Protein Data Bank Decoded: A Researcher's Guide to Structures, Analysis, and Drug Discovery
This guide provides researchers, scientists, and drug development professionals with a comprehensive framework for navigating and utilizing the Protein Data Bank (PDB).
Protein Data Bank Decoded: A Researcher's Guide to Structures, Analysis, and Drug Discovery
Abstract
This guide provides researchers, scientists, and drug development professionals with a comprehensive framework for navigating and utilizing the Protein Data Bank (PDB). It covers the foundational hierarchy of PDB entries, the application of experimental and computational methods for structure determination, strategies for troubleshooting common data interpretation challenges, and the critical evaluation and validation of structural models. By synthesizing current data and tools, this article aims to empower professionals to leverage the full potential of structural data in accelerating biomedical research and therapeutic development.
Navigating the PDB Archive: Understanding Data Hierarchy and Content
The Protein Data Bank (PDB) archive serves as the single global repository for experimentally determined three-dimensional structures of biological macromolecules, providing foundational data for researchers, educators, and students worldwide. Managed by the worldwide Protein Data Bank (wwPDB) consortium, this critical resource supports breakthroughs in structural biology, drug discovery, and biomedical research. The wwPDB ensures the archive's integrity through continuous curation, standardization, and remediation processes, maintaining a comprehensive collection of structures of proteins, nucleic acids, and complex assemblies determined by X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and electron microscopy (3DEM) techniques. This technical guide examines the scope, exponential growth, and sophisticated global management framework that make the PDB archive an indispensable resource for the scientific community.
Global Management Framework
The wwPDB Consortium Structure
The wwPDB consortium, established in 2003, maintains a unified PDB archive through an international partnership of data centers specializing in deposition, processing, and distribution of structural biology data. This distributed model ensures both data integrity and global accessibility.
The organizational structure and data flow of the wwPDB consortium can be visualized as follows:
The consortium's operations include several critical functions. Deposition and Biocuration involves expert review where each structure undergoes examination for self-consistency, standardization using controlled vocabularies, cross-referencing with other biological data resources, and validation for scientific and technical accuracy [1]. Archive Management requires maintaining data dictionaries and standardization while integrating PDB data with other available information resources. Distribution and Access is facilitated through multiple protocols including HTTPS and rsync, with the FTP protocol being deprecated as of November 2024 [2]. Outreach and Education includes developing resources for teachers, students, and the general public delivered via PDB-101 websites.
Data Processing and Quality Assurance
The wwPDB employs rigorous data processing workflows to ensure archive quality and consistency. The OneDep system provides a unified platform for deposition, validation, and biocuration of structures determined by all supported experimental methods. The validation process generates comprehensive reports assessing structural quality using metrics including geometry, steric clashes, and agreement with experimental data.
A critical component of quality assurance is the ongoing remediation program that addresses inconsistencies arising from evolving standards and practices. Key recent remediation projects include:
- 2026 Metalloprotein Remediation: Improving metal-containing ligand definitions and enhancing polyatomic metal-containing entry annotations using community tools for error correction and better metalloprotein annotation [3].
- 2025 Post-Translational Modification (PTM) Remediation: Updating 76,821 entries with new PTM/protein chemical modification annotations to the Chemical Component Dictionary (CCD) and PDB entries, with approximately 12,900 affected entries receiving additional corrections [3].
- 2023 Peptide Residues Chemical Component Dictionary Remediation: Standardizing atom naming and adding annotation of protein backbone and terminal atoms within peptide residues to improve data findability and interoperability [3].
- 2020 Carbohydrate Remediation: Standardizing atom nomenclature and providing uniform data representation and linear descriptors in collaboration with the glycoscience community [3].
- 2017 PDBx/mmCIF Files Update: Conforming the entire archive to data standards version 5.0 of the PDBx/mmCIF dictionary, which supports the global wwPDB OneDep system [3].
Archive Scope and Composition
Content and Data Types
The PDB archive contains atomic coordinates, crystallographic structure factors, NMR experimental data, and 3DEM maps. Each entry includes detailed metadata such as molecular names, primary and secondary structure information, sequence database references, ligand and biological assembly information, data collection details, and bibliographic citations [4]. The archive supports multiple file formats including the legacy PDB format, PDBx/mmCIF (the primary distribution format), and PDBML (XML variant) [2].
The Chemical Component Dictionary (CCD) provides standardized chemical descriptions for all monomer units and small molecule ligands in the archive. This reference dictionary includes model and idealized coordinates, chemical descriptors (SMILES and InChI), systematic names, stereochemical assignments, and standardized atom naming following IUPAC conventions for standard amino acids and nucleotides [5].
Versioning and Data Preservation
The PDB versioned archive, established in October 2017, maintains all major versions of each PDB entry, providing a complete revision history. Changes triggering major version increments include updates to atomic coordinates, polymer sequence, or chemical description in the coordinate file, while metadata modifications are considered minor revisions [6]. The versioned archive uses extended 8-character accession codes (e.g., "pdb_00001abc") and follows a structured naming scheme: <PDB_ID>_<content_type>_v<major_version>-<minor_version>.<file_format_type>.<file_compression_type> [6].
Quantitative Analysis of Archive Growth
Structure Growth Trends
The PDB archive has experienced exponential growth since its inception in 1971, accelerating significantly with methodological advances and structural genomics initiatives. The annual deposition rate has increased from single digits in the 1970s to over 15,000 structures in recent years [7].
Table 1: Annual Growth of Released PDB Structures (Selected Years)
| Year | Total Entries Available | Structures Released Annually |
|---|---|---|
| 1990 | 507 | 142 |
| 2000 | 13,583 | 2,624 |
| 2010 | 69,486 | 7,742 |
| 2020 | 172,815 | 14,006 |
| 2023 | 214,191 | 14,500 |
| 2024 | 229,662 | 15,471 |
| 2025* | 245,392 | 15,730 |
Projected value [7]
The cumulative growth trajectory demonstrates the archive's expanding importance to the research community, with the total number of structures approximately doubling every decade. This growth pattern reflects both technological advances in structure determination and increasing recognition of structural biology's importance in understanding biological mechanisms and drug development.
Data Storage Requirements
The increasing size and complexity of structural data, particularly from 3DEM techniques, has significantly increased the PDB archive's storage footprint. The core archive now exceeds 1TB of storage, with related holdings requiring substantially more space [2] [8].
Table 2: PDB Archive Storage Growth (2018-2024)
| Year | PDB Legacy Archive Snapshot | PDB Versioned Archive | EMDB Core Archive |
|---|---|---|---|
| 2018 | 441 GB (136,472 structures) | 85 GB | 592 GB (5,753 entries) |
| 2020 | 822 GB (173,005 structures) | 148 GB | 2.9 TB (13,731 entries) |
| 2022 | 1,086 GB (199,755 structures) | 218 GB | 7.5 TB (25,319 entries) |
| 2024 | 1,437 GB (229,564 structures) | 269 GB | 21 TB (41,282 entries) |
Data compiled from PDB statistics on data storage growth [8]
The Electron Microscopy Data Bank (EMDB) core archive shows particularly rapid expansion, growing from 592GB in 2018 to 21TB in 2024 – a 35-fold increase in six years. This reflects both the rising number of 3DEM structures and the increasing size of individual map files. The total RCSB PDB storage holdings, including all copies and related data, reached 279TB in 2024 [8].
Data Access and Retrieval Protocols
Access Methods and Distribution
The wwPDB provides multiple access protocols to accommodate diverse user needs. The primary distribution sites are updated every Wednesday at 00:00 UTC with new and modified entries [2]. For individual file downloads, HTTPS is recommended, while rsync is preferred for bulk transfers. The FTP protocol, previously used for archive access, was deprecated in November 2024 [2].
The major access points include:
- wwPDB: https://files.wwpdb.org, rsync://rsync.wwpdb.org
- RCSB PDB (US): https://files.rcsb.org, rsync://rsync.rcsb.org
- PDBe (UK): ftp.ebi.ac.uk/pub/databases/pdb/
- PDBj (Japan): ftp.pdbj.org, https://files.pdbj.org [2]
Directory Organization and File Structure
The PDB archive employs a hash-based directory structure for efficient data organization. Coordinate files in various formats (mmCIF, PDBML, legacy PDB) are distributed in divided directories based on the middle two characters of the four-digit PDB ID [2]. For example, files for entry 1ABC would be located in the 'ab' subdirectory.
The versioned archive uses a different organization, with all files for a particular entry stored in a single directory grouped under a 2-character hash from the two penultimate characters of the extended PDB code. For entry pdb_00001abc, files are stored in: ../pdb_versioned/data/entries/ab/pdb_00001abc/ [6].
Essential Research Tools and Reagents
Structural biology research relies on specialized tools and resources for data analysis and interpretation. The wwPDB provides several essential resources that function as the "research reagent solutions" for structural bioinformatics.
Table 3: Essential Research Reagent Solutions for PDB Data Analysis
| Resource Name | Type | Function | Access Method |
|---|---|---|---|
| Chemical Component Dictionary | Reference Data | Standardized chemical descriptions of small molecules and residues | HTTPS download [2] |
| Validation Reports | Quality Metrics | Assessment of structural quality using multiple geometric and experimental metrics | HTTPS/rsync [2] |
| Versioned Archive | Data Repository | Complete version history of all PDB entries with revision tracking | Versioned HTTPS/rsync [6] |
| NMR-STAR Format | Standardized Data | Unified NMR restraints and chemical shifts data in standard format | PDB FTP archive [3] |
| Biological Assembly Files | Processed Data | Pre-computed biological units based on crystallographic symmetry | Structure download directories [2] |
Programmatic Access and APIs
For advanced researchers, the wwPDB sites provide flexible APIs for programmatic access, enabling integration of structural data into bioinformatics pipelines and custom applications. These services support specialized searching for macromolecules and ligands, data mining, and bulk retrieval operations [9]. The REST-based APIs allow querying by sequence similarity, chemical structure, ligand properties, and structural motifs, facilitating high-throughput structural bioinformatics research.
Future Directions and Challenges
The wwPDB continues to evolve to meet emerging challenges in structural biology. Key initiatives include handling increasingly large and complex structures from integrative/hybrid methods, improving representation of dynamics and conformational heterogeneity, and enhancing interoperability with other biological data resources. The ongoing transition to PDBx/mmCIF as the primary data standard and the implementation of extended PDB accession codes address anticipated growth and FAIR data principles [6].
The wwPDB's remediation program will continue to enhance data quality, with planned updates for metalloprotein annotations (2026) and completion of PTM remediation (2025) [3]. These efforts ensure the PDB archive remains a robust, reliable foundation for scientific discovery and innovation in structural biology and drug development.
The PDB archive represents an extraordinary collaborative achievement in scientific data preservation and dissemination. Through the coordinated efforts of the wwPDB partners, this resource has grown from a small collection of structures to a comprehensive archive exceeding 229,000 entries, with sophisticated data management practices ensuring both quality and accessibility. The archive's continued growth and evolution reflect its fundamental importance to biomedical research, enabling breakthroughs in understanding biological mechanisms, drug discovery, and therapeutic development. As structural biology advances, the wwPDB's commitment to data integrity, standardization, and open access ensures the PDB archive will remain an indispensable resource for the global scientific community.
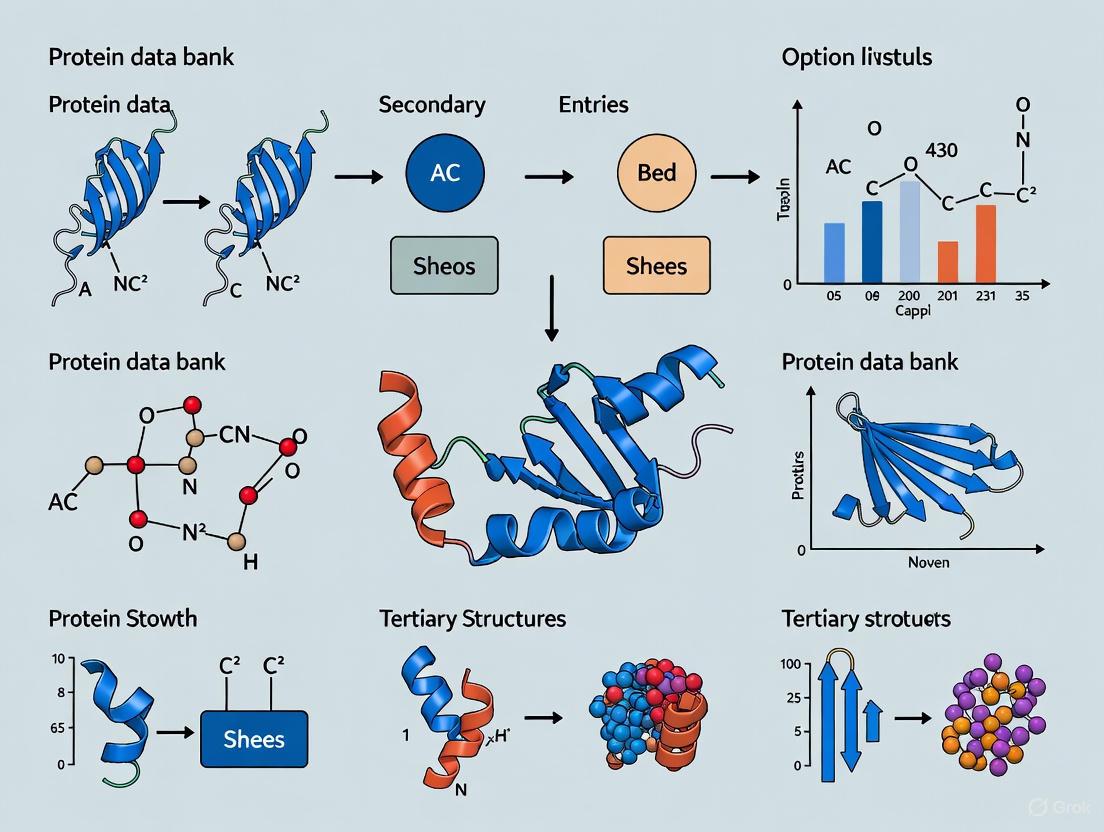
The Protein Data Bank (PDB) is a critical repository for three-dimensional structural data of biological macromolecules, serving as an indispensable resource for researchers, scientists, and drug development professionals worldwide. Understanding the organization of this data is fundamental to effective structural bioinformatics research. Biomolecules exhibit inherent hierarchical organization, from their basic chemical components to complex functional complexes. The PDB archive structures its data to reflect this biological reality, implementing a structured framework that simplifies searching, visualization, and analysis [10]. This technical guide examines the four core levels of the PDB structural hierarchy—Entry, Entity, Instance, and Assembly—providing a foundational framework for navigating structural data within the broader context of macromolecular research.
The rationale for this hierarchical system stems from the complexity of biomolecular structures. Proteins, for instance, are composed of linear chains of amino acids that fold into compact subunits, which can then associate into higher-order complexes with other proteins, nucleic acids, small molecule ligands, and solvent molecules [10]. Without a standardized system to describe these relationships, interpreting structural data would be prohibitively difficult. The hierarchy enables researchers to navigate seamlessly from the complete structural entry to specific chemical components, facilitating precise queries about particular aspects of a structure while maintaining the context of its biological function.
The Four-Level Hierarchical Framework
The PDB organizes structural data into four primary levels, each serving a distinct purpose in describing the components and organization of a macromolecular structure. These levels form a logical progression from the broadest container (Entry) to the most specific structural context (Assembly), enabling precise data annotation and retrieval.
Level 1: Entry
An ENTRY represents the fundamental container for all data pertaining to a particular structure deposited in the PDB. It serves as the top-level organizational unit and is designated with a unique PDB identifier (PDB ID), which is typically a 4-character alphanumeric code (e.g., 2hbs for sickle cell hemoglobin) [10]. Future extensions to eight characters prefixed by 'pdb' are planned to accommodate the growing number of structures [11]. Each entry encompasses all experimental data, metadata, and coordinate information for a single deposition, including:
- Atomic coordinates for all components
- Experimental methodology and conditions
- Citation information and author details
- Sequence and chemical descriptor data
- Validation reports and quality metrics
Every entry must contain at least one polymer entity or one branched entity (such as a linear or branched oligosaccharide) [10]. The entry serves as the primary access point for structural data and connects to various external database identifiers, including PubMed IDs for associated literature and EMDB IDs for electron microscopy maps [11].
