Optimizing Your Illumina Sequencer Selection for the AmpliSeq Childhood Cancer Panel: A Guide for MiSeq and NextSeq Users
This article provides a comprehensive guide for researchers and laboratory professionals on implementing the AmpliSeq for Illumina Childhood Cancer Panel across compatible Illumina sequencing platforms, with a focused comparison of...
Optimizing Your Illumina Sequencer Selection for the AmpliSeq Childhood Cancer Panel: A Guide for MiSeq and NextSeq Users
Abstract
This article provides a comprehensive guide for researchers and laboratory professionals on implementing the AmpliSeq for Illumina Childhood Cancer Panel across compatible Illumina sequencing platforms, with a focused comparison of MiSeq and NextSeq systems. It covers foundational knowledge of the panel's specifications and capabilities, detailed methodological protocols for library preparation and sequencing, best practices for troubleshooting and data optimization, and a review of the panel's technical validation and clinical utility in pediatric cancer research. The content is designed to empower scientists to generate high-quality, reliable genomic data to advance the understanding and treatment of childhood cancers.
Understanding the AmpliSeq Childhood Cancer Panel and Its Place in Your Lab
Targeted gene panels represent a powerful and efficient methodology in the genomic analysis of pediatric cancers, which are biologically distinct from adult malignancies. Unlike whole genome or exome sequencing, targeted panels concentrate sequencing efforts on a carefully selected subset of genomic regions known to be clinically relevant to disease pathogenesis. This focused approach enables researchers and clinicians to achieve exceptional sequencing depth at a fraction of the cost and computational resources required for broader sequencing methods, making comprehensive genomic profiling more accessible across healthcare settings [1]. The strategic design of these panels is particularly crucial for pediatric cancers, which often harbor different driver genes and variant types compared to adult cancers, including a higher prevalence of gene fusions and structural variants that require specialized detection capabilities [2].
The AmpliSeq for Illumina Childhood Cancer Panel exemplifies this targeted approach, offering a standardized solution for evaluating somatic variants across multiple pediatric cancer types, including leukemias, brain tumors, and sarcomas [3]. Similarly, the recently developed SJPedPanel from St. Jude Children's Research Hospital demonstrates the advanced capabilities of pediatric-specific design, covering critical coding and non-coding regions to identify subtype-defining fusions and other clinically relevant alterations [1] [2]. These panels address the unique molecular landscape of childhood cancers while providing the sensitivity required for analyzing challenging specimens with low tumor content, such as minimal residual disease monitoring samples [2].
Technical Specifications of Pediatric Cancer Panels
AmpliSeq for Illumina Childhood Cancer Panel
The AmpliSeq for Illumina Childhood Cancer Panel is a targeted resequencing solution specifically configured for comprehensive evaluation of somatic variants in childhood and young adult cancers. This integrated workflow employs amplicon-based sequencing to interrogate 203 genes associated with pediatric malignancies through a single, streamlined assay [3]. The panel simultaneously analyzes both DNA and RNA from patient samples, enabling detection of diverse variant types including single nucleotide polymorphisms (SNPs), insertions-deletions (indels), gene fusions, copy number variants (CNVs), and other somatic variants critical for molecular classification [3].
The technical workflow requires minimal input material, with only 10 ng of high-quality DNA or RNA needed per reaction, making it suitable for precious biopsy specimens and low-input samples [3]. Library preparation is remarkably efficient, requiring less than 1.5 hours of hands-on time and approximately 5-6 hours total assay time (excluding library quantification, normalization, and pooling steps) [3]. This rapid turnaround facilitates integration into research pipelines and clinical workflows where time-sensitive results are essential for therapeutic decision-making.
Table 1: AmpliSeq Childhood Cancer Panel Technical Specifications
| Parameter | Specification |
|---|---|
| Target Genes | 203 genes |
| Input Quantity | 10 ng DNA or RNA |
| Hands-On Time | < 1.5 hours |
| Total Assay Time | 5-6 hours (library prep only) |
| Nucleic Acid Types | DNA, RNA |
| Variant Types Detected | SNPs, indels, gene fusions, CNVs, somatic variants |
| Specialized Sample Types | Blood, bone marrow, FFPE tissue, low-input samples |
| Reactions per Kit | 24 reactions |
SJPedPanel: A Pediatric-Specific Design
The SJPedPanel developed at St. Jude Children's Research Hospital represents a significant advancement in targeted sequencing for childhood cancers, having been designed from inception specifically for pediatric malignancies rather than adapted from adult cancer panels. This panel exemplifies the evolution of pediatric cancer genomics, incorporating knowledge gained from large-scale initiatives like the Pediatric Cancer Genome Project to create an optimized testing approach [1]. The panel's design covers approximately 0.15% of the human genome, yet provides diagnostic information for >90% of pediatric cancer patients [1].
Unlike conventional panels focused primarily on coding regions, SJPedPanel incorporates comprehensive coverage of non-coding regions essential for detecting pediatric cancer drivers. The panel includes 5,275 coding exons plus 297 intronic regions for identifying fusion oncoproteins and structural variations, along with 7,590 polymorphic sites distributed across chromosomes for copy-number alteration analysis [2]. This sophisticated design enables detection of promoter/enhancer alterations such as those affecting TAL1 and TERT, and rearrangements responsible for fusion oncoproteins that are uniquely recurrent in pediatric cancers [2].
Table 2: Performance Comparison of Pediatric Cancer Gene Panels
| Performance Metric | SJPedPanel | Typical Commercial Panels |
|---|---|---|
| Coverage of Pediatric Cancer Driver Genes | ~90% | ~60% |
| Pathogenic Variant Coverage | 86% (of 485 variants in validation cohort) | Not specified |
| Rearrangement Detection | 82% (of 90 rearrangements in validation cohort) | Not specified |
| Detection Rate at AF 0.5% | ~95% | Varies |
| Detection Rate at AF 0.2% | ~80% | Varies |
| Specimens with Low Tumor Purity | Suitable for analysis | Challenging |
Compatible Sequencing Systems and Configurations
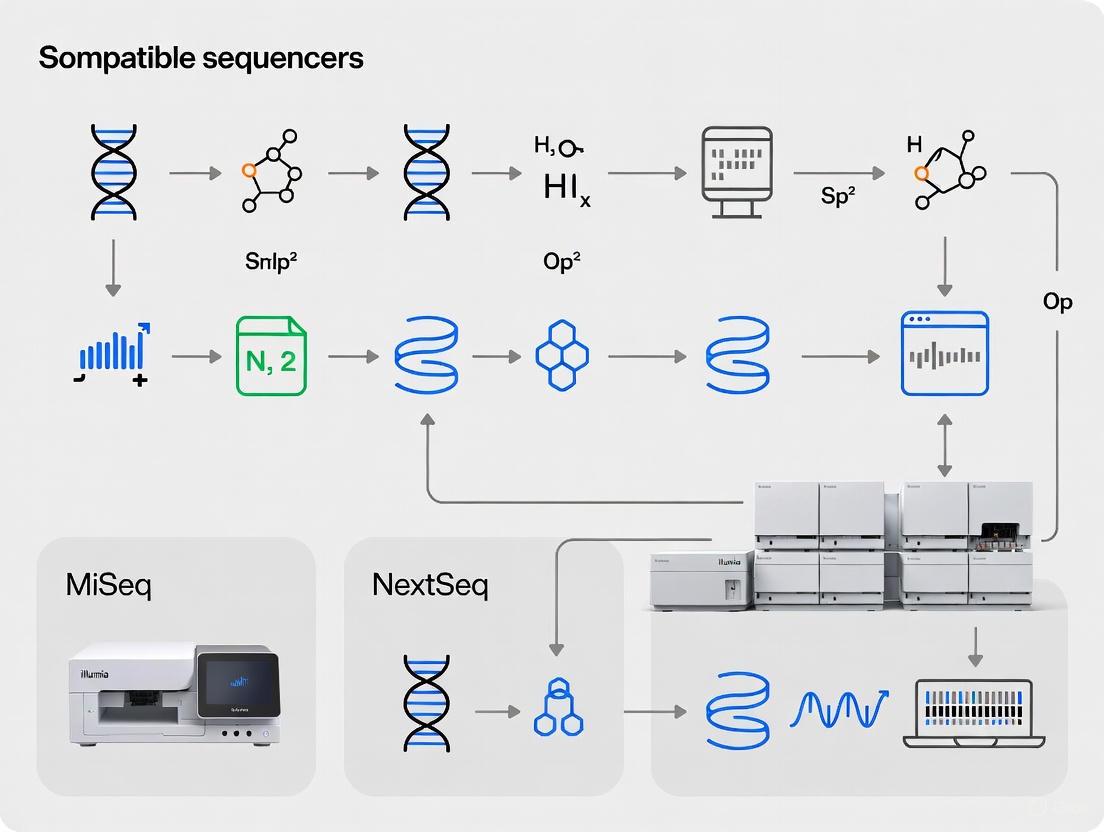
The AmpliSeq for Illumina Childhood Cancer Panel is compatible with multiple Illumina sequencing platforms, providing flexibility for laboratories with different infrastructure capabilities and throughput requirements. This compatibility enables researchers to select the most appropriate system based on their project scale, budget constraints, and turnaround time requirements [4].
The MiniSeq System offers an entry-level solution suitable for lower throughput applications, while the MiSeq System provides intermediate capacity with rapid turnaround times. For higher throughput needs, the NextSeq 1000/2000 Systems and NextSeq 550 System deliver enhanced sequencing capabilities with improved efficiency and scalability [3] [4]. This multi-platform support ensures that the childhood cancer panel can be implemented across diverse research environments, from individual laboratories to large core facilities.
Table 3: Sequencing Configuration Guidelines for AmpliSeq Childhood Cancer Panel
| Sequencing System | Reagent Kit | Max DNA Samples Per Run | Max RNA Samples Per Run | Max Combined Samples Per Run | DNA:RNA Pooling Ratio |
|---|---|---|---|---|---|
| MiniSeq | Mid Output | 1 | 8 | 1 | 5:1 |
| MiniSeq | High Output | 5 | 25 | 4 | 5:1 |
| MiSeq | Reagent Kit v2 | 3 | 15 | 2 | 5:1 |
| MiSeq | Reagent Kit v3 | 5 | 25 | 4 | 5:1 |
| NextSeq | Mid Output v2 | 27 | 96 | 22 | 5:1 |
| NextSeq | High Output v2 | 83 | 96 | 48 | 5:1 |
The recommended 5:1 DNA:RNA pooling ratio is based on optimal read coverage requirements for each nucleic acid type [4]. This balanced approach ensures sufficient depth for variant detection while maximizing sample throughput and cost-efficiency. For the SJPedPanel, sequencing depth requirements vary based on application: 2,500X for medium tumor purity (2.5%-10%), 5,000X for low tumor purity (0.5%-1%), and 10,000X for ultralow tumor purity (0.1%-0.2%) samples [2].
Essential Research Reagent Solutions
Implementing the AmpliSeq for Illumina Childhood Cancer Panel requires several complementary reagent systems to complete the workflow from sample to sequence data. These integrated solutions ensure optimal performance and reproducible results across applications.
Table 4: Essential Research Reagents for Childhood Cancer Panel Workflow
| Component | Function | Configuration Options |
|---|---|---|
| AmpliSeq Library PLUS | Library preparation reagents | 24-, 96-, 384-reaction kits |
| AmpliSeq CD Indexes | Sample multiplexing | Sets A-D (96 indexes each) |
| cDNA Synthesis Kit | RNA to cDNA conversion | Required for RNA analysis |
| Library Equalizer | Library normalization | Bead-based normalization |
| Direct FFPE DNA | DNA from FFPE tissue | 24 reactions, no purification needed |
| Sample ID Panel | Sample tracking | SNP-based sample identification |
The modular design of these reagent systems allows researchers to scale operations based on project requirements while maintaining consistency across experiments [3]. The AmpliSeq CD Indexes are particularly important for multiplexed sequencing, enabling efficient sample pooling and cost-effective processing through unique dual indexing that minimizes index hopping and cross-contamination [3]. For RNA analysis, the cDNA Synthesis Kit is essential for converting total RNA to cDNA compatible with the AmpliSeq workflow, ensuring comprehensive detection of fusion events and expression-based markers [3].
Experimental Protocol and Workflow
Library Preparation Methodology
The library preparation process for the AmpliSeq Childhood Cancer Panel follows a standardized workflow with specific quality control checkpoints. For DNA analysis, the process begins with 4X AmpliSeq Childhood Cancer DNA Panel pools, while RNA analysis utilizes 5X AmpliSeq Childhood Cancer RNA Panel pools [5]. The procedure involves several critical steps:
Template Preparation: Dilute DNA or RNA samples to appropriate concentrations in nuclease-free water. For RNA samples, first perform cDNA synthesis using the AmpliSeq cDNA Synthesis Kit according to manufacturer specifications [3].
Amplification Reaction: Combine the DNA or cDNA template with the AmpliSeq Childhood Cancer Panel pools and AmpliSeq HiFi Mix in a thermal cycler. Use the following cycling conditions: initial hold at 99°C for 2 minutes; 21 cycles of 99°C for 15 seconds and 60°C for 4 minutes; final hold at 10°C [3].
Partial Digest: Treat amplified products with FuPa reagent to partially digest primer sequences and prepare amplicons for adapter ligation. Incubate at 50°C for 10 minutes, 55°C for 10 minutes, then hold at 60°C [3].
Adapter Ligation: Add AmpliSeq CD Indexes and DNA Ligase to the reaction mixture. Incubate at 22°C for 30 minutes followed by 68°C for 5 minutes [3].
Library Amplification: Amplify the ligated products using the following protocol: 98°C for 1 minute; 9-12 cycles of 98°C for 15 seconds and 60°C for 1 minute; final hold at 10°C [3].
Library Normalization: Purify and normalize libraries using the AmpliSeq Library Equalizer according to manufacturer instructions to ensure balanced representation in sequencing [3].
Validation and Performance Assessment
The validation of pediatric cancer panels requires rigorous testing using clinically characterized samples and reference materials. The SJPedPanel validation approach provides a comprehensive framework for performance assessment:
Dilution Experiments: Prepare serial dilutions of cancer cell lines (e.g., ME1, 697, Rh30, EW8, K562, Molm13) in non-cancer cell line (GM12878) background to achieve tumor concentrations ranging from 0.1% to 10% [2]. Sequence these dilutions at appropriate depths: 10,000X for ultralow (0.1-0.2%), 5,000X for low (0.5-1%), and 2,500X for medium (2.5-10%) tumor concentrations [2].
Limit of Detection Determination: Calculate recall rates of known cell line-specific markers (SNVs, indels, SVs) across different dilutions to establish assay sensitivity and specificity [2]. Implement computational error suppression methods to achieve background error rates of approximately 10⁻⁶ to 10⁻⁴ for substitutions [2].
Coverage Uniformity Assessment: Evaluate capture efficiency and coverage uniformity across all targeted regions using control samples like COLO829BL cell line sequenced at both high-depth (~2,000X) and low-depth (~200X) on Illumina NovaSeq and NextSeq platforms [2].
In Silico Downsampling: Perform computational downsampling of sequencing data from dilution samples to determine optimal balance between sequencing depth, variant recall rate, and cost efficiency [2].
Data Analysis and Interpretation Framework
Bioinformatics Processing Pipeline
The analysis of data generated from pediatric cancer panels requires specialized bioinformatics approaches tailored to the unique variant spectrum of childhood malignancies. The SJPedPanel analysis framework demonstrates key considerations:
Variant Calling: Implement specialized algorithms for detecting diverse variant types including single nucleotide variants (SNVs), small insertions and deletions (indels), gene fusions, structural variations (SVs), and internal tandem duplications (ITDs) [2].
Copy Number Alteration Analysis: Utilize the panel's 7,590 evenly distributed SNP sites across chromosomes to detect large genomic structural rearrangements such as copy number variants (CNVs) and loss of heterozygosity (LOH) [2].
Error Suppression: Apply computational error correction methods specifically optimized for targeted sequencing data to achieve ultra-low error rates necessary for detecting low-frequency variants in minimal residual disease settings [2].
Annotation and Prioritization: Annotate identified variants using pediatric-specific knowledge bases incorporating data from initiatives like the Pediatric Cancer Genome Project and recent research discoveries such as the UBTF gene identified in 2022 [1].
Clinical and Research Applications
The implementation of targeted pediatric cancer panels enables multiple advanced applications in both clinical management and research settings:
Diagnostic Classification: Molecular subtyping of childhood cancers based on signature genetic alterations, with the SJPedPanel providing diagnoses for >90% of pediatric cancer patients through detection of subtype-defining fusions and mutations [1].
Minimal Residual Disease Monitoring: Ultra-deep sequencing capability enables detection of low-frequency driver alterations (as low as 0.2% allele fraction) in morphologic remission samples, providing sensitive monitoring for disease recurrence [2].
Therapeutic Target Identification: Detection of clinically actionable alterations guides targeted treatment approaches, with the comprehensive gene coverage ensuring identification of potentially targetable mutations across pediatric cancer types [1].
Specimens with Low Tumor Purity: Enhanced sensitivity compared to whole genome sequencing for analyzing samples with low cancer cell content or after bone marrow transplantation, addressing an important clinical gap in pediatric oncology [1].
Advancing Pediatric Cancer Research Through Data Sharing
The full potential of genomic profiling in pediatric oncology is realized through collaborative data sharing initiatives that aggregate information across institutions. The success of the International Neuroblastoma Risk Group (INRG) Data Commons demonstrates the power of this approach, having collected data on more than 26,000 neuroblastoma patients worldwide and enabled over 40 research studies that have directly influenced treatment approaches [6]. This model has expanded to the Pediatric Cancer Data Commons (PCDC), which now houses data on over 44,400 patients across 17 different childhood cancers and conditions [6].
Recent government initiatives further reinforce the importance of data sharing in pediatric oncology. The Childhood Cancer Data Initiative at the National Cancer Institute has seen funding increased to $100 million to accelerate the development of improved diagnostics, treatments, and prevention strategies through advanced artificial intelligence applications [7]. These collaborative frameworks, combined with targeted genomic technologies, create an powerful ecosystem for advancing our understanding of childhood cancers and developing more effective, personalized treatments for pediatric patients.
Targeted next-generation sequencing (NGS) panels have revolutionized oncology research by enabling simultaneous assessment of multiple genomic alteration types from limited sample material. The AmpliSeq for Illumina Childhood Cancer Panel represents a significant advancement for researchers investigating pediatric and young adult cancers, providing a comprehensive solution for evaluating 203 genes associated with various cancer types including leukemias, brain tumors, and sarcomas [3]. This integrated workflow addresses the critical need for efficient genomic profiling in childhood cancers, where sample availability is often limited and comprehensive data is essential for understanding disease mechanisms.
Traditional molecular testing approaches relying on sequential single-gene tests present significant limitations for pediatric cancer research. These methods consume precious biopsy material and may miss important biomarkers due to their limited content scope [8]. The Childhood Cancer Panel overcomes these challenges by consolidating multiple assay types into a single, efficient workflow that preserves sample integrity while maximizing data output. This technical guide provides researchers with comprehensive information on implementing this targeted resequencing solution within their experimental frameworks, with specific focus on compatibility across Illumina sequencing platforms.
Panel Specifications and Technical Parameters
Comprehensive Genomic Coverage
The AmpliSeq Childhood Cancer Panel employs a targeted resequencing approach specifically designed for somatic variant detection in pediatric and young adult cancer research. The panelinterrogates 203 genes with known associations to childhood cancers through a single optimized workflow [3]. This content was carefully selected to cover clinically relevant genomic regions while maintaining efficiency in library preparation and sequencing.
The panel's comprehensive design detects multiple variant classes simultaneously, providing researchers with a complete genomic profile from minimal input material. The technical specifications demonstrate the panel's versatility across sample types and experimental conditions, making it suitable for various research scenarios in pediatric oncology.
Technical Specifications and Performance Metrics
Table 1: Technical Specifications of the AmpliSeq Childhood Cancer Panel
| Parameter | Specification |
|---|---|
| Number of Genes | 203 genes associated with childhood and young adult cancers [3] |
| Variant Classes Detected | Single nucleotide variants (SNVs), Insertions-deletions (indels), Copy number variants (CNVs), Gene fusions, Somatic variants [3] |
| Input Requirements | 10 ng high-quality DNA or RNA [3] |
| Hands-on Time | < 1.5 hours [3] |
| Total Assay Time | 5-6 hours (library preparation only) [3] |
| Nucleic Acid Type | DNA, RNA [3] |
| Specialized Sample Types | Blood, Bone marrow, FFPE tissue, Low-input samples [3] |
The panel's technical profile demonstrates significant advantages for childhood cancer research. The minimal hands-on time of less than 1.5 hours streamlines laboratory workflows, while the 5-6 hour total assay time enables rapid turnaround for research applications [3]. The flexibility to use both DNA and RNA from various sample types, including challenging FFPE tissues, makes this panel particularly valuable for translational research utilizing archival specimens.
The low input requirement of only 10 ng of high-quality DNA or RNA is especially crucial for pediatric cancers, where biopsy material is often limited [3]. This efficient input utilization preserves precious samples for additional analyses while still generating comprehensive genomic data.
Compatible Sequencing Systems and Experimental Design
Supported Illumina Sequencing Platforms
The AmpliSeq Childhood Cancer Panel is compatible with multiple Illumina sequencing systems, providing researchers with flexibility in experimental design and throughput capacity. The panel has been validated for use with MiSeq System, NextSeq 550 System, NextSeq 2000 System, NextSeq 1000 System, MiSeqDx in Research Mode, and MiniSeq System [3]. This broad compatibility ensures that researchers can implement the panel regardless of their laboratory's specific Illumina instrumentation.
The selection of an appropriate sequencing platform depends on several factors, including project scale, desired throughput, and coverage requirements. Each system offers distinct advantages for different research scenarios, from smaller-scale investigations to higher-throughput studies.
Platform Selection Guidelines
Table 2: Compatible Illumina Sequencers and Application Guidelines
| Sequencing System | Recommended Application | Key Advantages |
|---|---|---|
| MiSeq System | Targeted sequencing projects, method validation | Benchtop convenience, fast turnaround time [3] |
| NextSeq 1000/2000 Systems | Medium to high-throughput projects | Production-scale power in a compact system [3] |
| NextSeq 550 System | Flexible DNA and RNA analysis | Dual functionality for array scanning and sequencing [3] |
| MiniSeq System | Low to medium-throughput studies | Most affordable entry point to Illumina sequencing [3] |
For researchers focusing specifically on childhood cancer research, the MiSeq and NextSeq series systems provide optimal performance for the AmpliSeq Childhood Cancer Panel. The MiSeq System offers benchtop convenience for smaller studies or validation work, while the NextSeq 1000/2000 Systems provide higher throughput for more comprehensive research projects [3]. The NextSeq 550 System adds flexibility with its dual functionality for both sequencing and array scanning applications.
Experimental Design Considerations
The following decision pathway illustrates the experimental design process for implementing the AmpliSeq Childhood Cancer Panel:
Figure 1: Experimental workflow for implementing the AmpliSeq Childhood Cancer Panel, showing key steps from sample preparation through data analysis and platform selection criteria.
Integrated Workflow and Research Reagent Solutions
End-to-End Experimental Workflow
The complete AmpliSeq Childhood Cancer Panel workflow integrates library preparation, sequencing, and data analysis into a streamlined process. The workflow begins with sample preparation and quality control, proceeds through library preparation and sequencing, and concludes with comprehensive data analysis and interpretation.
The following workflow diagram illustrates the complete experimental process from sample to analysis:
Figure 2: Complete end-to-end workflow for the AmpliSeq Childhood Cancer Panel, showing key steps and specialized processing options for challenging sample types like FFPE tissues.
Essential Research Reagent Solutions
Successful implementation of the AmpliSeq Childhood Cancer Panel requires several specialized reagents and companion products that optimize performance across various sample types and experimental conditions.
Table 3: Essential Research Reagents for Childhood Cancer Panel Implementation
| Reagent Solution | Function | Application Context |
|---|---|---|
| AmpliSeq Library PLUS | Provides core reagents for preparing sequencing libraries | Required for all library preparations; available in 24, 96, or 384 reactions [3] |
| AmpliSeq CD Indexes | Enables sample multiplexing through unique barcode sequences | Essential for pooling multiple samples; available in sets A-D with 96 indexes each [3] |
| AmpliSeq cDNA Synthesis for Illumina | Converts total RNA to cDNA for RNA sequencing applications | Required when working with RNA samples or RNA panels [3] |
| AmpliSeq for Illumina Direct FFPE DNA | Enables DNA preparation from FFPE tissues without deparaffinization | Streamlines workflow for archival tissue samples; eliminates DNA purification step [3] |
| AmpliSeq Library Equalizer for Illumina | Normalizes libraries for balanced sequencing representation | Improves sequencing efficiency and data quality [3] |
| AmpliSeq for Illumina Sample ID Panel | Provides SNP genotyping for sample identification | Generates unique IDs for each research sample; includes gender-determining pair [3] |
These specialized reagents address common challenges in childhood cancer research, particularly when working with limited or challenging sample types. The Direct FFPE DNA solution is particularly valuable for translational studies utilizing archival tissue resources, as it eliminates the need for deparaffinization or DNA purification steps [3]. The Library Equalizer ensures consistent coverage across samples, while the Sample ID Panel provides crucial sample tracking capabilities essential for research integrity.
Methodological Framework and Data Analysis
Comprehensive Variant Detection Methodology
The AmpliSeq Childhood Cancer Panel employs a multiplex PCR-based approach that enables simultaneous amplification of target regions across the 203-gene content. This method provides uniform coverage and highly efficient target enrichment, making it particularly suitable for analyzing degraded samples such as FFPE tissues [3]. The panel's design incorporates optimized primer sequences that ensure specific binding and minimal off-target amplification.
The technical methodology builds upon established NGS principles while incorporating specific enhancements for pediatric cancer genomics. The panel utilizes a dual-strand sequencing approach that minimizes artifacts and improves variant calling accuracy, particularly for low-frequency variants that may be relevant in heterogeneous tumor samples. The integrated DNA and RNA analysis capability provides complementary information for comprehensive genomic profiling.
Data Analysis and Interpretation Framework
The data analysis pipeline for the Childhood Cancer Panel follows best practices for somatic variant detection in targeted sequencing data. The process includes base calling, quality assessment, alignment to reference genome, variant identification, and functional annotation. Specialized algorithms are employed for different variant types: GATK tools for SNVs and indels [9], ExomeDepth or cn.MOPS for CNV detection [9], and fusion-aware aligners for gene fusion identification.
Variant annotation incorporates multiple biological databases to prioritize potentially clinically significant findings. The interpretation framework should consider population frequency data, functional prediction algorithms, and cancer-specific databases to distinguish driver mutations from passenger events. For childhood cancers, particular attention should be paid to germline variants that may indicate cancer predisposition syndromes, requiring matched normal tissue analysis when available.
Advancements in Childhood Cancer Genomics
Technical Advantages Over Conventional Methods
The AmpliSeq Childhood Cancer Panel represents a significant advancement over traditional biomarker detection methods such as FISH, PCR, and IHC. While these conventional techniques are limited in scope and require significant amounts of biopsy material, the NGS-based approach enables comprehensive assessment of hundreds of biomarkers simultaneously from minimal input [8]. This comprehensive profiling provides more opportunity to match research findings with biologically relevant mechanisms and potential therapeutic targets.
