Optimized ddPCR Workflow for Sensitive CCR5Δ32 Detection: A Guide for Clinical Research and Therapeutic Development
This article provides a comprehensive guide for researchers and drug development professionals on implementing droplet digital PCR (ddPCR) for the detection and quantification of the CCR5Δ32 mutation in clinical samples.
Optimized ddPCR Workflow for Sensitive CCR5Δ32 Detection: A Guide for Clinical Research and Therapeutic Development
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on implementing droplet digital PCR (ddPCR) for the detection and quantification of the CCR5Δ32 mutation in clinical samples. The CCR5Δ32 mutation confers resistance to HIV infection, and its accurate measurement is crucial for developing and monitoring gene therapies and stem cell transplants. We cover the foundational principles of ddPCR, detail a step-by-step optimized methodology, address common troubleshooting and optimization challenges, and present validation data comparing ddPCR to other molecular techniques. The content is designed to equip scientists with the knowledge to establish a robust, sensitive, and clinically applicable ddPCR assay for precise genotyping and monitoring of CCR5Δ32 in heterogeneous cell populations.
CCR5Δ32 and ddPCR: Foundations for a Functional HIV Cure
The Critical Role of CCR5Δ32 in HIV Resistance and Curative Strategies
The C-C chemokine receptor type 5 (CCR5) is a seven-transmembrane G-protein-coupled receptor expressed on the surface of immune cells including T lymphocytes, macrophages, and dendritic cells [1]. As a primary co-receptor for human immunodeficiency virus (HIV) entry, CCR5 facilitates viral attachment and membrane fusion alongside the CD4 receptor [1] [2]. The CCR5Δ32 mutation refers to a 32-base-pair deletion in the CCR5 gene coding region that results in a frameshift and premature stop codon, producing a truncated protein that is not expressed on the cell surface [1] [3]. Individuals homozygous for this mutation (CCR5Δ32/Δ32) exhibit substantial resistance to R5-tropic HIV-1 strains—the viral variants predominantly responsible for establishing new infections [1] [4]. This natural resistance mechanism has inspired multiple therapeutic strategies aimed at mimicking this protective effect in HIV-positive individuals [2] [5].
Biological Significance of CCR5Δ32 in HIV Resistance
Population Genetics and Protective Effects
The CCR5Δ32 polymorphism occurs with varying prevalence across different populations, being most common in Northern European descendants where approximately 10% of individuals are heterozygous and 1% are homozygous [3] [4]. Meta-analyses of case-control studies have quantitatively demonstrated the protective effect of this mutation against HIV-1 infection [4].
Table 1: CCR5Δ32 Genotype Association with HIV-1 Susceptibility
| Genotype | Effect on HIV-1 Susceptibility | Odds Ratio (95% Credible Interval) | Reference |
|---|---|---|---|
| Heterozygous (CCR5/Δ32) | Increased susceptibility* | 1.16 (1.02-1.32) | [4] |
| Homozygous (Δ32/Δ32) | Reduced susceptibility | 0.25 (0.09-0.68) | [4] |
| Δ32 allele carriers (vs. exposed uninfected) | Reduced susceptibility | 0.71 (0.54-0.94) | [4] |
Note: The observed increased susceptibility in heterozygous individuals requires further investigation and may reflect population-specific factors.
The profound protection afforded by the homozygous CCR5Δ32 genotype has been validated through clinical observations of the "Berlin," "London," and "Düsseldorf" patients—HIV-positive individuals who received CCR5Δ32/Δ32 allogeneic hematopoietic stem cell transplantation (HSCT) for hematological malignancies and subsequently achieved long-term HIV remission without antiretroviral therapy [2] [5]. These cases provide proof-of-concept that CCR5 ablation can lead to functional HIV cure.
CCR5 in HIV Entry and Signaling Pathways
The mechanism of CCR5-mediated HIV entry involves complex interactions between viral envelope proteins, CD4 receptors, and CCR5 coreceptors. The following diagram illustrates the key molecular events in this process and how the Δ32 mutation confers resistance:
Diagram 1: CCR5-mediated HIV entry pathway. Wild-type CCR5 enables viral fusion, while the truncated Δ32 mutant protein prevents HIV entry.
Beyond its role as an HIV coreceptor, CCR5 functions as a receptor for pro-inflammatory chemokines including CCL3 (MIP-1α), CCL4 (MIP-1β), and CCL5 (RANTES) [1]. These natural ligands can competitively inhibit HIV binding, suggesting complex immunoregulatory functions. The CCR5Δ32 mutation appears to have minimal deleterious effects on overall immune function, though its potential impact on responses to specific pathogens continues to be investigated [1] [6].
ddPCR Workflow for CCR5Δ32 Genotyping
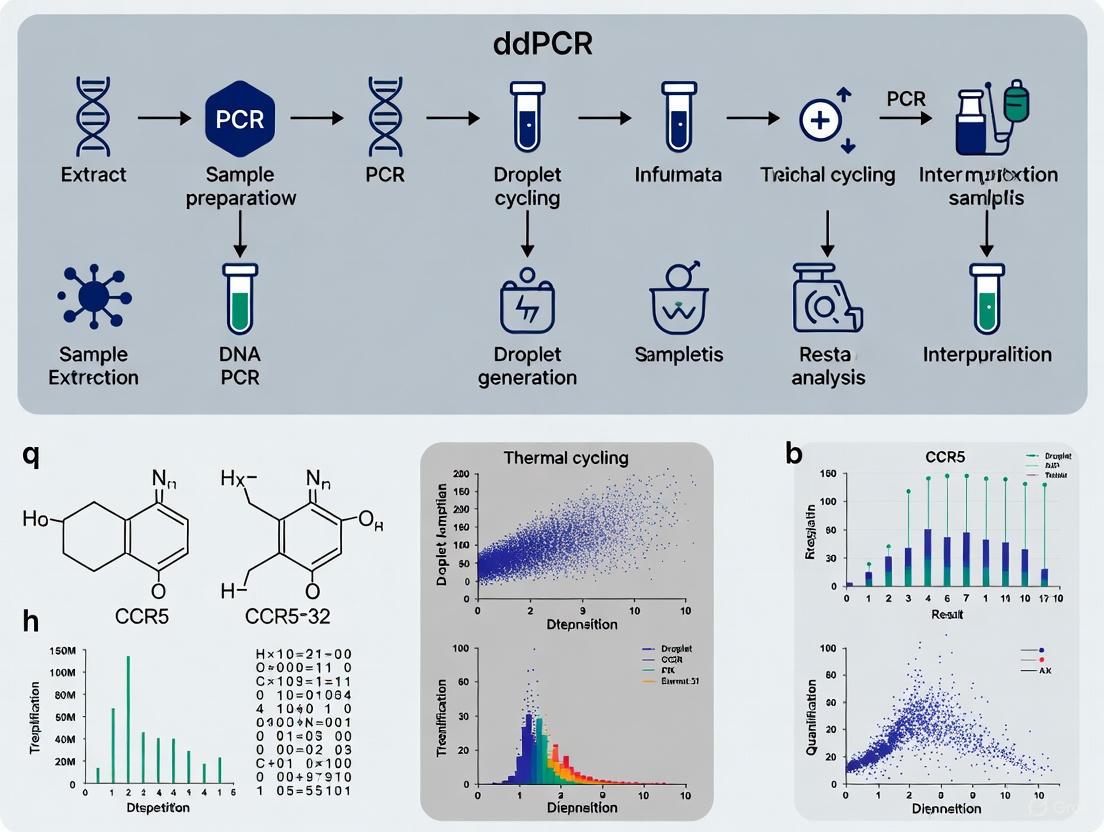
Droplet digital PCR (ddPCR) represents a transformative technology for precise quantification of the CCR5Δ32 allele fraction in heterogeneous cell populations. This absolute quantification method offers significant advantages for monitoring engraftment of CCR5-modified cells in therapeutic contexts [3] [7].
Principle of ddPCR
Unlike conventional quantitative PCR (qPCR) that relies on standard curves for relative quantification, ddPCR partitions samples into thousands of nanoliter-sized droplets, with each droplet functioning as an individual PCR reactor [8] [9]. After endpoint amplification, the fraction of positive droplets is counted and target concentration is calculated using Poisson statistics, enabling absolute quantification without reference standards [8] [9] [7]. This approach provides enhanced sensitivity, precision, and tolerance to PCR inhibitors compared to qPCR methods [7] [10].
Complete Experimental Protocol
Table 2: Research Reagent Solutions for CCR5Δ32 ddPCR
| Reagent/Category | Specific Product/Example | Function in Protocol |
|---|---|---|
| Nucleic Acid Extraction | ExtractDNA Blood and Cells Kit (Evrogen) | Genomic DNA isolation from patient samples |
| Target Amplification | QX200 ddPCR EvaGreen Supermix (Bio-Rad) | PCR reaction mixture for droplet-based amplification |
| Sequence-Specific Detection | CCR5-7 gRNA: CAGAATTGATACTGACTGTATGG [3] | Guides Cas9 to create Δ32 mutation in experimental systems |
| Droplet Generation | DG32 Cartridge (Bio-Rad) | Microfluidic generation of uniform droplets |
| Absolute Quantification | QX200 Droplet Reader (Bio-Rad) | Fluorescence detection and counting of positive/negative droplets |
Protocol: CCR5Δ32 Detection in Clinical Samples Using ddPCR
I. Sample Preparation and DNA Extraction
- Source Material: Collect peripheral blood mononuclear cells (PBMCs) or hematopoietic stem cells from clinical samples using standard venipuncture or apheresis procedures.
- DNA Extraction: Isolate genomic DNA using commercial extraction kits (e.g., ExtractDNA Blood and Cells Kit, Evrogen) following manufacturer protocols.
- Quality Assessment: Measure DNA concentration and purity using spectrophotometry (A260/A280 ratio of 1.8-2.0 indicates acceptable purity). Dilute samples to working concentration of 10-50 ng/μL in nuclease-free water.
II. ddPCR Reaction Setup
- Reaction Mixture (22 μL total volume):
- 11 μL of 2× ddPCR EvaGreen Supermix
- 1.1 μL of CCR5 Forward Primer (10 μM; sequence: 5'-CCCAGGAATCATCTTTACCA-3')
- 1.1 μL of CCR5 Reverse Primer (10 μM; sequence: 5'-GACACCGAAGCAGAGTTT-3')
- 5-50 ng of template DNA
- Nuclease-free water to 22 μL
- Droplet Generation:
- Transfer 20 μL of reaction mixture to DG32 cartridge wells
- Add 70 μL of Droplet Generation Oil
- Place cartridge in QX200 Droplet Generator
- Collect generated droplets in 96-well PCR plate
III. PCR Amplification
- Thermal Cycling Conditions:
- 95°C for 10 minutes (enzyme activation)
- 40 cycles of:
- 94°C for 30 seconds (denaturation)
- 60°C for 60 seconds (annealing/extension)
- 98°C for 10 minutes (enzyme deactivation)
- 4°C hold
- Ramp Rate: Set to 2°C/second for all steps
IV. Droplet Reading and Analysis
- Instrument Setup: Place PCR plate in QX200 Droplet Reader
- Data Acquisition: Measure fluorescence in each droplet using appropriate channel (FAM for CCR5 wild-type, HEX for Δ32 mutation if using probe-based detection)
- Data Analysis:
- Set threshold between positive and negative droplet populations
- Apply Poisson statistics to calculate absolute copy numbers of wild-type and Δ32 alleles
- Calculate allele frequency: [Δ32 count] / [Δ32 count + wild-type count] × 100%
The complete workflow from sample to result is visualized below:
Diagram 2: Complete ddPCR workflow for CCR5Δ32 detection, from sample collection to data analysis.
Analytical Validation
The developed ddPCR assay demonstrates a limit of detection of 0.8% for CCR5Δ32 mutant alleles in heterogeneous cell mixtures, enabling sensitive monitoring of CCR5-modified cell populations in therapeutic contexts [3]. This sensitivity is crucial for evaluating engraftment success in HSCT and gene therapy applications. Comparative studies show that ddPCR offers higher accuracy, precision, and reproducibility compared to qPCR, particularly at low target concentrations relevant to residual HIV reservoir quantification [7].
Therapeutic Applications and Clinical Translation
CCR5Δ32-Based Stem Cell Transplantation
Allogeneic hematopoietic stem cell transplantation from CCR5Δ32/Δ32 donors has proven to be a curative approach for HIV in several documented cases [5]. The detailed virological and immunological follow-up of the "Düsseldorf patient" (IciStem no. 19) provides insights into the correlates of cure: despite sporadic detection of HIV DNA traces by ddPCR and in situ hybridization, no replication-competent virus was recovered through extensive culture attempts, and the patient maintained aviremia for more than 4 years after treatment interruption [5]. Notably, declining HIV-specific immune responses indicated absence of ongoing antigen stimulation, further supporting cure [5].
Gene Editing Strategies
Gene editing technologies now enable recreation of the CCR5Δ32 phenotype in patient-derived cells, offering a promising alternative to allogeneic transplantation [2]. Several platforms have been developed for precise CCR5 disruption:
Table 3: Gene Editing Platforms for CCR5 Disruption
| Technology | Mechanism of Action | Advantages | Limitations |
|---|---|---|---|
| Zinc Finger Nucleases (ZFNs) | Custom-designed zinc finger proteins fused with FokI nuclease induce DNA cleavage | Early clinical trial data (SB-728-T) demonstrating safety and virological benefit | Complex design, higher off-target risk, potential immunogenicity |
| TALENs | Transcription activator-like effector proteins fused to FokI nuclease for DNA cleavage | Improved specificity over ZFNs, reduced off-target activity | Technically demanding construction, large size challenges viral delivery |
| CRISPR/Cas9 | Guide RNA directs Cas9 nuclease to specific genomic loci for cleavage | Easy design, high efficiency, multiplex editing capability | Off-target effects, PAM sequence dependency, potential immune responses to Cas9 |
| Base Editors | Cas protein fused with deaminase enables precise single-nucleotide changes without double-strand breaks | Avoids double-strand break risks (indels, translocations) | Potential off-target editing, limited editing window constraints |
Clinical trials using CRISPR/Cas9 for CCR5 editing in hematopoietic stem cells are underway (NCT03164135), demonstrating the feasibility of this approach [2]. Multiplex gene editing strategies that target both CCR5 and CXCR4 (alternative HIV coreceptor) or HIV proviral DNA are being developed to create comprehensive viral barriers and prevent viral escape through tropism switching [2].
The CCR5Δ32 mutation represents a naturally occurring resistance mechanism against HIV that has inspired multiple therapeutic strategies. ddPCR technology provides a highly sensitive and accurate method for detecting this mutation and quantifying allelic frequencies in clinical samples, enabling precise monitoring of CCR5-targeted interventions. Combined with advanced gene editing platforms, CCR5 disruption holds significant promise for achieving HIV remission or cure. Future directions include optimizing multiplex editing strategies, enhancing delivery efficiency, and addressing potential safety concerns to broaden the clinical applicability of these innovative approaches.
Absolute Quantification via Partitioning and Poisson Statistics
Droplet Digital PCR (ddPCR) represents a third-generation PCR technology that enables absolute quantification of nucleic acid targets without the need for a standard curve. This advanced method relies on sample partitioning and Poisson statistics to calculate target concentration directly from the ratio of positive to negative partitions, providing exceptional precision for detecting rare alleles and low-abundance targets in complex clinical samples [9]. The principle of partitioning a sample into thousands of individual reactions was conceptually established in the 1990s, but technological advances in microfluidics have now made it readily accessible for research and clinical applications [9].
In the context of CCR5Δ32 mutation detection, ddPCR offers significant advantages for monitoring transplanted hematopoietic stem cells in HIV patients or quantifying gene editing efficiency in experimental therapies [3]. The CCR5Δ32 mutation, a natural 32-base pair deletion in the CCR5 gene, confers resistance to HIV infection when homozygous, making it a critical biomarker in both natural immunity studies and emerging CRISPR/Cas9-based therapeutic approaches [3]. This application note details the theoretical framework and practical protocols for implementing ddPCR in CCR5Δ32 detection workflows.
Theoretical Foundation: Partitioning and Poisson Statistics
The Partitioning Process
The fundamental innovation of ddPCR lies in the physical partitioning of a PCR reaction mixture into thousands of nanoliter-sized droplets, typically ranging from 10,000 to 20,000 droplets per sample. This process creates discrete reaction chambers where individual nucleic acid molecules are randomly distributed according to Poisson statistics [8] [9]. Each droplet functions as an individual microreactor that may contain zero, one, or a few copies of the target DNA sequence [11] [8].
The partitioning process begins with a water-in-oil emulsion, where the aqueous PCR mixture (containing template DNA, primers, probes, and PCR master mix) is dispersed into uniform droplets within an immiscible oil phase containing surfactants for stabilization [9]. This microfluidic-based emulsification occurs at high speeds (1-100 kHz) using either passive methods like T-junction or flow-focusing geometries, or active methods utilizing external forces [8]. The resulting monodisperse droplets are then thermally cycled through conventional PCR amplification protocols.
Poisson Statistics for Absolute Quantification
Following PCR amplification, the droplets are analyzed one-by-one using a droplet reader that detects fluorescence signals in each channel. The binary readout (positive or negative) from thousands of individual reactions provides the fundamental data for absolute quantification through Poisson distribution mathematics [12] [13].
The Poisson model accounts for the random distribution of target molecules across partitions and corrects for the probability that any positive partition may have contained more than one target molecule. The fundamental Poisson equation for ddPCR is:
λ = -ln(1 - p)
Where:
- λ represents the average number of target molecules per partition
- p is the ratio of positive partitions to total partitions [12]
The absolute concentration of the target in the original sample (in copies/μL) is then calculated as:
Concentration = λ × (total partitions / volume analyzed)
This mathematical approach enables calibration-free quantification, eliminating the need for standard curves required by qPCR methods and providing superior accuracy, particularly at low target concentrations [11] [14] [13].
Figure 1: ddPCR Workflow Overview. The process begins with sample preparation and progresses through droplet generation, amplification, and analysis to achieve absolute quantification.
Comparative Analysis: ddPCR vs. qPCR
Performance Characteristics
Digital PCR offers distinct advantages for applications requiring high precision, absolute quantification, and detection of rare variants. The table below summarizes key performance characteristics compared to quantitative PCR (qPCR):
Table 1: Comparative Analysis of ddPCR and qPCR Performance Characteristics
| Parameter | ddPCR | qPCR |
|---|---|---|
| Quantification Method | Absolute (via Poisson) | Relative (standard curve) |
| Precision at Low Target Concentration | High (low variability) [11] [14] | Moderate to low (higher variability) [11] |
| Dynamic Range | Limited by partition count [14] | Wider dynamic range [14] |
| Sensitivity to Inhibitors | More tolerant [11] [8] | Highly sensitive [11] |
| Detection of Rare Alleles | Superior for rare variants [3] [9] | Limited by background signal |
| Throughput and Cost | Moderate throughput, higher cost per sample | High throughput, lower cost per sample [14] |
| Data Analysis Complexity | Simple binary interpretation | Complex curve analysis required |
Tolerance to PCR Inhibitors
ddPCR demonstrates superior performance with complex samples that may contain PCR inhibitors. The partitioning process effectively dilutes inhibitors across thousands of droplets, reducing their concentration in target-positive partitions and maintaining amplification efficiency. This characteristic is particularly valuable for clinical samples that may contain hemoglobin, heparin, or other substances that can inhibit PCR amplification [11] [8]. Studies have shown that ddPCR can maintain quantitative accuracy in samples where qPCR quantification fails due to inhibition, making it particularly suitable for direct analysis of crude extracts or challenging sample matrices [11].
Application in CCR5Δ32 Mutation Detection
Clinical Relevance
The CCR5Δ32 mutation represents a critical biomarker in HIV research and treatment. As a co-receptor for HIV entry into T-cells, the CCR5 protein serves as an essential gateway for viral infection. Individuals homozygous for the 32-base pair deletion in the CCR5 gene exhibit natural resistance to HIV-1 infection, while heterozygotes show delayed disease progression [3]. This biological significance has propelled CCR5Δ32 to the forefront of therapeutic development, including:
- Stem cell transplantation from CCR5Δ32 homozygous donors to HIV-positive patients, which has led to complete viral eradication in documented cases [3]
- CRISPR/Cas9 genome editing to introduce CCR5Δ32 mutations in autologous or immunocompatible hematopoietic stem cells [3]
- Patient stratification based on CCR5 genotype for targeted therapeutic approaches
Experimental Protocol for CCR5Δ32 Quantification
Sample Preparation and DNA Extraction
Materials:
- Patient samples (whole blood, purified cells, or stem cell preparations)
- DNA extraction kit (e.g., DNeasy PowerSoil Pro Kit, QIAamp DNA Blood Mini Kit)
- Spectrophotometer (NanoDrop) or fluorometer for DNA quantification
- Thermal cycler
Procedure:
- Extract genomic DNA from patient samples using manufacturer's protocol
- Determine DNA concentration and purity (A260/A280 ratio of ~1.8-2.0)
- Adjust DNA concentration to 10-100 ng/μL for ddPCR analysis
- Store extracted DNA at -20°C until use
Note: DNA quality is critical for assay performance. Assess DNA degradation by agarose gel electrophoresis if necessary.
ddPCR Reaction Setup
Reagent Solutions: Table 2: Essential Research Reagents for CCR5Δ32 ddPCR
| Reagent | Function | Working Concentration |
|---|---|---|
| ddPCR Supermix for Probes | Provides optimized buffer, polymerase, dNTPs | 1× concentration |
| CCR5 Wild-Type Probe (FAM-labeled) | Detects wild-type CCR5 allele | 0.25 μM |
| CCR5Δ32 Mutation Probe (HEX/VIC-labeled) | Detects Δ32 deletion allele | 0.25 μM |
| CCR5 Forward Primer | Amplifies CCR5 target region | 0.9 μM |
| CCR5 Reverse Primer | Amplifies CCR5 target region | 0.9 μM |
| Nuclease-Free Water | Adjusts reaction volume | - |
| DNA Template | Sample for analysis | 10-100 ng total |
Reaction Setup:
- Prepare reaction mix on ice according to the following formulation:
- 11 μL ddPCR Supermix for Probes (2×)
- 1.0 μL CCR5 Wild-Type Probe (FAM, 5 μM stock)
- 1.0 μL CCR5Δ32 Mutation Probe (HEX/VIC, 5 μM stock)
- 1.8 μL CCR5 Forward Primer (10 μM stock)
- 1.8 μL CCR5 Reverse Primer (10 μM stock)
- 2-4 μL DNA Template (10-100 ng)
- Adjust to 22 μL with nuclease-free water
- Gently mix by pipetting, avoid introducing bubbles
- Include appropriate controls:
- No-template control (nuclease-free water)
- Wild-type homozygous control
- Δ32 homozygous control (if available)
- Heterozygous control
Droplet Generation and Thermal Cycling
Materials:
- Droplet generator (e.g., QX200 Droplet Generator, Bio-Rad)
- DG8 cartridges and gaskets
- Droplet generation oil
- Thermal cycler with 96-well block
- Semi-skirted 96-well PCR plates
- Foil heat seals
Procedure:
- Load 20 μL of each reaction mixture into middle wells of DG8 cartridge
- Add 70 μL of droplet generation oil to bottom wells of cartridge
- Place gasket on cartridge and load into droplet generator
- Generate droplets according to manufacturer's protocol (~20,000 droplets/sample)
- Carefully transfer 40 μL of generated droplets to 96-well PCR plate
- Seal plate with foil heat seal using plate sealer (180°C for 5 seconds)
- Perform PCR amplification using the following protocol:
- Enzyme activation: 95°C for 10 minutes
- 40 cycles of:
- Denaturation: 94°C for 30 seconds
- Annealing/Extension: 60°C for 60 seconds
- Enzyme deactivation: 98°C for 10 minutes
- Hold: 4°C ∞
Note: Ramp rate should be set to 2°C/second for optimal results. Annealing temperature may require optimization for specific primer/probe combinations.
Droplet Reading and Data Analysis
Materials:
- Droplet reader (e.g., QX200 Droplet Reader, Bio-Rad)
- ddPCR data analysis software
Procedure:
- Load PCR plate into droplet reader
- Run droplet reading according to manufacturer's specifications
- Analyze data using companion software:
- Set appropriate fluorescence thresholds to distinguish positive and negative droplets
- Identify four droplet populations:
- Wild-type positive (FAM+ only)
- Δ32 mutation positive (HEX/VIC+ only)
- Heterozygous (FAM+ and HEX/VIC+)
- Negative (no fluorescence)
- Apply Poisson correction to calculate absolute copy numbers
- Calculate mutation frequency as:
- % CCR5Δ32 = [Δ32 copies / (Wild-type copies + Δ32 copies)] × 100
Figure 2: Poisson Statistics Workflow in ddPCR. The random distribution of targets followed by droplet classification and Poisson correction enables absolute quantification without standard curves.
