34]
Primer sequences should be verified using NCBI's Primer-BLAST tool to ensure specificity for SARS-CoV-2, and synthesized commercially with quality control certificates [7].
RNA Extraction and Purification
Proper RNA extraction is critical for successful nested PCR detection of SARS-CoV-2. The following protocol applies to various sample types including nasopharyngeal swabs, throat swabs, sputum, and blood samples [11].
Materials Needed:
- Sample material (swabs, sputum, blood)
- Nucleic acid extraction kit (e.g., Membrane Adsorption Kits)
- Nuclease-free water
- Centrifuge capable of 13,000 × g
- Sterile 1.5 mL microcentrifuge tubes
Procedure:
- Sample Preparation:
- For swab samples: Vortex swab in collection medium and centrifuge briefly
- For sputum: Add equal volume of dithiothreitol (DTT) or proteinase K, incubate at 56°C for 20 minutes, then centrifuge at 13,000 × g for 10 minutes
- For blood: Isolate peripheral blood mononuclear cells (PBMCs) using density gradient centrifugation
RNA Extraction:
- Transfer 200 μL of sample supernatant to a clean microcentrifuge tube
- Add recommended lysis buffer from extraction kit and incubate at room temperature for 10 minutes
- Add ethanol (70-100%) and mix thoroughly by pipetting
- Transfer mixture to spin column and centrifuge at 11,000 × g for 30 seconds
- Wash with wash buffer 1, centrifuge, then wash with wash buffer 2
- Elute RNA in 30-60 μL of nuclease-free water [11]
RNA Quantification and Quality Control:
- Measure RNA concentration using Nanodrop spectrophotometer
- Store extracted RNA at -80°C if not used immediately
One-Step Nested RT-PCR Amplification
The one-step nested approach combines both amplification rounds in a single tube, reducing contamination risk and streamlining the process [11].
Reaction Setup:
- Template RNA: 20 μL
- Reaction Buffer: 26 μL
- Enzyme Mixture: 4 μL
- Total Reaction Volume: 50 μL
Thermal Cycling Conditions:
- Reverse Transcription: 50°C for 30 minutes
- Initial Denaturation: 95°C for 1 minute
- First Amplification Round (20 cycles):
- Denaturation: 95°C for 30 seconds
- Annealing: 70°C for 40 seconds
- Extension: 72°C for 40 seconds
- Second Amplification Round (40 cycles):
- Denaturation: 95°C for 15 seconds
- Annealing: 60°C for 30 seconds
- Final Step: 25°C for 10 seconds [11]
Alternative Two-Step Protocol: For traditional two-step nested PCR:
- First PCR Round: Use outer primers with 35-40 cycles of amplification
- Product Dilution: Dilute first-round product 10-fold
- Second PCR Round: Use 1 μL of diluted product with inner primers for another 35-40 cycles [7]
Product Analysis and Interpretation
Agarose Gel Electrophoresis:
- Prepare 2.5% agarose gel in 1× TBE buffer with ethidium bromide or SYBR Safe
- Load 10 μL of PCR product mixed with loading dye
- Run gel at 100V for 45-60 minutes
- Visualize under UV transilluminator
- Expected results:
- Positive samples show clear bands at expected sizes
- Negative controls show no bands
- Positive controls confirm reaction success [34]
Alternative Detection Methods:
- SYBR Green-Based Real-Time Detection: Monitor fluorescence during amplification
- Sequencing Validation: Sequence positive products and compare with GenBank database using BLAST [8]
Comparative Performance Data
Sensitivity Comparison Across Molecular Detection Methods
Table 1: Analytical Sensitivity Comparison of SARS-CoV-2 Detection Methods
| Detection Method | Limit of Detection (copies/mL) | Clinical Positive Rate (%) | Reference |
|---|---|---|---|
| One-Step Nested qRT-PCR | 194.74 (ORF1ab), 189.1 (N) | 82.35% (28/34) | [11] |
| Digital PCR (ddPCR) | 401.8 (ORF1ab), 336.8 (N) | 67.65% (23/34) | [11] |
| Conventional qRT-PCR | 520.1 (ORF1ab), 528.1 (N) | 58.82% (20/34) | [11] |
| LAMP Assay | ~7-70 genomic copies | Varies by implementation | [8] [3] |
| Conventional PCR | 1000 trophozoite equivalents | ~30-60% for SARS-CoV-2 | [3] |
Method Comparison for Pathogen Detection
Table 2: Comprehensive Comparison of Nested PCR and LAMP Technologies
| Parameter | Nested PCR | LAMP | Conventional PCR |
|---|---|---|---|
| Sensitivity | 10-100× more sensitive than LAMP [6] | 10× more sensitive than conventional PCR [6] | Baseline sensitivity |
| Equipment Needs | Thermal cycler, electrophoresis system | Water bath or heating block, minimal equipment [3] | Thermal cycler |
| Amplification Time | ~2-3 hours | ~60 minutes [3] | ~1-2 hours |
| Technical Complexity | Moderate to high | Low [3] | Moderate |
| Cost | Moderate | Low to moderate [8] | Moderate |
| Contamination Risk | Higher (two-step format) | Lower | Moderate |
| Sample Throughput | Moderate | High | Moderate |
| Resistance to Inhibitors | Good [8] | Excellent [8] | Variable |
Research Reagent Solutions
Table 3: Essential Research Reagents for SARS-CoV-2 Nested PCR
| Reagent/Category | Specific Examples | Function/Purpose | Specifications |
|---|---|---|---|
| Primers | ORF1ab outer & inner primers, N gene primers [11] | Target-specific amplification | HPLC-purified, specific to SARS-CoV-2 conserved regions |
| Enzyme Systems | Bst DNA polymerase, Taq DNA polymerase [3] | DNA amplification | Reverse transcriptase activity for one-step protocols |
| Extraction Kits | QIAamp DNA Microbiome Kit, Membrane Adsorption Kits [8] [11] | RNA purification | Column-based or magnetic bead purification |
| Amplification Kits | OSN-qRT-PCR assay kit, QuantiFast SYBR Green PCR Kit [11] [3] | Nucleic acid amplification | Optimized buffer systems with detection chemistry |
| Detection Methods | Agarose gel electrophoresis, SYBR Green, lateral flow dipsticks [3] | Amplicon detection | Varying complexity and equipment requirements |
| Positive Controls | SARS-CoV-2 pseudovirus, cloned target sequences [11] | Assay validation | Quantified standards for calibration |
Experimental Workflow and Technical Diagrams
SARS-CoV-2 Nested PCR Workflow
Method Selection Decision Pathway
The step-by-step protocol for SARS-CoV-2 nested PCR presented herein provides researchers with a robust methodology for highly sensitive detection of the virus across various sample types. The comprehensive comparison data demonstrates that one-step nested qRT-PCR offers superior sensitivity (82.35% positive rate) compared to both ddPCR (67.65%) and conventional qRT-PCR (58.82%) when testing clinical samples from COVID-19 patients [11].
For research applications requiring maximal detection sensitivity, particularly when working with samples containing low viral loads, nested PCR represents the optimal choice despite its moderate technical complexity. The method's enhanced sensitivity stems from its two-stage amplification process which effectively dilutes inhibitors and reduces non-specific amplification [3]. However, for field applications or resource-limited settings where extreme sensitivity is less critical than operational simplicity, LAMP technology provides a viable alternative with faster results and minimal equipment requirements [8] [3].
The selection between nested PCR and alternative amplification technologies should be guided by specific research objectives, available instrumentation, and required detection thresholds. As SARS-CoV-2 continues to evolve with variants exhibiting different transmission patterns and viral load dynamics, the implementation of highly sensitive detection methods like nested PCR remains crucial for effective surveillance and research characterization of emerging strains.
Optimized Workflow for RT-LAMP Assay Implementation
The global COVID-19 pandemic created an unprecedented demand for rapid, accurate, and accessible diagnostic testing, highlighting the critical need for alternative molecular detection methods beyond traditional reverse transcription quantitative polymerase chain reaction (RT-qPCR). Reverse transcription loop-mediated isothermal amplification (RT-LAMP) has emerged as a powerful nucleic acid amplification technology (NAAT) that addresses many limitations of conventional PCR-based methods. This comparison guide objectively evaluates the performance of RT-LAMP against other molecular techniques, particularly within the context of SARS-CoV-2 diagnostic accuracy research, providing researchers and drug development professionals with comprehensive experimental data and implementation protocols.
RT-LAMP represents a significant methodological shift from PCR-based amplification, operating at a constant temperature (typically 60-65°C) through the use of strand-displacing DNA polymerase, eliminating the need for thermal cycling equipment. This fundamental difference enables faster reaction times, reduced instrumentation requirements, and greater flexibility in detection methods, making it particularly suitable for point-of-care and resource-limited settings.
Technical Comparison: RT-LAMP vs. RT-qPCR
Performance Characteristics and Diagnostic Accuracy
Table 1: Comparative analytical performance of RT-LAMP versus RT-qPCR for SARS-CoV-2 detection
| Parameter | RT-LAMP | RT-qPCR | Experimental Conditions |
|---|---|---|---|
| Reaction Time | 5-35 minutes [35] [16] | 60-120 minutes [1] | Constant temperature vs. thermal cycling |
| Reaction Temperature | 60-65°C (isothermal) [1] [36] | 40-95°C (thermal cycling) | Instrument complexity |
| Detection Limit | 6.7-50 copies/reaction [35] [1] [37] | Varies by protocol | Clinical sample validation |
| Sensitivity (Early Infection ≤9 days) | 92.8-100% [1] [37] | Gold standard | Nasopharyngeal swabs |
| Specificity | 97-100% [1] [36] [38] | Gold standard | Multiple sample types |
| Equipment Needs | Heating block or water bath [35] | Thermal cycler [1] | Infrastructure requirements |
| Sample Processing | Compatible with extraction-free protocols [39] [40] | Typically requires RNA extraction [36] | Workflow simplification |
The diagnostic accuracy of RT-LAMP shows particularly strong performance during the acute phase of infection. Research demonstrates that up to the 9th day after symptom onset, RT-LAMP showed a positivity rate of 92.8% with sensitivity and specificity of 100% compared to RT-qPCR [1] [37]. However, after the 10th day post-onset, the positivity rate decreased to less than 25%, with concordance between the methods dropping below 60% [1] [37]. This indicates that RT-LAMP performs comparably to RT-qPCR during the high viral load phase but may have reduced sensitivity in later stages of infection when viral loads decline.
Sample Type Considerations
Table 2: RT-LAMP performance across different sample types
| Sample Type | Processing Method | Sensitivity | Specificity | Implementation Considerations |
|---|---|---|---|---|
| Nasopharyngeal Swab | Traditional RNA extraction | 96% [36] | 97% [36] | High concordance with gold standard |
| Saliva | Traditional RNA extraction | 88-96% [36] | 95-100% [36] | Non-invasive collection |
| Saliva | Heat-induced RNA release (HIRR) | 56% [36] | Variable | Significant sensitivity reduction |
| Nasal Swab | Extraction-free | 100% (Ct<30) [40] | 100% [40] | Balanced sensitivity and convenience |
Sample selection and processing methods significantly impact RT-LAMP performance. While nasopharyngeal swabs processed through traditional RNA extraction show the highest concordance with gold standard methods, saliva samples offer a non-invasive alternative with good diagnostic performance when properly processed [36]. However, simplified processing methods such as heat-induced RNA release (HIRR) can substantially reduce sensitivity, highlighting the importance of RNA extraction for reliable results [36]. The acidity of some saliva samples (9-22%) may also affect colorimetric readouts, potentially requiring pH adjustment [36].
Experimental Protocols and Methodologies
Optimized RT-LAMP Workflow
Detailed Experimental Protocol
Primer Design and Optimization
Effective RT-LAMP begins with optimized primer design. A typical assay requires six primers (F3, B3, FIP, BIP, LF, LB) recognizing eight distinct regions of the target sequence [39]. Research indicates that primer length optimization significantly impacts assay efficiency, with optimal stem lengths of 12-17 bp and Tm >45°C providing the best performance [39]. Bioinformatics tools such as Primer Explorer V5 should be employed with additional specificity verification through BLAST analysis against relevant databases [16]. Multiplexing capability can be enhanced by designing primer sets targeting different genes (N, M, S, ORF1a) [40].
Reaction Setup and Optimization
A standard 25μL RT-LAMP reaction contains:
- 12.5μL of 2X master mix (containing buffer, dNTPs, MgSO₄)
- 1-2μL of primer mix (FIP/BIP: 40μM, F3/B3: 5μM, LF/LB: 20μM)
- 1-2μL of Bst DNA polymerase (8U/μL)
- 5μL of RNA template
- Nuclease-free water to volume [16] [40]
For improved sensitivity and specificity, newer enzyme formulations like Lyo-ready Bst DNA Polymerase demonstrate faster reaction speed (detection in as little as 10 minutes) and enhanced tolerance to inhibitors [35]. The SuperScript IV RT-LAMP Master Mix incorporates reverse transcriptase for one-step reactions, enabling detection of as low as 30 copies in under 10 minutes [35].
Amplification and Detection
Reactions are incubated at 60-65°C for 15-60 minutes, depending on the target copy number and enzyme formulation. Real-time detection can be performed using fluorescent intercalating dyes (SYTO 9, SYBR Green) with portable isothermal fluorimeters [35] [40]. For endpoint detection, colorimetric changes (pH-sensitive indicators) or turbidity measurements (magnesium pyrophosphate precipitation) enable visual interpretation without specialized equipment [36] [38].
Advanced Applications and Methodological Innovations
Internal Controls for Enhanced Reliability
Incorporation of internal amplification controls (IACs) is critical for distinguishing true negative results from assay failures. A novel approach uses human 18S ribosomal RNA in saliva as an IAC in a one-pot duplex RT-LAMP format [38]. This enables co-amplification of sample adequacy and pathogen targets, with results visualized on triple-line lateral flow immunoassays. Implementation of such controls significantly improves test reliability, particularly in point-of-care settings, demonstrating 95% sensitivity, 100% specificity, and 96% accuracy in clinical validation [38].
Digital RT-LAMP for Absolute Quantification
Recent advances in microfluidics have enabled the development of droplet digital RT-LAMP (ddRT-LAMP), allowing absolute quantification of viral load without standard curves. This technique partitions reactions into thousands of nanoliter droplets, with amplification occurring in individual partitions [41]. Critical parameters for optimal ddRT-LAMP include:
- Primer design and master mix composition
- Droplet diameter of ~105μm
- 30-minute incubation time
- Appropriate fluorescent dye selection (GelGreen recommended)
- Surfactant-stabilized droplets in fluorinated oil [41]
This approach achieves detection limits of 10² copies/μL and provides more accurate quantification than traditional LAMP, bridging the sensitivity gap with RT-qPCR while maintaining the advantages of isothermal amplification [41].
Research Reagent Solutions
Table 3: Essential reagents and materials for RT-LAMP implementation
| Reagent/Material | Function | Examples & Specifications | Performance Considerations |
|---|---|---|---|
| Bst DNA Polymerase | Strand-displacing enzyme for isothermal amplification | Lyo-ready Bst, Bst 2.0/3.0 | Faster reaction speed, inhibitor tolerance [35] |
| RT-LAMP Master Mix | Optimized reaction components | SuperScript IV RT-LAMP Master Mix | Integrated reverse transcriptase, rapid detection [35] |
| Primer Sets | Target-specific amplification | N2, E1, Orf1a primer sets [40] | Multi-gene targeting enhances detection reliability |
| Detection Reagents | Amplification signal generation | SYTO 9, GelGreen, colorimetric dyes | Compatibility with endpoint/real-time detection [35] [41] |
| RNA Extraction Kits | Nucleic acid purification | QIAamp Viral RNA Mini Kit [1] | Traditional extraction superior to HIRR for sensitivity [36] |
| Internal Controls | Process verification | Human 18S rRNA [38] | Distinguish true negatives from assay failures |
RT-LAMP technology represents a significant advancement in molecular diagnostic capabilities, offering speed, simplicity, and reliability that bridges the gap between laboratory-based testing and point-of-care applications. While RT-qPCR remains the gold standard for maximum sensitivity, particularly in low viral load scenarios, RT-LAMP provides comparable performance during the acute phase of infection with substantially reduced operational complexity.
The optimized workflows and experimental protocols detailed in this guide provide researchers and drug development professionals with evidence-based strategies for implementing RT-LAMP assays. Through appropriate primer design, reagent selection, and methodology optimization, RT-LAMP can achieve 92.8-100% sensitivity and 97-100% specificity compared to RT-qPCR during early infection, with results available in as little as 5-35 minutes.
As molecular diagnostics continue to evolve, RT-LAMP platforms offer versatile solutions for pandemic preparedness, outbreak management, and decentralized testing environments. The integration of internal controls, digital quantification methods, and extraction-free protocols further enhances the utility of this technology for diverse research and clinical applications.
Within molecular diagnostics, the initial sample processing step is a critical determinant of the success and accuracy of downstream nucleic acid amplification tests (NAATs). The prevailing gold standard, RNA extraction, purifies and concentrates nucleic acids prior to amplification but is often time-consuming, costly, and reliant on specialized reagents [42] [43]. In response, direct sample protocols that bypass nucleic acid purification have been developed to offer rapid, cost-effective alternatives, making them particularly attractive for point-of-care testing and resource-limited settings [44].
This guide objectively compares these two sample processing approaches within the specific context of a broader thesis on nested PCR versus LAMP for SARS-CoV-2 diagnostic accuracy. The performance of any amplification technique is intrinsically linked to the quality of its input material; thus, the choice between extracted RNA and direct sample input directly impacts assay sensitivity, specificity, and operational efficiency [45] [43]. We will summarize key experimental data, provide detailed methodologies from foundational studies, and outline essential research reagents to inform researchers, scientists, and drug development professionals.
Performance Comparison: Key Experimental Findings
The tables below synthesize quantitative data from comparative studies to illustrate the performance characteristics of RNA extraction and direct protocols in SARS-CoV-2 detection.
Table 1: Comparative Sensitivity of Different Sample Processing and Assay Combinations for SARS-CoV-2 Detection
| Sample Processing Method | Amplification Technique | Positive Agreement (Sensitivity) | Specificity | Key Findings | Source (Study) |
|---|---|---|---|---|---|
| RNA Extraction | RT-qPCR (Gold Standard) | 58.82%-100%* | N/R | Performance varies with viral load; lower sensitivity in low viral load samples. | [45] [11] |
| RNA Extraction | One-Step Nested (OSN)-qRT-PCR | 82.35% | N/R | Superior sensitivity for low viral load samples vs. standard RT-qPCR and ddPCR. | [11] |
| Direct Input (QuickExtract) | Direct RT-PCR | 98% (Ct <35) | >98% | Ct values ~2 cycles higher than diagnostic RT-qPCR; strong correlation (Rho=0.93). | [44] |
| Direct Input (Rapid Protocol 10: GITC+Triton X-100) | Direct RT-LAMP | 98.4% | 88.8% | Efficient RNA release and inhibitor removal for direct amplification. | [43] |
| Direct Input (QuickExtract) | Bead-LAMP | 93% (Ct <35) | >98% | High sensitivity without RNA extraction. | [44] |
| Direct Input (QuickExtract) | RT-LAMP | 82% (Ct <35) | >98% | Lower sensitivity than direct RT-PCR and bead-LAMP. | [44] |
N/R: Not Reported in the source data. The range reflects different studies and sample types.
Table 2: Operational and Technical Comparison of Sample Processing Methods
| Characteristic | RNA Extraction | Direct Sample Protocols |
|---|---|---|
| Total Processing Time | Several hours (including extraction) | ~70 minutes (5-min inactivation + 60-min assay) [44] |
| Relative Cost | High (specialized kits and equipment) | Low (fewer reagents, no purification kits) |
| Technical Complexity & Labor | High, requires trained personnel | Low to moderate, simplified workflow |
| Risk of Contamination | Moderate (multiple open-tube steps) | Lower (fewer handling steps) |
| Suitability for Point-of-Care | Low | High |
| Impact on Downstream Assays | Removes PCR inhibitors; consistent, high-quality input [42] | Susceptible to sample-derived inhibitors; may require optimized buffers [43] |
| Automation Potential | High (multiple automated platforms available) [42] | Moderate |
Experimental Protocols in Practice
To ensure reproducibility, this section details key experimental protocols from the cited literature.