Level 2: Entity
An ENTITY describes a chemically unique molecule within an entry. This level distinguishes molecules based on their distinct chemical composition, regardless of how many copies exist in the structure. Entities are categorized into several types [11]:
- Polymeric entities: Proteins and nucleic acids with defined sequences
- Branched entities: Oligosaccharides with linear or branched structures
- Non-polymeric entities: Small molecules such as ligands, inhibitors, ions, and cofactors
- Complex small molecules: Biologically Interesting molecules (BIRD) such as peptide-like inhibitors
Each entity is assigned a unique entity ID specific to its parent entry (e.g., 4HHB_1 refers to entity 1 in PDB entry 4HHB) [11]. Entities connect to external database identifiers, including UniProt accession codes for proteins and GenBank codes for gene sequences, providing critical links to complementary biological data. For small molecules, entities reference the Chemical Component Dictionary (CCD) with specific chemical IDs (e.g., ATP for adenosine triphosphate) [11].
Level 3: Instance
An INSTANCE represents a specific occurrence or copy of an entity within the crystallographic asymmetric unit of an entry. A single chemical entity may have multiple spatial instances in a structure. For example, a homooligomeric protein contains multiple instances of the same protein entity [10]. Instance identification follows specific conventions:
- Polymer instances are assigned unique chain identifiers (chain IDs), which are one or more alphanumeric characters (e.g., A, AA) [10]
- Small molecule instances adopt the chain ID of their closest neighboring polymer instance, with additional unique numbering (e.g., heme groups associated with chain A may be identified as A101 and A102) [10]
- Oligosaccharide instances receive unique chain IDs like polymers, unless they consist of a single sugar molecule covalently linked to a protein, in which case they adopt the protein's chain ID [11]
Chain ID assignment lacks a specific rationale and may differ between entries of the same protein, necessitating careful interpretation when comparing structures [10]. Each instance maintains specific coordinate data, including potential alternate locations for flexible residues identified with unique Alt IDs, and multiple models in NMR structures distinguished by Model IDs [11].
Level 4: Assembly
An ASSEMBLY represents a biologically functional unit composed of one or more instances arranged in a stable complex. This level reflects the native, functional state of the molecule as it exists in its biological context [10]. Assemblies provide critical insights into:
- Quaternary structure and subunit interactions
- Functional oligomerization states
- Biological mechanisms and allosteric regulation
Numerical assembly IDs are assigned to each biologically relevant assembly within an entry [11]. Some entries contain multiple assemblies, while others may require symmetry operations to generate the biological assembly from the asymmetric unit. For example, PDB entry 2hbs contains two complete sickle cell hemoglobin tetramers, each representing a distinct biological assembly [10]. The assembly represents the functional form of the molecule—in this case, the oxygen-binding tetramer found in blood.
Table 1: Summary of PDB Structural Hierarchy Levels
| Level | Definition | Identifier | Example |
|---|---|---|---|
| Entry | All data for a deposited structure | PDB ID (4-character) | 2hbs |
| Entity | Chemically unique molecule | Entity ID | Alpha chain entity |
| Instance | Specific occurrence of an entity | Chain ID | Chain A, Chain B |
| Assembly | Biologically functional unit | Assembly ID | Hemoglobin tetramer |
Table 2: Identifier Systems Across Hierarchy Levels
| Level | Identifier Type | Format | Purpose |
|---|---|---|---|
| Entry | PDB ID | 4-character alphanumeric | Unique structure identification |
| Entity | Entity ID | Number (e.g., 1, 2) | Distinguish chemical components |
| Instance | Chain ID | 1-2 character alphanumeric | Identify specific copies in structure |
| Residue | Residue Number | Integer | Position in polymer sequence |
| Atom | Atom Name | 1-4 characters (e.g., N, CA) | Specific atomic coordinates |
Visualizing the Structural Hierarchy
The relationships between the different levels of the PDB structural hierarchy can be visualized as a logical progression from the complete dataset to the functional biological unit. The following diagram illustrates these relationships and dependencies:
Structural Hierarchy Relationships
This diagram illustrates the containment relationships within the PDB structural hierarchy, showing how entries contain entities, which have instances that form biological assemblies. The color scheme follows the specified palette while maintaining sufficient contrast for readability.
Case Study: Hemoglobin (PDB ID 2hbs)
The structural hierarchy concepts are effectively illustrated by examining hemoglobin (PDB ID 2hbs), a well-characterized oxygen transport protein. This entry provides a concrete example of how the abstract hierarchy manifests in a real biological system:
- ENTRY Level: PDB ID 2hbs encompasses all data for the sickle cell hemoglobin structure, including coordinate data for two complete tetramers, heme cofactors, and surrounding water molecules [10]
- ENTITY Level: The entry contains three distinct chemical entities: (1) the alpha globin chain (a polymer entity), (2) the beta globin chain (a polymer entity), and (3) heme (a non-polymeric entity) [10]
- INSTANCE Level: The asymmetric unit contains multiple instances of these entities: two instances of the alpha chain entity, two instances of the beta chain entity, and four instances of the heme entity (each associated with a globin chain) [10]
- ASSEMBLY Level: The functional biological assembly is a hemoglobin tetramer composed of two alpha chain instances and two beta chain instances, each associated with a heme instance [10]. This tetramer represents the oxygen-binding unit in blood
This example demonstrates how a single entry can contain multiple entities, how one entity can have multiple instances, and how these instances assemble into a functional complex. The hemoglobin tetramer assembly exemplifies the biological relevance of this hierarchical organization, as the tetramer—not the individual chains—represents the physiologically functional form of the molecule.
Practical Applications in Research and Drug Development
Understanding PDB structural hierarchy enables researchers to design more effective queries, accurately interpret structural data, and extract biologically relevant information. Key applications include:
Structure Visualization and Analysis
Molecular graphics programs leverage the hierarchy to enable selective visualization and analysis. Researchers can:
- Display specific representations (wireframe, spacefilling, ribbon) appropriate for different tasks [12]
- Color by chain to distinguish individual instances in complexes [12]
- Focus on active sites using ball-and-stick representations while showing the overall fold with ribbon diagrams [12]
- Select specific entities or instances for distance measurements and interaction analysis
Visualization tools like Mol* provide intuitive interfaces that reflect the structural hierarchy, allowing users to select specific entities or instances and apply different representations to each component [13]. The Sequence Panel displays polymer sequences, providing quick access to specific residues and ligands [13].
Advanced Query Design
The hierarchical organization enables sophisticated database queries that would be impossible with a flat structure. Researchers can:
- Find all structures containing a specific chemical entity (e.g., ATP-binding proteins)
- Identify entries with particular assembly compositions (e.g., homodimers vs. heterodimers)
- Locate specific instances of entities across multiple structures
- Retrieve structures with specific interface types between instances
Drug Discovery Applications
In pharmaceutical research, the hierarchy facilitates:
- Binding site identification across multiple instances of the same entity
- Assessment of biological relevance by focusing on assemblies rather than asymmetric units
- Polypharmacology studies by tracking specific entities across different protein targets
- Selectivity analysis by comparing binding sites across related entities
Table 3: Experimental Protocols for Hierarchy Analysis
| Method | Purpose | Key Steps | Hierarchy Focus |
|---|---|---|---|
| Structure Determination | Determine atomic coordinates | Data collection, phasing, model building, refinement | Entry creation with full hierarchy annotation |
| Complex Assembly Analysis | Identify biological units | Symmetry operations, interface analysis, oligomer validation | Assembly identification and validation |
| Ligand Binding Studies | Characterize small molecule interactions | Density fitting, restraint generation, interaction analysis | Entity identification and instance localization |
| Comparative Structure Analysis | Compare related structures | Structure alignment, conserved feature identification | Entity matching across multiple entries |
Working effectively with PDB structures requires familiarity with key resources and tools designed to navigate the structural hierarchy:
Table 4: Essential Research Tools and Resources
| Resource | Type | Function | Hierarchy Application |
|---|---|---|---|
| RCSB PDB Website | Database portal | Structure search, visualization, and download | Navigation across all hierarchy levels |
| Mol* Viewer | Visualization tool | Interactive 3D structure exploration | Instance selection and assembly visualization |
| Chemical Component Dictionary | Reference database | Chemical descriptions of small molecules | Entity identification and standardization |
| PDBx/mmCIF Format | Data format | Comprehensive structure representation | Complete hierarchy representation in files |
| UniProt Database | Sequence database | Protein sequence and functional information | Entity-level sequence mapping and annotation |
The four-level hierarchical framework of Entry, Entity, Instance, and Assembly provides a powerful conceptual model for organizing and interpreting macromolecular structure data in the Protein Data Bank. This system moves beyond simple file management to reflect fundamental biological principles, enabling researchers to navigate efficiently from complete structures to specific chemical components while maintaining the context of biological function. For structural biologists and drug discovery researchers, mastery of this hierarchy is not merely academic—it enables precise query formulation, accurate data interpretation, and biologically relevant analysis, forming a cornerstone of effective structural bioinformatics practice. As structural biology continues to evolve with emerging techniques in cryo-EM and computational structure prediction, this foundational framework ensures that complex structural data remains accessible, interpretable, and biologically meaningful.
Within structural biology and drug development, the Protein Data Bank (PDB) serves as a fundamental repository for three-dimensional structural data of biological macromolecules. Effective navigation and precise interpretation of these structures hinge on a clear understanding of core identifiers: the PDB ID, Chain ID, and residue numbering system. This technical guide delineates the hierarchy, conventions, and practical applications of these identifiers, providing researchers with a formal framework for structural analysis. As the PDB archive evolves, with an ongoing transition from legacy formats to PDBx/mmCIF and the forthcoming exhaustion of 4-character PDB IDs, mastery of these concepts is critical for ensuring the continuity and reproducibility of structural research [11] [14] [15].
A PDB entry is organized as a structural hierarchy, with specific identifiers pin-pointing data at each level [11]:
- Entry: The entire deposited structure, identified by a PDB ID.
- Entity: Distinct chemical components (e.g., a protein polymer, a DNA strand, a ligand).
- Instance: A specific copy of an entity as positioned in the 3D coordinate space, identified by a Chain ID.
- Residue: A monomer within a polymer or a small molecule, identified by a residue number and name.
- Atom: The fundamental coordinate point, identified by an atom name.
This hierarchical organization enables the unique identification of every atom in a structure, which is a prerequisite for molecular visualization, interaction analysis, and computational modeling [11].
Core Identifier Specifications and Conventions
The table below summarizes the key identifiers, their roles, and formats.
Table 1: Key Identifiers in a PDB Entry
| Identifier Level | Identifier Name | Format & Examples | Primary Function |
|---|---|---|---|
| Entry | PDB ID | 4-character alphanumeric (e.g., 2hbs). Future: 12-character, prefixed (e.g., pdb_00002hbs) [11] [14]. |
Uniquely identifies a structure entry in the PDB archive. |
| Entity | Entity ID | Integer specific to an entry (e.g., 1 for the first entity in entry 4HHB) [11]. |
Tracks a unique chemical component (e.g., a specific protein sequence) throughout the PDB file. |
| Instance | Chain ID | 1- or 2-character alphanumeric (e.g., A, A1). Two systems exist: PDB-assigned (label_asym_id) and author-assigned (auth_asym_id) [11] [16]. |
Identifies a specific copy of an entity located in the 3D coordinate system. |
| Residue | Residue Number | A string combining a sequence number and an optional insertion code (e.g., 50, 50A) [17]. Two numbering schemes exist: PDB sequential (label_seq_id) and author (auth_seq_id) [11]. |
Specifies the position and identity of a residue (e.g., an amino acid) within a chain. |
| Atom | Atom Name | 4-character name per the Chemical Component Dictionary (e.g., N, CA, C, O for protein backbone atoms) [11] [18]. |
Identifies a specific atom within a residue. |
The PDB ID: Entry-Level Access
The PDB ID is the primary access key for any structure in the archive. The current 4-character system (e.g., 4HHB for human hemoglobin) is expected to be fully exhausted by 2028 [14]. Subsequently, all new entries will be assigned a 12-character extended PDB ID, formatted as pdb_########xxxx, where # is a digit and x is an alphanumeric character (e.g., pdb_00008y9m) [11] [14]. This change necessitates updates to software, scripts, and communication practices to ensure future compatibility. The associated DOI for structures will also transition to the format 10.2210/[Extended_PDB_ID]/pdb [14].
Chain IDs: Tracking Molecular Instances
A Chain ID specifies the location of a molecule in 3D space. A single entity (e.g., a protein sequence) can have multiple instances (chains) in an asymmetric unit, each with a unique Chain ID [11] [19]. Researchers must be aware of the dual labeling system:
label_asym_id: The PDB-assigned identifier, typically starting with 'A' [11].auth_asym_id: The identifier provided by the depositing scientist, which may match literature conventions [11].
For structure alignment tools on RCSB.org, the Chain ID input field is case-sensitive and must correspond to the label_asym_id when using PDBx/mmCIF format files [16].
Residue Numbering: Sequence and Structural Landmarks
Residue numbering pinpoints the location of amino acids or nucleotides. Two numbering schemes exist, which often but do not always align [11]:
label_seq_id: A sequential integer assigned by the PDB, starting from 1 for the first residue in the polymer chain.auth_seq_id: The residue numbering provided by the depositor, which may match the numbering in a related UniProt entry or publication.
Residue numbers are stored as 5-character strings to accommodate an insertion code (e.g., 50A), which is used to maintain a continuous sequence when an extra residue is inserted without renumbering the entire chain [17]. Gaps in residue numbering are common and typically indicate residues that are present in the full protein sequence but are not resolved in the experimental electron density map, often due to structural flexibility [20].
Experimental Protocols for Identifier Utilization
Protocol: Structure Retrieval and Inspection
Objective: To correctly retrieve a structure and identify its constituent chains and molecules.
- Access Structure: Navigate to
https://www.rcsb.org/structure/[PDB_ID](e.g.,4HHB). - Identify Macromolecules: On the Structure Summary page, scroll to the "Macromolecules" section to view the list of entities (proteins, nucleic acids) and their corresponding PDB-assigned Chain IDs.
- Visualize in 3D: Click "3D View" to open the Mol* visualization tool. Select specific chains to highlight them and observe their spatial arrangement.
- Check for Alternate Conformations: Within the 3D viewer, inspect residues for "alternate locations," indicated by Alt IDs, which represent discrete conformational states of side chains or ligands observed in the experimental data [11].
Protocol: Pairwise Structure Alignment
Objective: To quantitatively compare the three-dimensional structures of two protein chains.
- Access Tool: On RCSB.org, select "Analyze" from the menu and choose "Pairwise Structure Alignment" [16].
- Define Reference Structure: Under "Reference Structure," use the "Entry ID" option to input the first PDB ID (e.g.,
4HHB) and select the desired Chain ID (e.g.,A). - Define Target Structure: Under "Structure to Align," input the second PDB ID (e.g.,
1OJ6) and its Chain ID (e.g.,A). - Select Algorithm: Choose an alignment method based on your goal (see Table 2). For general comparison of closely related structures,
jFATCAT-rigidorjCEis recommended [16]. - Execute and Analyze: Click "Compare." Review the resulting metrics (RMSD, TM-score) and inspect the sequence-structure alignment in the interactive viewer to assess the quality and regions of variation [16].
Table 2: Selection Guide for Structure Alignment Algorithms on RCSB.org
| Algorithm | Type | Best Use Case |
|---|---|---|
| jFATCAT-rigid | Rigid-body | Identifying the largest structurally conserved core between proteins with similar conformations [16]. |
| jFATCAT-flexible | Flexible | Comparing proteins that undergo conformational changes (e.g., upon ligand binding) by introducing hinges between rigid domains [16]. |
| jCE | Rigid-body | Optimal rigid-body superposition for identifying substructural similarities [16]. |
| jCE-CP | Flexible/Topology-independent | Aligning proteins related by circular permutations or with different loop connectivities [16]. |
| TM-align | Topology-based | Fast, sensitive comparison of global protein fold, even with low sequence similarity [16]. |
Protocol: Mapping a Residue from UniProt to PDB
Objective: To locate the 3D coordinates of a specific residue of interest (e.g., a catalytic site residue known from biochemical studies) within a PDB structure.
- Obtain UniProt Alignment: On the PDB Structure Summary page, find the relevant protein entity and click "See Sequence Details." This displays the alignment between the PDB sequence and the UniProt reference sequence.
- Identify Numbering Scheme: Note the mapping between the UniProt residue number (often reflected in the
auth_seq_id) and the PDB's internallabel_seq_id. - Locate in 3D Viewer: Use the
label_seq_idto select and center the residue in the Mol* 3D viewer. If the residue is missing, the sequence alignment will typically indicate it as "unmodeled," often due to a lack of electron density [20].
The Scientist's Toolkit: Research Reagent Solutions
The following table details essential "reagents" for working with PDB structures—primarily data resources and software tools.