Troubleshooting and Quality Control
Common Technical Issues
Table 3: Troubleshooting Guide for CCR5Δ32 ddPCR
| Problem | Potential Cause | Solution |
|---|---|---|
| Low Droplet Count | Cartridge or gasket issues | Ensure proper cartridge loading and gasket placement |
| Poor Resolution Between Positive/Negative Droplets | Suboptimal probe concentration or thermal cycling conditions | Titrate probe concentrations; optimize annealing temperature |
| High Background Signal | Probe degradation or non-specific amplification | Use fresh probe aliquots; verify primer specificity |
| Rain Effect (Droplets with intermediate fluorescence) | Imperfect amplification or inhibitor presence | Optimize template quality; increase annealing temperature |
| Significant Well-to-Well Variation | Improper droplet generation or pipetting errors | Use reverse pipetting technique; ensure consistent droplet generation |
Quality Control Measures
- Threshold Setting: Establish consistent fluorescence thresholds based on negative control droplets while ensuring clear separation between positive and negative populations
- Limit of Detection: The described CCR5Δ32 ddPCR assay can reliably detect mutant alleles at frequencies as low as 0.8% in heterogeneous mixtures [3]
- Precision Assessment: Run replicate samples to evaluate intra-assay and inter-assay variability
- Dynamic Range: Verify linearity across expected target concentrations using reference standards if available
Droplet Digital PCR represents a powerful technological advancement for absolute quantification of nucleic acid targets, with particular utility in detecting rare mutations like CCR5Δ32 in heterogeneous clinical samples. The combination of sample partitioning, endpoint amplification, and Poisson statistical analysis provides a robust framework for precise molecular quantification without standard curves. For CCR5Δ32 detection specifically, ddPCR enables accurate monitoring of mutant allele frequency in stem cell transplantation settings and genome editing applications, supporting the development of next-generation HIV therapies. The protocols outlined in this application note provide researchers with a comprehensive framework for implementing this powerful technology in both basic research and clinical development contexts.
The C-C chemokine receptor type 5 (CCR5) serves as a crucial co-receptor for human immunodeficiency virus (HIV) entry into T-cells [3]. A natural 32-base pair deletion variant, CCR5Δ32, results in a non-functional receptor that confers resistance to HIV R5-tropism strains, the most common and contagious variants [3]. This biological phenomenon provides the foundation for innovative HIV treatment strategies utilizing hematopoietic stem and progenitor cell (HSPC) transplantation. Clinical proof-of-principle has been established through cases in Berlin and London, where HIV-positive patients with acute lymphoblastic leukemia received HSPC transplants from CCR5Δ32 homozygous donors, resulting in sustained viral remission [3]. Simultaneously, CRISPR/Cas9 genome editing technologies now enable artificial reproduction of the CCR5Δ32 mutation in autologous or immunocompatible cells, creating novel therapeutic cell products [3]. These advancing therapeutic approaches create an urgent clinical need for robust monitoring methodologies to precisely quantify CCR5Δ32 mutant alleles in heterogeneous cell populations, enabling accurate assessment of transplant engraftment and therapeutic potency.
Quantitative Monitoring Solutions: The Case for Digital PCR
Traditional quantitative PCR (qPCR) methods face significant limitations for monitoring CCR5Δ32 in clinical samples. While qPCR has been used for CCR5Δ32 screening, it requires standard curves for quantification and offers limited sensitivity for detecting rare mutant alleles in mixed cell populations [8]. Digital PCR (dPCR), particularly droplet digital PCR (ddPCR), represents a third-generation PCR technology that overcomes these constraints through absolute quantification without calibration curves [9].
ddPCR operates by partitioning a PCR reaction into thousands to millions of nanoliter-sized droplets, effectively creating individual micro-reactors [9] [8]. Following PCR amplification, each droplet is analyzed for fluorescence, and the target concentration is absolutely quantified using Poisson statistics based on the ratio of positive to negative partitions [9]. This approach provides exceptional sensitivity down to 0.8% for detecting CCR5Δ32 mutations in mixed cell populations [3], making it ideally suited for monitoring engraftment dynamics of CCR5-modified HSPCs. Furthermore, ddPCR demonstrates high tolerance to PCR inhibitors and offers superior reproducibility compared to qPCR methods [8], critical advantages for clinical monitoring applications.
Table 1: Performance Comparison of Nucleic Acid Quantification Methods
| Parameter | qPCR | Digital PCR | Next-Generation Sequencing |
|---|---|---|---|
| Quantification Type | Relative (requires standard curve) | Absolute (Poisson statistics) | Relative (requires standardization) |
| Sensitivity | Moderate | High (detects rare alleles <1%) | Variable |
| Multiplexing Capability | Limited (4-6 channels) | Moderate | Exceptional |
| Cost | Low | Moderate | High |
| Turnaround Time | Fast (< 4 hours) | Fast (< 4 hours) | Slow (days) |
| Instrument Base | Widely available | Growing availability | Limited |
| Quantitative Output | Cycle threshold (Ct) | Copies/μL | Read counts |
Experimental Protocol: CCR5Δ32 Quantification via ddPCR
Sample Preparation and DNA Extraction
Begin with the MT-4 human T-cell line or primary hematopoietic cells cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum, maintained at 37°C with 5% CO₂ [3]. Extract genomic DNA using phenol-chloroform methodology or commercial kits (e.g., ExtractDNA Blood and Cells Kit). Precisely quantify DNA concentration and assess purity using spectrophotometry (NanoPhotometer P-Class P360) [3]. For clinical samples, including blood, sputum, or bronchoalveolar lavage fluid, extract pathogen DNA using specialized kits (QIAamp UCP Pathogen Mini Kit) [15].
ddPCR Reaction Setup and Partitioning
Prepare ddPCR reactions using a master mix compatible with droplet generation (TaqPath ProAmp Master Mix) [15]. Design and validate primers and probes specific to both wild-type CCR5 and the CCR5Δ32 deletion variant. Include appropriate fluorescence labels (FAM, HEX) for multiplex detection. Assemble reactions according to the following formulation:
Table 2: ddPCR Reaction Components
| Component | Volume | Final Concentration |
|---|---|---|
| ddPCR Master Mix (2X) | 10 μL | 1X |
| CCR5 Wild-Type Probe/Primer Mix | 1 μL | Optimized concentration |
| CCR5Δ32 Probe/Primer Mix | 1 μL | Optimized concentration |
| Template DNA | Variable | 10-100 ng total |
| Nuclease-Free Water | To 20 μL | - |
Transfer the reaction mixture to a droplet generator cartridge. Generate droplets using a commercial system (Bio-Rad QX200 Droplet Digital) according to manufacturer specifications, typically producing ~20,000 droplets per sample [3] [8]. Transfer emulsified samples to a 96-well PCR plate and seal properly.
Thermal Cycling and Signal Acquisition
Perform PCR amplification using the following thermal cycling conditions:
- Enzyme activation: 95°C for 10 minutes
- 40-45 cycles of:
- Denaturation: 95°C for 30 seconds
- Annealing/Extension: 55-60°C for 60 seconds (optimize based on primer Tm)
- Enzyme deactivation: 98°C for 10 minutes
- Hold at 4°C
Following amplification, transfer the plate to a droplet reader which sequentially analyzes each droplet through a fluorescence detection system [9]. The reader identifies positive droplets (containing amplified target) and negative droplets (no amplification) for each fluorescence channel.
Data Analysis and Interpretation
Analyze raw fluorescence data using manufacturer-provided software (QuantaSoft for Bio-Rad systems). Set appropriate threshold(s) to distinguish positive from negative droplets for each target. The software automatically calculates target concentration using Poisson statistics:
[ \text{Target concentration (copies/μL)} = \frac{-\ln(1 - p)}{V} ]
Where ( p ) = fraction of positive partitions and ( V ) = partition volume [9]. Report results as copies/μL of wild-type CCR5, CCR5Δ32 mutant, and calculate the percentage of CCR5Δ32 alleles:
[ \%\text{CCR5Δ32} = \frac{[\text{CCR5Δ32}]}{[\text{CCR5Δ32}] + [\text{Wild-Type CCR5}]} \times 100 ]
Diagram Title: ddPCR Workflow for CCR5Δ32 Detection
Research Reagent Solutions
Table 3: Essential Reagents and Materials for CCR5Δ32 Monitoring
| Reagent/Material | Function | Example Products |
|---|---|---|
| DNA Extraction Kits | Isolation of high-quality genomic DNA from cells/tissues | ExtractDNA Blood and Cells Kit, QIAamp UCP Pathogen Mini Kit |
| ddPCR Master Mix | Provides optimized buffer, enzymes, dNTPs for amplification | TaqPath ProAmp Master Mix |
| Custom Primers/Probes | Target-specific amplification and detection of CCR5 variants | IDT PrimeTime qPCR Assays |
| Droplet Generation Oil | Creates stable water-in-oil emulsion for partitioning | DG Oil for Probes, Droplet Generation Oil |
| Microfluidic Cartridges | Facilitates nanodroplet formation | DG8 Cartridges, QX200 Droplet Generator Cartridge |
| PCR Plates | Holds samples during amplification and reading | Twin.tec 96-Well PCR Plates |
| Droplet Reader Oil | Enables sequential droplet analysis in reader | QX200 Droplet Reader Oil |
Data Quality Assurance and Analysis
Robust quality assurance protocols are essential for generating reliable clinical monitoring data. Implement systematic data cleaning procedures to identify and address anomalies, including verification that all fluorescence measurements fall within expected technical boundaries [16]. Establish pre-defined thresholds for data inclusion/exclusion, such as minimum droplet counts (>10,000 per sample) and acceptable ranges for technical controls [3].
For quantitative analysis, begin with descriptive statistics including mean, standard deviation, and coefficient of variation for replicate measurements [16]. Assess data distribution using normality tests (Kolmogorov-Smirnov, Shapiro-Wilk) and examine kurtosis and skewness values (±2 indicates normal distribution) [16]. Report both statistically significant and non-significant findings to prevent publication bias and inform future research directions [16].
Ensure proper management of missing data through rigorous documentation and appropriate statistical handling. When data are missing completely at random, advanced imputation methods may be employed, though clinical monitoring of CCR5Δ32 typically demands complete data sets for accurate patient assessment [16].
ddPCR technology provides an exceptionally powerful platform for monitoring CCR5Δ32 in HSPC transplants and gene-edited cell products, offering the sensitivity, precision, and absolute quantification required for critical clinical decision-making. As CCR5-directed therapies continue to evolve, emerging technologies like color cycle multiplex amplification (CCMA) may further enhance monitoring capabilities by dramatically increasing multiplexing capacity through fluorescence permutation strategies [15]. The growing clinical adoption of dPCR platforms, including QIAcuity and Digital LightCycler systems [9], will make these essential monitoring tools increasingly accessible. Implementation of the standardized protocols and quality assurance measures outlined in this application note will ensure reliable, reproducible quantification of CCR5Δ32 mutant alleles, ultimately supporting the safe and effective translation of these innovative HIV treatment strategies into clinical practice.
Droplet Digital PCR (ddPCR) represents a third-generation PCR technology that provides absolute quantification of nucleic acids without the need for a standard curve. [17] [8] This technology partitions a PCR reaction into thousands of nanoliter-sized water-in-oil droplets, effectively creating individual reaction chambers where amplification occurs. The principle of endpoint detection and Poisson statistical analysis enables direct counting of target DNA molecules, offering significant advantages over quantitative PCR (qPCR) for clinical applications requiring high precision. [17] In the context of CCR5Δ32 mutation detection for HIV research, these advantages translate to more reliable monitoring of gene editing efficiency and accurate quantification of mutant alleles in heterogeneous cell mixtures, which is crucial for developing stem cell therapies and monitoring transplanted cells in patients. [3]
Key Advantages of ddPCR Over qPCR
Absolute Quantification Without Standard Curves
Unlike qPCR, which relies on standard curves derived from reference samples for relative quantification, ddPCR provides absolute quantification by directly counting target molecules through binary endpoint detection (positive or negative partitions). [17] [8] This elimination of calibration curves removes a significant source of variability and potential inaccuracy, particularly important when reliable standards are unavailable. Studies have demonstrated that qPCR values can overestimate actual concentrations by up to 40% compared to ddPCR when using certain calibrants, highlighting the potential for miscalibration in qPCR methodologies. [8]
Enhanced Sensitivity and Precision
The partitioning process in ddPCR significantly enhances detection sensitivity by effectively concentrating low-abundance targets and reducing background noise. This enables precise detection of rare mutations present at frequencies as low as 0.1-0.8% in wild-type backgrounds. [17] [3] For CCR5Δ32 detection, specifically developed ddPCR assays can accurately quantify mutant alleles down to 0.8% in heterogeneous cell mixtures, a level of sensitivity crucial for monitoring gene editing efficiency and detecting minimal residual disease. [3] The high number of partitions (typically 20,000+ per reaction) provides exceptional precision for absolute quantification, making it superior for applications requiring exact copy number determination. [17] [18]
Superior Tolerance to PCR Inhibitors
ddPCR demonstrates markedly improved resistance to PCR inhibitors commonly found in clinical samples compared to qPCR. [8] [18] By partitioning the sample, inhibitors are diluted unevenly across droplets, ensuring that a sufficient number of amplification reactions proceed efficiently despite the presence of inhibitory substances. This advantage is particularly valuable when working with complex sample matrices such as dried blood spots (DBS), crude lysates, or samples with high mucopolysaccharide content. [8] [19] The reduced impact of inhibitors in ddPCR leads to more reliable results from suboptimal samples without the need for extensive purification.
Comparative Performance Data
Table 1: Quantitative Comparison of ddPCR vs. qPCR Performance Characteristics
| Performance Metric | ddPCR | Conventional qPCR | Clinical Significance |
|---|---|---|---|
| Quantification Method | Absolute counting via Poisson statistics | Relative to standard curve | Eliminates calibration bias and reference material variability |
| Detection Sensitivity | Can detect rare mutations at 0.1-0.8% frequency [3] | Typically limited to 1-5% mutant detection | Crucial for monitoring minimal residual disease and gene editing efficiency |
| Precision at Low Targets | Superior consistency for medium viral loads (RSV) [18] | Higher variability in mid-range Ct values (25.1-30) [18] | More reliable monitoring of treatment response and viral load dynamics |
| Inhibitor Tolerance | High resistance to common PCR inhibitors [8] [18] | Susceptible to inhibition affecting amplification efficiency | Better performance with complex clinical samples (blood, tissue) |
| Dynamic Range | Linear across 5 orders of magnitude with precise partitioning | Limited by standard curve quality and amplification efficiency | More accurate for both high and low abundance targets in same run |
Table 2: Experimental Validation in CCR5Δ32 Detection Context
| Experimental Parameter | ddPCR Performance | qPCR Performance | Reference Application |
|---|---|---|---|
| CCR5Δ32 Detection Limit | 0.8% in heterogeneous mixtures [3] | Not specifically reported for this application | Monitoring CRISPR/Cas9 editing efficiency in MT-4 cell line [3] |
| Accuracy in Cell Mixtures | Linear quantification from 0.8-100% mutant alleles [3] | Limited precision for rare allele quantification | Transplantation monitoring and chimerism analysis |
| Sample Type Flexibility | Effective with crude lysates and inhibited samples [8] | Requires high-quality purified nucleic acids | Suitable for direct clinical sample analysis |
| Multiplexing Capacity | 2-5 color channels available depending on platform [17] | Typically 2-4 targets with spectral overlap | Simultaneous detection of mutant and wild-type alleles |
Detailed Protocol: CCR5Δ32 Detection by ddPCR
Sample Preparation and DNA Extraction
Materials:
- Cell culture or patient samples (e.g., PBMCs, whole blood)
- Phenol-chloroform or commercial DNA extraction kits (e.g., ExtractDNA Blood and Cells Kit)
- NanoPhotometer for DNA quantification and purity assessment (A260/280 ratio 1.8-2.0)
Procedure:
- Extract genomic DNA using standard phenol-chloroform protocol or commercial kits according to manufacturer's instructions.
- Quantify DNA concentration using spectrophotometry (NanoPhotometer P-Class P360 or equivalent).
- Adjust DNA concentration to 10-50 ng/μL in nuclease-free water.
- Assess DNA purity via A260/A280 ratio (target: 1.8-2.0) and A260/A230 ratio (target: >2.0).
ddPCR Reaction Setup
Reagent Composition:
- 10-100 ng genomic DNA template
- 1× ddPCR Supermix for Probes (no dUTP)
- 900 nM forward and reverse primers (CCR5-specific)
- 250 nM HEX-labeled probe for CCR5Δ32 detection
- Nuclease-free water to final volume of 20-22 μL
Primer and Probe Sequences for CCR5Δ32 Detection:
- Forward Primer: 5'-CCCAGGAATCATCTTTACCA-3'
- Reverse Primer: 5'-GACACCGAAGCAGAGTTT-3'
- Wild-Type Probe: FAM-labeled (sequence not specified in sources)
- Δ32 Mutant Probe: HEX-labeled (sequence not specified in sources)
Procedure:
- Prepare reaction mix on ice by combining all components except DNA template.
- Add DNA template last and mix gently by pipetting.
- Centrifuge briefly to collect reaction mixture at tube bottom.
Droplet Generation and PCR Amplification
Materials:
- Automated Droplet Generator (e.g., QX200 Droplet Generator)
- DG8 Cartridges and Gaskets
- Droplet Generation Oil for Probes
Procedure:
- Transfer 20 μL of reaction mixture to DG8 Cartridge sample well.
- Add 70 μL of Droplet Generation Oil to oil well.
- Place gasket on cartridge and load into QX200 Droplet Generator.
- Generate droplets according to manufacturer's protocol (approximately 20,000 droplets per sample).
- Transfer 40 μL of generated droplets to a 96-well PCR plate.
- Seal plate with foil heat seal using plate sealer at 180°C for 5 seconds.
- Perform PCR amplification with following cycling conditions:
- 95°C for 10 minutes (enzyme activation)
- 40 cycles of:
- 94°C for 30 seconds (denaturation)
- 55-60°C for 60 seconds (annealing/extension)
- 98°C for 10 minutes (enzyme deactivation)
- 4°C hold
Droplet Reading and Data Analysis
Materials:
- Droplet Reader (e.g., QX200 Droplet Reader)
- QuantaSoft Software or equivalent
Procedure:
- Load PCR plate into droplet reader.
- Run analysis using appropriate software settings for HEX and FAM detection channels.
- Set threshold between positive and negative populations based on negative controls.
- Apply Poisson statistics to calculate absolute copy numbers:
- Concentration (copies/μL) = -ln(1 - p) × (1/partition volume)
- Where p = fraction of positive partitions
- Calculate mutant allele frequency:
- % Mutant = [Mutant copies/(Mutant copies + Wild-type copies)] × 100
Research Reagent Solutions
Table 3: Essential Reagents and Materials for CCR5Δ32 ddPCR Detection
| Reagent/Material | Function/Purpose | Specifications/Alternatives |
|---|---|---|
| ddPCR Supermix for Probes | Provides optimized buffer, enzymes, and dNTPs for probe-based digital PCR | No-dUTP formulation preferred; available from multiple vendors |
| CCR5-specific Primers | Amplify target region spanning Δ32 deletion | 900 nM final concentration; sequence-specific validation required |
| HEX-labeled Δ32 Probe | Specifically detects 32-bp deletion mutant allele | 250 nM final concentration; specific binding to mutant sequence |
| FAM-labeled WT Probe | Detects wild-type CCR5 sequence | 250 nM final concentration; competitive design with mutant probe |
| Droplet Generation Oil | Creates stable water-in-oil emulsion for partitioning | Surfactant-stabilized for thermal cycling stability |
| DG8 Cartridges & Gaskets | Microfluidic chambers for droplet generation | Single-use consumables compatible with automated systems |
| Nuclease-free Water | Solvent for reaction preparation without degradation | PCR-grade, certified nuclease-free |
| DNA Extraction Kits | Isolation of high-quality genomic DNA from cells/tissues | Phenol-chloroform or commercial silica-based methods |
Troubleshooting and Quality Control
Common Technical Issues and Solutions
Poor Droplet Generation:
- Cause: Improper oil-to-sample ratio or contaminated cartridge
- Solution: Ensure precise pipetting (20 μL sample + 70 μL oil) and use fresh cartridges
Low Positive Droplet Count:
- Cause: Insufficient DNA input or primer/probe degradation
- Solution: Verify DNA quality and concentration; prepare fresh primer/probe aliquots
High Background Signal:
- Cause: Non-specific amplification or probe degradation
- Solution: Optimize annealing temperature; protect fluorescent probes from light
Rain Effect (Intermediate Populations):
- Cause: Suboptimal PCR efficiency or inhibitor presence
- Solution: Improve DNA purification; adjust thermal cycling conditions
Quality Control Measures
- Include no-template controls (NTC) in every run to monitor contamination
- Use positive controls with known mutation frequency (e.g., 1%, 5%, 50% mutant)
- Monitor droplet count per sample (target >10,000 droplets)
- Assess separation between positive and negative populations (clear threshold setting)
- Verify DNA quality metrics (A260/280, A260/230 ratios) before analysis
Applications in Clinical Research Context
The superior technical capabilities of ddPCR make it particularly suitable for CCR5Δ32 detection in HIV therapy research and monitoring. The technology's precision in quantifying low-frequency mutations enables accurate assessment of gene editing efficiency in CRISPR/Cas9-modified cells and reliable monitoring of transplanted cell populations in patients. [3] As hematopoietic stem cell transplantation with CCR5Δ32 mutations emerges as a promising approach for HIV treatment, robust monitoring tools become increasingly critical for tracking therapeutic efficacy and patient outcomes. [3] The absolute quantification capability of ddPCR provides reliable data for regulatory submissions and clinical decision-making, while its resistance to inhibitors ensures consistent performance across diverse clinical sample types encountered in multicenter trials.
A Step-by-Step Protocol for CCR5Δ32 ddPCR in Clinical Samples
The accuracy of a droplet digital PCR (ddPCR) workflow for detecting the CCR5Δ32 mutation is fundamentally dependent on the quality of the input nucleic acids. Efficient and standardized sample preparation protocols for genomic DNA (gDNA) and cell-free DNA (cfDNA) are critical for reliable quantification of mutant alleles in heterogeneous clinical samples, such as blood and tissues [3] [9]. This application note provides detailed methodologies for the extraction of high-quality gDNA and cfDNA, framed within the context of clinical research on the CCR5Δ32 mutation, a co-receptor for the human immunodeficiency virus (HIV) [3].
Sample Collection, Storage, and Preprocessing
Proper handling of specimens before extraction is essential to preserve nucleic acid integrity and prevent pre-analytical variations.
Blood Samples
- Anticoagulants: Collect blood in EDTA tubes [20] [21]. Heparin is not recommended for molecular applications as it can inhibit downstream PCR reactions [22].
- Storage: For short-term storage (a few days), keep blood at 2–8°C. For long-term storage (several weeks), freeze at –20°C or –80°C. Repeated freezing and thawing must be avoided, as it leads to gDNA fragmentation and reduced yield [22].
- Processing for cfDNA: Process blood samples for cfDNA extraction while fresh. Centrifuge fresh blood at 1,900 × g for 10 minutes at 4°C to separate plasma. To remove residual cells, perform a second centrifugation of the plasma supernatant at 16,000 × g for 10 minutes at 4°C. This step is critical to avoid gDNA contamination [21].
Tissue Samples
- Storage: Freshly harvested tissue should be immediately frozen and stored at –80°C or in liquid nitrogen [22].
- Fixation: For formalin-fixed paraffin-embedded (FFPE) tissues, use neutral-buffered formalin with a formalin-to-tissue ratio of at least 10:1. Fixation time should not exceed 24 hours to avoid overfixation, which causes excessive biomolecular cross-linking and impairs DNA recovery [22].