High-Sensitivity RNA Extraction and Nested PCR Protocol
This protocol, adapted from SARS-CoV-2 validation studies, uses extracted RNA with a highly sensitive One-Step Nested quantitative RT-PCR (OSN-qRT-PCR) [11].
- Sample Collection and RNA Extraction: Throat swabs, nasopharyngeal swabs, or sputum samples are collected from patients. Total RNA is extracted using a membrane adsorption-based nucleic acid extraction kit according to the manufacturer's instructions (e.g., Di'an, Hangzhou, China) [11].
- OSN-qRT-PCR Reaction Setup: The reaction is prepared using a commercial OSN-qRT-PCR assay kit (e.g., Sansure, Changsha, China). A 50 μL reaction mix typically contains 20 μL of extracted RNA template, 26 μL of reaction buffer, and 4 μL of enzyme mixture [11].
- Amplification Protocol: The tube is placed in a real-time PCR instrument (e.g., LightCycler 480 II, Roche) and run under the following program [11]:
- Reverse Transcription: 50°C for 30 minutes.
- Enzyme Activation: 95°C for 1 minute.
- First-Stage Amplification (Outer Primers): 20 cycles of:
- Denaturation: 95°C for 30 seconds
- Annealing/Extension: 70°C for 40 seconds
- Second-Stage Amplification (Inner Primers): 40 cycles of:
- Denaturation: 95°C for 15 seconds
- Annealing/Extension: 60°C for 30 seconds
- Data acquisition at 25°C for 10 seconds.
This method's increased sensitivity stems from the two sequential amplification reactions with two primer pairs in a single tube, which reduces contamination risk compared to traditional two-tube nested PCR [11].
Direct RT-LAMP with Rapid RNA Release Protocol
This protocol outlines a direct method that uses a chemical treatment to release RNA directly from swab samples for RT-LAMP amplification, omitting the purification step [43].
- Sample Collection: Oropharyngeal and nasopharyngeal swabs are collected and placed in viral transport medium.
- Rapid RNA Release (Method 10): A 100 μL sample is mixed with a lysis buffer containing 4M Guanidinium Isothiocyanate (GITC) and 2% Triton X-100. The mixture is incubated at 85°C for 10 minutes. GITC is a potent chaotropic agent that denatures proteins and inactivates RNases, while Triton X-100 is a detergent that disrupts the viral envelope and cell membranes, facilitating RNA release [43].
- RT-LAMP Reaction: The RT-LAMP assay is performed in a 25 μL total volume. The reaction mixture includes:
- 12.5 μL of 2x WarmStart LAMP Kit (DNA and RNA)
- 0.5 μL LAMP fluorescent dye
- 2.5 μL of a 10x primer mix (targeting SARS-CoV-2 N, E, and RdRp genes)
- 9 μL PCR-grade water
- 1 μL of the heat-treated sample (without purification) [43]
- Amplification and Detection: The reaction is incubated at a constant temperature of 62°C for 40 minutes in a real-time PCR analyzer with continuous fluorescence detection. A positive result is indicated by a fluorescence signal that crosses a predetermined threshold within the incubation time [43].
Diagram 1: A comparison of the Direct Sample and RNA Extraction protocol workflows.
The Scientist's Toolkit: Key Research Reagent Solutions
The following table catalogues essential reagents and kits used in the featured studies for comparing RNA extraction and direct protocols.
Table 3: Essential Reagents for Sample Processing and Amplification
| Reagent/Kits | Primary Function | Example Use Case |
|---|---|---|
| MagMAX Viral RNA Isolation Kit | Efficient RNA purification from complex samples (e.g., stool). Manual and automated formats. | Identified as a top-performing method for poliovirus RNA extraction in a comparison of 11 methods [42]. |
| QuickExtract DNA/RNA Extraction Solution (Lucigen) | Rapid, single-step lysis and inactivation of samples. Inactivates RNases and renders virus non-infectious. | Used for direct RT-PCR and LAMP protocols with nasopharyngeal and gargle samples, enabling a 5-minute sample prep [44]. |
| Guanidinium Isothiocyanate (GITC) | Powerful chaotropic agent. Denatures proteins, inactivates nucleases, and promotes nucleic acid release. | A key component in the most efficient rapid RNA release protocol (Method 10) for direct RT-LAMP [43]. |
| Triton X-100 | Non-ionic detergent. Disrupts lipid membranes of viruses and cells to release nucleic acids. | Combined with GITC in a rapid RNA extraction method to facilitate direct detection in RT-LAMP [43]. |
| Direct RNA Sequencing Kit (SQK-RNA004) | Library preparation for nanopore sequencing of native RNA, bypassing cDNA synthesis and PCR. | Used for direct detection and characterization of viruses like poliovirus from extracted RNA [46]. |
| WarmStart LAMP Kit (DNA and RNA) | Isothermal amplification master mix. Used for sensitive detection of RNA/DNA targets at constant temperature. | The core enzyme mix for optimized direct and standard RT-LAMP assays in SARS-CoV-2 detection [43]. |
| One-Step Nested (OSN)-qRT-PCR Kit | Integrated assay for highly sensitive and specific detection of RNA targets in a single tube. | Used with extracted RNA to achieve superior sensitivity for SARS-CoV-2 compared to standard RT-qPCR and ddPCR [11]. |
Diagram 2: Logical relationships between sample processing methods and their key performance attributes.
The choice between RNA extraction and direct sample protocols involves a clear trade-off between analytical sensitivity and operational efficiency. For SARS-CoV-2 diagnostics, the evidence shows that RNA extraction followed by sophisticated amplification methods like OSN-qRT-PCR provides the highest sensitivity, which is crucial for reliably detecting low viral loads [11]. In contrast, optimized direct protocols using rapid lysis buffers coupled with robust techniques like RT-LAMP or direct RT-PCR offer a compelling alternative for rapid mass screening, with minimal loss of sensitivity for moderate to high viral loads and significant gains in speed and cost-effectiveness [43] [44]. The optimal choice depends on the specific diagnostic context, including the prevalence of the disease, available resources, and the required balance between sensitivity and throughput.
Primer Design Strategies for Specificity and Variant Coverage
The accurate detection of SARS-CoV-2 through molecular diagnostics has been pivotal in managing the COVID-19 pandemic. The high mutation rate of the virus, with an early estimate of approximately (1.12 \times 10^{-3}) mutations per site-year, has necessitated the continuous evolution of primer design strategies to maintain diagnostic accuracy against emerging variants of concern (VOCs) [47]. This comparison guide examines two principal molecular techniques—nested PCR and loop-mediated isothermal amplification (LAMP)—evaluating their respective primer design requirements, analytical performance, and applicability within diagnostic and research settings. The foundational thesis framing this analysis posits that while both methods offer superior sensitivity compared to conventional RT-qPCR, their optimal implementation requires distinctly different primer design philosophies and operational considerations, particularly when targeting diverse SARS-CoV-2 variants.
Fundamental Principles of Nested PCR and LAMP
Nested PCR: Mechanism and Primer Requirements
Nested PCR is a two-stage amplification method designed to enhance sensitivity and specificity. The initial amplification uses an outer primer pair targeting a larger genomic region, followed by a secondary reaction using inner primers that bind internal to the first amplicon. This sequential targeting significantly reduces non-specific amplification and enables detection of low viral loads. Conventional nested PCR requires separate reaction tubes, increasing contamination risk, whereas one-step nested quantitative RT-PCR (OSN-qRT-PCR) integrates both amplification stages into a single, closed-tube reaction, mitigating contamination concerns while preserving analytical advantages [11].
Primer design for nested PCR follows standard thermodynamic considerations but requires strategic positioning of two distinct primer pairs. The protocol typically involves designing external primers spanning several hundred base pairs, followed by internal primers that anneal to conserved regions within the initial amplicon. This approach is particularly valuable for detecting SARS-CoV-2 in samples with low viral loads, where conventional RT-qPCR may yield false negatives [11].
LAMP: Isothermal Amplification and Multi-Primer Systems
LAMP represents a fundamentally different amplification approach, utilizing strand-displacing DNA polymerase to amplify nucleic acids isothermally at 60-65°C. The reaction employs four to six primers recognizing six to eight distinct regions on the target genome, creating complex loop structures that enable exponential amplification without thermal cycling [48]. The standard primer set includes Forward Inner Primer (FIP), Backward Inner Primer (BIP), and outer primers (F3, B3), with the optional addition of loop primers (LF, LB) to accelerate reaction kinetics, reducing amplification time by approximately one-third [48].
RT-LAMP adapts this system for RNA targets by incorporating reverse transcriptase, enabling direct detection of SARS-CoV-2 RNA without separate cDNA synthesis. Primer design considerations for LAMP include maintaining appropriate spacing (40-60 bases) between primer binding regions, avoiding repetitive nucleotides, ensuring compatible melting temperatures across primer pairs, and preventing secondary structure formation at 3' ends [48]. The requirement for multiple primers binding simultaneously makes LAMP inherently highly specific, though designing effective primer sets for rapidly evolving viral targets presents distinct challenges.
Comparative Performance Analysis
Analytical Sensitivity and Detection Limits
Table 1: Analytical Sensitivity Comparison of SARS-CoV-2 Detection Methods
| Method | Limit of Detection (copies/mL) | Target Genes | Clinical Positive Rate |
|---|---|---|---|
| OSN-qRT-PCR | 194.74 (ORF1ab), 189.1 (N) | ORF1ab, N | 82.35% (28/34 samples) |
| ddPCR | 401.8 (ORF1ab), 336.8 (N) | ORF1ab, N | 67.65% (23/34 samples) |
| Conventional RT-qPCR | 520.1 (ORF1ab), 528.1 (N) | ORF1ab, N | 58.82% (20/34 samples) |
| RT-LAMP | 50-100 copies/reaction | ORF1ab, E, N | 90.6% sensitivity vs. RT-qPCR |
| Nested One-Step RT-PCR | Not specified | N gene | 100% concordance with LAMP |
OSN-qRT-PCR demonstrates superior sensitivity metrics, with limits of detection approximately 2.7-fold lower than conventional RT-qPCR for both ORF1ab and N gene targets [11]. This enhanced sensitivity translates to improved clinical detection rates, with OSN-qRT-PCR identifying 28 of 34 positive samples compared to only 20 for conventional RT-qPCR in a direct comparative study [11]. The clinical advantage is particularly evident in patients with low viral loads, where OSN-qRT-PCR detected positive cases missed by standard methods.
RT-LAMP shows variable performance across studies, with one validated colorimetric assay demonstrating 90.6% sensitivity and 100% specificity compared to RT-qPCR, detecting between 50-100 viral genome copies per reaction [49]. Another comparative analysis reported 100% sensitivity and specificity for RT-LAMP during the first 9 days after symptom onset, with performance declining thereafter as viral loads decreased [1]. The temporal dependency of LAMP sensitivity highlights the importance of sample collection timing for optimal performance.
Variant Coverage and Primer Design Adaptations
Table 2: Variant Coverage of Different Primer Design Approaches
| Method | Variant Coverage | Design Strategy | Validation Approach |
|---|---|---|---|
| AI-Based Primer Design | B.1.1.7 (Alpha), B.1.1.529 (Omicron) | Evolutionary algorithms screening GISAID database | Clinical testing on patient samples |
| Multi-Target LAMP | B.1.1.7, B.1.351, P.1, B.1.617.2, B.1.427/B.1.429, Omicron lineages | Three primer sets targeting ORF1aa, ORF1ab, E gene | In silico analysis with 100% homology |
| Updated Protocol Design | Continuously evolving variants | Regular primer evaluation against updated databases | Periodic in silico validation |
The rapid emergence of SARS-CoV-2 variants has necessitated primer design strategies that accommodate genomic diversity while maintaining detection capabilities. Artificial intelligence-based approaches using evolutionary algorithms to screen sequences from the GISAID repository have successfully generated variant-specific primers for Alpha (B.1.1.7) and Omicron (B.1.1.529) lineages, with clinical validation confirming accurate detection [47]. This automated pipeline can deliver optimized primer sets within hours, dramatically accelerating response to emerging variants.
Multiplexed primer targeting provides another strategy for maintaining variant coverage. Colorimetric RT-LAMP assays employing three primer sets targeting two ORF1ab regions and the E gene demonstrated 100% in silico recognition of multiple VOCs, including Alpha, Beta, Gamma, Delta, and Omicron lineages [49]. The multi-target approach reduces the probability of simultaneous mutations eliminating all primer binding sites, thereby conferring robustness against viral evolution.
A fundamental primer design protocol for rapidly evolving genomes emphasizes continuous evaluation against updated sequence databases, systematic identification of mutations affecting primer binding, and iterative redesign to maintain coverage [50]. This approach, while resource-intensive, provides a methodological framework for sustaining assay effectiveness amidst viral evolution.
Experimental Protocols and Methodologies
OSN-qRT-PCR Workflow
The OSN-qRT-PCR methodology employs a single-tube, two-stage amplification process. Reaction components include 20μL of template, 26μL of reaction buffer, and 4μL of enzyme mixture. Thermal cycling parameters comprise: reverse transcription at 50°C for 30 minutes; initial denaturation at 95°C for 1 minute; 20 cycles of denaturation at 95°C for 30 seconds, annealing at 70°C for 40 seconds, and extension at 72°C for 40 seconds; followed by 40 cycles of denaturation at 95°C for 15 seconds, annealing at 60°C for 30 seconds, and final extension at 25°C for 10 seconds [11]. This integrated protocol maintains the sensitivity advantages of nested amplification while reducing contamination risk through single-tube implementation.
OSN-qRT-PCR Experimental Workflow
RT-LAMP Implementation Protocol
The RT-LAMP assay protocol utilizes strand-displacing Bst polymerase with reverse transcriptase activity. A standardized 25μL reaction contains 5 pmol each of F3 and B3 primers, 40 pmol each of FIP and BIP primers, 20 pmol each of LF and LB loop primers, and 8U of Bst DNA/RNA Polymerase. Reactions proceed isothermally at 62-65°C for 30-60 minutes, with amplification detectable via real-time turbidimetry, fluorescence, or colorimetric change using hydroxynaphthol blue (HNB) or calcein dyes [1] [49]. The colorimetric detection format enables visual interpretation without instrumentation, making it suitable for point-of-care applications.
Sample preparation for direct RT-LAMP without RNA extraction involves heat inactivation of 10μL nasopharyngeal swab sample with 40μL of lysis buffer at 95°C for 8 minutes, followed by direct addition of 10μL of treated sample to the LAMP reaction mixture [49]. This streamlined processing reduces hands-on time and resource requirements compared to methods requiring nucleic acid purification.
RT-LAMP Experimental Workflow
Research Reagent Solutions
Table 3: Essential Research Reagents for SARS-CoV-2 Primer Evaluation
| Reagent Category | Specific Examples | Application Function |
|---|---|---|
| Polymerase Enzymes | Bst DNA/RNA Polymerase 3.0 (NEB), Warm Start RTx Reverse Transcriptase | Strand displacement for LAMP, reverse transcription |
| Primer Design Tools | Primer Explorer V5, MFEprimer, Vector NTI Advance | In silico primer design and specificity validation |
| Detection Chemistries | Hydroxynaphthol Blue (HNB), Calcein, SYBR Green | Visual or fluorescent detection of amplification |
| Reference Materials | Quantitative Synthetic SARS-CoV-2 RNA (ATCC VR-3276SD) | Assay standardization and limit of detection determination |
| RNA Extraction Kits | QIAamp Viral RNA Mini Kit (automated on QIAcube) | Nucleic acid purification for reference methods |
| Amplification Master Mixes | TaqMan Fast Virus 1-Step Master Mix, Loopamp SARS-CoV-2 Detection kit | Optimized reaction components for efficient amplification |
Operational Considerations and Implementation Contexts
Throughput, Infrastructure and Contamination Control
Nested PCR methods, particularly conventional two-tube formats, present significant contamination risks due to required transfer of initial amplification products. The OSN-qRT-PCR format mitigates this concern through single-tube implementation but requires sophisticated thermal cyclers capable of complex cycling protocols [11]. Equipment costs and technical expertise requirements generally limit nested PCR approaches to centralized laboratory settings with trained personnel.
RT-LAMP offers distinct advantages in resource-limited environments due to isothermal operation eliminating need for expensive thermal cyclers. The method's tolerance to inhibitors enables direct amplification from minimally processed samples, further simplifying workflow requirements [48] [49]. Colorimetric detection formats provide results interpretable without instrumentation, though quantitative capabilities are limited compared to real-time fluorescence detection systems.
Time-to-Result and Processing Efficiency
RT-LAMP typically delivers results within 30-60 minutes from sample receipt, significantly faster than nested PCR protocols requiring 2-4 hours for complete analysis [49] [16]. This rapid turnaround stems from isothermal amplification eliminating time-consuming thermal cycling and streamlined detection methods. The direct amplification capability of LAMP, bypassing RNA extraction, further reduces hands-on time and processing requirements.
Nested PCR protocols, while more time-intensive, provide quantitative data and higher throughput compatibility in automated systems. The OSN-qRT-PCR method enables processing of 94-96 samples per run in standard thermal cyclers, with continuous fluorescence monitoring throughout amplification [11]. This quantitative capability supports viral load monitoring applications beyond mere detection.
The selection between nested PCR and LAMP primer design strategies represents a trade-off between analytical sensitivity and operational practicality. Nested PCR approaches, particularly OSN-qRT-PCR, provide exceptional sensitivity and reliable quantitation, making them ideal for clinical reference applications and low viral load detection. Conversely, LAMP-based strategies offer rapid results, minimal infrastructure requirements, and robust performance against diverse variants through multi-target primer design, positioning them as valuable tools for point-of-care testing and resource-limited settings.
The evolving landscape of SARS-CoV-2 variants necessitates continuous primer design refinement regardless of platform. Artificial intelligence-driven pipelines, multi-target priming approaches, and systematic database monitoring represent promising strategies for maintaining diagnostic accuracy amidst viral evolution. Future developments will likely focus on streamlining nested PCR workflows and enhancing LAMP quantification capabilities, further blurring the distinctions between these complementary technologies while expanding their respective applications in research and clinical diagnostics.
The accurate detection of SARS-CoV-2 across diverse sample types remains critical for effective disease management and surveillance. While reverse transcription quantitative polymerase chain reaction (RT-qPCR) has established itself as the gold standard, diagnostic laboratories continue to evaluate alternative molecular methods that offer advantages in speed, cost, or simplicity. Among these alternatives, Reverse Transcription Loop-Mediated Isothermal Amplification (RT-LAMP) and nested PCR formats have demonstrated significant potential for detecting SARS-CoV-2 RNA across various specimen types, including nasopharyngeal swabs, saliva, and animal specimens [16] [11] [26]. This comparison guide objectively evaluates the performance of these methodological approaches within the context of a broader thesis on diagnostic accuracy for SARS-CoV-2 detection, providing researchers with experimental data to inform their methodological selections.
Performance Comparison Across Sample Types
Analytical Sensitivity and Detection Limits
Table 1: Analytical Sensitivity of SARS-CoV-2 Detection Methods
| Method | Target Gene | Limit of Detection (copies/mL) | 95% Confidence Interval | Reference |
|---|---|---|---|---|
| One-Step Nested qRT-PCR | ORF1ab | 194.74 | 139.7–430.9 | [11] |
| One-Step Nested qRT-PCR | N | 189.1 | 130.9–433.9 | [11] |
| ddPCR | ORF1ab | 401.8 | 284.8–938.3 | [11] |
| ddPCR | N | 336.8 | 244.6–792.5 | [11] |
| Conventional qRT-PCR | ORF1ab | 520.1 | 363.23–1145.69 | [11] |
| Conventional qRT-PCR | N | 528.1 | 347.7–1248.7 | [11] |
| Conventional Nested PCR | N | 0.015 ng/μL (RNA concentration) | Not specified | [26] |
The one-step nested qRT-PCR method demonstrates superior analytical sensitivity compared to both digital PCR (ddPCR) and conventional qRT-PCR, with limits of detection approximately 2-3 times lower for both ORF1ab and N gene targets [11]. This enhanced sensitivity is particularly valuable for detecting low viral loads in clinical and animal specimens.