Table 3: Essential Digital Tools and Resources for PDB Analysis
| Tool/Resource | Type | Function |
|---|---|---|
| RCSB PDB Website | Data Portal | Primary interface for searching, browsing, and downloading PDB structures and their metadata [11]. |
| PDBx/mmCIF Format | Data Format | The master and future-proof format for PDB data, required for all new entries with extended PDB IDs [21] [15]. |
| Mol* | Visualization Software | An interactive, web-based tool for high-performance 3D visualization and analysis of structures directly on RCSB.org [16]. |
| Chemical Component Dictionary | Reference Database | A curated resource defining standard chemical descriptions for ligands, residues, and modified amino acids found in PDB entries [11]. |
| Pairwise Structure Alignment Tool | Analysis Software | A suite of algorithms on RCSB.org for superposing and quantitatively comparing protein structures [16]. |
Logical Workflow for Structural Analysis
The diagram below outlines a standard workflow for accessing and analyzing a PDB structure, from entry retrieval to residue-level inspection.
Figure 1: A logical workflow for structural analysis using PDB identifiers.
Navigating Common Challenges and the Evolving PDB Landscape
Researchers frequently encounter several practical challenges:
- Residue Numbering Inconsistencies: The sequence in a PDB file often does not start at 1, has gaps, or is shorter than the canonical UniProt sequence. This is normal and results from N- or C-terminal residues, or internal loops, that are disordered and not visible in the experimental map [20].
- Multiple PDB IDs per UniProt ID: A single UniProt entry often has multiple associated PDB structures. These may represent the protein bound to different partners (e.g., drugs, DNA), captured in different conformational states, or from various experimental conditions. Their sequences might differ slightly due to cloning boundaries, point mutations, or unmodeled regions [20].
- Legacy PDB Format Limitations: Approximately 3.7% of the archive is no longer available in the legacy PDB file format due to structural complexity (e.g., >62 chains, >99999 atoms). For these entries, data is provided in PDBx/mmCIF format or as "best effort" bundles of truncated PDB files [21]. Users must transition to PDBx/mmCIF to ensure access to all current and future structures [21] [15].
The precise application of PDB ID, Chain ID, and residue numbering is foundational to rigorous structural biology research. These identifiers form a coordinate system that translates biological questions into actionable queries within three-dimensional models. As the PBD archive undergoes a significant transition in its foundational data format and identifier system, proactive adoption of the PDBx/mmCIF format and extended PDB IDs is no longer optional but a necessary step for all researchers and drug development professionals aiming to maintain the forefront of structural science.
The Protein Data Bank (PDB) archive serves as the global repository for experimentally-determined three-dimensional (3D) structures of biological macromolecules, operating as the first open-access digital data resource in biology since 1971 [22]. The archive has grown from just seven protein structures to nearly 200,000 experimentally-determined structures of proteins, nucleic acids (DNA and RNA), and their complexes with small-molecule ligands as of 2022 [22]. Managed by the Worldwide Protein Data Bank (wwPDB) partnership, this resource adheres to FAIR (Findability, Accessibility, Interoperability, and Reusability) and FACT (Fairness, Accuracy, Confidentiality, and Transparency) Principles, emblematic of responsible data stewardship in the modern era [22]. Understanding the composition of these entries is fundamental for researchers, scientists, and drug development professionals who rely on these structural data for insights into molecular interactions, function, and evolution.
Biomolecules in the PDB archive are organized using a hierarchical structure that reflects their biological organization [10]. This hierarchy consists of four primary levels: Entry, Entity, Instance, and Assembly. An Entry encompasses all data pertaining to a particular structure deposited in the PDB and is designated with a 4-character alphanumeric identifier called the PDB ID [10]. An Entity represents a chemically unique molecule, which may be polymeric (such as a protein chain or DNA strand) or non-polymeric (such as a small-molecule ligand) [10]. An Instance refers to a specific occurrence of an Entity within an Entry, and an Assembly constitutes a biologically relevant grouping of one or more Instances that form a stable complex and/or perform a function [10]. This organizational framework enables meaningful exploration, search, and visualization of structural data, providing researchers with a systematic approach to investigating complex biomolecular systems.
Table 1: Hierarchy of Organizational Levels in PDB Structures
| Level | Definition | Example |
|---|---|---|
| Entry | All data for a specific PDB structure | PDB ID 2hbs |
| Entity | Chemically unique molecule | Alpha chain protein, beta chain protein, heme |
| Instance | Specific occurrence of an Entity | Two copies of alpha chain in hemoglobin tetramer |
| Assembly | Biologically functional group of Instances | Hemoglobin tetramer (oxygen-carrying form) |
The composition of PDB structures has evolved significantly over time, with increasing complexity in terms of the number of residues, polymer chains, and ligands per structure [22]. As of mid-2022, the total number of amino acid and nucleotide residues in the archive exceeded 200 million, and the total number of atoms surpassed 1.5 billion [22]. This growth in complexity reflects advances in structural biology methods that now enable the determination of larger and more intricate macromolecular complexes. For drug development professionals, this expanding repository provides critical structural insights into molecular recognition, binding sites, and mechanisms of action that inform rational drug design.
Quantitative Analysis of PDB Archive Contents
The PDB archive has experienced exponential growth since its inception, with the number of released structures increasing dramatically year by year. As of mid-2022, the archive contained 166,894 structures determined by macromolecular crystallography (MX), 11,294 by 3D electron microscopy (3DEM), and 13,738 by nuclear magnetic resonance (NMR) spectroscopy [22]. The distribution of structural biology methods has shifted significantly over time, with MX structures plateauing at approximately 10,000 annually since 2016, NMR structure releases declining, and 3DEM structure releases growing exponentially—increasing approximately six-fold in just four years [22]. This methodological evolution reflects technological advances, particularly in cryo-EM, that have enabled structure determination of increasingly complex biological assemblies.
Protein-nucleic acid complexes represent a biologically crucial category of structures in the archive. As of 2025, the PDB contains 15,366 such complexes, with 1,407 released in that year alone [23]. The growth trajectory of these complexes has been steadily increasing, from just 3 structures in 1989 to over 15,000 by 2025, reflecting growing research interest in fundamental biological processes such as transcription, translation, and DNA repair [23]. This expansion provides researchers with an increasingly complete structural picture of how proteins and nucleic acids interact to execute cellular functions.
Table 2: Distribution of Experimental Methods in the PDB Archive (as of mid-2022)
| Experimental Method | Number of Structures | Percentage of Archive | Key Quality Indicators |
|---|---|---|---|
| Macromolecular Crystallography (MX) | 166,894 | ~87% | Resolution, R-factor, R-free, RSR |
| Nuclear Magnetic Resonance (NMR) | 13,738 | ~7% | Restraint violations, RCI, chemical shift validation |
| 3D Electron Microscopy (3DEM) | 11,294 | ~6% | Resolution (FSC), Q-score, atom inclusion |
| Other Methods | ~8,000 | ~4% | Method-specific validation metrics |
Ligands represent another crucial component of PDB structures, with over 70% of structures containing one or more small-molecule ligands (excluding water molecules) [24]. These ligands are classified as either "functional" (playing biological/biochemical roles such as co-factors, activators, inhibitors, substrates, or products) or "non-functional" (typically solvents, salts, ions, or crystallization agents) [24]. The wwPDB Chemical Component Dictionary (CCD) defines each unique small-molecule ligand found in the PDB with a distinct identifier (CCD ID) and detailed chemical description [24]. The quality of ligand structures is particularly important for drug development applications, where accurate molecular representations of binding interactions are essential for structure-based drug design.
The complexity of structures in the PDB archive has increased substantially over time, as evidenced by the rising average number of polymer chains per structure and average number of ligands per structure [22]. This trend reflects methodological advances that enable determination of larger macromolecular complexes, such as ribosomes, polymerases, and viral capsids, providing researchers with more complete structural understanding of complex cellular machinery. For drug development professionals, these complex structures offer insights into polypharmacology and allosteric regulation that can inform the design of more selective and effective therapeutics.
Methodologies for Structural Determination and Analysis
Experimental Structure Determination Methods
Structural biologists employ several principal methods for determining biomolecular structures, each with distinct methodologies and quality assessment protocols. Macromolecular crystallography (MX) remains the most prevalent method, comprising approximately 87% of the archive as of August 2022 [25]. The MX structure determination process involves growing crystals of the biomolecule, collecting X-ray diffraction data, solving the phase problem, building an atomic model, and refining this model against the experimental data [22]. Key technical innovations that accelerated MX include the development of molecular replacement (MR) by Michael Rossmann for structure determination and the adoption of multiple-wavelength anomalous dispersion (MAD) and single-wavelength anomalous dispersion (SAD) methods for solving the phase problem [22].
Nuclear magnetic resonance (NMR) spectroscopy represents the second major structural biology method, particularly suited for studying protein dynamics and smaller proteins that prove difficult to crystallize. NMR structure determination involves measuring chemical shifts and conformational restraints (such as NOEs, J-couplings, and residual dipolar couplings) from which 3D structures are calculated [25]. The methodology produces an ensemble of structures that satisfy the experimental restraints, providing insights into molecular flexibility and dynamics [25]. Quality assessment focuses on chemical shift validation, analysis of random coil index (RCI) to identify disordered regions, and quantification of restraint violations [25].
Three-dimensional electron microscopy (3DEM) has emerged as the fastest-growing method for structure determination, particularly for large macromolecular complexes that defy crystallization. This method involves collecting images of individual molecules frozen in vitreous ice, classifying these images, generating 3D reconstructions, and building atomic models into the resulting density maps [22] [25]. The resolution of 3DEM structures is estimated using Fourier-Shell Correlation (FSC), while quality assessment includes visual inspection of map-model fit, calculation of atom inclusion fractions, and computation of Q-scores that measure how well atoms in the structure can be resolved [25]. Technological advances in direct electron detectors, image processing software, and phase plate technology have enabled 3DEM to achieve near-atomic resolution for many biological specimens.
Ligand Structure Quality Assessment
The quality of small-molecule ligands in PDB structures is assessed using specialized methodologies that evaluate both agreement with experimental data and geometric parameters [24]. For X-ray crystal structures, ligand quality assessment focuses on two principal composite indicators: PC1-fitting (which aggregates real space R factor (RSR) and real space correlation coefficient (RSCC) to measure how well the ligand model fits the electron density) and PC1-geometry (which aggregates Root-Mean-Squared deviation Z-scores for bond lengths and bond angles to measure geometric accuracy) [24]. These composite ranking scores are uniformly distributed from 0% (worst) to 100% (best), simplifying interpretation and comparison across different ligands and structures.
The ligand validation process employs principal component analysis (PCA) to reduce correlated quality indicators into unidimensional metrics. For the electron density fit indicators (RSR and RSCC), the first principal component (PC1-fitting) explains 84% of the variance of both parameters [24]. Similarly, for the geometry indicators (RMSZ-bond-length and RMSZ-bond-angle), PC1-geometry explains 82% of the total variance [24]. This statistical approach enables comprehensive assessment of ligand quality while maintaining interpretability. Currently, ligand quality analysis focuses on X-ray co-crystal structures with complete validation data, excluding structures not solved by X-ray diffraction, single-atom ions, ligands in structures lacking associated structure factor data (typically deposited before 2008), and branched oligosaccharides [24].
For drug development applications, the concept of "Ligands of Interest" (LOI) identifies functional ligands designated as the focus of research by structure authors or by RCSB PDB based on specific criteria: formula weight > 150 Da and exclusion from a list of likely non-functional ligands [24]. This classification helps researchers quickly identify biologically relevant small molecules for further analysis. The ligand quality assessment enables researchers to select the best instances of specific ligands for visualization, analysis, and molecular design, crucial for structure-based drug discovery efforts targeting specific binding sites.
Visualization and Analysis of Structural Components
Structure Visualization Tools and Techniques
The RCSB PDB provides powerful visualization tools for exploring and analyzing structural components, with Mol* serving as the default web-based tool that requires no software installation [26]. The Mol* interface simultaneously displays molecules in 3D and the sequences of polymers present in the structure, along with any ligands, ions, and water molecules [26]. Key components of the interface include the 3D canvas (for rotating, translating, and zooming into structures), the sequence panel (for selecting specific amino acids or regions), and the Controls panel (for modifying representations and coloring schemes) [26]. This integrated visualization environment enables researchers to comprehensively analyze structural features and interactions.
Coloring schemes in molecular visualization follow specific conventions that convey structural information. For experimental structures with a single protein chain, the default coloring follows a rainbow scheme from the N-terminus (blue) to the C-terminus (red) [27]. Computed Structure Models (CSMs) with single protein chains are colored by model confidence score (pLDDT), with high-confidence regions in dark blue and lower-confidence regions in yellow or orange [27]. Structures with multiple chains employ distinct colors for each polymer chain to facilitate differentiation [27]. Effective colorization of biological data visualization should consider the nature of the data (nominal, ordinal, interval, or ratio), select appropriate color spaces (preferably perceptually uniform spaces like CIE Luv and CIE Lab), and ensure accessibility for color-deficient users [28].
The Structure Summary page on RCSB PDB provides multiple visualization options tailored to different analytical needs [27]. The "Structure" option visualizes the entire structure or assembly in Mol*, while "Ligand Interaction" opens the structure zoomed in and focused on a specific ligand [27]. Specialized visualization options include "Predict Membrane" (which draws predicted membrane location for membrane protein structures) and "Electron Density" (which displays electron density for X-ray structures, enabling researchers to visualize a structure within its experimental density map) [27]. These context-specific visualization modes support diverse research applications from binding site analysis to membrane protein characterization.
Structure Comparison and Analysis Methods
Advanced structure comparison tools enable researchers to analyze variations in interactions, distances, and properties across single structures, sets of structures, or between two sets of structures [29]. These tools employ multiple data browsers that present information on contact position pairs (residue-residue interactions), contact position-AA pairs (interaction frequencies for specific amino acid pairs), residue backbone and sidechain movement (rotamer angles, solvent accessibility), and residue helix types, bulges, and constrictions (backbone conformation and secondary structure) [29]. This comprehensive analysis framework supports diverse research questions from conformational changes to conserved interaction networks.
Visualization of comparative analysis results employs multiple plotting modalities tailored to different analytical perspectives. Flare plots depict interacting residues in a circular fashion, showing interactions between consecutive segments on the outside and other interactions on the inside [29]. Heatmaps provide an all-against-all residue interaction overview, while network plots (2D and 3D) display interconnected residues in subnetworks [29]. Specialized plots for GPCR structures include segment movement diagrams that depict the movement and rotation of transmembrane helices at extracellular, membrane-middle, and cytosolic positions [29]. These diverse visualization approaches enable researchers to extract structural insights at multiple scales from atomic interactions to domain movements.
Structure similarity trees offer another powerful approach for comparing the overall conformation of a selected set of structures [29]. These trees are generated by calculating distances from all Cα atoms to all other Cα atoms for residues in shared regions (typically the seven transmembrane helices for GPCRs), normalizing these distances, computing pairwise similarities using summed absolute differences, and performing hierarchical clustering with average linkage [29]. The resulting trees are enriched with additional structural and receptor data, with internal nodes colored according to the Silhouette index that indicates separation of structures in that node from structures in the nearest neighboring node [29]. This approach enables systematic analysis of conformational diversity and phylogenetic relationships among related structures.
Table 3: Essential Research Tools for PDB Structure Analysis
| Tool/Resource | Function | Application Context |
|---|---|---|
| Mol* | Web-based 3D structure visualization | Interactive exploration of molecular structures, ligands, and interactions [26] |
| wwPDB Validation Reports | Structure quality assessment | Evaluating global and local quality of experimental structures [25] |
| Chemical Component Dictionary (CCD) | Ligand chemical reference | Standardized chemical descriptions of small molecules in PDB structures [24] |
| Structure Comparison Tool | Multi-structure analysis | Comparing interactions, distances, and properties across structure sets [29] |
| OneDep Deposition System | Structure submission | Unified system for depositing, validating, and curating PDB structures [22] |
| Ligand Quality Assessment | Small-molecule validation | Evaluating fit-to-density and geometry of ligands in X-ray structures [24] |
The RCSB PDB offers comprehensive programmatic access to structural data through its Data API, enabling researchers to programmatically retrieve and analyze structural information [27]. This interface supports complex queries and data retrieval tasks, facilitating large-scale bioinformatic analyses and integration of structural data with other biological data resources. The API provides access to the full hierarchy of structural data, from entry-level information to atomic coordinates, supporting diverse research applications from structural genomics to drug discovery.
Specialized validation tools address the unique requirements of different structure determination methods. For X-ray structures, validation includes analysis of electron density fit using real space R (RSR) values and real space correlation coefficients (RSCC) [25]. NMR structure validation focuses on restraint violations and chemical shift analysis [25]. 3DEM validation employs Fourier Shell Correlation (FSC) for resolution estimation and Q-scores for map-model fit assessment [25]. Computed Structure Models are evaluated using predicted Local Distance Difference Test (pLDDT) scores that estimate confidence in different regions of the model [25]. These method-specific validation approaches ensure appropriate quality assessment across the diverse methodological landscape of structural biology.