Table 1: Sample Storage Guidelines Prior to DNA Extraction
| Sample Type | Short-Term Storage | Long-Term Storage | Key Considerations |
|---|---|---|---|
| Whole Blood | 2–8°C for a few days [22] | –20°C or –80°C for a few weeks [22] | Use EDTA anticoagulant; avoid heparin [22]. |
| Plasma/Serum | 2–8°C for several hours [22] | –20°C or –80°C [22] | Double-centrifugation is critical for clean plasma [21]. |
| Animal/Human Tissue | N/A | –80°C or liquid nitrogen [22] | Avoid repetitive freeze-thaw cycles. |
| FFPE Tissue | Room temperature (after processing) | Room temperature (after processing) | Limit formalin fixation to <24 hours [22]. |
Genomic DNA (gDNA) Extraction from Whole Blood
This protocol is adapted from the CDC procedure for extracting DNA from whole blood collected in EDTA tubes, utilizing the QIAamp Blood Kit [20]. The yielded gDNA (3-12 µg from 200 µL of blood) is suitable for ddPCR analysis of CCR5Δ32 [20] [3].
Materials and Equipment
- QIAamp DNA Blood Mini Kit (Qiagen) or equivalent [20] [23].
- QIAGEN Protease [20].
- Microcentrifuge tubes (1.5 ml, low-binding) [20].
- Water bath or heat block (set to 56°C) [20].
- Ethanol (96–100%) [20].
- Vortex mixer and microcentrifuge [20].
Step-by-Step Protocol
- Lysis and Digestion: Pipet 200 µL of whole blood, 20 µL of QIAGEN Protease, and 200 µL of Buffer AL into a 1.5 mL microcentrifuge tube. Mix thoroughly by vortexing [20].
- Incubation: Incubate the mixture at 56°C for 10 minutes to lyse cells and digest proteins. Briefly spin down the tube to remove drops from the inside of the lid [20].
- Binding: Add 200 µL of ethanol (96-100%) to the mixture and vortex again. Carefully apply the entire volume to a QIAamp spin column seated in a 2 mL collection tube. Centrifuge at 15,000 × g for 1 minute. Discard the flow-through and place the column in a clean 2 mL collection tube [20].
- Washing:
- First Wash: Add 500 µL of Buffer AW1 to the column. Centrifuge at 15,000 × g for 1 minute. Discard the flow-through and place the column in a new 2 mL collection tube [20].
- Second Wash: Add 500 µL of Buffer AW2 to the column. Centrifuge at full speed for 3 minutes. Discard the flow-through and collection tube [20].
- Final Spin: Place the column in a new 2 mL collection tube and centrifuge at full speed for 1 minute to ensure all residual ethanol is removed [20].
- Elution: Transfer the spin column to a clean 1.5 mL microcentrifuge tube. Add 200 µL of Buffer AE or distilled water to the center of the membrane. Incubate at room temperature for 5 minutes, then centrifuge at 15,000 × g for 1 minute to elute the DNA [20].
- Storage: Store the purified gDNA at 4°C for immediate use or at –20°C/–80°C for long-term storage [20].
Quality Control for gDNA
- Quantification: Use a fluorometer (e.g., Qubit with dsDNA HS Assay Kit) for accurate concentration measurement [21] [23].
- Purity: Assess via spectrophotometry (e.g., NanoPhotometer). Optimal A260/A280 ratio is ~1.8, indicating minimal protein contamination [3] [24].
- Use in ddPCR: For a 50 µL ddPCR reaction, 1 µL of extracted gDNA is typically used [20]. The ddPCR assay for CCR5Δ32 mutation can accurately quantify its content down to 0.8% in a background of wild-type sequences [3].
Cell-Free DNA (cfDNA) Extraction from Plasma
This protocol is adapted from the Oxford Nanopore Technologies method for extracting human blood cfDNA using the QIAamp MinElute ccfDNA Midi Kit, yielding 15-30 ng of cfDNA from 3.5-4 mL of plasma [21]. High-quality cfDNA is crucial for sensitive liquid biopsy applications.
Materials and Equipment
- QIAamp MinElute ccfDNA Midi Kit (Qiagen) [21].
- Magnetic rack for 15 mL and 2 mL tubes [21].
- Isopropanol and ethanol (100% and 80%) [21].
- Hula mixer or gentle rotation mixer [21].
- Refrigerated centrifuge with swing-out and fixed-angle rotors [21].
- Qubit fluorometer and dsDNA HS Assay Kit [21].
Step-by-Step Protocol
- Plasma Preparation: Centrifuge 10 mL of fresh blood in an EDTA tube at 1,900 × g for 10 minutes at 4°C. Transfer the supernatant (plasma) to a new tube. Perform a second centrifugation of the plasma at 16,000 × g for 10 minutes at 4°C to remove any residual cells. Transfer the final supernatant to a fresh 15 mL tube [21].
- Binding to Magnetic Beads: For 4 mL of plasma, mix it with 120 µL of Magnetic Bead Suspension, 220 µL of Proteinase K, and 600 µL of Bead Binding Buffer in a 15 mL tube. Incubate for 10 minutes at room temperature with slow end-over-end mixing [21].
- Bead Capture and Washing:
- Place the tube on a magnetic rack for 1 minute until the solution clears. Discard the supernatant [21].
- Remove the tube from the magnet. Add 200 µL of Bead Elution Buffer to resuspend the bead pellet. Transfer the mixture to a Bead Elution Tube and incubate for 5 minutes at room temperature with shaking at 300 rpm [21].
- Place the tube back on the magnetic rack. Once clear, transfer the supernatant (which contains the cfDNA) to a new Bead Elution Tube, avoiding bead carryover [21].
- Column Purification:
- Add 300 µL of Buffer ACB to the supernatant and vortex to mix. Apply this mixture to a QIAamp UCP MinElute column and centrifuge at 6,000 × g for 1 minute [21].
- Wash the column with 500 µL of Buffer ACW2 by centrifuging at 6,000 × g for 1 minute. Place the column in a new collection tube and perform a "dry" spin at 20,000 × g for 3 minutes [21].
- Elution:
- Place the column in a clean 1.5 mL elution tube and incubate with the lid open at 56°C for 3 minutes in a shaker to dry the membrane completely [21].
- Pipette 50 µL of ultra-clean water onto the center of the membrane. Incubate for 1 minute at room temperature, then centrifuge at 20,000 × g for 1 minute to elute the cfDNA. For maximum yield, reload the eluate onto the membrane and repeat the elution step [21].
- Storage: Store purified cfDNA at –80°C to maintain stability [25].
Quality Control for cfDNA
- Quantification: Use a highly sensitive fluorometer (Qubit) due to low cfDNA concentrations [25] [21].
- Fragment Size Profiling: Analyze using a high-sensitivity system (e.g., Agilent Femto Pulse). A characteristic profile shows a dominant peak at ~167 bp (mononucleosomal fragment) with minimal high-molecular-weight gDNA contamination [21].
- Purity: Check A260/A280 and A260/230 ratios. Ratios close to 1.8 and 2.0, respectively, indicate minimal contamination from proteins or solvents [25].
Table 2: Key Differences in gDNA and cfDNA Extraction
| Parameter | Genomic DNA (gDNA) | Cell-Free DNA (cfDNA) |
|---|---|---|
| Primary Source | White blood cells (from whole blood) [20] | Plasma fraction of blood [21] |
| Extraction Focus | Isolation from within cells (requires lysis) [20] | Isolation from acellular fluid (requires careful plasma prep) [21] |
| Typical Yield | 3-12 µg from 200 µL whole blood [20] | 15-30 ng from 4 mL plasma [21] |
| Fragment Size | High molecular weight (>10 kb) [26] | Short fragments (~167 bp peak) [27] [21] |
| Critical Step | Proteinase K digestion and lysis [20] | Double-centrifugation to remove all cells [21] |
The ddPCR Workflow for CCR5Δ32 Detection
The following diagram illustrates the complete experimental workflow, from sample collection to data analysis for CCR5Δ32 mutation detection.
Diagram 1: The integrated ddPCR workflow for CCR5Δ32 detection in clinical samples.
The Scientist's Toolkit: Essential Reagents and Kits
Table 3: Research Reagent Solutions for DNA Extraction and Analysis
| Product Name | Supplier | Function/Application |
|---|---|---|
| QIAamp DNA Blood Mini Kit | Qiagen | Silica-membrane based purification of genomic DNA from whole blood [20] [23]. |
| QIAamp MinElute ccfDNA Midi Kit | Qiagen | Purification of cell-free DNA from large-volume plasma/serum samples using magnetic bead technology [21]. |
| ExtractDNA Blood and Cells Kit | Evrogen | gDNA extraction via phenol-chloroform method, used in CCR5Δ32 research [3]. |
| MagMAX DNA Multi-Sample Ultra 2.0 | Thermo Fisher | Bead-based chemistry for automated, high-throughput DNA extraction from various sample types [24]. |
| Nanobind PanDNA Kit | PacBio | Extraction of high-molecular-weight DNA for long-read sequencing applications [26]. |
| QX100/QX200 Droplet Digital PCR System | Bio-Rad | Instrumentation for absolute quantification of nucleic acids, used for CCR5Δ32 allele quantification [3] [9]. |
Robust and reproducible sample preparation is the foundation of a reliable ddPCR assay for detecting the CCR5Δ32 mutation. Adherence to the protocols outlined here for the collection, storage, and extraction of gDNA and cfDNA from blood and tissues ensures the integrity of the genetic material, thereby maximizing the sensitivity and accuracy of downstream molecular analyses. These standardized methods are critical for advancing clinical research and therapeutic development in HIV and other fields utilizing precise nucleic acid quantification.
The C-C chemokine receptor type 5 (CCR5) serves as a crucial co-receptor for human immunodeficiency virus (HIV) entry into T-cells [3] [28]. A naturally occurring 32-base pair deletion (CCR5Δ32) results in a non-functional receptor that confers resistance to HIV infection in homozygous individuals [29] [30]. This mutation has paved the way for novel HIV therapeutic strategies, including CCR5Δ32/Δ32 hematopoietic stem cell transplantation and autologous cell therapies using CRISPR/Cas9-edited cells [3] [23]. The efficacy of these approaches depends on accurate assessment of editing success, necessitating precise quantification of the Δ32 mutant allele among wild-type sequences [3]. Droplet Digital PCR (ddPCR) enables absolute quantification of mutant alleles in mixed cell populations with superior sensitivity and precision compared to conventional qPCR [9] [8] [10]. This application note details the design and optimization of primers and probes for specific discrimination and quantification of wild-type CCR5 and Δ32 mutant alleles using ddPCR, providing a critical tool for advancing HIV cure research.
Key Principles of ddPCR Assay Design
The fundamental advantage of ddPCR lies in its partitioning technology, which separates a PCR reaction into thousands of nanoliter-sized droplets, effectively diluting the sample to a single template molecule per droplet [9] [8]. This allows for binary endpoint detection (positive or negative for the target) followed by absolute quantification using Poisson statistics, eliminating the need for standard curves [8] [10]. This partitioning enhances sensitivity for rare alleles (e.g., in heterogeneous cell mixtures) and improves tolerance to PCR inhibitors [8].
For CCR5 genotyping assays, optimal design requires careful consideration of several factors to ensure specific and efficient amplification. The Δ32 deletion must be strategically positioned within the amplicon to create a significant difference in probe binding or amplicon length between wild-type and mutant sequences. Assays typically utilize a multiplex approach with two probe-based assays distinguishing wild-type and Δ32 alleles, plus an internal reference gene assay for normalization and DNA quality control [31].
Primer and Probe Sequences for CCR5Δ32 Detection
The following sequences and concentrations have been optimized for specific detection of wild-type CCR5 and the Δ32 mutant allele in a duplex ddPCR reaction [3] [23].
Table 1: Primer and Probe Sequences for CCR5 Genotyping
| Component | Sequence (5' → 3') | Final Concentration | Label | Target |
|---|---|---|---|---|
| Forward Primer | CCCAGGAATCATCTTTACCA [3] | 900 nM | - | CCR5 (WT & Δ32) |
| Reverse Primer | GACACCGAAGCAGAGTTT [3] | 900 nM | - | CCR5 (WT & Δ32) |
| Wild-Type Probe | Designed to span Δ32 deletion region | 250 nM | FAM | Wild-Type CCR5 |
| Δ32 Mutant Probe | Designed to span deletion junction | 250 nM | HEX/VIC | Δ32 Mutant CCR5 |
Table 2: Reference Gene Assay for Normalization
| Component | Sequence (5' → 3') | Final Concentration | Label | Purpose |
|---|---|---|---|---|
| Reference Assay | Commercially available (e.g., RPP30) | As per manufacturer | HEX/VIC | Copy number control |
The wild-type probe is designed to bind the sequence encompassing the 32-bp region, producing fluorescence only in droplets containing wild-type DNA. The Δ32 mutant probe is designed to bind the novel sequence junction created by the deletion, ensuring it fluoresces only when the mutant allele is present [31].
Detailed Experimental Protocol
Sample Preparation and DNA Extraction
- Source: Use genomic DNA from patient peripheral blood mononuclear cells (PBMCs), hematopoietic stem cells, or edited cell lines [3] [23].
- Extraction: Employ silica-membrane based kits (e.g., QIAamp DNA Blood Mini Kit) or phenol-chloroform extraction [3] [23].
- Quality Control: Assess DNA concentration and purity using a spectrophotometer (e.g., NanoPhotometer). Acceptable samples have A260/A280 ratios of ~1.8-2.0 [3].
ddPCR Reaction Setup
Prepare Reaction Mix: Combine components in a 20 µL total volume as specified below. Use the Bio-Rad QX200 ddPCR system or equivalent [31]. Table 3: ddPCR Reaction Master Mix
Component Final Volume/Reaction ddPCR Supermix for Probes (no dUTP) 1X Forward Primer (CCR5) 900 nM Reverse Primer (CCR5) 900 nM Wild-Type Probe (FAM) 250 nM Δ32 Mutant Probe (HEX) 250 nM Restriction Enzyme (e.g., HaeIII) 4 units Genomic DNA Template 5-125 ng Nuclease-Free Water To 20 µL Droplet Generation: Transfer the 20 µL reaction mix to a DG8 cartridge. Add 70 µL of droplet generation oil and generate droplets using the QX200 Droplet Generator [31].
PCR Amplification: Transfer 40 µL of generated droplets to a 96-well PCR plate. Seal the plate and run on a thermal cycler using the following protocol: Table 4: Thermal Cycling Conditions
Step Temperature Time Cycles Enzyme Activation 95°C 10 minutes 1 Denaturation 94°C 30 seconds 40 Annealing/Extension 55-60°C (optimize) 60 seconds Enzyme Deactivation 98°C 10 minutes 1 Hold 4°C ∞ Droplet Reading and Analysis: Read the plate on the QX200 Droplet Reader. Analyze data using QuantaSoft software, which automatically assigns droplets as FAM-positive (wild-type), HEX-positive (Δ32 mutant), double-positive (heterozygous), or negative [31]. The software calculates the absolute copy concentration (copies/µL) for each target using Poisson statistics.
Assay Validation and Optimization
- Primer/Probe Concentration Optimization: Perform a matrix of reactions with varying primer (e.g., 50-900 nM) and probe (e.g., 50-250 nM) concentrations to determine the combination yielding the highest amplitude and clearest cluster separation [32].
- Annealing Temperature Optimization: Test a gradient from 55°C to 60°C to determine the optimal temperature for specificity.
- Sensitivity and Limit of Detection (LOD): Serially dilute DNA heterozygous for CCR5Δ32 into wild-type DNA to demonstrate reliable detection down to 0.8% mutant allele frequency [3] [29].
Workflow and Detection Strategy
The following diagram illustrates the complete ddPCR workflow for CCR5Δ32 detection, from sample preparation to final analysis:
Diagram 1: ddPCR Workflow for CCR5Δ32 Genotyping. The process involves sample preparation, reaction partitioning, amplification, and fluorescence analysis to achieve absolute quantification.
The core detection mechanism relies on specific probe binding to distinct sequence features of each allele, as shown below:
Diagram 2: Allele-Specific Detection Mechanism. Probes are designed to discriminate alleles based on the presence (wild-type) or absence (Δ32 mutant) of the 32-bp sequence, generating distinct fluorescent signals.
The Scientist's Toolkit: Essential Reagents and Equipment
Table 5: Key Research Reagent Solutions for CCR5 ddPCR
| Category | Specific Product/Kit | Function in Workflow |
|---|---|---|
| Nucleic Acid Extraction | QIAamp DNA Blood Mini Kit [23] | High-quality genomic DNA isolation from blood/cells. |
| ddPCR Master Mix | ddPCR Supermix for Probes (no dUTP) [31] | Optimized buffer, enzymes, and dNTPs for probe-based ddPCR. |
| Restriction Enzyme | HaeIII or MseI [31] | Digests genomic DNA to reduce viscosity and improve partitioning efficiency. |
| Droplet Generation | DG8 Cartridges & Droplet Generation Oil [31] | Creates stable, monodisperse water-in-oil emulsions for partitioning. |
| Thermal Cycling | Standard 96-Well Thermal Cycler | Executes precise PCR amplification protocol. |
| Droplet Reading | QX200 Droplet Reader [31] | Measures endpoint fluorescence in each droplet. |
| Analysis Software | QuantaSoft & QuantaSoft Analysis Pro [31] | Analyzes droplet data, assigns clusters, and calculates concentrations. |
This application note provides a detailed framework for designing and implementing a robust ddPCR assay for the quantification of wild-type and Δ32 mutant CCR5 alleles. The outlined primer and probe sequences, optimized protocol, and validation steps enable researchers to achieve highly sensitive and accurate genotyping, critical for monitoring the efficacy of CCR5-targeted gene therapies and understanding the population genetics of this important HIV-resistance mutation. The absolute quantification capability of ddPCR without external standards makes it an indispensable tool for translating CCR5 research into clinical applications.
The detection of the CCR5Δ32 mutation is of significant interest in clinical research, particularly in the development of novel therapies for HIV. The 32-base pair deletion in the CCR5 gene confers natural resistance to HIV-1 infection, and its accurate quantification is essential for monitoring therapeutic interventions, such as hematopoietic stem cell transplantations and CRISPR/Cas9-based gene editing approaches [3]. Droplet Digital PCR (ddPCR) has emerged as a powerful tool for this application, enabling the precise, absolute quantification of mutant allele fractions in heterogeneous cell mixtures with a sensitivity down to 0.8% [3]. This application note details a standardized ddPCR protocol for CCR5Δ32 detection, guiding researchers through the critical phases of partitioning, thermal cycling, and endpoint fluorescence reading.
ddPCR Workflow Principles
The ddPCR workflow partitions a single PCR reaction into thousands of nanoliter-sized water-in-oil droplets, effectively creating a massive array of individual reaction vessels [9]. Following a standard PCR amplification, the fluorescence of each droplet is read in an endpoint analysis. A fundamental principle of this technology is that the random distribution of DNA molecules into partitions follows a Poisson distribution [9] [17]. The target concentration in the original sample is then calculated based on the fraction of positive (fluorescent) and negative (non-fluorescent) partitions, allowing for absolute quantification without the need for a standard curve [9] [17] [33]. This method provides high sensitivity, accuracy, and reproducibility, making it ideal for detecting rare mutations like CCR5Δ32 [9] [3].
Experimental Protocol for CCR5Δ32 Detection
Sample Preparation and DNA Extraction
- Cell Culture & Genomic DNA Extraction: The protocol was validated using the human T-cell line MT-4 [3]. Cells are cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum under standard conditions (37°C, 5% CO2). Genomic DNA is extracted using a phenol-chloroform method or commercial kits (e.g., ExtractDNA Blood and Cells Kit). DNA concentration and purity (A260/A280 ratio) should be measured using a spectrophotometer [3].
ddPCR Reaction Setup
The following table outlines the key reagents and their functions for the ddPCR assay.
Table 1: Research Reagent Solutions for CCR5Δ32 ddPCR Assay
| Reagent | Function | Final Concentration/Quantity |
|---|---|---|
| ddPCR Supermix | Provides optimized buffer, dNTPs, and hot-start DNA polymerase for robust amplification. | 1X |
| Wild-Type CCR5 Probe (e.g., HEX-labeled) | Detects the wild-type CCR5 allele. | Optimized (e.g., 250 nM) |
| CCR5Δ32 Mutant Probe (e.g., FAM-labeled) | Specifically detects the 32-bp deletion mutant allele. | Optimized (e.g., 250 nM) |
| Forward/Reverse Primers | Amplify a region flanking the CCR5Δ32 deletion. | Optimized (e.g., 900 nM each) |
| Nuclease-Free Water | Solvent to achieve the desired reaction volume. | Variable |
| Template DNA | The sample containing wild-type and/or mutant CCR5 genes. | 10-100 ng per reaction |
- Prepare the reaction mix on ice. A typical 20-22 µL reaction volume is recommended for the QX200 system [3] [34].
- Gently vortex and briefly centrifuge the mixture before droplet generation.
The Partitioning Phase
- Droplet Generation: Transfer the reaction mix to the sample well of a DG8 cartridge. Add droplet generation oil to the appropriate oil well. Place the cartridge into the droplet generator. This instrument partitions the aqueous PCR reaction into approximately 20,000 nanoliter-sized droplets [33] [35].
- Transfer: After droplet generation, carefully transfer the emulsified sample (~40 µL) to a 96-well PCR plate. Seal the plate with a foil heat seal to prevent evaporation and cross-contamination during thermal cycling. Ensure a tight seal to maintain droplet integrity.
The Thermal Cycling Phase
- Place the sealed PCR plate into a thermal cycler and run the following profile:
- Enzyme Activation: 95°C for 10 minutes.
- Amplification (40-45 cycles):
- Denaturation: 94°C for 30 seconds.
- Annealing/Extension: 55-60°C for 60 seconds (optimize temperature based on primer design).
- Enzyme Deactivation: 98°C for 10 minutes.
- Hold: 4°C ∞.
- Use a heated lid (105°C) throughout the run. After cycling, the plate can be stored at 4°C for several hours before reading.
The Endpoint Fluorescence Reading Phase
- Droplet Reading: Place the PCR plate into the droplet reader. The instrument aspirates the droplets from each well, streams them single-file past a two-color optical detection system, and classifies each droplet as FAM-positive (mutant), HEX-positive (wild-type), double-positive, or negative [35].
- Data Acquisition: The reader software counts the number of positive and negative droplets for each fluorescent channel.
Data Analysis and Interpretation
Quantification and Statistical Analysis
The ddPCR software automatically calculates the concentration of wild-type and mutant targets in copies/µL based on Poisson statistics using the formula: [ \text{Concentration} = -\ln(1 - p) / V ] where 'p' is the fraction of positive partitions and 'V' is the volume of each partition [9].
- The mutant allele frequency (in percentage) is calculated as: [ \text{Allele Frequency} = \frac{[\text{Mutant DNA}]}{[\text{Mutant DNA}] + [\text{Wild-type DNA}]} \times 100\% ]
Table 2: Typical Performance Metrics for a CCR5Δ32 ddPCR Assay
| Parameter | Performance Value | Notes |
|---|---|---|
| Limit of Detection (LOD) | As low as 0.8% mutant fraction [3] | Varies with total DNA input. |
| Precision (CV) | <5% [34] | Coefficient of Variation for replicate measurements. |
| Dynamic Range | 1 to >100,000 copies/reaction [34] | Linearity may degrade at extreme highs. |
| Partition Number | ~20,000 droplets/reaction [33] [35] | Higher counts improve precision. |
Visualization of Detection Principle
The following diagram illustrates the core principle of endpoint fluorescence detection and droplet classification in a duplex ddPCR assay for CCR5Δ32.
Droplet Digital PCR (ddPCR) represents a transformative third-generation PCR technology that enables absolute quantification of nucleic acids without the need for standard curves. This calibration-free technology provides powerful advantages including high sensitivity, absolute quantification, high accuracy and reproducibility, as well as rapid turnaround time [9]. In the context of clinical research, ddPCR has emerged as a particularly valuable tool for detecting rare genetic mutations within a background of wild-type genes, a breakthrough that paved the way for tumor heterogeneity analysis and liquid biopsy applications [9].
Within the scope of a thesis focusing on ddPCR workflow for CCR5Δ32 detection in clinical samples, understanding data analysis principles becomes paramount. The CCR5Δ32 mutation, a 32-base pair deletion in the C-C chemokine receptor type 5 gene, confers resistance to HIV infection and represents a critical therapeutic target. Recent research has demonstrated that using the modern CRISPR/Cas9 genome editing method, researchers can effectively reproduce the CCR5Δ32 mutation in any wild-type cells, creating a need for accurate quantification systems in heterogeneous cell mixtures [3]. The ddPCR system developed in recent studies allows researchers to quickly and accurately measure the content of cells with the CCR5Δ32 mutation, down to 0.8% sensitivity, making it an indispensable tool for clinical research and therapeutic development [3].