Clinical Performance in Human Sample Types
Table 2: Clinical Performance Across Different Sample Types
| Method | Sample Type | Positive Detection Rate | Agreement with Reference | Clinical Context | Reference |
|---|---|---|---|---|---|
| RT-LAMP | Saliva | 92/342 (26.9%) | 93% (Cohen's κ) | Symptomatic & Asymptomatic | [16] |
| RT-LAMP | Nasopharynx | 94/342 (27.4%) | 94% (Cohen's κ) | Symptomatic & Asymptomatic | [16] |
| RT-qPCR | Saliva | 86/342 (25.1%) | Reference | Symptomatic & Asymptomatic | [16] |
| RT-qPCR | Nasopharynx | 93/342 (27.1%) | Reference | Symptomatic & Asymptomatic | [16] |
| Nested PCR | Human & Cat Samples | 100% Sensitivity & Specificity | Validated against IVD assays | Symptomatic | [26] |
The performance of different sample types varies depending on clinical context. Saliva demonstrates excellent concordance with nasopharyngeal samples in broad populations [16], though the optimal sample type may depend on symptom status, with evidence suggesting saliva performs better in asymptomatic individuals while nasopharyngeal samples may be more sensitive for symptomatic cases [51].
Methodological Comparison and Throughput
Table 3: Technical Comparison of Detection Methods
| Parameter | RT-qPCR | RT-LAMP | Nested PCR |
|---|---|---|---|
| Amplification Temperature | Multiple temperatures (e.g., 50°C, 95°C) | Isothermal (~65°C) | Multiple temperatures (e.g., 94°C, 54°C, 72°C) |
| Amplification Time | 1.5-2 hours | 30-60 minutes | 2-3 hours (including two rounds) |
| Instrument Requirements | Thermal cycler with fluorescence detection | Water bath or simple block | Standard thermal cycler |
| Primer Complexity | 2 primers + probe | 4-6 primers | 2 pairs of primers (external + internal) |
| Sensitivity | High | Moderate to High | Very High |
| Cost per Test | High | Moderate | Low |
| Sample Throughput | High | Moderate to High | Moderate |
RT-LAMP offers significant advantages in procedural simplicity and speed, requiring only a single temperature incubation, which simplifies instrument requirements [16] [10]. Conversely, nested PCR provides exceptional sensitivity through its two-stage amplification process, though at the cost of longer processing time and increased risk of contamination [11] [26].
Experimental Protocols
One-Step RT-LAMP Protocol for SARS-CoV-2 Detection
The following protocol is adapted from the evaluation of one-step real-time PCR and one-step RT-LAMP methods for detection of SARS-CoV-2 [16]:
Sample Processing:
- Collect nasopharyngeal or saliva samples in viral transport media (VTM)
- Extract RNA using commercial extraction kits (e.g., Biorexfars SARS-CoV-2 RNA Extraction Kit)
- Assess RNA purity and quantity using spectrophotometry (260/280 ratio ~2.0)
RT-LAMP Reaction Setup:
- Prepare 25 μL reaction mixture containing:
- 5 pmol each of F3 and B3 external primers
- 40 pmol each of FIP and BIP internal primers
- 20 pmol each of LF and LB loop primers
- 8U (1 μL) of Bst DNA/RNA Polymerase 3.0
- Appropriate buffer components
- 5 μL of RNA template
- Incubate reaction at 65°C for 30-60 minutes
- Visualize results through colorimetric change or fluorescence detection
Primer Design Considerations:
- Target the N gene (NC_045512.2) positions 28274-29533
- Design primers using Primer Explorer V5 software
- Verify specificity using Primer BLAST against SARS-CoV-2 database
- Confirm alignment with multiple SARS-CoV-2 sequences using CLC Genomics Workbench
One-Step Nested qRT-PCR Protocol
This protocol is adapted from the validation of a highly sensitive one-step nested quantitative real-time PCR assay [11]:
Reaction Setup:
- Prepare 50 μL reaction mixture containing:
- 20 μL of template RNA
- 26 μL of reaction buffer
- 4 μL of enzyme mixture
- Amplification conditions:
- Reverse transcription: 50°C for 30 minutes
- Initial denaturation: 95°C for 1 minute
- First amplification: 20 cycles of:
- 95°C for 30 seconds
- 70°C for 40 seconds
- 72°C for 40 seconds
- Second amplification: 40 cycles of:
- 95°C for 15 seconds
- 60°C for 30 seconds
- Final step: 25°C for 10 seconds
Detection and Analysis:
- Utilize dual-labeled probes for real-time detection
- Analyze amplification curves using appropriate software
- Determine positivity based on cycle threshold (Ct) values
Conventional Nested PCR for Animal Specimens
This protocol is adapted from the development and validation of nested PCR for SARS-CoV-2 detection in humans and cats [26]:
First Round PCR:
- Prepare 25 μL reaction mixture:
- 12.5 μL My Taq HS red mix
- 4 μL cDNA template
- 1 μL each external primer (10 pmol/μL)
- 6.5 μL PCR grade water
- Cycling conditions:
- Initial denaturation: 95°C for 1 minute
- 35 cycles of:
- 95°C for 15 seconds
- 54.6°C for 15 seconds
- 72°C for 10 seconds
Second Round PCR:
- Prepare 25 μL reaction mixture:
- 12.5 μL My Taq HS red mix
- 0.5 μL first PCR product
- 1 μL each internal primer (10 pmol/μL)
- 10 μL PCR grade water
- Use identical cycling conditions as first round
- Analyze products by gel electrophoresis (2% agarose)
Methodological Workflow Visualization
Research Reagent Solutions
Table 4: Essential Research Reagents for SARS-CoV-2 Detection Studies
| Reagent Category | Specific Examples | Function & Application | Reference |
|---|---|---|---|
| Reverse Transcriptase Enzymes | Bst DNA/RNA Polymerase 3.0, SensiFAST cDNA synthesis kit | RNA-to-cDNA conversion; isothermal amplification | [16] [26] |
| Primer Sets | N gene-specific LAMP primers (F3, B3, FIP, BIP, LF, LB); Nested PCR external/internal primers | Target-specific amplification | [16] [26] |
| RNA Extraction Kits | Biorexfars SARS-CoV-2 RNA Extraction Kit, ISOLATE II RNA Mini Kit | Nucleic acid purification from clinical samples | [16] [26] |
| Amplification Master Mixes | My Taq HS red mix, COVID-19 RT-qPCR kits (Pishtaz Teb Co.) | Provides optimized buffer conditions for amplification | [16] [26] |
| Detection Systems | Intercalating dyes, TaqMan probes, Gel electrophoresis reagents | Amplification product visualization and quantification | [16] [11] [26] |
| Sample Collection Materials | Viral Transport Media (VTM), FLOQSwabs, sterile collection cups | Sample integrity maintenance during collection and transport | [16] [52] |
The comparative analysis of SARS-CoV-2 detection methods across diverse sample types reveals a complex landscape where methodological selection depends heavily on specific application requirements. RT-LAMP technology offers compelling advantages in settings requiring rapid results with minimal equipment, demonstrating excellent agreement (93-94%) with RT-qPCR standards in both nasopharyngeal and saliva samples [16]. The isothermal nature of RT-LAMP and its procedural simplicity make it particularly suitable for point-of-care testing and resource-limited environments.
Nested PCR formats, whether in conventional or real-time configurations, provide exceptional sensitivity that surpasses even digital PCR approaches [11]. This enhanced detection capability makes nested PCR particularly valuable for applications requiring identification of low viral loads, such as surveillance of animal reservoirs [26] or detection during convalescent phases when viral concentrations diminish. The two-stage amplification process, while increasing analytical sensitivity, does require additional procedural controls to prevent amplicon contamination.
The choice of sample type represents another critical consideration. While nasopharyngeal samples remain the clinical standard, saliva has emerged as a reliable alternative that enables self-collection and reduces healthcare worker exposure risk [52] [53]. Evidence suggests that optimal sample type may vary with clinical presentation, with saliva potentially offering advantages in asymptomatic individuals while nasopharyngeal samples may be more sensitive for symptomatic cases [51]. This distinction has important implications for screening programs versus diagnostic testing of symptomatic individuals.
For researchers working with animal specimens or conducting surveillance studies in susceptible species, the validated nested PCR approach provides a sensitive and specific option that can be implemented with conventional laboratory equipment [26]. The ability to detect SARS-CoV-2 across species barriers is increasingly important for understanding viral transmission dynamics and potential reservoir hosts.
In conclusion, the selection of SARS-CoV-2 detection methodology should be guided by specific application requirements, including sensitivity needs, available resources, sample type, and intended use context. RT-LAMP offers speed and simplicity, nested PCR provides maximal sensitivity, and conventional RT-qPCR remains the reference standard for clinical diagnosis. As SARS-CoV-2 continues to evolve and circulate globally, this methodological diversity will remain essential for effective surveillance and outbreak management across human and animal populations.
Overcoming Technical Challenges and Enhancing Assay Performance
Addressing Sensitivity Limitations in Low Viral Load Samples
The diagnostic accuracy of SARS-CoV-2 testing faces significant challenges when detecting low viral loads, which are common during the initial and final stages of infection. Low viral load samples often produce false-negative results with conventional molecular tests, hampering effective disease control and surveillance. This comparison guide objectively evaluates the performance of two prominent amplification techniques—Nested PCR and Loop-Mediated Isothermal Amplification (LAMP)—for detecting SARS-CoV-2 in low viral load scenarios. Based on comprehensive experimental data, we provide researchers and drug development professionals with critical insights to inform diagnostic strategies and assay selection.
Nested PCR is a two-stage amplification method that significantly enhances sensitivity and specificity. The initial amplification round uses an outer primer pair to generate a primary amplicon, which then serves as the template for a second amplification with an inner primer pair. This sequential targeting reduces non-specific amplification and enables detection of minimal target sequences [54] [55].
LAMP is an isothermal amplification technique that utilizes 4-6 primers recognizing 6-8 distinct regions of the target DNA. The reaction proceeds at a constant temperature (typically 60-65°C) through a strand displacement mechanism, generating abundant amplification products within 30-60 minutes. Results can be visualized through colorimetric changes, turbidity, or fluorescence without sophisticated equipment [3] [56].
Performance Comparison in Low Viral Load Detection
Sensitivity and Limit of Detection
Table 1: Comparative Sensitivity of Molecular Detection Methods
| Detection Method | Target Pathogen | Limit of Detection (LoD) | Comparison Reference |
|---|---|---|---|
| Nested PCR | SARS-CoV-2 | ~50 copies/μL [54] | |
| One-Step Nested qRT-PCR | SARS-CoV-2 | 194.74 copies/mL (ORF1ab); 189.1 copies/mL (N) [11] | More sensitive than conventional qRT-PCR and ddPCR [11] |
| Colorimetric RT-LAMP | SARS-CoV-2 | 19.3-200 copies/μL [56] [57] | |
| LAMP | Entamoeba histolytica | 1 trophozoite [3] | 10-1000x more sensitive than PCR methods [3] |
| LAMP | Alternaria solani | 100 fg genomic DNA [6] | 10x more sensitive than conventional PCR [6] |
| Nested PCR | Alternaria solani | 1 fg genomic DNA [6] | 100x more sensitive than LAMP [6] |
| qPCR | Fusarium tricinctum | 3.1 fg/μL [7] | 10x more sensitive than LAMP and nested PCR [7] |
Clinical Performance with SARS-CoV-2 Samples
Table 2: Clinical Validation Results for SARS-CoV-2 Detection
| Detection Method | Sample Type | Sensitivity | Specificity | Key Findings |
|---|---|---|---|---|
| Nested PCR | Animal samples (dogs/cats) | 95% [54] | 100% [54] | Detected samples with Ct values 17-31.5; excellent agreement (κ=0.829) with reference methods [54] |
| Nested PCR | Human and cat samples | 100% [23] | 100% [23] | Detected RNA as low as 0.015 ng/μL; validated against commercial LAMP and real-time RT-PCR kits [23] |
| Colorimetric RT-LAMP | Clinical samples | 98% (for Ct<32) [57] | 100% [57] | Reliable for samples with RT-qPCR Ct<30; indeterminate results (orange color) at low viral loads [56] |
| One-Step Nested qRT-PCR | Clinical samples | 82.35% [11] | 100% [11] | Superior to ddPCR (67.65%) and qRT-PCR (58.82%) for positive detection [11] |
Experimental Protocols for SARS-CoV-2 Detection
Nested PCR Protocol for SARS-CoV-2 N Gene
Primer Design:
- External primers: Ext2019nCorVF (5'-GGCAGTAACCAGAATGGAGA-3') and Ext2019nCorVR (5'-CTCAGTTGCAACCCATATGAT-3') generating 335 bp product [23]
- Internal primers: intF (5'-CACCGCTCTCACTCAACAT-3') and intR (5'-CATAGGGAAGTCCAGCTTCT-3') generating 212 bp product [23]
Reaction Setup:
- First round: 25 μL reaction with 12.5 μL My Taq HS red mix, 4 μL cDNA, 1 μL each external primer (10 pmol/μL), and 6.5 μL PCR grade water [23]
- Second round: 25 μL reaction with 12.5 μL My Taq HS red mix, 0.5 μL first PCR product, 1 μL each internal primer (10 pmol/μL), and 10 μL PCR grade water [23]
Thermal Cycling Conditions:
- First round: Initial denaturation at 95°C for 3 min; 35 cycles of denaturation (95°C for 15s), annealing (49°C for 15s), extension (72°C for 15s); final extension at 72°C for 1 min [55]
- Second round: Same as first round with annealing temperature of 51°C [55]
Detection: Analyze 5 μL of second-round product by 2% agarose gel electrophoresis with ethidium bromide staining [23].
Colorimetric RT-LAMP Protocol for SARS-CoV-2
Primer Design: Primers targeting SARS-CoV-2 N and E genes designed using PrimerExplorer V5 software [56] [57]
Reaction Setup:
- 25 μL reaction containing: 15 μL isothermal amplification buffer, 1 μL dye, 1 μL enzyme mix, 2 μL primer mix (FIP/BIP: 1.6 μM each; F3/B3: 0.2 μM each; LF/LB: 0.4 μM each), and 6 μL RNA template [56]
- Alternative: WarmStart Colorimetric LAMP 2X Master Mix (New England Biolabs) with 1.5 μL primer mix and 5 μL template in 25 μL reaction [57]
Amplification Conditions:
Result Interpretation:
- Positive: Color change from pink to yellow
- Negative: Remains pink
- Indeterminate: Orange color (recommend retesting or confirmation by RT-qPCR) [56]
Visualizing Diagnostic Workflows
Diagnostic Workflow Comparison: Nested PCR vs. LAMP
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Molecular Detection
| Reagent/Kit | Function | Application Examples |
|---|---|---|
| Bst DNA Polymerase | Strand-displacing DNA polymerase for isothermal amplification | LAMP reactions [3] [7] |
| My Taq HS Red Mix | Ready-to-use master mix for high-specificity PCR | Nested PCR reactions [23] |
| SensiFAST cDNA Synthesis Kit | Efficient reverse transcription for cDNA generation | First-step in nested PCR after RNA extraction [23] |
| ISOLATE II RNA Mini Kit | RNA purification from clinical samples | Nucleic acid extraction prior to amplification [23] |
| WarmStart Colorimetric LAMP Master Mix | All-in-one mix with pH indicator for visual detection | Colorimetric RT-LAMP without separate dye addition [57] |
| Hydroxy Naphthol Blue (HNB) | Colorimetric metal indicator for visual LAMP detection | Pre-added to LAMP reactions for color change visualization [7] |
| QuantiFast SYBR Green PCR Kit | Sensitive qPCR detection with SYBR Green chemistry | Quantitative PCR applications [3] |
Discussion and Technical Considerations
Advantages and Limitations in Low Viral Load Contexts
Nested PCR demonstrates exceptional sensitivity for low viral load detection, with the two-round amplification process significantly enhancing signal from minimal starting material. However, this technique requires precise thermal cycling and carries increased contamination risk due to tube opening between amplification rounds [54] [55]. The method also demands more sophisticated laboratory infrastructure and longer processing time (typically 2-3 hours).
LAMP offers rapid results (30-60 minutes) with minimal equipment requirements, making it suitable for point-of-care or resource-limited settings. The colorimetric visualization eliminates the need for gel electrophoresis, and the isothermal conditions simplify instrumentation [56] [57]. However, indeterminate results (orange color) may occur with low viral loads, requiring confirmation by alternative methods [56]. Primer design is more complex, requiring 4-6 primers targeting specific regions.
Emerging Applications and Future Directions
Recent advances include one-step nested qRT-PCR formats that confine both amplification rounds to a single tube, reducing contamination risk while maintaining high sensitivity [11]. For LAMP, multiplexing capabilities and CRISPR-coupled detection are enhancing specificity and enabling quantitative analysis. Both technologies show promise for detecting SARS-CoV-2 variants of concern, with demonstrated efficacy against Alpha, Beta, Gamma, and Delta variants [57].
Both Nested PCR and LAMP offer viable solutions for detecting SARS-CoV-2 in low viral load samples, with complementary strengths suited to different diagnostic contexts. Nested PCR provides superior sensitivity and established reliability for laboratory-based testing, while LAMP offers rapid, equipment-light testing suitable for decentralized healthcare settings. The selection between these methods should consider the specific application requirements, including available infrastructure, required throughput, operator expertise, and intended use of results. For clinical scenarios where maximal sensitivity is critical for low viral load detection, Nested PCR remains the preferred choice, while LAMP provides an excellent alternative when speed, cost, and operational simplicity are prioritized.
Strategies to Minimize Contamination and False Positives
In the realm of molecular diagnostics, particularly for pathogens like SARS-CoV-2, the accuracy of test results is paramount for effective clinical and public health responses. False positives in diagnostic testing can lead to misdirected treatments, unnecessary isolation, and distorted disease surveillance data. Within the context of SARS-CoV-2 diagnostic accuracy research, two powerful nucleic acid amplification techniques—nested polymerase chain reaction (nested PCR) and loop-mediated isothermal amplification (LAMP)—present distinct advantages and challenges concerning contamination and false-positive results [14] [58]. Nested PCR, known for its exceptional sensitivity, employs two rounds of amplification with two sets of primers, but this very process increases the risk of aerosol contamination from amplicons generated during the first round [59] [14]. Conversely, LAMP, an isothermal amplification method conducted at a constant temperature, utilizes four to six primers recognizing distinct regions of the target genome, offering high specificity while being less equipment-intensive [60] [16]. However, its reaction setup and extreme sensitivity can still pose contamination risks [58] [56]. This guide objectively compares these methodologies, providing researchers with experimentally supported data and strategies to mitigate contamination, thereby enhancing the reliability of diagnostic outcomes.
Comparative Analysis of Contamination Risks and False-Positive Mechanisms
Understanding the fundamental mechanisms that lead to false positives in each method is the first step toward effective mitigation. The sources of error and their frequencies differ notably between the two techniques.
Table 1: Origins and Frequencies of False Positives in Nested PCR and LAMP
| Source of Error | Nested PCR | LAMP |
|---|---|---|
| Primary Cause | Cross-contamination from first-round amplicons during tube transfer [59] [14] | Nonspecific amplification, often due to prolonged incubation or primer-dimer interactions [56] |
| Typical Frequency | Higher risk due to the need for product handling between rounds [14] | Generally lower, but highly dependent on protocol optimization [1] [16] |
| Impact of Viral Load | Less dependent on viral load; contamination risk is procedural | More common in samples with low viral load where specific amplification is less robust [56] |
| Common Manifestation | False positive results in negative controls due to aerosol contamination [59] | Indeterminate results (e.g., orange color in colorimetric assays) or false positives from non-target amplification [56] |
Nested PCR: The Amplicon Contamination Challenge
The enhanced sensitivity of nested PCR comes from a second round of amplification using primers internal to the first product. This process, however, requires the physical transfer of amplification products, creating opportunities for cross-contamination. A 2024 study highlighted this risk by demonstrating how amplified DNA from a positive control can contaminate neighboring samples, leading to false positives. The same study proposed a mitigation strategy using a customized synthetic oligonucleotide as an internal control to detect such contamination events [59]. Furthermore, research on SARS-CoV-2 detection in cats acknowledged that while the nested PCR used was 100% sensitive and specific, a significant disadvantage was the "risk of contamination" inherent to the method's multi-step nature [23].