Educational resources and documentation support effective use of PDB data resources by researchers at all career stages. The RCSB PDB provides detailed documentation on understanding the organization of 3D structures in the PDB, assessing structure quality, and utilizing visualization tools [27] [10] [25]. These resources include explanations of key concepts such as biological assemblies, structure validation metrics, and the hierarchical organization of structural data [10]. For drug development professionals, specialized guidance on assessing ligand structure quality supports informed selection of structural data for structure-based drug design applications [24].
The Protein Data Bank (PDB) archive serves as the global repository for experimentally-determined three-dimensional structures of biological macromolecules, enabling breakthroughs in scientific research and drug development. This technical guide provides researchers and drug development professionals with a comprehensive overview of the core file formats—PDBx/mmCIF (master format), legacy PDB, and PDBML (XML)—and the practical methodologies for accessing these critical data resources. As the archive undergoes a significant transition from legacy formats to more robust and scalable solutions, understanding these foundational concepts is paramount for effective structural bioinformatics and computational drug discovery. The wwPDB strongly recommends transitioning to PDBx/mmCIF format, as legacy PDB format files will be completely phased out once all four-character PDB IDs are exhausted, which is expected to occur before 2028 [21] [30].
Legacy PDB Format
The legacy PDB format, often called the "flat-file" format, is an ASCII text file format consisting of 80-column records that has been used since the inception of the PDB archive [18] [31]. This format organizes structural information into specific record types that describe atomic coordinates, secondary structure, connectivity, and metadata. Despite its historical importance, this format suffers from inherent limitations in representing complex modern structural data and is being progressively phased out.
Key Record Types in Legacy PDB Files:
| Record Type | Data Provided |
|---|---|
ATOM |
Atomic coordinates for atoms in standard residues (amino acids, nucleic acids) |
HETATM |
Atomic coordinates for atoms in nonstandard residues (inhibitors, cofactors, ions, solvent) |
TER |
Indicates the end of a chain of residues |
HELIX |
Location and type of protein helices |
SHEET |
Location and sense of beta strands |
SSBOND |
Defines disulfide bond linkages |
Table: Essential record types in the legacy PDB file format [18].
The technical limitations of the legacy format have become increasingly apparent with modern structural biology data. As of April 2025, 3.7% of the PDB archive is not available in legacy PDB format due to structural complexity that exceeds the format's specifications [21]. Specific cases where legacy files are unavailable include entries containing multiple character chain IDs, more than 62 chains, more than 99,999 ATOM coordinates, complex beta sheet topology, B-factors exceeding 999.99, or chemical IDs for ligands and chemical components that are 5 characters long [21].
PDBx/mmCIF Format
PDBx/mmCIF (macromolecular Crystallographic Information File) is the master data format for the PDB archive, based on the STAR (Self-defining Text Archive and Retrieval) format [21] [31]. This format overcomes the limitations of the legacy format through its flexible, dictionary-defined structure that supports virtually unlimited data fields and values. The mmCIF format provides a more robust framework for representing complex structural data, including large macromolecular assemblies, intricate structural features, and comprehensive metadata.
The transition to mmCIF as the primary format began in 2014, and it now serves as the foundation for all current and future PDB data representation [31]. The format's dictionary-driven approach ensures consistent data representation and enables more sophisticated querying and analysis capabilities essential for drug development research.
PDBML (XML) Format
PDBML is the canonical XML representation of PDB data, adapted from the PDBx/mmCIF specification [31]. This format provides the same comprehensive data content as mmCIF but structured according to XML conventions, making it accessible to XML-aware tools and libraries. The PDBML format follows the same mmCIF dictionary, ensuring data consistency across representations [21].
Three variants of PDBML are available: the complete format including atom records, a version without atom records ("no-atom"), and a version with extended atom records ("extatom") [31]. This flexibility allows researchers to choose the appropriate data subset for their specific applications, optimizing download times and processing requirements.
Quantitative Format Comparison and Selection Framework
Technical Specifications and Limitations
| Feature | Legacy PDB | PDBx/mmCIF | PDBML (XML) |
|---|---|---|---|
| Underlying Technology | Fixed-column text | STAR-based dictionary | XML schema |
| Chain Limit | 62 chains | Unlimited | Unlimited |
| Atom Record Limit | 99,999 per file | Unlimited | Unlimited |
| Chain ID Length | Single character | Multiple characters | Multiple characters |
| B-factor Range | < 999.99 | Unlimited | Unlimited |
| Chemical ID Length | 3 characters | 5+ characters | 5+ characters |
| Data Integrity | Prone to formatting errors | Dictionary-validated | Schema-validated |
| Metadata Richness | Limited | Comprehensive | Comprehensive |
Table: Technical comparison of PDB file formats highlighting limitations of the legacy format [21] [31].
Format Selection Decision Framework
The following workflow provides a systematic approach for selecting the appropriate file format based on research requirements and technical constraints:
Diagram: Decision workflow for selecting appropriate PDB file formats based on research requirements.
Download Protocols and Access Methods
The PDB archive provides multiple access protocols optimized for different use cases. The FTP protocol has been deprecated since November 2024, with HTTPS and rsync recommended for current applications [2] [32].
Primary Download Protocols:
| Protocol | Use Case | Example Command/URL |
|---|---|---|
| HTTPS | Individual file downloads, scripted access | https://files.rcsb.org/download/4hhb.cif.gz |
| Rsync | Bulk downloads, archive mirroring | rsync -rlpt -v -z --delete --port=33444 rsync.rcsb.org::ftp_data/structures/divided/mmCIF/ ./mmCIF |
| AWS S3 | Large-scale data integration | s3://pdbsnapshots/ |
Table: Recommended protocols for accessing PDB data [2] [32].
Programmatic Access URLs
The following table provides standardized URL patterns for programmatic access to PDB data files, essential for automated pipelines in drug discovery workflows:
| File Format | Compression | Example URL Pattern |
|---|---|---|
| PDBx/mmCIF | Compressed | https://files.rcsb.org/download/4hhb.cif.gz |
| PDBx/mmCIF | Uncompressed | https://files.rcsb.org/download/4hhb.cif |
| Legacy PDB | Compressed | https://files.rcsb.org/download/4hhb.pdb.gz |
| Legacy PDB | Uncompressed | https://files.rcsb.org/download/4hhb.pdb |
| PDBML/XML | Compressed | https://files.rcsb.org/download/4hhb.xml.gz |
| PDBML/XML | Uncompressed | https://files.rcsb.org/download/4hhb.xml |
| Biological Assembly (mmCIF) | Uncompressed | https://files.rcsb.org/download/5a9z-assembly1.cif |
| Validation Report | Compressed | https://files.rcsb.org/pub/pdb/validation_reports/ |
Table: Standardized URL patterns for programmatic access to PDB files [32].
For batch downloading large datasets, researchers are encouraged to use provided shell scripts or rsync for efficient data transfer [33] [32]. The archive is substantial, requiring over 1TB of storage and growing with weekly updates every Wednesday at 00:00 UTC [2].
Download Workflow and Source Selection
The following diagram illustrates the systematic process for accessing PDB data through various sources and protocols:
Diagram: Systematic workflow for downloading PDB data through appropriate protocols and sources.
| Tool/Resource | Function | Access Method |
|---|---|---|
| RCSB PDB REST API | Programmatic access to individual structure files | HTTPS requests to files.rcsb.org |
| Rsync Mirroring | Maintain local copy of entire or partial archive | Rsync protocol on port 33444 |
| Batch Download Script | Automated download of multiple structures | Custom scripts using wget/curl |
| mmCIF Parser Libraries | Read and interpret mmCIF files programmatically | Various programming languages |
| Archive Snapshots | Stable datasets for reproducible research | Annual snapshots via HTTPS/AWS |
| Validation Reports | Assess structure quality and experimental data | Separate download directory |
Table: Essential tools and resources for effective PDB data access and processing [2] [34] [32].
Transition Timeline and Strategic Recommendations
The wwPDB has established a clear timeline for transitioning from legacy formats to modern data representations. Key milestones include:
- Immediate: wwPDB strongly encourages all users to adopt PDBx/mmCIF file format and extended PDB ID format [30]
- Early 2026: Beta PDB Archive organized by extended PDB ID will be available [30]
- Before 2028: All four-character PDB IDs expected to be fully assigned [21]
- 2028 and beyond: New entries will only receive extended PDB IDs; data will not be provided in legacy PDB file format [21] [30]
For drug development professionals and researchers, the following strategic recommendations ensure seamless continuity of research workflows:
- Update analytical pipelines to prioritize PDBx/mmCIF format over legacy PDB
- Implement extended PDB ID support in databases and referencing systems
- Utilize archive snapshots for reproducible, long-term research projects
- Leverage validation reports for quality assessment of structural data used in drug discovery
- Adopt bulk download methodologies for large-scale structural bioinformatics analyses
The PDB archive continues to evolve with emerging structural biology techniques, including integrative/hybrid methods (IHM) and computed structure models, making format flexibility and robust data access strategies essential components of modern structural bioinformatics research [35] [32].
From Experiment to Model: Methodologies and Research Applications
Structural biology integrates techniques from molecular biology, biochemistry, and biophysics to elucidate the molecular structures and dynamics of biologically significant molecules. Understanding the three-dimensional structures of proteins and protein complexes offers profound insights into the mechanisms of life and disease, facilitating the rational design of novel diagnostic and therapeutic agents [36]. The core experimental techniques of X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and cryo-electron microscopy (cryo-EM) form the foundation for the atomic-resolution structures deposited in the Protein Data Bank (PDB). According to recent PDB statistics, X-ray crystallography remains the dominant technique, accounting for approximately 66% of structures released in 2023, while cryo-EM has seen a dramatic rise to about 31.7%. NMR contributes a smaller but vital portion at 1.9% [36] [37]. This whitepaper provides an in-depth technical examination of these three foundational methods, detailing their principles, methodologies, and applications within the context of modern biomedical research and drug discovery.
The three major techniques each possess distinct strengths and limitations, making them uniquely suited for different types of biological questions and samples. The following table provides a structured, quantitative comparison of their key characteristics.
Table 1: Comparative Analysis of Core Structural Biology Techniques
| Feature | X-Ray Crystallography | NMR Spectroscopy | Cryo-Electron Microscopy |
|---|---|---|---|
| Typical Resolution | Atomic (≤ 2.0 Å) | Atomic (for defined regions) | Near-atomic to Atomic (3.0 - 1.8 Å) |
| Sample State | Crystalline solid | Solution or solid state | Vitrified solution (Vitreous ice) |
| Sample Requirement | High-quality, ordered crystals | Isotopically labeled, soluble | Purified complex, no crystal needed |
| Typical Size Range | No upper limit, lower limit ~10 kDa | ~5 - 50 kDa (Solution NMR) | > ~50 kDa (for single particle) |
| Key Output | Single, static atomic model | Ensemble of models, dynamics data | 3D electrostatic potential map |
| Throughput | High (once crystallized) | Medium to Low | Medium (data collection & processing) |
| Key Advantage | High throughput, atomic resolution | Studies dynamics & interactions | Handles large complexes & flexibility |
| Primary Limitation | Requires crystallization | Size limitation, sample complexity | Small proteins remain challenging |
X-Ray Crystallography
Principles and History
X-ray crystallography is a powerful technique for determining the three-dimensional structures of biological macromolecules at atomic resolution. The technique is based on the diffraction of X-rays by the electron density of crystallized molecules [36] [38]. It originated in the early 20th century following Wilhelm Conrad Röntgen's discovery of X-rays in 1895 and Max von Laue's demonstration of X-ray diffraction by crystals in 1912. Sir William Henry Bragg and Sir William Lawrence Bragg later developed the fundamental method for determining crystal structure and formulated Bragg's Law (nλ = 2d sinϑ), which relates the angles of diffracted X-rays to the spacing between crystal planes, earning them the Nobel Prize in Physics in 1915 [36]. This method was famously used in the determination of the DNA double helix structure by Watson and Crick using Rosalind Franklin's diffraction data [36] [39].
Experimental Workflow and Protocol
The process of X-ray crystallography involves several key, sequential steps [36] [40] [38].
- Protein Purification and Crystallization: The target molecule must be purified to homogeneity and crystallized. This is often the most significant hurdle. The principle is to take a high concentration of protein (e.g., ~10 mg/mL) and slowly induce it to come out of solution in a controlled manner that promotes crystal growth rather than precipitation. This involves extensive screening of variables like precipitant, buffer, pH, protein concentration, and temperature. For membrane proteins, specialized methods like lipidic cubic phase (LCP) crystallization are used [36] [37].
- Data Collection: A high-quality crystal is mounted and exposed to an intense, monochromatic X-ray beam, typically from a synchrotron radiation source. The crystal may be rotated to capture diffraction from all angles, and the resulting diffraction pattern is recorded on a detector [36] [37].
- Data Processing: The recorded diffraction spots are indexed, and their intensities are measured. This data is scaled and merged to produce a set of structure factors that describe the amplitude of each diffracted beam [36] [37].
- Phasing: A critical step where the phase information for each structure factor, which is not directly measurable, is estimated. Common methods include:
- Model Building and Refinement: An initial atomic model is built into the experimental electron density map. This model is iteratively refined by adjusting atomic positions to improve the fit to the data while satisfying chemical restraints [36] [40].
Key Research Reagents and Materials
Table 2: Essential Reagents for X-Ray Crystallography
| Reagent/Material | Function |
|---|---|
| Crystallization Screens | Pre-formulated solutions to screen conditions for crystal formation by varying precipitant, pH, and buffer. |
| Cryoprotectants | Chemicals (e.g., glycerol, ethylene glycol) to protect crystals from ice formation during flash-cooling in liquid N₂. |
| Heavy Atom Compounds | Atoms like selenium (in Se-Met labeled protein) or compounds for soaking to provide phasing information via anomalous scattering. |
| Synchrotron Beamtime | Access to high-intensity X-ray radiation sources for data collection on microcrystals or weakly diffracting samples. |
Nuclear Magnetic Resonance (NMR) Spectroscopy
Principles
Nuclear Magnetic Resonance (NMR) spectroscopy is a non-destructive technique that exploits the magnetic properties of atomic nuclei to determine the structure and dynamics of molecules in solution [37] [41]. When placed in a strong magnetic field, certain nuclei (such as ¹H, ¹⁵N, ¹³C) absorb and re-emit electromagnetic radiation at characteristic frequencies. These frequencies are exquisitely sensitive to the local chemical environment, providing information on inter-atomic distances, dihedral angles, and dynamics [42] [41]. Unlike crystallography, which provides a single static model, NMR can capture an ensemble of conformations and study dynamic processes and molecular interactions under physiological conditions [43] [37].
Experimental Workflow and Protocol
The standard workflow for protein structure determination by solution NMR is as follows [37]:
- Sample Preparation: The protein of interest must be produced recombinantly in a host (typically E. coli) grown in a medium enriched with stable isotopes (¹⁵N and/or ¹³C). This isotopic labeling is essential for resolving signals and assigning them to specific atoms in larger proteins. The protein must be soluble and stable at high concentrations (e.g., > 200 µM) for several days [37].
- Data Collection: A series of multi-dimensional NMR experiments are performed on a high-field spectrometer (typically ≥ 600 MHz). Key experiments include:
- HSQC: A 2D fingerprint spectrum that correlates ¹H and ¹⁵N atoms, used to assess protein folding and stability.
- NOESY: Critical for measuring through-space ¹H-¹H distances, which are the primary data for calculating the 3D structure.
- TOCSY and HCCH-TOCSY: Used to identify spin systems and through-bond correlations for sequence-specific backbone and sidechain assignments [37].
- Resonance Assignment: The signals in the NMR spectra are systematically assigned to specific atoms in the protein sequence. This is done by analyzing through-bond connectivity in triple-resonance experiments (e.g., HNCA, HNCOCA, CBCACONH) [37].
- Restraint Collection and Structure Calculation: Distance restraints are derived from NOESY spectra. Torsion angle restraints are obtained from chemical shift analysis (e.g., using TALOS). These experimental restraints are used in a computational process (such as simulated annealing) to calculate an ensemble of structures that satisfy all the restraints [43] [37].
- Validation: The final ensemble of structures is validated for its agreement with the experimental data and its stereochemical quality.