Fundamental Principles of ddPCR Data Analysis
The Partitioning Principle and Poisson Distribution
Digital PCR operates on the fundamental principle of sample partitioning, where a PCR mixture supplemented with the sample is divided into a large number of parallel reactions so that each partition contains either 0, 1, or a few nucleic acid targets according to a Poisson distribution [9]. Following PCR amplification, the fraction of positive partitions is extracted from an end-point measurement, allowing computation of the target concentration based on Poisson statistics.
The mathematical foundation of ddPCR quantification relies on the Poisson distribution, which describes the probability of a given number of events occurring in a fixed interval of time or space if these events occur with a known constant mean rate and independently of the time since the last event. In ddPCR, this translates to the random distribution of DNA molecules across thousands to millions of partitions.
ddPCR Workflow and Data Generation
The modern ddPCR protocol follows four key steps that generate the data requiring interpretation:
- Partitioning: The PCR mixture containing the sample is partitioned into thousands to millions of compartments (droplets or microchambers)
- Amplification: Individual target-containing partitions undergo PCR amplification
- Endpoint Analysis: Fluorescence of each partition is measured after amplification
- Quantification: Target concentration is computed using Poisson statistics based on the fraction of positive and negative partitions [9]
This process generates two primary types of data plots that researchers must interpret: one-dimensional (1D) amplitude plots and two-dimensional (2D) droplet plots, which form the basis for calculating mutation frequencies in clinical samples.
Interpreting 1D and 2D ddPCR Plots
One-Dimensional (1D) Amplitude Plots
One-dimensional plots in ddPCR display fluorescence amplitude for a single detection channel. These plots are particularly useful for singleplex assays or when analyzing one target at a time.
Interpretation Guidelines:
- Droplet clusters: Identify distinct populations of droplets based on fluorescence amplitude
- Threshold setting: Establish a clear threshold between negative and positive droplets
- Cluster separation: Evaluate the quality of separation between populations
- Rain effect: Identify and account for intermediate fluorescence droplets
In CCR5Δ32 detection research, 1D plots can be used to visualize wild-type versus mutant alleles when using a single fluorescent probe, though this approach provides less information than 2D plots for multiplex applications.
Two-Dimensional (2D) Droplet Plots
Two-dimensional plots represent the core of multiplex ddPCR analysis, displaying fluorescence amplitudes for two different detection channels simultaneously. These plots are essential for detecting multiple targets in a single reaction, such as distinguishing wild-type CCR5 from CCR5Δ32 mutations.
Key Populations in 2D Plots:
- Double-negative population: Droplets containing no target DNA (both channels negative)
- Single-positive populations: Droplets containing only one target type (FAM-positive/HEX-negative or HEX-positive/FAM-negative)
- Double-positive population: Droplets containing both targets simultaneously
For CCR5Δ32 detection, a well-designed assay would show four distinct clusters corresponding to: (1) empty droplets, (2) wild-type CCR5 only, (3) CCR5Δ32 only, and (4) potentially heterozygous or mixed samples.
Calculating Mutation Frequency with Poisson Correction
Poisson Statistics Fundamentals
The application of Poisson statistics is essential for accurate quantification in ddPCR because it accounts for the random distribution of molecules and the possibility of multiple targets occupying a single partition. The fundamental Poisson equation used in ddPCR is:
λ = -ln(1 - p)
Where:
- λ is the average number of target molecules per partition
- p is the fraction of positive partitions
- ln is the natural logarithm
This calculation corrects for the fact that some partitions may contain more than one target molecule, which would still register as a single positive partition, potentially leading to underestimation of the true concentration.
Mutation Frequency Calculation Protocol
Step-by-Step Calculation:
Determine positive fractions:
- pmutant = Nmutant / N_total
- pwildtype = Nwildtype / N_total
- ptotal = (Nmutant + Nwildtype + Ndouble) / N_total
Apply Poisson correction:
- λmutant = -ln(1 - pmutant)
- λwildtype = -ln(1 - pwildtype)
- λtotal = -ln(1 - ptotal)
Calculate mutation frequency:
- Mutation Frequency = λmutant / (λmutant + λ_wildtype) × 100%
Adjust for total DNA content:
- Account for input DNA mass and extraction efficiency
- Normalize to reference genes if needed
Example Calculation for CCR5Δ32 Detection: For a clinical sample with:
- 15,000 total partitions
- 2,000 mutant-positive partitions
- 10,000 wild-type-positive partitions
- 500 double-positive partitions
- 2,500 negative partitions
pmutant = (2,000 + 500) / 15,000 = 0.1667 λmutant = -ln(1 - 0.1667) = 0.1823
pwildtype = (10,000 + 500) / 15,000 = 0.7000 λwildtype = -ln(1 - 0.7000) = 1.2040
Mutation Frequency = 0.1823 / (0.1823 + 1.2040) × 100% = 13.15%
Confidence Interval Estimation
For clinical applications, it's essential to calculate confidence intervals for mutation frequency estimates:
Variance estimation:
- Var(λ) = λ / [N × (1 - p)^2]
- Where N is the total number of partitions
Standard error calculation:
- SE(λ) = √[Var(λ)]
Confidence interval:
- 95% CI = λ ± 1.96 × SE(λ)
This statistical rigor is particularly important when detecting low-frequency mutations near the detection limit, such as monitoring residual disease or early treatment response.
Essential Research Reagent Solutions
Table 1: Key Research Reagents for ddPCR-Based CCR5Δ32 Detection
| Reagent/Category | Specific Example | Function/Application |
|---|---|---|
| ddPCR Systems | QX200 Droplet Reader (Bio-Rad) [9] | Measures fluorescence of individual droplets for target quantification |
| Partitioning Technology | DG8 Cartridges and Droplet Generation Oil [9] | Creates water-in-oil emulsion droplets for sample partitioning |
| Detection Chemistry | TaqMan Probes (FAM/HEX) [36] | Target-specific fluorescent probes for multiplex detection |
| DNA Polymerase | ddPCR Supermix for Probes [3] | Optimized enzyme mix for digital PCR applications |
| gDNA Extraction | TIANamp Bacteria DNA Kit [36] | High-quality genomic DNA isolation for accurate quantification |
| Reference Assays | RNase P Reference Assay [3] | Reference gene for normalization of DNA input |
| Positive Controls | CRISPR/Cas9-edited MT-4 cells [3] | Controls with known CCR5Δ32 mutation status |
| Primer/Probe Design | ttrA/ttrC, GltS FMN-binding domain probes [36] | Target-specific reagents for mutation detection |
Comprehensive Experimental Protocol
Sample Preparation and DNA Extraction
Materials Required:
- Clinical samples (whole blood, tissue biopsies, or cellular samples)
- TIANamp Bacteria DNA Kit or equivalent [36]
- Nuclease-free water
- Spectrophotometer (NanoPhotometer P-Class P360) [3]
Procedure:
- Process clinical samples to obtain cell pellets
- Extract genomic DNA using commercial kits according to manufacturer's instructions
- Quantify DNA concentration and purity using spectrophotometry
- Assess DNA quality (A260/A280 ratio of 1.7-1.9) [36]
- Adjust DNA concentration to working dilution (10-100 ng/μL) for ddPCR reactions
ddPCR Reaction Setup and Droplet Generation
Reaction Mixture (20μL total volume):
- 10μL of 2× ddPCR Supermix for Probes
- 1.8μL of CCR5 wild-type primer/probe mix (FAM-labeled)
- 1.8μL of CCR5Δ32 primer/probe mix (HEX-labeled)
- 2μL of template DNA (10-100 ng)
- 4.4μL of nuclease-free water
Thermal Cycling Conditions:
- Enzyme activation: 10 min at 95°C
- 40 cycles of:
- Denaturation: 30 sec at 94°C
- Annealing/Extension: 60 sec at 60°C
- Enzyme deactivation: 10 min at 98°C
- Hold at 4°C
Droplet Generation and Reading:
- Generate droplets using DG8 Cartridges and Droplet Generation Oil
- Transfer droplets to 96-well PCR plate
- Seal plate with foil heat seal
- Perform PCR amplification as described above
- Read plate using QX200 Droplet Reader [9]
Data Analysis and Quality Control
Quality Assessment Parameters:
- Minimum of 10,000 droplets per sample
- Clear separation between positive and negative populations
- Limited "rain" (intermediate fluorescence droplets)
- Reference gene amplification for DNA quality control
Analysis Steps:
- Import data into analysis software (QuantaSoft)
- Set fluorescence thresholds for each channel
- Apply Poisson correction to calculate absolute copy numbers
- Calculate mutation frequency as described in Section 4
- Generate reports with confidence intervals
Troubleshooting Common Data Analysis Issues
Table 2: Troubleshooting Guide for ddPCR Data Analysis
| Problem | Potential Causes | Solutions |
|---|---|---|
| High "Rain" | Suboptimal primer/probe design, poor DNA quality, improper thermal cycling | Redesign assays, repurify DNA, optimize annealing temperature |
| Poor Cluster Separation | Probe concentration too high, spectral cross-talk, enzyme inhibitors | Titrate probe concentrations, adjust gain settings, clean DNA extraction |
| Low Droplet Count | Sample viscosity, improper droplet generation, cartridge issues | Dilute sample, ensure proper droplet generation technique, replace cartridges |
| Inconsistent Replicates | Pipetting errors, incomplete mixing, droplet coalescence | Use reverse pipetting, mix thoroughly, fresh droplet generation oil |
| Abnormal Negative Control Signal | Contamination, probe degradation, improper threshold setting | Prepare fresh reagents, store probes properly, validate threshold placement |
Applications in Clinical Research and Drug Development
The methodology described for interpreting ddPCR plots and calculating mutation frequency with Poisson correction has direct applications in clinical research and pharmaceutical development. Specifically, for CCR5Δ32 detection in clinical samples:
Therapeutic Monitoring:
- Track engraftment of CCR5Δ32-modified cells in HIV patients
- Monitor mutation frequency in heterogeneous cell populations
- Assess treatment efficacy in gene therapy approaches
Clinical Trial Applications:
- Stratify patients based on CCR5Δ32 mutation status
- Monitor minimal residual disease in hematological malignancies
- Evaluate off-target effects in gene editing therapies
The sensitivity of ddPCR to detect mutant alleles at frequencies as low as 0.8% makes it particularly valuable for monitoring low-level mutations in clinical samples, providing crucial data for drug development and personalized medicine approaches [3].
The robust data analysis framework presented here, combining precise plot interpretation with statistical rigor through Poisson correction, ensures reliable mutation frequency calculations that meet the stringent requirements of clinical research and regulatory submissions.
The C-C chemokine receptor type 5 (CCR5) serves as a critical co-receptor for human immunodeficiency virus (HIV) entry into host cells. The naturally occurring CCR5Δ32 mutation, a 32-base pair deletion, confers resistance to HIV-1 infection when homozygous. This application note demonstrates a methodology for the generation of an artificial CCR5Δ32 mutation using CRISPR/Cas9 genome editing followed by accurate quantification of mutant allele content in heterogeneous cell mixtures via multiplex droplet digital PCR (ddPCR). The developed system enables precise measurement of cells carrying the CCR5Δ32 mutation at frequencies as low as 0.8%, providing a valuable tool for monitoring transplanted or genome-edited cells in HIV therapeutic development [3] [29].
The CCR5Δ32 mutation results in a frameshift and premature stop codons, producing a non-functional receptor that prevents R5-tropic HIV-1 entry. Transplantations with CCR5Δ32/Δ32 hematopoietic stem cells have demonstrated complete cure of HIV-1 in documented cases, passing the proof-of-principle stage [3] [37] [38]. Concurrently, CRISPR/Cas9 genome editing enables introduction of this protective mutation into wild-type cells [39]. These advances create an urgent need for sensitive methods to quantify CCR5Δ32 mutant alleles in mixed cell populations for both basic research and clinical monitoring [3].
Droplet digital PCR represents an ideal platform for this application, providing absolute quantification of target sequences without calibration curves and exhibiting enhanced sensitivity for rare variant detection compared to traditional qPCR [9]. This case study details optimized experimental protocols for detecting CCR5Δ32 mutations in heterogeneous cell mixtures, enabling researchers to monitor minimal residual disease or engraftment of edited cells with exceptional precision.
Materials and Methods
Key Research Reagent Solutions
Table 1: Essential research reagents for CCR5Δ32 detection workflow
| Reagent Category | Specific Product/Description | Function in Workflow |
|---|---|---|
| Cell Culture | Roswell Park Memorial Institute medium (RPMI-1640) with 10% FBS | Maintenance of human T-cell lines (e.g., MT-4) [3] |
| Genomic DNA Isolation | Phenol-chloroform method or Commercial DNA Extraction Kits | High-quality genomic DNA preparation for downstream applications [3] |
| CRISPR/Cas9 Components | pCas9-IRES2-EGFP plasmid, pU6-gRNA vectors with CCR5-targeting gRNAs (CCR5-7: CAGAATTGATACTGACTGTATGG, CCR5-8: AGATGACTATCTTTAATGTCTGG) | Introduction of CCR5Δ32 mutation via genome editing [3] |
| ddPCR Master Mix | ddPCR Supermix for Probes | Optimized reaction mixture for droplet digital PCR [40] |
| Fluorescent Probes & Primers | Target-specific primers and FAM/VIC-labeled TaqMan MGB probes | Multiplex detection of wild-type and CCR5Δ32 alleles [41] |
| Droplet Generation | Droplet Generator Cartridges and Gaskets | Creation of nanoliter-sized reaction partitions [40] |
Cell Culture and Genomic DNA Extraction
The MT-4 human T-cell line was maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum at 37°C in a 5% CO₂ humidified incubator [3]. Genomic DNA was extracted using either:
- Phenol-chloroform method followed by ethanol precipitation
- Commercial extraction kits (e.g., ExtractDNA Blood and Cells Kit)
DNA concentration and purity were assessed using spectrophotometry (NanoPhotometer P-Class P360), with optimal A260/A280 ratios of 1.8-2.0 indicating minimal protein contamination [3].
CRISPR/Cas9 Genome Editing for CCR5Δ32 Generation
To create an artificial CCR5Δ32 mutation:
gRNA preparation: Two CCR5-specific gRNAs (CCR5-7 and CCR5-8) were designed based on previously published sequences [3]. Oligonucleotides were annealed, phosphorylated with T4 polynucleotide kinase, and cloned into the BsmBI-digested pU6-gRNA vector using T7 DNA ligase.
Plasmid verification: Successful cloning was confirmed by Sanger sequencing of the constructed plasmids.
Electroporation: 6 × 10⁶ MT-4 cells were co-electroporated with 10 μg of pCas9-IRES2-EGFP plasmid and 5 μg each of pU6-gRNA-CCR5-7 and pU6-gRNA-CCR5-8 using a Gene Pulser Xcell system (settings: 275 V, 5 ms, three pulses) [3].
Cell sorting and cloning: After 48 hours, EGFP-positive cells were isolated using fluorescence-activated cell sorting (FACS). Cells were subsequently cloned by limiting dilution into 96-well plates to generate monoclonal cell lines.
Mutation screening: Monoclonal expansions were screened for CCR5Δ32 alleles using PCR amplification of the target region (primers: forward CCCAGGAATCATCTTTACCA and reverse GACACCGAAGCAGAGTTT) followed by TA-cloning and sequencing [3].
Multiplex Droplet Digital PCR Analysis
Reaction Setup
The ddPCR reaction mixture was assembled with the following components [3] [40]:
- 1× ddPCR Supermix for Probes
- Primers and probes at optimized concentrations (typically 900 nM primers, 250 nM probes)
- 5 μL template DNA (approximately 50-100 ng)
- Nuclease-free water to a final volume of 20-22 μL
Table 2: Optimal ddPCR assay parameters for CCR5Δ32 detection
| Parameter | Wild-Type Allele Detection | CCR5Δ32 Mutant Detection | Purpose |
|---|---|---|---|
| Fluorophore | FAM-labeled probe | VIC/HEX-labeled probe | Multiplex discrimination |
| Probe Binding Site | Sequence spanning deletion region | Junction created by 32-bp deletion | Specific mutation detection |
| Annealing/Extension Temperature | 56-60°C (optimized gradient) | 56-60°C (optimized gradient) | Enhanced specificity |
| Oligonucleotide Concentration | Standard (per validated protocol) or High (900 nM primers, 250 nM probes) | Standard (per validated protocol) or High (900 nM primers, 250 nM probes) | Signal optimization |
Droplet Generation and PCR Amplification
- 20 μL of the reaction mixture was loaded into droplet generator cartridges
- Droplets were generated using the QX200 Droplet Generator
- Emulsified samples were transferred to a 96-well PCR plate
- PCR amplification was performed with the following cycling conditions [3] [40]:
- Enzyme activation: 95°C for 10 minutes
- 40 cycles of:
- Denaturation: 95°C for 30 seconds
- Annealing/Extension: 56-60°C for 60 seconds (temperature optimized via gradient)
- Enzyme deactivation: 98°C for 10 minutes
- Hold: 4°C
Data Acquisition and Analysis
- Droplets were read using the QX200 Droplet Reader
- Analysis of fluorescence amplitude data was performed using QuantaSoft software
- Threshold setting between positive and negative droplets was guided by control samples (wild-type only and mutant only)
- Target concentration was calculated using Poisson statistics based on the fraction of positive droplets [9]
Results and Discussion
Sensitivity and Accuracy of CCR5Δ32 Detection
The optimized ddPCR assay demonstrated robust detection of CCR5Δ32 mutant alleles with sensitivity down to 0.8% in heterogeneous cell mixtures [3] [29]. This exceptional sensitivity enables reliable monitoring of rare mutant cells in complex biological samples, a critical requirement for evaluating the efficacy of stem cell transplantation or genome editing therapies.
Key performance characteristics include:
- Precise absolute quantification without requirement for standard curves
- High partitioning efficiency (>10,000 droplets per reaction) enabling rare allele detection
- Minimal "rain" (intermediate fluorescence droplets) through optimized thermal cycling conditions and oligonucleotide concentrations
- Excellent signal separation between wild-type and mutant clusters due to optimized probe design
Technical Optimization Strategies
Several parameters critically influenced assay performance and required optimization:
Annealing temperature: Testing a gradient (56-60°C) identified the optimal temperature for maximal fluorescence separation and minimal rain [40].
Oligonucleotide concentrations: Increased primer (900 nM) and probe (250 nM) concentrations enhanced fluorescence amplitude compared to standard qPCR concentrations [40].
Probe selection: TaqMan MGB probes provided superior mismatch discrimination compared to conventional probes, crucial for distinguishing the highly similar wild-type and Δ32 sequences [41].
Droplet separation value: An objective metric incorporating both absolute fluorescence signal distance and population variation enabled quantitative assessment of assay performance and guided optimization [40].
Application in HIV Cure Strategies
The ability to precisely quantify CCR5Δ32 alleles addresses a critical need in developing HIV curative strategies. Following allogeneic hematopoietic stem-cell transplantation with CCR5Δ32/Δ32 cells, this ddPCR assay enables:
- Monitoring engraftment of donor cells in the recipient
- Tracking mutant cell expansion during immune reconstitution
- Detecting minimal residual wild-type cells that could potentially support viral replication
- Evaluating the efficacy of CCR5Δ32/Δ32 stem-cell transplantation for achieving HIV-1 remission [38]
For CRISPR-edited cell products, the method provides quality control assessment of editing efficiency and stability of the mutation in expanded cell populations.
This application note details a robust methodology for sensitive detection and quantification of CCR5Δ32 mutant alleles in heterogeneous cell mixtures using droplet digital PCR. The described protocols enable researchers to reliably monitor mutant frequencies as low as 0.8%, providing a valuable tool for advancing HIV cure strategies based on CCR5 disruption. The optimized ddPCR approach offers superior sensitivity and absolute quantification compared to traditional methods, making it particularly suitable for tracking rare genome-edited cells or monitoring engraftment following transplantation with CCR5Δ32/Δ32 stem cells.
The integration of CRISPR/Cas9 for generating reference materials and ddPCR for detection creates a powerful synergy that accelerates the development of CCR5-targeted therapies, ultimately contributing to the quest for an HIV cure.
Maximizing Sensitivity and Specificity: ddPCR Troubleshooting and Optimization
Droplet Digital PCR (ddPCR) represents a third-generation PCR technology that enables absolute quantification of nucleic acids without the need for a standard curve [17] [42]. This calibration-free approach partitions a PCR reaction into thousands of nanoliter-sized droplets, effectively creating individual microreactors where amplification occurs. Following end-point thermal cycling, the system counts positive and negative droplets to provide absolute target quantification based on Poisson statistics [17] [9]. For clinical research applications involving CCR5Δ32 mutation detection, ddPCR offers significant advantages in sensitivity and precision, allowing researchers to accurately quantify the proportion of edited cells in heterogeneous mixtures with demonstrated sensitivity down to 0.8% [28]. This technical note provides detailed protocols and optimization strategies for calculating copy number and avoiding over-partitioning in ddPCR workflows specifically tailored for CCR5Δ32 detection in clinical samples.
Theoretical Foundations of Sample Input Optimization
Principles of Partitioning and Poisson Statistics
The fundamental principle underlying ddPCR quantification is that nucleic acid molecules are randomly distributed into partitions according to Poisson statistics. The relationship between the fraction of positive partitions and the initial target concentration is defined by the equation λ = -ln(1-p), where λ represents the average number of target molecules per partition and p is the fraction of positive partitions [42]. This statistical foundation allows for absolute quantification without external calibrators. The precision of ddPCR quantification depends directly on the number of partitions analyzed, with confidence intervals that can be calculated using methods such as the Wilson score interval to account for the binomial nature of the data [42].
The dynamic range of ddPCR is constrained by the total number of partitions available. Commercial ddPCR systems typically generate between 10,000 and 20,000 droplets per reaction, though this number can vary between platforms [17] [43]. This finite number of partitions establishes practical boundaries for optimal target quantification, making understanding of Poisson distribution parameters essential for experimental design.
Defining Optimal Partition Occupancy
Optimal quantification in ddPCR requires careful consideration of partition occupancy to maximize precision while avoiding issues associated with over-partitioning or under-partitioning. Theoretical and empirical studies have demonstrated that precision is maximized when approximately 20% of partitions remain negative (λ ≈ 1.6) [42]. At this optimal occupancy, the confidence interval for concentration estimation is minimized relative to the number of partitions available.
Table 1: Guidelines for Optimal Target Concentration Ranges in ddPCR
| Partition Number | Optimal Target Range (Copies/Reaction) | Minimum Detectable Copies | Saturation Threshold |
|---|---|---|---|
| 10,000 | 8,000-16,000 | 3 | >40,000 |
| 15,000 | 12,000-24,000 | 3 | >60,000 |
| 20,000 | 16,000-32,000 | 3 | >80,000 |
Under-partitioning occurs when target concentrations are too high, leading to multiple target molecules occupying individual partitions. This saturation effect compromises quantification accuracy as the relationship between positive partitions and target concentration becomes non-linear [42]. Conversely, while less problematic for accuracy, over-partitioning (excessively dilute samples) reduces measurement precision and increases the relative impact of background signals.
Practical Implementation for CCR5Δ32 Detection
Sample Input Calculation Protocol
Implementing optimal sample input for CCR5Δ32 detection requires a systematic approach to template quantification and dilution. The following step-by-step protocol ensures accurate copy number calculation and appropriate partitioning:
Initial Template Quantification: Pre-quantify genomic DNA (gDNA) samples using fluorometric methods (e.g., Qubit dsDNA BR Assay) to determine total DNA concentration [28] [23]. Assess purity via spectrophotometric ratios (A260/A280 ≈ 1.8-2.0).
Copy Number Calculation: Calculate the theoretical diploid genome copy number using the formula: [ \text{Genome copies/μL} = \frac{\text{DNA concentration (ng/μL)} \times 6.022 \times 10^{23}}{(\text{Genome size in bp}) \times (1 \times 10^9) \times 650} ] For human gDNA (∼3.3 × 10^9 bp), this simplifies to approximately 91.5 diploid genome copies per ng of gDNA [43].
Dilution Factor Determination: Based on the optimal λ value of 1.6 and the partition volume of your ddPCR system, calculate the appropriate dilution factor. For a system generating 20,000 droplets of 1 nL each (total reaction volume = 20 μL): [ \text{Optimal copies/μL} = \frac{1.6}{\text{Partition volume in μL}} ] [ \text{Dilution Factor} = \frac{\text{Initial copies/μL}}{\text{Optimal copies/μL}} ]
Sample Dilution: Prepare diluted gDNA samples in nuclease-free water or TE buffer to achieve the calculated target concentration. Include both positive controls (CCR5Δ32 homozygous DNA) and negative controls (wild-type DNA) [28] [23].