The panel's ability to detect multiple variant classes—including SNVs, indels, CNVs, and fusions—in a single assay is particularly valuable for childhood cancers, where diverse genomic alterations may contribute to pathogenesis [3]. This multi-analyte approach conserves precious sample material that would otherwise be divided among multiple single-analyte tests, while providing a more complete genomic profile from limited material.
Research Applications and Future Directions
The implementation of comprehensive genomic profiling in childhood cancer research enables several advanced applications beyond routine variant detection. The panel's data can support assessment of complex genomic signatures such as tumor mutational burden (TMB) and microsatellite instability (MSI), which are increasingly relevant for immunotherapy research [8]. The simultaneous DNA and RNA analysis capability also facilitates detection of expressed mutations and alternative splicing events that may have functional significance.
Future applications may include longitudinal monitoring of treatment response and resistance mechanisms, particularly when applied to liquid biopsy samples. The panel's efficient design and compatibility with multiple Illumina platforms make it suitable for adaptation to emerging sequencing technologies that may offer further improvements in throughput, cost-effectiveness, and analytical sensitivity for childhood cancer research.
Targeted next-generation sequencing (NGS) has revolutionized oncogenomics by enabling comprehensive molecular profiling of tumors. For pediatric and young adult cancers, the AmpliSeq for Illumina Childhood Cancer Panel provides a specialized tool for evaluating somatic variants across 203 genes associated with childhood cancers [3]. This technical guide examines the compatibility of Illumina sequencing systems with this panel, providing researchers and drug development professionals with essential information for experimental planning and platform selection. Understanding the capabilities of each sequencing system—from the MiSeq to NextSeq platforms—is crucial for optimizing throughput, coverage, and resource allocation in childhood cancer research.
The AmpliSeq for Illumina Childhood Cancer Panel is a targeted resequencing solution designed specifically for comprehensive evaluation of somatic variants in childhood and young adult cancers [3]. The panel employs amplicon sequencing to analyze multiple variant types including single nucleotide polymorphisms (SNPs), gene fusions, somatic variants, insertions-deletions (indels), and copy number variants (CNVs) [3]. This multi-variant approach provides researchers with a comprehensive view of the genomic alterations driving pediatric cancers.
The panel simultaneously analyzes both DNA and RNA from patient samples, requiring separate library preparations for each nucleic acid type. The DNA component targets 3,069 amplicons with an average length of 114 base pairs, while the RNA component targets 1,701 amplicons with an average length of 122 base pairs [4]. The library preparation process requires approximately 5-6 hours of assay time with less than 1.5 hours of hands-on time, and utilizes a minimal input of 10 ng of high-quality DNA or RNA [3]. This streamlined workflow enables rapid preparation of libraries for various sample types including blood, bone marrow, and FFPE tissue [3].
Compatible Sequencing Systems and Performance Specifications
The AmpliSeq Childhood Cancer Panel is compatible with multiple Illumina sequencing platforms, each offering different throughput capacities suitable for varying project scales. The table below summarizes the key performance metrics for each compatible system:
Sequencing System Specifications for Childhood Cancer Panel
| System | Reagent Kit | Maximum DNA Samples Per Run | Maximum RNA Samples Per Run | Maximum Combined* Samples Per Run | Recommended DNA:RNA Pooling Ratio | Run Time |
|---|---|---|---|---|---|---|
| MiniSeq System | MiniSeq Mid Output Kit | 1 | 8 | 1 | 5:1 | 17 hours |
| MiniSeq High Output Kit | 5 | 25 | 4 | 5:1 | 24 hours | |
| MiSeq System | MiSeq Reagent Kit v2 | 3 | 15 | 2 | 5:1 | 24 hours |
| MiSeq Reagent Kit v3 | 5 | 25 | 4 | 5:1 | 32 hours | |
| NextSeq System | NextSeq Mid Output v2 Kit | 27 | 96 | 22 | 5:1 | 26 hours |
| NextSeq High Output v2 Kit | 83 | 96 | 48 | 5:1 | 29 hours |
*Combined samples refer to paired DNA and RNA from the same sample, generating two separately indexed libraries [4].
The selection of an appropriate sequencing system depends on several factors including project scale, required throughput, and time constraints. The MiniSeq System offers a lower-throughput solution suitable for smaller projects or individual samples, while the MiSeq System provides moderate throughput with run times extending up to 32 hours. For larger studies, the NextSeq System delivers significantly higher throughput, capable of processing up to 83 DNA-only samples or 48 combined DNA-RNA samples per run [4]. The recommended 5:1 DNA:RNA pooling ratio is based on optimal read coverage requirements for each nucleic acid type [4].
Experimental Design and Workflow
Library Preparation and Sample Requirements
The experimental workflow for the Childhood Cancer Panel begins with library preparation, which requires several specialized reagents and kits. The following table outlines the essential components needed for library construction:
Research Reagent Solutions for Library Preparation
| Item | Function | Configuration Options |
|---|---|---|
| AmpliSeq for Illumina Childhood Cancer Panel | Core primer pools for targeting cancer-associated genes | Fixed configuration for 24 samples |
| AmpliSeq Library PLUS for Illumina | Reagents for library preparation | 24-, 96-, or 384-reaction configurations |
| AmpliSeq CD Indexes | Sample barcoding for multiplexing | Set A, B, C, or D (96 indexes each) |
| AmpliSeq cDNA Synthesis for Illumina | Converts total RNA to cDNA for RNA panels | Required for RNA analysis |
| AmpliSeq Library Equalizer | Normalizes libraries for sequencing | Included in library prep workflow |
The Childhood Cancer Panel generates one DNA and one RNA library per sample, with each library requiring separate barcoding [4]. For a standard set of 24 samples, this results in 48 total libraries (24 DNA and 24 RNA) [4]. The required number of kits scales accordingly, with 24 samples typically requiring one Childhood Cancer Panel, two 24-reaction Library PLUS kits, one AmpliSeq CD Set A (96 indexes), and one cDNA Synthesis kit [4].
The following diagram illustrates the complete workflow from sample preparation through data analysis:
Sequencing and Data Analysis
For sequencing, the prepared libraries are pooled at the recommended 5:1 DNA:RNA ratio based on read coverage requirements [4]. The pooled libraries are then loaded onto the chosen Illumina sequencing system. Following sequencing, data analysis can be performed using several Illumina bioinformatics solutions. The DNA Amplicon Analysis App and RNA Amplicon Analysis App are available on BaseSpace Sequence Hub, with similar analysis modules available in Local Run Manager [10]. These workflows perform alignment, variant calling (for DNA), and fusion calling (for RNA). For additional CNV analysis, the OncoCNV caller BaseSpace Lab App is available [10].
Technical Validation and Performance Metrics
Independent technical validation of the AmpliSeq Childhood Cancer Panel demonstrates its robustness for clinical research applications. A 2022 study published in Frontiers in Molecular Biosciences validated the panel for acute leukemia diagnostics, reporting a mean read depth greater than 1000× across targets [11]. The panel demonstrated high sensitivity for DNA (98.5% for variants with 5% variant allele frequency) and RNA (94.4%), with 100% specificity and reproducibility for DNA and 89% reproducibility for RNA [11].
In terms of clinical utility, the validation study found that 49% of mutations and 97% of the fusions identified had clinical impact, with 41% of mutations refining diagnosis and 49% considered targetable [11]. For RNA, fusion genes were particularly impactful, refining diagnosis in 97% of cases [11]. Overall, the panel provided clinically relevant results in 43% of patients tested in the cohort, demonstrating its utility in pediatric hematology practice [11].
Platform Selection Guidance
Considerations for System Choice
Selecting the appropriate Illumina sequencing system depends on several factors:
Project Scale: For small-scale studies or pilot projects (1-5 samples), the MiniSeq or MiSeq systems offer appropriate throughput. For larger studies requiring higher sample throughput, the NextSeq systems provide substantially greater capacity [4].
Turnaround Time: When time-to-results is critical, the MiniSeq with Mid Output kit provides the fastest turnaround at 17 hours, while larger capacity runs on MiSeq or NextSeq systems require 24-32 hours [4].
Multiplexing Flexibility: Researchers can run up to three different AmpliSeq designs with barcodes on the same sequencing run, provided the target amplicon size and required coverage can be achieved [10].
Coverage Requirements: Coverage can be manipulated by increasing sequencing throughput (using a larger flow cell output) or reducing the number of samples pooled per run [10].
Application in Cancer Research
The Childhood Cancer Panel enables multiple research applications in pediatric oncology. The panel's design covers genes associated with various pediatric cancer types including leukemias, brain tumors, and sarcomas [3]. By combining DNA and RNA analysis in a single workflow, researchers can detect multiple variant types including single nucleotide variants, insertions-deletions, copy number variants, and gene fusions, providing a comprehensive molecular profile of childhood tumors.
The panel's technical performance characteristics, including its sensitivity for low-frequency variants and ability to work with challenging sample types like FFPE tissue, make it particularly valuable for translational research applications [3] [11]. The integration of this panel into research workflows provides valuable genetic information that can redefine diagnostic, prognostic, and therapeutic strategies for the management of childhood cancers [11].
The AmpliSeq for Illumina Childhood Cancer Panel represents a specialized tool for genomic investigation of pediatric and young adult cancers, compatible across Illumina's sequencing portfolio from MiSeq to NextSeq systems. Each platform offers distinct advantages in throughput, run time, and sample capacity, allowing researchers to select systems based on their specific project requirements. The comprehensive nature of the panel, combined with its validated performance characteristics, enables researchers to obtain crucial molecular insights into childhood cancers. By understanding the capabilities and specifications of each compatible sequencing system, research and drug development professionals can optimize their experimental designs to advance our understanding and treatment of childhood cancers.
This technical guide provides a comprehensive analysis of the workflow efficiency for the AmpliSeq for Illumina Childhood Cancer Panel, with a specific focus on hands-on time requirements and total assay duration. Targeted at researchers, scientists, and drug development professionals, this whitepaper synthesizes experimental protocols and performance metrics to facilitate informed planning and optimization of childhood cancer genomics research. The data presented herein is contextualized within a broader evaluation of compatible Illumina sequencing platforms, particularly MiSeq and NextSeq systems, to provide a complete picture of the integrated workflow from library preparation to final sequencing output.
Workflow Timing and Process Breakdown
The AmpliSeq for Illumina Childhood Cancer Panel features a streamlined workflow designed to maximize efficiency while maintaining data quality. The process can be divided into distinct phases with specific time requirements.
Core Timing Metrics
| Metric | Specification |
|---|---|
| Total Hands-On Time | < 1.5 hours [3] |
| Total Library Prep Assay Time | 5-6 hours [3] |
| Input Quantity | 10 ng high-quality DNA or RNA [3] |
| Library Prep Method | Amplicon sequencing [3] |
Table 1: Core workflow efficiency metrics for the Childhood Cancer Panel
It is important to note that the 5-6 hour library preparation time does not include additional time required for library quantification, normalization, or pooling operations [3]. These ancillary steps must be factored into overall project planning.
Sequencing Time Considerations
Sequencing duration varies significantly based on the Illumina platform and reagent kit selection, creating important trade-offs between throughput and turnaround time.
| System | Reagent Kit | Run Time | Max DNA Samples Per Run | Max RNA Samples Per Run |
|---|---|---|---|---|
| MiSeq System | MiSeq Reagent Kit v2 | ~24 hours [4] | 3 [4] | 15 [4] |
| MiSeq System | MiSeq Reagent Kit v3 | ~32 hours [4] | 5 [4] | 25 [4] |
| NextSeq System | NextSeq Mid Output v2 Kit | ~26 hours [4] | 27 [4] | 96 [4] |
| NextSeq System | NextSeq High Output v2 Kit | ~29 hours [4] | 83 [4] | 96 [4] |
Table 2: Sequencing platform performance characteristics for Childhood Cancer Panel applications
Experimental Protocols and Methodologies
Library Preparation Workflow
The AmpliSeq for Illumina Childhood Cancer Panel employs a PCR-based targeted resequencing approach that enables comprehensive evaluation of somatic variants across 203 genes associated with childhood and young adult cancers [3]. The protocol generates separate DNA and RNA libraries for each sample, requiring coordinated processing of both nucleic acid types.
Diagram 1: Library preparation workflow
Key Experimental Notes:
- The panel utilizes two primer pools for DNA (3,069 amplicons) and two pools for RNA (1,701 amplicons) to comprehensively cover the 203 target genes [4]
- For RNA samples, an initial cDNA synthesis step is required using the AmpliSeq cDNA Synthesis for Illumina kit [3]
- The protocol supports multiple sample types including blood, bone marrow, and FFPE tissue, with specialized solutions available for challenging sample types like the AmpliSeq for Illumina Direct FFPE DNA kit that eliminates need for deparaffinization or DNA purification [3]
- Library normalization is streamlined using the AmpliSeq Library Equalizer for Illumina, which provides bead-based normalization to reduce hands-on time [3]
Platform Selection Logic
The choice between MiSeq and NextSeq systems involves careful consideration of project scale, required throughput, and turnaround time requirements.
Diagram 2: Sequencing platform selection logic
Research Reagent Solutions
Successful implementation of the Childhood Cancer Panel workflow requires specific reagent systems designed for compatibility and efficiency.
| Component | Product Name | Function | Specifications |
|---|---|---|---|
| Core Panel | AmpliSeq for Illumina Childhood Cancer Panel | Target enrichment for 203 childhood cancer genes | 24 reactions; detects SNPs, indels, CNVs, fusions [3] |
| Library Prep | AmpliSeq Library PLUS | PCR-based library construction | Available in 24-, 96-, 384-reaction configurations [3] |
| Indexing | AmpliSeq CD Indexes | Sample multiplexing | 8bp indexes; available in sets A-D (96 indexes each) [3] |
| RNA Conversion | AmpliSeq cDNA Synthesis | RNA-to-cDNA conversion for RNA panels | Required for RNA input; compatible with total RNA [3] |
| Normalization | AmpliSeq Library Equalizer | Library concentration normalization | Bead-based normalization; reduces pipetting steps [3] |
| FFPE Optimization | AmpliSeq for Illumina Direct FFPE DNA | Direct library prep from FFPE tissue | Eliminates deparaffinization and DNA purification [3] |
| Sample Tracking | AmpliSeq for Illumina Sample ID Panel | Sample identification and tracking | 8 SNP primer pairs + gender determination [3] |
Table 3: Essential research reagent solutions for Childhood Cancer Panel workflow
Sequencing Platform Performance Characteristics
The compatibility of the Childhood Cancer Panel with multiple Illumina platforms provides flexibility for different laboratory needs and project scales.
Output and Capacity Specifications
| System | Reagent Kit | Read Length | Total Output | Reads Passing Filter |
|---|---|---|---|---|
| MiSeq | MiSeq Reagent Kit v2 | 2 × 150 bp | 4.5–5.1 Gb [12] | 24–30M paired-end reads [12] |
| MiSeq | MiSeq Reagent Kit v3 | 2 × 300 bp | 13.2–15 Gb [12] | 44–50M paired-end reads [12] |
| NextSeq | NextSeq Mid Output v2 Kit | Not specified | Not specified in results | Not specified in results |
| NextSeq | NextSeq High Output v2 Kit | Not specified | Not specified in results | Not specified in results |
Table 4: Sequencing output specifications for compatible platforms
Data Quality and Performance Metrics
The MiSeq system demonstrates exceptional data quality across various run configurations, with Q30 scores exceeding 70% even at the maximum read length of 2 × 300 bp [12]. This high accuracy is particularly valuable for somatic variant detection in cancer research, where confidence in variant calls is critical.
The integrated workflow from AmpliSeq library preparation through Illumina SBS sequencing provides a seamless pathway from sample to results, with the Childhood Cancer Panel specifically optimized for this ecosystem [3]. The panel's design enables detection of multiple variant types including single nucleotide polymorphisms (SNPs), insertions-deletions (indels), copy number variants (CNVs), and gene fusions from both DNA and RNA inputs [3], making it particularly comprehensive for pediatric cancer genomics applications where multiple variant types drive oncogenesis.
Targeted next-generation sequencing (NGS) has become indispensable in oncology research, enabling comprehensive evaluation of somatic variants from diverse sample types. The AmpliSeq for Illumina Childhood Cancer Panel provides a targeted resequencing solution designed specifically for investigating childhood and young adult cancers, supporting input from both DNA and RNA derived from blood, bone marrow, and Formalin-Fixed Paraffin-Embedded (FFPE) tissues [3]. This technical guide details the specific sample and input requirements for this panel, framed within the context of compatible Illumina sequencing systems such as MiSeq and NextSeq platforms to facilitate robust cancer research.
The AmpliSeq for Illumina Childhood Cancer Panel is a targeted research panel that enables comprehensive evaluation of somatic variants across 203 genes with known associations to cancer in children and young adults [3]. This ready-to-use panel detects variants within multiple pediatric cancer types, including leukemias, brain tumors, and sarcomas, while conserving laboratory resources by eliminating the time and effort associated with custom target identification and primer optimization [3].
The panel utilizes an efficient amplicon sequencing method with a rapid workflow, requiring approximately 5-6 hours for library preparation with less than 1.5 hours of hands-on time [3]. A key strength of this panel is its compatibility with multiple sample types, including challenging specimens such as FFPE tissues, bone marrow, and blood, making it particularly valuable for translational research where sample availability and quality may be limiting factors [3].
Technical Specifications
| Parameter | Specification |
|---|---|
| Target Genes | 203 genes associated with childhood and young adult cancers [3] |
| Assay Time | 5-6 hours (library preparation only) [3] |
| Hands-on Time | <1.5 hours [3] |
| Input Quantity | 10 ng high-quality DNA or RNA [3] |
| Nucleic Acid Type | DNA, RNA [3] |
| Method | Amplicon sequencing [3] |
| Variant Classes Detected | Single nucleotide polymorphisms (SNPs), Gene fusions, Somatic variants, Insertions-deletions (indels), Copy number variants (CNVs) [3] |
| Compatible Instruments | MiSeq System, NextSeq 550 System, NextSeq 2000 System, NextSeq 1000 System, MiSeqDx in Research Mode, MiniSeq System [3] |
Sample Input Requirements and Guidelines
The AmpliSeq Childhood Cancer Panel is designed to work with minimal input material while maintaining assay robustness, which is particularly important when dealing with precious pediatric cancer samples where material may be limited.
Universal Input Specifications
The panel requires only 10 ng of high-quality DNA or RNA per reaction, making it suitable for samples with limited starting material [3]. For RNA samples, the AmpliSeq cDNA Synthesis for Illumina kit is required to convert total RNA to cDNA before library preparation [3]. Each sample generates both DNA and RNA libraries when both nucleic acid types are processed, requiring appropriate indexing strategies for multiplexed sequencing [4].
Sample Type-Specific Considerations
Blood and Bone Marrow
Hematological samples, including blood and bone marrow, are commonly used for childhood cancer research, particularly for leukemias. The panel supports these sample types with the following considerations:
- Input Material: The panel is validated for DNA and RNA extracted from blood and bone marrow samples [3]
- Nucleic Acid Extraction: Magnetic bead-based purification methods, such as those employed in MagMAX kits, offer advantages for these sample types, including higher purity and yields due to exceptional binding capacity, thorough exposure to target molecules during mixing and washing steps, and efficient capture and release of nucleic acids [13]
- Sequential DNA/RNA Isolation: For comprehensive analysis, the MagMAX Sequential DNA/RNA Kit uses magnetic bead purification technology to sequentially isolate high-quality genomic DNA and total RNA from a single whole blood or bone marrow sample, maximizing information obtained from limited specimens [13]
FFPE Tissues
FFPE tissues present unique challenges for NGS due to nucleic acid degradation and crosslinking caused by formalin fixation. The Childhood Cancer Panel includes specific solutions to address these challenges:
- Input Material: The panel supports DNA and RNA extracted from FFPE tissue samples with the standard 10 ng input requirement [3]
- Specialized Extraction: The AmpliSeq for Illumina Direct FFPE DNA product allows for DNA preparation and library construction from unstained, slide-mounted FFPE tissues without the need for deparaffinization or DNA purification, streamlining the workflow [3]
- Alternative Extraction Methods: The MagMAX FFPE DNA/RNA Ultra Kit allows for sequential isolation of DNA and RNA from the same FFPE tissue sample, yielding high-quality separate eluates. This kit is compatible with solvent-free deparaffinization systems that minimize tissue loss [13]
- FFPE-Specific Challenges: DNA from FFPE samples is often fragmented and may contain formalin-induced artifacts such as cytosine deamination, which can lead to C>T/G>A substitution artifacts. Careful DNA extraction and bioinformatics analysis can minimize these issues [14]
Input Quality Assessment
Proper quality control of input DNA and RNA is essential for successful sequencing:
- DNA Quality: For FFPE-derived DNA, fragmentation is common. While the amplicon-based approach of the Childhood Cancer Panel is more tolerant of fragmentation than other NGS methods, highly degraded samples may still exhibit reduced performance
- RNA Quality: RNA integrity number (RIN) or similar metrics should be assessed for RNA samples. Partially degraded RNA from FFPE samples may still yield usable data for targeted sequencing, but extensive degradation will impact results
- QC Recommendations: Illumina provides specific quality control recommendations for FFPE samples to determine whether they are viable input material for library preparation kits [15]
Library Preparation and Sequencing Configuration
Library Preparation Workflow
AmpliSeq Childhood Cancer Panel - Library Preparation Workflow
Kit Requirements for Different Sample Throughputs
The table below outlines the complete kit requirements for processing different numbers of samples, recognizing that each sample produces both DNA and RNA libraries.
| Component | 24 Samples(48 Libraries) | 96 Samples(192 Libraries) | 384 Samples(768 Libraries) |
|---|---|---|---|
| AmpliSeq Childhood Cancer Panel | 1 panel | 4 panels | 16 panels [4] |
| AmpliSeq Library PLUS | 2 × 24-reaction kits | 2 × 96-reaction kits | 2 × 384-reaction kits [4] |
| AmpliSeq CD Indexes Set A | 1 set (96 indexes) | 2 sets (192 indexes) | 8 sets (768 indexes) [4] |
| cDNA Synthesis Kit | 1 kit | 1 kit | 4 kits [4] |
Sequencing System Specifications and Performance
When planning experiments, researchers must consider the sequencing platform and configuration to ensure adequate coverage for both DNA and RNA libraries.
| System | Reagent Kit | Max DNA-Only Samples | Max RNA-Only Samples | Max Combined Samples | DNA:RNA Pooling Ratio | Run Time |
|---|---|---|---|---|---|---|
| MiniSeq | Mid Output | 1 | 8 | 1 | 5:1 | 17 hours [4] |
| MiniSeq | High Output | 5 | 25 | 4 | 5:1 | 24 hours [4] |
| MiSeq | Reagent Kit v2 | 3 | 15 | 2 | 5:1 | 24 hours [4] |
| MiSeq | Reagent Kit v3 | 5 | 25 | 4 | 5:1 | 32 hours [4] |
| NextSeq | Mid Output v2 | 27 | 96 | 22 | 5:1 | 26 hours [4] |
| NextSeq | High Output v2 | 83 | 96 | 48 | 5:1 | 29 hours [4] |
Essential Research Reagent Solutions
The following reagents and kits are essential for implementing a complete workflow from sample to sequence with the AmpliSeq Childhood Cancer Panel.
| Product Name | Function | Application Notes |
|---|---|---|
| AmpliSeq for Illumina Childhood Cancer Panel | Targeted amplification of 203 cancer-associated genes | Ready-to-use panel; detects SNPs, indels, CNVs, fusions [3] |
| AmpliSeq Library PLUS for Illumina | Library preparation reagents | Available in 24-, 96-, and 384-reaction configurations [3] |
| AmpliSeq CD Indexes | Sample multiplexing | Unique 8-base indexes for sample identification; available in sets A-D [3] |
| AmpliSeq cDNA Synthesis for Illumina | RNA-to-cDNA conversion | Required for RNA samples before library prep [3] |
| AmpliSeq for Illumina Direct FFPE DNA | DNA preparation from FFPE | Enables library construction without deparaffinization or DNA purification [3] |
| MagMAX DNA/RNA Kits | Nucleic acid extraction | Magnetic bead-based purification for blood, bone marrow, FFPE [13] |
| MagMAX Sequential DNA/RNA Kit | Co-isolation of DNA/RNA | Isolates both nucleic acids from single sample of blood or bone marrow [13] |
| MagMAX FFPE DNA/RNA Ultra Kit | Nucleic acids from FFPE | Sequential isolation of DNA and RNA from same FFPE sample [13] |
The AmpliSeq for Illumina Childhood Cancer Panel offers researchers a robust, targeted sequencing solution with exceptional sample flexibility. By supporting DNA and RNA from blood, bone marrow, and FFPE tissues with minimal input requirements, this panel enables comprehensive molecular profiling of childhood cancers even from limited or challenging specimens. The optimized workflow, combined with compatible Illumina sequencing systems and specialized extraction methods, provides a complete solution for investigating somatic variants in pediatric and young adult cancer research. Proper attention to sample-specific preparation guidelines, quality control measures, and appropriate sequencing configurations ensures successful implementation of this panel in diverse research settings.
A Step-by-Step Protocol from Library Prep to Sequencer Setup
Library preparation is a critical first step in next-generation sequencing (NGS), converting raw genetic material into a format compatible with sequencing platforms. For clinical and research applications requiring rapid turnaround, such as childhood cancer research, optimized workflows are essential. This guide details the essentials of a proven 5-6 hour library preparation workflow, focusing on the AmpliSeq for Illumina Childhood Cancer Panel, and frames it within the broader context of compatible Illumina sequencers for AmpliSeq-based research.
Core Library Preparation Workflow: A 5-6 Hour Process
The AmpliSeq for Illumina Childhood Cancer Panel features a core library preparation process of 5-6 hours [3]. This targeted resequencing solution is designed for the comprehensive evaluation of somatic variants in childhood and young adult cancers, including leukemias, brain tumors, and sarcomas [3]. It is part of an integrated workflow that utilizes PCR-based library preparation and Illumina sequencing-by-synthesis (SBS) technology.
It is important to distinguish the core assay time from the total hands-on time and the complete process duration. The 5-6 hour metric refers specifically to the library preparation component. The total hands-on time is less than 1.5 hours, offering efficiency for laboratory personnel [3]. However, this 5-6 hour period does not include subsequent, necessary steps such as library quantification, normalization, or pooling, which must be accounted for in overall project planning [3].
Workflow Visualization
The following diagram illustrates the key stages and decision points in the AmpliSeq library preparation and sequencing workflow, from sample input to data analysis.