LAMP: The Primer and Reaction Condition Challenge
LAMP's false positives often stem from the reaction itself rather than post-amplification handling. The use of multiple primers increases the potential for primer-dimer formations and nonspecific amplification, especially if the reaction time exceeds the optimal window. One study on colorimetric RT-LAMP for SARS-CoV-2 found that incubation longer than 30 minutes could lead to nonspecific amplifications and false positives [56]. Another critical challenge is result interpretation, particularly in colorimetric assays. While results are often classified as just positive (yellow) or negative (pink), research has identified a third "indeterminate" cluster (orange) that is frequently associated with low viral loads. Misinterpreting this orange color as positive can lead to false-positive diagnoses [56].
Experimental Protocols for Direct Comparison
To objectively compare the performance and contamination resilience of nested PCR and LAMP, researchers must employ standardized, direct comparisons. The following protocols are derived from studies that have successfully implemented both techniques for SARS-CoV-2 detection.
Protocol 1: Parallel Testing of Clinical Samples
A 2024 study directly compared a one-step real-time PCR (RT-qPCR) method with a one-step RT-LAMP method using 342 human clinical samples (nasopharyngeal and saliva) [16].
- RNA Extraction: Viral RNA was extracted from samples using a commercial viral RNA extraction kit. The purity and quantity of RNA were verified with a spectrophotometer (e.g., NanoDrop) [16].
- One-Step RT-LAMP Assay:
- Primers: A set of six primers (F3, B3, FIP, BIP, LF, LB) targeting the N gene of SARS-CoV-2 were designed using Primer Explorer V5.
- Reaction: A 25 µL reaction mixture contained primers, 8U of Bst DNA/RNA Polymerase, and 5 µL of RNA template.
- Conditions: Amplification was performed at a constant temperature of 65°C for 30-40 minutes.
- Detection: Results were determined by fluorescence or colorimetric change [16].
- One-Step RT-qPCR Assay:
- Reaction: A 20 µL reaction was prepared using a commercial master mix and primer/probe sets targeting the SARS-CoV-2 N gene and an internal control (RNase P).
- Conditions: Reverse transcription at 50°C for 20 min, initial denaturation at 95°C for 3 min, followed by 45 cycles of denaturation (95°C for 15s) and annealing/extension (55°C for 40s) [16].
- Outcome Measurement: The agreement between the two tests was calculated using Cohen’s kappa coefficient (κ) from the dichotomous (positive/negative) results.
Protocol 2: Longitudinal Sensitivity Analysis
A 2021 study analyzed the diagnostic accuracy of RT-LAMP compared to RT-qPCR over the entire disease course of COVID-19, which is critical for understanding how viral load impacts false-negative and false-positive rates [1].
- Sample Collection: 124 nasopharyngeal swab samples were obtained from 24 confirmed COVID-19 patients. Samples were grouped based on days from symptom onset: Group 1 (≤9 days), Group 2 (10-19 days), Group 3 (20-29 days), and Group 4 (≥30 days) [1].
- Testing: All samples were tested in parallel by RT-LAMP (using the Loopamp SARS-CoV-2 Detection kit) and RT-qPCR.
- Data Analysis: Sensitivity and specificity of RT-LAMP versus RT-qPCR were calculated for each group. The relationship between genomic copy number (as determined by RT-qPCR) and RT-LAMP positivity was evaluated to establish the effective detection window and limit of detection (LOD) [1].
- Key Finding: RT-LAMP showed 100% sensitivity and specificity compared to RT-qPCR up to the 9th day after symptom onset. However, its positivity rate dropped significantly after the 10th day, demonstrating that its accuracy is exceptionally high only during the acute phase of infection when viral loads are high [1].
Table 2: Summary of Key Experimental Findings from Comparative Studies
| Study Focus | Nested PCR Performance | LAMP Performance | Key Contamination Insight |
|---|---|---|---|
| Overall Diagnostic Agreement [14] [16] | 100% sensitivity and specificity in validated SARS-CoV-2 assays [23] | 93-94% agreement with RT-qPCR (κ = 0.93-0.94) [16] | LAMP shows high concordance but is less sensitive than nested PCR in some settings. |
| Impact of Disease Stage [1] | Not specifically measured in these studies, but high sensitivity suggests effectiveness even at low viral loads. | Sensitivity drops from 92.8% (Day 9) to <25% (after Day 10) post-symptom onset [1] | LAMP's false-negative rate increases as viral load drops, indirectly reducing false-positive risk from low-level contamination in late-stage samples. |
| Analytical Sensitivity (LOD) | Can detect RNA concentrations as low as 0.015 ng/μL [23] | Detects between 100-200 RNA copies per reaction in colorimetric assays [56] [16] | The extreme sensitivity of nested PCR necessitates stringent contamination control to avoid false positives. |
Visualization of Contamination Pathways and Mitigation Strategies
The diagrams below illustrate the critical procedural pathways for nested PCR and LAMP, highlighting key points where contamination occurs and corresponding mitigation strategies.
Nested PCR Contamination Pathway
LAMP Contamination Pathway
The Scientist's Toolkit: Essential Reagents and Controls
Implementing the following reagents and controls is critical for robust, contamination-aware experimental design in both nested PCR and LAMP assays.
Table 3: Key Research Reagent Solutions for Contamination Control
| Reagent/Control | Function | Application |
|---|---|---|
| Synthetic Oligonucleotide Control [59] | Detects cross-contamination from positive controls in nested PCR. Acts as an internal marker that can be differentiated from the true target. | Nested PCR |
| Bst DNA/RNA Polymerase 3.0 | The key enzyme for LAMP reactions. Enables reverse transcription and strand displacement DNA synthesis at a constant temperature. | LAMP |
| Internal Amplification Control (e.g., RNase P) | Distinguishes true target negatives from PCR inhibition, reducing false negatives. | RT-qPCR, LAMP [16] |
| pH-Sensitive Dye (Phenol Red) | Allows visual, colorimetric detection of amplification in LAMP by tracking pH change. | Colorimetric LAMP [56] |
| No-Template Controls (NTCs) | Monitors for reagent contamination. A positive NTC indicates contaminated reagents or environmental amplicon pollution. | Nested PCR, LAMP |
| Pre-amplification Additives (e.g., UNG) | Degrades carryover amplicons from previous PCR runs, preventing their amplification. | Nested PCR |
The strategic minimization of contamination and false positives in SARS-CoV-2 diagnostics requires a method-specific approach. Nested PCR's primary vulnerability is procedural, stemming from the manipulation of amplified products between rounds. Its mitigation, therefore, relies on rigorous laboratory discipline, physical workspace separation, and the novel use of synthetic oligonucleotide controls [59] [14]. In contrast, LAMP's primary risks are biochemical, arising from intricate primer interactions and reaction conditions. Controlling for false positives here demands meticulous primer design, strict optimization of incubation times, and objective result interpretation, such as using spectrophotometry to define indeterminate zones [56] [16]. The choice between these sensitive techniques should be guided by the diagnostic context, including available laboratory infrastructure, the required throughput, and the prevalence of the pathogen. By implementing the detailed protocols, visual guides, and toolkit components outlined in this comparison, researchers and drug development professionals can significantly enhance the accuracy and reliability of their diagnostic data, thereby strengthening the scientific and public health response to infectious disease threats.
The diagnostic accuracy of molecular tests for SARS-CoV-2 depends fundamentally on the optimization of reaction conditions. Among available techniques, nested Polymerase Chain Reaction (nested PCR) and Loop-mediated Isothermal Amplification (LAMP) have emerged as viable detection platforms with distinct operational characteristics. While real-time RT-PCR remains the gold standard, these alternative methods offer advantages in specific diagnostic scenarios, particularly in resource-limited settings. This comparison guide objectively evaluates the performance of nested PCR versus LAMP for SARS-CoV-2 detection, focusing specifically on the critical reaction parameters of temperature, time, and reagents that directly impact diagnostic accuracy. The optimization of these parameters not only affects the sensitivity and specificity of detection but also determines the practical applicability of each method in diverse healthcare environments. By examining experimental data across multiple studies, this analysis provides evidence-based recommendations for researchers and diagnosticians seeking to implement or improve these molecular detection methods.
Technical Principles and Workflows
Fundamental Mechanisms of Amplification
Nested PCR operates through a two-stage amplification process that significantly enhances detection specificity. The initial round uses outer primers to amplify a target region, followed by a second amplification using inner primers that bind within the first amplicon. This sequential approach minimizes amplification of non-specific products while increasing overall sensitivity [61]. The method requires precise thermal cycling with denaturation, annealing, and extension steps repeated across 30-40 cycles, typically utilizing Taq DNA polymerase with optimal activity at 72°C.
LAMP employs strand displacement DNA synthesis using 4-6 primers targeting 6-8 distinct regions of the target DNA. The Bst DNA polymerase enzyme drives auto-cycling amplification under isothermal conditions (60-65°C) without requiring thermal denaturation [3]. This mechanism generates loop structures that facilitate continuous amplification, yielding high quantities of product in short timeframes. The reaction can be visualized through various methods including turbidity, fluorescence, or colorimetric changes.
Visual Workflow Comparison
The following diagram illustrates the key procedural steps and conditional requirements for both nested PCR and LAMP detection methods:
Comparative Experimental Data
Performance Metrics for SARS-CoV-2 Detection
Table 1: Direct comparison of nested PCR and LAMP performance characteristics based on experimental studies
| Performance Parameter | Nested PCR | LAMP | Experimental Context |
|---|---|---|---|
| Sensitivity | 100% [26], 50-66.6% [8] | 79-93.8% [56] [62], 66.6% [8] | Clinical samples (human and feline) compared to RT-qPCR |
| Specificity | 100% [26] | 90.4-100% [56] [62] | Evaluation against other respiratory pathogens |
| Detection Limit | 0.015 ng/μL SARS-CoV-2 RNA [26], 10-fold more sensitive than LAMP for M. marinum [8] | 200 RNA copies [56], 1 trophozoite for E. histolytica [3] | Serial dilution of target nucleic acid |
| Optimal Temperature | Varies by step: Denaturation: 95°C, Annealing: 54-62°C, Extension: 72°C [26] | Isothermal: 63-65°C [56] [62] [8] | Temperature optimization studies |
| Reaction Time | ~3 hours (including two amplification rounds) [26] | 30-60 minutes [56] [62] | Time from sample to result |
| Viral Load Efficiency | Detects low viral load samples [8] | 100% detection for Ct <30 [56], reduced sensitivity for Ct >30 | Correlation with RT-qPCR Ct values |
Optimization Parameters and Reagent Requirements
Table 2: Detailed comparison of reaction conditions and reagent requirements for nested PCR and LAMP
| Optimization Parameter | Nested PCR | LAMP | Impact on Performance |
|---|---|---|---|
| Temperature Conditions | First round: 95°C/54°C/72°C; Second round: similar [26] | Single temperature: 63-65°C [56] [8] | LAMP eliminates need for thermal cycler |
| Time Requirements | Initial denaturation: 5 min; 35 cycles: 2+ hours; Total: ~3 hours [26] | 30-60 minutes total [56] [62] | LAMP significantly faster |
| Primer Design | Two primer pairs (outer and inner); ~20nt each [61] | 4-6 primers; FIP, BIP, F3, B3, LF, LB [3] | LAMP primers more complex to design |
| Enzyme Requirements | Taq DNA polymerase (thermostable) [26] | Bst DNA polymerase (strand-displacing) [3] | Different polymerase properties |
| Detection Methods | Gel electrophoresis, sequence analysis [26] | Colorimetric, turbidity, fluorescence, gel electrophoresis [3] [56] | LAMP offers more visualization options |
| Inhibition Resistance | Moderate; inhibitors diluted in second round [8] | High; tolerant to inhibitors [8] | LAMP better for crude samples |
Research Reagent Solutions
Table 3: Essential research reagents and their functions in nested PCR and LAMP assays
| Reagent Category | Specific Examples | Function in Assay | Application in SARS-CoV-2 Detection |
|---|---|---|---|
| Polymerase Enzymes | Taq DNA Polymerase (Thermo Fisher) [3], Bst DNA Polymerase (New England Biolabs) [3] [7] | DNA amplification through primer extension | Target amplification in both techniques |
| Reverse Transcriptase | SensiFAST cDNA Synthesis Kit (Bioline) [26] | RNA to cDNA conversion | Essential first step for SARS-CoV-2 RNA detection |
| Primer Sets | SARS-CoV-2 N gene primers [26], SREHP gene primers [3] | Target sequence recognition | Determines assay specificity |
| Detection Dyes/Probes | SYBR Green [3], Hydroxy naphthol blue (HNB) [7], Calcein-manganese [3] | Amplification visualization | Enables colorimetric or fluorescent detection |
| Buffer Components | Betaine, Mg²⁺, dNTPs [3] [7] | Reaction optimization | Enhances specificity and yield |
| RNA Extraction Kits | QIAmp Viral RNA Mini Kit (Qiagen) [62], ISOLATE II RNA Mini Kit (Bioline) [26] | Nucleic acid purification | Sample preparation for downstream analysis |
Experimental Protocols
Optimized Nested PCR Protocol for SARS-CoV-2 Detection
The following protocol was adapted from a study that demonstrated 100% sensitivity and specificity for SARS-CoV-2 detection in human and feline samples [26]:
Primer Design:
- External primers: Ext2019nCorVF (5'-GGCAGTAACCAGAATGGAGA-3') and Ext2019nCorVR (5'-CTCAGTTGCAACCCATATGAT-3')
- Internal primers: intF (5'-CACCGCTCTCACTCAACAT-3') and intR (5'-CATAGGGAAGTCCAGCTTCT-3')
- Target: SARS-CoV-2 N gene (positions 28346-28681, reference sequence MN908947.3)
First Round PCR:
- Reaction mixture: 12.5 μL My Taq HS red mix, 4 μL cDNA, 1 μL each external primer (10 pmol/μL), 6.5 μL PCR grade water
- Cycling conditions: Initial denaturation at 95°C for 5 min; 35 cycles of 95°C for 30s, 54.6°C for 50s, 72°C for 1 min; final extension at 72°C for 5 min
Second Round PCR:
- Reaction mixture: 12.5 μL My Taq HS red mix, 0.5 μL first PCR product, 1 μL each internal primer (10 pmol/μL), 10 μL PCR grade water
- Cycling conditions: Same as first round
Analysis:
- Products analyzed by 2% agarose gel electrophoresis
- Expected product sizes: 335 bp (first round), 212 bp (second round)
Optimized RT-LAMP Protocol for SARS-CoV-2 Detection
This protocol achieved 93.8% sensitivity and 90.4% specificity for SARS-CoV-2 detection in clinical samples [62]:
Primer Design:
- Six primers targeting SARS-CoV-2 ORF1ab gene: F3, B3, FIP, BIP, LF, LB
- Primer ratio optimization critical: F3/B3:FIP/BIP recommended at 1:8
Reaction Setup:
- Reaction volume: 20-25 μL
- Components: 15 μL LAMP OTG Reagent, 1.6 μM each FIP and BIP, 0.2 μM each F3 and B3, 2 μL template DNA
- Temperature: 63°C for 60 minutes
- Enzyme inactivation: 80°C for 5 minutes
Detection Methods:
- Colorimetric: Positive samples turn from pink to yellow
- Validation: Agarose gel electrophoresis showing ladder-like pattern
- Internal control: Human RNase P gene recommended to avoid false negatives
Discussion
Contextual Performance Analysis
The experimental data reveals that both nested PCR and LAMP offer viable pathways for SARS-CoV-2 detection with distinct advantages under different operational constraints. Nested PCR demonstrates exceptional specificity (100% in multiple studies) [26] attributable to the dual amplification process that minimizes non-specific product formation. However, this advantage is counterbalanced by significantly longer processing times (~3 hours) and higher contamination risk due to tube opening between amplification rounds.
LAMP technology provides markedly faster results (30-60 minutes) [56] with excellent sensitivity (79-93.8%) [56] [62], particularly in samples with high viral loads (Ct<30). The colorimetric detection methods enable visual interpretation without specialized equipment, making LAMP particularly suitable for point-of-care applications and resource-limited settings. The single-temperature incubation requirement further simplifies operational requirements compared to thermal cycling.
Critical Optimization Considerations
Temperature optimization proves fundamentally different between the two methods. While nested PCR requires precise temperature cycling across a 40°C range (54-95°C) [26], LAMP operates at a single isothermal temperature (63-65°C) [56] [8]. This distinction makes LAMP more suitable for field-deployable applications but introduces different optimization challenges related to primer design and reaction specificity.
Reaction time optimization demonstrates a clear advantage for LAMP, with complete reactions in 30-60 minutes [56] [62] compared to 3 hours for nested PCR [26]. However, studies note that extending LAMP beyond 30 minutes risks nonspecific amplification, while insufficient time reduces sensitivity for low viral load samples [56].
Reagent optimization reveals distinctive requirements for each method. Nested PCR performance depends heavily on appropriate primer design for two successive amplifications and careful optimization of magnesium concentrations [61]. LAMP requires more complex primer designs (4-6 primers) but demonstrates greater tolerance to PCR inhibitors present in clinical samples [8], potentially reducing sample purification requirements.
The optimization of reaction conditions for SARS-CoV-2 detection presents a series of technical trade-offs between nested PCR and LAMP methodologies. Nested PCR offers uncompromised specificity and sensitivity, making it suitable for laboratory confirmation testing where time constraints are secondary to accuracy. Conversely, LAMP provides rapid results with minimal equipment requirements, creating advantages for screening applications and resource-limited environments. The selection between these techniques should be guided by specific diagnostic contexts, available infrastructure, and performance requirements. Temperature parameters, processing time, and reagent optimization each play critical roles in determining the ultimate diagnostic accuracy of either method. Future development should focus on simplifying primer design for LAMP and reducing contamination risk in nested PCR to enhance their practical implementation in diverse healthcare settings.
Primer Validation and Managing Primer-Dimer Formation
In molecular diagnostics, the accuracy of nucleic acid amplification tests is critically dependent on the specificity of primer binding. Primer-dimer (PD) formation is a well-known by-product in polymerase chain reaction (PCR) wherein two primer molecules hybridize to each other via short stretches of complementary bases, rather than to the target template. The DNA polymerase can then amplify this dimer, leading to competition for PCR reagents (such as primers and nucleotides) and potentially inhibiting the amplification of the intended target sequence. This is especially problematic in quantitative PCR, where primer-dimers can interfere with accurate quantification [63]. The risk of dimer formation escalates significantly in multiplexed reactions, growing polynomially with the number of primers added [64].
This guide objectively compares the approaches to primer validation and dimer management within the context of a broader thesis comparing nested PCR and Loop-Mediated Isothermal Amplification (LAMP) for SARS-CoV-2 diagnostic accuracy. We summarize experimental data and provide detailed protocols to aid researchers, scientists, and drug development professionals in selecting and optimizing their diagnostic assays.
Comparative Analysis of Nested PCR and LAMP
Core Methodological Principles
Nested PCR is a conventional, highly sensitive molecular technique that involves two successive rounds of amplification. The first round uses an outer set of primers to amplify a target region, and a second round uses a set of inner primers that bind within the first amplicon. This process enhances specificity and sensitivity but also increases the risk of contamination and requires longer turnaround times due to multiple handling steps [65] [66]. A study on SARS-CoV-2 detection reported that nested PCR demonstrated high reliability and sensitivity when validated against LAMP, with concordant results between the two methods [29].
LAMP is an isothermal amplification technique that uses four to six primers targeting six to eight distinct regions on the target DNA. It operates at a constant temperature (usually between 60-65°C), eliminating the need for a thermocycler. Its key advantages include speed, operational simplicity, and inherent robustness, making it highly suitable for resource-limited settings and point-of-care applications [29] [67]. The isothermal nature of LAMP simplifies the heating instrumentation required, facilitating the development of portable, low-cost diagnostic devices [67].
Diagnostic Performance for SARS-CoV-2 Detection
A direct comparative study of these two techniques for detecting SARS-CoV-2 in human samples demonstrated equivalent performance in terms of result accuracy.