CCR5Δ32-Specific Optimization Considerations
Detection of the CCR5Δ32 mutation presents specific challenges that require optimization of sample input parameters. The 32-base pair deletion in the CCR5 gene creates a distinct target that can be discriminated from wild-type sequences using specific probe systems [28]. Research demonstrates successful CCR5Δ32 quantification in heterogeneous cell mixtures with sensitivity to 0.8% mutant alleles, highlighting the importance of optimal sample input for rare allele detection [28].
When designing ddPCR assays for CCR5Δ32, implement a duplex reaction that simultaneously targets both wild-type and mutant alleles using different fluorescent probes (e.g., FAM for mutant, HEX/VIC for wild-type) [28] [23]. This approach enables direct calculation of the mutation frequency from a single reaction, reducing technical variability. For clinical samples with limited material, such as patient-derived cells, the high tolerance of ddPCR to inhibitors allows for direct analysis without extensive purification [42] [43].
Table 2: Research Reagent Solutions for CCR5Δ32 ddPCR
| Reagent Category | Specific Product | Function in Workflow |
|---|---|---|
| Nucleic Acid Isolation | QIAamp DNA Blood Mini Kit [23] | High-quality gDNA extraction from blood and cells |
| Quantification | Qubit dsDNA BR Assay Kit [23] | Accurate DNA concentration measurement |
| ddPCR Supermix | ddPCR Supermix for Probes (Bio-Rad) | Optimized reaction buffer for droplet generation |
| CCR5 Probes | FAM-labeled CCR5Δ32 probe, HEX/VIC-labeled wild-type CCR5 probe [28] | Mutation-specific detection in duplex assay |
| Droplet Generation Oil | DG8 Cartridges and Droplet Generation Oil | Stable water-in-oil emulsion formation |
| Reference Assay | RPP30 Reference Assay [43] | Diploid copy number control for normalization |
Experimental Protocols
Complete ddPCR Workflow for CCR5Δ32 Quantification
Figure 1: Complete ddPCR workflow for CCR5Δ32 detection, highlighting critical optimization points for sample input and partitioning.
Detailed Step-by-Step Protocol
Reaction Setup:
- Prepare a 20-22 μL reaction mix containing:
- 10 μL of 2× ddPCR Supermix for Probes
- 1 μL of CCR5Δ32 FAM-labeled assay (final concentration: 900 nM primers, 250 nM probe)
- 1 μL of wild-type CCR5 HEX-labeled assay (final concentration: 900 nM primers, 250 nM probe)
- X μL of template DNA (optimally 10-100 ng, calculated for λ ≈ 1.6)
- Nuclease-free water to final volume
- Gently mix by pipetting, avoiding bubble formation [28] [23].
- Prepare a 20-22 μL reaction mix containing:
Droplet Generation:
- Transfer 20 μL of the reaction mix to the sample well of a DG8 cartridge.
- Add 70 μL of Droplet Generation Oil to the oil well.
- Place the gasket on the cartridge and load into the droplet generator.
- Once droplet generation is complete (approximately 1-2 minutes), carefully transfer the emulsified sample to a 96-well PCR plate [43].
PCR Amplification:
- Seal the plate with a pierceable foil heat seal.
- Perform amplification using the following thermal cycling conditions:
Droplet Reading and Analysis:
- Place the PCR plate in the droplet reader.
- Analyze droplets using the appropriate channel settings (FAM for CCR5Δ32, HEX/VIC for wild-type CCR5).
- Set the fluorescence threshold using positive and negative controls to distinguish positive from negative droplets.
- Export data for Poisson analysis using the instrument software [28] [43].
Data Analysis and Interpretation
Following droplet reading, analyze data using the instrument's proprietary software (e.g., QuantaSoft for Bio-Rad systems). The concentration of wild-type and mutant targets will be automatically calculated based on Poisson statistics. Calculate the CCR5Δ32 allele frequency using the formula:
[ \text{CCR5Δ32 Frequency (\%)} = \frac{[\text{CCR5Δ32}]}{[\text{CCR5Δ32}] + [\text{Wild-type CCR5}]} \times 100 ]
For absolute quantification of edited cells in a population, apply the following calculations based on binomial distribution principles:
- Heterozygous editing rate = (CCR5Δ32 concentration / Total CCR5 concentration) × 100
- Biallelic editing rate = (CCR5Δ32 concentration)^2 / (Total CCR5 concentration)^2 × 100 [23]
Figure 2: Troubleshooting guide for partitioning issues in ddPCR experiments, highlighting pathways to optimal detection sensitivity and quantification accuracy.
Optimal sample input calculation and avoidance of over-partitioning are critical success factors in ddPCR-based CCR5Δ32 detection. By applying Poisson distribution principles to target approximately 1.6 copies per partition (λ = 1.6), researchers can maximize quantification precision while maintaining sensitivity for rare mutant alleles. The protocols and guidelines presented here provide a framework for implementing robust ddPCR assays in HIV immunotherapy research, enabling accurate monitoring of CCR5 gene editing efficiencies in clinical samples. As ddPCR technology continues to evolve with increased partition numbers and multiplexing capabilities, these fundamental principles of sample optimization will remain essential for generating reliable, reproducible data in both research and clinical settings.
The accuracy of a Droplet Digital PCR (ddPCR) workflow for the sensitive detection of the CCR5Δ32 mutation in clinical samples is critically dependent on sample purity. PCR inhibitors are substances that co-extract with nucleic acids and can significantly impair the efficiency of the amplification reaction, leading to false-negative results or inaccurate quantification. In the context of clinical research, particularly for applications such as quantifying the frequency of CCR5Δ32 mutant alleles in heterogeneous cell mixtures for HIV cure research, these inhibitors pose a substantial challenge [3]. Common sources of inhibitors in human clinical samples include hemoglobin from blood, immunoglobulins, urea, bile salts, and complex polysaccharides from tissues [44] [8]. These compounds can interfere with the PCR reaction through various mechanisms, such as inhibiting the DNA polymerase, chelating essential metal ions like Mg²⁺, or binding directly to the nucleic acids, preventing their denaturation or primer annealing [44].
While ddPCR is generally more tolerant of inhibitors than traditional quantitative PCR (qPCR) due to the partitioning of the reaction mixture into thousands of nanoliter-sized droplets, it is not immune to their effects [45] [11]. Partitioning can effectively dilute inhibitors in some droplets, allowing amplification to proceed in those compartments. However, at high concentrations, inhibitors can still cause a significant underestimation of the target concentration and reduce the assay's sensitivity [11]. Therefore, robust strategies for identifying and mitigating PCR inhibition are a non-negotiable step in developing a reliable ddPCR protocol for critical clinical research applications.
Identifying PCR Inhibition in ddPCR Assays
Recognizing the signs of inhibition in a ddPCR experiment is the first step toward remediation. The following indicators, accessible through standard ddPCR software output, can signal the presence of interfering substances:
- Reduced Amplitude Separation: A clear, distinct cluster of positive droplets is a hallmark of a robust ddPCR assay. Inhibitors often cause a decrease in the fluorescence amplitude of positive droplets, leading to a less distinct separation between positive and negative populations. This can manifest as a "shift" of the positive cluster toward the negative cluster or an increase in the spread (width) of the positive cloud.
- Decreased Total Droplet Count: A lower-than-expected number of generated droplets can indicate issues with the droplet generation step, which viscous or surfactant-like inhibitors can disrupt.
- Abnormal Cluster Patterns: The presence of "rain" (a scattering of droplets between the clear negative and positive clusters) can be a symptom of partial inhibition, where amplification is inefficient and inconsistent across droplets.
It is crucial to include appropriate controls in every run. A no-template control (NTC) checks for contamination, while a positive control with a known copy number of the target sequence helps assess overall assay performance. When testing new sample types, a "spike-in" control is highly recommended. This involves adding a known quantity of a synthetic control template or DNA from a different species to the sample reaction. A significant drop in the measured concentration of the spike-in control in the sample compared to a clean background (e.g., nuclease-free water) provides direct evidence of inhibition [11].
Strategies for Mitigating PCR Inhibition
A multi-faceted approach is most effective for overcoming PCR inhibition. The strategies below can be employed individually or in combination, with the optimal choice often depending on the specific sample type and inhibitor.
Sample Preparation and Purification
The most effective strategy is to prevent inhibitors from entering the PCR reaction through optimized nucleic acid extraction.
- Inhibitor Removal Kits: Several commercially available kits are specifically designed to remove common inhibitors (e.g., polyphenolic compounds, humic acids, tannins) from complex matrices. These kits often use a column matrix with a specialized chemistry that binds impurities while allowing DNA to pass through [44].
- Modified Extraction Protocols: Techniques such as silica-based membrane purification in the presence of chaotropic salts are highly effective. For particularly challenging samples, incorporating a pre-wash step or using a kit optimized for the specific sample type (e.g., blood, stool, formalin-fixed tissues) is advisable.
Dilution of Nucleic Acid Extracts
A simple and effective first-line approach is to dilute the extracted DNA. This dilutes the inhibitors to a sub-critical concentration while ideally retaining enough target molecules for detection. However, this method must be used judiciously, as excessive dilution can push the target concentration below the limit of detection, particularly for rare targets like low-frequency CCR5Δ32 alleles [44].
PCR Enhancers and Additives
The addition of specific compounds to the PCR master mix can counteract the effect of inhibitors. These enhancers work through various mechanisms, such as stabilizing the polymerase, competing with the template for inhibitor binding, or altering DNA melting behavior. The following table summarizes the most common and effective PCR enhancers based on recent studies:
Table 1: Efficacy of Common PCR Enhancers in Mitigating Inhibition
| Enhancer | Common Working Concentration | Proposed Mechanism of Action | Effectiveness & Notes |
|---|---|---|---|
| Bovine Serum Albumin (BSA) | 0.1 - 1.0 μg/μL | Binds to and neutralizes inhibitors like phenolics and humic acids [44]. | Highly effective for a wide range of inhibitors; a versatile first-choice additive. |
| T4 Gene 32 Protein (gp32) | 0.1 - 0.5 μM | Binds single-stranded DNA, preventing denaturation and polymerase stalling; binds humic acids [44]. | Shows high efficacy, particularly in complex environmental and plant samples [44]. |
| Dimethyl Sulfoxide (DMSO) | 1 - 5% (v/v) | Lowers DNA melting temperature (Tm), destabilizes secondary structures [44]. | Effective but requires optimization of annealing temperature; can be toxic to polymerase at high concentrations. |
| Formamide | 1 - 5% (v/v) | Similar to DMSO, lowers Tm and destabilizes DNA helix [44]. | Can improve specificity and reduce inhibition. |
| TWEEN-20 | 0.1 - 1% (v/v) | Non-ionic detergent that counteracts inhibitory effects on Taq DNA polymerase [44]. | Useful for inhibitors found in fecal and wastewater samples. |
| Glycerol | 1 - 10% (v/v) | Stabilizes enzymes, protecting them from denaturation [44]. | Improves efficiency and specificity of PCR. |
ddPCR-Specific Optimizations
Leveraging the inherent advantages of the ddPCR platform itself provides another powerful mitigation strategy.
- Platform Selection: The compartmentalization in ddPCR effectively reduces the inhibitor concentration in each individual droplet. This makes ddPCR fundamentally more tolerant to inhibitors than qPCR, a fact confirmed by studies on complex plant [45] and environmental samples [11]. One study on grapevine Bois noir phytoplasma detection found that while qPCR was inhibited in root samples, ddPCR was not affected, allowing for reliable detection [45].
- Chemistry Choice: Both TaqMan probe-based and EvaGreen dye-based ddPCR assays are widely used. While TaqMan probes offer higher specificity, EvaGreen chemistry can be a cost-effective alternative, and its performance in ddPCR is less affected by non-specific amplification due to partitioning [45].
Experimental Protocol: Evaluation of PCR Enhancers for CCR5Δ32 ddPCR
This protocol provides a step-by-step method for systematically testing the efficacy of different PCR enhancers in a CCR5Δ32 ddPCR assay, using spiked clinical samples as a model for inhibitor presence.
Materials and Equipment
Table 2: Research Reagent Solutions for Inhibitor Mitigation
| Reagent / Equipment | Function / Application | Example / Specification |
|---|---|---|
| ddPCR Supermix | Provides optimized buffers, nucleotides, and polymerase for the partitioned reaction. | ddPCR Supermix for Probes (No dUTP) or QX200 ddPCR EvaGreen Supermix (Bio-Rad) [45] [11]. |
| PCR Enhancers | Counteract the effect of inhibitors in the reaction mix. | BSA, gp32, DMSO, Formamide, TWEEN-20, Glycerol (molecular biology grade) [44]. |
| Primers & Probes | For specific amplification and detection of wild-type CCR5 and CCR5Δ32 alleles. | Validated assays for duplex ddPCR [3] [23]. |
| Droplet Generator | Partitions the PCR reaction into ~20,000 nanoliter-sized droplets. | QX200 Droplet Generator (Bio-Rad) [3] [11]. |
| Droplet Reader | Performs fluorescence detection of each droplet post-PCR. | QX200 Droplet Reader (Bio-Rad) [3] [23]. |
| Inhibitor-Rich Matrix | A representative clinical sample known to contain PCR inhibitors. | Extracted DNA from whole blood, stool, or tissue samples. |
Step-by-Step Procedure
Sample Preparation and Spiking:
- Obtain DNA extracted from a challenging clinical matrix (e.g., whole blood).
- Create a reference standard by mixing wild-type genomic DNA with DNA from a cell line heterozygous or homozygous for the CCR5Δ32 mutation (e.g., an edited MT-4 cell line [3]) to a known ratio (e.g., 5% mutant allele frequency).
- Aliquot this standard mixture and spike it into the test clinical DNA samples.
ddPCR Reaction Setup with Enhancers:
- Prepare separate master mixes for each enhancer to be tested (see Table 1 for concentrations). A negative control (nuclease-free water) and a positive control (reference standard in clean buffer) must be included.
- Example Master Mix for a TaqMan Probe Assay (22 μL final volume):
- 11 μL of 2x ddPCR Supermix for Probes
- Forward and Reverse Primer (CCR5 wt and CCR5Δ32-specific), optimal concentration (e.g., 900 nM each)
- FAM-labeled probe (for CCR5Δ32) and HEX-labeled probe (for wild-type CCR5), optimal concentration (e.g., 250 nM each) [3]
- PCR enhancer (e.g., 1 μg/μL BSA)
- Nuclease-free water to 20 μL
- Add 2 μL of the spiked DNA sample (or control) to each mix.
Droplet Generation and PCR Amplification:
- Following the manufacturer's instructions, load the reaction mixes and droplet generation oil into the DG8 cartridges of the droplet generator.
- After generation, carefully transfer the emulsion (~40 μL) to a 96-well PCR plate and seal it.
- Perform PCR amplification on a thermal cycler using a validated protocol. An example cycling condition is:
- 95°C for 10 minutes (enzyme activation)
- 40 cycles of: 94°C for 30 seconds (denaturation) and 55-60°C for 60 seconds (annealing/extension) [3]
- 98°C for 10 minutes (enzyme deactivation)
- 4°C hold.
Droplet Reading and Data Analysis:
- Place the plate in the droplet reader for fluorescence measurement in each droplet.
- Analyze the data using the instrument's software (e.g., QuantaSoft). The software will apply Poisson statistics to calculate the absolute concentration (copies/μL) of both wild-type and mutant alleles in each reaction.
- Key Metrics for Evaluation:
- Measured Mutant Allele Frequency: Compare the calculated frequency to the known 5% spike-in value. The condition closest to 5% is the most effective.
- Total Copy Number: The sum of wild-type and mutant concentrations should be consistent across reactions unless the enhancer itself is inhibitory.
- Cluster Separation: Visually assess the quality of the 2D amplitude plot for clear separation of four droplet populations (double-negative, FAM-positive, HEX-positive, double-positive).
The workflow for this systematic evaluation is summarized in the following diagram:
Managing PCR inhibitors is not merely a troubleshooting exercise but a fundamental component of a robust ddPCR assay for sensitive clinical applications like CCR5Δ32 detection. A systematic approach is paramount. We recommend:
- Prioritize Purification: Begin with a high-quality, inhibitor-removing nucleic acid extraction kit validated for your specific sample type.
- Validate with Spike-Ins: Always use a spike-in control when working with new sample batches to diagnostically check for inhibition.
- Empirically Test Enhancers: If inhibition is detected, empirically test a panel of enhancers. BSA and T4 gp32 are excellent starting points due to their broad efficacy [44].
- Consider Dilution: If the target concentration is high enough, a simple dilution of the DNA extract (e.g., 1:5 or 1:10) can be a very effective and straightforward solution.
- Leverage ddPCR's Robustness: Remember that the ddPCR platform itself is more resilient to inhibition than qPCR. In many cases, optimization of the sample preparation and reaction mix alone will yield highly accurate and precise results for quantifying CCR5Δ32 mutant alleles, even in complex clinical samples [3] [45].
By integrating these strategies into the standard ddPCR workflow, researchers can ensure the integrity of their data, which is crucial for advancing clinical research in areas such as HIV cure strategies utilizing CCR5Δ32-modified cells.
The detection of the CCR5Δ32 mutation, a 32-base pair deletion in the C-C chemokine receptor type 5 gene, represents a critical tool in HIV research and potential cure strategies [3]. The analysis of this mutation in clinical samples using droplet digital PCR (ddPCR) requires the highest level of precision and sensitivity, as it often involves quantifying rare mutant alleles within a vast background of wild-type genomic DNA (gDNA) [3] [9]. The integrity and preparation of the gDNA template are foundational to assay success. Restriction enzyme digestion of gDNA serves as a powerful preparatory step to overcome challenges posed by the complex, heterogeneous nature of genomic templates, ensuring that the subsequent ddPCR assay achieves the accuracy required for reliable clinical interpretation [46].
The Scientific Rationale: Why Digest gDNA for ddPCR?
Droplet digital PCR operates by partitioning a PCR mixture into thousands of nanoliter-sized droplets, each functioning as an individual amplification reactor [9]. The absolute quantification of targets, such as the CCR5Δ32 allele, is then determined by counting positive and negative droplets and applying Poisson statistics [9]. The use of restriction-digested gDNA directly enhances this process by addressing several key challenges:
- Reducing Sample Complexity: Genomic DNA is a long, entangled polymer. Restriction digestion cuts this polymer into smaller, more manageable fragments. This prevents the co-partitioning of multiple target molecules into a single droplet, which can lead to underestimation of the target concentration [46].
- Improving Amplification Efficiency: Smaller, linearized DNA fragments are more accessible to PCR primers and polymerase than large, supercoiled gDNA. This can lead to more consistent and efficient amplification within each droplet, reducing the number of failed or delayed amplifications and improving the clarity of the positive/negative endpoint readout [46].
- Ensuring Uniform Template Distribution: Digesting gDNA into a more homogeneous population of fragments promotes the random distribution of targets across droplets according to a Poisson distribution, a fundamental assumption for accurate ddPCR quantification [9].
Table 1: Benefits of Restriction Digestion for gDNA in ddPCR Workflows
| Challenge of Intact gDNA | Effect of Restriction Digestion | Impact on ddPCR Assay |
|---|---|---|
| Large molecular size & complexity | Generates smaller, discrete fragments | Reduces target co-partitioning; improves quantification accuracy |
| Variable secondary structures | Linearizes DNA templates | Enhances PCR efficiency and consistency across droplets |
| Non-homogeneous template mix | Creates a more uniform population of molecules | Ensures random distribution critical for Poisson statistics |
Protocol: Restriction Enzyme Digestion of gDNA for CCR5Δ32 ddPCR
This protocol is optimized for the preparation of human gDNA prior to ddPCR analysis of the CCR5Δ32 locus.
Reagents and Equipment
Table 2: Essential Reagents and Materials for gDNA Restriction Digest
| Item | Function/Description | Example/Note |
|---|---|---|
| Genomic DNA | The template for analysis. | Isolated from whole blood or PBMCs; quantify via spectrophotometry. |
| Restriction Enzyme(s) | Enzyme that cleaves DNA at specific recognition sequences. | Select an enzyme that does not cut within the CCR5Δ32 amplicon (e.g., HindIII, EcoRI). |
| 10x Reaction Buffer | Provides optimal conditions (pH, salt) for enzyme activity. | Use the buffer recommended by the enzyme manufacturer. |
| Molecular Biology Grade Water | Nuclease-free water to bring reaction to final volume. | Ensures no enzymatic degradation of the reaction components. |
| Thermal Cycler or Heated Block | For precise incubation at the enzyme's optimal temperature. | Typically 37°C for most common restriction enzymes. |
Step-by-Step Procedure
- Select Restriction Enzymes: Choose one or more enzymes that recognize 6-base pair or longer sequences to generate appropriately sized fragments (500 bp to 5 kbp is often suitable). Critically, the enzyme must not have a recognition site within the region targeted by your CCR5Δ32 ddPCR assay [46]. In silico analysis of the CCR5 gene sequence is mandatory.
- Prepare the Reaction Mixture: In a sterile, nuclease-free microcentrifuge tube, combine the components in the following order. It is recommended to create a Master Mix when digesting multiple samples [47].
1 µgGenomic DNA5 µL10x Reaction Buffer1 µL(10-20 units) of each Restriction Enzyme- Nuclease-free water to a final volume of
50 µL
- Incubate: Mix the reaction gently by pipetting and briefly centrifuging. Incubate the tube at the enzyme's optimal temperature (commonly 37°C) for 4 hours to overnight. Longer incubation times are recommended for complete digestion of complex gDNA, especially when using a single enzyme [47].
- Enzyme Inactivation: After digestion, the restriction enzyme(s) can be inactivated by heating at 70°C for 15 minutes [47]. Alternatively, the digested DNA can be purified using a DNA clean-up kit according to the manufacturer's instructions. Purification is often preferred for ddPCR to remove enzymes, salts, and buffers that might interfere with the subsequent partitioning or amplification steps.
- Quality Control: Analyze an aliquot (e.g., 100 ng) of the digested and purified DNA by agarose gel electrophoresis. A successful digest should show a smear of DNA fragments, confirming the reduction in genomic DNA size. The undigested control gDNA should appear as a high-molecular-weight band.
Integration with the CCR5Δ32 ddPCR Workflow
The restriction-digested gDNA is now an optimized template for the ddPCR assay. In the study by PMC8898955, a similar approach using CRISPR/Cas9 to generate the CCR5Δ32 mutation was followed by multiplex ddPCR to accurately quantify the mutant allele content in mixed cell populations, achieving sensitivity down to 0.8% [3]. This level of precision in detecting minor allele fractions is contingent on proper sample preparation, where restriction digestion mitigates the risk of assay failure due to inefficient amplification of large genomic fragments. The digested DNA is used directly in the ddPCR reaction mix, which contains primers and probes specific to the wild-type CCR5 sequence and the Δ32 deletion, enabling the absolute quantification of both alleles in a single, highly sensitive reaction [3] [9].
Table 3: Troubleshooting Common Issues in gDNA Restriction Digest for ddPCR
| Problem | Potential Cause | Solution |
|---|---|---|
| Incomplete Digestion | Insufficient enzyme units or incubation time. | Increase enzyme concentration; extend incubation time to overnight. |
| No Digestion | Enzyme inhibited; incorrect buffer; DNA is methylated. | Ensure DNA is clean; use correct buffer; check for Dam/Dcm methylation sensitivity of the enzyme [47]. |
| Unexpected Banding Pattern | Star activity (non-specific cutting). | Avoid high glycerol concentrations; use high-fidelity (HF) enzymes; do not exceed recommended reaction volume [47]. |
| Poor ddPCR Amplification | Enzyme or buffer carryover inhibits PCR. | Implement a DNA purification step post-digestion instead of heat inactivation alone. |
The accurate detection of the CCR5Δ32 mutation is a critical component of advanced therapeutic strategies for HIV, including stem cell transplantation and gene therapy approaches. Within these workflows, droplet digital PCR (ddPCR) has emerged as a powerful tool for the precise quantification of mutant alleles in heterogeneous cell mixtures, capable of detecting mutant allele content as low as 0.8% [3]. The reliability of this sensitive detection, however, is profoundly dependent on the optimal performance of the primers and probes used in the assay. Sub-optimal reagent concentrations, improper storage conditions, and significant background noise can severely compromise data quality, leading to insufficient separation between positive and negative droplet populations and hindering accurate threshold setting [40] [48]. This application note provides detailed protocols and data-driven recommendations for optimizing primer and probe usage within a ddPCR workflow tailored for CCR5Δ32 detection in clinical samples, ensuring that researchers can achieve the highest standards of accuracy and sensitivity required for drug development and clinical research.