Key Research Reagents and Materials
Table 3: Essential Reagents for NMR Spectroscopy
| Reagent/Material | Function |
|---|---|
| Isotopically Labeled Media | ¹⁵N-ammonium chloride/ sulfate and ¹³C-glucose for incorporation of NMR-active nuclei into recombinant proteins. |
| NMR Tubes | High-quality, precision glass tubes designed for specific spectrometer field strengths to hold the sample. |
| Deuterated Solvents | Solvents (e.g., D₂O) used to prepare the sample to minimize the strong signal from solvent protons. |
| NMR Spectrometer | High-field instrument with cryoprobes for enhanced sensitivity, required for biomolecular NMR. |
Cryo-Electron Microscopy (Cryo-EM)
Principles
Cryo-electron microscopy (cryo-EM) has undergone a "resolution revolution," transforming it into a dominant technique for determining high-resolution structures of large and dynamic macromolecular complexes [42]. In single-particle cryo-EM, a purified protein solution is applied to a grid and rapidly vitrified in liquid ethane, embedding the particles in a thin layer of amorphous ice that preserves their native structure [41]. The grid is then imaged in a high-powered electron microscope under cryo-conditions. Thousands to millions of 2D particle images are collected, computationally sorted and aligned, and then used to reconstruct a 3D electrostatic potential map [44] [42]. A key advantage is that it does not require crystallization, making it ideal for membrane proteins, large complexes, and proteins with inherent flexibility [37] [41].
Experimental Workflow and Protocol
The standard workflow for single-particle cryo-EM structure determination is [44] [42]:
- Sample Preparation: The macromolecular complex must be purified to homogeneity and biochemically stable. For small proteins (< ~50 kDa), strategies like fusion to a scaffold protein (e.g., a coiled-coil module) or encapsulation in a DARPin cage may be necessary to increase the particle size and facilitate analysis [44].
- Grid Preparation and Vitrification: A small volume (~3 µL) of the sample is applied to an EM grid coated with a holey carbon film. The grid is blotted with filter paper to create a thin liquid film and is then plunged into a cryogen (liquid ethane) so rapidly that the water vitrifies instead of forming crystalline ice [42].
- Data Collection: The vitrified grid is loaded into a cryo-electron microscope, maintained at liquid nitrogen temperatures. Using a direct electron detector, thousands of micrograph movies are collected automatically. Each micrograph contains images of thousands of individual protein particles in random orientations [42].
- Image Processing and 3D Reconstruction: This computationally intensive step involves several sub-steps:
- Particle Picking: Automated software identifies and extracts the individual particle images from the micrographs.
- 2D Classification: Extracted particles are aligned and averaged into 2D class averages to remove non-particle images and sort particles by conformational or compositional heterogeneity.
- Initial Model Generation: An initial low-resolution 3D model is created ab initio or from a existing model.
- 3D Classification and Refinement: Particles are assigned to different 3D classes to isolate homogeneous subsets, which are then iteratively refined to produce a high-resolution 3D reconstruction (map) [44] [42].
- Atomic Model Building and Refinement: An atomic model is built into the cryo-EM map, either de novo or by fitting and refining a known homologous structure, using computational tools similar to those in crystallography [42].
Key Research Reagents and Materials
Table 4: Essential Reagents for Cryo-Electron Microscopy
| Reagent/Material | Function |
|---|---|
| Holey Carbon Grids | EM grids with a perforated carbon support film that allows the sample to span the holes for optimal imaging. |
| Vitrification System | Instrument (plunge freezer) for reproducible and rapid freezing of samples to form vitreous ice. |
| Direct Electron Detector | Advanced camera capable of counting individual electrons with high sensitivity, crucial for high-resolution reconstruction. |
| Scaffold Proteins | For small proteins: fusion partners (e.g., coiled-coil modules, DARPins) to increase effective particle size for analysis [44]. |
X-ray crystallography, NMR spectroscopy, and cryo-electron microscopy constitute the essential triad of experimental techniques for determining protein structures in the PDB. Each method offers a unique perspective: crystallography provides high-throughput atomic detail from crystals, NMR reveals dynamic behavior in solution, and cryo-EM visualizes large complexes in near-native states. The ongoing evolution of these technologies—such as serial crystallography at XFELs, ultra-fast spinning NMR probes [45], and enhanced cryo-EM detectors—continues to push the boundaries of what is possible. Furthermore, the integration of these experimental data with powerful AI-based structure prediction tools like AlphaFold is creating a new paradigm in structural biology [42]. For researchers in drug discovery and basic science, a deep understanding of these core techniques' principles, capabilities, and methodologies is fundamental to designing effective experiments and interpreting structural data within a broader biological context.
The field of structural biology has undergone a revolutionary transformation with the advent of computed structure models (CSMs), driven by advances in deep learning and artificial intelligence. This whitepaper provides an in-depth technical examination of machine learning methodologies, with a focused analysis on the AlphaFold system that has enabled the accurate, atomic-resolution prediction of protein structures from amino acid sequences. We explore the architectural innovations, performance benchmarks, and practical applications of these technologies, framing them within the broader context of protein data bank research and their implications for drug discovery and development. The integration of these CSMs with established experimental structural biology databases creates a powerful synergy, accelerating research across the life sciences.
Proteins are essential macromolecules that undertake vital activities in living organisms, including material transport, energy conversion, and catalytic reactions [46]. A protein's function is largely determined by its unique three-dimensional (3D) structure, which is encoded in its linear amino acid sequence [46]. The challenge of predicting a protein's 3D structure solely from its amino acid sequence—known as the protein folding problem—has been one of the most important open research problems in biochemistry for over 50 years [47].
The significance of this challenge is highlighted by the growing gap between known protein sequences and experimentally determined structures. As of 2022, the TrEMBL database contained over 200 million protein sequence entries, while the Protein Data Bank (PDB) contained only approximately 200,000 experimentally solved structures [46]. This massive disparity—less than 0.1% structural coverage—has created an urgent need for computational approaches to bridge the sequence-structure gap.
Table 1: The Sequence-Structure Gap in Protein Data (as of 2022)
| Data Type | Database | Number of Entries | Reference |
|---|---|---|---|
| Protein Sequences | TrEMBL | Over 200 million | [46] |
| Experimentally Determined Structures | Protein Data Bank (PDB) | ~200,000 | [46] |
| Computed Structure Models | AlphaFold Database | Over 200 million | [48] |
Historical Context and Methodological Evolution
Traditional Computational Approaches
Protein structure prediction approaches have historically been classified into three main categories:
Template-Based Modeling (TBM): Relies on identifying and using known protein structures as templates, typically through sequence or structural homology [46]. Key tools include MODELLER and SwissPDBViewer [46]. This approach requires at least 30% sequence identity between target and template sequences for reliable modeling.
Template-Free Modeling (TFM): Predicts structure directly from sequence without using global template information, utilizing only amino acid sequence information and without reference to any protein template [46]. Modern AI-based approaches like AlphaFold represent advanced forms of TFM.
Ab Initio Methods: Based purely on physicochemical principles and do not rely on existing structural information, attempting to predict structure through physical simulation of folding forces [46].
The Machine Learning Revolution
The past decade has witnessed a dramatic "neuralization" of structure prediction pipelines, whereby computations previously based on energy models and sampling procedures have been replaced by neural networks [49]. This transformation has resulted in algorithms that can now predict single protein domains with a median accuracy of 2.1 Å, setting the stage for a foundational reconfiguration of the role of biomolecular modeling within the life sciences [49].
AlphaFold: Architectural Innovations and Technical Methodology
AlphaFold is an AI system developed by Google DeepMind that predicts a protein's 3D structure from its amino acid sequence with accuracy competitive with experimental methods [48]. In the 14th Critical Assessment of protein Structure Prediction (CASP14), AlphaFold was the top-ranked method by a large margin, producing predictions with high accuracy [48] [47].
The system demonstrated remarkable performance metrics, achieving a median backbone accuracy of 0.96 Å RMSD₉₅ (Cα root-mean-square deviation at 95% residue coverage), compared to 2.8 Å for the next best performing method [47]. As a reference point for this accuracy, the width of a carbon atom is approximately 1.4 Å [47].
Table 2: AlphaFold Performance Metrics from CASP14 Assessment
| Accuracy Metric | AlphaFold Performance | Next Best Method | Improvement Factor |
|---|---|---|---|
| Backbone Accuracy (Median RMSD₉₅) | 0.96 Å | 2.8 Å | ~3x |
| All-Atom Accuracy (RMSD₉₅) | 1.5 Å | 3.5 Å | ~2.3x |
| Side-Chain Accuracy | Highly accurate when backbone is correct | Less accurate | Significant |
Neural Network Architecture and Training Methodology
The AlphaFold network employs a novel architecture that directly predicts the 3D coordinates of all heavy atoms for a given protein using the primary amino acid sequence and aligned sequences of homologues as inputs [47]. The system comprises two main stages:
Evoformer Processing: The trunk of the network processes inputs through repeated layers of a novel neural network block termed "Evoformer" to produce representations of the multiple sequence alignment (MSA) and residue pairs [47]. The Evoformer blocks incorporate innovative attention-based mechanisms to exchange information between the MSA and pair representations, enabling direct reasoning about spatial and evolutionary relationships.
Structure Module: This module introduces an explicit 3D structure in the form of a rotation and translation for each residue of the protein [47]. Key innovations include breaking the chain structure to allow simultaneous local refinement of all parts of the structure, a novel equivariant transformer, and a loss term that places substantial weight on the orientational correctness of residues.
Diagram Title: AlphaFold Architecture and Information Flow
Key Algorithmic Innovations
AlphaFold incorporates several groundbreaking technical approaches:
Iterative Refinement (Recycling): The network repeatedly applies the final loss to outputs and feeds them recursively into the same modules, significantly enhancing accuracy [47].
Triangle Multiplicative Updates: These operations enforce geometric constraints within the pair representation by reasoning about triangles of edges involving three different nodes, ensuring physical plausibility of the predicted structures [47].
Confidence Estimation: The model provides precise, per-residue estimates of reliability (pLDDT - predicted Local Distance Difference Test) that enable researchers to confidently use these predictions in downstream applications [47].
The AlphaFold Ecosystem and Database Infrastructure
AlphaFold Protein Structure Database
Google DeepMind and EMBL's European Bioinformatics Institute (EMBL-EBI) have partnered to create the AlphaFold Database (AFDB) to make protein structure predictions freely available to the scientific community [48]. The latest database release contains over 200 million entries, providing broad coverage of UniProt, the standard repository of protein sequences and annotations [48].
The database provides individual downloads for the human proteome and for the proteomes of 47 other key organisms important in research and global health, plus a download for the manually curated subset of UniProt (Swiss-Prot) [48]. All data is available for both academic and commercial use under a CC-BY-4.0 license [48].
Recent Enhancements and Capabilities
The 2025 update to the AlphaFold Database introduced a redesigned interface and updated structural coverage aligned with the UniProt 2025_03 release [50]. Key enhancements include:
Custom Annotations: New functionality enables users to integrate and visualize custom sequence annotations through a protein feature web visualization component [48].
Enhanced Visualization: Annotations are visible on both 2D and 3D tracks, alongside the predicted Local Distance Difference Test (pLDDT) score track [48].
Structural Coverage Expansion: The update includes isoforms plus underlying multiple sequence alignments, broadening the database's research utility [50].
Extending to Protein Complexes and Challenges in Multimer Prediction
The Protein Complex Challenge
While AlphaFold2 made revolutionary breakthroughs in predicting protein monomeric structures, accurately capturing inter-chain interaction signals and modeling the structures of protein complexes remains a formidable challenge [51]. Determining protein complex structures is crucial for understanding cellular processes, as proteins perform key functions by interacting to form complexes [51].
Advanced Approaches for Complex Prediction
Recent methodologies have extended deep learning approaches to protein complexes:
DeepSCFold Pipeline: This approach uses sequence-based deep learning models to predict protein-protein structural similarity and interaction probability, providing a foundation for identifying interaction partners and constructing deep paired multiple-sequence alignments (MSAs) for protein complex structure prediction [51].
Key Innovations in DeepSCFold:
- Predicts structural similarity (pSS-score) purely from sequence information
- Estimates interaction probability (pIA-score) based solely on sequence-level features
- Leverages structural complementarity between protein chains rather than relying solely on co-evolutionary signals
Table 3: Performance Comparison of Protein Complex Prediction Methods
| Method | TM-score Improvement on CASP15 | Antibody-Antigen Interface Success Rate | Key Innovation |
|---|---|---|---|
| DeepSCFold | 11.6% over AlphaFold-Multimer10.3% over AlphaFold3 | 24.7% over AlphaFold-Multimer12.4% over AlphaFold3 | Sequence-derived structure complementarity |
| AlphaFold-Multimer | Baseline | Baseline | Extension of AlphaFold2 for multimers |
| AlphaFold3 | Not specified | Not specified | Integrated complex prediction |
Paired Multiple Sequence Alignment Construction
A critical innovation in protein complex prediction is the development of methods to construct paired MSAs, which enable the identification of inter-chain co-evolutionary signals between interacting partners [51]. These approaches include:
DeepMSA2: Performs iterative alignment searches across genomic and metagenomic sequence databases, followed by filtering using AlphaFold2/AlphaFold-Multimer [51].
ESMPair: Ranks monomeric MSAs using ESM-MSA-1b and integrates species information to construct paired MSAs [51].
DiffPALM: Employs an MSA transformer to estimate amino acid probabilities, creating a permutation matrix to pair protein sequences [51].
Experimental Protocols and Validation Methodologies
Validation Metrics and Benchmarks
The accuracy of computed structure models is rigorously assessed using multiple complementary metrics:
Global Distance Test (GDT): A measure of the percentage of residues that can be superimposed under a given distance cutoff.
Template Modeling Score (TM-score): A metric for measuring the similarity of protein structures that is more sensitive to global fold similarity than local deviations.
Root-Mean-Square Deviation (RMSD): Measures the average distance between atoms of superimposed proteins.
Local Distance Difference Test (lDDT): A local quality estimation method that does not require superposition, evaluating the reliability of individual residues.
Cross-Validation Framework
The performance of computational methods is typically evaluated using carefully designed cross-validation protocols:
Temporal Split Validation: Models are trained on data available before a specific cutoff date and tested on structures solved after that date, ensuring no data leakage [51].
CASP Assessment: The Critical Assessment of protein Structure Prediction is a biennial blind trial that serves as the gold-standard evaluation for structure prediction methods [47].
K-Fold Cross-Validation: Datasets are partitioned into k subsets, with each subset serving as a test set while the remaining k-1 subsets form the training data [52].
Table 4: Key Research Reagents and Computational Resources for CSM Research
| Resource Name | Type | Primary Function | Access Method |
|---|---|---|---|
| AlphaFold Database | Database | Open access to over 200M protein structure predictions | Web interface, FTP, API [48] |
| AlphaFold Codebase | Software | Generate custom structure predictions from sequences | Open source download [48] |
| DeepSCFold | Software Pipeline | Protein complex structure modeling | Research implementation [51] |
| Evoformer | Algorithm | Neural network block for MSA and pair representation processing | Part of AlphaFold codebase [47] |
| pLDDT | Metric | Per-residue confidence estimate for predicted structures | Integrated in AlphaFold output [47] |
| Cutoff Scanning Matrix (CSM) | Method | Structural classification and function prediction | Custom implementation [52] |
| Paired MSA Constructors | Tools | Build multiple sequence alignments for protein complexes | Various implementations (DeepMSA2, ESMPair) [51] |
Diagram Title: Protein Structure Research Workflow
Future Directions and Research Challenges
Despite the remarkable progress in computed structure models, several important challenges remain:
Dynamic and Multi-State Structures: Current CSMs primarily predict static structures, while proteins often exist in multiple conformational states that are critical for their function.
Ligand and Small Molecule Interactions: Accurately predicting how proteins interact with small molecules, drugs, and other ligands remains an active area of development.
Condition-Specific Structures: Protein structures can be influenced by environmental conditions, post-translational modifications, and cellular context—factors not currently captured in standard predictions.
Very Large Complexes and Assemblies: Scaling these methods to accurately model massive cellular machinery, such as ribosomes and nuclear pores, presents computational and methodological challenges.
The field continues to evolve rapidly, with ongoing research focused on integrating physical constraints more explicitly, incorporating time-resolved dynamics, and expanding beyond natural amino acid sequences to engineered proteins and designed biomolecules.
The rise of computed structure models, particularly through deep learning systems like AlphaFold, represents a paradigm shift in structural biology and bioinformatics. By providing accurate, accessible protein structure predictions at an unprecedented scale, these tools have democratized structural insights and accelerated research across diverse fields from basic molecular biology to drug discovery. The integration of these computational approaches with traditional experimental methods creates a powerful synergistic relationship, each informing and validating the other. As the field continues to advance, we anticipate further innovations that will expand the scope, accuracy, and biological relevance of computed structure models, solidifying their role as foundational tools in life sciences research.
The Protein Data Bank (PDB) represents an indispensable, open-access digital resource for modern, structure-guided drug discovery. Established in 1971 as the first open-access digital-data resource in the biological sciences, the PDB archive now holds over 155,000 atomic-level three-dimensional structures of biomolecules determined experimentally using macromolecular X-ray crystallography (MX), nuclear magnetic resonance (NMR) spectroscopy, and electron microscopy (3DEM) [53]. The impact of this archive on medicine is profound; analyses reveal that publicly available PDB data facilitated the discovery of approximately 90% of the 210 new drugs approved by the US Food and Drug Administration (FDA) between 2010 and 2016 [53]. Structure-guided drug discovery is a well-established tool that leverages these 3D structural studies to optimize small-molecule ligand affinity and selectivity for target proteins, as exemplified by drugs like vemurafenib for metastatic melanoma [53]. This guide provides a comprehensive technical framework for exploiting PDB entries to analyze drug targets and their complexes with therapeutic molecules, framing this process within the broader context of foundational PDB research.