The Scientist's Toolkit: Essential Research Reagent Solutions
Executing the 5-6 hour library preparation requires a specific set of compatible reagents and kits. The table below details the essential components for building a functional workflow with the AmpliSeq Childhood Cancer Panel [3] [4].
| Component Category | Product Name | Function & Key Specifications |
|---|---|---|
| Target Enrichment Panel | AmpliSeq for Illumina Childhood Cancer Panel | Targeted panel investigating 203 genes associated with pediatric and young adult cancers. Sufficient for 24 samples [3]. |
| Core Library Prep Kit | AmpliSeq Library PLUS for Illumina | Contains reagents for preparing sequencing libraries. Available in 24-, 96-, and 384-reaction configurations [3] [4]. |
| Index Adapters | AmpliSeq CD Indexes (e.g., Set A, B, C, D) | Unique dual indexes for multiplexing, allowing sample pooling and identification post-sequencing. Each set labels 96 samples [3]. |
| RNA-to-cDNA Conversion | AmpliSeq cDNA Synthesis for Illumina | Converts total RNA to cDNA, a mandatory step when using the RNA component of the panel [3] [4]. |
| Library Normalization | AmpliSeq Library Equalizer for Illumina | Provides beads and reagents for normalizing library concentrations prior to pooling, simplifying workflow [3]. |
| Specialized Sample Input | AmpliSeq for Illumina Direct FFPE DNA | Enables DNA preparation and library construction from FFPE tissues without deparaffinization or DNA purification [3]. |
Compatible Illumina Sequencing Platforms
The prepared libraries are compatible with several benchtop Illumina sequencers, allowing labs to choose a system that matches their throughput needs. Key performance metrics for the most common systems are summarized below [4].
| Sequencing System | Reagent Kit | Max Combined* Samples/Run | Recommended DNA:RNA Pooling Ratio | Run Time |
|---|---|---|---|---|
| MiniSeq System | Mid Output Kit | 1 sample | 5:1 | 17 hours |
| MiniSeq System | High Output Kit | 4 samples | 5:1 | 24 hours |
| MiSeq System | MiSeq Reagent Kit v3 | 4 samples | 5:1 | 32 hours |
| NextSeq 550 System | High Output v2 Kit | 48 samples | 5:1 | 29 hours |
Note: *"Combined" refers to paired DNA and RNA from the same sample, which generates two separately indexed libraries [4].
Sequencer Selection Logic
The choice of sequencer depends on your project's scale and desired turnaround time. The following diagram outlines the decision-making logic for selecting the most appropriate Illumina system for a childhood cancer panel study.
Detailed Experimental Protocol
This section provides a step-by-step methodological breakdown for library preparation and sequencing using the AmpliSeq Childhood Cancer Panel.
Sample Preparation and Quality Control
The protocol requires 10 ng of high-quality DNA or RNA as input [3]. Specialized sample types including blood, low-input samples, bone marrow, and FFPE tissue are supported [3]. For FFPE samples, the use of the AmpliSeq for Illumina Direct FFPE DNA kit is recommended to bypass the need for deparaffinization or DNA purification [3]. When RNA is the starting material, the first mandatory step is its conversion to cDNA using the AmpliSeq cDNA Synthesis for Illumina kit [3] [4].
Library Preparation and AmpliSeq Workflow
The core library construction process is PCR-based and optimized for amplicon sequencing [3].
- Amplicon Generation: The panel consists of two pools for DNA (3,069 amplicons) and two pools for RNA (1,701 amplicons) to comprehensively cover the 203 target genes [4].
- Library Construction: The fragmented and repaired DNA or cDNA is ligated to platform-specific adapters. For the AmpliSeq workflow, this involves using the AmpliSeq Library PLUS kit and AmpliSeq CD Indexes for sample multiplexing [3] [4].
- Purification and Normalization: Post-ligation clean-up is performed using SPRI beads. Libraries are then normalized to equimolar concentrations using the AmpliSeq Library Equalizer to ensure balanced representation in the final pool [3].
Sequencing and Data Analysis
Following the 5-6 hour library prep, the normalized and pooled libraries are loaded onto a compatible Illumina sequencer [4].
- Sequencing Configuration: The workflow is optimized for paired-end sequencing. The recommended read length depends on the instrument but is typically 2x150 bp or 2x300 bp to adequately cover the amplicons, which have an average length of 114-122 bp [16] [4].
- Data Analysis: The integrated workflow includes automated analysis pipelines. For the Childhood Cancer Panel, the primary output is a variant report detailing somatic mutations, single nucleotide polymorphisms (SNPs), insertions-deletions (indels), copy number variants (CNVs), and gene fusions present in the sample [3].
The 5-6 hour library preparation workflow for the AmpliSeq Childhood Cancer Panel represents a robust and efficient pipeline for targeted genomic analysis in pediatric oncology research. Its integration with a range of Illumina sequencers, from the MiSeq to the NextSeq series, provides laboratories with scalable solutions to generate timely and comprehensive data on somatic variants. This enables researchers and drug development professionals to accelerate the discovery of diagnostic, prognostic, and therapeutic targets for childhood cancers.
In the field of pediatric oncology research, comprehensive genomic profiling is essential for uncovering the molecular drivers of childhood cancers. The AmpliSeq for Illumina Childhood Cancer Panel enables targeted evaluation of 203 genes associated with cancers in children and young adults across multiple cancer types, including leukemias, brain tumors, and sarcomas [3]. To maximize throughput and minimize costs while processing these valuable samples, researchers must implement effective sample multiplexing strategies—the practice of pooling multiple uniquely indexed libraries together for simultaneous sequencing during a single instrument run [17].
Combining DNA and RNA libraries from the same patient specimens presents both opportunities and challenges. While it streamlines workflow and conserves precious sample material, it requires careful consideration of library compatibility, indexing strategies, and sequencing platform specifications to ensure data integrity. This technical guide examines optimal pooling strategies within the context of Illumina sequencing systems compatible with the AmpliSeq Childhood Cancer Panel, specifically the MiSeq and NextSeq series [3] [16].
Core Principles of Library Multiplexing and Indexing
The Role of Index Adapters in Sample Multiplexing
Sample multiplexing, also known as multiplex sequencing, allows large numbers of libraries to be pooled and sequenced simultaneously during a single run on Illumina instruments [17]. The fundamental enabling technology for this approach is the incorporation of unique "barcode" sequences (index adapters) to each DNA or RNA fragment during next-generation sequencing (NGS) library preparation. These indexes allow bioinformatic identification and sorting of reads to their original samples after sequencing through a process called demultiplexing [17].
The AmpliSeq for Illumina workflow utilizes specific indexing systems that are integral to the multiplexing process. The panel requires separate purchases of index adapters, with options including AmpliSeq CD Indexes Sets A-D, each containing 96 unique 8-base pair indexes sufficient for labeling 96 samples [3]. This extensive indexing capacity enables researchers to create highly multiplexed sequencing runs while maintaining sample identity throughout the analytical process.
Addressing Index Hopping with Unique Dual Indexing
A known challenge in multiplexed sequencing is index hopping (or index switching), a phenomenon where indexes are incorrectly assigned, causing reads to be misattributed to the wrong sample [18]. This rare occurrence (typically 0.1–2% on patterned flow cell systems) can be mitigated through specific library preparation strategies:
- Use Unique Dual Indexing (UDI): UDI employs unique combinations of i5 and i7 indexes, allowing bioinformatic filtering of unexpected index combinations that result from index hopping [18].
- Minimize Free Adapters: Libraries with higher levels of free adapters experience elevated index hopping rates [18].
- Proper Library Storage: Store libraries individually at -20°C before pooling [18].
- Pool Libraries Strategically: Pool libraries just prior to sequencing rather than long in advance [18].
For the AmpliSeq Childhood Cancer Panel, Illumina recommends using unique dual indexes over combinatorial dual indexes to maximize data integrity in multiplexed runs [18].
Platform Selection for AmpliSeq Childhood Cancer Panel
Compatible Sequencing Systems
The AmpliSeq Childhood Cancer Panel is compatible with several Illumina sequencing platforms, each offering different throughput capacities suitable for varying project scales [3]. The selection of an appropriate platform depends on several factors, including sample volume, required sequencing depth, and turnaround time requirements.
Table 1: Compatible Sequencing Platforms for AmpliSeq Childhood Cancer Panel
| Platform | Maximum Output | Run Time Range | Maximum Read Length | Ideal Use Case |
|---|---|---|---|---|
| MiSeq System | 30 Gb [16] | ~4–24 hours [16] | 2 × 500 bp [16] | Low-to-moderate throughput projects; method development |
| MiniSeq System | 7.5 Gb (not in search results, but known compatibility) | ~5–24 hours (not in search results) | 2 × 150 bp (not in search results) | Small-scale projects; teaching environments |
| NextSeq 550 System | 120 Gb [16] | ~11–29 hours [16] | 2 × 150 bp [16] | Medium-scale research studies |
| NextSeq 1000/2000 Systems | 540 Gb [16] | ~8–44 hours [16] | 2 × 300 bp [16] | Large-scale projects requiring high throughput |
Platform Selection Guidance
For combined DNA and RNA library pooling, the NextSeq 1000/2000 Systems offer optimal flexibility due to their higher throughput capacity and support for 2 × 300 bp read lengths, which can be beneficial for covering longer amplicons [16]. The MiSeq System remains valuable for smaller-scale studies or protocol optimization due to its faster turnaround times and longer read capabilities [16].
When planning a pooled run, calculate the required sequencing depth per sample for both DNA and RNA analyses, then multiply by the total number of samples to determine the total data output needed. This calculation will guide appropriate platform selection and flow cell configuration.
Experimental Design and Workflow Integration
Sample Preparation and Quality Control
The AmpliSeq Childhood Cancer Panel requires only 10 ng of high-quality DNA or RNA as input, making it suitable for precious pediatric cancer samples, including those from FFPE tissue, blood, or bone marrow [3]. For RNA samples, the AmpliSeq cDNA Synthesis for Illumina kit is required to convert total RNA to cDNA before library preparation [3].
Key considerations for sample preparation:
- FFPE Samples: The AmpliSeq for Illumina Direct FFPE DNA kit allows for DNA preparation and library construction from FFPE tissues without the need for deparaffinization or DNA purification [3].
- Quality Control: Implement rigorous QC checks after library preparation using fluorometric or electrophoretic methods to ensure proper library size distribution and concentration before pooling.
- Library Normalization: Use AmpliSeq Library Equalizer for Illumina to normalize libraries before pooling, ensuring balanced representation of each sample in the final pool [3].
Workflow Visualization: Combined DNA and RNA Library Preparation and Pooling
The following diagram illustrates the integrated workflow for processing DNA and RNA samples through library preparation, indexing, pooling, and sequencing:
Diagram 1: DNA and RNA Library Prep and Pooling Workflow
Indexing Strategy Visualization for Demultiplexing
The following diagram illustrates how unique dual indexing enables correct sample identification and prevents issues from index hopping:
Diagram 2: Unique Dual Indexing for Demultiplexing
Quantitative Considerations for Library Pooling
Library Pooling Calculations and Normalization
Successful pooling of DNA and RNA libraries requires precise quantification and normalization to ensure balanced representation across data types. The following table outlines key reagents and their functions in the pooling workflow:
Table 2: Essential Research Reagent Solutions for Library Pooling
| Reagent Solution | Function in Workflow | Specifications |
|---|---|---|
| AmpliSeq CD Indexes Sets A-D [3] | Unique sample identification | 96 indexes per set; 8 bp indexes |
| AmpliSeq Library Equalizer for Illumina [3] | Library normalization | Normalizes libraries before pooling |
| AmpliSeq Library PLUS for Illumina [3] | Library preparation | 24, 96, or 384 reactions |
| AmpliSeq cDNA Synthesis for Illumina [3] | RNA-to-cDNA conversion | Required for RNA panels |
| AmpliSeq for Illumina Direct FFPE DNA [3] | FFPE DNA preparation | 24 reactions; no deparaffinization needed |
Pooling Strategy and Sequencing Depth Considerations
When combining DNA and RNA libraries from the Childhood Cancer Panel, consider these key parameters:
- Library Balance: Adjust molar ratios to account for different amplification efficiencies between DNA and RNA targets.
- Sequencing Depth: For somatic variant detection in DNA, aim for minimum coverage of 500-1000x, while RNA may require 50-100 million reads per sample for expression analysis.
- Pooling Calculator: Use Illumina's pooling calculator to determine appropriate loading concentrations for optimal cluster density [17].
- Loading Concentration: For MiSeq systems, denature and dilute libraries to a loading concentration of 6–20 pM [19].
The total number of samples that can be pooled depends on the sequencing platform selected and the required depth of coverage per sample. For example, a NextSeq 1000 system with 540 Gb output could theoretically sequence approximately 180 samples at 30 Gb per sample (assuming 15 Gb each for DNA and RNA), though practical considerations may reduce this number.
Technical Protocols and Best Practices
Detailed Protocol: Library Pooling for Combined DNA and RNA Sequencing
This protocol assumes completion of initial library preparation using the AmpliSeq Childhood Cancer Panel with AmpliSeq Library PLUS reagents.
Pre-Pooling Quality Control
- Quantify Libraries: Use fluorometric methods (Qubit) for accurate concentration measurement.
- Assess Library Size Distribution: Analyze 1 μL of each library on Bioanalyzer or TapeStation to confirm expected size profile (~250-350 bp for AmpliSeq libraries).
- Verify Library Integrity: Ensure absence of adapter dimers or other contamination.
Library Normalization
- Dilute Libraries: Dilute all DNA and RNA libraries to the same concentration (e.g., 10 nM) in low-EDTA TE buffer or nuclease-free water.
- Normalize with AmpliSeq Library Equalizer: Follow manufacturer's instructions for bead-based normalization to ensure equimolar representation [3].
- Confirm Normalization: Re-quantify a subset of normalized libraries to verify equal concentration.
Experimental Pooling Strategy
- Create Sub-Pools: For large studies, create smaller sub-pools of 24-48 libraries each to minimize pipetting errors.
- Combine Sub-Pools: Mix sub-pools in equimolar ratios to create the final sequencing pool.
- Final QC: Quantify the final pool and assess size distribution to confirm proper distribution.
Sequencing Setup
- Denature and Dilute: Denature the pooled library with NaOH following Illumina's "Denature and Dilute Libraries Guide" [19].
- Adjust Loading Concentration: Dilute to appropriate loading concentration (6–20 pM for MiSeq) [19].
- Sequence: Load onto compatible Illumina instrument (MiSeq, NextSeq 550, NextSeq 1000/2000) and initiate run with appropriate cycle configuration.
Troubleshooting Common Pooling Issues
- Uneven Coverage: If certain samples show consistently lower coverage, check initial library quality and consider increasing their representation in the pool.
- Excessive Undetermined Reads: This may indicate index hopping or poor library quality. Verify use of unique dual indexes and check library QC metrics [18].
- Low Cluster Density: Increase loading concentration or verify library concentration measurements.
- Failed Demultiplexing: Confirm index sequences are correct and compatible with the sequencing kit being used.
Combining DNA and RNA libraries from the AmpliSeq Childhood Cancer Panel in multiplexed sequencing runs represents an efficient approach to comprehensive pediatric cancer genomics. Successful implementation requires:
- Strategic Platform Selection based on project scale and throughput requirements [16]
- Rigorous Quality Control throughout library preparation and pooling processes [3]
- Implementation of Unique Dual Indexing to mitigate index hopping effects [18]
- Precise Normalization and Pooling to ensure balanced representation [3]
By following the strategies and protocols outlined in this technical guide, researchers can maximize sequencing efficiency while maintaining data quality, ultimately accelerating discoveries in childhood cancer genomics. The integrated workflow enables comprehensive profiling of DNA and RNA from limited patient samples, making it particularly valuable for rare pediatric cancer studies where sample material is often precious and limited.
Selecting the appropriate sequencing platform and reagent kit is a critical step in designing targeted resequencing studies. For researchers using the AmpliSeq for Illumina Childhood Cancer Panel, which investigates 203 genes associated with cancer in children and young adults, understanding the specific capabilities of the MiSeq and NextSeq systems ensures efficient experimental design and resource utilization [3]. This guide provides a detailed comparison of sample throughput and reagent kit selection for these two platforms, framed within the context of optimizing workflows for somatic variant detection in pediatric cancer research.
Sequencing System Performance and Throughput
The choice between MiSeq and NextSeq systems significantly impacts project scalability. The table below summarizes the maximum sample throughput and key run parameters for the AmpliSeq Childhood Cancer Panel on each system and their compatible reagent kits [4].
Table 1: Sequencing Throughput for the Childhood Cancer Panel on MiSeq and NextSeq Systems
| System | Reagent Kit | Max DNA-Only Samples | Max RNA-Only Samples | Max Combined* Samples | Recommended DNA:RNA Pooling Ratio | Run Time |
|---|---|---|---|---|---|---|
| MiSeq System | MiSeq Reagent Kit v2 | 3 | 15 | 2 | 5:1 | 24 hours |
| MiSeq Reagent Kit v3 | 5 | 25 | 4 | 5:1 | 32 hours | |
| NextSeq System | NextSeq Mid Output v2 Kit | 27 | 96 | 22 | 5:1 | 26 hours |
| NextSeq High Output v2 Kit | 83 | 96 | 48 | 5:1 | 29 hours |
*A "Combined" sample refers to paired DNA and RNA from the same source, generating two separate libraries [4].
Key Selection Criteria
- Project Scale: The MiSeq system is ideal for lower-throughput projects, processing up to 5 DNA-only or 4 combined samples per run. The NextSeq system enables medium- to high-throughput studies, handling up to 83 DNA-only or 48 combined samples [4].
- Run Time vs. Throughput: MiSeq runs require 24-32 hours, suitable for rapid turnaround. NextSeq runs take 26-29 hours but provide substantially higher data output, offering greater efficiency for large batches [4].
- Read Requirements: The differing DNA:RNA pooling ratios reflect coverage requirements. The 5:1 ratio for combined samples ensures optimal read depth for both DNA and RNA targets [4].
Experimental Workflow and Protocol
The following diagram illustrates the integrated workflow from sample to data, highlighting the parallel paths for DNA and RNA analysis.
Detailed Methodologies
1. Library Preparation The protocol uses the AmpliSeq for Illumina Library PLUS kit. The panel generates two library pools per sample: one for DNA (3,069 amplicons) and one for RNA (1,701 amplicons after cDNA synthesis). Total hands-on time for library prep is under 1.5 hours, with a total assay time of 5-6 hours [3] [4].
2. cDNA Synthesis For RNA samples, the AmpliSeq cDNA Synthesis for Illumina kit is required to convert total RNA to cDNA before library preparation. Input quantity is 10 ng of high-quality DNA or RNA [3].
3. Indexing and Pooling Unique AmpliSeq CD Indexes (e.g., Set A, B, C, or D) are used to label each library, enabling sample multiplexing. Libraries are then pooled based on the recommended DNA:RNA pooling volume ratio of 5:1 for combined runs [3] [4].
4. Sequencing Denatured and diluted libraries are loaded onto the sequencer. For MiSeq systems, ensure the MiSeq Control Software (MCS) version is compatible with your flow cell (e.g., MCS v2.4 or later for PGS Flow Cells) [19]. Each MiSeq reagent kit is a single-use package containing a flow cell and prefilled reagent cartridge [20].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Essential Components for the AmpliSeq Childhood Cancer Panel Workflow
| Component | Function | Key Specifications |
|---|---|---|
| AmpliSeq Childhood Cancer Panel | Ready-to-use primer panel for targeted amplification of 203 cancer-associated genes. | 24 reactions per panel; detects SNPs, indels, CNVs, fusions [3]. |
| AmpliSeq Library PLUS Kit | Core reagents for preparing sequencing libraries from amplicons. | Available in 24-, 96-, and 384-reaction configurations [3]. |
| AmpliSeq CD Indexes | Unique 8-base pair indexes for sample multiplexing. | Sold in sets (A-D); each set contains 96 indexes [3]. |
| AmpliSeq cDNA Synthesis Kit | Converts total RNA to cDNA for RNA library preparation. | Required when processing RNA samples [3]. |
| MiSeq Reagent Kits (v2/v3) | Single-use cartridge with flow cell and sequencing reagents for MiSeq. | Kit version determines read length and output [19] [20]. |
| NextSeq Mid/High Output Kits | Reagent kits for medium- and high-throughput runs on NextSeq. | Determines maximum sample throughput per run [4]. |
Selecting between MiSeq and NextSeq systems for the AmpliSeq Childhood Cancer Panel involves a direct trade-off between project scale and operational flexibility. The MiSeq system serves as a robust solution for focused studies and rapid validation, while the NextSeq system provides the throughput necessary for larger cohort studies and drug development pipelines. By aligning the experimental goals with the detailed throughput guidelines and ensuring the use of compatible reagent kits, researchers can optimize their sequencing strategy to effectively investigate the genomic landscape of childhood cancers.
Targeted sequencing panels, such as the AmpliSeq for Illumina Childhood Cancer Panel, are powerful tools for the comprehensive evaluation of somatic variants associated with pediatric and young adult cancers. A key challenge in maximizing the efficiency and data quality of these workflows is the simultaneous sequencing of libraries derived from different nucleic acid types—DNA and RNA—from the same research sample. This technical guide delves into the application and critical importance of the 5:1 DNA:RNA pooling volume ratio, a strategy recommended by Illumina to achieve balanced coverage when processing paired DNA and RNA libraries on compatible sequencers within a broader thesis on optimizing sequencing workflows for childhood cancer research.
The AmpliSeq for Illumina Childhood Cancer Panel is a targeted resequencing solution designed to evaluate 203 genes linked to childhood and young adult cancers, including leukemias, brain tumors, and sarcomas [3]. This panel streamlines the workflow by eliminating the need for researchers to identify targets, design primers, and optimize panels independently.
A unique feature of this panel is that it generates two separate libraries from a single patient sample: one from DNA (comprising 3,069 amplicons) and one from RNA (comprising 1,701 amplicons) [4]. These libraries are uniquely indexed, allowing them to be pooled together for a single, multiplexed sequencing run. The entire library preparation process is efficient, with an assay time of 5-6 hours and less than 1.5 hours of hands-on time [3]. The following diagram illustrates the integrated workflow from sample to data analysis.
Compatible Sequencers and Pooling Guidelines
The 5:1 DNA:RNA pooling ratio is specifically calibrated for use across several Illumina sequencing systems. The table below summarizes the maximum number of samples and key parameters for sequencing the Childhood Cancer Panel on different instruments, as per Illumina's guidelines [4].
Table 1: Sequencing and Pooling Guidelines for the Childhood Cancer Panel
| Sequencing System | Reagent Kit | Max DNA-Only Samples | Max RNA-Only Samples | Max Combined* Samples | Recommended DNA:RNA Ratio | Run Time |
|---|---|---|---|---|---|---|
| MiniSeq System | Mid Output | 1 | 8 | 1 | 5:1 | 17 hours |
| High Output | 5 | 25 | 4 | 5:1 | 24 hours | |
| MiSeq System | v2 | 3 | 15 | 2 | 5:1 | 24 hours |
| v3 | 5 | 25 | 4 | 5:1 | 32 hours | |
| NextSeq System | Mid Output v2 | 27 | 96 | 22 | 5:1 | 26 hours |
| High Output v2 | 83 | 96 | 48 | 5:1 | 29 hours |
Combined samples refer to paired DNA and RNA from the same source, generating two separately indexed libraries [4].
Technical Rationale Behind the 5:1 Pooling Ratio
The recommendation for a 5:1 DNA:RNA pooling volume ratio is not arbitrary; it is a calculated decision based on the characteristics of the panel and the goal of achieving uniform sequence coverage.
- Coverage Balance: The ratio is designed to produce balanced read coverage between the DNA and RNA libraries. This ensures that both libraries are sequenced to a sufficient depth for reliable variant detection without one library type consuming a disproportionate share of the sequencing data [4].
- Amplicon Load: The panel generates a greater number of DNA amplicons (3,069) compared to RNA amplicons (1,701) [4]. The 5:1 ratio accounts for this difference in target load, effectively normalizing the representation of each library type in the final pool to ensure efficient and comprehensive sequencing of all targets.
Critical Implementation Protocols
Library Pooling and Index Balancing
Successful sequencing relies on proper library preparation and indexing. The use of Unique Dual Indexes (UDIs) is critical for accurate sample demultiplexing, while "color balance" prevents sequencing failures, especially on modern two-channel instruments like the NextSeq and NovaSeq series [21] [22].
- Unique Dual Indexing (UDI): Always use UDIs to provide a unique combination of an i7 and an i5 index for each library. This minimizes index hopping and ensures that each sequence read can be accurately assigned to its correct sample of origin [21].
- Index Color Balance: The index sequences themselves must be "color balanced." On two-channel SBS chemistry instruments, each cycle of the index read must contain signal in at least one channel. Pools where all indexes start with two consecutive G bases (which appear as "dark" bases with no signal) can cause cluster registration to fail, leading to poor data output or aborted runs [22]. Commercial index kits are designed to avoid this, but researchers must ensure their selected index combinations maintain balance [21].
- Automated Checking: Utilize software tools like Illumina Experiment Manager (IEM) or the
--validate-balanceoption inbcl-convertto verify the color balance of your chosen index combinations before starting a sequencing run [21].
Sample Preparation and Quality Control
Robust library construction begins with high-quality input material. The Childhood Cancer Panel requires only 10 ng of input DNA or RNA, but sample purity is paramount [3]. For DNA samples, a 260/280 ratio of 1.8-2.0 and a 260/230 ratio greater than 2.0 are recommended. RNA samples should have a 260/280 ratio of 1.8-2.1 and a 260/230 ratio greater than 1.5 [23]. For RNA work, the protocol requires a separate cDNA synthesis step using the AmpliSeq cDNA Synthesis for Illumina kit [3] [4]. Library purification and size selection can be effectively performed using Solid-Phase Reversible Immobilization (SPRI) beads, which also help remove unwanted adapter dimers [24].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for the AmpliSeq Childhood Cancer Workflow
| Item | Function | Example Product |
|---|---|---|
| Core Panel | Targeted primer pool for amplifying 203 childhood cancer genes. | AmpliSeq for Illumina Childhood Cancer Panel [3] |
| Library Prep Kit | Reagents for PCR-based library construction from amplicons. | AmpliSeq Library PLUS for Illumina [3] |
| cDNA Synthesis Kit | Converts input RNA to cDNA for subsequent amplification. | AmpliSeq cDNA Synthesis for Illumina [3] |
| Unique Dual Indexes | 8 bp sequences for multiplexing samples and demultiplexing data. | AmpliSeq CD Indexes Sets A-D [3] |
| Library Normalization | Streamlines the normalization of multiple libraries for pooling. | AmpliSeq Library Equalizer for Illumina [3] |
| FFPE Direct Prep | Enables library construction from FFPE tissues without DNA purification. | AmpliSeq for Illumina Direct FFPE DNA [3] |
The 5:1 DNA:RNA pooling volume ratio is a finely tuned parameter essential for the effective use of the AmpliSeq for Illumina Childhood Cancer Panel. By adhering to this guideline and integrating robust laboratory practices—including rigorous sample QC, the use of color-balanced UDIs, and following validated protocols—researchers can achieve uniform coverage and high-quality data. This optimized approach ensures that sequencing capacity is used efficiently, ultimately supporting the reliable detection of somatic variants across multiple nucleic acid types to advance research into childhood cancers.
Efficient run planning is a critical component of high-throughput genomic research, particularly in the field of pediatric oncology where targeted sequencing panels are employed. For researchers using the AmpliSeq for Illumina Childhood Cancer Panel, precise calculation of maximum sample outputs for Mid and High-Output reagent kits directly impacts experimental design, resource allocation, and cost efficiency. This technical guide provides a comprehensive framework for maximizing throughput across compatible Illumina sequencing systems, including MiniSeq, MiSeq, and NextSeq platforms. We present detailed methodologies, sample calculations, and optimized pooling strategies to assist researchers in achieving optimal coverage while minimizing reagent waste. Within the broader context of compatible Illumina sequencers for AmpliSeq Childhood Cancer Panel research, proper run planning ensures that precious clinical samples from pediatric cancer patients are utilized to their fullest potential, accelerating discoveries in drug development and molecular pathology.