The Scientist's Toolkit: Research Reagent Solutions
The following table details key reagents and their optimized roles within the CCR5Δ32 ddPCR assay, as drawn from established protocols.
Table 1: Essential Research Reagents for CCR5Δ32 ddPCR
| Reagent | Function & Explanation | Key Considerations |
|---|---|---|
| ddPCR Supermix for Probes | Provides the core biochemical environment for amplification, optimized for probe-based assays in partitioned droplets [40] [49]. | Essential for robust signal generation. Use the version specified by the manufacturer of your ddPCR system. |
| Primers (Wild-type & Mutant-specific) | Amplify the target CCR5 locus and specifically discriminate the wild-type from the Δ32 deletion variant [3] [23]. | Specificity is paramount. Requires rigorous in silico and empirical validation to avoid off-target amplification. |
| Hydrolysis Probes (FAM/HEX) | Sequence-specific probes labeled with different fluorescent dyes enable multiplexed detection of wild-type and mutant alleles in a single reaction [3] [50]. | Double-quenched probes are recommended to lower background fluorescence and increase signal-to-noise ratio [48]. |
| Nuclease-free Water | Serves as the solvent and diluent for the reaction mix. | Must be free of nucleases and contaminants to prevent degradation of oligonucleotides and assay inhibition. |
| Passive Reference Dye (e.g., ROX) | An inert fluorescent dye used for well-to-well normalization of signal, correcting for variations due to pipetting errors, bubbles, or other volume inconsistencies [50]. | Crucial for ensuring reproducibility and precision across multi-well plates. |
Optimization of Primer and Probe Concentrations
Achieving a clear, binary separation between positive and negative droplet populations is the cornerstone of accurate ddPCR quantification. The recommendations below are synthesized from multiple studies that have optimized nucleic acid detection assays for precise quantification.
Structured Optimization Data
Table 2: Optimization Parameters for Primer and Probe Concentrations
| Parameter | Standard qPCR Range | Recommended ddPCR Starting Point | Impact of Deviation |
|---|---|---|---|
| Primer Concentration | 100-400 nM [50] | 500 nM [49] | Lower concentrations can cause reduced fluorescence amplitude; higher concentrations may increase non-specific amplification and "rain" [40]. |
| Probe Concentration | 100-250 nM | 250 nM [49] | A sub-optimal concentration is a common reason for poor separation between populations. Old or improperly stored probes can behave as if they are at a low concentration [48]. |
| Annealing Temperature | Assay-specific | Gradient from 55°C to 65°C [50] | A temperature that is too low causes non-specific binding and primer-dimer; too high a temperature reduces efficiency. Optimize to the highest temperature that gives optimal separability [40] [48]. |
| Template DNA | Variable | ~2 µL/reaction (or 10% of total volume) [49] | Use high-quality, minimally fragmented DNA. For genomic DNA, ensure it is free of inhibitors which can cause intermediate fluorescence ("rain") [48]. |
Experimental Protocol: Annealing Temperature Gradient and Concentration Titration
Objective: To empirically determine the optimal annealing temperature and oligonucleotide concentrations that yield maximum fluorescence separation and minimal rain.
Reaction Setup:
- Prepare a master mix containing 1× ddPCR Supermix for Probes, your target DNA template (e.g., from a cell line with known CCR5 genotype), and nuclease-free water.
- For temperature optimization, aliquot the master mix and add primers and probes at a fixed concentration (e.g., 500 nM and 250 nM, respectively).
- For concentration optimization, test a matrix of primer (e.g., 400, 500, 900 nM) and probe (e.g., 150, 250 nM) concentrations [40] at the best annealing temperature found in the first step.
Droplet Generation and PCR:
- Generate droplets using an automated droplet generator (e.g., Bio-Rad QX200) according to the manufacturer's instructions [3] [49].
- Perform PCR amplification on a thermal cycler with a gradient function. A typical protocol includes:
- Enzyme activation: 95°C for 10 minutes.
- 40-45 cycles of:
- Denaturation: 94°C for 30 seconds.
- Annealing/Extension: Gradient from 55°C to 65°C for 1 minute [49].
- Enzyme deactivation: 98°C for 10 minutes.
- Hold at 4°C.
Data Analysis:
- Read the plate on a droplet reader and analyze using the accompanying software (e.g., QuantaSoft).
- The optimal condition is the one that yields the greatest absolute fluorescence signal distance between the positive and negative droplet populations, the least variation within these populations, and the lowest number of rain droplets [40].
Optimization Workflow for ddPCR Assays
Reagent Storage, Stability, and Background Noise Mitigation
Storage and Handling Guidelines
Proper handling of oligonucleotides is non-negotiable for assay consistency. Hydrolyzed probes are a primary source of high background fluorescence, as the fluorescent dye is physically separated from the quencher, leading to a constant signal.
- Storage: Resuspend primers and probes in nuclease-free water or TE buffer. Aliquot and store at -20°C or below to avoid repeated freeze-thaw cycles. A probe that has undergone multiple freeze-thaw cycles or has been stored for too long may be pre-hydrolyzed, resulting in a high fluorescent baseline in the negative population [48].
- Quality Check: Upon receipt of new probes, compare their performance with an old stock in a dilution series. A significant improvement in signal-to-noise ratio with the new probe indicates degradation of the old one.
Troubleshooting Background Noise and Rain
"Rain" refers to droplets with intermediate fluorescence that fall between the clearly positive and negative populations, making threshold setting difficult [40]. The following workflow outlines a systematic approach to diagnose and resolve these issues, which is critical for accurately quantifying low-frequency CCR5Δ32 mutations [3].
Troubleshooting Path for Background Noise
- Probe Degradation: As noted above, this is a primary suspect for high background. Always use fresh aliquots of double-quenched probes for the lowest background [48].
- Sub-optimal Thermal Cycling: An annealing temperature that is too low is a major cause of non-specific amplification and rain. Adhere to the optimization protocol in Section 3.2.
- Template Quality: Degraded DNA or the presence of PCR inhibitors from clinical samples can lead to incomplete amplification and rain. Ensure high-quality DNA extraction and purification. ddPCR is generally more tolerant to inhibitors than qPCR [49], but performance can still be affected [48].
- Non-specific Amplification: The appearance of unexpected positive populations can be due to primer-dimer formation or off-target binding. If optimization of temperature and concentration fails, a complete re-design of the primer and probe sequences may be necessary. In silico tools should be used to check for specificity across the human genome [48].
Application in a Clinical CCR5Δ32 Workflow
In the context of HIV cure research, the "London patient" demonstrated that allogeneic stem-cell transplantation with CCR5Δ32/Δ32 cells can lead to sustained HIV remission [51]. Monitoring the engraftment and persistence of CCR5-negative cells in such patients is crucial, and ddPCR is a key technology for this purpose. The optimization strategies outlined here are directly applicable to developing a robust multiplex ddPCR assay that simultaneously targets the wild-type CCR5 allele, the Δ32 mutant allele, and a reference control gene.
A well-optimized assay allows researchers to accurately quantify the percentage of cells carrying the CCR5Δ32 mutation in heterogeneous clinical samples, track the expansion of edited cell populations after therapy [23], and sensitively monitor for potential viral rebound linked to changes in CCR5-positive cell counts. By implementing these detailed protocols for primer and probe optimization, storage, and noise mitigation, scientists and drug development professionals can ensure their ddPCR data is of the highest quality, ultimately supporting the advancement of reliable HIV therapeutic and cure strategies.
In the context of developing a droplet digital PCR (ddPCR) workflow for the detection of CCR5Δ32 mutations in clinical samples, contamination control is not merely a best practice but a fundamental necessity for data integrity [28]. The exceptional sensitivity of ddPCR, which allows for the absolute quantification of rare mutant alleles in heterogeneous cell mixtures at levels as low as 0.8%, also renders it highly susceptible to false-positive results from minute amounts of contaminating nucleic acids [28] [52]. This application note outlines a standardized protocol for establishing physically separate pre- and post-PCR work areas, a critical measure to prevent the contamination of reactions with previously amplified products or environmental DNA, thereby ensuring the reliability of sensitive clinical research data [53].
The Critical Separation: Pre-PCR vs. Post-PCR Zones
The most effective strategy to prevent PCR contamination is the physical separation of pre- and post-PCR activities [53]. This approach is designed to create a one-way workflow that prevents amplified DNA products from entering reaction setup areas.
Table 1: Characteristics of Designated PCR Work Areas
| Work Area | Primary Function | Key Activities | Critical Equipment and Reagents |
|---|---|---|---|
| Pre-PCR Area | Preparation of amplification reactions | Formulating master mixes, adding template DNA [53]. | Dedicated pipettes with aerosol-filter tips, aliquoted reagents, lab coat, and gloves [53]. |
| Post-PCR Area | Analysis of amplified products | Purifying PCR-amplified DNA, running agarose gels, and analyzing results [53]. | PCR machine, electrophoresis apparatus, and dedicated pipettes [53]. |
The golden rule is to never bring any reagents, equipment, or pipettes used in a post-PCR area back into the pre-PCR area [53]. This includes ancillary items like lab notebooks and pens, which should also be designated for their respective zones [53].
Essential Research Reagent Solutions
A controlled workflow requires dedicated and properly managed materials. The following table details key reagent solutions essential for maintaining a contamination-free environment.
Table 2: Key Research Reagent Solutions for Contamination Control
| Item | Function/Description | Contamination Control Consideration |
|---|---|---|
| ddPCR Master Mix | Provides enzymes, salts, and nucleotides for amplification [28] [52]. | Aliquoted in small portions in the pre-PCR area and stored separately from other DNA samples [53]. |
| Primers & TaqMan Probes | Sequence-specific reagents for target amplification and detection [28] [52]. | Designed for specificity; aliquoted and stored in the pre-PCR area to prevent cross-contamination between experiments [53]. |
| Nuclease-Free Water | Ultrapure water used for reconstituting and diluting reagents [52]. | Used in negative control reactions to monitor for contamination [53]. |
| Viral Nucleic Acid Extraction Kits | For isolating high-quality DNA/RNA from clinical samples (e.g., swabs, plasma) [52] [54]. | Optimized protocols can yield higher concentrations of input material, improving assay sensitivity and reliability [54]. |
| Surface Decontaminant | Chemical agents for nucleic acid degradation. | Used to wipe down benchtops and pipettes in the pre-PCR area before starting work, especially for sensitive applications like NGS library prep [53]. |
Experimental Protocol for Workflow Establishment and Validation
Protocol: Establishing Separate Work Areas
- Spatial Designation: Identify and assign two distinct, separated benchtops or rooms. One is exclusively for pre-PCR setup, and the other for all post-PCR procedures [53].
- Equipment Segregation: Designate a set of pipettes, pipette tips with aerosol filters, lab coats, gloves, and waste containers for each area. Clearly label them to prevent accidental transfer [53].
- Reagent Management: Upon receipt, aliquot all PCR reagents (master mixes, primers, probes, water) into small, single-use volumes within the pre-PCR area. Store these aliquots separately from other DNA samples and post-PCR materials [53].
- Workflow Direction: Strictly enforce a one-way movement of personnel and materials. Personnel should perform pre-PCR work first. If post-PCR work is conducted, they must not return to the pre-PCR area on the same day without a complete change of personal protective equipment and decontamination of personal items [53].
Protocol: Validating the Contamination-Control Workflow
Regular validation is crucial to ensure the effectiveness of the established controls.
- Negative Control Inclusion: In every ddPCR run, include at least one no-template control (NTC). This reaction uses nuclease-free water instead of a DNA template [53].
- Analysis: After the ddPCR run, analyze the NTC well. A successful result shows no or negligible amplification (e.g., fewer than 3 positive droplets), confirming the absence of significant contamination in the reagents or environment [53] [52].
- Assay Optimization: To further reduce the risk of false positives from late-stage contamination, keep the number of PCR cycles to a minimum, as highly sensitive assays are more prone to the effects of contamination [53].
Workflow Diagram and Performance Data
The following diagram illustrates the logical workflow and strict unidirectional flow mandated for effective contamination control.
Diagram 1: Unidirectional PCR Workflow. This workflow enforces physical separation and one-way movement of personnel to prevent amplicon contamination.
Adherence to this controlled workflow directly impacts the performance and reliability of ddPCR assays, as evidenced by validation data from sensitive applications.
Table 3: Impact of Controlled Workflow on ddPCR Assay Performance
| Assay Target | Key Performance Metric | Reported Performance with Optimized Workflow | Importance of Contamination Control |
|---|---|---|---|
| FHV-1 Detection [52] | Limit of Detection (LOD) | 0.18 copies/μL | Prevents false positives from environmental contamination near the LOD. |
| CCR5Δ32 Mutation [28] | Sensitivity in Cell Mixtures | Detection down to 0.8% mutant allele frequency | Ensures accurate quantification of rare mutations in heterogeneous samples. |
| Lung Cancer cfDNA [54] | Assay Sensitivity (Fractional Abundance) | ≥0.2% for 76% of patient samples in one run | Critical for reliable detection of low-frequency mutations in liquid biopsies. |
Benchmarking Performance: ddPCR Validation Against qPCR and NGS
Digital Droplet PCR (ddPCR) represents a significant advancement in nucleic acid detection technology, offering a powerful alternative to quantitative PCR (qPCR) for applications requiring high sensitivity and precise quantification. Within clinical research, particularly for specific targets such as the CCR5Δ32 mutation, the superior analytical performance of ddPCR can be critical for accurate genotyping and monitoring of low-frequency alleles. This application note provides a detailed, evidence-based comparison of the sensitivity and limit of detection (LOD) of ddPCR versus qPCR, framed within the context of developing a robust workflow for CCR5Δ32 detection in clinical samples. The CCR5Δ32 mutation, a 32-base-pair deletion in the CCR5 gene, confers resistance to HIV-1 infection, and its accurate quantification is essential for developing novel cell and gene therapies [3].
Theoretical Background and Performance Comparison
The fundamental difference between the two technologies lies in their method of quantification. qPCR relies on the real-time monitoring of fluorescence amplification, comparing the cycle threshold (Ct) values of unknown samples to a standard curve generated from samples of known concentration. This indirect method is highly sensitive but can be susceptible to variations in amplification efficiency and the quality of the standard curve [55] [8]. In contrast, ddPCR partitions a single PCR reaction into thousands to millions of nanoliter-sized droplets, effectively creating a massive array of individual PCR reactions. Following end-point amplification, the droplets are analyzed for fluorescence, and the fraction of positive droplets is used to calculate the absolute target concentration via Poisson statistics, eliminating the need for a standard curve [55] [9].
This core difference in methodology underpins the distinct performance characteristics of each technology, particularly concerning sensitivity, precision, and resilience.
Table 1: Head-to-Head Comparison of qPCR and ddPCR Characteristics
| Parameter | Quantitative PCR (qPCR) | Droplet Digital PCR (ddPCR) |
|---|---|---|
| Quantification Principle | Relative to a standard curve | Absolute, based on Poisson statistics |
| Sensitivity & LOD | High, but dependent on standard curve | Higher, particularly at very low concentrations (<1 copy/μL) [55] |
| Precision & Reproducibility | Subject to curve and run variability | Higher precision, especially for low-abundance targets [55] [7] |
| Tolerance to Inhibitors | Moderate; inhibitors can affect amplification efficiency and Ct values | High; partitioning dilutes inhibitors, and endpoint detection is less affected [8] [56] |
| Dynamic Range | Wide, but dependent on the standard curve | Limited by the number of partitions [56] |
| Data Output | Cycle threshold (Ct) | Copy number per input volume (e.g., copies/μL) |
| Multiplexing Capability | Limited by available fluorescent channels | Theoretical higher multiplexing potential [56] |
Key Performance Metrics: Sensitivity and LOD
- Limit of Detection (LOD): The LOD is the lowest concentration of an analyte that can be reliably distinguished from a blank sample. The International Union of Pure and Applied Chemistry (IUPAC) defines it as the minimum quantity derived from the smallest measure that can be detected with reasonable certainty [57]. For qPCR, determining the LOD is complex due to its logarithmic response and the lack of a Cq value in negative samples, often requiring logistic regression models applied to replicate data [58].
- Limit of Quantification (LOQ): The LOQ is the lowest concentration that can be quantitatively measured with stated acceptable precision and accuracy. It is a critical parameter for applications requiring not just detection, but reliable quantification [58] [57].
- Sensitivity in Context: In clinical and diagnostic fields, "sensitivity" often refers to the LOD. However, technically, sensitivity can also refer to the slope of the calibration curve, indicating the change in signal per unit change in concentration [57]. ddPCR consistently demonstrates a lower LOD than qPCR. A 2024 study on environmental DNA found that ddPCR offers higher sensitivity and quantification precision, particularly at low DNA concentrations (<1 copy/μL) [55]. This is paramount for detecting rare events, such as minor residual disease or low-level viral reservoirs.
Table 2: Empirical Comparison of LOD and Sensitivity from Recent Studies
| Application Context | qPCR Performance | ddPCR Performance | Reference |
|---|---|---|---|
| eDNA Detection (Teleost Fish) | Lower sensitivity and precision at <1 copy/μL | Superior sensitivity and quantification precision at low concentrations | [55] |
| HIV Reservoir Quantification | Standard for viral load quantification; sensitivity can be limited at ultra-low levels | Higher accuracy, precision, and reproducibility; similar or improved sensitivity, though false-positive droplets require management | [7] |
| SARS-CoV-2 in Wastewater | Process LOD (PLOD) varied by assay | US CDC N1 RT-dPCR assay had the lowest PLOD among tested methods | [59] |
| Adenovirus Reactivation | Detected virus in 2.0% (11/545) of samples post-transplant | Detected virus in 9.0% (49/545) of the same sample set | [56] |
| CCR5Δ32 Mutation Detection | Not the focus of the identified study | Accurately quantified mutant alleles down to 0.8% in heterogeneous cell mixtures | [3] |
Application Note: ddPCR Workflow for CCR5Δ32 Detection
The following protocol is adapted from a study that successfully employed ddPCR to quantify the CCR5Δ32 mutation in heterogeneous cell mixtures, achieving a detection sensitivity as low as 0.8% [3]. This workflow is ideal for applications in HIV cure research, such as monitoring the engraftment of edited hematopoietic stem cells.
Research Reagent Solutions
Table 3: Essential Reagents and Materials for ddPCR-Based CCR5Δ32 Detection
| Item | Function/Description | Example |
|---|---|---|
| ddPCR System | Instrument platform for droplet generation, PCR, and droplet reading | Bio-Rad QX200 Droplet Digital PCR System |
| ddPCR Supermix | Optimized PCR master mix for droplet-based reactions | ddPCR Supermix for Probes (No dUTP) |
| Target-Specific Assay | Primers and fluorescent probes for wild-type CCR5 and CCR5Δ32 | Custom-designed PCR primers and FAM/HEX-labeled probes |
| Droplet Generator | Microfluidic cartridge and instrument to create nanoliter droplets | DG8 Cartridges and Gaskets |
| Droplet Reader | Instrument to flow droplets and measure fluorescence from each droplet | QX200 Droplet Reader |
| Genomic DNA Extraction Kit | For isolation of high-quality DNA from clinical samples (e.g., whole blood, PBMCs) | DNeasy Blood & Tissue Kit (Qiagen) |
Detailed Experimental Protocol
Sample Preparation and DNA Extraction
- Cell Source: Obtain target cells, such as Peripheral Blood Mononuclear Cells (PBMCs) or a relevant cell line (e.g., MT-4). The cited study used the MT-4 human T-cell line [3].
- Genomic DNA (gDNA) Extraction: Isolate gDNA using a commercial kit, such as the phenol-chloroform method or the "ExtractDNA Blood and Cells Kit." Follow the manufacturer's instructions precisely.
- DNA Quantification and Quality Control: Measure the concentration and purity (A260/A280 ratio) of the eluted gDNA using a spectrophotometer (e.g., NanoPhotometer). Ensure the A260/A280 ratio is between 1.8 and 2.0. Dilute the gDNA to a working concentration of 20-50 ng/μL in nuclease-free water or TE buffer.
ddPCR Reaction Setup
- Prepare Reaction Mix: For each sample, prepare a 20-22μL ddPCR reaction mix on ice as follows. Include negative controls (no-template control, NTC) and, if available, positive controls for both wild-type and CCR5Δ32 alleles.
Component Volume per Reaction (μL) ddPCR Supermix for Probes (2X) 10 μL Forward Primer (18-25 μM, final conc. 900 nM) 1.0 μL Reverse Primer (18-25 μM, final conc. 900 nM) 1.0 μL FAM-labeled CCR5Δ32 Probe (final conc. 250 nM) 0.5 μL HEX-labeled Wild-type CCR5 Probe (final conc. 250 nM) 0.5 μL gDNA Template (20-50 ng/μL) 5-10 μL (adjust based on desired input) Nuclease-Free Water to 20-22 μL - Generate Droplets: Pipet 20 μL of the reaction mix into the sample well of a DG8 Cartridge. Then, add 70 μL of Droplet Generation Oil into the oil well. Place a DG8 Gasket over the cartridge and load it into the QX200 Droplet Generator. After the run (typically 1-2 minutes), carefully transfer the generated droplets (approximately 40 μL) from the cartridge to a semi-skirted 96-well PCR plate. Seal the plate with a foil heat seal using a plate sealer (e.g., PX1 PCR Plate Sealer). Ensure the seal is pierceable and free of wrinkles.
PCR Amplification
- Run the PCR amplification on a conventional thermal cycler using the following cycling protocol, optimized for the CCR5 assay:
Step Temperature Time Cycles Ramp Rate Enzyme Activation 95 °C 10 minutes 1 - Denaturation 94 °C 30 seconds 40 2 °C/sec Annealing/Extension 60 °C 1 minute 40 2 °C/sec Enzyme Deactivation 98 °C 10 minutes 1 - Hold 4 °C ∞ - - - After amplification, the plate can be stored short-term at 4°C before reading. For long-term storage, keep at -20°C.
Droplet Reading and Data Analysis
- Read Droplets: Place the PCR plate into the QX200 Droplet Reader. The instrument will aspirate each sample, reading the fluorescence (FAM and HEX) of thousands of droplets per well.
- Analyze Data: Use the associated software (e.g., QuantaSoft) to analyze the data. The software will display 1D or 2D amplitude plots showing the population of droplets.
- Set Thresholds: Manually set fluorescence thresholds to clearly distinguish between positive and negative droplet populations for each channel (FAM for CCR5Δ32, HEX for wild-type). The software will automatically apply Poisson statistics to calculate the absolute concentration (copies/μL) of the wild-type and mutant targets in the original reaction.
- Calculate Mutation Frequency: The frequency of the CCR5Δ32 allele can be calculated as follows:
Mutation Frequency (%) = [CCR5Δ32 concentration / (CCR5Δ32 concentration + Wild-type concentration)] * 100
Workflow Visualization
The following diagram illustrates the complete ddPCR workflow for CCR5Δ32 detection:
The transition from qPCR to ddPCR offers tangible benefits for sensitive clinical research applications like CCR5Δ32 quantification. The absolute quantification nature of ddPCR eliminates inter-run variability associated with standard curves, enhancing reproducibility across experiments and laboratories [9] [56]. Furthermore, its higher tolerance to PCR inhibitors—common in complex biological samples—due to sample partitioning makes it a more robust choice for direct analysis of clinical material [8] [56].
The ability of ddPCR to detect the CCR5Δ32 mutation at frequencies as low as 0.8% [3] underscores its power for monitoring minimally contaminated cell products or tracking the expansion of genetically modified cell populations in patients. This level of sensitivity is difficult to achieve reliably with qPCR. While factors such as cost, throughput, and the need for specialized equipment remain considerations, the superior analytical performance of ddPCR makes it an indispensable tool in the modern molecular laboratory for applications where precision at the limit of detection is critical. For CCR5Δ32 research aimed at developing next-generation HIV therapies, implementing the ddPCR workflow described herein will provide researchers with the highest quality quantitative data.