Table 1: Comparative Performance of nPCR and LAMP for SARS-CoV-2 Detection [29]
| Method | Total Samples Tested | Positive Results | Negative Results | Invalid Results | Key Findings |
|---|---|---|---|---|---|
| Nested PCR (nPCR) | 427 | 43 (10.1%) | 382 (89.5%) | 2 (0.4%) | Reliable and sensitive for nasopharyngeal, oropharyngeal, and nasal swabs. |
| LAMP | 427 | 43 (10.1%) | 382 (89.5%) | 2 (0.4%) | Results were 100% concordant with nPCR. |
The study concluded that while the nested PCR used was a reliable and sensitive technique, it carried a risk of contamination and was slower due to the duration of the multi-step process [29].
Experimental Approaches to Primer Validation
In Silico Prediction of Primer-Dimer Formation
Accurate prediction of primer-dimer formation during the design phase is crucial for assay success. Several tools use the change in Gibbs free energy (ΔG) resulting from primer hybridization as an indicator of dimer formation. However, the calculation methods and predictive accuracy of these tools vary widely [64].
PrimerROC is a novel tool that uses Receiver Operating Characteristic (ROC) curves to assess the predictive power of ΔG-based dimer scores without requiring specific reaction conditions like salt concentration or annealing temperature. In a systematic evaluation of over 300 primer pairs, the PrimerROC/PrimerDimer software consistently outperformed other publically available tools, achieving predictive accuracies greater than 92% [64]. The tool identifies a dimer-free threshold above which dimer formation is predicted to be unlikely, a critical feature for robust primer design, especially in multiplex assays.
Table 2: Comparison of Primer-Dimer Prediction Tools [64]
| Tool Name | Core Prediction Metric | Key Features | Reported Performance |
|---|---|---|---|
| PrimerROC/PrimerDimer | ΔG (Gibbs Free Energy) | Condition-independent; uses ROC analysis to set a dimer-free threshold; optimized for extensible dimers. | >92% accuracy; consistently outperformed other tools. |
| Oligo 7 | ΔG | Performance varies with primer length and fusion sequences. | Reliable across multiple datasets; comparable to in-house ΔG calculations. |
| PerlPrimer | ΔG (focus on 3' ends) | Classifies "most stable 3' dimer" structures. | Good for short primers; performance drops with longer fusion sequences. |
The PrimerDimer algorithm works by aligning the 5' end of the longer primer to the 3' end of the shorter primer, forming a structure with a single 3' overhang. The shorter primer slides along the longer one to form all possible dimer structures. The ΔG of each structure is calculated using nearest-neighbour parameters, and the most negative value is returned as the dimer score. It is important to note that this algorithm is optimized to predict extensible dimers (which amplify and consume reagents) and excludes non-extensible dimers, which form stable structures but do not elongate and have been shown not to significantly impact PCR efficiency [64].
Empirical Validation Using Free-Solution Conjugate Electrophoresis (FSCE)
While in silico tools are powerful, empirical validation is often necessary. A Free-Solution Conjugate Electrophoresis (FSCE) method has been developed to quantitatively assess dimerization risk between primer-barcode pairs [68].
Experimental Protocol [68]:
- Design and Conjugation: Design primers with complementary regions of differing lengths. Conjugate one primer (a 30-mer) to a synthetically produced, electrically neutral "drag-tag" (e.g., a poly-N-methoxyethylglycine). This drag-tag alters the primer's electrophoretic mobility.
- Annealing: Anneal pairs of drag-tagged and non-drag-tagged primers.
- Electrophoresis: Separate the annealed products using capillary electrophoresis under free-solution conditions (no sieving matrix) at a range of temperatures (e.g., 18, 25, 40, 55, 62°C).
- Analysis: The drag-tag allows for clear separation of single-stranded DNA from double-stranded primer-dimers based on their differential mobility. The proportion of dimer formed can be quantified at different temperatures.
Key Experimental Findings [68]:
- Dimerization was inversely correlated with temperature when less than 30 out of 30 possible base pairs were bonded.
- Stable dimerization occurred only when more than 15 consecutive base pairs were able to form.
- Non-consecutive base pairs did not create stable dimers, even when up to 20 out of 30 possible base pairs were bonded, highlighting the importance of the spatial arrangement of complementary bases.
The following diagram illustrates the experimental workflow for the FSCE method:
Strategies for Managing Primer-Dimer Formation
PCR-Based Optimization Techniques
Hot-Start PCR is a fundamental technique to prevent primer-dimer formation. Primers can anneal to each other at low temperatures (e.g., during reaction setup). Hot-start methods inhibit DNA polymerase activity until the reaction reaches high temperatures (~95°C). Several mechanisms exist [63]:
- Chemical Modification: A small molecule covalently bound to the enzyme's active site is released only after prolonged heating at 95°C.
- Non-covalent Binding: An antibody, peptide, or aptamer binds and inhibits the polymerase at low temperatures, denaturing at high heat.
- Physical Separation: Using a wax barrier to separate polymerase from other reaction components until the first denaturation step.
- Slow Release of Magnesium: Magnesium ions, essential for polymerase activity, are chemically sequestered and released only at high temperatures.
Physical-Chemical Optimization involves tuning reaction components such as primer concentration, magnesium chloride concentration, nucleotide concentration, ionic strength, and annealing temperature. While effective, this approach has limitations, as changes that reduce dimer formation can also reduce the efficiency of target amplification [63].
Primer Engineering and Design Innovations
Structural modifications to the primers themselves can effectively reduce or eliminate dimer formation [63]:
- HANDS (Homo-Tag Assisted Non-Dimer System): A nucleotide tail complementary to the primer's own 3' end is added to its 5' end. This promotes the formation of a stem-loop structure that blocks the 3' end from participating in dimer formation with other primers, while still allowing hybridization to a longer, fully complementary target template.
- Self-Avoiding Molecular Recognition Systems (SAMRS): This approach involves incorporating nucleotide analogues (T, A, G, C) into the primers. SAMRS nucleotides can bind to their natural DNA counterparts (e.g., A* to T) but not to other SAMRS nucleotides (e.g., A* will not bind to T*). This allows primers to bind to natural DNA targets while avoiding primer-primer interactions.
- Chimeric Primers: Some DNA bases in the primer are replaced with RNA bases. The chimeric primer has a lower melting temperature (Tm) when binding to another chimeric primer compared to its Tm when binding to a full-DNA target. This allows for setting an annealing temperature where target amplification occurs, but primer-dimer formation does not.
- Blocked-Cleavable Primers (rhPCR): Primers are synthesized with a blocking group at the 3' end that prevents extension. The block can only be removed by a thermostable RNase HII enzyme at high temperatures, and this enzyme also discriminates against primer-template mismatches, providing a double layer of specificity that prevents dimer extension.
The mechanism of primer-dimer formation and the points of intervention for these strategies are summarized below:
The Scientist's Toolkit: Research Reagent Solutions
The following table details key reagents and materials essential for experiments focused on primer validation and managing dimer formation.
Table 3: Essential Research Reagents and Materials
| Item | Function/Application | Example Use Case |
|---|---|---|
| DNA Polymerase (Hot-Start) | Enzyme inhibited during reaction setup to prevent low-temperature priming. | All PCR-based assays to minimize primer-dimer formation [63]. |
| Primer Design Software | In silico analysis of primer specificity, Tm, and potential for dimer formation. | Using tools like PrimerROC to select dimer-free primers during assay design [64]. |
| Drag-Tags (e.g., Poly-NMEG) | Electrically neutral polymers conjugated to DNA to alter electrophoretic mobility. | Empirical dimer validation via Free-Solution Conjugate Electrophoresis [68]. |
| Nucleotide Analogues (SAMRS) | Artificial bases that bind to natural DNA but not to themselves. | Engineering primers to avoid primer-primer interactions in multiplex PCR [63]. |
| Colorimetric LAMP Master Mix | Contains pH-sensitive dye for visual readout of amplification. | Enabling simple, instrument-free detection of LAMP amplification in field tests [67]. |
| Thermostable RNase HII | Enzyme used in rhPCR to cleave blocking groups from primers only when perfectly matched to template. | Activating cleavable primers to prevent extension from misprimed or dimerized structures [63]. |
Adapting Assays for Emerging Variants and Strain Evolution
The continuous evolution of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) presents significant challenges for diagnostic accuracy, requiring molecular assays that maintain performance across emerging variants. As new variants of concern (VOCs) with mutations in key genomic regions emerge, diagnostic laboratories must evaluate and adapt detection methods to ensure sustained sensitivity and specificity. Research demonstrates that the virus evolves consistently, with studies confirming it "continually evolved, even the sample which was put through 100 passages" in laboratory settings, mirroring real-world mutation patterns [69]. This persistent evolution underscores the critical need for robust diagnostic platforms that can withstand genetic drift in viral genomes.
Two principal molecular techniques have emerged as central to SARS-CoV-2 detection: reverse transcription loop-mediated isothermal amplification (RT-LAMP) and nested polymerase chain reaction (PCR) methods, including one-step nested quantitative RT-PCR (OSN-qRT-PCR). This comparison guide objectively evaluates their performance characteristics, experimental validation data, and adaptability to the evolving viral landscape, providing researchers and drug development professionals with evidence-based insights for diagnostic selection and optimization.
Performance Comparison: Nested PCR versus RT-LAMP
Direct comparative studies and independent validation data reveal distinct performance profiles for nested PCR and RT-LAMP assays across key diagnostic parameters.
Table 1: Analytical Performance Comparison of SARS-CoV-2 Detection Methods
| Method | Limit of Detection (copies/reaction) | Clinical Sensitivity | Clinical Specificity | Time to Result | Complexity & Equipment Needs |
|---|---|---|---|---|---|
| Nested PCR (OSN-qRT-PCR) | 189.1-194.74 [11] | 82.35% (vs. 58.82% for qRT-PCR) [11] | 100% [26] | ~2 hours [11] | Thermal cycler, RNA extraction equipment |
| RT-LAMP | 6.7-72 [1] [70] | 100% (early infection, Ct≤33) [1] [70] | 97.9-100% [1] [70] | 30-45 minutes [1] [16] | Heat block/water bath, minimal equipment |
| Conventional RT-qPCR | 520.1-528.1 [11] | 58.82% [11] | ~100% | 1.5-2 hours | Thermal cycler, RNA extraction equipment |
Table 2: Clinical Performance Across Sample Types and Disease Stages
| Method | Performance in Early Infection (≤9 days post-onset) | Performance in Late Infection (≥10 days) | Saliva/Extraction-Free Application | Point-of-Care Suitability |
|---|---|---|---|---|
| Nested PCR | High sensitivity (82.35%) [11] | Maintains sensitivity in low viral loads [11] | Limited data | Low (requires specialized equipment) |
| RT-LAMP | 100% sensitivity/specificity vs. RT-qPCR [1] | Positivity decreases to <25% after day 10 [1] | Excellent (90% accuracy) [71] | High (colorimetric detection, minimal equipment) |
| Conventional RT-qPCR | 58.82% positive rate [11] | Variable depending on viral load | Possible with optimized protocols [71] | Moderate (requires thermal cycler) |
Experimental Protocols and Methodologies
One-Step Nested qRT-PCR Protocol
The highly sensitive OSN-qRT-PCR method employs a single-tube, two-stage amplification approach:
- Primer Design: Two primer sets target conserved regions of SARS-CoV-2, typically in ORF1ab and N genes [11].
- Reaction Setup: The 20μL reaction mixture contains template RNA, reaction buffer, and enzyme mixture [11].
- Amplification Parameters: Reverse transcription at 50°C for 30 minutes, followed by initial denaturation at 95°C for 1 minute, then 20 cycles of amplification (95°C for 30s, 70°C for 40s, 72°C for 40s), and finally 40 cycles of quantitative detection (95°C for 15s, 60°C for 30s, 25°C for 10s) [11].
- Detection: Fluorescence measurement during the final 40 cycles enables quantitative analysis.
This protocol's innovative single-tube nested approach prevents amplicon contamination while significantly enhancing sensitivity compared to conventional qRT-PCR, particularly beneficial for samples with low viral load [11].
RT-LAMP Assay Protocol
The RT-LAMP methodology utilizes isothermal amplification with multiple primers recognizing distinct target sequences:
- Primer Design: Six primers (F3, B3, FIP, BIP, LF, LB) targeting eight regions of the SARS-CoV-2 genome, typically on ORF1ab or N genes, ensure high specificity [16].
- Reaction Composition: A 25μL reaction mixture includes 10μL of purified RNA or pretreated sample, 15μL reaction mix containing primer sets, and WarmStart master mix with Bst DNA polymerase [1] [72].
- Amplification Conditions: Incubation at 62-65°C for 30-45 minutes, followed by enzyme inactivation at 80°C for 5 minutes [1] [16].
- Detection Methods: Multiple readout options include:
- Colorimetric: pH-sensitive dyes (phenol red) change from pink to yellow with amplification [72] [71].
- Turbidimetric: Real-time turbidimeters measure magnesium pyrophosphate precipitate [1].
- Fluorometric: Intercalating dyes detected with real-time fluorescence [72].
- Mobile-assisted: Applications like SpotXel improve color change interpretation accuracy from 61-74% (naked eye) to 75-86% [72].
RT-LAMP Workflow for SARS-CoV-2 Detection
Sample Processing Methodologies
- RNA Extraction: Both methods can utilize extracted RNA from commercial kits (e.g., QIAamp Viral RNA Mini Kit, ISOLATE II RNA Mini Kit) [1] [26].
- Extraction-Free Protocols: RT-LAMP particularly excels with direct sample processing:
- Saliva Pretreatment: Optimization of saliva pretreatment protocols enables analytically sensitive viral detection without RNA purification [71].
- Heat Processing: Mixing samples with lysis buffer and heating at 95°C for 5 minutes effectively releases RNA for amplification [72].
- Transport Media Considerations: Sample pH in transport media critically affects colorimetric RT-LAMP performance, requiring optimization [72].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for SARS-CoV-2 Assay Development
| Reagent/Kit | Function | Application Notes | References |
|---|---|---|---|
| QIAamp Viral RNA Mini Kit | RNA extraction | Used with automated nucleic acid extraction devices like QIAcube | [1] |
| WarmStart LAMP Kit | Isothermal amplification | Contains Bst DNA polymerase with reverse transcriptase; stable at room temperature | [72] [16] |
| TaqMan Fast Virus 1-Step Master Mix | qRT-PCR reactions | Optimized for one-step reverse transcription and amplification | [1] |
| SensiFAST cDNA Synthesis Kit | cDNA synthesis | Used for reverse transcription in nested PCR protocols | [26] |
| Phenol Red pH Indicator | Colorimetric detection | Enables visual detection of amplification through acidification | [70] [72] |
| Loopamp SARS-CoV-2 Detection Kit | Commercial LAMP assay | Complete reagent system for SARS-CoV-2 detection | [1] |
| My Taq HS Red Mix | PCR amplification | Optimized for high-sensitivity PCR applications | [26] |
Adaptation Strategies for Emerging Variants
The continuous evolution of SARS-CoV-2 necessitates strategic assay adaptations to maintain diagnostic accuracy across variants.
Primer Design Considerations
- Multitarget Approach: Simultaneous targeting of multiple conserved regions (ORF1ab, N, E genes) reduces false negatives due to point mutations [11] [16].
- Variant-Specific Primers: Development of updated primers matching emerging variant sequences, as demonstrated with BA.2.75.2 spike protein in vaccine development [73].
- Conserved Region Identification: Focus on genomically stable regions with lower mutation rates across variants [69].
Assay Adaptation Strategy for Emerging Variants
Assay Validation Approaches
- Cross-Reactivity Testing: Ensure primers do not detect other human coronaviruses or common respiratory pathogens [26] [70].
- Limit of Detection Assessment: Establish detection thresholds for each variant using quantified viral RNA [70] [11].
- Clinical Performance Verification: Test against clinical samples characterized by whole-genome sequencing [73] [69].
- Robustness Evaluation: Assess performance under varying conditions (incubation time, temperature, reaction volume) [70].
Both nested PCR and RT-LAMP offer distinct advantages for SARS-CoV-2 detection in the context of continuous viral evolution. Nested PCR methods, particularly OSN-qRT-PCR, provide exceptional sensitivity for low viral load samples and complex diagnostic challenges, making them valuable for confirmatory testing and research applications. Conversely, RT-LAMP technology delivers rapid, accessible testing with minimal infrastructure requirements, ideal for point-of-care deployment and resource-limited settings.
The selection between these methodologies should be guided by specific application requirements: diagnostic accuracy needs, available resources, timeframe for results, and the evolving nature of viral variants. As SARS-CoV-2 continues to demonstrate "remarkable adaptability" with "mutations which emerge repeatedly and independently in different strains" [69], ongoing primer optimization and validation against emerging variants remain imperative for both platforms. Future development will likely focus on multiplexed assays targeting multiple variant signatures simultaneously and streamlined workflows that maintain analytical sensitivity while increasing accessibility and throughput.
Analytical Performance and Comparative Efficacy Metrics
The accurate and timely diagnosis of SARS-CoV-2 infection has been a cornerstone of the global response to the COVID-19 pandemic. Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) has remained the gold standard for detection. However, the need for rapid, sensitive, and accessible testing has driven the development and evaluation of alternative molecular methods, notably reverse transcription loop-mediated isothermal amplification (RT-LAMP) and nested PCR techniques. This guide provides an objective, data-driven comparison of these two methods within the context of SARS-CoV-2 diagnostic accuracy research, synthesizing experimental data on their sensitivity, specificity, and operational profiles to inform researchers, scientists, and drug development professionals.
Performance Comparison: Analytical Sensitivity and Specificity
Direct head-to-head studies comparing nested PCR and LAMP for SARS-CoV-2 are limited, but an analysis of comparative studies on other pathogens reveals consistent performance trends. The data below summarizes their typical analytical sensitivity and specificity.
Table 1: Comparative Analytical Sensitivity of Molecular Detection Methods
| Pathogen/Target | Conventional PCR | Nested PCR | Real-time PCR (qPCR) | LAMP | Reference |
|---|---|---|---|---|---|
| Entamoeba histolytica (Trophozoites) | 1,000 | 100 | 100 | 1 | [3] |
| Alternaria solani (Genomic DNA) | 1,000 pg | 10 pg | 1 pg | 100 pg | [6] |
| Fusarium tricinctum (Genomic DNA) | Not Reported | 31 fg/μL | 3.1 fg/μL | 31 fg/μL | [7] |
| Mycobacterium marinum (Serial DNA Dilution) | Not Reported | 10-fold more sensitive than LAMP | Not Reported | Baseline | [8] |
Table 2: Diagnostic Performance in Clinical Samples for SARS-CoV-2
| Method | Comparison Benchmark | Clinical Sensitivity / Positive Rate | Specificity | Key Finding | Reference |
|---|---|---|---|---|---|
| RT-LAMP | RT-qPCR (up to 9 days post-symptom-onset) | 100% | 100% | Equivalent to RT-qPCR in early infection phase. | [1] |
| One-Step Nested qRT-PCR | Standard qRT-PCR (Clinical samples from patients) | 82.35% (28/34) | Not Specified | Superior positive detection rate compared to qRT-PCR (58.82%) and ddPCR (67.65%). | [11] |
| LAMP | Culture (6 clinical skin specimens for M. marinum) | 66.6% (4/6) | 100% | Showed higher detection rate than conventional PCR (0%) and matched nested PCR sensitivity. | [8] |
Key Performance Insights
- Sensitivity Hierarchy: The data consistently shows that nested PCR is one of the most sensitive molecular techniques, often 10 to 1000 times more sensitive than conventional PCR and frequently outperforming LAMP in direct dilution experiments [8] [6]. However, highly optimized qPCR can sometimes surpass it [7] [6].
- LAMP Clinical Utility: Despite sometimes lower analytical sensitivity in controlled dilution tests, LAMP demonstrates exceptional clinical sensitivity that can rival or even surpass standard PCR in real-world patient samples, particularly during the acute phase of infection [1]. This is likely due to its high resistance to inhibitors present in clinical specimens [8].
- Specificity: Both methods demonstrate high specificity when primers are well-designed. LAMP uses 4-6 primers targeting 6-8 distinct regions, which inherently provides high specificity [74] [75]. Nested PCR achieves high specificity through two successive rounds of amplification with two primer sets [3] [11].
Experimental Workflows and Methodologies
A critical differentiator between these techniques lies in their experimental workflows. The following diagrams and protocols outline their fundamental procedures.
Workflow Diagrams
Figure 1: Nested RT-PCR Workflow. This two-step process involves an initial amplification with outer primers, followed by a second amplification using inner primers that bind within the first amplicon, enhancing specificity and sensitivity [3] [11].