Foundational Concepts: Navigating PDB Entries
The Architecture of a PDB Entry
A PDB entry is an experimentally determined macromolecular structure that provides a 3D atomic coordinate model of a biological sample. The contents can be broadly categorized into polymers (biological macromolecules like proteins, DNA, and RNA) and non-polymers (including bound small molecules, ligands, ions, and water) [54]. To navigate an entry effectively, one must understand the critical distinction between an entity and an instance. An entity is a distinct chemical component, such as a protein with a unique sequence or a small molecule with a specific chemical structure. An instance is a distinct copy of that entity found within the structure. For example, a homodimeric protein structure comprises one protein entity but two chain instances (e.g., chains A and B) [54].
Key Identifiers and Their Meanings
Identifiers (IDs) are used at all levels of the structural hierarchy to uniquely locate and specify atoms, residues, molecules, and entire entries [11]. The most common identifiers are summarized in the table below.
Table 1: Key Identifiers in a PDB Entry
| Identifier Level | Identifier Name | Format & Example | Purpose |
|---|---|---|---|
| Entry | PDB ID | 4-character alphanumeric (e.g., 4MBS) [11] [54] |
Uniquely identifies the entire structure. |
| Entity | Entity ID | Number specific to the entry (e.g., 1) [11] |
Identifies a unique chemical component (e.g., a specific protein sequence). |
| Instance | Chain ID (for polymers) | 1- or 2-character alphanumeric (e.g., A, BC) [11] |
Identifies a specific copy of a polymer entity in the structure. |
| Instance | Residue Number & Author Chain ID (for ligands) | Number and chain ID (e.g., A101) [11] |
Locates a specific small molecule/ligand instance. |
| Chemical Component | Chemical ID (CCD ID) | 3-character code (e.g., ATP) [11] |
Standardized name for a residue or small molecule from the PDB's Chemical Component Dictionary. |
It is crucial to note that two chain ID systems may be present: one assigned by the PDB (label_asym_id) and another provided by the depositing author (auth_asym_id). These usually match but can differ, which is important when referencing specific residues from a publication [11]. Similarly, residue numbering can follow a PDB-assigned sequential scheme (label_seq_id) or an author-defined scheme (auth_seq_id) that may match the numbering in related database entries (e.g., UniProt) [11].
Accessing Drug and Drug Target Information in the PDB
The PDB provides specialized tools to access structures relevant to drug discovery directly. The RCSB PDB website features dedicated tables accessible from the Tools menu that are updated weekly with drug and drug target information mapped from the DrugBank database [55].
- Drugs Bound to Primary Targets Table: This resource lists drugs that are co-crystallized with their primary target(s) or a homolog of their primary target. It is the primary resource for finding structures where the drug molecule is physically bound to its protein target [55].
- Primary Drug Targets Table: This table lists primary drug targets found in the PDB, regardless of whether the drug molecule itself is present in the entry. This includes, for example, apo forms of the target (without any bound ligand) or the target bound to different ligands, enabling studies on conformational changes and binding site plasticity [55].
These tables can be searched by generic or brand drug name and then filtered and sorted to find relevant structures. Complementary resources, such as the DrugPort database maintained by the EBI, also provide analyses of structural information in the PDB relating to drugs and their protein targets, offering additional search options like sequence-based queries [56].
A Methodological Framework for Analysis
For any PDB entry, the Structure Summary page on the RCSB PDB website serves as the central hub for information and further analysis [27]. Key sections for drug discovery include:
- Snapshot of the Structure: This section provides a visual overview of the biological assembly. It allows users to interactively visualize the structure in Mol*, zoom in on specific ligands ("Ligand Interaction"), and view predicted membrane locations for membrane proteins [27].
- Header: This section contains the PDB ID, structure title, source organism, and critical quality assessments. The wwPDB Validation slider provides a percentile-based assessment of the overall structure quality. For structures with small-molecule ligands, the Ligand Structure Quality Assessment slider indicates the goodness of fit of the ligand to the experimental data [27].
- Sequence Tab: Provides the sequences of all polymers and shows how they map to external sequence databases like UniProt, which is essential for understanding residue numbering and the functional context of the protein [27].
- Ligands Tab: Details all small molecules in the structure, including their chemical identities, interactions, and structural fit to the experimental data [27].
- Experiment Tab: Offers detailed information on the experimental method (MX, NMR, 3DEM), sample preparation, data collection, and refinement procedures. Understanding these details is vital for critically assessing the reliability and limitations of the structural model [57].
- 3D View Tab: The gateway to the interactive Mol* visualizer, where in-depth structural analysis is performed [27].
Experimental Protocols for Structural Analysis
The core methodology for analyzing a drug-target complex involves a multi-step process of data retrieval, visualization, and interaction analysis, underpinned by an understanding of the experimental data.
Table 2: Essential Research Reagents and Resources
| Resource Category | Specific Tool / Resource | Function in Analysis |
|---|---|---|
| Primary Data Archive | RCSB PDB (rcsb.org) [53] | Central repository for retrieving PDB entries, validation reports, and integrated external annotations. |
| Specialized Drug Search | Drugs Bound to Primary Targets Table [55] | Locates co-crystal structures of drugs with their targets. |
| 3D Visualization Software | Mol* (via RCSB) [27], PyMOL, Chimera [58] | Interactive visualization of structures, measurement of distances/angles, and creation of publication-quality images. |
| 2D Interaction Diagram | PoseView [59], LigPlot+ [58] | Automatically generates schematic 2D diagrams of protein-ligand interactions from 3D coordinates. |
| Surface Generation | EDTSurf Algorithm [58] | Computes molecular surfaces (e.g., solvent-accessible surface) to visualize ligand binding pockets and molecular recognition. |
| Structure Validation | wwPDB Validation Report [27] | Assesses the quality and reliability of an experimental structural model. |
Workflow for Analyzing a Drug-Target Complex:
- Data Retrieval and Validation: Begin by retrieving your target PDB entry (e.g.,
4MBS, a CCR5 chemokine receptor complexed with the drug maraviroc). Immediately consult the wwPDB Validation Report and the Ligand Quality Slider in the Header to assess global structure and ligand fit quality [27]. - Visualization of the Biological Assembly: In the 3D View tab, load the biological assembly, not just the asymmetric unit, to understand the physiologically relevant quaternary structure [27].
- Focus on the Binding Site: Use the "Ligand Interaction" view or manually select the drug molecule to center and zoom in on the binding pocket.
- Analysis of Non-covalent Interactions: Within the 3D visualizer (e.g., Mol*), configure the display to reveal key interactions. This typically involves:
- Representing the protein binding pocket as a cartoon or ribbon to show secondary structure.
- Showing amino acid side chains within ~5Å of the ligand as sticks.
- Displaying the drug ligand as sticks or ball-and-stick.
- Enabling the calculation and display of hydrogen bonds and hydrophobic contacts.
- (Optional) Generating a molecular surface (e.g., solvent-accessible surface) around the binding pocket to visualize its topology and properties [58].
- Generation of a 2D Interaction Diagram: To create a publication-ready schematic, use a tool like PoseView, which automatically generates a 2D diagram from the 3D coordinates, depicting hydrogen bonds, ionic interactions, and hydrophobic contacts [59].
- Comparative Analysis: If analyzing a series of complexes (e.g., the same target with different inhibitors), employ methods that generate a consistent 2D layout for all residues across the complex series. This preserves the spatial arrangement of residues across diagrams, dramatically simplifying the visual comparison of binding modes between different ligands [59].
The following diagram illustrates the logical workflow for this analytical process:
Visualization Techniques and Tools
Effective visualization is a cornerstone of structure-based analysis. Tools range from interactive 3D visualizers to automated 2D diagram generators.
- Interactive 3D Visualization with Mol*: Integrated into the RCSB PDB site, Mol* allows for real-time, hardware-accelerated exploration of structures. Users can create multiple representations (cartoon, stick, surface) simultaneously, measure distances and angles, and map properties like model confidence onto the structure [27].
- Advanced 3D Features with iview: The
iviewWebGL visualizer exemplifies advanced features, including support for virtual reality settings (anaglyph, parallax barrier) and the real-time calculation of four types of macromolecular surfaces: Van der Waals surface, solvent-excluded surface, solvent-accessible surface, and molecular surface [58]. Surface representation is vital for understanding the shape and properties of binding pockets. - Consistent 2D Visualization for Complex Series: As described by, comparing a series of ligands bound to the same protein active site is greatly facilitated by a consistent 2D layout of the interacting protein residues [59]. The algorithm behind tools like PoseView achieves this by computing a global two-dimensional layout for all residues of the complex ensemble based on their 3D adjacencies, resulting in a circular arrangement of residues around the ligand that is uniform across all diagrams in the series [59]. This eliminates the layout heterogeneity that arises from independent diagram generation and substantially simplifies the visual analysis of large compound series.
The technical process for creating these consistent visualizations can be summarized as follows:
The Protein Data Bank provides the foundational structural data that powers contemporary, structure-guided drug discovery. By understanding the architecture of a PDB entry, leveraging specialized drug-target search tools, systematically applying rigorous analytical methodologies, and utilizing advanced visualization techniques, researchers can deeply interrogate the molecular interactions between drugs and their targets. This structured approach, firmly rooted in the public domain data of the PDB, continues to accelerate the rational design of new and more effective therapeutic agents, underscoring the critical role of open-access structural biology in advancing biomedical research and human health.
The Protein Data Bank (PDB) archive serves as the foundational repository for experimentally-determined three-dimensional structures of proteins, nucleic acids, and complex molecular assemblies, enabling breakthroughs in structural biology, drug discovery, and biomedical research. Established in 1971, the PDB has experienced exponential growth, with over 214,000 experimentally-determined structures available as of 2023, and an annual growth rate exceeding 14,000 new structures [7]. This wealth of structural data provides critical insights into the relationship between molecular form and biological function. The RCSB PDB portal (RCSB.org) operates as a key member of the Worldwide PDB, providing open access to these structural data alongside integrated computational tools for visualization, analysis, and exploration. The visualization tools available through RCSB.org have evolved significantly, transitioning from physical models to sophisticated web-based applications that leverage GPU-accelerated rendering and interactive 3D visualization [60]. This technical guide examines the core visualization technologies—RCSB.org's integrated platform, the deprecated NGL Viewer, and external resources—within the context of foundational protein data bank research, providing researchers and drug development professionals with methodologies for effective structural analysis.
The RCSB.org Structural Biology Platform
Platform Architecture and Core Capabilities
The RCSB.org portal provides a comprehensive ecosystem for structural biology research, integrating data from multiple sources including the core PDB archive of experimentally-determined structures, integrative/hybrid structures, and computed structure models (CSMs) from AlphaFold DB and ModelArchive [35]. The platform's architecture enables simultaneous access to structural data and analytical tools, creating a unified research environment. A key feature is the Mol* viewer, which has superseded the NGL Viewer as the primary visualization tool on RCSB.org as of June 2024 [61]. This transition reflects the ongoing evolution of visualization technologies to handle increasingly complex structural data and multi-scale representations.
The platform supports the entire research workflow from structure discovery to analysis. Researchers can initiate investigations using various entry points: PDB ID codes for known structures, sequence similarity searches for homologous structures, or keyword searches for functional annotations. The system provides seamless access to complementary data types including sequence information, functional annotations, biological assemblies, and literature references. This integrated approach eliminates the need for researchers to navigate between disparate resources, significantly accelerating the pace of structural analysis.
Mol* Viewer: Next-Generation Visualization
Mol* has been architected to address the limitations of previous visualization tools, offering enhanced performance, improved rendering capabilities, and specialized features for analyzing complex structural data. The viewer implements multiple rendering algorithms optimized for different representation models including skeletal models (line, stick, ball-and-stick), cartoon models (ribbons, tubes), and surface models (van der Waals, solvent-accessible, Gaussian surfaces) [60]. This multi-representation approach enables researchers to visualize structural features at different scales—from atomic-level interactions to domain organization and molecular surfaces.
The component-based logic in Mol* provides sophisticated selection and grouping capabilities essential for complex analyses. Researchers can create persistent selections of specific structural elements (residues, chains, ligands) as named components, which can be independently manipulated—shown/hidden, recolored, or represented differently [62]. This functionality is particularly valuable for studying ligand-binding sites, mutation sites, or specific domains within large macromolecular complexes. The selection system supports multiple picking levels (atom, residue, chain) and set operations (union, intersection, difference), enabling precise selection of structural elements based on spatial relationships or chemical properties.
Table 1: Core Visualization Capabilities of Mol*
| Feature Category | Specific Capabilities | Research Applications |
|---|---|---|
| Structure Representation | Cartoon, ball-and-stick, surface, molecular density | Domain organization, ligand interaction analysis, surface property mapping |
| Selection System | Multi-level picking (atom/residue/chain), set operations, component creation | Binding site analysis, mutation studies, comparative anatomy of structures |
| Color Encoding | Sequential, diverging, qualitative palettes; pLDDT coloring for CSMs | Conservation analysis, uncertainty visualization, functional annotation |
| Movement & Focus | Rotation, translation, zoom, focus on selection, animation modes | Spatial relationship analysis, publication-quality image creation, educational materials |
| Measurement Tools | Distances, angles, intermolecular contacts | Interaction quantification, mutagenesis planning, drug design |
Biological Assemblies and Experimental Data Integration
A critical functionality of the RCSB.org platform is the accurate representation of biological assemblies—the functional quaternary structures that exist in biological contexts. Many protein structures are determined in non-physiological states (crystal asymmetry units), and the platform applies symmetry operations to reconstruct the biologically relevant oligomers [62]. This capability is essential for understanding molecular mechanisms, as the functional form often requires specific quaternary interactions. The platform provides explicit controls for switching between asymmetric units and biological assemblies, with visual examples demonstrating different assembly states of insulin, proinsulin NMR ensembles, and viral capsids [62].
The platform also integrates experimental metadata and validation reports, enabling researchers to assess structure quality and experimental parameters. This includes resolution statistics for crystallographic structures, map validation metrics for cryo-EM structures, and restraint analyses for NMR structures. The introduction of the 3DEM Model-Map percentile slider based on Q-score validation metrics enhances the evaluation of cryo-EM structures [35]. These integrated validation tools help researchers identify potential limitations in structural models and make informed decisions about their suitability for specific research applications.
Computed Structure Models: Expanding the Structural Universe
Methodological Foundations of CSMs
Computed Structure Models represent a paradigm shift in structural biology, complementing experimental methods with computationally-predicted structures. CSMs are generated through two primary methodological approaches: template-based modeling and template-free modeling. Template-based modeling leverages the evolutionary principle that proteins with similar sequences fold into similar structures, using experimentally-determined structures of homologous proteins as templates [63]. This approach is effective when template structures with >30% sequence identity are available, with homology modeling typically successful above 40% sequence identity. Template-free modeling, in contrast, uses co-evolutionary analysis from multiple sequence alignments to identify correlated mutations that indicate spatial proximity in the folded structure [63].
The revolutionary advances in CSM accuracy stem from artificial intelligence and machine learning approaches, particularly AlphaFold2 and RoseTTAFold. These systems employ iterative processes that analyze multiple sequence alignments, refine predicted 3D contacts, and computationally reassemble protein structures in ways consistent with evolutionary and physical constraints [63]. The AlphaFold2 algorithm specifically breaks initial protein models into individual amino acids and computationally recombines them according to predicted contacts, resulting in models with accuracy comparable to low-resolution experimental structures for compact globular domains.
Validation and Interpretation of CSMs
The interpretation of Computed Structure Models requires careful assessment of confidence metrics, particularly the predicted Local Distance Difference Test (pLDDT) score, which ranges from 0-100 and estimates per-residue prediction reliability [63]. Regions with pLDDT > 70 are generally considered confident predictions, while lower scores indicate either intrinsically disordered regions, regions requiring binding partners for folding, or prediction uncertainties. This confidence metric is visually encoded in Mol* through color schemes, enabling immediate assessment of model quality.
CSMs exhibit particular strengths and limitations that researchers must consider. They perform exceptionally well for well-folded, single-domain proteins without large conformational changes. However, they struggle with multi-domain proteins with flexible linkers, membrane proteins, and structures requiring ligand-induced folding [63]. The case study of the Src oncoprotein exemplifies these limitations—while individual SH2, SH3, and kinase domains are well-predicted, the flexible linkers between domains and regulation by phosphorylation are not accurately captured [63]. Researchers should prioritize experimental structures when available, as approximately 95% of crystallographic structures in the PDB exceed the accuracy of current CSMs [63].