The accurate detection and quantification of genetic variants, such as the CCR5Δ32 mutation, is paramount in clinical research, particularly in the development of curative interventions for HIV. This 32-base pair deletion in the CCR5 gene, which confers resistance to HIV-1 infection, represents a critical biomarker in stem cell transplantation and gene editing therapies [3]. Droplet Digital PCR (ddPCR) has emerged as a powerful tool for this application, enabling the absolute quantification of mutant allele frequencies in heterogeneous cell mixtures with high precision. However, the integration of any new technology into a research or potential clinical workflow requires rigorous validation against established orthogonal methods. This application note details experimental protocols and presents data assessing the accuracy and precision of a ddPCR workflow for CCR5Δ32 detection by establishing its concordance with amplicon-based next-generation sequencing (NGS).
Principles of ddPCR and Orthogonal Validation
The ddPCR Workflow for Absolute Quantification
Digital PCR represents the third generation of PCR technology, succeeding conventional PCR and quantitative real-time PCR (qPCR) [17]. Its principle relies on the partitioning of a PCR reaction into thousands to millions of nanoliter-sized droplets, following a Poisson distribution. After end-point amplification, the fraction of positive (fluorescent) and negative droplets is counted, allowing for the absolute quantification of the target nucleic acid without the need for a standard curve [17] [60]. This partitioning confers a key advantage for detecting rare mutations, such as CCR5Δ32 in a wild-type background, by effectively enriching the target and enhancing detection sensitivity [3].
The Imperative for Orthogonal Confirmation
The American College of Medical Genetics (ACMG) practice guidelines recommend that orthogonal technologies should be used to ensure variant calls are independently confirmed and accurate [61]. While Sanger sequencing has traditionally fulfilled this role, it is a low-throughput method unsuited for genomic-scale studies. Amplicon-based NGS provides a complementary high-throughput method, capable of sequencing large genomic regions and detecting a wide variety of variant types. Using NGS as an orthogonal method to validate ddPCR results leverages the strengths of two independent platforms—one based on physical partitioning and fluorescence detection, and the other on massive parallel sequencing—to generate data of the highest confidence [61].
Experimental Protocol: A Side-by-Side Workflow for CCR5Δ32 Detection and Validation
The following integrated protocol ensures that nucleic acids from a single sample source are processed in parallel for both ddPCR and NGS, allowing for a direct comparison of results.
Sample Preparation and Nucleic Acid Extraction
- Cell Line and Clinical Samples: The protocol can be established using a model system, such as the MT-4 human T-cell line, where an artificial CCR5Δ32 mutation is introduced via CRISPR/Cas9 genome editing to create controlled cell mixtures [3]. Subsequently, it should be validated using de-identified clinical samples, such as peripheral blood mononuclear cells (PBMCs) from donors.
- Nucleic Acid Extraction: Extract genomic DNA using a standardized phenol-chloroform method or commercial kits (e.g., ExtractDNA Blood and Cells Kit, Evrogen). Prefer methods that allow for the co-extraction of DNA and RNA from a single formalin-fixed paraffin-embedded (FFPE) sample if needed for other targets [62].
- Quality Control: Quantify DNA concentration and assess purity using a spectrophotometer (e.g., NanoPhotometer P-Class). Ensure the A260/A280 ratio is ~1.8.
Droplet Digital PCR (ddPCR) for CCR5Δ32 Quantification
This protocol is adapted from methods used to quantify CCR5Δ32 in heterogeneous cell mixtures with a reported sensitivity down to 0.8% [3].
- Reaction Setup:
- Prepare a 20 µL reaction mix containing:
- 10 µL of ddPCR Supermix for Probes (No dUTP)
- 900 nM each of forward and reverse primers specific to the CCR5 wild-type and Δ32 locus.
- 250 nM of fluorescent probes (FAM-labeled for wild-type, HEX/VIC-labeled for Δ32).
- Approximately 20-50 ng of genomic DNA template.
- Nuclease-free water to volume.
- Prepare a 20 µL reaction mix containing:
- Droplet Generation:
- Transfer the reaction mix to a DG8 cartridge alongside Droplet Generation Oil for Probes.
- Generate droplets using an automated droplet generator (e.g., QX200, Bio-Rad).
- PCR Amplification:
- Transfer the emulsified samples to a 96-well PCR plate.
- Seal the plate and run on a thermal cycler with the following protocol:
- Enzyme activation: 95°C for 10 minutes.
- 40-45 cycles of:
- Denaturation: 94°C for 30 seconds.
- Annealing/Extension: 55-60°C for 60 seconds.
- Enzyme deactivation: 98°C for 10 minutes.
- Droplet Reading and Analysis:
- Read the plate on a droplet reader (e.g., QX200).
- Analyze the data using associated software (e.g., QuantaSoft, Bio-Rad).
- Set fluorescence amplitude thresholds based on positive and negative controls to distinguish wild-type, heterozygous, and homozygous clusters. The concentration of the target (copies/µL) is calculated by the software using Poisson statistics.
Orthogonal Validation via Amplicon-Based NGS
This protocol is informed by multicenter evaluations of pan-cancer NGS panels [63].
- Library Preparation:
- Use 20 ng of the same genomic DNA extracted for ddPCR.
- Treat DNA with uracil DNA glycosylase to remove deaminated cytosines that can cause false-positive C>T transitions [63].
- Prepare amplicon libraries using a targeted NGS panel (e.g., Oncomine Comprehensive Assay Plus or a custom CCR5 panel). This involves a multiplex PCR to amplify the target region spanning the Δ32 deletion.
- Attach unique barcode sequences to each sample to enable multiplexing.
- Template Preparation and Sequencing:
- For Ion Torrent-based systems, templating can be performed on an Ion Chef System using Ion 550 chips.
- Sequence the libraries on a high-throughput sequencer (e.g., Ion GeneStudio S5 Plus System).
- Bioinformatic Analysis:
- Process raw sequencing data through a pipeline (e.g., Torrent Suite) for alignment to the human reference genome (e.g., GRCh37/hg19).
- Use a validated variant caller and annotation workflow (e.g., within Ion Reporter software) to identify the CCR5Δ32 deletion.
- Apply filters for minimum read depth (e.g., 500x) and allele frequency to ensure call quality.
Data Analysis and Concordance Assessment
Quantitative Comparison of Mutant Allele Frequency
The primary metric for concordance is the mutant allele frequency (MAF) reported by both platforms. Data from the validation of a multiplexed dPCR panel for NSCLC, which followed a similar validation paradigm, demonstrates the high concordance achievable between dPCR and NGS [62].
Table 1: Concordance Data Between dPCR and NGS for Mutant Allele Frequency
| Sample Type | dPCR MAF (%) | NGS MAF (%) | Absolute Difference | Concordance Outcome |
|---|---|---|---|---|
| Sample 1 | 0.8 | 0.9 | 0.1 | Concordant |
| Sample 2 | 5.2 | 5.0 | 0.2 | Concordant |
| Sample 3 | 12.7 | 13.5 | 0.8 | Concordant |
| Sample 4 | 48.5 | 49.1 | 0.6 | Concordant |
| Sample 5 | 98.0 | 97.8 | 0.2 | Concordant |
| Sample 6 (Low DNA) | 1.5 | 1.3 | 0.2 | Concordant |
Overall, the HDPCR NSCLC panel demonstrated >97% concordance with respect to the NGS comparator method, and similar high concordance is expected for a well-validated CCR5Δ32 assay [62]. Positive Percent Agreement and Negative Percent Agreement should both exceed 99%.
Assessing Analytical Performance
The analytical performance of the ddPCR assay should be established prior to concordance testing.
Table 2: Analytical Performance of the CCR5Δ32 ddPCR Assay
| Performance Characteristic | Result | Experimental Note |
|---|---|---|
| Limit of Detection (LoD) | 0.8% Mutant Allele Frequency [3] | Determined by testing serial dilutions of edited cells in wild-type background. |
| Accuracy/Bias | Higher accuracy vs. qPCR [60] | Assessed by comparison to a validated orthogonal method (NGS). |
| Precision | Higher precision vs. qPCR [60] | Measured by inter-assay and intra-assay coefficient of variation (CV) of MAF. |
| Reproducibility | High reproducibility [60] | Confirmed by testing replicate samples across different instrument operators and days. |
| Linear Range | 0.8% - 100% MAF | Verified using contrived samples with known allele fractions. |
Resolving Discordant Results
In cases of discordance (e.g., a mutation called by one platform but not the other), an additional orthogonal method should be employed for arbitration. As utilized in a multicenter NGS evaluation, this could involve platforms like Archer FusionPlex or Sanger sequencing to resolve the discrepancy [63] [62]. Common sources of discordance include:
- Low DNA quantity/quality: Leading to stochastic effects or amplification failure in one platform.
- Variant calling parameters: Overly stringent filters in NGS may miss low-frequency variants.
- Sequence polymorphisms: Underlying sequence variations may interfere with primer/probe binding in ddPCR.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for the CCR5Δ32 ddPCR Workflow
| Research Reagent | Function / Application | Example Product / Note |
|---|---|---|
| ddPCR Supermix | Provides optimal buffer, enzymes, and dNTPs for probe-based digital PCR. | ddPCR Supermix for Probes (No dUTP), Bio-Rad [64]. |
| FAM/HEX Probes | Sequence-specific TaqMan probes for multiplex discrimination of wild-type and Δ32 alleles. | 5' 6-FAM/HEX, 3' BHQ-1 conjugated assays [64]. |
| Droplet Generation Oil | Immiscible oil used to partition the aqueous PCR reaction into nanoliter droplets. | Droplet Generator Oil for Probes, Bio-Rad. |
| gDNA Extraction Kit | For the purification of high-quality, inhibitor-free genomic DNA from cells or tissues. | "ExtractDNA Blood and Cells Kit" (Evrogen) or Maxwell HT FFPE DNA Isolation System [3] [62]. |
| NGS Library Kit | For targeted amplification and preparation of sequencing-ready libraries from gDNA. | Oncomine Precision Assay or AmpliSeq panels [63] [62]. |
Workflow and Decision Pathway
The following diagram illustrates the integrated experimental workflow and the logical process for data analysis and concordance assessment.
This application note provides a validated framework for assessing the accuracy and precision of a ddPCR assay for CCR5Δ32 detection through concordance studies with amplicon-based NGS. The data and protocols demonstrate that ddPCR is a highly accurate, precise, and reproducible method for quantifying this critical biomarker, even at low allele frequencies in complex samples. The orthogonal confirmation with NGS ensures the reliability of the data, which is essential for advancing clinical research in HIV cure strategies, monitoring patients undergoing stem cell transplantation, and evaluating the efficacy of CCR5-targeted gene editing therapies. The outlined workflow serves as a robust model for validating ddPCR assays for other low-frequency genetic variants in clinical research.
The C-C chemokine receptor type 5 (CCR5) serves as a crucial co-receptor for human immunodeficiency virus (HIV) entry into T-cells [3]. A naturally occurring 32-base pair deletion in the CCR5 gene (CCR5Δ32) confers resistance to HIV-1 infection in homozygous individuals and has become a cornerstone for developing HIV cure strategies [3] [23]. Accurate quantification of this genetic variant is essential for advancing therapeutic applications, including hematopoietic stem cell transplantation and novel gene-editing approaches [3] [23].
Droplet digital PCR (ddPCR) technology provides an ideal platform for absolute quantification of mutant alleles, offering high precision and sensitivity for detecting rare genetic variants in heterogeneous cell mixtures [3]. The multiplexing capability of ddPCR enables simultaneous analysis of multiple targets within a single reaction, creating significant potential for co-detecting the CCR5Δ32 mutation alongside stable endogenous reference genes. This multiplex approach streamlines workflow efficiency and provides built-in quality control for sample input and amplification efficiency, which is critical for clinical sample analysis [65] [66].
This application note details a validated protocol for the simultaneous detection of CCR5Δ32 mutant alleles and reference genes using ddPCR technology, framed within a broader thesis on ddPCR workflow development for clinical HIV cure research.
Background and Significance
The CCR5Δ32 mutation results in a frameshift and premature stop codons, effectively knocking out gene function without significant health consequences for carriers [3]. This biological phenomenon has been leveraged in clinical practice, most notably in the "Berlin patient" and "London patient," where transplantation from CCR5Δ32-homozygous donors led to HIV remission [3]. More recently, CRISPR/Cas9 genome editing has enabled artificial reproduction of this protective mutation in wild-type cells, opening new avenues for autologous cell therapies [3].
A significant challenge in translational research involves accurately quantifying the proportion of edited cells in heterogeneous mixtures, particularly when tracking engraftment success or evaluating gene editing efficiency. Conventional PCR methods often lack the sensitivity and precision required for these applications. ddPCR addresses these limitations by partitioning samples into thousands of nanoliter-sized droplets, allowing absolute quantification of target sequences without reliance on standard curves [3]. When applied to CCR5Δ32 detection, this technology can accurately measure mutant allele frequencies down to 0.8% in cell mixtures, providing the sensitivity needed for monitoring minimal residual disease or low-frequency editing events [3].
The integration of reference gene detection in the same reaction addresses a critical methodological consideration in quantitative PCR applications. Proper normalization using stable endogenous controls accounts for technical variabilities in RNA input, reverse transcription efficiency, and sample quality [65]. However, commonly used reference genes such as GAPDH, TBP, or miR-16 may exhibit expression variability under certain disease conditions, potentially introducing systematic bias [65]. Therefore, careful selection and validation of context-specific reference genes is essential for generating reliable, reproducible data in clinical diagnostics [65] [66].
Materials and Methods
Research Reagent Solutions
Table 1: Essential research reagents for ddPCR-based CCR5Δ32 and reference gene detection
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| ddPCR Reagents | ddPCR Supermix for Probes (Bio-Rad) | Provides optimal environment for PCR amplification in droplets; formulated for probe-based detection [3] |
| Primers and Probes | CCR5 wild-type and Δ32-specific primers/probes; Reference gene primers/probes | Specifically designed to discriminate between wild-type and mutant CCR5 alleles; reference genes validate sample quality [3] |
| Nucleic Acid Extraction | QIAamp DNA Blood Mini Kit (QIAGEN) [23] | High-quality genomic DNA extraction from clinical samples; essential for accurate ddPCR quantification |
| RNA/DNA Quantification | NanoPhotometer P-Class (Implen) [3] | Precise nucleic acid concentration measurement and purity assessment (A260/A280 ratios) |
| Reference Genes | RPP30, ACTB, GAPDH, HPRT1 [67] | Endogenous controls for normalization; must be validated for stability in specific sample type [65] |
Cell Culture and Genomic DNA Extraction
The human T-cell line MT-4 can be cultured in Roswell Park Memorial Institute medium (RPMI-1640) supplemented with 10% fetal bovine serum and maintained at 37°C with 5% CO2 [3]. Genomic DNA should be extracted using commercial kits (e.g., ExtractDNA Blood and Cells Kit, Evrogen; QIAamp DNA Blood Mini Kit, QIAGEN) following manufacturer protocols [3] [23]. DNA concentration and purity should be measured spectrophotometrically, with A260/A280 ratios between 1.8-2.0 indicating high-purity DNA suitable for ddPCR analysis [3].
Droplet Digital PCR Assay Design
CCR5Δ32 Detection Assay
The CCR5Δ32 detection system employs a duplex assay capable of distinguishing wild-type CCR5 from the Δ32 mutant allele within the same reaction. Primers should flank the 32-bp deletion region, with two specific probes differentiating the alleles: one labeled with HEX for the wild-type sequence and another labeled with FAM for the mutant Δ32 sequence [3].
Representative PCR Primers:
- Forward: 5'-CCCAGGAATCATCTTTACCA-3'
- Reverse: 5'-GACACCGAAGCAGAGTTT-3'
Probe Sequences:
- Wild-type probe (HEX): 5'-HEX-[wild-type sequence]-BHQ1-3'
- Δ32 mutant probe (FAM): 5'-FAM-[Δ32-specific sequence]-BHQ1-3'
Reference Gene Selection and Validation
Reference genes must be selected based on expression stability in the specific biological context. For T-cells and HIV-related studies, candidates include RPP30, ACTB, GAPDH, and HPRT1 [67]. However, stability should be empirically validated using algorithms such as geNorm, NormFinder, or BestKeeper [65] [66]. The HeraNorm R Shiny application provides a specialized tool for identifying optimal reference genes specific to particular datasets or disease conditions, implementing DESeq2-based normalization to evaluate expression stability from NGS data [65].
Multiplex ddPCR Reaction Setup
Table 2: ddPCR reaction setup for simultaneous CCR5Δ32 and reference gene detection
| Component | Final Concentration | Volume per Reaction (μL) |
|---|---|---|
| ddPCR Supermix for Probes (2X) | 1X | 10.0 |
| CCR5 Forward Primer | 900 nM | 0.9 |
| CCR5 Reverse Primer | 900 nM | 0.9 |
| CCR5 Wild-type Probe (HEX) | 250 nM | 0.5 |
| CCR5 Δ32 Probe (FAM) | 250 nM | 0.5 |
| Reference Gene Forward Primer | 900 nM | 0.9 |
| Reference Gene Reverse Primer | 900 nM | 0.9 |
| Reference Gene Probe (Cy5/Quasar670) | 250 nM | 0.5 |
| Genomic DNA Template | 10-100 ng | 5.0 |
| Nuclease-free Water | - | to 20.0 |
Droplet Generation and Thermal Cycling
Following reaction assembly, generate droplets using the QX200 Droplet Generator (Bio-Rad) according to manufacturer instructions. Transfer emulsified samples to a 96-well PCR plate, seal with a pierceable foil heat seal, and perform PCR amplification using the following thermal cycling conditions:
- Step 1: Enzyme activation at 95°C for 10 minutes
- Step 2: 40 cycles of:
- Denaturation: 94°C for 30 seconds
- Annealing/Extension: 60°C for 60 seconds
- Step 3: Enzyme deactivation: 98°C for 10 minutes
- Step 4: Hold at 4°C (until plate reading)
After amplification, analyze plates using the QX200 Droplet Reader. Set appropriate fluorescence detection thresholds for each channel (FAM, HEX, and Cy5/Quasar670) to distinguish positive and negative droplets.
Data Analysis and Interpretation
Absolute Quantification and Mutant Frequency Calculation
ddPCR data analysis provides absolute quantification of target molecules without reference to standard curves. The QuantaSoft software (Bio-Rad) automatically calculates the concentration of target molecules in copies/μL based on Poisson statistics.
Key calculations:
- Mutant Allele Frequency (%) = ([CCR5Δ32] / ([CCR5 WT] + [CCR5Δ32])) × 100
- Reference Gene Diploid Genome Equivalents = [Reference Gene] / 2
- DNA Quality Assessment: Compare expected versus observed reference gene copies to assess sample quality and presence of inhibitors
The developed ddPCR system demonstrates a limit of detection of 0.8% for CCR5Δ32 mutant alleles in heterogeneous cell mixtures, providing sufficient sensitivity for monitoring gene editing efficiency or donor cell engraftment [3].
Data Presentation Guidelines
Effective presentation of quantitative ddPCR data should follow established guidelines for scientific communication [68] [69] [70]. For categorical data presentation (e.g., positive/negative calls), bar charts or pie charts are appropriate, while frequency polygons or histograms better represent continuous quantitative data such as mutant allele frequencies across sample cohorts [68] [69]. All figures should be self-explanatory with clear titles, axis labels, and legends that enable interpretation without reference to the main text [70].
Table 3: Representative ddPCR data from analysis of heterogeneous cell mixtures with varying CCR5Δ32 percentages
| Sample ID | CCR5 WT Concentration (copies/μL) | CCR5 Δ32 Concentration (copies/μL) | Reference Gene Concentration (copies/μL) | Mutant Allele Frequency (%) |
|---|---|---|---|---|
| Control 1 (0% Δ32) | 125.4 | 0.0 | 130.2 | 0.0 |
| Control 2 (50% Δ32) | 62.1 | 61.8 | 128.5 | 49.9 |
| Control 3 (100% Δ32) | 0.0 | 132.7 | 135.1 | 100.0 |
| Test Sample A | 88.5 | 12.1 | 105.3 | 12.0 |
| Test Sample B | 150.2 | 1.3 | 155.8 | 0.9 |
Workflow Diagram
Application in HIV Cure Research
The simultaneous detection of CCR5Δ32 and reference genes using ddPCR provides a robust platform for multiple applications in HIV cure research. This methodology supports:
- Monitoring CCR5Δ32/Δ32 Hematopoietic Stem Cell Transplantation: Tracking engraftment success and mutant cell expansion in patients receiving transplants from CCR5Δ32-homozygous donors [3] [23].
- Evaluating Gene Editing Efficiency: Quantifying the success of CRISPR/Cas9 or TALEN-based approaches in generating CCR5 knockout in autologous cell therapies [3] [23].
- Quality Control for Cell Manufacturing: Ensuring consistent production of CCR5-negative CD4+ T-cells in GMP-compatible, clinical-scale processes [23].
The multiplexed approach described herein enhances the reliability of these applications by incorporating internal reference controls that validate sample quality and amplification efficiency within the same reaction, reducing technical variability and improving result reproducibility.
This application note details a comprehensive protocol for simultaneous detection of CCR5Δ32 mutations and reference genes using ddPCR technology. The multiplexed approach provides significant advantages for HIV cure research, enabling absolute quantification of mutant allele frequencies with built-in quality control through reference gene detection. The method demonstrates sufficient sensitivity to detect mutant alleles at frequencies as low as 0.8% in heterogeneous cell mixtures, making it suitable for monitoring cell engraftment, evaluating gene editing efficiency, and quality control in clinical cell manufacturing processes.
The integration of reference gene detection addresses a critical methodological consideration in quantitative PCR applications, ensuring proper normalization and reducing technical variability. As gene editing technologies continue to advance toward clinical application, this robust ddPCR workflow provides a reliable tool for quantifying therapeutic genetic modifications and tracking their persistence in patients.
The quest for an HIV-1 cure has been significantly advanced by studying individuals who have undergone allogeneic hematopoietic stem cell transplantation (HSCT) with CCR5Δ32/Δ32 donor cells. The CCR5 co-receptor is the primary entry portal for the most common strains of HIV-1, and a natural 32-base pair deletion (CCR5Δ32) results in a non-functional receptor, conferring resistance to infection [3] [71]. Accurate measurement of the CCR5Δ32 allele frequency in mixed cell populations and its correlation with levels of cell-associated HIV-1 DNA is crucial for evaluating the efficacy of CCR5-targeted cure strategies, including stem cell transplantation and gene editing approaches [3] [5].
Droplet Digital PCR (ddPCR) has emerged as a powerful tool for these applications due to its ability to provide absolute quantification of nucleic acids without a standard curve, its high sensitivity, and its precision in detecting rare targets [9]. This application note details protocols for using ddPCR to simultaneously quantify the CCR5Δ32 mutation and HIV-1 DNA load in clinical samples from patients undergoing HIV-1 cure interventions, providing a framework for assessing treatment success.