The AmpliSeq for Illumina Childhood Cancer Panel provides a targeted resequencing solution for comprehensive evaluation of somatic variants associated with childhood and young adult cancers [3]. This ready-to-use panel investigates 203 genes associated with pediatric cancers, including leukemias, brain tumors, and sarcomas, enabling researchers to detect single nucleotide polymorphisms (SNPs), gene fusions, somatic variants, insertions-deletions (indels), and copy number variants (CNVs) [3]. The panel utilizes amplicon sequencing technology with a remarkably low input requirement of just 10 ng of high-quality DNA or RNA, making it particularly valuable for precious pediatric cancer samples where material is often limited [3].
A unique aspect of this panel is its dual approach to genomic profiling: it generates both DNA and RNA libraries from each patient sample. Specifically, the panel consists of two component pools—a DNA pool with 3,069 amplicons (average length 114 bp) and an RNA pool with 1,701 amplicons (average length 122 bp) [4]. This dual nucleic acid approach provides complementary information about genetic alterations and their transcriptional consequences, offering a more comprehensive molecular portrait of each pediatric cancer case. For research purposes, a "sample" is defined as a specimen of nucleic acid from one source, while a "library" refers to nucleic acids from a sample after preparation [4]. When processing paired DNA and RNA from the same source, this generates two separate libraries—one DNA and one RNA—that are individually indexed [4].
Compatible Sequencing Systems and Their Specifications
The AmpliSeq for Illumina Childhood Cancer Panel is compatible with multiple Illumina sequencing systems, each offering distinct advantages in terms of throughput, run time, and operational costs. Understanding the specifications of each system is fundamental to effective run planning and maximizing sample outputs. The compatible systems include the MiniSeq System, MiSeq System, and NextSeq System, each with Mid and High-Output reagent options that substantially impact maximum sample throughput [4].
The MiniSeq System represents the entry point for Illumina sequencing, with the Mid Output Kit generating up to 2.4 Gb and the High Output Kit delivering up to 7.5 Gb of data [25]. The MiSeq System offers enhanced throughput with reagent kits that support longer read lengths, making it suitable for applications requiring greater coverage depth. The NextSeq 550 System provides significantly higher throughput, with the Mid-Output Kit generating 32.5-39 Gb and the High-Output Kit delivering 100-120 Gb for 2 × 150 bp read configurations [26]. This substantial increase in data output directly translates to higher sample multiplexing capabilities, making the NextSeq platform particularly advantageous for larger studies. The NextSeq 550 System leverages 2-channel sequencing by synthesis (SBS) technology for faster data generation than the original Illumina 4-channel SBS technology, while maintaining the same high data accuracy [26].
Table 1: Sequencing System Specifications for AmpliSeq Childhood Cancer Panel
| System | Reagent Kit | Maximum Output | Quality Scores | Run Time |
|---|---|---|---|---|
| MiniSeq | MiniSeq Mid Output | 2.4 Gb | >75% bases >Q30 at 2×150 bp | 26 hr |
| MiniSeq | MiniSeq High Output | 7.5 Gb | >75% bases >Q30 at 2×150 bp | 24 hr |
| MiSeq | MiSeq Reagent Kit v3 | 8.5 Gb | >80% bases >Q30 | 32 hr |
| NextSeq 550 | NextSeq Mid Output v2 Kit | 32.5-39 Gb | >75% bases >Q30 at 2×150 bp | 26 hr |
| NextSeq 550 | NextSeq High Output v2 Kit | 100-120 Gb | >75% bases >Q30 at 2×150 bp | 29 hr |
Calculating Maximum Sample Outputs
Key Parameters for Sample Output Calculations
Calculating maximum sample outputs for the AmpliSeq Childhood Cancer Panel requires consideration of several interdependent parameters. The fundamental factors include: (1) total sequencing output of the reagent kit, (2) required coverage depth for reliable variant detection, (3) number of amplicons in the panel (3,069 for DNA, 1,701 for RNA), (4) average amplicon length (114 bp for DNA, 122 bp for RNA), and (5) read configuration (paired-end vs. single-read) [4]. For the AmpliSeq Childhood Cancer Panel, Illumina provides specific guidelines for DNA:RNA pooling volume ratios, recommending a 5:1 ratio based on recommended read coverage [4]. This optimized ratio ensures both DNA and RNA libraries receive sufficient sequencing coverage despite their different amplicon counts.
The total data requirement per sample can be calculated using the formula: Total Data per Sample = (DNA Amplicon Count × DNA Amplicon Length × Coverage Depth) + (RNA Amplicon Count × RNA Amplicon Length × Coverage Depth). For example, at 500× coverage for DNA and 300× coverage for RNA, the calculation would be: (3,069 × 114 × 500) + (1,701 × 122 × 300) = approximately 175 Mb + 62 Mb = 237 Mb per sample. This per-sample data requirement can then be divided into the total output of a sequencing kit to determine maximum sample throughput.
Sample Outputs by Sequencing System
Table 2: Maximum Sample Throughput for AmpliSeq Childhood Cancer Panel
| System | Reagent Kit | Max DNA-Only Samples | Max RNA-Only Samples | Max Combined Samples* | Recommended DNA:RNA Pooling Ratio |
|---|---|---|---|---|---|
| MiniSeq | Mid Output | 1 | 8 | 1 | 5:1 |
| MiniSeq | High Output | 5 | 25 | 4 | 5:1 |
| MiSeq | v3 Reagent Kit | 5 | 25 | 4 | 5:1 |
| NextSeq 550 | Mid Output v2 | 27 | 96 | 22 | 5:1 |
| NextSeq 550 | High Output v2 | 83 | 96 | 48 | 5:1 |
Note: Combined samples refer to paired DNA and RNA from the same sample that generates two libraries, one from each nucleic acid and separately indexed [4].
The calculations reveal substantial differences in multiplexing capacity across platforms. The NextSeq 550 System with High Output v2 Kit provides the highest throughput, supporting up to 48 combined samples (96 libraries) per run, making it ideal for large-scale studies [4]. For smaller batches or individual samples, the MiSeq System offers a balanced approach with reasonable throughput and faster turnaround times. The MiniSeq System serves as a cost-effective option for laboratories with lower throughput needs or for pilot studies.
Experimental Protocol for Run Planning
Library Preparation Workflow
The library preparation process for the AmpliSeq Childhood Cancer Panel follows a standardized protocol with minimal hands-on time of less than 1.5 hours, though total assay time ranges from 5-6 hours (excluding library quantification, normalization, and pooling time) [3]. The workflow begins with quality assessment of input DNA and RNA, requiring 10 ng of high-quality nucleic acids from specimen types including blood, bone marrow, or FFPE tissue [3]. For RNA samples, an initial cDNA synthesis step is performed using the AmpliSeq cDNA Synthesis for Illumina kit, which converts total RNA to cDNA for subsequent amplification [3].
The core amplification step utilizes the AmpliSeq for Illumina Childhood Cancer Panel with the AmpliSeq Library PLUS for Illumina kit, which is available in 24-, 96-, and 384-reaction configurations to match different throughput needs [4]. Following amplification, libraries are uniquely indexed using the AmpliSeq CD Indexes system, which enables sample multiplexing during sequencing [3]. Set A provides 96 indexes sufficient for labeling 96 samples, with additional Sets B, C, and D available for larger studies [3]. Library normalization is performed using the AmpliSeq Library Equalizer for Illumina, which simplifies the pooling process by ensuring equimolar representation of each library [3]. For studies involving FFPE tissues, the AmpliSeq for Illumina Direct FFPE DNA kit allows for DNA preparation without the need for deparaffinization or DNA purification [3].
Library Pooling and Loading Strategy
For combined DNA and RNA sequencing, libraries are pooled at the recommended 5:1 DNA:RNA ratio based on read coverage requirements [4]. This ratio accounts for the different numbers of amplicons between the DNA and RNA components and ensures balanced coverage across both nucleic acid types. The pooled libraries are then diluted to appropriate concentrations for clustering on the selected sequencing system. The following workflow diagram illustrates the complete experimental process from sample preparation through data analysis:
Workflow for AmpliSeq Childhood Cancer Panel Sequencing
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Essential Research Reagent Solutions for AmpliSeq Childhood Cancer Panel
| Product Name | Function | Specifications |
|---|---|---|
| AmpliSeq for Illumina Childhood Cancer Panel | Targeted amplification of 203 childhood cancer genes | 24 reactions; detects SNPs, indels, CNVs, fusions |
| AmpliSeq Library PLUS for Illumina | Library preparation reagents | 24-, 96-, 384-reaction configurations |
| AmpliSeq CD Indexes | Unique sample barcoding for multiplexing | 96 indexes per set; multiple sets available (A-D) |
| AmpliSeq cDNA Synthesis for Illumina | Converts total RNA to cDNA for RNA panels | Required for RNA samples; included in workflow |
| AmpliSeq Library Equalizer for Illumina | Normalizes libraries for balanced representation | Bead-based normalization; minimal hands-on time |
| AmpliSeq for Illumina Direct FFPE DNA | DNA preparation from FFPE tissues | 24 reactions; no deparaffinization needed |
| NextSeq 550 Mid/High Output Kits | Sequencing reagents for NextSeq platform | Mid Output: 32.5-39 Gb; High Output: 100-120 Gb |
Discussion and Best Practices
Optimizing Run Planning for Different Study Designs
Effective run planning must align with specific research objectives and sample availability. For drug development professionals conducting large-scale screening studies, the NextSeq 550 System with High Output v2 Kits offers the most efficient solution, enabling analysis of up to 48 patients (96 libraries) in a single 29-hour run [4]. This high-throughput approach significantly reduces per-sample costs and processing time. For clinical researchers validating specific biomarkers or conducting longitudinal monitoring of smaller patient cohorts, the MiSeq System provides an excellent balance of throughput, flexibility, and rapid turnaround (32 hours for v3 reagent kits) [4]. For individual investigators or pilot studies with limited sample numbers, the MiniSeq System represents a cost-effective option, particularly when using the High Output Kit to sequence up to 4 combined samples [4].
A critical consideration in run planning is the inclusion of appropriate controls and balancing samples across experimental groups within the same sequencing run to minimize batch effects. When processing multiple runs for a single study, using the same lot of reagents and maintaining consistent library preparation protocols across all samples enhances data reproducibility. Researchers should also consider implementing the AmpliSeq for Illumina Sample ID Panel, which includes eight SNP-targeting primer pairs and one gender-determining pair to generate unique IDs for each research sample, providing an additional quality control measure [3].
Troubleshooting Common Challenges
Several technical challenges may arise during run planning and execution. Inadequate coverage often results from inaccurate library quantification or suboptimal pooling ratios. This can be addressed by using the AmpliSeq Library Equalizer for more consistent normalization and verifying library quality with appropriate quantification methods. Sample mix-ups can be mitigated through the implementation of the Sample ID Panel and maintaining rigorous sample tracking throughout the workflow. Low data yield from sequencing runs may indicate issues with library quality, overloading or underloading of the flow cell, or problems with sequencing reagents. Regular maintenance of instrumentation and adherence to recommended quality control checkpoints throughout the library preparation process can prevent these issues.
For studies involving FFPE-derived nucleic acids, which often yield degraded material, the AmpliSeq for Illumina Direct FFPE DNA kit provides a specialized solution that bypasses the need for DNA purification, potentially improving success rates with challenging samples [3]. When working with limited sample quantities, which is common in pediatric cancer research, the panel's low input requirement (10 ng) enables profiling even from minute tissue samples, though additional caution should be exercised during library preparation to ensure representative amplification.
Maximizing sample outputs for the AmpliSeq Childhood Cancer Panel requires careful consideration of sequencing platform capabilities, reagent kit specifications, and experimental design requirements. The Mid and High-Output kits available for MiniSeq, MiSeq, and NextSeq systems offer researchers flexibility in scaling their pediatric cancer studies from targeted investigations to large-scale genomic profiling. By following the calculated throughput guidelines, optimized pooling strategies, and standardized protocols outlined in this technical guide, researchers and drug development professionals can effectively plan sequencing runs that maximize data quality while controlling costs. The comprehensive approach to run planning presented here supports the broader research objective of accelerating discoveries in childhood cancer genomics through efficient, reproducible, and scalable targeted sequencing methodologies.
Achieving Peak Performance and Overcoming Common Challenges
The AmpliSeq for Illumina Childhood Cancer Panel provides a targeted resequencing solution for the comprehensive evaluation of somatic variants associated with childhood and young adult cancers [4] [3]. This ready-to-use panel is designed to detect variants within multiple pediatric cancer types, including leukemias, brain tumors, and sarcomas, interrogating 203 genes of interest [3]. The panel requires only 10 ng of high-quality DNA or RNA input and has a hands-on time of less than 1.5 hours [3]. Each sample processed with this panel generates two separate libraries—one for DNA and one for RNA—which are uniquely indexed to allow for multiplexed sequencing [4]. The integrity and quality of these final library preparations are paramount to the success of the subsequent sequencing run and the reliability of the generated data. Consequently, rigorous quality control (QC) using tools like the BioAnalyzer and Fragment Analyzer is not merely a recommended step but a critical one, ensuring that only libraries of the highest quality are loaded onto compatible Illumina sequencers such as the MiSeq, NextSeq 550, NextSeq 1000, and NextSeq 2000 systems [4] [3].
Failure to perform adequate QC can lead to sequencing failures, reduced data quality, and inaccurate variant calls. Common issues detectable during QC include the presence of adapter dimers, primer dimers, and other by-products that can consume precious sequencing capacity and reduce the yield of usable reads [27]. Furthermore, accurate quantification of the library is essential for achieving an even read distribution across all multiplexed samples, a gold-standard for most RNA-Seq experiments that ensures comparability during downstream analysis [27]. This guide details the methodologies for employing microcapillary electrophoresis platforms, specifically the BioAnalyzer and Fragment Analyzer, to perform this essential pre-sequencing QC within the context of the AmpliSeq Childhood Cancer Panel workflow.
Core Principles of Library Quality Control
The Role of Microcapillary Electrophoresis in QC
Microcapillary electrophoresis has become the standard method for quality control in next-generation sequencing (NGS) laboratories. Platforms like the BioAnalyzer and Fragment Analyzer analyze a small aliquot of the amplified library to provide vital information on several key parameters [27]. This analysis generates an electrophoretogram, or trace, which visually represents the library's size distribution, concentration, and overall profile. The primary goals of this analysis are to:
- Determine Library Size Distribution: Confirm that the final library fragments are within the expected size range. For the AmpliSeq Childhood Cancer Panel, the average library lengths are approximately 254 bp for DNA and 262 bp for RNA libraries [4].
- Assess Library Concentration: Obtain a quantitative measure of the library's concentration, which is necessary for accurate normalization and pooling.
- Identify Undesired By-products: Detect the presence of artifacts such as adapter dimers (short fragments, typically ~100-150 bp, resulting from ligation of adapters without an insert) or primer dimers, which can significantly reduce sequencing efficiency if present in substantial quantities (>3% of the total library) [27].
- Evaluate Library Profile and Shape: A clean, single-peaked trace indicates a high-quality library, while multiple peaks or a broad, smeared distribution can indicate degradation, contamination, or PCR artifacts.
Comparison of BioAnalyzer and Fragment Analyzer
The choice between the BioAnalyzer and Fragment Analyzer often depends on the laboratory's throughput needs and specific requirements. While both platforms serve the same fundamental purpose, they differ in their operational mechanics and capabilities [27].
Table 1: Comparison of Microcapillary Electrophoresis Platforms
| Feature | BioAnalyzer | Fragment Analyzer |
|---|---|---|
| Format | Chip-based | Plate-based (96-well) |
| Sample Throughput | 11-12 samples per chip [27] | Multiple 96-well plates per run [27] |
| Ideal Use Case | Low to medium-throughput laboratories | High-throughput NGS laboratories [27] |
| Resolution & Dynamic Range | High | High, with potential for higher sensitivity depending on the assay |
| Visual Output | Distinct electropherogram trace (see Figure 1A) | Distinct electropherogram trace (see Figure 1B) |
It is important to note that the same library may appear slightly different when run on these different instruments due to variations in their resolution, sensitivity, and dye chemistry (see Figure 2). Therefore, consistency in the platform used for a given project is recommended for reliable comparative analysis.
Experimental Protocols for Library QC
Protocol for Library QC Using BioAnalyzer/Fragment Analyzer
This protocol outlines the steps for analyzing a final, amplified AmpliSeq Childhood Cancer Panel library using a microcapillary electrophoresis system.
Key Research Reagent Solutions:
- AmpliSeq for Illumina Childhood Cancer Panel: The core targeted resequencing panel [3].
- AmpliSeq Library PLUS for Illumina: Reagents for preparing the sequencing libraries [4] [3].
- AmpliSeq CD Indexes: Unique index adapters for multiplexing samples [4] [3].
- High Sensitivity DNA Kit/Assay: The specific dye and gel matrix kit required for the instrument (e.g., Bioanalyzer High Sensitivity DNA Kit or Fragment Analyzer HS NGS Fragment Kit).
Methodology:
- Library Preparation: Prepare the DNA and RNA libraries from your samples according to the official AmpliSeq for Illumina Childhood Cancer Panel protocol [4]. This involves target amplification, PCR cleanup, partial adapter ligation, index ligation, and a final library amplification PCR.
- Instrument Preparation: Prime the instrument and prepare the proprietary gel-dye mix as specified by the manufacturer's instructions for your chosen platform and the appropriate sensitivity kit (e.g., High Sensitivity DNA for Bioanalyzer).
- Sample and Ladder Preparation: Pipette 1 µL of the provided molecular weight ladder into the designated well. For each library, combine 1 µL of the library with the appropriate amount of deionized water (as per the kit protocol, typically 4 µL) to achieve the recommended concentration range.
- Loading and Run: Load the ladder and diluted samples onto the chip or plate. Start the run protocol. The instrument will automatically perform electrophoresis, data collection, and trace generation.
- Data Analysis: Inspect the generated electropherogram traces for each library.
Interpreting Results and Troubleshooting Common Issues
The analysis of the electropherogram is a critical skill. The following workflow outlines the logical process for interpreting results and diagnosing common problems.
Figure 1: A decision workflow for interpreting BioAnalyzer/Fragment Analyzer traces and troubleshooting common library preparation issues.
Troubleshooting Based on Trace Outputs:
- Residual Primers or Adapter Dimers: A small peak at ~100-150 bp indicates adapter-linker by-products (see Figure 1B, purple highlight) [27]. If this peak accounts for more than 3% of the total area under the curve, the library should be re-purified using solid-phase reversible immobilization (SPRI) beads to selectively remove these short fragments before sequencing.
- Overamplification (Overcycling): A characteristic high molecular weight "bump" or a broad, smeared profile can indicate the formation of aberrant "bubble products" due to excessive PCR cycles (see Figure 6) [27]. This occurs when the PCR reaction depletes critical components, leading to incomplete fragments and chimera formation. Libraries suffering from overcycling are difficult to quantify accurately and often yield data with higher duplication rates and reduced complexity. The solution is to re-prepare the library using a lower number of amplification cycles, ideally determined by a qPCR assay (see Section 3.3).
- Undercycling: A very low library yield, making accurate quantification and size estimation difficult. This requires additional PCR amplification cycles, which uses more resources and can potentially introduce bias.
The Critical Role of qPCR in Accurate Quantification
While microcapillary devices provide excellent qualitative data on library size and purity, they should be combined with a sensitive quantification assay for optimal sequencing results. Fluorometric methods like the Qubit dsDNA HS Assay are sensitive but quantify all double-stranded DNA, including non-amplifiable fragments and by-products.
qPCR-based quantification assays use primers targeting the adapter sequences and are therefore specific for fully functional, amplifiable library molecules [27]. This provides a more accurate measure of the library concentration that will actually cluster on the flow cell. The concentration is determined by comparing the Cycle Quantification (Cq) values of the library to a standard curve of known concentrations.
Key Consideration: qPCR assays use intercalating dyes whose signal strength is proportional to fragment length. Therefore, the molarity estimated by qPCR must be normalized according to the average library length obtained from the BioAnalyzer or Fragment Analyzer trace for absolute accuracy [27].
Integration with the AmpliSeq Childhood Cancer Panel Workflow
The QC data obtained from the BioAnalyzer/Fragment Analyzer and qPCR is directly used to guide the final steps of the sequencing workflow. The quantified libraries are normalized to equimolar concentrations and pooled in the recommended DNA:RNA ratio. For the AmpliSeq Childhood Cancer Panel, a 5:1 DNA to RNA pooling volume ratio is recommended to achieve optimal read coverage for both nucleic acid types [4].
This pooled library is then loaded onto a compatible Illumina sequencer. The maximum number of samples per run depends on the sequencing system and the type of libraries (DNA-only, RNA-only, or combined). The table below summarizes the key sequencing parameters for different Illumina systems.
Table 2: Compatible Illumina Sequencers and Run Specifications for the Childhood Cancer Panel
| Sequencing System | Reagent Kit | Max Combined* Samples per Run | Recommended DNA:RNA Pooling Ratio | Approximate Run Time |
|---|---|---|---|---|
| MiniSeq System | Mid Output Kit | 1 | 5:1 | 17 hours |
| High Output Kit | 4 | 5:1 | 24 hours | |
| MiSeq System | MiSeq Reagent Kit v3 | 4 | 5:1 | 32 hours |
| NextSeq 550/1000/2000 | Mid Output v2 Kit | 22 | 5:1 | 26 hours |
| High Output v2 Kit | 48 | 5:1 | 29 hours |
*Combined means paired DNA and RNA from the same sample, generating two libraries [4].
Robust pre-sequencing quality control is a non-negotiable step in ensuring the success of targeted NGS studies using the AmpliSeq Childhood Cancer Panel. The combined use of the BioAnalyzer or Fragment Analyzer for qualitative assessment of library integrity and size, together with qPCR for accurate quantification of amplifiable fragments, provides researchers with the confidence that their libraries are of the highest possible quality. This rigorous approach minimizes the risk of sequencing failures, ensures even coverage across samples, and lays the foundation for robust and reliable somatic variant detection in childhood cancer research. By adhering to these detailed QC protocols and integrating the results with the specified sequencing parameters for Illumina platforms, researchers can maximize the output and value of their valuable genomic studies.
Best Practices for Preventing PCR Contamination in the NGS Workflow
Next-generation sequencing (NGS) has revolutionized biomedical research, providing unprecedented insights into the genetic basis of diseases like childhood cancers. The AmpliSeq for Illumina Childhood Cancer Panel exemplifies this progress, enabling comprehensive evaluation of somatic variants in pediatric and young adult cancers through targeted resequencing. However, the exquisite sensitivity of PCR-based NGS library preparation methods makes these workflows particularly vulnerable to contamination, potentially compromising data integrity and leading to false discoveries. This technical guide outlines a systematic approach to preventing PCR contamination, ensuring the reliability of NGS data generated from sensitive applications such as cancer panel sequencing on Illumina platforms including MiSeq and NextSeq systems.
Understanding PCR Contamination Risks in NGS
In NGS workflows, contamination primarily arises from two sources: foreign nucleic acids (such as genomic DNA from other samples or environmental sources) and carryover amplicons from previous amplification reactions. The latter is particularly problematic in NGS library preparation because a single PCR can generate as many as 10⁹ copies of target sequences, creating significant contamination potential. Aerosolized droplets from these reactions can contain up to 10⁶ amplification products, which can permeate laboratory environments, contaminating reagents, equipment, and ventilation systems if uncontrolled [28]. For sensitive applications like the AmpliSeq Childhood Cancer Panel, which requires only 10 ng of input DNA or RNA and targets numerous low-frequency variants, even minimal contamination can yield misleading results with serious implications for research conclusions [3].
Strategic Spatial Separation and Workflow Design
Physical Laboratory Segregation
Implementing strict physical separation of laboratory areas is the foundational principle for contamination control. This requires designating distinct, isolated spaces for specific workflow stages:
Pre-PCR Area: Dedicated to PCR reaction setup, master mix preparation, and sample processing. This area must remain free of amplified DNA and should contain dedicated equipment, reagents, and consumables that never enter post-PCR areas [29] [28].
Post-PCR Area: Located separately for activities involving amplified DNA, including PCR product purification, quantification, gel electrophoresis, and library analysis. Equipment such as PCR machines and electrophoresis apparatus should remain permanently in this area [29].
Unidirectional Workflow: Researchers must maintain a strict unidirectional workflow moving from pre-PCR to post-PCR areas without backtracking. This prevents the introduction of amplicons into clean pre-PCR spaces [28] [30].
Dedicated Equipment and Supplies
Cross-contamination is often mediated through shared equipment. Prevention strategies include:
- Maintaining separate sets of pipettes with aerosol-filter tips for pre-PCR and post-PCR work [29] [30]
- Using dedicated lab coats, gloves, and waste containers for each area [29]
- Storing reagents for PCR separately in small aliquots to minimize repeated use and exposure [29] [30]
The following diagram illustrates the recommended laboratory workflow and spatial organization:
Procedural Controls and Decontamination Protocols
Environmental Decontamination
Regular decontamination of workspaces is essential for degrading contaminating DNA. Effective methods include:
Surface treatment with 10% sodium hypochlorite (bleach) solution, which causes oxidative damage to nucleic acids, followed by ethanol removal to prevent equipment corrosion [28]. Bleach is particularly effective because it renders nucleic acids unamplifiable.
UV irradiation of workstations, equipment, and consumables. UV light induces thymidine dimers and other covalent modifications in DNA, rendering it unsuitable as a PCR template. This method is most effective when implemented consistently before each use and for sterilizing equipment like pipettes and plasticware [28].
Aseptic Technique
Meticulous technique minimizes the risk of introducing contaminants:
- Using positive displacement or filter tips to prevent aerosol transfer between samples [30]
- Preparing PCR master mixes in a template-free clean room before adding template DNA in a separate area [30]
- Aliquoting reagents in single-use portions to prevent repeated exposure to potential contaminants [29] [30]
Molecular and Enzymatic Contamination Control Methods
Uracil-N-Glycosylase (UNG) System
The UNG system is the most widely implemented method for preventing carryover contamination in diagnostic PCR and NGS workflows. This enzymatic approach employs a straightforward but effective mechanism:
dUTP Incorporation: During PCR amplification, dUTP is substituted for dTTP in the reaction mix, resulting in amplicons that contain uracil instead of thymine [28].
Contaminant Degradation: Prior to the next PCR, the UNG enzyme is added to the reaction mix and incubates at room temperature. During this step, UNG recognizes and catalyzes the hydrolysis of uracil-containing DNA from previous amplifications, rendering them unamplifiable [28].
Enzyme Inactivation: The subsequent 95°C incubation step inactivates UNG, preventing degradation of newly synthesized amplicons in the current reaction [28].
The UNG system is highly effective for thymine-rich amplification products but has reduced activity with G+C-rich targets. Additionally, U-containing DNA may not hybridize as efficiently in some downstream applications, and certain restriction enzymes cleave it with reduced efficiency [28].
K-Box Method for Two-Step PCR NGS Libraries
For two-step PCR procedures commonly used in NGS library preparation, the K-box method provides a sophisticated contamination control system. This approach is particularly valuable for applications requiring high sensitivity, such as the AmpliSeq Childhood Cancer Panel, where detecting rare variants is crucial [31].
The K-box architecture incorporates three synergistic sequence elements:
K1 Elements: Sample-specific sequences (typically 7 nucleotides) in both first and second amplification primers that must match for amplification to occur. Mismatched K1 sequences prevent amplification of carryover contaminants [31].
K2 Elements: Sequences (typically 3 nucleotides) present only in first amplification primers that enable detection of residual contaminations during bioinformatics analysis [31].