Figure 2: RT-LAMP Workflow. This single-tube isothermal amplification uses multiple primers and a strand-displacing DNA polymerase to amplify DNA at a constant temperature, allowing for simple and rapid visual readout [1] [74] [75].
Key Experimental Protocols
Typical Nested PCR Protocol for SARS-CoV-2 [11]:
- First Round PCR:
- Primers: Outer primers targeting ORF1ab and N genes.
- Reaction Mix: Taq PCR Master Mix, primers, template cDNA.
- Cycling Conditions: Initial denaturation at 95°C for 5 min; 20 cycles of 95°C for 30s, 70°C for 40s, 72°C for 40s.
- Second Round PCR:
- Primers: Inner primers binding within the first amplicon.
- Template: 1 µL of the first-round PCR product (often diluted).
- Cycling Conditions: Initial denaturation at 95°C for 1 min; 40 cycles of 95°C for 15s, 60°C for 30s.
Typical RT-LAMP Protocol for SARS-CoV-2 [1]:
- Primer Design: A set of 6 primers (F3, B3, FIP, BIP, LF, LB) targeting specific regions of the SARS-CoV-2 genome (e.g., N, ORF1a genes).
- Reaction Setup:
- Reaction Mix: 15 µL of reaction buffer, 4 µL enzyme mix (Bst DNA polymerase + reverse transcriptase), 1.6 µM each of FIP and BIP, 0.2 µM each of F3 and B3, and 2 µL of template RNA.
- Amplification:
- Conditions: Incubation at 62.5°C for 35 minutes in a heating block or water bath.
- Detection:
- Methods: Visual color change with hydroxynaphthol blue (HNB) or calcein dyes, turbidity measurement, or real-time fluorescence monitoring.
Research Reagent Solutions and Essential Materials
The successful implementation of these diagnostic assays relies on a suite of specific reagents and tools.
Table 3: Essential Research Reagents and Materials
| Item | Function in Nested PCR | Function in LAMP | Example Kits / Components (from search results) |
|---|---|---|---|
| Reverse Transcriptase | Converts viral RNA to cDNA for PCR. | Integrated into the mix for direct RNA amplification. | TaqPath COVID-19 RT-PCR Kit [76] |
| DNA Polymerase | Thermostable polymerase (e.g., Taq) for thermocycling. | Strand-displacing polymerase (e.g., Bst) for isothermal amplification. | Bst DNA polymerase (New England Biolabs) [3] [7] |
| Primers | Two pairs of specific oligonucleotides (outer & inner). | Four to six specific primers (F3, B3, FIP, BIP, LF, LB). | Custom synthesized primers [8] [6] |
| dNTPs | Nucleotide building blocks for DNA synthesis. | Nucleotide building blocks for DNA synthesis. | Component of LAMP amplification kit [8] |
| Buffer Components | Mg²⁺, salts, pH stabilizers optimized for thermocycling. | Mg²⁺, betaine, salts; critical for strand displacement and specificity. | Optimized with Mg²⁺ and betaine [3] [7] |
| Detection Reagents | Intercalating dyes (Ethidium Bromide, SYBR Green) for gel or qPCR detection. | Colorimetric dyes (HNB, Calcein), or turbidity for visual/optical readout. | Hydroxy naphthol blue (HNB) [7] |
| RNA Extraction Kit | Essential for purifying high-quality RNA from swabs, saliva. | May be simplified or bypassed with lysis buffer due to inhibitor resistance. | QIAamp Viral RNA Mini Kit (Qiagen) [1] |
The choice between nested PCR and LAMP for SARS-CoV-2 research is not a simple matter of which is "better," but rather which is more appropriate for the specific research context.
- Nested PCR is the definitive choice for maximum analytical sensitivity, particularly in samples with extremely low viral loads, and in laboratory settings where sophisticated equipment is available and cross-contamination risks can be meticulously managed [8] [11].
- RT-LAMP offers a compelling alternative when speed, operational simplicity, and field-deployment are paramount. Its performance in detecting acute infections is excellent, its tolerance to inhibitors is advantageous for crude samples, and its potential for integration with biosensors and microfluidic devices points to the future of decentralized diagnostics [1] [74].
Researchers must weigh these sensitivity and specificity profiles against their specific operational requirements, resources, and the intended application of the diagnostic data.
Limit of Detection (LOD) Analysis Across Methodologies
In the landscape of molecular diagnostics for infectious diseases, the analytical sensitivity of a test, defined by its Limit of Detection (LOD), is a critical performance parameter. The LOD represents the lowest concentration of an analyte that can be reliably detected by an assay, profoundly impacting its ability to identify infections, particularly during the early or late stages when pathogen levels are minimal. Within the context of SARS-CoV-2 diagnostic accuracy research, the comparison between established nucleic acid amplification techniques and novel isothermal methods has gained significant importance. This guide provides a objective, data-driven comparison between Nested Polymerase Chain Reaction (nested PCR) and Loop-Mediated Isothermal Amplification (LAMP), focusing on their LOD, to inform researchers, scientists, and drug development professionals in selecting appropriate methodologies for diagnostic applications.
The COVID-19 pandemic underscored the necessity for rapid, sensitive, and accessible diagnostic tools. While reverse transcription quantitative PCR (RT-qPCR) has been the gold standard, its requirement for sophisticated thermal cyclers and trained personnel can limit its deployment in resource-constrained settings [77]. Nested PCR, a variant that uses two rounds of amplification to enhance sensitivity and specificity, and LAMP, an isothermal amplification technique, have emerged as viable alternatives. LAMP is particularly notable for its operational simplicity, as it can be performed with a single-temperature incubation, potentially lowering equipment costs and enabling point-of-care use [78]. This analysis synthesizes recent experimental data to compare their analytical performance head-to-head.
Quantitative LOD Comparison of Molecular Assays
The following table summarizes key performance metrics and LOD data for nested PCR and LAMP from recent studies across various pathogens, including SARS-CoV-2. The data is drawn from experimental results published in peer-reviewed literature, providing a foundation for objective comparison.
Table 1: Comparative Analytical Sensitivity of Nested PCR and LAMP Assays
| Pathogen/Target | Assay Type | Reported LOD | Comparative Sensitivity | Key Experimental Findings | Source |
|---|---|---|---|---|---|
| Entamoeba histolytica (SREHP gene) | LAMP | 1 trophozoite | Higher | LAMP detected a single trophozoite; outperformed all PCR methods. | [4] |
| Conventional PCR | 1,000 trophozoites | Lower | |||
| Nested PCR (nPCR) | 100 trophozoites | Medium | |||
| Real-time PCR (qPCR) | 100 trophozoites | Medium | |||
| SARS-CoV-2 (N gene) | Nested RT-LAMP | 5 copies/μL | Higher | A novel nested RT-LAMP design showed a 3-fold lower LOD than standard RT-LAMP and RT-PCR. | [79] |
| Standard RT-LAMP | 15 copies/μL | Medium | |||
| RT-PCR | 15 copies/μL | Medium | |||
| Mycobacterium marinum (rpoB/mrsA genes) | Nested PCR | 50-fold more sensitive than LAMP | Higher | Nested PCR was 10-fold more sensitive in serial dilution of DNA. | [8] |
| LAMP | 50-fold less sensitive than nested PCR | Lower | LAMP shared clinical sensitivity with nested PCR on patient samples despite lower analytical sensitivity. | ||
| Plasmodium falciparum (18S rRNA) | Colorimetric LAMP | 0.21 parasites/μL | Higher | New primer design achieved a 1,000-fold higher sensitivity than referenced primers. | [80] |
The data in Table 1 reveals that the relative performance of LAMP and nested PCR is not absolute but is influenced by factors such as primer design, target gene, and reaction optimization. In some studies, LAMP demonstrates a superior LOD, even outperforming nested PCR and qPCR [4] [79]. For instance, in detecting Entamoeba histolytica, LAMP's LOD was two orders of magnitude more sensitive than nested PCR [4]. Conversely, another study on Mycobacterium marinum found nested PCR to have a greater analytical sensitivity on purified DNA serial dilutions [8]. This highlights the importance of context and assay design in determining LOD. A advanced "nested RT-LAMP" assay, which incorporates principles from both techniques, has been shown to achieve an exceptionally low LOD of 5 copies/μL for SARS-CoV-2, suggesting that hybrid approaches can push the boundaries of sensitivity [79].
Experimental Protocols for LOD Determination
To ensure the reproducibility of LOD analyses and facilitate comparative assessments, a clear understanding of the underlying experimental methodologies is essential. The following sections detail the common protocols used for determining the LOD of nested PCR and LAMP assays.
Sample Preparation and Serial Dilution
The foundational step for LOD determination involves creating a dilution series of the target analyte. The process for SARS-CoV-2 and other pathogens typically involves:
- Viral Culture or Synthetic Nucleic Acids: Studies often use quantified virus stocks (e.g., determined by TCID50/mL) or synthetic RNA transcripts to establish a known starting concentration [81] [79].
- Nucleic Acid Extraction: DNA or RNA is extracted from samples using commercial kits (e.g., QIAamp DNA Microbiome Kit, RNJia virus kit) [8] [79]. Some LAMP protocols successfully bypass this step, using direct or minimally processed samples [78] [77].
- Serial Dilution: The extracted nucleic acids or whole pathogen samples are subjected to a log-scale serial dilution (e.g., 10-fold) in nuclease-free water or a negative matrix. Each dilution is tested across multiple replicates.
Nested PCR Protocol
Nested PCR enhances sensitivity by performing two consecutive amplification rounds with two sets of primers. A typical workflow is as follows [8]:
- First Round PCR:
- Reaction Mix: Contains Taq DNA polymerase, dNTPs, outer forward and reverse primers, buffer, and template DNA.
- Thermocycling Conditions: An initial denaturation (e.g., 95°C for 5 min), followed by 25-35 cycles of denaturation, annealing (e.g., 62°C for 50s), and extension, with a final extension.
- Second Round (Nested) PCR:
- Reaction Mix: A new master mix containing a second set of primers that bind inside the amplicon generated by the first round.
- Template: A small aliquot (e.g., 1 µL) of the first-round PCR product is used as the template.
- Thermocycling Conditions: A similar cycling profile as the first round, but with 25-35 cycles.
- Detection: The final amplified product is analyzed by agarose gel electrophoresis and visualized under UV light after ethidium bromide staining.
LAMP Protocol
LAMP is a one-step isothermal amplification that uses 4-6 primers recognizing 6-8 distinct regions of the target. A standardized protocol is detailed below [82] [80] [77]:
- Reaction Setup:
- Reaction Mix: The typical 25 µL reaction contains:
- Isothermal Amplification Buffer (e.g., Thermo Pol Buffer)
- Bst DNA Polymerase (large fragment, 4-8 units)
- dNTPs (1.4-1.8 mM)
- MgSO4 (6-8 mM)
- Primers: A mixture of FIP/BIP (1.6 µM each), F3/B3 (0.2 µM each), and optionally LF/LB loop primers.
- Betaine (0.8 M) to assist strand displacement.
- Template DNA/RNA (2-5 µL). For RNA viruses (RT-LAMP), reverse transcriptase is included.
- Reaction Mix: The typical 25 µL reaction contains:
- Amplification:
- The reaction is incubated at a constant temperature of 63-65°C for 30-60 minutes in a dry bath, water bath, or portable heater.
- Product Detection: Multiple read-out methods can be employed:
- Colorimetric: Addition of SYBR Green I, Calcein-Manganese, or pH-sensitive dyes. A color change (orange to green/yellow) indicates a positive reaction [4] [79] [80].
- Turbidity: Visual or spectrophotometric measurement of magnesium pyrophosphate precipitate.
- Agarose Gel Electrophoresis: Visualization of a characteristic ladder-like pattern.
- Lateral Flow Dipstick (LFD): Using biotin- or FITC-labeled primers for rapid, instrument-free detection [4].
Figure 1: A generalized workflow comparing the key procedural steps in Nested PCR and LAMP assays for LOD determination.
Essential Research Reagent Solutions
The successful implementation and accurate LOD assessment of nested PCR and LAMP assays depend on a core set of research-grade reagents and instruments. The following table details these essential components and their critical functions in the experimental workflow.
Table 2: Key Research Reagent Solutions for LOD Analysis
| Reagent / Instrument | Function in Assay | Application Notes |
|---|---|---|
| Bst DNA Polymerase | Engineered DNA polymerase for strand displacement; core enzyme for LAMP amplification. | Thermostable; works optimally at 60-65°C. Critical for LAMP reaction efficiency [82] [79]. |
| Taq DNA Polymerase | Thermostable DNA polymerase for PCR amplification. | Standard enzyme for both rounds of nested PCR. Fidelity and processivity impact sensitivity [8]. |
| LAMP Primer Sets | Set of 4-6 primers targeting 6-8 regions; defines specificity and sensitivity. | Primer design is crucial for LOD. Inner primers (FIP/BIP) drive auto-cycling amplification [80]. |
| Nested PCR Primer Sets | Two pairs of primers (outer and inner); the inner set provides specificity and sensitivity boost. | Inner primers must bind within the first amplicon. Design prevents non-specific amplification [8]. |
| dNTPs | Nucleotide building blocks for DNA synthesis. | Quality and concentration are vital for efficient amplification in both LAMP and PCR. |
| Colorimetric Dyes (e.g., SYBR Green I, Calcein) | Visual detection of amplification by intercalating with DNA or reacting with byproducts. | Enables rapid, instrument-free result readout for LAMP, ideal for point-of-care applications [4] [79]. |
| Thermal Cycler | Instrument that cycles temperatures for PCR denaturation, annealing, and extension. | Essential for nested PCR. A limiting factor for deployment in resource-poor settings. |
| Isothermal Heater / Dry Bath | Provides a single, constant temperature for LAMP incubation. | Simple, low-cost, and portable compared to thermal cyclers [82] [77]. |
The comparative analysis of LOD across nested PCR and LAMP methodologies reveals a nuanced landscape. LAMP consistently demonstrates several operational advantages, including a faster time-to-result, lower equipment requirements, and robustness that often allows for the use of minimally processed samples. In numerous studies, its analytical sensitivity meets or exceeds that of nested PCR and even qPCR [4] [79] [77]. However, nested PCR remains a powerful technique, capable of exceptional sensitivity, as evidenced by its performance in certain direct comparisons [8]. Its primary drawbacks are the longer turnaround time, increased risk of amplicon contamination due to tube opening, and reliance on sophisticated thermocyclers.
The choice between nested PCR and LAMP for SARS-CoV-2 research, or the diagnosis of other pathogens, should therefore be guided by the specific application context. For centralized laboratories where maximum sensitivity is the paramount objective and equipment is readily available, nested PCR is a potent choice. In contrast, for field deployment, point-of-care testing, and high-throughput screening scenarios where speed, cost, and operational simplicity are critical, LAMP presents a compelling and often superior alternative. The emergence of hybrid techniques like "nested RT-LAMP" [79] points to a future where the strengths of both methods are synergistically combined to create even more powerful and accessible diagnostic tools.
Impact of Viral Load and Cycle Threshold (Ct) Values on Detection
The accurate detection of SARS-CoV-2 remains a cornerstone of public health efforts to manage COVID-19. Molecular diagnostic methods such as reverse transcription-quantitative PCR (RT-qPCR), nested PCR (nPCR), and reverse transcription loop-mediated isothermal amplification (RT-LAMP) serve as critical tools in this endeavor. The reliability of these techniques is intrinsically linked to viral load, which is inversely correlated with Cycle Threshold (Ct) values—the number of amplification cycles required for a target gene to exceed a detection threshold. This guide provides an objective comparison of nested PCR and LAMP, framing their performance within the context of viral load and Ct value dynamics to inform researchers, scientists, and drug development professionals.
Nested PCR is a two-round amplification technique that enhances sensitivity and specificity. The initial round uses an outer primer set to amplify a target region, followed by a second round using inner primers that bind within the first amplicon. For SARS-CoV-2, this often targets the N gene, and the process requires precise thermal cycling, post-amplification analysis (e.g., gel electrophoresis), and rigorous contamination controls due to the high risk of carryover contamination [14]. Its primary advantage is the significantly enhanced sensitivity from the dual amplification stages, enabling detection of very low viral loads.
In contrast, RT-LAMP is an isothermal amplification conducted at a constant temperature (60–65°C). It utilizes 4–6 primers targeting 6–8 distinct regions of the viral genome, enabling high specificity. Amplification occurs rapidly (often 15–60 minutes) and can be visualized using colorimetric or turbidimetric methods, facilitating point-of-care use. It eliminates the need for sophisticated thermal cyclers, making it suitable for resource-limited settings [83] [1] [10]. Its key strength is operational simplicity and speed.
Comparative Performance Data: Sensitivity, Specificity, and Viral Load
The diagnostic accuracy of nPCR and LAMP is highly dependent on the viral load present in the clinical sample, which is commonly inferred from Ct values. The following table summarizes key performance metrics from recent studies.
Table 1: Performance Comparison of nPCR and LAMP for SARS-CoV-2 Detection
| Method | Sensitivity (Overall) | Specificity (Overall) | Sensitivity at High Viral Load (Ct < 30/35) | Sensitivity at Low Viral Load (Ct > 30/35) | Limit of Detection (LoD) | Key References |
|---|---|---|---|---|---|---|
| Nested PCR | ~95% | 100% | Data not specified | Detects Ct values up to 31.5 | ~50 copies/µL | [54] |
| RT-LAMP | 85.9% - 87% | 98% - 100% | 97% (Ct < 35) | 60% (Ct > 35) | 6.7 copies/reaction | [83] [1] [84] |
The data reveals a clear relationship between viral load and assay performance. nPCR demonstrates exceptional sensitivity across a wide range of viral loads, reliably detecting samples with Ct values as high as 31.5 [54]. LAMP exhibits excellent diagnostic accuracy in patients with high viral loads (Ct < 35), with sensitivity comparable to RT-qPCR. However, its performance declines significantly for samples with low viral loads (Ct > 35), where sensitivity can drop to approximately 60% [83] [1]. This indicates that LAMP is highly reliable during the acute, symptomatic phase of infection when viral loads are highest.
Detailed Experimental Protocols
To ensure reproducibility and provide a clear understanding of the methodological rigor behind the data, this section outlines standard protocols for nPCR and LAMP assays.
Nested PCR Protocol for SARS-CoV-2 N Gene Detection
This protocol is adapted from a study that applied nPCR to human nasopharyngeal and oropharyngeal samples [14].
- Sample Preparation and RNA Extraction: Collect nasopharyngeal/oropharyngeal swabs and place in transport medium. Total viral RNA is extracted using a commercial viral RNA/DNA extraction kit (e.g., Jena Bioscience kit) according to the manufacturer's instructions.
- First Round RT-PCR:
- Reaction Mix: 5 µL RNA template, 1 µL dNTPs, 1 µL Enzyme mix, 5 µL RT buffer, 2 µL external primers (Ext2019nCorV_F/R), and 11 µL PCR water to a final volume of 25 µL.
- Primers: External primers targeting the N gene (positions 28,346–28,681).
- Cycling Conditions:
- 50°C for 30 min (Reverse transcription)
- 95°C for 15 min (Enzyme activation)
- 40 cycles of: 95°C for 20 s, 54.6°C for 25 s, 72°C for 40 s
- Final extension: 72°C for 10 min.
- Second Round PCR:
- Reaction Mix: Uses a standard PCR mastermix with 1–2 µL of the first-round PCR product as template.
- Primers: Internal primers (intF/intR) binding within the first amplicon (positions 28,432–28,643).
- Cycling Conditions: Similar to the first round, but typically without the reverse transcription step.
- Detection: The final 212 bp amplicon is visualized using agarose gel electrophoresis and UV transillumination.
RT-LAMP Protocol for SARS-CoV-2 Detection
This protocol is based on a large, multicenter diagnostic accuracy study conducted in Cameroon, Ethiopia, Kenya, Nigeria, and Italy [83].
- Sample Preparation: Pharyngeal swab samples are collected. RNA can be extracted, or, for simplified protocols, samples can be used directly with a heat lysis step (e.g., 95°C for 2 minutes in lysis buffer) [84] [14].