Table 2: Comparison of Experimental Structure Determination vs. Computed Structure Models
| Parameter | Experimental Structures | Computed Structure Models |
|---|---|---|
| Data Source | X-ray crystallography, NMR, cryo-EM | AI/ML prediction from sequence & evolutionary data |
| Coverage | ~200,000 protein structures | Millions of models available |
| Accuracy | Atomic resolution to lower resolution | Near-experimental for well-folded domains |
| Confidence Metrics | Resolution, R-factors, validation scores | pLDDT scores (0-100 per residue) |
| Limitations | Technical challenges, crystallization requirements | Poor performance on flexible regions, multi-domain proteins |
| Typical Applications | Detailed mechanism studies, drug design | Hypothesis generation, template for experimental design |
Research Applications of CSMs
Computed Structure Models serve three primary research applications within structural biology and drug discovery. First, they enable hypothesis generation for molecular and cellular biologists studying proteins without experimental structures, allowing identification of potential functional domains and key amino acids [63]. Second, they support structure-based drug discovery through identification of conserved binding pockets and active sites, even when experimental structures are unavailable. Third, they accelerate integrative structural biology by providing models that can be fit into experimental maps of larger complexes [63].
The integration of CSMs with experimental data represents a powerful approach for studying complex biological systems. Researchers can use CSMs of individual proteins or domains as building blocks for modeling larger assemblies, fitting them into cryo-EM density maps or SAXS envelopes. This hybrid approach leverages the strengths of both computational and experimental methods, enabling structural characterization of complexes that resist direct experimental determination.
Visualization Methodologies and Technical Implementation
Representation Models for Biomolecular Visualization
Molecular visualization employs multiple representation models, each optimized for highlighting specific structural features. These representations can be categorized into three primary classes: skeletal models, cartoon models, and surface models [60]. Skeletal models include wireframe, stick, and ball-and-stick representations, which depict atomic connectivity and are ideal for analyzing ligand binding, catalytic sites, and chemical interactions. Cartoon models abstract secondary structure elements into ribbons, tubes, and arrows, providing intuitive visualization of protein folding, domain architecture, and topological relationships. Surface models compute the outer boundaries of molecules, revealing shape complementarity, electrostatic potentials, and interaction interfaces.
The technical implementation of these representations has evolved significantly, with modern viewers employing GPU-accelerated algorithms for real-time rendering of complex molecular scenes. Recent advances include HyperBall representations using hyperboloids to connect atoms [60], signed distance fields with sphere tracing for cartoon representations [60], and dynamic visibility-driven surface visualization [60]. These technical innovations enable interactive visualization of massive structural datasets while maintaining visual quality and performance.
Color Theory and Molecular Representation
Color serves as a critical semantic tool in molecular visualization, conveying structural, functional, and quantitative information. Effective color palettes follow established color harmony rules: monochromatic (single hue with varying saturation/lightness), analogous (adjacent hues on color wheel), and complementary (opposite hues) schemes [64]. Mol* implements these principles through its standardized color palettes, which include sequential scales for ordered continuous data (e.g., occupancy, B-factors), diverging scales for data with critical midpoints, and qualitative scales for categorical data (e.g., chain differentiation) [62].
The psychological and cultural associations of color must be considered when creating visualizations for specific audiences. Western cultures often associate red with danger or activation and blue with calmness, though these associations vary across cultures [64]. For standardized communication, certain color conventions have emerged—CPK coloring for atoms (oxygen red, nitrogen blue, carbon gray), red blood cells as red, and immune cells in cool colors [64]. Researchers should employ high luminance colors for focus molecules and desaturated colors for context, establishing clear visual hierarchy in complex scenes.
Experimental Protocols for Structural Analysis
Protocol 1: Ligand-Binding Site Analysis
Structure Preparation: Access the target structure via RCSB.org using its PDB ID. Select the biological assembly for physiological relevance. Focus visualization on the ligand of interest by clicking on it in the 3D canvas or sequence panel.
Binding Site Characterization: Create a component for residues within 5Å of the ligand using the selection tools. Represent these residues in ball-and-stick format to visualize atomic interactions. Represent the ligand in space-filling mode to assess steric constraints.
Interaction Analysis: Identify hydrogen bonds, hydrophobic interactions, and salt bridges through visual inspection and measurement tools. Use the distance measurement tool to quantify specific atomic distances. Employ complementary coloring for the ligand and contrasting colors for different interaction types.
Conservation Mapping: Color binding site residues by evolutionary conservation using available conservation scores. Correlate conserved residues with key interactions to identify functionally critical regions.
Protocol 2: Computed Structure Model Validation
Model Sourcing: Retrieve CSMs from AlphaFold DB or ModelArchive through RCSB.org. Download the corresponding PDB file and associated metadata, including pLDDT confidence scores.
Confidence Assessment: Visualize the model in Mol* with coloring by pLDDT score. Identify low-confidence regions (pLDDT < 70) that may represent disordered regions or prediction inaccuracies.
Experimental Comparison: When available, compare CSMs with experimental structures of homologs. Use the pairwise alignment tool to assess structural similarity in well-folded domains.
Functional Annotation: Integrate CSM analysis with functional data from sequence annotations and literature. Correlate high-confidence predicted structures with known functional domains and motifs.
Essential Research Reagents and Computational Tools
Table 3: Research Reagent Solutions for Structural Biology Studies
| Reagent/Tool Category | Specific Examples | Function and Application |
|---|---|---|
| Experimental Structure Determination | X-ray crystallography, NMR spectroscopy, Cryo-EM | Generate experimental 3D structures at atomic or near-atomic resolution |
| Structure Prediction Services | AlphaFold DB, RoseTTAFold, SWISS-MODEL | Provide computed structure models for proteins without experimental structures |
| Visualization Software | Mol*, PyMOL, ChimeraX | Render 3D molecular structures with multiple representation options |
| Validation Metrics | pLDDT scores, Q-scores, Ramachandran plots | Assess quality and reliability of structural models |
| Specialized Databases | PDB archive, AlphaFold DB, ModelArchive | Repository for structural models with associated metadata |
| Sequence Analysis Tools | BLAST, Clustal Omega, HMMER | Identify homologous sequences and generate alignments for evolutionary analysis |
The ecosystem for protein structure visualization and analysis has matured into an integrated framework combining experimental data, computational predictions, and advanced visualization tools. The transition from NGL to Mol* on RCSB.org reflects the ongoing evolution toward more powerful, accessible, and specialized tools for structural biology research. The emergence of Computed Structure Models as complementary resources to experimental structures has dramatically expanded the structural universe available for research and drug discovery. Effective utilization of these resources requires understanding their methodological foundations, appropriate application contexts, and interpretation guidelines. As structural biology continues to advance toward multi-scale modeling and time-resolved dynamics, the visualization tools will increasingly focus on representing structural flexibility, uncertainty, and complexity. The integration of these tools into research workflows empowers scientists to translate structural information into biological insights and therapeutic advances, fulfilling the promise of structural biology to illuminate the molecular mechanisms of life and disease.
Integrative/Hybrid Methods for Studying Large Complexes and Dynamics
Integrative/Hybrid Methods (IHM) represent a paradigm shift in structural biology, enabling the determination of macromolecular complex architectures that are intractable to any single experimental approach. By combining spatial restraints from diverse biochemical, biophysical, and computational techniques, IHM provides a powerful framework for modeling large, flexible, and dynamic biological assemblies. This technical guide examines the core principles, methodological workflows, and data standards underpinning integrative structural biology, with specific emphasis on applications for studying complex dynamics within the context of Protein Data Bank (PDB) entries research. The synthesis of complementary data sources allows researchers to construct multi-scale and multi-state models that capture biological heterogeneity and functional mechanisms, significantly expanding the structural coverage of the interactome for therapeutic discovery.
Integrative/Hybrid Methods (IHM) refer to computational modeling approaches that determine macromolecular structures by combining multiple sources of experimental information and theoretical principles. These methods have become essential for characterizing complex biological assemblies that are too large, flexible, or heterogeneous for traditional structure determination techniques like X-ray crystallography, NMR spectroscopy, or cryo-electron microscopy (EM) alone [65]. The fundamental premise of integrative structural biology is that no single experimental method may provide sufficient information at the desired resolution, but the combination of complementary datasets can yield reliable structural models when integrated with computational modeling.
The biological significance of IHM stems from its ability to elucidate structures of essential macromolecular machines that govern cellular function, including nuclear pore complexes, chromatin remodelers, viral capsids, and large protein-RNA complexes [65]. These assemblies often exhibit inherent dynamics, existing in multiple conformational states that are crucial for their biological activity. By leveraging partial and lower-resolution datasets from multiple sources, IHM broadens the range of macromolecular systems that can be structurally characterized, thereby filling critical gaps in our structural understanding of cellular processes.
Within the PDB ecosystem, integrative structures follow community-developed data standards based on the IHMCIF dictionary, a modular extension of the PDBx/mmCIF dictionary used for archiving atomic structures [65]. This standardized framework promotes reproducibility and aligns with FAIR (Findable, Accessible, Interoperable, Reusable) data principles, which are crucial for modern collaborative bioscience. The wwPDB accepts integrative structures of biological macromolecules that are at least partly based on experimental data via the PDB-IHM system, with mandatory deposition of spatial restraints, modeling protocols, and relevant metadata [66].
Key Concepts and Methodological Framework
Core Principles of Integrative Modeling
Integrative modeling operates on several fundamental principles that distinguish it from single-method approaches. The process typically involves: (1) gathering diverse experimental and computational data that provide spatial information about the system; (2) converting these data into spatial restraints; (3) generating structural models that satisfy these restraints; and (4) analyzing and validating the resulting models to assess their uncertainty and accuracy [65]. The spatial restraints can include distance restraints (e.g., from crosslinking mass spectrometry), shape information (e.g., from small-angle X-ray scattering), density maps (e.g., from electron microscopy), and proximity data (e.g., from FRET spectroscopy).
A critical advantage of integrative approaches is their ability to handle multi-scale representations, where different components of a complex are represented at different resolutions appropriate to the available data [65]. For instance, a well-structured domain might be represented at atomic resolution, while a flexible region might be modeled as coarse-grained beads. Similarly, IHM supports multi-state models that capture structural heterogeneity, representing a system as an ensemble of structures that collectively satisfy the experimental restraints [65]. This is particularly valuable for describing dynamic processes such as conformational changes, binding events, and enzymatic reactions.
Experimental Techniques in IHM
Integrative modeling incorporates data from a wide spectrum of experimental methods, each providing unique and complementary information about the system under study. The PDB accepts structures that incorporate data from traditional structure determination methods alongside other biophysical and proteomics approaches [66] [65].
Table: Experimental Techniques Used in Integrative/Hybrid Methods
| Technique | Type of Information | Spatial Resolution | Key Applications |
|---|---|---|---|
| Crosslinking-MS | Distance restraints between residues | Low (~Ångström) | Proximity mapping, subunit interaction interfaces |
| Small Angle Scattering (SAS) | Overall shape and dimensions | Low (~10 Å) | Complex shape, oligomeric state |
| FRET Spectroscopy | Inter-probe distances | Medium (10-100 Å) | Conformational changes, dynamics |
| Cryo-EM | 3D density maps | Medium to High (3-10 Å) | Complex architecture, subunit arrangement |
| NMR Spectroscopy | Distance restraints, chemical shifts | High (Atomic) | Local structure, dynamics |
| HDX-MS | Solvent accessibility, dynamics | Low (Residue-level) | Flexible regions, binding interfaces |
| AFM | Topographical imaging | Low (Nanometer) | Surface structure, mechanical properties |
The power of integrative modeling lies in combining these techniques to overcome their individual limitations. For example, while crosslinking mass spectrometry provides distance restraints but not 3D coordinates, and cryo-EM provides 3D density but may lack atomic detail, their combination can yield precise atomic models of large complexes [65]. Similarly, FRET efficiency measurements can guide the modeling of conformational ensembles when combined with shape information from SAS.
Experimental Protocols and Workflows
Data Collection and Restraint Generation
The initial phase of any integrative modeling project involves systematic collection of experimental data and their conversion into spatial restraints. For crosslinking mass spectrometry, the protocol involves: (1) crosslinking the native complex using chemical crosslinkers; (2) digesting the crosslinked complex with proteases; (3) identifying crosslinked peptides by mass spectrometry; and (4) converting identified crosslinks into distance restraints (typically 0-30 Å depending on linker length) for modeling. For FRET spectroscopy, measurements of energy transfer efficiency between donor and acceptor fluorophores are converted into distance restraints using the Förster relationship, typically in the range of 10-100 Å.
Small-angle X-ray scattering (SAXS) data processing involves: (1) collecting scattering curves from the sample and buffer control; (2) subtracting buffer scattering to obtain the macromolecular scattering profile; (3) calculating the pair distribution function to estimate overall dimensions; and (4) generating shape restraints either as volumetric envelopes or as spatial restraints against calculated scattering profiles from models. Cryo-EM data processing follows standard single-particle analysis workflows to obtain 3D density maps, which are then used as volumetric restraints during modeling.
Modeling Protocols and Structure Calculation
Structure calculation in integrative modeling typically employs sampling algorithms that generate models satisfying the composite set of spatial restraints. The modeling protocol generally follows these steps [67]:
Preparing starting structures: Known structures of components or subunits are obtained from the PDB or generated by homology modeling.
Defining representation: The system is represented at appropriate resolution levels—atomic, coarse-grained, or multi-scale—based on available data.
Sampling conformational space: Using methods like molecular dynamics, Monte Carlo sampling, or genetic algorithms, models are generated that satisfy the spatial restraints.
Selecting and validating models: The resulting models are assessed based on satisfaction of restraints, and a representative ensemble is selected.
The LZerD docking suite exemplifies a computational approach used in integrative modeling, particularly for protein-protein docking [67]. LZerD represents protein surface shape using 3D Zernike descriptors (3DZDs)—rotational invariants derived from a moment expansion of a 3D shape function—that act as a soft representation of the molecular surface. This approach allows for efficient sampling of binding orientations while accommodating flexibility and uncertainty in the input data.
For more complex systems, specialized software like Multi-LZerD enables assembly of complexes from three or more subunits using genetic algorithm-based methods [67]. In recent years, deep learning approaches such as AlphaFold-multimer have been integrated into these pipelines, substantially enhancing prediction accuracy for certain classes of complexes [67].
Validation and Uncertainty Quantification
A critical aspect of integrative modeling is the validation of final models and quantification of their uncertainty. Unlike high-resolution structures where validation metrics like R-factors are well-established, integrative models require specialized validation approaches. These include: (1) assessing the satisfaction of experimental restraints; (2) cross-validation by excluding subsets of data during modeling; (3) assessing model precision through the variability in the ensemble; and (4) comparing with experimental data not used in modeling.
The variability among models in an ensemble reflects the uncertainty of the modeling process and the completeness of input data [65]. For multi-state models, the relative populations of different states can be estimated based on how well each state explains the experimental data, providing insights into the energy landscape and dynamics of the system.
Visualization and Computational Implementation
Workflow Diagram for Integrative Modeling
The integrative modeling process follows a systematic workflow that cycles between data acquisition, model generation, and validation. The diagram below illustrates this iterative process:
Multi-Scale Representation in IHM
Integrative structures often employ multi-scale representations to optimally encode molecular complexity. The following diagram illustrates how different components can be represented at varying resolutions:
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of integrative/hybrid methods requires specialized computational tools, experimental reagents, and data resources. The following table catalogues essential resources for researchers in this field:
Table: Essential Research Resources for Integrative/Hybrid Methods
| Resource Category | Specific Tools/Reagents | Function/Purpose | Application Context |
|---|---|---|---|
| Modeling Software | LZerD, Multi-LZerD [67] | Protein-protein docking using 3D Zernike descriptors | Rigid-body docking of subunits |
| AlphaFold-multimer [67] | Deep learning-based complex structure prediction | Ab initio complex modeling | |
| MODELLER [67] | Homology modeling of subunits | Template-based structure prediction | |
| CHARMM, NAMD [67] | Molecular dynamics simulation | Model refinement and flexibility | |
| Experimental Techniques | Chemical Crosslinkers (BS3, DSS) | Covalent linking of proximal residues | Distance restraint generation for MS |
| Fluorophore Pairs (FRET) | Distance measurement via energy transfer | Conformational dynamics analysis | |
| Hydrogen-Deuterium Exchange | Solvent accessibility profiling | Flexible region identification | |
| Data Resources | PDB-IHM [66] [65] | Archive for integrative structures | Model deposition and retrieval |
| EMDB, SASBDB, BMRB [65] | Specialized experimental data repositories | Restraint source data | |
| IHMCIF Dictionary [65] | Data standard for integrative models | Model representation and metadata |
Data Management and PDB Integration
Data Standards and Deposition Requirements
The integrative structure determination pipeline produces heterogeneous data that must be standardized for effective archiving and sharing. The wwPDB requires specific information for depositing integrative structures to ensure reproducibility and adherence to FAIR principles [66]. The mandatory deposition elements include:
- Atomic and/or coarse-grained coordinates of the modeled system in PDBx/mmCIF format
- Starting models used in the modeling process
- Spatial restraints derived from experimental data
- Modeling protocols and software details
- Comprehensive metadata including citations, authors, software, and reference sequence information
The IHMCIF dictionary provides the data framework for representing integrative models, supporting multi-scale, multi-state, and ordered-state representations [65]. This dictionary is developed as a collaborative community project and is freely available through a public GitHub repository, ensuring transparency and ongoing development based on researcher needs.