S Elements: Separator sequences designed as mismatches with the template that prevent PCR bias by separating the template-matching primer region from the 5' tail [31].
This method effectively suppresses even high rates of artificial contaminations and provides a mechanism to identify any breakthrough contamination in subsequent data analysis [31].
Verification and Quality Control Measures
Control Reactions
Implementing appropriate controls is essential for monitoring contamination:
Negative Controls: Always include template-free controls (using ultrapure water instead of template DNA) in every PCR run to detect reagent or environmental contamination [29].
No-RT Controls: For RNA-based NGS applications, include controls omitting reverse transcriptase to identify genomic DNA contamination in RNA preparations [30].
Assay Design Considerations
Strategic assay design can minimize contamination impact:
Designing assays to span exon junctions helps ensure that amplification of contaminating genomic DNA can be distinguished from target cDNA in RNA sequencing applications [30].
Keeping PCR cycle numbers to a minimum reduces amplicon generation, thereby decreasing contamination potential [29].
Application to AmpliSeq Childhood Cancer Panel Workflow
Implementing these contamination control practices is particularly critical when using targeted panels like the AmpliSeq for Illumina Childhood Cancer Panel. This panel enables simultaneous analysis of 203 genes associated with pediatric cancers through a PCR-based library preparation method requiring minimal input material (10 ng DNA or RNA) [3]. The panel's high sensitivity makes it vulnerable to contamination effects, necessitating rigorous preventive measures throughout the workflow.
Essential Research Reagent Solutions
The following table outlines key components for implementing contamination-controlled NGS workflows with the Childhood Cancer Panel:
| Component Type | Specific Product Examples | Function in Contamination Control |
|---|---|---|
| Library Prep Kits | AmpliSeq Library PLUS for Illumina [3] | Provides optimized reagents for targeted amplification with minimal side products |
| Enzymatic Controls | Uracil-N-Glycosylase (UNG) [28] | Degrades uracil-containing carryover contaminants from previous reactions |
| Decontamination Reagents | Sodium hypochlorite (10% bleach) [28] | Oxidatively degrades nucleic acids on surfaces and equipment |
| Specialized Tips | Aerosol-resistant filter barrier tips [29] [30] | Prevents cross-contamination between samples during pipetting |
| Nucleic Acid Modification | AmpliSeq for Illumina Direct FFPE DNA [3] | Enables direct library prep from FFPE tissues without DNA purification, reducing handling |
Platform-Specific Considerations for Illumina Sequencers
The AmpliSeq Childhood Cancer Panel is compatible with various Illumina sequencing systems, each with different throughput capacities that influence contamination management strategies:
| Sequencing System | Reagent Kit | Maximum Combined* Samples/Run | Contamination Control Implications |
|---|---|---|---|
| MiniSeq System | Mid Output Kit | 1 sample | Lower throughput reduces potential cross-contamination between samples |
| MiniSeq System | High Output Kit | 4 samples | Moderate throughput requires careful sample indexing and tracking |
| MiSeq System | MiSeq Reagent Kit v3 | 4 samples | Moderate throughput with established contamination controls |
| NextSeq System | High Output v2 Kit | 48 samples | High throughput necessitates rigorous sample segregation and indexing |
*Combined means paired DNA and RNA from the same sample generating two libraries [4]
Preventing PCR contamination in NGS workflows requires a comprehensive, multi-layered approach combining physical segregation, procedural discipline, molecular safeguards, and rigorous quality control. For researchers using the AmpliSeq Childhood Cancer Panel on Illumina platforms, implementing these evidence-based practices is essential for generating reliable, reproducible data. As NGS technologies continue to evolve toward even greater sensitivity, maintaining vigilance against contamination will remain fundamental to advancing our understanding of childhood cancers and developing more effective targeted therapies.
Amplicon sequencing is a powerful, cost-effective method for targeted genomic analysis, widely used in applications from microbiome studies to cancer research. A significant technical challenge in this domain is the occurrence of low-diversity libraries, which can severely compromise data quality on Illumina sequencing platforms. This guide details optimized experimental and computational protocols to overcome these challenges, with a specific focus on workflows utilizing the AmpliSeq for Illumina Childhood Cancer Panel.
Low-diversity libraries, common in targeted amplicon sequencing, fail to meet Illumina's requirement for nucleotide heterogeneity in the initial sequencing cycles. This results in poor cluster identification and color matrix calibration, leading to decreased quality scores and increased error rates. Within the context of childhood cancer research, where detecting low-frequency somatic variants is critical, ensuring the highest data quality is not just beneficial—it is essential for reliable results.
Understanding the Low-Diversity Challenge and a Primer-Free Solution
Illumina's sequencing-by-synthesis technology relies on balanced representation of all four nucleotides during the initial cycles (particularly the first 11 bases) to accurately identify clusters on the flow cell and calibrate the optical system for base calling [32]. Amplicon libraries, especially those from a single genetic locus, are inherently homogeneous in their starting sequences, leading to synchronized nucleotide incorporation and low signal diversity. Traditionally, this has been mitigated by spiking in a high-diversity library, such as PhiX [32]. However, this practice consumes sequencing capacity that could otherwise be used for sample data.
A robust alternative is the use of 'N' spacer-linked primers. This method incorporates a pool of primers with random nucleotide sequences (0-10 'N' spacers) at their 5' ends during the target amplification step [32]. This simple modification introduces frameshifts at the beginning of every read, creating the necessary nucleotide diversity from the very first cycle without the need for PhiX.
Experimental Protocol: Implementing 'N' Spacer-Linked Primers
The following protocol is adapted for use with the AmpliSeq Childhood Cancer Panel, which requires DNA and/or RNA input [3].
Key Reagent Solutions:
- AmpliSeq for Illumina Childhood Cancer Panel: Ready-to-use panel targeting 203 genes associated with childhood and young adult cancers [3].
- AmpliSeq Library PLUS for Illumina: reagents for PCR-based library preparation [3].
- 'N' Spacer-Linked Primer Pool: A custom-synthesized, equimolar pool of forward and reverse primers where each primer is appended with 0 to 10 random nucleotides (Ns) at the 5' end [32].
- AmpliSeq CD Indexes: Unique dual indexes for sample multiplexing [3].
Procedure:
- Library Amplification: For each sample, set up the amplification reaction as per the standard AmpliSeq protocol, but replace the standard primer mix with the custom 'N' Spacer-Linked Primer Pool [32].
- Post-Amplification Cleanup: Perform cleanup following the standard AmpliSeq protocol.
- Indexing PCR: Incorporate Illumina CD indexes during the indexing PCR step as usual [3].
- Library Normalization & Pooling: Normalize libraries using a tool like the AmpliSeq Library Equalizer and pool them for sequencing [3].
This workflow integrates the 'N' spacer method into the standard AmpliSeq procedure, as illustrated below:
Pre-Sequencing Quality Control and Platform Selection
Quality Metrics and Sequencing Platform Guidelines
After library preparation, accurate quantification is essential for optimal loading on the sequencer. Use fluorometric methods like Qubit for precise DNA concentration measurement. For the AmpliSeq Childhood Cancer Panel, which generates both DNA and RNA libraries from a single sample, careful calculation of library pooling ratios is required to achieve balanced coverage [4].
The choice of sequencing platform and kit determines the throughput and number of samples that can be multiplexed per run. The table below summarizes the key specifications for compatible Illumina systems when using the Childhood Cancer Panel.
Table 1: Compatible Illumina Sequencers and Sample Throughput for the AmpliSeq Childhood Cancer Panel
| Sequencing System | Reagent Kit | Maximum Combined* Samples per Run | Recommended DNA:RNA Pooling Ratio | Run Time |
|---|---|---|---|---|
| MiSeq System | MiSeq Reagent Kit v3 | 4 | 5:1 | 32 hours |
| NextSeq 550 System | NextSeq Mid Output v2 Kit | 22 | 5:1 | 26 hours |
| NextSeq 550 System | NextSeq High Output v2 Kit | 48 | 5:1 | 29 hours |
| MiniSeq System | MiniSeq High Output Reagent Kit | 4 | 5:1 | 24 hours |
Combined samples refer to paired DNA and RNA from the same sample, generating two libraries [4].
Evaluating the Success of the 'N' Spacer Method
The effectiveness of the 'N' spacer method can be evaluated before a full production run. In a pilot experiment, libraries prepared with the 'N' (0-10) spacer pool should be sequenced on a rapid-run mode (e.g., HiSeq 2500 Rapid-V2). Analysis of the generated FASTQ files with a base composition script (e.g., in Python) will confirm heterogeneous distribution of A, C, G, and T across all initial cycles, successfully mimicking a high-diversity library [32].
Data Analysis and Post-Processing Workflow
Once high-quality sequencing data is obtained, proper bioinformatic processing is crucial.
Pre-Processing and Trimming 'N' Spacers
The first analysis step is the removal of the artificially added 'N' spacers. This can be accomplished using a purpose-built tool like MetReTrim, a Python-based command-line software [32].
Protocol: Trimming with MetReTrim
- Installation: Download MetReTrim from GitHub (
https://github.com/Mohak91/MetReTrim). - Execution: Run the software on your raw FASTQ files. The algorithm identifies and trims the variable-length 'N' spacers from the 5' end of the reads.
- Output: The process typically results in ≥97% of total reads being successfully trimmed and prepared for downstream analysis [32].
From Reads to Results: A Bioinformatics Pipeline
After spacer trimming, data can be processed through a standard amplicon sequencing pipeline. The following workflow, which can be executed using tools like LotuS2, outlines the key steps from raw data to biological insights [33].
Detailed Steps:
- Quality Filtering and Denoising: Use stringent quality filters to remove reads with low-quality scores, ambiguous bases, or excessive homopolymer repeats. Denoising algorithms (e.g., DADA2, integrated in LotuS2) correct sequencing errors and infer the true biological Amplicon Sequence Variants (ASVs) [33].
- Sequence Clustering: Group identical sequences into ASVs. LotuS2 supports multiple clustering algorithms (DADA2, UNOISE3, VSEARCH) for high-resolution data analysis [33].
- Taxonomic or Variant Assignment: For microbiome data, assign taxonomy using reference databases (SILVA, Greengenes). For cancer panels, align ASVs to reference genomes to identify somatic mutations, insertions/deletions (indels), and copy number variants (CNVs) [3] [33].
- Data Normalization for Diversity Analysis: For comparative diversity metrics, normalize the data to account for varying library sizes between samples. Rarefaction is a commonly used technique, though it is subject to debate [34].
- Protocol: Repeated Rarefying: Instead of a single subsampling step, perform repeated rarefying (e.g., 100-1000 iterations). In each iteration, randomly subsample all samples to the same library size and calculate the desired diversity metric (e.g., Shannon index). The final result is a distribution of values that characterizes the stability of the diversity estimate and the variation introduced by the normalization process itself [34].
Optimizing amplicon sequencing data requires a holistic approach that integrates experimental wet-lab techniques with robust bioinformatic analysis. The implementation of the 'N' spacer-linked primer method provides a reliable and cost-effective strategy to eliminate the low-diversity problem, thereby enhancing data quality and maximizing sample throughput on Illumina platforms like MiSeq and NextSeq. When combined with the targeted power of the AmpliSeq Childhood Cancer Panel and careful post-processing, researchers can generate highly reliable data crucial for advancing our understanding of childhood and young adult cancers.
For researchers and drug development professionals utilizing powerful tools like the AmpliSeq for Illumina Childhood Cancer Panel, technical proficiency is paramount. This targeted resequencing solution enables comprehensive evaluation of somatic variants across 203 genes associated with pediatric and young adult cancers, including leukemias, brain tumors, and sarcomas [3]. However, the full potential of this panel—from library preparation through sequencing and data analysis—can only be realized through proper training. A structured continuous education program ensures that your team can generate accurate, reliable data, ultimately accelerating cancer research discoveries.
The Illumina Training Ecosystem: Formats and Offerings
Illumina provides multiple training pathways tailored to different learning preferences, schedules, and operational requirements. These opportunities range from hands-on, in-person sessions to flexible online courses, all led by certified Illumina instructors [35].
Hands-On and Instructor-Led Training
For teams seeking immersive learning experiences, Illumina offers several formats for direct engagement with expert instructors:
Training at Illumina Solutions Centers (ISCs): Located in multiple countries, ISCs provide hands-on NGS training with expert instructors. These courses offer best practices for optimal sequencing results, guidance on each workflow step, and comprehensive troubleshooting tips. Courses require a minimum of four participants [35].
On-Site Training at Your Facility: For organizations preferring in-house training, certified Illumina trainers can conduct sessions in your own lab. This option includes hands-on NGS training for up to four participants, with course fees covering the instructor's time and travel expenses. Organizations must provide required library prep products, sequencing reagents, and lab equipment [35].
Live Online Training: Certain courses are available as live online sessions led by certified Illumina trainers. This format provides direct access to instructor expertise for question-and-answer sessions and crucial workflow tips while avoiding travel and reagent costs [35].
For researchers requiring flexibility or seeking to address specific knowledge gaps, Illumina offers supplementary educational resources:
Self-Paced Online Training: The Illumina website hosts free online training, recorded webinars, and other educational resources accessible at any time. These resources cover key NGS topics to help plan experiments and optimize workflows [35].
Specialized Technical Resources: Beyond formal courses, Illumina provides method guides, eBooks, and technical notes focused on specific applications. For cancer research, these include comprehensive resources like the Cancer Research Methods Guide (40+ pages) and Liquid Biopsy Methods Guide (20+ pages), which provide detailed workflows for thorough sample characterization [36].
Core Training Courses for Targeted Sequencing Applications
While search results do not list a course specifically named for the AmpliSeq Childhood Cancer Panel, several relevant instructor-led courses cover essential techniques and workflows for targeted sequencing. The most applicable offerings include product-specific training that builds foundational skills necessary for running this panel effectively [35].
Table: Key Instructor-Led Training Courses for Targeted Sequencing
| Course Name | Duration | Primary Focus | Relevant Applications | Catalog Number |
|---|---|---|---|---|
| Illumina DNA Prep Training | 1 day | Complete workflow from sample input to library prep, QC, troubleshooting, sequencing, and data analysis | Foundational skills for DNA library preparation | 20022900 |
| TruSight Oncology 500 Training | 3 days | Sample and library preparation, enrichment, sequencing, and data analysis | Comprehensive oncology panel training | 20031667 |
| Illumina Stranded Total RNA Prep Training | 2 days | RNA quantification, best practices, troubleshooting, and analysis tools | RNA sequencing components of cancer panels | 20044764 |
These courses emphasize hands-on instruction with best practices, troubleshooting tips, and training on Illumina-supported analysis tools specific to each application workflow [35]. The TruSight Oncology 500 Training is particularly relevant as it familiarizes researchers with essential steps in a comprehensive oncology workflow, including sample and library preparation, enrichment, sequencing, and data analysis [35].
Technical Framework: AmpliSeq Childhood Cancer Panel Specifications
Understanding the technical requirements of the AmpliSeq Childhood Cancer Panel provides context for the specialized training needed to implement it successfully. This panel offers a targeted resequencing solution for comprehensive evaluation of somatic variants across 203 cancer-associated genes [3].
Essential Research Reagent Solutions
The following reagents and kits are essential for implementing the Childhood Cancer Panel workflow in a research setting:
Table: Essential Research Reagents for AmpliSeq Childhood Cancer Panel Workflow
| Component | Function | Key Specifications |
|---|---|---|
| AmpliSeq for Illumina Childhood Cancer Panel [3] | Core panel targeting 203 cancer-associated genes | Detects SNVs, indels, CNVs, gene fusions; sufficient for 24 samples |
| AmpliSeq Library PLUS for Illumina [3] | Library preparation reagents | Available in 24-, 96-, and 384-reaction configurations |
| AmpliSeq CD Indexes [3] | Sample multiplexing | Enable sample pooling; available in sets of 24, 96, or 384 indexes |
| AmpliSeq cDNA Synthesis for Illumina [3] | Converts total RNA to cDNA for RNA sequencing | Required for RNA workflows with the panel |
| AmpliSeq for Illumina Direct FFPE DNA [3] | DNA preparation from FFPE tissues | Eliminates need for deparaffinization or DNA purification |
Compatible Sequencing Systems and Configurations
The Childhood Cancer Panel is compatible with multiple Illumina sequencing systems, with varying throughput capacities depending on the instrument and reagent configuration:
Table: Sequencing Guidelines for Combined DNA:RNA Samples (5:1 Pooling Ratio)
| Sequencing System | Reagent Kit | Max Combined Samples per Run | Run Time |
|---|---|---|---|
| MiniSeq System [4] | MiniSeq High Output Reagent Kit | 4 | 24 hours |
| MiSeq System [4] | MiSeq Reagent Kit v3 | 4 | 32 hours |
| NextSeq System [4] | NextSeq High Output v2 Kit | 48 | 29 hours |
The panel requires both DNA and RNA library preparation from each sample, generating two separate libraries that are separately indexed [4]. The workflow has an assay time of 5-6 hours (library prep only) with less than 1.5 hours of hands-on time, and requires 10 ng of high-quality DNA or RNA input [3].
Implementing a Continuous Education Strategy
Structured Learning Pathway for Research Teams
The following workflow diagram outlines a strategic approach to building and maintaining technical proficiency with the AmpliSeq Childhood Cancer Panel:
Practical Implementation Guide
To effectively implement a continuous education program for your research team:
Schedule Regular Training Assessments: Conduct quarterly evaluations to identify knowledge gaps and training needs specific to the Childhood Cancer Panel workflow, from library preparation through data analysis.
Combine Multiple Training Formats: Begin with self-paced online courses to establish foundational knowledge, then progress to instructor-led sessions for technical application, and finally implement hands-on practice in your lab environment [35].
Leverage Technical Resources: Utilize the free methodological guides and technical documents provided by Illumina, such as the Cancer Research Methods Guide, which provides simple workflows for a broad range of cancer research applications [36].
Establish a Training Calendar: Plan training sessions to coincide with new instrument acquisitions, protocol updates, or when onboarding new team members to maintain consistent operational standards.
For research teams utilizing the AmpliSeq for Illumina Childhood Cancer Panel, a strategic approach to continuous education is essential for maximizing research output and data quality. By leveraging Illumina's comprehensive training ecosystem—from hands-on product training to self-paced online resources—scientists and drug development professionals can maintain proficiency with this sophisticated technology. The structured learning pathway outlined in this guide provides a framework for building and sustaining the technical expertise necessary to advance cancer research and contribute to improved outcomes for childhood and young adult cancer patients.
Next-generation sequencing (NGS) has revolutionized oncology research, particularly in the pediatric cancer domain where comprehensive genomic profiling is essential for diagnosis, prognosis, and therapeutic decision-making. The AmpliSeq for Illumina Childhood Cancer Panel represents a sophisticated tool designed specifically for this purpose, enabling investigators to simultaneously assess 203 genes associated with childhood and young adult cancers through targeted resequencing [3]. This panel detects multiple variant types—including single nucleotide polymorphisms (SNPs), insertions-deletions (indels), copy number variants (CNVs), and gene fusions—from minimal input material (as little as 10 ng of DNA or RNA) [3]. However, despite its optimized design, researchers often encounter technical challenges that can compromise data quality and variant calling accuracy.
Suboptimal results in NGS workflows can arise from numerous sources, spanning the entire process from library preparation to final bioinformatic analysis. Issues such as low read depth, poor coverage uniformity, high duplicate read rates, and inaccurate variant calling can significantly impact the sensitivity and specificity of detecting clinically relevant variants [37] [11]. For the Childhood Cancer Panel, which is frequently used on Illumina platforms including the MiSeq and NextSeq systems, understanding these challenges within the specific context of the technology is paramount for obtaining reliable results [4]. This guide provides a comprehensive troubleshooting framework to address these issues, with particular emphasis on the AmpliSeq Childhood Cancer Panel workflow, ensuring that researchers can maximize the quality and utility of their genomic data in pediatric cancer research.
Understanding Sequencing Metrics and Performance Benchmarks
Key Quality Metrics and Their Significance
Evaluating the success of an NGS run requires careful assessment of multiple performance metrics. Each metric provides distinct insights into different aspects of data quality and potential sources of technical issues:
Coverage Depth: Refers to the average number of reads covering a given genomic position. The AmpliSeq Childhood Cancer Panel typically achieves mean coverage depths exceeding 1000× when optimized, which provides high sensitivity for variant detection [11]. In a recent validation study, this depth enabled detection of DNA variants with 98.5% sensitivity at a 5% variant allele frequency (VAF) and RNA fusions with 94.4% sensitivity [11]. Inadequate coverage (<200×) can result in missed variants, particularly in regions with inherent technical challenges.
Coverage Uniformity: Measures the consistency of read distribution across targeted regions. Poor uniformity, characterized by significant variation in coverage across amplicons, creates coverage gaps that may obscure clinically relevant variants. The AmpliSeq panel generates 3069 DNA amplicons and 1701 RNA amplicons, making uniform coverage challenging to maintain throughout [4].
Cluster Density: A critical parameter determined during sequencing initialization that directly impacts data quality. For MiSeq and NextSeq systems, optimal cluster densities are system-specific. Excessive density can lead to overlapping clusters and impaired base calling, while insufficient density reduces overall throughput and efficiency [38].
Q30 Score: Represents the percentage of bases with a base call accuracy of 99.9% or higher. This metric reflects the overall sequencing accuracy and is particularly important for confident variant calling, especially for heterozygous single nucleotide variants.
Duplicate Read Rate: Indicates the percentage of PCR-amplified duplicate fragments. Elevated duplication rates suggest limited library complexity, often resulting from insufficient input material or over-amplification during library preparation.
Expected Performance Benchmarks for the Childhood Cancer Panel
The following table summarizes key performance benchmarks established through technical validation of the AmpliSeq Childhood Cancer Panel:
Table 1: Performance Benchmarks for AmpliSeq Childhood Cancer Panel
| Performance Metric | DNA Analysis | RNA Analysis | Notes |
|---|---|---|---|
| Sensitivity | 98.5% (for variants at 5% VAF) | 94.4% (for fusion detection) | VAF = Variant Allele Frequency [11] |
| Specificity | 100% | 100% | [11] |
| Reproducibility | 100% | 89% | [11] |
| Mean Read Depth | >1000× | Targeted depth achieved with proper pooling | Varies by sequencing system [11] |
| Input Requirement | 10 ng (high-quality DNA) | 10 ng (high-quality RNA) | FFPE-compatible protocols available [3] |
For different Illumina sequencing systems, the maximum sample throughput varies significantly. The table below outlines the recommended specifications for combining DNA and RNA libraries from the same samples:
Table 2: Sequencing System Specifications for Childhood Cancer Panel
| Sequencing System | Reagent Kit | Max Combined Samples per Run | Recommended DNA:RNA Pooling Ratio | Run Time |
|---|---|---|---|---|
| MiSeq System | MiSeq Reagent Kit v3 | 4 | 5:1 | 32 hours |
| NextSeq System | NextSeq High Output v2 Kit | 48 | 5:1 | 29 hours |
| MiniSeq System | MiniSeq High Output Reagent Kit | 4 | 5:1 | 24 hours |
The 5:1 DNA:RNA pooling ratio is based on recommended read coverage requirements for each data type [4]. Deviation from this ratio may result in inadequate coverage for one nucleic acid type and excessive coverage for the other, reducing overall efficiency.
Systematic Troubleshooting of Common Issues
Low Read Depth and Coverage Gaps
Insufficient coverage remains one of the most frequent challenges in targeted sequencing and can severely impact variant calling accuracy. Research has demonstrated that modest increases in sequencing depth can result in rapid gains in total variant recovery, particularly for heterozygous variants [37]. Several factors can contribute to low read depth:
Insufficient Sequencing Saturation: The most straightforward cause is simply not generating enough total reads for the number of samples multiplexed. For the Childhood Cancer Panel, the required depth per sample must be calculated based on the total output of the specific Illumina instrument and reagent combination being used.
Suboptimal Library Quantification: Inaccurate quantification of libraries prior to pooling and sequencing can lead to disproportionate representation of samples. This is particularly problematic when employing the recommended 5:1 DNA:RNA pooling ratio [4]. Fluorometric methods (e.g., Qubit) are preferred over spectrophotometric approaches for accurate library quantification.
PCR Amplification Biases: The AmpliSeq panel employs PCR-based library preparation, which can introduce amplification biases particularly in GC-rich regions [3]. These biases can result in significant coverage dropouts in certain genomic regions, potentially obscuring clinically relevant variants.
Nucleic Acid Quality: While the panel requires only 10 ng of input DNA or RNA, sample quality profoundly impacts coverage uniformity. Degraded samples, particularly FFPE-derived material, may exhibit substantial coverage gaps [3]. The panel offers specific solutions for FFPE tissues without requiring deparaffinization or DNA purification [3].
Remedial Strategies:
- Recalculate pooling concentrations using highly accurate fluorometric methods
- Incorporate molecular barcodes to account for amplification biases
- For FFPE samples, utilize the AmpliSeq for Illumina Direct FFPE DNA protocol to improve coverage [3]
- Consider increasing sequencing depth by 20-30% for regions with historically poor coverage
Poor Variant Calling Accuracy
Variant calling inaccuracies can manifest as both false positives and false negatives, potentially leading to incorrect research conclusions. A study of 4577 molecularly characterized families found numerous scenarios where variant identification and interpretation proved challenging, estimating a 34.3% probability of encountering at least one such challenge in Mendelian disease research [39]. Specific issues include:
Low-Frequency Variants: Detection of variants with low VAF (<5%) remains challenging, particularly in heterogeneous tumor samples. The technical validation of the Childhood Cancer Panel demonstrated 98.5% sensitivity for DNA variants at 5% VAF, but performance decreases substantially below this threshold [11].
RNA-Specific Artifacts: Variant calling from RNA-Seq data presents unique challenges including difficulties distinguishing true mutations from RNA-editing events, reverse transcription artifacts, and strand-specific biases [40]. The most common RNA editing in humans (adenosine-to-inosine editing) appears as A-to-G changes in sequencing data and can be misinterpreted as genuine genomic variants without matched DNA sequencing [40].
Post-Mortem Damage Patterns: While more common in ancient DNA studies, degradation patterns similar to post-mortem damage can occur in clinical samples, featuring elevated C-T and G-A transitions at sequence ends that confound accurate SNP identification [37].
Alignment Issues: Increased divergence between sample and reference genomes decreases alignment success, particularly for longer read lengths [37]. This can be especially problematic in populations with genetic backgrounds divergent from the reference genome.
Remedial Strategies:
- Implement duplex sequencing methods for enhanced low-frequency variant detection
- For RNA variants, utilize databases of known editing sites and characteristic sequence motifs to distinguish true mutations
- Apply damage pattern correction algorithms for degraded samples
- Consider population-specific reference genomes or graph-based alignment approaches to improve mapping efficiency
Library Preparation and Quality Control Issues
Library preparation represents a critical phase where multiple issues can be introduced, ultimately affecting final data quality:
Adapter Dimer Formation: During library preparation, adapter dimers can form and sequester sequencing capacity if not adequately removed [41]. These dimers amplify efficiently but provide no meaningful data, reducing useful sequence output.
RNA Integrity Issues: For RNA component analysis, integrity is paramount for accurate fusion detection. The Childhood Cancer Panel validation utilized tape station or Labchip analysis to confirm RNA integrity prior to library preparation [11].
cDNA Synthesis Artifacts: The reverse transcription step in RNA library preparation can introduce artifacts, including template switching that generates chimeric cDNA molecules and sequence-specific pausing that creates coverage gaps [40].