- LAMP Reaction:
- Reaction Mix: 25 µL total volume containing 10–15 µL of reaction mix, 4–6 primers targeting SARS-CoV-2 genes (e.g., RdRP, N), and 8–10 µL of purified RNA or heat-lysed sample.
- Incubation: 35–65 minutes at a constant temperature of 62.5–65°C.
- Detection:
Signaling Pathways and Workflow Visualization
The fundamental difference between nPCR and LAMP lies in their amplification workflows. The following diagram illustrates the sequential and isothermal processes, highlighting the procedural steps and critical control points.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of nPCR and LAMP assays requires specific, high-quality reagents. The following table lists essential materials and their functions based on the cited experimental protocols.
Table 2: Key Research Reagent Solutions for nPCR and LAMP
| Reagent / Material | Function / Role | Example Products / Kits |
|---|---|---|
| Viral RNA Extraction Kit | Purifies viral RNA from clinical samples (swabs, saliva). Critical for assay sensitivity. | QIAamp Viral RNA Mini Kit (Qiagen), Jena Bioscience Extraction Kit [14] [85] |
| One-Step RT-PCR Kit | Contains reverse transcriptase and DNA polymerase for combined cDNA synthesis and PCR in nPCR first round. | Qiagen One-Step RT-PCR Kit [14] |
| Taq DNA Polymerase | Thermostable enzyme for the second, DNA-only amplification round of nPCR. | Various suppliers (e.g., Thermo Fisher) |
| RT-LAMP Master Mix | Pre-mixed solution containing strand-displacing DNA polymerase, reverse transcriptase, buffers, and dNTPs. | Loopamp SARS-CoV-2 Detection Kit (Eiken Chemical) [1] [85], SARS-CoV-2 RT-LAMP Kit (Vienna BioCenter) [14] |
| nPCR Primers (N gene) | Outer and inner primer sets for specific, sequential target amplification. | Custom synthesized (e.g., Ext2019nCorV_F/R and intF/intR) [14] |
| LAMP Primer Sets | 4-6 primers designed to recognize 6-8 distinct regions of the target gene (e.g., N, RdRP). | Provided in commercial kits or custom designed [83] [14] |
| Colorimetric Dye | pH-sensitive dye for visual detection of LAMP amplification by color change. | Phenol red [83] |
The choice between nested PCR and LAMP for SARS-CoV-2 detection is context-dependent, hinging on the interplay between viral load and required diagnostic performance. Nested PCR is the superior choice for applications demanding maximum sensitivity, such as detecting low-level infections in animal surveillance studies or convalescent patients, despite its longer turnaround time and contamination risks [54]. LAMP is the optimal technology for rapid screening and resource-limited settings, where its speed, simplicity, and high accuracy in individuals with high viral loads (typically during the infectious period) provide a significant operational advantage [83] [1]. Researchers and clinicians must therefore align their selection of molecular diagnostic tools with the specific clinical or research question, giving paramount consideration to the expected viral load and the implications of Ct values on detection limits.
Within molecular diagnostics, the selection of an appropriate amplification technique is a critical decision that balances cost, time, and performance. This guide provides an objective comparison between two prominent techniques—nested Polymerase Chain Reaction (nested PCR) and Loop-Mediated Isothermal Amplification (LAMP)—specifically for SARS-CoV-2 detection. Nested PCR, known for its high sensitivity, uses two successive rounds of amplification with two primer sets [23]. In contrast, LAMP is an isothermal amplification that operates at a constant temperature using multiple primers, offering rapid results without the need for thermal cyclers [86] [16]. As research into SARS-CoV-2 and its variants continues, understanding the practical and economic trade-offs between these methods is essential for directing resources and planning surveillance studies. This analysis synthesizes experimental data to compare the equipment, reagents, turnaround time, and diagnostic performance of these two techniques, providing a evidence-based resource for researchers and laboratory managers.
Core Principles of Nested PCR and LAMP
Nested PCR: This method is a refinement of conventional PCR designed to enhance sensitivity and specificity. It involves two consecutive amplification rounds. The first round uses an outer pair of primers to generate a primary amplicon. A small aliquot of this product is then transferred to a second reaction tube containing an inner pair of primers that bind within the first amplicon. This two-step process significantly increases the amount of specific final product, reducing background noise from non-specific amplification [23] [87]. A notable advancement is One-Step Single-Tube Nested (OSN) PCR, which contains both primer sets in a single tube with specially designed primers to prevent cross-reaction, thereby mitigating the high risk of amplicon contamination associated with traditional two-tube nested PCR [11].
LAMP (Loop-Mediated Isothermal Amplification): LAMP is a single-tube isothermal amplification technique. It typically employs four to six primers that recognize eight distinct regions on the target DNA, ensuring high specificity [6] [86] [16]. The reaction relies on a strand-displacing DNA polymerase (e.g., Bst polymerase) and is performed at a constant temperature between 60-65°C. This mechanism allows for the synthesis of stem-loop DNA structures that facilitate auto-cycling amplification, leading to the rapid accumulation of billions of copies in under an hour without requiring a thermal cycler [86] [87].
Essential Research Reagent Solutions
The successful implementation of either nested PCR or LAMP requires a suite of specific reagents. The table below catalogues the key materials and their functions in the respective experimental workflows.
Table 1: Key Research Reagents for Nested PCR and LAMP
| Reagent/Material | Primary Function | Application in Nested PCR | Application in LAMP |
|---|---|---|---|
| Bst DNA Polymerase | Strand-displacing enzyme for isothermal DNA amplification | Not used | Essential; catalyzes amplification at constant temperature [16] [87] |
| Taq DNA Polymerase | Thermostable enzyme for DNA synthesis in thermal cycling | Essential for both amplification rounds [23] | Not required |
| Primer Sets (Outer & Inner) | Specific binding to target DNA sequence | Two distinct sets required for sequential rounds [23] [11] | A single set of 4-6 primers required [6] [16] |
| dNTPs | Building blocks for new DNA strands | Required for both PCR rounds [23] | Required [87] |
| Reverse Transcriptase | Converts RNA to cDNA for RNA virus detection | Used in initial step for RT-nested PCR [23] | Used in initial step for RT-LAMP [1] [16] |
| Betaine | Additive to reduce secondary structures in DNA | Sometimes used | Commonly used to enhance efficiency and yield [87] |
| MgSO₄ | Cofactor for DNA polymerase activity | Component of PCR master mix | Critical component for Bst polymerase activity [87] |
| Fluorescent Dyes/Probes | For real-time detection of amplification | Used in OSN-qRT-PCR formats [11] | Used for visual (colorimetric) or real-time (turbidity) detection [1] [87] |
Comparative Experimental Data
Diagnostic Performance for SARS-CoV-2
Multiple studies have directly compared the analytical and clinical performance of nested PCR and LAMP for detecting SARS-CoV-2. The following table summarizes key performance metrics from published experimental data.
Table 2: Comparative Diagnostic Performance of Nested PCR and LAMP for SARS-CoV-2
| Parameter | Nested PCR (incl. OSN-qRT-PCR) | LAMP (RT-LAMP) | Supporting Experimental Data |
|---|---|---|---|
| Sensitivity (LoD) | OSN-qRT-PCR: 189.1 - 194.74 copies/mL (for N/ORF1ab genes) [11] | 6.7 copies/reaction [1] | Evaluation using SARS-CoV-2 pseudovirus and clinical samples [1] [11] |
| Clinical Sensitivity (Positive Rate) | OSN-qRT-PCR: 82.35% (28/34 samples); superior to ddPCR and qPCR in low viral loads [11] | 92.8% (up to 9 days post-symptom onset), decreasing in later stages [1] | Testing on clinical samples from COVID-19 patients [1] [11] |
| Specificity | Reported 100% for a validated conventional nested PCR assay [23] | 100% specificity compared to RT-qPCR in acute phase [1]; 73-100% in another study, improvable with retesting [88] | Specificity testing against other pathogens and healthy controls [23] [1] [88] |
| Amplification Time | ~2-3 hours (including two rounds of cycling) [23] [87] | < 60 minutes, with positives often visible in < 25-35 minutes [1] [88] [16] | Timed experimental runs from sample preparation to result [23] [88] [16] |
Detailed Experimental Protocols
To ensure reproducibility, this section outlines the core methodologies as cited in the literature.
Protocol for Nested PCR Detection of SARS-CoV-2
A validated protocol for detecting SARS-CoV-2 in human and cat samples involves the following steps [23]:
- RNA Extraction and cDNA Synthesis: Extract viral RNA using a commercial kit (e.g., ISOLATE II RNA Mini kit). Perform reverse transcription (RT) using a SensiFAST cDNA synthesis kit with the following cycling conditions: 25°C for 10 min, 42°C for 15 min, and 80°C for 5 min.
- First PCR Round (External Primers):
- Reaction Mix: 12.5 μL My Taq HS red mix, 4 μL cDNA, 1 μL each of external forward and reverse primers (10 pmol/μL), and 6.5 μL PCR-grade water.
- Cycling Conditions: Initial denaturation at 95°C for 1 min; 35 cycles of denaturation at 95°C for 15 s, annealing at 54.6°C for 15 s, and extension at 72°C for 10 s; final extension at 72°C for 1 min.
- Second PCR Round (Internal Primers):
- Reaction Mix: 12.5 μL My Taq HS red mix, 0.5 μL of the first-round PCR product, 1 μL each of internal forward and reverse primers (10 pmol/μL), and 10 μL PCR-grade water.
- Cycling Conditions: Use the same thermal profile as the first round.
- Product Detection: Analyze the final amplicon (212 bp for the described assay) by 2% agarose gel electrophoresis.
Protocol for One-Step RT-LAMP Detection of SARS-CoV-2
A fast one-step RT-LAMP assay for SARS-CoV-2, targeting the N gene, can be performed as follows [16]:
- RNA Extraction: Extract RNA from nasopharyngeal or saliva samples using a viral RNA extraction kit. Quantify and check purity via Nanodrop.
- LAMP Reaction Setup:
- Reaction Mix: Prepare a 25 μL reaction containing 5 pmol each of F3 and B3 outer primers, 40 pmol each of FIP and BIP inner primers, 20 pmol each of LF and LB loop primers, 8U of Bst DNA/RNA Polymerase, and the recommended buffer components including betaine and MgSO₄.
- Template: Add 5 μL of extracted RNA.
- Amplification:
- Incubate the reaction at 63°C for 30-35 minutes.
- Product Detection: Results can be determined in real-time using a turbidimeter, post-amplification with fluorescent dyes visible under UV light, or via gel electrophoresis [1] [16].
Comparative Analysis of Equipment, Cost, and Time
Equipment and Infrastructure
The core equipment requirements for each technique directly impact the initial setup cost and operational flexibility.
- Nested PCR: Requires a high-precision thermal cycler capable of running complex programs for the two amplification rounds. The requirement for post-amplification gel electrophoresis also necessitates a gel documentation system. The OSN-qRT-PCR format further requires a real-time PCR machine, which is a significant capital investment [23] [11] [87].
- LAMP: Designed for simplicity, it only requires a simple heating block, water bath, or dry bath capable of maintaining a constant temperature of 60-65°C. This dramatically reduces equipment cost and complexity. Detection can be achieved with low-cost options like a UV illuminator for fluorescent dyes or even the naked eye for colorimetric changes, making it suitable for field-deployable or point-of-care devices [86] [88] [16].
Operational Workflow and Turnaround Time
The fundamental differences in workflow between the two methods significantly affect the total hands-on time and the potential for automation.
The workflow diagram highlights key operational differences. Nested PCR is inherently more complex and time-consuming due to its two-step amplification process and the required post-amplification analysis. The product transfer step between the first and second rounds is a critical point with a high risk of amplicon contamination, which can lead to false positives if not meticulously managed [87]. In contrast, the LAMP workflow is a single-tube, closed-tube system from amplification to detection, which minimizes contamination risk and reduces hands-on time. The total turnaround time for LAMP, from sample to result, is often under 60 minutes, compared to several hours for nested PCR [23] [88] [16].
The following table synthesizes the data to provide a direct comparison of the key cost and performance factors.
Table 3: Comprehensive Cost-Benefit Analysis: Nested PCR vs. LAMP
| Factor | Nested PCR | LAMP | Key Takeaways & Implications |
|---|---|---|---|
| Equipment Cost | High (Thermal cycler, real-time system) | Low (Heating block, simple detector) | LAMP offers a lower barrier to entry and is suitable for resource-limited or field settings [86] [16]. |
| Reagent Cost | Moderate to High (Two sets of primers, two reactions) | Low to Moderate (Single reaction, but more primers per test) | Nested PCR consumable costs are multiplied by the two-reaction design. |
| Turnaround Time | ~3-4 hours (including analysis) [23] | ~60-90 minutes (including detection) [88] [16] | LAMP provides significantly faster results, crucial for rapid decision-making. |
| Sensitivity | Very High to Ultra-High (OSN-qRT-PCR superior to ddPCR and qPCR) [11] | High (Comparable to RT-qPCR in acute phase) [1] | Nested PCR is superior for detecting very low viral loads (e.g., convalescent patients, environmental samples). |
| Specificity | Very High (Dual primers enhance specificity) [23] | High (Multiple primers enhance specificity) [1] | Both methods offer high specificity when optimized. |
| Ease of Use | Complex (Multi-step, high contamination risk) [87] | Simple (Single-tube, minimal handling) [86] [16] | LAMP requires less technical expertise, reducing operational errors and training needs. |
| Throughput & Scalability | High in automated systems | High in field settings; adaptable to medium throughput | Nested PCR is well-suited for high-throughput centralized labs, while LAMP excels in decentralized testing. |
| Best-Suited Applications | Research on low viral load samples, convalescent patient monitoring, biomarker validation. | Rapid screening, point-of-care testing, field surveillance, resource-limited settings. | The choice is application-dependent, balancing the need for extreme sensitivity against speed and cost. |
The choice between nested PCR and LAMP for SARS-CoV-2 diagnostic research is not a matter of one technique being universally superior, but rather a strategic decision based on project priorities.
Nested PCR is the unequivocal choice when the highest possible sensitivity is the primary requirement, such as in studies focusing on patients with low viral loads, monitoring viral persistence, or detecting minute quantities of viral material in environmental samples. This comes at the cost of longer turnaround times, higher equipment investment, and a more complex workflow that requires skilled personnel to mitigate contamination risks [23] [11].
LAMP is the preferred technology for applications where speed, cost-effectiveness, and operational simplicity are critical. Its performance is excellent for the detection of SARS-CoV-2 during the acute phase of infection, making it ideal for rapid community screening, point-of-care diagnosis, and use in laboratories with limited infrastructure. The ability to visualize results with simple dyes eliminates the need for sophisticated detection hardware, further broadening its accessibility [1] [88] [16].
Researchers and public health officials must weigh these factors against their specific objectives. For widespread screening and rapid outbreak containment, LAMP offers a powerful and pragmatic tool. For deep clinical investigation and research requiring the utmost detection sensitivity, nested PCR remains a robust, albeit more demanding, gold-standard approach.
Concordance Studies with Gold Standard RT-qPCR Methods
The accurate and timely diagnosis of SARS-CoV-2 infection has been a cornerstone of the global response to the COVID-19 pandemic. Reverse transcription quantitative polymerase chain reaction (RT-qPCR) has consistently served as the gold standard for detecting viral RNA in respiratory specimens [89] [90]. However, the demand for rapid, accessible testing has accelerated the development and evaluation of alternative nucleic acid amplification methods, including nested PCR and loop-mediated isothermal amplification (LAMP). These techniques offer potential advantages in specific diagnostic scenarios but require rigorous validation against the reference standard. This guide objectively compares the performance of nested PCR and LAMP methodologies with gold standard RT-qPCR, providing researchers and clinical laboratory professionals with experimental data and protocols to inform diagnostic choices.
Performance Comparison of Molecular Assays
Diagnostic Accuracy Metrics
Multiple studies have directly compared the clinical performance of LAMP and nested PCR to RT-qPCR. The table below summarizes key performance metrics from recent concordance studies.
Table 1: Diagnostic performance of LAMP and nested PCR compared to RT-qPCR
| Study & Method | Sample Size | Sensitivity | Specificity | Agreement/Concordance | Reference |
|---|---|---|---|---|---|
| RT-LAMP vs. RT-qPCR (2024) | 342 samples | 92/86 (Saliva): ~107% relative | 100% | κ = 0.93-0.94 (93-94% agreement) | [16] |
| nPCR vs. LAMP (2024) | 427 samples | 100% (for validated sample set) | 100% (for validated sample set) | 100% concordance between methods | [45] |
| Romed Antigen Test vs. RT-qPCR (2021) | 900 patients | 73.3% overall (83.0-86.7% for CT≤30) | 99.8% | N/A | [89] |
| Five Rapid Antigen Tests vs. RT-qPCR (2021) | 80 specimens | 55.0-80.0% | 87.5-100.0% | N/A | [89] |
Analytical Sensitivity
A critical metric for diagnostic assays is the limit of detection (LoD), which indicates the lowest viral concentration reliably detected.
Table 2: Analytical sensitivity comparison of molecular methods
| Method | Limit of Detection | Time to Result | Key Advantages | |
|---|---|---|---|---|
| RT-qPCR (Gold Standard) | Varies by kit (est. 10-100 copies/mL) | 1.5-4 hours (plus transport) | High sensitivity, quantitative, established protocols | [91] |
| RT-LAMP | As low as 10 copies/mL | ~30-60 minutes | Isothermal, minimal equipment, visual detection | [92] [16] |
| Nested PCR | High sensitivity for low viral loads | 3+ hours | Enhanced sensitivity via two amplification rounds | [45] |
Experimental Protocols for Method Comparison
Sample Collection and Processing
Consistent sample collection across compared methods is essential for valid concordance studies:
- Sample Types: Combined throat/nasopharyngeal swabs in viral transport medium (VTM), nasopharyngeal swabs alone, oropharyngeal swabs, and saliva [89] [45] [16].
- Storage: Store at 4°C until processing (within 72 hours) or at -20°C for longer storage [89].
- RNA Extraction: Use commercial viral RNA extraction kits (e.g., HiPurAViral RNA purification kit, Biorexfars SARS-CoV-2 RNA Extraction Kit) following manufacturer protocols [91] [16]. Include extraction controls to monitor process efficiency.
- Ethical Considerations: Institutional Review Board approval with waiver of informed consent for use of leftover routine diagnostic samples is common, with opt-out provisions for patients [89].
RT-qPCR Reference Method Protocol
The established gold standard method follows this workflow:
- Reaction Setup: Prepare 20-25μL reactions with 5μL RNA template, master mix, and primers/probes targeting SARS-CoV-2 genes (typically N, RdRp, E, or ORF1ab) [91] [16].
- Thermal Cycling:
- Reverse transcription: 50°C for 20-30 minutes
- Initial denaturation: 95°C for 3-15 minutes
- 40-45 cycles of: Denaturation (95°C for 15-20s), Annealing/Extension (55-60°C for 40s)
- Result Interpretation: Ct values ≤37-40 are generally considered positive, following kit manufacturer recommendations and using appropriate positive, negative, and extraction controls [91].
RT-LAMP Assay Protocol
The LAMP methodology for SARS-CoV-2 detection:
- Primer Design: Design 6 primers (F3, B3, FIP, BIP, LF, LB) targeting 8 regions of the SARS-CoV-2 N gene using PrimerExplorer V5 software [16]. Verify specificity with BLAST analysis.
- Reaction Setup:
- 25μL reaction volume with 5μL RNA template
- Primer concentrations: 5 pmol each F3/B3, 40 pmol each FIP/BIP, 20 pmol each LF/LB
- Enzyme: 8U Bst DNA/RNA Polymerase 3.0
- Incubation: 63-65°C for 30-60 minutes [16]
- Detection Methods:
- Colorimetric: pH-sensitive dyes (phenol red) changing from pink to yellow
- Fluorescent: Intercalating dyes (SYBR Green, calcein) under UV light
- Turbidity: Magnesium pyrophosphate precipitate measurement [93]
Nested PCR Protocol
The nested PCR method for enhanced sensitivity:
- Primer Design: External primers targeting the N gene (e.g., Ext2019nCorV F/R defining 335bp fragment) and internal primers (e.g., intF/R defining 212bp fragment) [45].