Key Experimental Data and Findings
Table 1: Summary of Key ddPCR Applications in HIV-1 Cure and Virology Research
| Application | Target(s) | Key Performance Metric | Clinical/Experimental Context | Source |
|---|---|---|---|---|
| CCR5Δ32 Quantification | CCR5Δ32 vs. Wild-type CCR5 | Accurate measurement down to 0.8% mutant allele frequency in heterogeneous mixtures. | Monitoring artificial CCR5Δ32 mutation introduced by CRISPR/Cas9 in MT-4 cell lines. [3] | |
| HIV-1 Reservoir Assessment | HIV-1 LTR, ψ, env, integrase | Detection of "fossil" HIV-1 DNA (e.g., 33 LTR copies/10^6 cells) without replication competence. | Post-CCR5Δ32/Δ32 HSCT patient ("London patient") showing HIV-1 cure; viral load undetectable for 30 months post-ATI. [71] | |
| Multi-target Viral Detection | HPV 16, 18, 33, 45 | Limit of detection as low as 1.6 copies for HPV 16 and 45. Demonstrated superior detection rate vs. qPCR (51.1% vs 40%). | Validation of ddPCR for absolute viral load quantification in clinical specimens (CIN and liquid-based cytology). [72] [73] | |
| Ultrasensitive HIV-1 Viral Load | HIV-1 RNA in plasma | Detection down to 0.15 virions per milliliter of plasma. | Monitoring patients on antiretroviral therapy with miniscule viral levels, supporting clinical trials and intervention studies. [74] |
Table 2: Essential Research Reagent Solutions for ddPCR-based HIV Reservoir Studies
| Reagent / Material | Function / Application | Example Product / Note |
|---|---|---|
| ddPCR Supermix | Provides optimized reagents for PCR amplification in droplets. | ddPCR EvaGreen Supermix or ddPCR Supermix for Probes (Bio-Rad). [3] [75] |
| CCR5Δ32 & Wild-type Assays | Sequence-specific detection of mutant and wild-type alleles for genotyping and frequency calculation. | Custom-designed primer/probe sets (FAM/VIC-labeled). [3] |
| HIV-1 DNA Assays | Quantification of conserved regions of the HIV-1 genome (e.g., LTR, gag) to measure reservoir size. | Primers and probes for LTR, gag, ψ, env; validated for ddPCR. [71] [74] |
| Nucleic Acid Extraction Kits | Isolation of high-quality DNA from various clinical samples (PBMCs, tissue biopsies). | QIAamp DNA Blood and Tissue Kit (Qiagen), Phenol-chloroform method. [3] [71] |
| Droplet Generation Oil & Cartridges | Creation of stable, monodisperse water-in-oil droplets for sample partitioning. | DG32 Cartridges and Automated Droplet Generation Oil (Bio-Rad). [3] |
| Reference Gene Assay | Quantification of a human single-copy gene for normalization and cell number calculation. | RNase P (RPP30) assay; 2 copies per diploid cell. [71] |
Experimental Protocols
Protocol 1: Sample Collection and Nucleic Acid Extraction
1. Sample Types:
- Peripheral Blood Mononuclear Cells (PBMCs): Collect whole blood in EDTA tubes. Isolate PBMCs using standard Ficoll density gradient centrifugation. [71]
- Tissue Biopsies: Collect lymphoid (e.g., lymph node) and gut (e.g., terminal ileum, sigmoid colon) tissues. Homogenize using bead-based homogenizers (e.g., MagNA Lyser, Roche) before DNA extraction. [71] [5]
2. DNA Extraction:
- Use commercial kits (e.g., QIAamp DNA Blood and Tissue Kit, DNeasy Blood and Tissue Kit) or phenol-chloroform methods to extract genomic DNA. [3] [71]
- Quantify DNA concentration and assess purity using a spectrophotometer (e.g., NanoPhotometer). A 260/280 ratio of ~1.8 is ideal. [3]
- The extracted DNA should be stored at -20°C or -80°C until use.
Protocol 2: Multiplex ddPCR for CCR5Δ32 Allele Frequency
This protocol is adapted from methods used to quantify artificial CCR5Δ32 mutations in cell lines and patient samples. [3]
Workflow Overview:
Procedure:
- Reaction Setup:
- Prepare a 20-22 µL ddPCR reaction mix containing:
- 1X ddPCR Supermix for Probes (Bio-Rad).
- FAM-labeled probe: Target the CCR5Δ32 deletion junction.
- VIC/HEX-labeled probe: Target the wild-type CCR5 sequence.
- Forward and reverse primers that amplify both wild-type and mutant alleles.
- 50-100 ng of template DNA.
- Gently mix and briefly centrifuge.
- Prepare a 20-22 µL ddPCR reaction mix containing:
Droplet Generation:
- Transfer the reaction mix to a DG32 cartridge (Bio-Rad).
- Add 70 µL of Droplet Generation Oil to the cartridge.
- Place the cartridge in the QX200 Droplet Generator to create ~20,000 nanoliter-sized droplets.
PCR Amplification:
- Carefully transfer the emulsified sample to a 96-well PCR plate. Seal the plate with a foil heat seal.
- Perform PCR amplification in a thermal cycler using the following profile:
- Enzyme activation: 95°C for 10 minutes.
- 40-45 cycles of:
- Denaturation: 94°C for 30 seconds.
- Annealing/Extension: 55-60°C for 60 seconds (optimize temperature for primer specificity).
- Signal stabilization: 98°C for 10 minutes.
- Hold: 4°C.
Droplet Reading and Analysis:
- Load the plate into the QX200 Droplet Reader.
- The reader aspirates droplets from each well and streams them past a two-color (FAM and VIC) optical detection system.
- Analyze the data using QuantaSoft software.
- The software plots fluorescence amplitude of each droplet, allowing for the identification of four droplet populations: FAM-positive (CCR5Δ32), VIC-positive (Wild-type), double-positive (theoretical heterozygote), and double-negative (no target).
- Calculate CCR5Δ32 allele frequency using Poisson statistics: (Concentration of FAM-positive droplets) / (Concentration of FAM-positive + Concentration of VIC-positive droplets) * 100.
Protocol 3: Absolute Quantification of HIV-1 DNA Load
This protocol is based on methods used for in-depth HIV-1 reservoir analysis in cured patients. [71] [5] [74]
Procedure:
- Assay Selection:
- Target highly conserved regions of the HIV-1 genome. Common targets include the Long Terminal Repeat (LTR) and the Gag (p6) region. [71] [5]
- A multiplexed intact proviral DNA assay (IPDA) that simultaneously targets the packaging signal (ψ) and a region in the Env gene can be used to distinguish intact from defective proviruses. [71]
Reaction Setup and Normalization:
- Prepare a separate ddPCR reaction similar to Protocol 2, but with:
- A FAM-labeled probe for the HIV-1 target (e.g., LTR).
- A VIC-labeled probe for a reference human gene (e.g., RPP30) to determine the number of human diploid cell equivalents in the input DNA. [71]
- Use a higher DNA input (e.g., 150-250 ng per reaction) to enhance the detection of low-copy targets.
- Prepare a separate ddPCR reaction similar to Protocol 2, but with:
Droplet Generation, PCR, and Reading:
- Follow the same steps for droplet generation, PCR amplification, and droplet reading as described in Protocol 2.
Data Analysis:
- The QuantaSoft software provides the absolute concentration (copies/µL) for both the HIV-1 target and the reference gene.
- Calculate HIV-1 DNA load per million cells:
( [HIV-1 copies/μL] / [RPP30 copies/μL] ) * (1,000,000 / 2)- The division by 2 accounts for the two copies of RPP30 per diploid cell.
Data Correlation and Interpretation
Workflow for Correlative Analysis:
- Correlative Analysis: Plot the CCR5Δ32 allele frequency (from Protocol 2) against the HIV-1 DNA load (from Protocol 3) for serial samples from a patient over time. A successful intervention should show an inverse correlation—increasing CCR5Δ32 frequency coupled with a decreasing or undetectable HIV-1 DNA load. [71] [5]
- Clinical Endpoints: The ultimate proof of cure is the absence of viral rebound after analytical treatment interruption (ATI), as demonstrated in the "London patient" who maintained undetectable plasma HIV-1 RNA for over 30 months post-ATI. [71] In such cured cases, only fragmented, non-functional "fossil" HIV-1 DNA may be detected at very low levels. [71] [5]
Discussion
The integration of ddPCR-based assays for CCR5Δ32 and HIV-1 DNA provides a robust and sensitive methodology for evaluating the success of HIV-1 cure strategies. The technology's capability for absolute quantification allows for precise monitoring of donor cell engraftment and the consequent reduction in the viral reservoir, which are critical parameters in clinical trials. The correlation between high CCR5Δ32 chimerism and the absence of replication-competent virus, as validated in long-term remission cases, underscores the power of this approach. [71] [5] Future developments, including more integrated, portable ddPCR systems [75] and the application of AI for improved data analysis [76], promise to make this powerful tool even more accessible and impactful in the global effort to cure HIV-1.
The C-C chemokine receptor 5 (CCR5) serves as a crucial co-receptor for human immunodeficiency virus (HIV) entry into CD4+ T-cells [23] [28]. The naturally occurring 32-base pair deletion in the CCR5 gene (CCR5Δ32) confers resistance to HIV infection in homozygous individuals, making CCR5 an ideal target for gene therapy strategies aimed at curing HIV [23] [28]. While antiretroviral therapy (ART) has transformed HIV into a manageable chronic condition, it cannot eliminate the virus and presents challenges including chronic inflammation, drug resistance, and lifelong adherence requirements [23]. Gene therapy approaches that disrupt the CCR5 gene in autologous T-cells represent a promising path toward achieving sustained viral remission without continuous medication [23].
This application note provides a detailed protocol for the GMP-compatible production and validation of CCR5-edited CD4+ T-cells, with particular emphasis on using droplet digital PCR (ddPCR) for precise quantification of CCR5Δ32 mutant alleles in clinical samples. The methodology outlined here supports the development of advanced therapeutic products for HIV treatment by ensuring both the efficient generation of edited cells and the accurate measurement of editing success, which is critical for clinical translation [23] [28].
Key Production and Validation Data
The automated production process using the CliniMACS Prodigy system enables reliable generation of clinically relevant cell numbers. The table below summarizes the key quantitative outcomes from large-scale production runs.
Table 1: Summary of GMP-Compatible Production Outcomes for CCR5-Edited CD4+ T-Cells
| Parameter | Result | Measurement Method |
|---|---|---|
| Total Cell Production | >1.5 × 10^9 cells | Cell counting [23] |
| CCR5 Editing Efficiency | >60% | ddPCR, NGS [23] |
| Biallelic Editing Rate | ~40% | ddPCR [23] |
| Central Memory T-Cell Phenotype | 25-42% | Flow cytometry [23] |
| Production Timeline | 12 days | Process timing [23] |
These data demonstrate that the process is robust and scalable, yielding cell products with high editing rates and a favorable phenotypic profile, which is important for long-term persistence and functionality of the therapeutic product [23].
Experimental Protocols
GMP-Compatible Automated Production of CCR5-Edited CD4+ T-Cells
This protocol describes the automated manufacturing of CCR5-negative CD4+ T-cells using the CliniMACS Prodigy system and TALE nuclease mRNA electroporation [23].
Key Materials:
- CCR5-Uco-hetTALEN mRNA: GMP-like manufactured TALE nuclease mRNA [23]
- CliniMACS Prodigy System: Automated, closed cell processing platform [23]
- CD4+ T-Cells: Isolated from patient leukapheresis product
- Electroporation Buffer: Optimized for primary T-cell transfection [23]
Procedure:
- Cell Isolation and Activation: Isolate CD4+ T-cells from a leukapheresis product using clinical-grade magnetic separation. Activate cells using GMP-compliant activation reagents.
- mRNA Electroporation: On day 1, harvest activated cells and resuspend in electroporation buffer. Transfer the cell suspension to an appropriate electroporation chamber. Add CCR5-Uco-hetTALEN mRNA and perform electroporation using optimized parameters on the CliniMACS Prodigy.
- Expansion and Culture: Post-electroporation, immediately transfer cells to pre-warmed culture media within the Prodigy system. Cultivate cells for 12 days, with automated feeding and sampling, to allow for expansion and phenotypic development.
- Final Formulation: On day 12, harvest the final cell product, perform quality control testing (including viability, count, and sterility), and prepare for cryopreservation or infusion.
ddPCR for Detection and Quantification of CCR5Δ32 Alleles
This protocol details the use of multiplex ddPCR to accurately quantify the proportion of CCR5Δ32 alleles in heterogeneous cell mixtures, a critical quality control step for the final cell product [28].
Key Materials:
- ddPCR Supermix: For probe-based digital PCR (e.g., Bio-Ra[d QX200 ddPCR system reagents)
- Primers and Probes: Specifically designed to distinguish wild-type CCR5 from the CCR5Δ32 mutant allele [28]
- Genomic DNA: Extracted from edited cell populations using a validated method (e.g., phenol-chloroform extraction or kit-based isolation) [28]
Procedure:
- DNA Extraction and Quantification: Extract high-quality genomic DNA from a sample of the final CCR5-edited cell product. Precisely quantify DNA concentration using a fluorometric method.
- ddPCR Reaction Setup: Prepare a multiplex ddPCR reaction mix containing:
- 1x ddPCR Supermix
- Target-specific primers and FAM-labeled probe for CCR5Δ32
- Reference gene primers and HEX/VIC-labeled probe
- Approximately 20-50 ng of template genomic DNA
- Droplet Generation: Load the reaction mixture into a droplet generator to create thousands of nanoliter-sized water-in-oil droplets, effectively partitioning the sample.
- PCR Amplification: Transfer the droplets to a PCR plate and run the thermal cycling protocol with optimized annealing/extension temperatures for the primer-probe sets.
- Droplet Reading and Analysis: Read the plate on a droplet reader. Analyze the data to classify droplets as positive for wild-type, mutant, or both (heterozygous) based on fluorescence amplitude. Calculate the mutant allele frequency using the formula: Mutant Allele Frequency (%) = [Num. of mutant-positive droplets / (Num. of mutant-positive droplets + Num. of wild-type-positive droplets)] × 100 This system can reliably detect mutant alleles present at frequencies as low as 0.8% [28].
Research Reagent Solutions
The table below catalogues the essential reagents and materials required for the production and validation of CCR5-edited T-cell therapies.
Table 2: Essential Research Reagents for CCR5 Gene Editing and Analysis
| Reagent/Material | Function | Example/Note |
|---|---|---|
| CCR5-Targeting Nuclease | Mediates targeted DNA double-strand break in the CCR5 gene. | CCR5-Uco-hetTALEN [23] or CRISPR/Cas9 with gRNAs (e.g., CCR5-7, CCR5-8) [28]. |
| Nuclease Delivery Vector | Introduces nuclease encoding sequence into cells. | In-vitro transcribed mRNA for TALENs [23] or plasmid DNA for CRISPR/Cas9 system [28]. |
| Cell Culture Media | Supports the growth and expansion of T-cells. | RPMI-1640 supplemented with 10% FBS and cytokines [28]. |
| Electroporation System | Enforces transient cell membrane permeability for nuclease delivery. | CliniMACS Prodigy (automated) [23] or Gene Pulser Xcell (bench-top) [28]. |
| ddPCR Reagents | Enables absolute quantification of gene editing efficiency. | Assays with primers and probes specific to wild-type CCR5 and CCR5Δ32 alleles [23] [28]. |
| Cell Separation Reagents | Isulates target CD4+ T-cell population from PBMCs. | GMP-grade magnetic beads for clinical production [23]. |
Workflow Visualization
The following diagrams illustrate the integrated workflow for the production and quality control of CCR5-edited T-cells.
Diagram 1: Automated GMP Production of CCR5-Edited T-Cells.
Diagram 2: ddPCR Workflow for CCR5Δ32 Allele Quantification.
Conclusion
The integration of a optimized ddPCR workflow for CCR5Δ32 detection represents a significant advancement in the toolkit for HIV research and therapy development. This technique provides the sensitivity and absolute quantification necessary to monitor the low-frequency mutant alleles critical for the success of stem cell transplants and autologous gene-edited cell therapies. As the field moves towards multi-target editing strategies and combination immunotherapies, the role of precise molecular monitoring will only grow. Future directions should focus on further workflow automation, standardization across laboratories, and the application of direct detection methods in liquid biopsies to enable non-invasive therapy monitoring. By adopting this robust ddPCR framework, researchers can significantly contribute to the progress of achieving a functional cure for HIV.
References
- 1. CCR5: From Natural Resistance to a New Anti-HIV Strategy [https://pmc.ncbi.nlm.nih.gov/articles/PMC3185609/]
- 2. CCR5 gene editing and HIV immunotherapy - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12213807/]
- 3. Detection of CCR5Δ32 Mutant Alleles in Heterogeneous ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8898955/]
- 4. The CCR5-Delta32 Genetic Polymorphism and HIV-1 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6227735/]
- 5. In-depth virological and immunological characterization of ... [https://www.nature.com/articles/s41591-023-02213-x]
- 6. Innate resistance to HIV [https://en.wikipedia.org/wiki/Innate_resistance_to_HIV]
- 7. Diagnostic utility of droplet digital PCR for HIV reservoir ... [https://www.sciencedirect.com/science/article/pii/S205566402030460X]
- 8. Droplet digital PCR of viral DNA/RNA, current progress ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8013307/]
- 9. Digital PCR: from early developments to its future application ... [https://pubs.rsc.org/en/content/articlehtml/2025/lc/d5lc00055f]
- 10. Comparative Analysis of Viral Load Quantification Using ... [https://www.mdpi.com/1422-0067/26/2/725]
- 11. Comparative Study Between Droplet Digital PCR and Real ... [https://www.mdpi.com/2076-2607/13/11/2426]
- 12. Digital Assays Part I: Partitioning Statistics and Digital PCR [https://www.sciencedirect.com/science/article/pii/S2472630322013334]
- 13. Droplet digital PCR for absolute quantification of pathogens [https://pubmed.ncbi.nlm.nih.gov/25981265/]
- 14. Comparison of Quantitative PCR and Droplet Digital PCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4495218/]
- 15. Advancing quantitative PCR with color cycle multiplex ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11417387/]
- 16. Quantitative Data Quality Assurance, Analysis and Presentation [https://pmc.ncbi.nlm.nih.gov/articles/PMC12056448/]
- 17. Digital PCR: from early developments to its future application ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12278255/]
- 18. Comparative Performance of Digital PCR and Real-Time ... [https://www.mdpi.com/1999-4915/17/9/1259]
- 19. Droplet Digital PCR (ddPCR) Does Not Enhance the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10954086/]
- 20. Extraction of DNA from Blood Specimens [https://www.cdc.gov/dpdx/diagnosticprocedures/blood/dnaextraction.html]
- 21. Human blood cell-free DNA (cfDNA) extraction for ... [https://nanoporetech.com/document/extraction-method/singleplex-hb-cfdna]
- 22. Sample storage prior to extraction of genomic DNA T [https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/dna/handling-dna/sample-storage-prior-to-extraction-of-genomic-dna]
- 23. Automated production of CCR5-negative CD4 + -T cells in ... [https://www.nature.com/articles/s41434-021-00259-5]
- 24. DNA Extraction Guide: Mastering 7 Essential Sample Types [https://www.thermofisher.com/blog/life-in-the-lab/dna-extraction-guide-mastering-7-essential-sample-types/]
- 25. Exploring Cell free DNA extraction and testing | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/cfdna-extraction-here-s-what-you-need-to-know/]
- 26. DNA extraction tips and best practices for HiFi sequencing [https://www.pacb.com/blog/dna-extraction-tips-and-best-practices-for-hifi-sequencing/]
- 27. A hitchhiker's guide to cell-free DNA biology - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9650475/]
- 28. Detection of CCR5Δ32 Mutant Alleles in Heterogeneous ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2022.805931/full]
- 29. Detection of CCR5Δ32 Mutant Alleles in Heterogeneous ... [https://pubmed.ncbi.nlm.nih.gov/35265670/]
- 30. The evolutionary history of the CCR5-Δ32 HIV-resistance ... [https://www.sciencedirect.com/science/article/pii/S1286457904003636]
- 31. Ultra-Sensitive Quantification of Genome Editing Events ... [https://www.bioradiations.com/ultra-sensitive-quantification-of-genome-editing-events-using-droplet-digital-pcr/]
- 32. Optimizing Primer and Probe Concentrations for Use in ... [https://pubmed.ncbi.nlm.nih.gov/30275074/]
- 33. Digital droplet PCR is an accurate and precise method to ... [https://www.nature.com/articles/s41598-025-20944-4]
- 34. Comparing the precision of two digital PCR applications for ... [https://www.nature.com/articles/s41598-025-13143-8]
- 35. Development and application of a highly sensitive ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1612740/full]
- 36. A ddPCR method for simultaneous detection and ... [https://www.nature.com/articles/s41598-025-17272-y]
- 37. Evidence for the cure of HIV infection by CCR5Δ32/Δ32 ... [https://www.sciencedirect.com/science/article/pii/S0006497120431672]
- 38. HIV-1 remission following CCR / 5 haematopoietic... | Nature Δ 32 Δ 32 [https://www.nature.com/articles/s41586-019-1027-4?error=cookies_not_supported&code=886aeb2d-dab1-4edf-9f82-14bacc3fe5c4]
- 39. Inducing CCR5Δ32/Δ32 Homozygotes in the Human Jurkat ... [https://www.sciencedirect.com/science/article/pii/S2162253118301094]
- 40. Optimization of digital droplet polymerase chain reaction for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4827695/]
- 41. TaqMan Real-Time PCR Assays [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-assays.html]
- 42. dPCR: A Technology Review [https://www.mdpi.com/1424-8220/18/4/1271]
- 43. Application of Droplet Digital PCR for Estimating Vector Copy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5111482/]
- 44. Evaluation of PCR-enhancing approaches to reduce ... [https://www.sciencedirect.com/science/article/abs/pii/S0048969724069250]
- 45. Droplet Digital PCR Assay for Detection and Quantification ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12467802/]
- 46. Restriction Enzyme Digestion [https://www.neb.com/en-us/applications/cloning-and-synthetic-biology/dna-preparation/restriction-enzyme-digestion?srsltid=AfmBOorry_Y-njVn3bbsh2OleAQZnxpphjwJrjAdrh4jP3b7JKf9obFe]
- 47. Protocols Restriction Digest of Plasmid DNA [https://www.addgene.org/protocols/restriction-digest/]
- 48. Digital PCR Multiplex Assay Optimization [https://www.stillatechnologies.com/digital-pcr/primer-design/multiplex-assay-optimization/]
- 49. Development and validation of a droplet digital PCR assay ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1573949/full]
- 50. Deep Dive: qPCR [https://blog.addgene.org/deep-dive-qpcr]
- 51. Evidence for HIV-1 cure after CCR5Δ32/Δ32 allogeneic ... [https://www.sciencedirect.com/science/article/pii/S2352301820300692]
- 52. Development and Validation of a Droplet Digital PCR Assay for Detection of Feline Herpesvirus Type-1 [https://www.mdpi.com/2306-7381/12/11/1107]
- 53. Seven tips for avoiding DNA contamination in your PCR [https://www.takarabio.com/about/bioview-blog/tips-and-troubleshooting/avoid-dna-contamination-in-pcr?srsltid=AfmBOorbLFpLAKC7c35dMcN1fAFjHl1C_lQumiK2bPWmppehsIsuFI8Q]
- 54. Optimized (Pre) Analytical Conditions and Workflow for Droplet Digital PCR Analysis of Cell-Free DNA from Patients with Suspected Lung Carcinoma - PubMed [https://pubmed.ncbi.nlm.nih.gov/31229652/]
- 55. Quantifying the Detection Sensitivity and Precision of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11634988/]
- 56. Present and Future Applications of Digital PCR in ... [https://www.mdpi.com/2075-4418/14/9/931]
- 57. The difference between sensitivity and detection range in ... [https://www.bosterbio.com/blog/post/the-difference-between-sensitivity-and-the-minimum-level-of-assay-range-in-elisa-testing?srsltid=AfmBOorWHdsxYfE_b5huz4UyEeJvBvYeZSUecvIhASidHax8rSPpDMe2]
- 58. Methods to determine limit of detection and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5496743/]
- 59. Evaluation of process limit of detection and quantification ... [https://www.sciencedirect.com/science/article/pii/S0043135422000951]
- 60. Diagnostic utility of droplet digital PCR for HIV reservoir ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4967968/]
- 61. Orthogonal NGS for High Throughput Clinical Diagnostics [https://www.nature.com/articles/srep24650]
- 62. Analytical Performance and Concordance with Next ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10650055/]
- 63. Multicenter In-House Evaluation of an Amplicon-Based Next ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11860996/]
- 64. Validation of a One-Step Reverse Transcription-Droplet ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7934789/]
- 65. HeraNorm: an R shiny application for identifying optimal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12596163/]
- 66. Quantitative Evaluation of Endogenous Reference Genes ... [https://www.mdpi.com/2076-2607/13/8/1721]
- 67. Reference Gene Evaluation for digital PCR; applications ... [https://www.sciencedirect.com/science/article/pii/S0166093425001612]
- 68. Presenting Quantitative Data Graphically [https://courses.lumenlearning.com/waymakermath4libarts/chapter/presenting-quantitative-data-graphically/]
- 69. Presentation of Quantitative Data [https://ihatepsm.com/blog/presentation-quantitative-data]
- 70. Presenting data in tables and charts - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4008059/]
- 71. Evidence for HIV-1 cure after CCR5Δ32/Δ32 allogeneic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7606918/]
- 72. Simultaneous Detection and Viral DNA Load Quantification of Different Human Papillomavirus Types in Clinical Specimens by the High Analytical Droplet Digital PCR Method - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7710522/]
- 73. Digital droplet PCR (ddPCR) for the detection and quantification of HPV 16, 18, 33 and 45 - a short report - PubMed [https://pubmed.ncbi.nlm.nih.gov/28748500/]
- 74. HIV Molecular Monitoring Core | Frederick National Laboratory [https://frederick.cancer.gov/node/506]
- 75. An Integrated ddPCR Lab-on-a-Disc Device for Rapid Screening of Infectious Diseases [https://www.mdpi.com/2079-6374/14/1/2]
- 76. Digital PCR-dPCR Market Insights & Growth Outlook 2025 ... [https://www.congruencemarketinsights.com/report/digital-pcr-dpcr-market]