Remedial Strategies:
- Implement rigorous size selection using solid-phase reversible immobilization (SPRI) beads to remove adapter dimers
- Establish minimum RNA quality thresholds (e.g., RIN >7) for inclusion in sequencing
- Incorporate unique molecular identifiers (UMIs) to account for PCR duplicates and reverse transcription artifacts
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of the AmpliSeq Childhood Cancer Panel requires several specialized reagents and kits that facilitate optimal performance. The following table details these essential components:
Table 3: Essential Research Reagent Solutions for Childhood Cancer Panel
| Reagent/Kits | Function | Usage Notes |
|---|---|---|
| AmpliSeq Library PLUS for Illumina | Provides reagents for preparing sequencing libraries | Available in 24-, 96-, and 384-reaction configurations [3] |
| AmpliSeq CD Indexes | Enables sample multiplexing through barcoding | Sets A-D provide 384 unique indexes; critical for sample tracking [3] |
| AmpliSeq cDNA Synthesis for Illumina | Converts total RNA to cDNA for RNA analysis | Required when working with RNA components of the panel [3] |
| AmpliSeq Library Equalizer for Illumina | Normalizes libraries for balanced representation | Improves coverage uniformity across samples [3] |
| AmpliSeq for Illumina Direct FFPE DNA | Enables DNA preparation from FFPE tissues | Eliminates need for deparaffinization or DNA purification [3] |
| SeraSeq Tumor Mutation DNA Mix | Positive control for DNA variant detection | Multiplex biosynthetic mixture with variants at known VAF [11] |
| SeraSeq Myeloid Fusion RNA Mix | Positive control for RNA fusion detection | Contains synthetic RNA fusions for assay validation [11] |
Experimental Protocols for Key Validation Experiments
Protocol for Determining Limit of Detection (LOD)
Establishing the limit of detection for variant calling is essential for validating panel performance, particularly for low-frequency variants in heterogeneous tumor samples.
Materials:
- SeraSeq Tumor Mutation DNA Mix (v2 AF10 HC) or similar reference standard
- NA12878 as negative control
- AmpliSeq Library PLUS for Illumina
- AmpliSeq CD Indexes
- MiSeq or NextSeq sequencing system
Methodology:
- Serially dilute the positive control material with negative control DNA to create samples with variant allele frequencies spanning 1%-10%
- Process diluted samples through the standard Childhood Cancer Panel workflow [11]
- Sequence libraries using recommended instrument parameters [4]
- Analyze variants at each dilution level to determine sensitivity and specificity
- Apply statistical models to determine the VAF at which 95% detection sensitivity is achieved
This experimental approach enabled the validation study to establish that the Childhood Cancer Panel achieves 98.5% sensitivity for DNA variants at 5% VAF [11].
Protocol for Assessing Reproducibility
Technical reproducibility is fundamental for establishing assay reliability across multiple runs and operators.
Materials:
- Representative patient samples or commercial controls covering different variant types
- Multiple library preparation operators (if assessing inter-operator variability)
Methodology:
- Split each sample into multiple aliquots for parallel processing
- Have different operators prepare libraries independently on different days [11]
- Sequence libraries across different flow cells or instruments
- Compare variant calls across all replicates
- Calculate concordance rates for SNVs, indels, and fusions
In the technical validation, this approach demonstrated 100% reproducibility for DNA variants and 89% reproducibility for RNA fusion detection [11].
Advanced Troubleshooting: Specialized Scenarios
Addressing RNA-Seq Specific Challenges
RNA sequencing introduces unique complexities that require specialized troubleshooting approaches:
Allelic Dropout in Low-Expressed Genes: RNA-Seq coverage directly correlates with gene expression levels, potentially resulting in insufficient read depth for confident variant calling in lowly expressed genes [40]. This can lead to false homozygous calls when one allele fails to be represented. Solution: Implement expression-based filtering thresholds and consider integrating information across genes with similar expression patterns.
Strand-Specific Biases: Library preparation protocols can create asymmetric coverage patterns between forward and reverse strands [40]. Solution: Incorporate strand bias metrics into variant filtering pipelines and utilize strand-balanced protocols when possible.
Fusion Detection Validation: While the Childhood Cancer Panel demonstrated 94.4% sensitivity for fusion detection, certain complex rearrangements may be missed [11]. Solution: Orthogonal validation using qRT-PCR with specific primers and probes as implemented in the Europe Against Cancer Program [11].
Overcoming Bioinformatics Challenges
Bioinformatic analysis represents a potential bottleneck where properly optimized pipelines are essential for accurate variant calling:
Reference Genome Biases: Traditional linear reference genomes can introduce mapping biases, particularly for populations with genetic backgrounds divergent from the reference [40]. Solution: Explore graph-based aligners that incorporate known genetic variations to improve alignment accuracy.
Machine Learning-Enhanced Variant Calling: Tools like DeepVariant employ convolutional neural networks to analyze "images" of aligned reads, demonstrating superior performance for variant calling [40]. These approaches are particularly valuable for distinguishing true low-frequency variants from technical artifacts.
Copy Number Variant Detection: CNV calling from targeted panels requires specialized approaches due to uneven coverage patterns. Solution: Implement robust normalization algorithms that account for GC content and other sequence-specific biases.
Successful implementation of the AmpliSeq Childhood Cancer Panel on Illumina platforms requires meticulous attention to each step of the workflow, from initial library preparation through final bioinformatic analysis. By understanding the common pitfalls and their remedial strategies outlined in this guide, researchers can significantly improve data quality and variant calling accuracy. The structured troubleshooting approach presented here—addressing specific issues with read depth, variant calling, library preparation, and specialized scenarios—provides a comprehensive framework for optimizing performance of this important pediatric cancer research tool. As NGS technologies continue to evolve, maintaining rigorous quality control standards and implementing appropriate validation protocols will remain essential for generating reliable, clinically actionable genomic data.
Evidence and Context: Validating Panel Performance and Exploring Alternatives
Technical validation is a critical component of leukemia research, ensuring that diagnostic and prognostic assays produce reliable, accurate, and reproducible results. In the context of genomic studies utilizing targeted panels like the Illumina AmpliSeq Childhood Cancer Panel, rigorous assessment of analytical performance parameters—sensitivity, specificity, and reproducibility—is fundamental to generating clinically actionable data. The integration of these panels with compatible Illumina sequencing platforms (MiSeq, NextSeq) creates a standardized workflow for comprehensive genomic profiling in leukemias. This technical guide examines validation methodologies across multiple detection platforms, provides detailed experimental protocols, and establishes a framework for quality assurance within leukemia research settings, with particular emphasis on applications for the AmpliSeq Childhood Cancer Panel.
Core Principles of Technical Validation
Defining Key Validation Parameters
In leukemia studies, technical validation requires precise quantification of three fundamental assay characteristics:
Analytical Sensitivity: The lowest level at which a target (e.g., genetic variant, leukemia-associated phenotype) can be reliably detected. Sensitivity is typically expressed as a limit of detection (LOD), often ranging from 10⁻⁴ to 10⁻⁵ (1 in 10,000 to 1 in 100,000 cells) in optimized assays [42] [43].
Analytical Specificity: The ability of an assay to correctly distinguish the target of interest from similar but distinct entities, minimizing false positives resulting from cross-reactivity or background interference [44].
Reproducibility: The degree of concordance between results when the same sample is tested repeatedly under varying conditions, including different operators, instruments, laboratories, or time points [45] [44].
Importance in Leukemia Research and Clinical Applications
Robust technical validation is particularly crucial in leukemia research and clinical practice for several key applications:
Measurable Residual Disease (MRD) Monitoring: Detection of residual leukemia cells after treatment at sensitivity thresholds of 10⁻⁴ to 10⁻⁵ is strongly associated with clinical outcomes and can guide therapeutic decisions [42] [43].
Prognostic Model Development: Machine learning algorithms for outcome prediction require high-quality, reproducible input data to generate reliable risk stratification models [46] [47].
Therapeutic Target Identification: Accurate detection of somatic variants directs targeted therapy selection, necessitating high specificity to avoid inappropriate treatment.
Table 1: Technical Performance Requirements for Key Leukemia Applications
| Application | Required Sensitivity | Required Specificity | Primary Technologies |
|---|---|---|---|
| MRD Detection | 10⁻⁴ to 10⁻⁵ [42] [43] | >99% [43] | Multiparameter Flow Cytometry, NGS [42] [43] |
| Somatic Variant Detection | 1-5% variant allele frequency [4] [3] | >99% [4] | Targeted NGS Panels (e.g., AmpliSeq Childhood Cancer) [4] [3] |
| Gene Expression Profiling | Detection of 1.5-fold changes [44] | Balance with reproducibility [44] | Microarrays, RNA-Seq [47] [44] |
| Prognostic Model Development | Variable based on endpoint [46] | Variable based on endpoint [46] | Multiple platforms integrated via machine learning [46] [47] |
Validation Methodologies Across Platforms
Multiparameter Flow Cytometry (MPFC)
MPFC enables detection of leukemia-associated phenotypes (LAPs) based on surface antigen expression patterns. A standardized approach to validation includes:
Panel Design: CD45 gating strategy with 5-color staining using monoclonal antibodies against CD2, CD56, CD11b, CD7, and CD19, which represent the most sensitive and reliable markers for MRD detection in AML [42].
Sensitivity Determination: Serial dilution experiments using leukemia cell lines spiked into normal bone marrow samples establish detection limits between 10⁻⁴ and 10⁻⁵ [42].
Specificity Assessment: Comparison of LAP frequency in leukemic versus normal/regenerating bone marrow samples using maximum log difference statistics [42].
The technical workflow for MPFC validation can be visualized as follows:
Next-Generation Sequencing (NGS) Platforms
The AmpliSeq for Illumina Childhood Cancer Panel provides targeted resequencing for comprehensive evaluation of somatic variants across 203 genes associated with pediatric and young adult cancers, including leukemias [4] [3]. Technical validation for this panel involves:
Library Preparation: The protocol requires 10 ng of high-quality DNA or RNA input, with 5-6 hours for library preparation (excluding quantification and normalization) [3]. The panel generates both DNA and RNA libraries from each sample, with separate indexing.
Platform Compatibility: The panel is compatible with multiple Illumina sequencing systems, each with specific throughput characteristics as detailed in Table 2.
Sensitivity Verification: For mutation detection, sensitivity is typically validated using commercially available reference standards with known variant allele frequencies.
Table 2: Illumina Sequencing Platform Specifications for Childhood Cancer Panel
| Sequencing System | Reagent Kit | Max DNA Samples Per Run | Max RNA Samples Per Run | Recommended DNA:RNA Pooling Ratio | Run Time |
|---|---|---|---|---|---|
| MiniSeq System | Mid Output | 1 | 8 | 5:1 | 17 hours |
| MiniSeq System | High Output | 5 | 25 | 5:1 | 24 hours |
| MiSeq System | MiSeq Reagent Kit v3 | 5 | 25 | 5:1 | 32 hours |
| NextSeq System | Mid Output v2 Kit | 27 | 96 | 5:1 | 26 hours |
| NextSeq System | High Output v2 Kit | 83 | 96 | 5:1 | 29 hours |
Molecular Techniques (PCR-based)
PCR-based methods, particularly quantitative real-time PCR (qRT-PCR) with allele-specific oligonucleotides (ASOs), provide high sensitivity for detecting leukemia-specific genetic alterations:
Sensitivity Controls: Incorporation of well-characterized cell line DNA in each assay adds a critical quality-control parameter to ensure assay-to-assay reproducibility [45].
Reproducibility Assessment: Inter-laboratory comparison studies and standardization of sample processing, DNA extraction, and amplification conditions [43].
Specificity Optimization: Primer and probe design to minimize cross-reactivity with similar gene sequences or pseudogenes.
Experimental Protocols for Validation Studies
Protocol 1: Establishing Sensitivity Limits for MRD Detection
Purpose: To determine the limit of detection (LOD) for leukemia cells using multiparameter flow cytometry.
Materials and Reagents:
- Leukemia cell lines with characterized immunophenotype
- Normal bone marrow mononuclear cells
- Monoclonal antibodies: CD2, CD5, CD7, CD11b, CD19, CD56, CD45
- Staining buffer, flow cytometer with 5-color capability
Procedure:
- Prepare serial dilutions of leukemia cells in normal bone marrow mononuclear cells at ratios of 10⁻², 10⁻³, 10⁻⁴, 10⁻⁵, and 10⁻⁶.
- Aliquot 1×10⁶ cells per tube and stain with antibody cocktails according to established protocols [42].
- Acquire a minimum of 500,000 events per sample using a standardized CD45 gating strategy.
- Analyze LAP populations using differential expression patterns compared to normal cells.
- Determine LOD as the lowest dilution at which the LAP is consistently detected (≥95% of replicates).
Validation Criteria: The assay should demonstrate a sensitivity of at least 10⁻⁴ with a coefficient of variation <20% across replicates [42].
Protocol 2: Reproducibility Assessment for Gene Expression Studies
Purpose: To evaluate inter-platform and inter-site reproducibility of gene expression measurements in leukemia samples.
Materials and Reagents:
- Leukemia patient RNA samples (n≥20)
- Microarray platforms or RNA-Seq protocols
- TaqMan assays for reference genes
- Standardized RNA extraction and quality control reagents
Procedure:
- Distribute identical RNA aliquots to multiple testing sites or analyze using different platforms.
- Process samples according to standardized protocols for each platform.
- Identify differentially expressed genes (DEGs) using both statistical significance (p-value) and fold-change (FC) ranking [44].
- Calculate concordance between DEG lists using Cohen's kappa coefficient.
- Compare microarray/RNA-Seq results with TaqMan (reference standard) for key genes.
Analysis: Use FC-ranking with non-stringent p-value cutoff (e.g., p<0.05) to maximize reproducibility while balancing sensitivity and specificity [44].
The relationship between statistical stringency and reproducibility in gene expression studies follows a predictable pattern:
Quality Control Frameworks
Integrated QC Strategy for Leukemia Studies
A comprehensive quality control framework for leukemia studies incorporates multiple checkpoints throughout the experimental workflow:
Pre-analytical Controls: Standardized sample collection, processing, and nucleic acid extraction protocols to minimize pre-analytical variability.
Analytical Controls: Inclusion of positive, negative, and sensitivity controls in each assay batch to monitor performance [45].
Post-analytical Controls: Implementation of standardized analysis pipelines, reference datasets for benchmarking, and data quality metrics.
Machine Learning Validation Approaches
Modern prognostic model development in leukemia employs sophisticated validation frameworks:
Nested Cross-Validation: Implementation of outer-loop performance estimation with inner-loop hyperparameter tuning to prevent optimism bias [46].
External Validation: Assessment of model performance on temporally and geographically distinct datasets to evaluate generalizability [46].
Calibration Assessment: Evaluation of agreement between predicted probabilities and observed outcomes using calibration slopes, intercepts, and Hosmer-Lemeshow tests [46].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Leukemia Study Validation
| Reagent/Kit | Primary Function | Application in Validation | Specifications |
|---|---|---|---|
| AmpliSeq for Illumina Childhood Cancer Panel [4] [3] | Targeted resequencing of 203 cancer-associated genes | Detection of somatic variants in leukemia samples | 24 reactions, 10 ng DNA/RNA input, 5-6 hour prep time |
| AmpliSeq Library PLUS for Illumina [3] | Library preparation reagents | Construction of sequencing libraries | Available in 24-, 96-, and 384-reaction configurations |
| AmpliSeq CD Indexes [3] | Sample multiplexing | Allows pooling of multiple samples in one run | 96 indexes per set, 8 bp indexes |
| AmpliSeq cDNA Synthesis for Illumina [3] | RNA to cDNA conversion | Required for RNA analysis using AmpliSeq panels | Compatible with RNA panels |
| Monoclonal Antibody Panels for MPFC [42] | Detection of LAPs | MRD monitoring in AML | CD2, CD56, CD11b, CD7, CD19 most sensitive markers |
| Sensitivity Control Cell Lines [45] | Assay quality control | Ensures reproducibility in gene rearrangement studies | Well-characterized DNA for batch-to-batch comparison |
Case Studies in Leukemia Validation
Case Study 1: Machine Learning Prognostic Model
A recent large-scale study developed and validated a machine learning model to predict severe complications in acute leukemia patients following induction chemotherapy [46]:
- Dataset: 2,870 adults with newly diagnosed AML or ALL across three tertiary hematology centers.
- Validation Approach: Temporal and geographical external validation with 861 patients.
- Performance: LightGBM algorithm achieved AUROC of 0.801 (95% CI 0.774-0.827) in external validation with excellent calibration.
- Key Predictors: CRP, absolute neutrophil count, cytogenetic-risk tier, age, and ferritin were top-performing variables.
- Validation Rigor: Adherence to TRIPOD-AI and PROBAST-AI guidelines for transparent reporting.
Case Study 2: Immune Microenvironment Profiling
An integrated analysis combining bulk RNA-seq, single-cell RNA-seq, and machine learning identified critical immune regulatory genes in leukemia [47]:
- Technical Validation: RT-PCR confirmation of significant upregulation of key genes (TLR2, TLR4, CCR7, IL18) in leukemia cell lines compared to normal controls.
- Single-Cell Validation: CellChat algorithm mapped intercellular communication networks within the leukemia microenvironment.
- Reproducibility: Cross-platform validation using TCGA and GEO datasets.
- Predictive Performance: The prognostic model demonstrated exceptional performance with AUC values of 0.874, 0.891, and 0.925 for 1-, 2-, and 3-year survival endpoints.
Technical validation of sensitivity, specificity, and reproducibility forms the foundation of reliable leukemia research. The integration of standardized technological platforms like the Illumina AmpliSeq Childhood Cancer Panel with rigorous validation protocols enables robust genomic profiling that can inform both biological insights and clinical applications. As leukemia research increasingly incorporates multi-omic approaches and machine learning algorithms, adherence to comprehensive validation frameworks becomes ever more critical. Future directions will likely include greater standardization of validation protocols across laboratories, development of more sensitive single-cell technologies, and implementation of artificial intelligence-based quality control systems to further enhance reproducibility and reliability in leukemia studies.
Targeted next-generation sequencing (NGS) panels have revolutionized diagnostic precision in oncology, particularly for childhood cancers characterized by low mutational burden but high clinical relevance of genetic alterations. This technical guide examines the demonstration of clinical utility for the AmpliSeq for Illumina Childhood Cancer Panel, a targeted NGS solution for investigating 203 genes associated with pediatric and young adult cancers. We present comprehensive validation metrics, detailed experimental protocols, and clinical impact data demonstrating how panel findings refine diagnostic classification, inform therapeutic targeting, and ultimately improve patient management. Within the context of compatible Illumina sequencing platforms including MiSeq, NextSeq, and MiniSeq systems, we provide researchers and drug development professionals with frameworks for assessing clinical utility through analytical validation, clinical impact quantification, and implementation pathways that bridge laboratory findings to therapeutic applications.
Clinical utility represents the likelihood that a diagnostic test will, by prompting clinical intervention, result in improved health outcomes [48]. Beyond mere technical performance, demonstrating clinical utility requires evidence that testing provides information leading to changed patient management, superior therapeutic decisions, and ultimately better health results. The hierarchical model of diagnostic efficacy positions clinical utility as the critical bridge between analytical validity (technical performance) and patient outcomes [48]. For pediatric cancers, where genetic alterations frequently drive diagnostic classification, risk stratification, and therapeutic selection, targeted NGS panels offer unprecedented opportunities for comprehensive molecular profiling.
The AmpliSeq for Illumina Childhood Cancer Panel provides a targeted resequencing solution specifically designed for childhood and young adult cancers, detecting single nucleotide variants (SNVs), insertions-deletions (indels), copy number variants (CNVs), and gene fusions across 203 genes [3]. This panel addresses the distinctive genetic landscape of pediatric malignancies, which differ from adult cancers in their lower mutational burden but higher clinical actionability of identified variants [11]. This technical guide explores the demonstration of clinical utility through analytical validation, clinical impact assessment, and therapeutic decision-making support within the framework of compatible Illumina sequencing systems.
AmpliSeq Childhood Cancer Panel Fundamentals
The AmpliSeq for Illumina Childhood Cancer Panel employs PCR-based library preparation to target genes associated with childhood and young adult cancers, including leukemias, brain tumors, and sarcomas [3]. The panel simultaneously analyzes DNA and RNA from minimal input material (10 ng), generating comprehensive genomic profiles while saving the substantial time and effort associated with custom panel development [3]. The integrated workflow encompasses library preparation, Illumina sequencing by synthesis (SBS) technology, and automated analysis, creating a standardized pipeline for clinical research applications.
The panel's component structure separates DNA and RNA analyses, with distinct amplicon pools for each nucleic acid type, as detailed in Table 1 [4]. This dual approach enables detection of diverse variant types from limited specimen quantities, a critical advantage in pediatric settings where sample material is often scarce.
Table 1: AmpliSeq Childhood Cancer Panel Technical Components
| Component | Number of Pools | Concentration | Number of Amplicons | Average Amplicon Length | Average Library Length |
|---|---|---|---|---|---|
| DNA | 2 | 4X | 3,069 | 114 bp | 254 bp |
| RNA | 2 | 5X | 1,701 | 122 bp | 262 bp |
Compatible Sequencing Systems and Configurations
The AmpliSeq Childhood Cancer Panel is compatible with multiple Illumina sequencing platforms, offering flexibility for different throughput requirements and laboratory settings. The panel supports sequencing on MiSeq, NextSeq 550, NextSeq 2000, NextSeq 1000, MiSeqDx (in Research Mode), and MiniSeq systems [3]. Table 2 outlines the sequencing guidelines, including maximum sample capacities and recommended DNA:RNA pooling ratios for optimal coverage across different instrument configurations.
Table 2: Sequencing Guidelines for Compatible Illumina Systems
| System | Reagent Kit | Max DNA Samples Per Run | Max RNA Samples Per Run | Max Combined Samples Per Run | Recommended DNA:RNA Pooling Ratio | Run Time |
|---|---|---|---|---|---|---|
| MiniSeq | Mid Output | 1 | 8 | 1 | 5:1 | 17 hours |
| MiniSeq | High Output | 5 | 25 | 4 | 5:1 | 24 hours |
| MiSeq | v2 | 3 | 15 | 2 | 5:1 | 24 hours |
| MiSeq | v3 | 5 | 25 | 4 | 5:1 | 32 hours |
| NextSeq | Mid Output v2 | 27 | 96 | 22 | 5:1 | 26 hours |
| NextSeq | High Output v2 | 83 | 96 | 48 | 5:1 | 29 hours |
The recommended 5:1 DNA:RNA pooling ratio is based on optimal read coverage requirements, with DNA generally requiring higher coverage for confident variant calling, particularly for somatic mutations at low variant allele frequencies [4].
Experimental Protocol and Validation Methodology
Library Preparation Workflow
The standardized protocol for the AmpliSeq Childhood Cancer Panel begins with quality assessment of input nucleic acids. DNA and RNA purity should demonstrate OD260/280 ratio >1.8, with integrity confirmed through systems such as Labchip or TapeStation [11]. Fluorometric quantification using Qubit Fluorimetry with appropriate assay kits ensures accurate input measurement.
Library preparation follows a PCR-based approach using the AmpliSeq for Illumina Childhood Cancer Panel kit according to manufacturer instructions [11]. The process involves:
- DNA Processing: 100 ng of DNA generates 3,069 amplicons covering coding regions of targeted genes.
- RNA Processing: 100 ng of RNA is reverse transcribed to cDNA using the AmpliSeq cDNA Synthesis kit, then used to generate 1,701 amplicons targeting fusion genes.
- Amplification: Consecutive PCRs create amplicon libraries with sample-specific barcodes.
- Library Cleanup: Purification removes contaminants and reaction components.
- Quality Control: Assessment of library quality and quantity.
- Normalization: Using AmpliSeq Library Equalizer for consistent library representation.
- Pooling: Combining DNA and RNA libraries at 5:1 ratio based on recommended coverage requirements.
The complete library preparation requires 5-6 hours assay time with less than 1.5 hours hands-on time, enabling efficient processing of multiple samples [3].
Sequencing and Data Analysis
Pooled libraries are diluted to appropriate concentrations (typically 17-20 pM) and loaded onto compatible Illumina sequencing platforms [11]. The MiSeq System was utilized in the validation study referenced, providing sufficient coverage for confident variant calling. Following sequencing, data analysis proceeds through:
- Demultiplexing: Assignment of reads to specific samples based on barcode sequences.
- Alignment: Mapping of reads to reference genome.
- Variant Calling: Identification of SNVs, indels, CNVs, and gene fusions using appropriate algorithms.
- Annotation: Functional interpretation of variants using clinical databases.
- Reporting: Formatting of clinically actionable findings.
The following workflow diagram illustrates the complete experimental process from sample to analysis:
Validation Framework and Performance Metrics
Comprehensive validation of the AmpliSeq Childhood Cancer Panel followed established guidelines for diagnostic test evaluation, assessing analytical sensitivity, specificity, reproducibility, and limit of detection (LOD) [11]. The validation methodology included:
- Reference Materials: Commercial controls including SeraSeq Tumor Mutation DNA Mix and SeraSeq Myeloid Fusion RNA Mix for sensitivity and specificity determination.
- Clinical Samples: 76 pediatric patients with acute leukemia (B-ALL, T-ALL, AML) with available conventional molecular testing results for comparison.
- Performance Assessment: Comparison against established methods including Sanger sequencing, fragment analysis, and quantitative RT-PCR.
Validation results demonstrated robust performance characteristics, with the panel achieving mean read depth >1000×, significantly exceeding minimum requirements for confident variant calling [11]. The panel showed 98.5% sensitivity for DNA variants at 5% variant allele frequency (VAF) and 94.4% sensitivity for RNA fusions, with 100% specificity for DNA and 89% reproducibility for RNA [11]. These metrics establish the technical reliability necessary for clinical implementation.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of the AmpliSeq Childhood Cancer Panel requires specific reagents and components that constitute the essential research toolkit. Table 3 details the required products and their functions in the experimental workflow.
Table 3: Research Reagent Solutions for AmpliSeq Childhood Cancer Panel
| Component Category | Product Name | Function | Recommended Quantity for 24 Samples |
|---|---|---|---|
| Panel | AmpliSeq for Illumina Childhood Cancer Panel | Contains primer pools for targeting 203 cancer-associated genes | 1 panel (24 reactions) |
| Library Prep | AmpliSeq Library PLUS | Provides reagents for library preparation including enzymes and buffers | 2 × 24-reaction kits |
| Index Adapters | AmpliSeq CD Indexes | Sample-specific barcodes for multiplexing | 1 set (96 indexes) |
| cDNA Synthesis | AmpliSeq cDNA Synthesis | Converts RNA to cDNA for RNA fusion analysis | 1 kit |
| Library Normalization | AmpliSeq Library Equalizer | Normalizes libraries for balanced representation | 1 kit |
| Specialized Sample Processing | AmpliSeq for Illumina Direct FFPE DNA | Enables DNA preparation from FFPE tissues without deparaffinization | As needed |
| Sample Tracking | AmpliSeq for Illumina Sample ID Panel | SNP genotyping panel for sample identification and tracking | 1 panel |
The number of reactions and kits required scales with sample throughput, with configurations available for 24, 96, and 384 samples [3] [4]. For comprehensive sample processing, additional nucleic acid extraction and quantification reagents are necessary, including quality assessment tools such as spectrophotometers, fluorometers, and fragment analyzers.