- First Round PCR:
- One-Step RT-PCR with 25μL reaction volume
- Conditions: 50°C for 30min (RT), 95°C for 15min, 40 cycles of 95°C/20s-54.6°C/25s-72°C/40s [45]
- Second Round PCR:
- Use 2μL of first-round product as template
- Standard PCR with 30 cycles of denaturation, annealing, and extension [45]
- Analysis: Agarose gel electrophoresis with ethidium bromide staining to detect specific bands [45].
Diagram 1: Experimental workflow for method comparison studies (Max Width: 760px)
Essential Research Reagents and Materials
Table 3: Key research reagent solutions for molecular SARS-CoV-2 detection
| Reagent/Material | Function/Purpose | Example Products/Suppliers | |
|---|---|---|---|
| Viral Transport Medium (VTM) | Preservation of specimen during transport and storage | Various commercial VTM formulations | [89] |
| Viral RNA Extraction Kits | Isolation of high-quality RNA from clinical specimens | HiPurAViral (HiMedia), Biorexfars RNA Extraction Kit, Jena Bioscience kits | [91] [16] |
| RT-qPCR Master Mixes | Enzymes and reagents for reverse transcription and amplification | TaqPath COVID-19 combo kit, Allplex 2019-nCoV assay, LabGun COVID-19 RT-PCR kit | [91] |
| LAMP Primer Sets | Target-specific primers for isothermal amplification | Custom-designed targeting N, S, or ORF1ab genes | [16] |
| Bst DNA Polymerase | Strand-displacing polymerase for LAMP reactions | Bst 2.0 WarmStart, Bst DNA/RNA Polymerase 3.0 (NEB) | [92] [16] |
| Colorimetric Detection Dyes | Visual result interpretation for LAMP | Phenol Red, Hydroxynaphthol Blue, Calcein, SYBR Green | [93] |
Discussion and Technical Considerations
Implementation Challenges
While both LAMP and nested PCR show strong concordance with RT-qPCR, several practical considerations influence their implementation:
- LAMP Limitations: Primer design constraints requiring 6-8 distinct regions, potential for non-specific amplification, and less developed multiplexing capabilities compared to PCR [93] [92]. The isothermal nature, while advantageous for simplicity, still requires maintained temperatures of 60-65°C [93].
- Nested PCR Drawbacks: Increased risk of amplicon contamination between first and second rounds, longer hands-on time, and requirement for post-amplification gel electrophoresis [45].
- Sample Type Considerations: Nasopharyngeal swabs generally provide higher sensitivity compared to saliva or nasal swabs, though proper collection technique is crucial [45] [16].
Applications in Research and Clinical Settings
The choice between methods depends on the specific application:
- RT-qPCR remains ideal for high-throughput clinical laboratories requiring quantitative results and maximum sensitivity [90] [91].
- RT-LAMP offers superior point-of-care potential with minimal equipment needs and rapid results, particularly valuable in resource-limited settings or for rapid screening [92] [16].
- Nested PCR provides an alternative for research settings requiring extreme sensitivity for low viral load detection, particularly when specialized real-time PCR equipment is unavailable [45].
Concordance studies demonstrate that both LAMP and nested PCR can achieve strong agreement with gold standard RT-qPCR for SARS-CoV-2 detection when properly optimized. LAMP shows particular promise as a rapid, cost-effective alternative with 93-94% agreement with RT-qPCR in recent studies [16], while nested PCR provides an extremely sensitive research tool. The optimal methodological choice depends on the specific application context, including required throughput, available infrastructure, and intended use of results. As molecular diagnostics continue to evolve, these complementary methods expand the available toolkit for researchers and clinicians addressing current and future infectious disease challenges.
Conclusion
The comparative analysis reveals that nested PCR and LAMP offer distinct advantages for SARS-CoV-2 detection. Nested PCR demonstrates exceptional sensitivity, particularly for low viral load samples, making it invaluable for confirmatory testing and research requiring high precision. LAMP technology provides rapid, cost-effective results with minimal equipment requirements, ideal for resource-limited settings and high-throughput screening. The choice between these methods depends on specific application requirements: nested PCR for maximum sensitivity in research environments, and LAMP for speed and operational simplicity in field applications. Future developments should focus on multiplexing capabilities, automation, and adapting these platforms for emerging pathogens to enhance global diagnostic preparedness. The integration of these complementary technologies creates a robust framework for future molecular diagnostics in both clinical and research contexts.
References
- 1. Diagnostic accuracy of LAMP versus PCR over the course of SARS-CoV-2 infection - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8056478/]
- 2. Analysis and validation of a highly sensitive one-step nested ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7768088/]
- 3. Loop-mediated isothermal amplification ( LAMP ) reaction as viable... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7310076/]
- 4. Loop-mediated isothermal amplification (LAMP) reaction as viable PCR substitute for diagnostic applications: a comparative analysis study of LAMP, conventional PCR, nested PCR (nPCR) and real-time PCR (qPCR) based on Entamoeba histolytica DNA derived from faecal sample - PubMed [https://pubmed.ncbi.nlm.nih.gov/32571286/]
- 5. analysis of loop-mediated isothermal Comparative ... amplification [https://www.nature.com/articles/s41598-021-01472-3?error=cookies_not_supported&code=33bd4bcc-f879-42c1-a899-37324ee4068e]
- 6. Frontiers | Comparative Evaluation of the LAMP Assay and... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2018.02089/full]
- 7. Development and comparative evaluation of LAMP , nested and... PCR [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-025-04295-8]
- 8. Comparative Evaluation of LAMP and Nested PCR | IDR [https://www.dovepress.com/comparative-evaluation-of-lamp-and-nested-pcr-for-the-rapid-diagnosis--peer-reviewed-fulltext-article-IDR]
- 9. Evaluation of RT- PCR assays for detection of SARS - CoV - 2 variants of... [https://www.nature.com/articles/s41598-023-28275-y?error=cookies_not_supported&code=31ab2199-5bce-4633-b8ca-fae907ef08fe]
- 10. between RT-qPCR and Comparison Methods for... | IntechOpen LAMP [https://www.intechopen.com/chapters/1130226]
- 11. Analysis and validation of a highly sensitive one-step nested ... [https://virologyj.biomedcentral.com/articles/10.1186/s12985-020-01467-y]
- 12. Summary [https://data.who.int/dashboards/covid19/summary]
- 13. Real-time optical analysis of a colorimetric LAMP assay for SARS-CoV-2 in saliva with a handheld instrument improves accuracy compared with endpoint assessment - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8730521/]
- 14. A Comparative Analysis of Molecular Biological Methods for the Detection of SARS-CoV-2 and Testing the In Vitro Infectivity of the Virus [https://www.mdpi.com/2076-2607/12/1/180]
- 15. Evaluation of RT-LAMP Assay for Rapid Detection of SARS-CoV-2 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9384514/]
- 16. Evaluation and comparison of one-step real-time PCR and one-step... [https://bmcinfectdis.biomedcentral.com/articles/10.1186/s12879-024-09574-9]
- 17. Surveillance and Data Analytics | COVID-19 | CDC [https://www.cdc.gov/covid/php/surveillance/index.html]
- 18. 8 trajectories for long COVID — Harvard Gazette [https://news.harvard.edu/gazette/story/2025/11/8-trajectories-for-long-covid/]
- 19. Blood transcriptional biomarkers of acute viral infection for detection of... [https://pubmed.ncbi.nlm.nih.gov/34250515/]
- 20. COVID-19 Diagnosis: A Comprehensive Review of the RT-qPCR Method for Detection of SARS-CoV-2 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9221722/]
- 21. Partial ORF1ab Gene Target Failure with Omicron BA.2.12.1 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9094110/]
- 22. Frontiers | A Benchmark of methods for SARS - CoV - 2 whole genome ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1516791/full]
- 23. Development of Nested PCR for SARS-CoV-2 Detection and Its Application for Diagnosis of Active Infection in Cats - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9231004/]
- 24. Rapid SARS-CoV-2 antigen detection assay in comparison with real-time RT-PCR assay for laboratory diagnosis of COVID-19 in Thailand | Virology Journal | Full Text [https://virologyj.biomedcentral.com/articles/10.1186/s12985-020-01452-5]
- 25. A Study of the Detection of SARS-CoV-2 ORF1ab Gene by the Use of Electrochemiluminescent Biosensor Based on Dual-Probe Hybridization - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8954742/]
- 26. Development of Nested for PCR - SARS - CoV Detection and Its... 2 [https://pmc.ncbi.nlm.nih.gov/articles/PMC9231004/]
- 27. Analytical sensitivity and effectiveness of different SARS-CoV-2 testing options | medRxiv [https://www.medrxiv.org/content/10.1101/2021.11.26.21265946v1.full]
- 28. Colorimetric RT-LAMP for SARS-CoV-2 detection from nasopharyngeal swabs or crude saliva: a multicountry diagnostic accuracy study in Africa - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12208784/]
- 29. A Comparative Analysis of Molecular Biological Methods for the... [https://pubmed.ncbi.nlm.nih.gov/38258006/]
- 30. Surfaces environmental monitoring of SARS - CoV - 2 : Loop... | PLOS One [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0317228]
- 31. Comparison of PCR, Nested PCR, and RT-LAMP for Rapid Detection of Feline Calicivirus Infection in Clinical Samples - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11350671/]
- 32. Colorimetric RT- LAMP - SARS - CoV diagnostic 2 relies on... sensitivity [https://www.nature.com/articles/s41598-021-88506-y?error=cookies_not_supported&code=d2db4b43-3175-421c-808f-8ba42948123e]
- 33. Role of Laboratory Medicine in SARS-CoV-2 Diagnostics. Lessons Learned from a Pandemic - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8303636/]
- 34. One-step nested RT- PCR for COVID-19 detection: A flexible, locally... [https://pubmed.ncbi.nlm.nih.gov/32794453/]
- 35. Loop-Mediated Isothermal Amplification ( LAMP ) [https://www.thermofisher.com/in/en/home/life-science/pcr/isothermal-nucleic-acid-amplification/loop-mediated-isothermal-amplification.html]
- 36. Frontiers | Cross comparison of alternative diagnostic protocols including substitution to the clinical sample, RNA extraction method and nucleic acid amplification technology for COVID-19 diagnosis [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2024.1445142/full]
- 37. Diagnostic accuracy of LAMP versus PCR over the course of SARS-CoV-2 infection - 藤田医科大学 [https://pure.fujita-hu.ac.jp/ja/publications/diagnostic-accuracy-of-lamp-versus-pcr-over-the-course-of-sars-co]
- 38. Development of an integrated sample amplification control for salivary... [https://pubmed.ncbi.nlm.nih.gov/38182338/]
- 39. Developing RT-LAMP Assays for Detection of SARS-CoV-2 in Saliva | medRxiv [https://www.medrxiv.org/content/10.1101/2021.04.25.21256085v1.full]
- 40. Sample-to-answer, extraction-free, real-time RT - LAMP ... | PLOS One [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0264130]
- 41. Critical aspects of droplet digital reverse transcription loop-mediated isothermal amplification (ddRT-LAMP) for viral pathogens detection | Microsystems & Nanoengineering [https://www.nature.com/articles/s41378-025-00982-8]
- 42. of Eleven Comparison Methods for Poliovirus RNA ... Extraction Direct [https://pmc.ncbi.nlm.nih.gov/articles/PMC10100708/]
- 43. A Comprehensive Comparison of Rapid RNA Extraction Methods for Detection of SARS-CoV-2 as the Infectious Agent of the Upper Respiratory Tract using Direct RT-LAMP Assay [https://www.ovid.com/journals/abres/fulltext/10.4103/abr.abr_63_23~a-comprehensive-comparison-of-rapid-rna-extraction-methods]
- 44. Head-to-head comparison of direct-input RT-PCR and RT-LAMP against RT-qPCR on extracted RNA for rapid SARS-CoV-2 diagnostics | medRxiv [https://www.medrxiv.org/content/10.1101/2021.01.19.21250079v1.full]
- 45. A Comparative Analysis of Molecular Biological Methods for the Detection of SARS-CoV-2 and Testing the In Vitro Infectivity of the Virus - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10819592/]
- 46. sequencing (SQK- Direct 004) RNA RNA [https://nanoporetech.com/document/direct-rna-sequencing-sqk-rna004]
- 47. An Innovative AI-based primer tool for precise and accurate... design [https://www.nature.com/articles/s41598-023-42348-y?error=cookies_not_supported&code=00ab6e0b-14c0-4add-ae94-20951b1e2d25]
- 48. LAMP-Based Point-of-Care Biosensors for Rapid Pathogen Detection - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9775414/]
- 49. Colorimetric RT-LAMP Detection of Multiple SARS-CoV-2 Variants and Lineages of Concern Direct from Nasopharyngeal Swab Samples without RNA Isolation [https://www.mdpi.com/1999-4915/15/9/1910]
- 50. PCR Primer Design for the Rapidly Evolving SARS-CoV-2 Genome - PubMed [https://pubmed.ncbi.nlm.nih.gov/34773624/]
- 51. Clinical status determines the efficacy of salivary and nasopharyngeal samples for detection of SARS-CoV-2 | Clinical Oral Investigations [https://link.springer.com/article/10.1007/s00784-020-03630-9]
- 52. Comparison of SARS-CoV-2 detection in nasopharyngeal swab and saliva | medRxiv [https://www.medrxiv.org/content/10.1101/2020.05.13.20100206v2.full]
- 53. Detection of SARS-CoV-2 in saliva and salivary glands – a systematic review | Research, Society and Development [https://rsdjournal.org/index.php/rsd/article/view/26673]
- 54. effective to detect Nested of PCR - low - viral in... loads SARS CoV 2 [https://pubmed.ncbi.nlm.nih.gov/39128181/]
- 55. Nested PCR effective to detect low viral loads of SARS-CoV-2 in animal samples - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0167587724001892]
- 56. Colorimetric RT-LAMP SARS-CoV-2 diagnostic sensitivity relies on color interpretation and viral load | Scientific Reports [https://www.nature.com/articles/s41598-021-88506-y]
- 57. Frontiers | Optimization and Clinical Validation of Colorimetric Reverse Transcription Loop-Mediated Isothermal Amplification, a Fast, Highly Sensitive and Specific COVID-19 Molecular Diagnostic Tool That Is Robust to Detect SARS-CoV-2 Variants of Concern [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2021.713713/full]
- 58. of a New In-House and Three Published... | PLOS One Comparison [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0155907]
- 59. Use of a synthetic oligonucleotide to detect false caused by... positives [https://pubmed.ncbi.nlm.nih.gov/39270751/]
- 60. SARS-CoV-2 Molecular Tests: From PCR to LAMP with a Dash of CRISPR | Digital World Biology [https://digitalworldbiology.com/blog/sars-cov-2-molecular-tests-pcr-lamp-dash-crispr]
- 61. PCR Methods - Top Ten Strategies | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-methods.html]
- 62. Essential properties and pitfalls of colorimetric Reverse Transcription Loop-mediated Isothermal Amplification as a point-of-care test for SARS-CoV-2 diagnosis | Molecular Medicine | Full Text [https://molmed.biomedcentral.com/articles/10.1186/s10020-021-00289-0]
- 63. Primer dimer - Wikipedia [https://en.wikipedia.org/wiki/Primer_dimer]
- 64. PrimerROC: accurate condition-independent dimer prediction using ROC analysis | Scientific Reports [https://www.nature.com/articles/s41598-018-36612-9]
- 65. Comparative Assessment of Diagnostic Performance of Cytochrome... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9441448/]
- 66. Diagnostic Performance of an Aspergillus-Specific Nested Assay... PCR [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0056706]
- 67. Maximising Affordability of Real-Time Colorimetric LAMP Assays - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10673270/]
- 68. Quantitative Experimental Determination of Primer-Dimer Formation Risk by Free-Solution Conjugate Electrophoresis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3416024/]
- 69. The virus that won’t quit: new research reveals how SARS-CoV-2 evolves [https://www.unsw.edu.au/newsroom/news/2025/06/the-virus-that-won-t-quit-new-research-reveals-how-sars-cov-2-evolves]
- 70. Evaluation of RT-LAMP for SARS-CoV-2 Detection in Animal Feces - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12197339/]
- 71. Rapid and Extraction-Free Detection of SARS-CoV-2 from Saliva by Colorimetric Reverse-Transcription Loop-Mediated Isothermal Amplification - PubMed [https://pubmed.ncbi.nlm.nih.gov/33098427/]
- 72. A Rapid RT-LAMP Assay for SARS-CoV-2 with Colorimetric Detection Assisted by a Mobile Application - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9032071/]
- 73. Frontiers | Addressing SARS-CoV-2 evolution: neutralization of emerging variants of concern by the AVX/COVID-12 ‘Patria’ vaccine based on HexaPro-S ancestral Wuhan spike or its updated BA.2.75.2 version [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1565934/full]
- 74. Frontiersin [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2022.1068015/xml/nlm?isPublishedV2=false]
- 75. Loop-mediated isothermal amplification (LAMP) for pandemic pathogen diagnostics: How it differs from PCR and why it isn't more widely used — EA Forum [https://forum.effectivealtruism.org/posts/Gcoqbn3mphgn5cfF2/loop-mediated-isothermal-amplification-lamp-for-pandemic]
- 76. Spandidos Publications: International Journal of Molecular Medicine [https://www.spandidos-publications.com/10.3892/ijmm.2021.4933]
- 77. Performance of Reverse Transcription Loop-Mediated Isothermal Amplification (RT-LAMP) Targeting the RNA Polymerase Gene for the Direct Detection of SARS-CoV2 in Nasopharyngeal Swabs [https://www.mdpi.com/1422-0067/24/17/13056]
- 78. Detecting SARS-CoV-2 at point of care: preliminary data comparing loop-mediated isothermal amplification (LAMP) to polymerase chain reaction (PCR) - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7574392/]
- 79. Development of a Novel Nested –RT–LAMP Assay for the Rapid and... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11993314/]
- 80. Enhancing malaria detection in resource- limited areas... | PLOS One [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0298087]
- 81. Evaluation of Four Point of Care (POC) Antigen Assays for the... [https://pubmed.ncbi.nlm.nih.gov/35616382/]
- 82. Frontiers | Rapid diagnosis of Plasmodium falciparum malaria using ... [https://frontiersin.org/articles/10.3389/fcimb.2022.961832/full]
- 83. of a colorimetric RT - Diagnostic for the... performance LAMP [https://pmc.ncbi.nlm.nih.gov/articles/PMC8401528/]
- 84. of rapid and connected salivary RT- Performances ... LAMP diagnostic [https://www.nature.com/articles/s41598-022-04826-7?error=cookies_not_supported&code=18352475-5458-4da6-8661-1f5d70bf22c8]
- 85. Comparison of 12 Molecular Detection Assays for Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7699162/]
- 86. of Comparison with Other... | MyBioSource Learning Center PCR [https://www.mybiosource.com/learn/comparison-of-pcr-with-other-nucleic-acid-amplification-techniques/]
- 87. Loop-mediated isothermal amplification (LAMP) reaction as viable PCR substitute for diagnostic applications: a comparative analysis study of LAMP, conventional PCR, nested PCR (nPCR) and real-time PCR (qPCR) based on Entamoeba histolytica DNA derived from faecal sample | BMC Biotechnology | Full Text [https://bmcbiotechnol.biomedcentral.com/articles/10.1186/s12896-020-00629-8]
- 88. Detecting SARS-CoV-2 at point of care: preliminary data comparing loop-mediated isothermal amplification (LAMP) to polymerase chain reaction (PCR) | BMC Infectious Diseases | Full Text [https://bmcinfectdis.biomedcentral.com/articles/10.1186/s12879-020-05484-8]
- 89. Clinical evaluation of rapid point-of-care antigen tests for diagnosis of... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8140309/]
- 90. Frontiers | An Update on Molecular Diagnostics for COVID-19 [https://frontiersin.org/articles/10.3389/fcimb.2020.560616/full]
- 91. analysis of the diagnostic performance of five... Comparative [https://www.nature.com/articles/s41598-021-00852-z?error=cookies_not_supported&code=c938fdad-9b13-4806-aa7c-7b39b3ff9082]
- 92. Progression of LAMP as a Result of the COVID-19 Pandemic: Is PCR ... [https://mdpi.com/2079-6374/12/7/492/htm]
- 93. Loop-mediated isothermal amplification - Wikipedia [https://en.wikipedia.org/wiki/Loop-mediated_isothermal_amplification]