Demonstrating Clinical Utility: Evidence and Impact Metrics
Clinical Impact on Diagnostic Refinement
The ultimate measure of a diagnostic test's value lies in its ability to influence patient management and outcomes. In a validation study of 76 pediatric acute leukemia patients, the AmpliSeq Childhood Cancer Panel demonstrated substantial clinical impact, with 49% of identified mutations and 97% of detected fusions having clear clinical implications [11]. Specifically, the panel refined diagnosis in 41% of patients based on mutation findings and 97% based on fusion gene identification [11]. These metrics quantify the panel's capacity to provide clinically actionable information beyond conventional diagnostic approaches.
The panel identified targetable alterations in 49% of mutations, directly enabling precision medicine approaches [11]. Overall, 43% of patients tested received clinically relevant results that informed their management, demonstrating significant clinical utility in a real-world pediatric hematology setting [11].
Analytical Performance Supporting Clinical Implementation
The clinical utility of any diagnostic test depends fundamentally on its analytical performance. The AmpliSeq Childhood Cancer Panel validation established robust performance characteristics across critical parameters as summarized in Table 4.
Table 4: Analytical Performance Metrics of AmpliSeq Childhood Cancer Panel
| Performance Parameter | DNA Analysis | RNA Analysis | Method of Assessment |
|---|---|---|---|
| Mean Read Depth | >1000× | >1000× | Sequencing metrics |
| Sensitivity | 98.5% (at 5% VAF) | 94.4% | Commercial controls |
| Specificity | 100% | 100% | Commercial controls |
| Reproducibility | 100% | 89% | Replicate experiments |
| Limit of Detection | 5% VAF | Established for fusions | Serial dilutions |
The high sensitivity for DNA variants at 5% VAF enables detection of subclonal alterations potentially relevant to therapy resistance and disease progression [11]. The comprehensive nature of the panel, assessing multiple variant types simultaneously, provides a more complete molecular profile than sequential single-gene tests traditionally used in leukemia diagnostics.
Clinical Utility in the Context of Evolving Cancer Therapeutics
The clinical utility of genomic panels extends beyond diagnosis to therapeutic decision-making, particularly with the expanding landscape of targeted therapies. In oncology, next-generation sequencing technologies provide critical information for identifying patients who may benefit from molecularly targeted treatments [49]. The field is advancing toward "drugging the undruggable" with next-generation mutant-specific molecules targeting previously inaccessible oncogenes like KRAS variants [49].
The following diagram illustrates how panel findings translate to clinical utility through multiple pathways:
The integration of circulating tumor DNA (ctDNA) monitoring further expands clinical utility by enabling response assessment and detection of minimal residual disease [49]. As noted by experts, "We will continue to see drugs moving into earlier disease treatment settings, because this is where we would make the biggest difference in increasing cancer cures" [49].
The demonstration of clinical utility for the AmpliSeq Childhood Cancer Panel encompasses a comprehensive evidence framework spanning analytical validation, diagnostic refinement, and therapeutic guidance. With demonstrated impact on patient management in 43% of cases and ability to refine diagnosis in up to 97% of patients based on fusion identification, this panel represents a significant advancement in pediatric cancer genomics [11]. The compatibility with multiple Illumina sequencing platforms provides flexibility for implementation across diverse laboratory settings.
Future directions in pediatric cancer molecular profiling include the integration of whole transcriptome sequencing, minimal residual disease monitoring, and comprehensive genomic profiling through expanded panels. The evolving landscape of targeted therapies, including next-generation inhibitors, antibody-drug conjugates, and cellular therapies, will further enhance the clinical utility of comprehensive molecular profiling [49]. As the field advances, the demonstration of clinical utility will increasingly require prospective validation of improved outcomes through randomized trials and real-world evidence generation [50] [51].
For researchers and drug development professionals, the AmpliSeq Childhood Cancer Panel provides a validated, comprehensive solution for molecular profiling of pediatric cancers, with demonstrated clinical utility across multiple dimensions including diagnostic refinement, prognostic stratification, and therapeutic targeting. Its implementation within compatible Illumina sequencing workflows enables standardized, reproducible genomic analysis to advance precision medicine in childhood cancers.
Next-generation sequencing (NGS) has revolutionized pediatric cancer diagnostics by enabling comprehensive molecular profiling of childhood malignancies [52]. Unlike adult cancers, pediatric cancers are characterized by a relatively low mutational burden but a higher prevalence of structural variants, gene fusions, and copy number alterations [53]. This distinct genetic architecture necessitates specialized testing approaches tailored specifically to childhood cancers. Targeted sequencing panels have emerged as a critical tool in this landscape, offering a balance between comprehensive genomic assessment and practical clinical implementation. The AmpliSeq for Illumina Childhood Cancer Panel represents one such solution, designed specifically for investigating 203 genes associated with cancer in children and young adults [3]. This technical guide examines how the AmpliSeq platform complements and enhances the broader ecosystem of pediatric cancer genomic analysis, with particular focus on its integration within research environments utilizing MiSeq and NextSeq sequencing systems.
Core Specifications and Design Principles
The AmpliSeq Childhood Cancer Panel employs a targeted resequencing approach specifically optimized for pediatric and young adult cancers [3]. The panel utilizes multiplex PCR-based library preparation with remarkably low input requirements—only 10 ng of high-quality DNA or RNA—making it particularly suitable for precious pediatric tumor samples often limited in quantity [3]. The technical workflow is efficient, with a total assay time of 5-6 hours for library preparation and less than 1.5 hours of hands-on time, enabling rapid turnaround critical in pediatric oncology decision-making [3].
This panel demonstrates exceptional versatility in variant detection, capable of identifying single nucleotide polymorphisms (SNPs), gene fusions, somatic variants, insertions-deletions (indels), and copy number variants (CNVs) across a curated set of childhood cancer-associated genes [3]. The panel's design encompasses 203 genes specifically selected for their relevance in pediatric cancers, including those involved in leukemias, brain tumors, and sarcomas [3]. This focused approach provides researchers with a comprehensive yet cost-effective solution for evaluating known childhood cancer drivers without the computational overhead of whole-genome approaches.
Compatible Sequencing Platforms and Integration
The AmpliSeq Childhood Cancer Panel is optimized for seamless integration with Illumina sequencing systems, including MiSeq, NextSeq 550, NextSeq 2000, NextSeq 1000, and MiniSeq systems [3]. This platform flexibility allows research laboratories to implement the panel across various throughput needs and infrastructure capabilities. For MiSeq systems, the panel provides focused yet comprehensive analysis suitable for smaller batch processing, while the NextSeq platform compatibility enables higher-throughput applications for larger research cohorts [54].
Table 1: AmpliSeq Childhood Cancer Panel Sequencing Specifications by Platform
| Sequencing System | Recommended Reagent Kit | Typical Samples Per Run | Optimal Use Cases |
|---|---|---|---|
| MiSeq System | MiSeq Reagent Kit v3 | 3 samples | Low-to-medium throughput research projects |
| NextSeq 550 System | NextSeq Mid Output Kit v2 | 16 samples | Medium-scale research cohorts |
| NextSeq 550 System | NextSeq High Output Kit v2 | 48 samples | Larger research studies |
| NextSeq 2000 System | Compatible reagent kits | 48-96 samples | High-throughput research programs |
Experimental Protocol and Methodology
The standard experimental protocol for implementing the AmpliSeq Childhood Cancer Panel follows a streamlined workflow:
Library Preparation Protocol:
- Input Material Qualification: DNA and/or RNA samples are quantified and qualified using fluorometric methods (e.g., Qubit Fluorimeter) and quality control systems (e.g., TapeStation) to ensure adequate quality and concentration [11].
- cDNA Synthesis (for RNA targets): For fusion detection, 100 ng of RNA is reverse transcribed to cDNA using the AmpliSeq cDNA Synthesis for Illumina kit [3] [11].
- Multiplex PCR Amplification: Simultaneous amplification of 3,069 DNA amplicons (average size: 114 bp) and 1,701 RNA amplicons (average size: 122 bp) covering the targeted genes and fusion partners [11].
- Library Construction: Amplicons are barcoded with specific indexes using the AmpliSeq Library PLUS kit, followed by purification and normalization steps [3] [55].
- Library Pooling and Quantification: DNA and RNA libraries are pooled at an optimal 5:1 ratio (DNA:RNA), quantified, and diluted to appropriate loading concentrations (17-20 pM) [11].
- Sequencing: Loaded onto compatible Illumina sequencers using manufacturer-recommended reagent kits and sequencing parameters [54].
Comparative Analysis with Alternative Pediatric Panels
Performance Comparison with Commercially Available Panels
Recent independent validation studies demonstrate that the AmpliSeq Childhood Cancer Panel delivers exceptional performance characteristics. In analytical validation studies focused on pediatric acute leukemia, the panel demonstrated a mean read depth greater than 1000×, with 98.5% sensitivity for DNA variants at 5% variant allele frequency (VAF) and 94.4% sensitivity for RNA fusions [11]. The assay showed 100% specificity and reproducibility for DNA variants and 89% reproducibility for RNA detection, establishing its reliability for research applications [11].
When compared to other commercially available panels, the AmpliSeq Childhood Cancer Panel shows distinct advantages for pediatric-specific applications. However, it is important to note that newly developed pediatric-specific panels such as the SJPedPanel from St. Jude Children's Research Hospital have demonstrated the ability to cover approximately 90% of pediatric cancer driver genes, outperforming some commercially available panels that cover closer to 60% of relevant genes [56]. This highlights the importance of panel selection based on specific research objectives and target genes of interest.
Table 2: Comparative Analysis of Pediatric Cancer Panel Approaches
| Parameter | AmpliSeq Childhood Cancer Panel | Custom DNA Panel Alternative | SJPedPanel (St. Jude) |
|---|---|---|---|
| Number of Genes | 203 genes | 12 to >12,000 amplicons (customizable) | Not specified, but covers ~90% of pediatric drivers |
| Variant Types Detected | SNPs, fusions, indels, CNVs | SNPs, indels, CNVs | Multiple variant types |
| Input Requirements | 10 ng DNA or RNA | 1-100 ng DNA (10 ng recommended) | Not specified |
| Hands-on Time | <1.5 hours | 1.5 hours | Not specified |
| Sample Types | Blood, bone marrow, FFPE tissue | Blood, FFPE tissue | Optimized for pediatric samples |
| Key Advantage | Standardized content, validated workflow | Complete customization | Pediatric-specific optimization |
Complementary Role in the Pediatric Panel Ecosystem
The AmpliSeq Childhood Cancer Panel occupies a strategic position in the landscape of pediatric genomic analysis, complementing both broader whole-genome approaches and more focused single-gene tests:
Compared to Whole-Genome Sequencing (WGS): While WGS provides comprehensive genomic coverage, the AmpliSeq panel offers practical advantages for targeted analysis, including lower cost, reduced computational requirements, and higher sequencing depth for detecting low-frequency variants [56]. This is particularly valuable for analyzing samples with low tumor purity or after procedures like bone marrow transplantation, where WGS approaches may struggle with sensitivity [56].
Compared to Single-Gene Tests: The multiplexed nature of the AmpliSeq panel enables simultaneous evaluation of hundreds of targets in a single workflow, replacing multiple single-analyte tests and conserving precious sample material [57]. This comprehensive approach is especially valuable in pediatric cancers where multiple driver alterations may coexist and guide targeted treatment strategies [53].
Compared to Custom Panels: The AmpliSeq Custom DNA Panel alternative offers researchers complete flexibility in target selection through the DesignStudio portal, supporting content from 12 to over 12,000 amplicons across any species [58]. While custom panels provide ultimate flexibility, the pre-designed Childhood Cancer Panel offers a validated, ready-to-use solution specifically curated for pediatric malignancies, reducing development time and optimization requirements.
Research Applications and Clinical Utility
Validation in Pediatric Leukemia and Solid Tumors
The AmpliSeq Childhood Cancer Panel has been rigorously validated across various pediatric cancer types. In a comprehensive study of pediatric acute leukemia, the panel demonstrated significant clinical utility, identifying clinically impactful mutations in 49% of cases and fusion genes with clinical relevance in 97% of positive cases [11]. Overall, the panel provided clinically relevant results in 43% of patients tested in this cohort, refining diagnosis in 41% of mutated cases and identifying targetable alterations in 49% of mutations [11].
The panel's performance with challenging sample types is particularly noteworthy. It maintains sensitivity and specificity even with formalin-fixed, paraffin-embedded (FFPE) tissues, with specialized solutions like the AmpliSeq for Illumina Direct FFPE DNA available to streamline library construction from archived samples without requiring deparaffinization or DNA purification [3]. This capability is essential for translational research utilizing historical patient samples and biobank resources.
Integration with Broader Research Initiatives
Large-scale pediatric cancer genomics initiatives have demonstrated the powerful synergies between comprehensive sequencing and targeted approaches. The SickKids Cancer Sequencing (KiCS) program, which integrated deep sequencing of 864 cancer-associated genes with complete genomes and transcriptomes, found that 54% of pediatric patients had therapeutically targetable variants, with over 20% derived from germline sources [53]. The AmpliSeq Childhood Cancer Panel complements such comprehensive approaches by providing a cost-effective solution for routine screening and validation of prioritized targets identified in larger discovery studies.
Furthermore, integrative genomic analyses have revealed that comprehensive sequencing at multiple points in the care trajectory can identify changes in therapeutically targetable drivers in over one-third of patients, supporting the value of re-evaluation at disease relapse [53]. The efficiency and throughput of the AmpliSeq platform make it well-suited for such longitudinal studies in pediatric oncology research.
Essential Research Reagent Solutions
Successful implementation of the AmpliSeq Childhood Cancer Panel requires several key reagent components that form the complete research workflow:
Table 3: Essential Research Reagent Solutions for AmpliSeq Implementation
| Component | Function | Key Specifications |
|---|---|---|
| AmpliSeq Childhood Cancer Panel | Core primer pool for targeting 203 childhood cancer genes | 24 reactions; covers 3069 DNA and 1701 RNA amplicons |
| AmpliSeq Library PLUS Kit | Reagents for preparing sequencing libraries | Available in 24-, 96-, and 384-reaction configurations |
| AmpliSeq CD Indexes | Sample barcoding for multiplex sequencing | Available in sets A-D (96 indexes each) for 384 total samples |
| AmpliSeq cDNA Synthesis for Illumina | Converts RNA to cDNA for fusion detection | Required for RNA-based fusion analysis |
| AmpliSeq for Illumina Direct FFPE DNA | Prepares DNA from FFPE tissues without purification | 24 reactions; eliminates deparaffinization requirements |
| AmpliSeq Library Equalizer | Normalizes libraries for balanced sequencing | Bead-based normalization for improved data quality |
The AmpliSeq Childhood Cancer Panel represents a strategically important solution in the evolving landscape of pediatric cancer genomics. Its targeted design, analytical performance, and integration flexibility with Illumina sequencing platforms make it a valuable tool for researchers investigating childhood malignancies. While emerging pediatric-specific panels like the SJPedPanel offer alternative approaches with potentially enhanced coverage of pediatric cancer drivers, the AmpliSeq platform maintains distinct advantages in workflow standardization, validation data, and platform integration.
As pediatric oncology continues to embrace precision medicine approaches, the complementary roles of comprehensive genomic characterization and focused targeted sequencing will remain essential. The AmpliSeq Childhood Cancer Panel effectively bridges these approaches, providing researchers with a practical yet powerful tool for unlocking the molecular drivers of childhood cancer and accelerating the development of more effective, targeted therapies for young patients.
AmpliSeq Childhood Cancer Panel Workflow
Next-generation sequencing (NGS) has fundamentally transformed the landscape of clinical oncology, enabling comprehensive genomic profiling that informs personalized treatment strategies. The AmpliSeq for Illumina Childhood Cancer Panel exemplifies this transformation, providing a targeted resequencing solution for the comprehensive evaluation of somatic variants associated with childhood and young adult cancers [3]. This panel targets 203 genes specifically associated with pediatric and young adult cancers, including leukemias, brain tumors, and sarcomas, delivering a standardized approach that eliminates the time and effort typically required for target identification, primer design, and panel optimization [3].
Concurrently, Molecular Tumor Boards (MTBs) have emerged as critical multidisciplinary platforms for interpreting complex genomic data within a clinical context. These expert committees, comprising clinical and research specialists including oncologists, geneticists, molecular biologists, and bioinformaticians, collaboratively review patients' clinical histories and molecular profiling data to identify optimal therapeutic options, including standard-of-care drugs, off-label therapies, or clinical trial matches [59]. Real-world survey data from healthcare professionals across the UK demonstrates that 97.7% of respondents felt MTBs increased awareness of available clinical trials matched to genomic alterations, while 84% reported greater confidence in interpreting complex genomic data through this collaborative process [59]. The integration of structured NGS panel results, such as those generated by the Childhood Cancer Panel, into MTB workflows represents a cornerstone of modern precision oncology, bridging the gap between genomic findings and clinically actionable recommendations.
Technical Specifications and Workflow of the Childhood Cancer Panel
Panel Configuration and Performance Metrics
The AmpliSeq Childhood Cancer Panel is engineered to simultaneously analyze both DNA and RNA from minimal input material, facilitating comprehensive genomic profiling even from challenging sample types commonly encountered in pediatric oncology. The panel's design employs a two-pool amplification strategy with optimized primer concentrations to ensure uniform coverage across all targeted regions [4].
Table 1: Technical Specifications of the AmpliSeq Childhood Cancer Panel
| Component | Number of Pools | Concentration | Number of Amplicons | Average Amplicon Length | Average Library Length |
|---|---|---|---|---|---|
| DNA | 2 | 4X | 3069 | 114 bp | 254 bp |
| RNA | 2 | 5X | 1701 | 122 bp | 262 bp |
The panel requires only 10 ng of high-quality DNA or RNA input, making it particularly valuable for pediatric cases where biopsy material may be limited. It supports various specialized sample types, including blood, bone marrow, and FFPE tissue, with the AmpliSeq for Illumina Direct FFPE DNA product available to enable library construction from FFPE tissues without requiring deparaffinization or DNA purification [3]. The complete library preparation process requires approximately 5-6 hours of assay time with less than 1.5 hours of hands-on time, enabling rapid turnaround suitable for clinical decision-making contexts [3].
Compatible Sequencing Systems and Configurations
To ensure optimal performance across different laboratory setups, the Childhood Cancer Panel is compatible with multiple Illumina sequencing platforms. The pooling strategy and run configurations vary depending on the specific sequencing system and whether DNA, RNA, or combined libraries are being sequenced.
Table 2: Recommended Sequencing Configuration by System
| System | Reagent Kit | Max DNA Samples Per Run | Max RNA Samples Per Run | Max Combined Samples Per Run | DNA:RNA Pooling Ratio | Run Time |
|---|---|---|---|---|---|---|
| MiniSeq | Mid Output | 1 | 8 | 1 | 5:1 | 17 hours |
| MiniSeq | High Output | 5 | 25 | 4 | 5:1 | 24 hours |
| MiSeq | v2 | 3 | 15 | 2 | 5:1 | 24 hours |
| MiSeq | v3 | 5 | 25 | 4 | 5:1 | 32 hours |
| NextSeq | Mid Output v2 | 27 | 96 | 22 | 5:1 | 26 hours |
| NextSeq | High Output v2 | 83 | 96 | 48 | 5:1 | 29 hours |
The 5:1 DNA:RNA pooling volume ratio is optimized to achieve recommended read coverage for both nucleic acid types when processing paired samples from the same patient [4]. This balanced approach ensures sufficient sensitivity for detecting variants across different variant classes, including single nucleotide polymorphisms (SNPs), insertions-deletions (indels), copy number variants (CNVs), and gene fusions [3].
Molecular Tumor Boards: Structure, Workflow, and Clinical Impact
MTB Composition and Operational Framework
Molecular Tumor Boards function as specialized multidisciplinary teams that convene regularly to interpret complex molecular profiling data in the context of individual patient cases. The core participants typically include medical oncology consultants (72.7% of respondents in the UK survey), clinical fellows (22.7%), alongside nurses, scientists, genetic counselors, and bioinformaticians [59]. These diverse expertise areas collectively enable comprehensive assessment of genomic alterations and their potential clinical implications.
The operational structure of MTBs varies by institution, with meetings typically occurring weekly or monthly. Larger initiatives, such as the TARGET National study in the UK, implement virtual national MTBs that leverage cloud-based platforms like eTARGET to facilitate discussion of patients across multiple institutions [59]. This digital infrastructure seamlessly integrates clinical and genomic sequencing data, enabling collaborative review regardless of geographic location. Survey data indicates that approximately 34.9% of MTB participants had been attending for less than one year, while 44.2% had 1-3 years of experience, reflecting the relatively recent but growing establishment of these forums in clinical practice [59].
MTB Clinical Decision-Making Workflow
The MTB review process follows a structured pathway to ensure systematic evaluation of each case. The general workflow, as described by Hamamoto et al. and implemented in programs like TARGET National and CUP-COMP, encompasses six critical steps [59]:
- Assign biological significance to identified genetic abnormalities
- Interpret genetic evidence for diagnosis and prognosis
- Identify specific candidate drugs and evidence corresponding to the genetic abnormalities
- Discuss potential germline implications of gene abnormalities
- Review relevant clinical trials based on the patient's molecular profile and history
- Consider patient-specific factors that might influence treatment suitability
To support trial matching, MTBs utilize resources such as ClinicalTrials.gov and specialized digital matching tools like the ECMC experimental cancer trial finder and Digital Cancer Research trial finder [59]. These platforms extract relevant study information from a corpus of clinical trials, enabling clinicians to search using various criteria including cancer type, molecular alteration, trial location, and trial phase.
Diagram 1: MTB Clinical Decision-Making Workflow
Documented Impact on Patient Care
Evidence increasingly demonstrates the tangible impact of MTBs on clinical decision-making and patient outcomes. Published studies indicate that between 35.7% and 87% of patients referred to MTBs have actionable genetic alterations with therapeutic implications [59]. Furthermore, between 7% and 15% of discussed patients are enrolled into matched targeted clinical trials, with some studies reporting that up to 41% receive genomics-based therapeutic recommendations [59].
A prospective phase II clinical trial conducted by Miller et al. provided compelling evidence for the clinical utility of MTB-directed therapy, demonstrating that MTB-recommended treatments improved progression-free survival (PFS) over immediate prior therapy in patients with advanced malignancies [59]. The study reported that the probability of PFS ratio (targeted therapy PFS/previous standard of care PFS) >1.3 was 0.59 (95% CI 0.47-0.75), indicating a significant clinical benefit [59].
Beyond direct patient matching to therapies, survey data reveals that 95.4% of healthcare professionals value MTBs as educational opportunities that enhance their understanding of genomic medicine and available targeted therapies [59].
Integration of Panel Results into MTB Deliberations: A Technical Workflow
Pre-MTB Data Processing and Analysis
The integration of AmpliSeq Childhood Cancer Panel results into MTB discussions requires systematic data processing and interpretation. The pathway from raw sequencing data to clinically actionable insights involves multiple analytical steps and quality control checkpoints.
Diagram 2: NGS Data Analysis Workflow for MTB
The initial quality assessment evaluates coverage metrics, with the Childhood Cancer Panel requiring sufficient depth to reliably detect somatic variants across the 203-gene target space. Following variant identification, comprehensive annotation incorporates population frequency databases (e.g., gnomAD), cancer-specific databases (e.g., COSMIC), functional prediction algorithms, and therapeutic evidence resources such as Clinical Knowledgebase (CIViC) and OncoKB.
For the Childhood Cancer Panel specifically, the simultaneous assessment of DNA and RNA enhances fusion detection and provides orthogonal validation of findings. The integration of both nucleic acid types requires specialized analysis pipelines that can reconcile findings across platforms and generate a unified clinical report for MTB consideration.
Multimodal Data Integration in MTBs
Contemporary MTBs are increasingly moving beyond exclusive reliance on NGS data to incorporate complementary molecular and clinical information for comprehensive patient assessment. Emerging evidence supports the integration of proteomic profiling to complement genomic findings, as proteins represent the functional effectors of cellular processes and primary targets of most therapeutic agents.
A 2025 feasibility study demonstrated that incorporating real-time laser microdissection (LMD) enrichment of tumor epithelium with reverse phase protein array (RPPA) proteomic profiling could be completed within a therapeutically permissible timeframe (median dwell time of nine days) [60]. This hyphenated LMD-RPPA workflow quantified the abundance of 32 proteins and/or phosphoproteins with known cancer significance and provided additional therapeutic considerations for 54% of profiled patients following MTB review [60].
The integration of proteomic data addresses a critical limitation of NGS-based approaches, as genomic variation and transcriptomic expression show only loose correlation with protein activity and abundance due to post-translational modifications, protein isoform diversity, and post-translational stability [60]. This multimodal approach enables more comprehensive therapeutic targeting, particularly for protein-directed therapies.
Table 3: Essential Research Reagent Solutions for Childhood Cancer Panel Implementation
| Product Category | Specific Product | Function | Compatibility |
|---|---|---|---|
| Library Prep Kit | AmpliSeq Library PLUS (24, 96, 384 reactions) | Provides reagents for preparing sequencing libraries | Required for all configurations |
| Index Adapters | AmpliSeq CD Indexes Sets A-D | Enables sample multiplexing through unique barcodes | 96 indexes per set |
| cDNA Synthesis Kit | AmpliSeq cDNA Synthesis for Illumina | Converts total RNA to cDNA for RNA sequencing | Required for RNA analysis |
| Library Normalization | AmpliSeq Library Equalizer for Illumina | Normalizes libraries for balanced sequencing | Beads and reagents included |
| Sample Identification | AmpliSeq for Illumina Sample ID Panel | SNP genotyping for sample tracking | 96 reactions |
| FFPE Optimization | AmpliSeq for Illumina Direct FFPE DNA | Enables library construction from FFPE without DNA purification | 24 reactions |
Implementation Challenges and Optimization Strategies
Operational Hurdles in MTB Integration
The successful integration of Childhood Cancer Panel results into MTB workflows faces several operational challenges that require strategic solutions. Survey data from healthcare professionals identifies frequency and capacity of MTBs as significant hurdles, particularly for liquid biopsy (ctDNA) cases, alongside issues with sample collection and laboratory turnaround time for tissue samples [59]. Approximately one-third of respondents encountered challenges attending MTBs regularly due to clinical workload demands [59].
Sample quality and sufficiency represent additional critical barriers. The feasibility study incorporating proteomic profiling reported that 34% of patient failures were related to insufficient tumor tissue for LMD enrichment prior to RPPA analysis [60]. Specific tissue types obtained via fine needle aspirates (FNAs), particularly from lung, lymph nodes, liver, pancreas, and prostate, demonstrated the highest failure rates due to minimal tissue depth within specimen blocks or inadequate tumor area for microdissection [60].
Turnaround time from sample acquisition to MTB discussion remains another significant challenge. The multimodal profiling study reported a median of 14 days from patient consent to specimen receipt by the proteomics team, with an additional median of 9 days for the proteomic arm of the study during which specimens were unavailable for standard clinical testing [60]. These timelines highlight the logistical complexities inherent in comprehensive molecular profiling for precision oncology.
Strategic Optimization Approaches
Several strategies have emerged to address these implementation challenges and optimize the integration of panel results into MTB workflows:
Workflow Efficiency Enhancements: Streamlined laboratory processes, including the use of the AmpliSeq Library Equalizer for Illumina for library normalization and the Direct FFPE DNA protocol, can reduce hands-on time and processing bottlenecks [3].
Digital Solutions Implementation: Cloud-based platforms like eTARGET, which integrates clinical and genomic sequencing data to facilitate virtual MTB discussions, enable collaborative review without geographical constraints [59].
Multimodal Data Integration: As demonstrated by the LMD-RPPA workflow, incorporating proteomic data can expand therapeutic options, with the 2025 study showing additional considerations for 54% of patients [60].
MTB Structure Standardization: Including MTB participation in healthcare professionals' job plans, ensuring appropriate meeting frequency, and utilizing reliable trial matching tools can enhance participation and effectiveness [59].
The continued refinement of these approaches will be essential for maximizing the clinical utility of the AmpliSeq Childhood Cancer Panel within MTB contexts, ultimately improving outcomes for children and young adults with cancer through more precise and personalized therapeutic interventions.
The management of pediatric cancers is undergoing a profound transformation, moving from a one-size-fits-all approach to precision medicine strategies centered on comprehensive genomic profiling. Next-generation sequencing (NGS) technologies now enable researchers and clinicians to delve into the unique molecular drivers of childhood malignancies, revealing a genetic landscape distinct from adult cancers. Targeted NGS panels, such as the AmpliSeq for Illumina Childhood Cancer Panel, represent a critical technological advancement by providing a standardized, efficient method for analyzing hundreds of cancer-associated genes simultaneously with minimal sample input [3]. This evolution is particularly crucial in pediatric oncology, where cancers are often driven by distinct genetic alterations such as fusion genes, epigenetic changes, and fewer somatic mutations compared to adult cancers [61]. The integration of these molecular tools into clinical research pipelines is refining diagnostic classification, uncovering new therapeutic targets, and ultimately creating a more nuanced understanding of the biology of childhood cancers, setting the stage for more effective and less toxic treatments.
The Molecular Landscape of Pediatric Cancers
Distinct Genetic Alterations in Childhood Malignancies
Unlike adult cancers, which frequently arise from an accumulation of environmental exposures over time, pediatric malignancies are primarily driven by specific, often developmentally-linked, genetic aberrations. The molecular profile of pediatric hematologic and solid tumors reveals a complex array of chromosomal translocations, gene fusions, and point mutations that are critical for diagnosis, risk stratification, and treatment selection [62]. In pediatric acute lymphoblastic leukemia (ALL), recurrent genetic abnormalities such as ETV6-RUNX1 fusions (associated with favorable prognosis) and BCR-ABL1 fusions (indicating high-risk disease) exemplify how molecular markers directly inform clinical decision-making [62]. Similarly, in solid tumors, alterations in genes like BRAF, ALK, and MYCN serve as key drivers and therapeutic targets across various diagnostic categories [63] [61].
The genetic heterogeneity observed within histologically similar tumors underscores the limitations of traditional diagnostic methods alone. For instance, comprehensive molecular profiling has revealed that pediatric cancers frequently harbor actionable alterations, with one large-scale study of 888 pediatric tumors finding that 33% had at least one genomic variant matching a precision oncology trial protocol [63]. This molecular heterogeneity necessitates sophisticated diagnostic approaches that can detect the full spectrum of genomic alterations, from single nucleotide variants to complex structural rearrangements, to fully realize the potential of precision medicine in pediatric oncology.
Technical Foundations of Targeted NGS in Pediatric Oncology
The AmpliSeq for Illumina Childhood Cancer Panel represents a specialized targeted sequencing solution designed specifically for the genomic profiling of pediatric and young adult cancers. This ready-to-use panel enables comprehensive evaluation of somatic variants across 203 genes associated with childhood cancers, including leukemias, brain tumors, and sarcomas [3]. The panel utilizes a highly multiplexed PCR-based amplification approach, requiring only 10 ng of high-quality DNA or RNA as input, making it particularly suitable for the small biopsy samples often available in pediatric cases [3]. The complete library preparation process requires approximately 5-6 hours with less than 1.5 hours of hands-on time, facilitating rapid integration into research workflows [3].
The panel simultaneously interrogates multiple variant classes, including single nucleotide variants (SNVs), insertions-deletions (indels), copy number variants (CNVs), and gene fusions [3]. This comprehensive approach is essential in pediatric cancers where gene fusions are frequent drivers of oncogenesis. The technical specifications of the panel include two separate pools for DNA and RNA analysis, generating 3,069 and 1,701 amplicons respectively, with average amplicon lengths of 114-122 base pairs [4]. This design is optimized for performance with challenging sample types common in pediatric oncology, including formalin-fixed paraffin-embedded (FFPE) tissue, bone marrow, and blood samples [3].
Compatible Sequencing Systems and Configurations
The AmpliSeq Childhood Cancer Panel is compatible with multiple Illumina sequencing platforms, offering researchers flexibility in throughput and scale. The table below summarizes the key sequencing parameters for different Illumina systems:
Table 1: Compatible Illumina Sequencing Systems for AmpliSeq Childhood Cancer Panel
| Sequencing System | Reagent Kit | Max DNA Samples Per Run | Max RNA Samples Per Run | Max Combined Samples Per Run | Recommended DNA:RNA Pooling Ratio | Run Time |
|---|---|---|---|---|---|---|
| MiniSeq System | Mid Output | 1 | 8 | 1 | 5:1 | 17 hours |
| MiniSeq System | High Output | 5 | 25 | 4 | 5:1 | 24 hours |
| MiSeq System | v2 | 3 | 15 | 2 | 5:1 | 24 hours |
| MiSeq System | v3 | 5 | 25 | 4 | 5:1 | 32 hours |
| NextSeq System | Mid Output v2 | 27 | 96 | 22 | 5:1 | 26 hours |
| NextSeq System | High Output v2 | 83 | 96 | 48 | 5:1 | 29 hours |
Data sourced from [4]
This compatibility range allows research laboratories to select the appropriate sequencing scale based on their project needs, from smaller-scale studies on the MiniSeq or MiSeq systems to larger cohort analyses on the NextSeq platform. The NextSeq 1000 and 2000 Systems offer additional application flexibility, supporting not only targeted panels but also whole-exome and transcriptome sequencing, enabling more comprehensive genomic studies when needed [64].
Essential Research Reagent Solutions
Implementing the AmpliSeq Childhood Cancer Panel requires several complementary reagent systems to complete the library preparation and sequencing workflow. The following table outlines the essential components:
Table 2: Essential Research Reagent Solutions for AmpliSeq Childhood Cancer Panel Workflow
| Component Category | Product Name | Function | Key Specifications |
|---|---|---|---|
| Library Preparation | AmpliSeq Library PLUS for Illumina | Provides reagents for preparing sequencing libraries | Available in 24-, 96-, and 384-reaction configurations |
| Index Adapters | AmpliSeq CD Indexes Sets A-D | Enables sample multiplexing by attaching unique barcode sequences | Each set contains 96 unique 8-base indexes |
| RNA Processing | AmpliSeq cDNA Synthesis for Illumina | Converts total RNA to cDNA for RNA sequencing | Required for working with RNA panels |
| Library Normalization | AmpliSeq Library Equalizer for Illumina | Normalizes libraries before pooling for sequencing | Simplifies library quantification and pooling |
| Sample Tracking | AmpliSeq for Illumina Sample ID Panel | Provides unique SNP-based sample identification | Includes 8 SNP-targeting primer pairs and 1 gender-determining pair |
| FFPE Optimization | AmpliSeq for Illumina Direct FFPE DNA | Prepares DNA from FFPE tissues without deparaffinization | Enables library construction from 24 FFPE samples |
Data compiled from [3] [5] [65]
These integrated solutions ensure a streamlined workflow from sample to sequencing data, with particular optimizations for the challenging sample types frequently encountered in pediatric cancer research.
Experimental Protocol: Implementing Targeted NGS in Pediatric Cancer Research
Sample Preparation and Library Construction
The standard workflow for the AmpliSeq Childhood Cancer Panel begins with nucleic acid extraction from patient samples. For comprehensive analysis, both DNA and RNA are extracted from matched tumor samples. The protocol requires 10 ng of high-quality DNA or RNA per sample, making it feasible even with limited biopsy material [3]. For FFPE samples, the specialized AmpliSeq for Illumina Direct FFPE DNA kit can be used to bypass traditional deparaffinization and DNA purification steps [3].
The library construction process involves several key steps:
- cDNA Synthesis (for RNA samples): Total RNA is converted to cDNA using the AmpliSeq cDNA Synthesis for Illumina kit, which includes reaction mix and enzyme blend specifically formulated for compatibility with AmpliSeq panels [3].
- Multiplex PCR Amplification: The childhood cancer panel primer pools are combined with the extracted DNA or synthesized cDNA. The panel consists of two DNA pools and two RNA pools, which are amplified separately through a highly multiplexed PCR reaction [4] [5].
- Partial Digestion and Barcoding: The amplified products undergo enzymatic partial digestion to generate compatible ends for adapter ligation. Subsequently, Illumina-specific adapters and unique molecular barcodes (from AmpliSeq CD Indexes Sets) are ligated to each sample, enabling sample multiplexing in sequencing runs [3] [65].
- Library Normalization and Pooling: The barcoded libraries are normalized using the AmpliSeq Library Equalizer, which employs bead-based normalization to ensure equimolar pooling of libraries. For combined DNA and RNA sequencing from the same sample, a 5:1 DNA:RNA pooling ratio is recommended based on optimal read coverage requirements [4].
Diagram 1: Targeted NGS Experimental Workflow. This diagram illustrates the key steps in the AmpliSeq Childhood Cancer Panel workflow, from nucleic acid extraction to sequencing data generation.
Sequencing and Data Analysis
Following library preparation and quality control, pooled libraries are loaded onto compatible Illumina sequencing systems. The selection of specific sequencing platform and reagent kit depends on the project scale and desired throughput, as detailed in Table 1. The sequencing data generated undergoes a comprehensive bioinformatic analysis pipeline:
- Base Calling and Demultiplexing: Raw sequencing data is processed to identify bases and assign reads to specific samples based on their unique barcodes.
- Alignment to Reference Genome: Processed reads are aligned to the human reference genome (GRCh37/hg19) using alignment algorithms such as a modified Burrows-Wheeler transform (BWT) method [66].
- Variant Identification and Annotation: Multiple variant types are identified through specialized algorithms:
- Single nucleotide variants and small indels are detected using tools that typically require a minimum of 10 reads supporting the variant with a minimum variant allelic fraction of 10% for tumor samples [66].
- Copy number variations are identified from the targeted sequencing data by analyzing depth of coverage ratios compared to control samples [66].
- Gene fusions and structural variants are detected through analysis of the RNA sequencing data, identifying chimeric transcripts resulting from chromosomal rearrangements [66].
- Clinical Interpretation and Reporting: Identified variants are reviewed by a multidisciplinary molecular tumor board, comprising molecular pathologists, pediatric oncologists, and cancer biologists. Variants are tiered based on their known or potential clinical significance, evidence for actionability, and association with disease [66].
Clinical Implementation and Research Applications
Global Precision Medicine Initiatives for Pediatric Cancers
The implementation of targeted NGS in pediatric oncology has catalyzed the development of numerous precision medicine platforms worldwide. These collaborative initiatives have demonstrated the feasibility of large-scale molecular profiling and its impact on clinical decision-making. Major programs include:
- MAPPYACTS (Europe): This prospective, multicenter trial utilizes WES, RNAseq, and/or panel sequencing to identify targetable mutations in children with relapsed or refractory cancers. The study reported that 69% of patients had potentially actionable targets, with 30% of patients with follow-up receiving matched targeted therapies [67].
- GAIN/iCat2 (USA): A multicenter study employing targeted DNA and RNA NGS panels on FFPE tissue to reflect typical clinical practice. The study found that 86% of successfully sequenced patients had clinically impactful alterations, with 70% receiving precision therapy recommendations [67].
- INFORM (Germany): This multinational registry uses WES, low-coverage WGS, DNA methylation analysis, and RNAseq to inform treatment approaches. Patients receiving matched targeted therapies for ALK, BRAF, or NTRK alterations showed statistically significant improvement in progression-free survival (p=0.012) and overall survival (p=0.036) compared to those who did not receive matched therapy [67].
- ZERO Childhood Cancer (Australia): This program employs WGS (paired tumor-germline), RNA-seq, and DNA methylation profiling for children with high-risk cancers. The study reported that 67% of patients received at least one precision therapy recommendation based on molecular findings [67].
These initiatives collectively demonstrate that comprehensive molecular profiling can identify actionable alterations in a substantial proportion of pediatric cancer patients, creating opportunities for targeted therapeutic interventions.
Clinical Impact and Outcome Assessment
The integration of targeted NGS into pediatric oncology research has demonstrated measurable clinical impact across multiple dimensions. The table below summarizes key outcome metrics from major precision medicine initiatives:
Table 3: Clinical Outcomes from Major Pediatric Precision Oncology Initiatives
| Precision Medicine Initiative | Actionable Alteration Rate | PGT Uptake Rate | Objective Response Rate (ORR) | Key Findings |
|---|---|---|---|---|
| MAPPYACTS (n=624) | 69% | 30% | 17% (38% for "ready for routine use" recommendations) | Responses highest when treatment based on high-level evidence |
| GAIN/iCat2 (n=345) | 70% | 12% | 17% | 24% overall clinical benefit rate observed |
| INFORM (n=519) | Not specified | 28% | Not specified | Significant PFS and OS improvement with matched therapy for ALK, BRAF, NTRK alterations |
| Dana-Farber/Boston Children's (n=888) | 33% | 14% | Not specified | 88% of treated patients received therapy off-label or via single patient protocol |
The clinical impact of molecular profiling extends beyond direct matching to targeted therapies. Comprehensive sequencing can provide diagnostic clarification, prognostic information, and identification of germline cancer predisposition syndromes. One study reported that 20% of pediatric patients undergoing sequencing had clinically relevant germline alterations, with 14% having mutations in cancer predisposition genes [66]. Furthermore, RNA sequencing has proven particularly impactful, with one analysis finding that it provided clinically relevant information in 57% of cases, including diagnostic/prognostic insights (26%) and identification of therapeutic targets (23%) [66].
Diagram 2: Clinical Implementation Pathway. This diagram outlines the pathway from molecular sequencing to clinical impact through multidisciplinary molecular tumor board (MTB) review and interpretation.
Challenges and Future Directions
Current Limitations in Pediatric Precision Oncology
Despite the promising advances, several significant challenges impede the widespread implementation and effectiveness of targeted NGS in pediatric oncology. Low uptake rates of precision-guided therapies (ranging from 10-33% across studies) remain a persistent issue, often attributable to logistical barriers, limited drug access, and rapid clinical deterioration of patients with advanced disease [67]. The rarity of many pediatric cancers creates challenges for drug development, as commercial incentives for developing targeted therapies for small patient populations are limited [61]. Additionally, disparities in resource availability between high-income and low-income settings create significant barriers to implementing molecular profiling technologies, limiting global access to precision medicine approaches [62].
Beyond access issues, biological challenges also exist. Pediatric cancers often display molecular heterogeneity, with co-occurring alterations that may confer resistance to single-agent targeted therapies. Furthermore, a substantial proportion of driver alterations in pediatric cancers are currently considered "undruggable" with existing therapeutic modalities, necessitating continued development of novel therapeutic approaches [61].
Emerging Opportunities and Strategic Priorities
The future evolution of targeted NGS in pediatric oncology will likely focus on several key areas:
- Integration of Multi-Omics Technologies: Future approaches will combine DNA sequencing with transcriptomics, epigenomics, and proteomics to provide a more comprehensive understanding of tumor biology and identify novel therapeutic vulnerabilities [67].
- Standardization of Bioinformatic Pipelines and Data Sharing: Harmonization of analytical approaches and increased data sharing through initiatives like the National Cancer Institute's Childhood Cancer Data Initiative (CCDI) will enhance the power of genomic datasets for rare pediatric cancers [63].
- Development of Pediatric-Specific Clinical Trials: The implementation of the RACE for Children Act is expected to accelerate the development of targeted therapies for pediatric cancers by requiring earlier pediatric assessment of drugs directed against molecular targets relevant to childhood cancers [61].
- Implementation Science for Global Access: Innovative strategies such as establishing regional diagnostic hubs, strengthening molecular cancer registries, and fostering international collaborations will be critical for expanding access to precision medicine approaches in resource-limited settings [62].
- Longitudinal Monitoring and Adaptive Approaches: The application of NGS for minimal residual disease monitoring and tracking clonal evolution during treatment will enable more dynamic treatment adaptations and earlier intervention for relapsing disease [62].
Targeted NGS technologies, exemplified by the AmpliSeq for Illumina Childhood Cancer Panel, have fundamentally transformed the research landscape in pediatric oncology. These tools have enabled comprehensive molecular characterization of childhood cancers, revealing distinct biological drivers and creating unprecedented opportunities for precision medicine approaches. The integration of these technologies into global research initiatives has demonstrated substantial clinical impact, including diagnostic refinement, identification of therapeutic targets, and improved outcomes for specific molecular subgroups.
Looking forward, the evolving role of targeted NGS will be shaped by both technological advances and implementation strategies. The integration of multi-omics approaches, development of pediatric-focused therapeutics, and expansion of global access will be critical to realizing the full potential of precision medicine for all children with cancer. As these technologies become more accessible and their clinical utility continues to expand, targeted NGS is poised to transition from a research tool to an integral component of standard pediatric oncology practice worldwide, ultimately driving improvements in survival and quality of life for children affected by cancer.
Conclusion
The AmpliSeq for Illumina Childhood Cancer Panel, when paired with the appropriate MiSeq or NextSeq system, provides a robust, validated, and efficient solution for uncovering clinically relevant somatic variants in pediatric cancers. By understanding the foundational specifications, adhering to optimized methodological protocols, implementing proactive troubleshooting, and appreciating the panel's demonstrated clinical utility, research and clinical labs can reliably integrate this tool into their workflow. As precision medicine continues to transform pediatric oncology, this targeted NGS approach will play an increasingly critical role in refining diagnoses, stratifying risk, and identifying actionable therapeutic targets to improve outcomes for children with cancer.
References
- 1. St. Jude gene panel for pediatric cancers increases access ... [https://www.stjude.org/media-resources/news-releases/2024-medicine-science-news/gene-panel-for-pediatric-cancers-increases-access-to-high-quality-testing.html]
- 2. SJPedPanel: A Pan-Cancer Gene Panel for Childhood ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11393547/]
- 3. AmpliSeq for Illumina Childhood Cancer Panel [https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/ampliseq-childhood-cancer-panel.html]
- 4. AmpliSeq for Illumina Childhood Cancer Panel [https://support.illumina.com/sequencing/sequencing_kits/ampliseq-for-illumina-childhood-cancer/product-compatibility.html]
- 5. AmpliSeq for Illumina Childhood Cancer Panel [https://support.illumina.com/sequencing/sequencing_kits/ampliseq-for-illumina-childhood-cancer/contents-storage.html]
- 6. Data-sharing efforts create breakthroughs in pediatric cancer ... [https://biologicalsciences.uchicago.edu/news/data-sharing-pediatric-cancer-research]
- 7. HHS Doubles AI-Backed Childhood Cancer Research ... [https://www.hhs.gov/press-room/hhs-doubles-ai-backed-childhood-cancer-research-funding.html]
- 8. Cancer Biomarkers | NGS detection [https://www.illumina.com/areas-of-interest/cancer/ngs-in-oncology/biomarkers.html]
- 9. A method to comprehensively identify germline SNVs, ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11023157/]
- 10. AmpliSeq for Illumina Childhood Cancer Panel Support [https://support.illumina.com/sequencing/sequencing_kits/ampliseq-for-illumina-childhood-cancer/questions1.html]
- 11. Technical Validation and Clinical Utility of an NGS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9021638/]
- 12. MiSeq Sequencing System specifications [https://www.illumina.com/systems/sequencing-platforms/miseq/specifications.html]
- 13. Automated Nucleic Acid Extraction with MagMAX Kits [https://www.thermofisher.com/us/en/home/life-science/dna-rna-purification-analysis/automated-purification-extraction/automated-magmax-kits-nucleic-acid-extraction.html]
- 14. Applications of FFPE samples in modern research [https://www.lexogen.com/blog/applications-of-ffpe-samples-in-modern-research/]
- 15. FFPE Sample Analysis with Sequencing and Arrays [https://www.illumina.com/science/education/ffpe-sample-analysis.html]
- 16. Sequencing Platforms | Illumina NGS platforms [https://www.illumina.com/systems/sequencing-platforms.html]
- 17. Sample Multiplexing | Multiplex sequencing with indexes [https://www.illumina.com/techniques/sequencing/ngs-library-prep/multiplexing.html]
- 18. Index Hopping | Intro & how to minimize it [https://www.illumina.com/techniques/sequencing/ngs-library-prep/multiplexing/index-hopping.html]
- 19. MiSeq Compatible Products [https://support.illumina.com/sequencing/sequencing_instruments/miseq/compatibility.html]
- 20. MiSeq Reagent Kits [https://support.illumina.com/sequencing/sequencing_instruments/miseq/kit_contents.html]
- 21. Color Balance in Illumina Sequencing: Why It Still Matters [https://www.revvity.com/blog/color-balance-illumina-sequencing-why-it-still-matters]
- 22. Index color balancing for the NovaSeq 6000 system [https://knowledge.illumina.com/instrumentation/novaseq-6000/instrumentation-novaseq-6000-reference_material-list/000001931]
- 23. Short-read (Illumina) Library Construction Services [https://dnatech.ucdavis.edu/short-read-illumina-library-construction-services]
- 24. Improved Protocols for Illumina Sequencing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3849550/]
- 25. MiniSeq Reagent Kit | Simple, cost-efficient sequencing [https://www.illumina.com/products/by-type/sequencing-kits/cluster-gen-sequencing-reagents/miniseq-reagent-kit.html]
- 26. NextSeq 550 Sequencing System specifications [https://www.illumina.com/systems/sequencing-platforms/nextseq/specifications.html]
- 27. Library Preparation Quality Control and Quantification [https://www.lexogen.com/blog/rna-lexicon-library-preparation-quality-control-and-quantification/]
- 28. Preventing PCR Amplification Carryover Contamination in ... [https://www.annclinlabsci.org/content/34/4/389.long]
- 29. Seven tips for avoiding DNA contamination in your PCR [https://www.takarabio.com/about/bioview-blog/tips-and-troubleshooting/avoid-dna-contamination-in-pcr?srsltid=AfmBOoorNY3HkFq68mzCae9MbYCJKNMAf6kBr-0wgGkX6TeD7MsmG5RP]
- 30. Could your PCR be affected by contamination? | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/could-your-pcr-be-affected-by-contamination/]
- 31. A new method to prevent carry-over contaminations in two ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4787772/]
- 32. High-quality single amplicon sequencing method for illumina ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-023-09233-4]
- 33. LotuS2: an ultrafast and highly accurate tool for amplicon ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9580208/]
- 34. Enhancing diversity analysis by repeatedly rarefying next ... [https://www.nature.com/articles/s41598-021-01636-1]
- 35. NGS & Microarray Training [https://www.illumina.com/services/training.html]
- 36. Applications of NGS [https://www.illumina.com/science/technology/next-generation-sequencing/beginners/applications.html]
- 37. Impacts of low coverage depths and post-mortem DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4312461/]
- 38. Troubleshooting [https://knowledge.illumina.com/instrumentation/miseq/instrumentation-miseq-troubleshooting-list]
- 39. Diagnostic implications of pitfalls in causal variant ... [https://www.nature.com/articles/s41467-023-40909-3]
- 40. RNA-Seq Variant Calling: Key Challenges and Emerging ... [https://www.cd-genomics.com/resource-variant-calling-rna-seq-challenges-solutions.html]
- 41. Troubleshooting [https://knowledge.illumina.com/library-preparation/general/library-preparation-general-troubleshooting-list]
- 42. Incidence, sensitivity, and specificity of leukemia-associated phenotypes in acute myeloid leukemia using specific five-color multiparameter flow cytometry - PubMed [https://pubmed.ncbi.nlm.nih.gov/18480011/]
- 43. Measurable residual disease testing in chronic lymphocytic leukaemia: hype, hope neither or both? | Leukemia [https://www.nature.com/articles/s41375-021-01419-7]
- 44. The balance of reproducibility, sensitivity, and specificity of lists of differentially expressed genes in microarray studies - PubMed [https://pubmed.ncbi.nlm.nih.gov/18793455/]
- 45. Inclusion of a sensitivity control to add a quality-control parameter and improve reproducibility in BCR, immunoglobulin, and T-cell receptor gene-rearrangement studies - PubMed [https://pubmed.ncbi.nlm.nih.gov/7866638/]
- 46. Construction and validation of a risk prediction model for ... [https://www.nature.com/articles/s41598-025-24478-7]
- 47. Development and validation of a leukemia prognostic model ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12514105/]
- 48. The concept of clinical utility for diagnostic tests [https://www.thermofisher.com/blog/clinical-conversations/the-concept-of-clinical-utility-for-diagnostic-tests/]
- 49. Experts Forecast Cancer Research and Treatment ... [https://www.aacr.org/blog/2025/01/10/experts-forecast-cancer-research-and-treatment-advances-in-2025/]
- 50. Methods for determining clinical utility [https://www.sciencedirect.com/science/article/abs/pii/S0009912023002023]
- 51. The Clinical Utility of Diagnostic Tests [https://pubmed.ncbi.nlm.nih.gov/22730450/]
- 52. Next-Generation Sequencing Technology: Current Trends ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10376292/]
- 53. The clinical utility of integrative genomics in childhood ... [https://www.nature.com/articles/s43018-022-00474-y]
- 54. AmpliSeq for Illumina Comprehensive Panel v3 [https://support.illumina.com/sequencing/sequencing_kits/ampliseq-for-illumina-comprehensive-panel-v3/product-compatibility.html]
- 55. AmpliSeq for Illumina Library Prep, Indexes, and Accessories [https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/ampliseq-library-plus-adapters-accessories.html]
- 56. Gene panel for paediatric cancers increases access to high ... [https://ecancer.org/en/news/25106-gene-panel-for-paediatric-cancers-increases-access-to-high-quality-testing]
- 57. The Changing Landscape of NGS Implementation in ... [https://www.illumina.com/science/customer-stories/icommunity-customer-interviews-case-studies/patton-jennings-tan-yan-ngs-oncology.html]
- 58. AmpliSeq for Illumina Custom DNA Panel [https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/ampliseq-custom-dna-panel.html]
- 59. Real-world experience of Molecular Tumour Boards for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11937402/]
- 60. Real-time functional proteomics enhances therapeutic ... [https://www.nature.com/articles/s41698-025-00868-y]
- 61. Next-Generation Sequencing of Pediatric Cancer Patients ... [https://www.precisionmedicineonline.com/cancer/next-generation-sequencing-pediatric-cancer-patients-could-transform-treatment-paradigms]
- 62. Early detection and better outcomes: molecular approaches in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12577831/]
- 63. Molecular profiling of 888 pediatric tumors informs future ... [https://www.nature.com/articles/s41467-024-49944-0]
- 64. Exome sequencing - NextSeq 1000 & 2000 Systems [https://www.illumina.com/systems/sequencing-platforms/nextseq-1000-2000/applications.html]
- 65. AmpliSeq CD Indexes for Illumina Compatible Products [https://support.illumina.com/sequencing/sequencing_kits/ampliseq-cd-indexes/compatibility.html]
- 66. Implementation of next generation sequencing into ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5180407/]
- 67. The Growing Role of Precision Medicine in Paediatric ... [https://link.springer.com/article/10.1007/s11523-025-01178-w]