Accessing Integrative Structures in the PDB
Integrative structures are fully integrated into the RCSB PDB database and can be accessed through multiple approaches [65]:
Keyword Search: Using the Basic Search function with specific protein names (e.g., "BBSome") or PDB IDs for known integrative structures (e.g., 8ZZE, 9A03)
Advanced Search: Selecting "Integrative/Hybrid Method Details" under Structure Attributes to filter by specific model features (multi-scale, multi-state) or experimental datasets
Programmatic Access: Retrieving IHM entries using RCSB Search and Data APIs (search.rcsb.org, data.rcsb.org) for large-scale analyses
Integrative structures are visually distinguished in search results by a dedicated IHM icon, and structure summary pages provide overviews of key information about representative models [65]. Due to their complexity, these structures are available in mmCIF format but not in legacy PDB format, as the traditional PDB format cannot adequately represent the multi-scale nature of IHM models.
Integrative/Hybrid Methods have fundamentally expanded the scope of structural biology, enabling characterization of biological complexes that defy analysis by single approaches. As these methods continue to evolve, several emerging trends promise to further enhance their capabilities. Deep learning approaches are increasingly being integrated with classical docking methods, providing powerful hybrid pipelines that leverage both evolutionary information and physical principles [67]. The growing emphasis on dynamics and multi-state representations allows researchers to move beyond static snapshots to capture functional trajectories and energy landscapes.
For the structural biology community, the comprehensive archiving of integrative structures in the PDB following FAIR principles ensures that these complex models remain accessible, interpretable, and reusable. As methods for collecting diverse experimental data continue to advance, and as computational power grows, integrative approaches will likely become increasingly central to mechanistic studies of macromolecular complexes, with profound implications for understanding cellular function and designing therapeutic interventions.
Overcoming Common Challenges in PDB Data Interpretation
Identifying Biologically Relevant Assemblies vs. Asymmetric Units
For researchers, scientists, and drug development professionals, a precise understanding of the three-dimensional structures of biomolecules is paramount. When utilizing the Protein Data Bank (PDB), the primary repository for such data, a critical conceptual distinction must be made between the asymmetric unit and the biological assembly. The asymmetric unit is the fundamental building block of a crystal, defined by crystallographic symmetry, which is used to generate the entire crystal lattice [68]. However, this unit may not represent the functional form of the molecule in vivo. The biological assembly (or biological unit) is the macromolecular complex that has been demonstrated or is hypothesized to be the functional, biologically active state of the molecule [68] [69]. For example, the functional form of hemoglobin is a tetramer, but in various PDB entries, the asymmetric unit may contain only a dimer or even multiple tetramers [68]. Understanding and correctly identifying the biological assembly is therefore a foundational step in structural biology research, with direct implications for interpreting function, mechanism, and interactions in drug discovery.
Defining the Core Concepts
The Asymmetric Unit
The asymmetric unit is a crystallographic concept. It is the smallest portion of the crystal structure to which crystallographic symmetry operations (rotations, translations, screw axes) are applied to generate the complete unit cell, which then repeats infinitely to form the crystal [68]. The asymmetric unit contains the unique set of atomic coordinates refined against experimental data. Its contents are determined by the packing of molecules within the crystal and may bear no direct relationship to biological function. An asymmetric unit can contain [68]:
- One biological assembly
- A portion of a biological assembly (e.g., a single subunit of a multimeric complex)
- Multiple biological assemblies
The Biological Assembly
The biological assembly represents the structure of the molecule or complex as it is believed to function in a biological context [68] [69]. This is the structure that is typically of greatest interest to researchers studying mechanistic biology or developing therapeutics. The PDB requires depositors to provide the Cartesian coordinates for this assembly, which may be constructed from [68]:
- One copy of the asymmetric unit (no operations needed)
- Multiple copies of the asymmetric unit (by applying crystallographic symmetry operations)
- A portion of the asymmetric unit (by selecting a subset of the deposited coordinates)
The assembly is characterized by its stoichiometry (subunit composition, e.g., A₂B₂), its interfaces (the specific atomic contacts between subunits), and its symmetry (e.g., cyclic C₂, dihedral D₂, or icosahedral) [69].
Key Differences Summarized
Table 1: Core differences between the Asymmetric Unit and the Biological Assembly.
| Feature | Asymmetric Unit | Biological Assembly |
|---|---|---|
| Definition | Smallest unique crystallographic unit | Functional form of the molecule in vivo |
| Primary Purpose | To generate the crystal lattice via symmetry operations | To represent the biologically active structure |
| Content Determinant | Crystal packing | Biological evidence (experimental or computational) |
| Relationship to Crystal | Unique set of atoms; the deposited coordinates | May require application of symmetry operations to the asymmetric unit |
| Researcher's Focus | Often relevant for crystallographic methodology | Essential for functional analysis and drug design |
Quantitative Prevalence and Relationship Types
The biological assembly differs from the asymmetric unit in either symmetry or stoichiometry (or both) in approximately 42% of crystal structures in the PDB [69]. This high frequency underscores the importance of always verifying which structure is being analyzed. The relationship between the asymmetric unit and the biological assembly can be categorized, with hemoglobin providing classic illustrative examples [68].
Table 2: Relationship types between the Asymmetric Unit and the Biological Assembly, exemplified by hemoglobin structures.
| Relationship Type | Description | Example PDB Entry |
|---|---|---|
| Asymmetric Unit = Biological Assembly | The deposited coordinates already represent the functional oligomer. | 2HHB (One hemoglobin tetramer in the ASU) |
| Biological Assembly from Multiple Asymmetric Units | Symmetry operations must be applied to multiple copies of the ASU to build the functional oligomer. | 1OUT (A dimer in the ASU; a 180° rotation generates the tetramer) |
| Biological Assembly is a Subset of the Asymmetric Unit | The ASU contains multiple copies of the functional oligomer; a subset of chains must be selected. | 1HV4 (Two hemoglobin tetramers in the ASU; one tetramer is the biological assembly) |
Figure 1: A workflow for determining the relationship between the asymmetric unit and the biological assembly in a PDB entry.
Methodologies for Identifying the Biological Assembly
Author and Computational Annotation
The biological assembly provided in a PDB entry is annotated in one of two ways. The "author provided" assembly is based on the depositor's knowledge of the molecule's biology and supporting experimental evidence. The "software determined" assembly is predicted computationally by programs like PISA (Protein Interfaces, Surfaces and Assemblies), which analyzes buried surface area, interaction energies, and interface properties to identify stable complexes [68]. On the RCSB PDB website, downloaded biological assembly files are marked as (A) for author-provided or (S) for software-determined [68]. In some cases, both may be provided if there is a discrepancy, requiring researcher judgment.
Experimental Protocols for Validation
While computational methods are vital, the definitive identification of a biological assembly often requires experimental validation. The following are key methodologies cited in the literature:
- Analytical Ultracentrifugation (AUC): This technique measures the sedimentation velocity or equilibrium of a molecule in solution, directly providing information about its molecular weight and oligomeric state in near-native conditions, thereby validating the proposed assembly's stoichiometry [69].
- Dynamic Light Scattering (DLS): DLS estimates the hydrodynamic radius of particles in solution, which can be used to infer the oligomeric state and monodispersity of a sample, supporting the presence of a specific assembly.
- Cross-linking Mass Spectrometry (XL-MS): This method introduces covalent cross-links between proximate amino acids in a protein complex. By identifying the cross-linked peptides, spatial constraints are obtained that can validate the subunit interfaces present in the proposed biological assembly model [69].
- Site-Directed Mutagenesis of Interface Residues: A functional approach where residues at a putative interface are mutated. A significant disruption of function or a reduction in stability without affecting the folded state of individual subunits provides strong evidence for the biological relevance of that interface [69].
A Computational Toolkit for Assembly Analysis
The Scientist's Toolkit for handling biological assemblies involves both web resources and software for advanced analysis.
Table 3: Key research reagents and computational tools for assembly analysis.
| Tool / Resource | Type | Primary Function |
|---|---|---|
| RCSB PDB Website | Web Portal | Download biological assembly coordinates; view author vs. software annotations [68]. |
| PISA (Software Determined) | Algorithm | Predicts stable quaternary structures from crystal symmetry and interface properties [68]. |
| Reduce | Software | Adds missing hydrogen atoms to PDB structures and optimizes side-chain amide orientations [70]. |
| Analytical Ultracentrifuge | Laboratory Instrument | Empirically determines molecular weight and oligomeric state in solution [69]. |
Advanced Considerations and Special Cases
Viral Capsids and Large Symmetric Assemblies
Viral capsids represent a special case where the deposited coordinates are often a minimal asymmetric unit of the massive icosahedral shell. For example, in PDB entry 1qqp, the deposited coordinates represent one icosahedral asymmetric unit [68]. Generating the biological assembly—the entire icosahedral capsid—requires applying a set of non-crystallographic symmetry operators, which are distinct from the crystallographic symmetry operators used to build the crystal lattice. These operations are defined in the entry's data files (mmCIF or PDB format) and are used by visualization software to display the complete virus particle.
The Challenge of Crystal Packing vs. Biological Interfaces
A significant challenge in crystallography is distinguishing biologically relevant interfaces from those induced merely by crystal packing. Biologically relevant interfaces tend to be larger, more hydrophobic, and exhibit greater evolutionary sequence conservation than crystal packing interfaces [69]. Furthermore, the conservation of an interface across multiple crystal forms of the same or homologous proteins is a powerful indicator of biological relevance. Computational methods like PISA leverage these principles by calculating interaction energies and buried surface areas to score and rank potential assemblies [68] [69].
Data and Annotation Quality
Researchers must be aware that the deposited biological assembly is not always correct. Occasionally, depositors may not specify an assembly different from the asymmetric unit, or the software prediction may be inaccurate [69] [71]. A critical evaluation is always recommended. Furthermore, the PDB is transitioning from legacy 4-character accession codes (e.g., 2HHB) to extended 12-character identifiers (e.g., pdb_00002hhb) and is phasing out the legacy PDB file format in favor of the more robust PDBx/mmCIF format [14]. Researchers should ensure their software and scripts are updated to handle these changes.
For researchers relying on PDB data, the distinction between the asymmetric unit and the biological assembly is non-negotiable. The asymmetric unit is a crystallographic construct, while the biological assembly is a biological hypothesis. With over 40% of entries displaying a difference between the two, failing to select the correct structure risks a fundamentally flawed biological interpretation. By leveraging the available data on the PDB website, understanding the annotation sources, and applying critical judgment supported by experimental and computational validation methods, scientists can confidently identify the true functional structure, thereby ensuring the integrity of their research in biochemistry, molecular biology, and drug development.
Protein structures archived in the Protein Data Bank (PDB) serve as fundamental resources for understanding biological function and guiding drug discovery. However, these 3D models are imperfect representations of biological reality. Both experimentally determined structures and computed structure models contain inherent limitations that create data gaps—regions where atomic coordinates are missing, poorly resolved, or of limited reliability [25]. These gaps, including missing loops and residues, along with low-resolution regions, present significant challenges for researchers relying on these structures for detailed analysis and molecular design.
For experimental structures, limitations may stem from mismatches between the model and experimental data, regions of local disorder causing lack of experimental data, distortions in atomic geometry, or inappropriate atom-atom clashes [25]. Computed structure models face different challenges, with regions of low confidence due to limitations in the supporting data used for predictions [25] [72]. Understanding these limitations is crucial for proper interpretation of structural data, particularly for drug development professionals who require accurate molecular interaction information.
Identifying and Assessing Structural Gaps
Quality Metrics for Experimental Structures
The Worldwide PDB partnership has established comprehensive validation protocols to assess structure quality. For X-ray crystallography structures (~87% of the experimental PDB archive), several key metrics help identify problematic regions [25]:
- Resolution: Measures how well adjacent atoms can be distinguished, with lower values (e.g., 1.8 Å) indicating better quality. This is a global measure that doesn't highlight local disorder [25].
- R-factor and R-free: Describe agreement between experimental data and the model. R-free is particularly important as it measures agreement with experimental data not used during structure determination. A large difference between R-factor and R-free may indicate model errors [25].
- Real Space Correlation Coefficient (RSCC): Reflects agreement between atomic coordinates and experimental electron density for individual residues. Residues with RSCC in the lowest 1% should not be trusted, while those between 1-5% should be considered with caution [25].
- Clashscore: Measures inappropriate atom-atom clashes, with higher scores indicating more steric conflicts [73].
The wwPDB Validation Report provides summary illustrations of these measures using five graphical sliders that show how a structure compares to all archived structures and those at similar resolution [73].
Table 1: Key Quality Metrics for Experimental Structures
| Metric | Optimal Values | Concerning Values | Interpretation |
|---|---|---|---|
| Resolution | <2.0 Å | >3.0 Å | Lower values indicate better quality |
| R-free | <0.25 | >0.30 | Higher values indicate poorer fit to experimental data |
| RSCC | >0.9 | <0.8 | Values <0.8 indicate poor electron density fit |
| Clashscore | <5 | >20 | Higher values indicate more atom-atom clashes |
| Ramachandran outliers | <1% | >5% | Higher percentages indicate problematic backbone geometry |
Confidence Measures for Computed Structure Models
For computed structure models, different confidence measures are employed:
- Predicted Local Distance Difference Test (pLDDT): Ranges from 0-100, with values ≥90 indicating high confidence, 70-90 indicating good confidence, 50-70 indicating low confidence, and <50 indicating very low confidence that should be interpreted with caution [25].
- Predicted Aligned Error (PAE): Used in multi-chain modeling to estimate positional uncertainty between residues [72].
These AI-predicted models have limitations in capturing protein dynamics, predicting multi-chain structures accurately, and representing ligands, cofactors, and post-translational modifications [72].
Ligand-Specific Quality Considerations
Small-molecule ligands present particular challenges for structure quality. The RCSB PDB provides specialized ligand quality assessment using composite ranking scores that aggregate correlated quality indicators into unidimensional measures [24]. The ligand quality analysis focuses on:
- PC1-fitting: Composite indicator of how well ligand atomic coordinates fit experimental electron density.
- PC1-geometry: Composite indicator of how well ligand structure conforms to known chemical geometry [24].
These assessments are visualized through 1D sliders and 2D ligand quality plots on the RCSB PDB Structure Summary pages, enabling researchers to quickly identify the best-quality ligand instances for their analyses [24].
Methodologies for Addressing Structural Gaps
Experimental Approaches for Gap Resolution
Several experimental strategies can help resolve structural gaps:
- Construct Optimization: Modifying protein constructs to remove flexible regions that cause disorder, potentially improving crystallization and resolution.
- Ligand Binding: Co-crystallization with binding partners can stabilize flexible regions, allowing previously disordered areas to become structured and visible in electron density.
- Alternative Techniques: Using cryo-electron microscopy for larger complexes or NMR to study dynamic regions that resist crystallization.
The PEPBI database exemplifies rigorous criteria for including high-quality protein-peptide complexes, requiring structure resolution ≤2.0 Å and peptides composed of only the 20 common amino acids to ensure reliable structural information [74].
Computational Protocols for Model Completion
When experimental data is insufficient, computational methods can fill structural gaps:
Figure 1: Computational Workflow for Addressing Structural Gaps
For regions with missing residues in experimental structures, the following protocol is recommended based on the PEPBI database methodology [74]:
- Gap Identification: Determine regions with unresolved residues in the experimental structure.
- Template Selection: For homology modeling, identify suitable templates with high sequence similarity.
- Model Building:
- For gaps with 5 or fewer missing residues at termini: Manual insertion using molecular visualization tools like UCSF Chimera.
- For internal gaps or longer terminal missing fragments: Use comparative modeling tools like Modeller integrated in UCSF Chimera.
- For extensive missing regions: Employ deep learning tools like RoseTTAFold to predict complete structures, then splice missing regions into the experimental structure.
- Energy Minimization: Perform structural refinement to relieve steric clashes and optimize geometry.
- Validation: Assess completed models using geometric validation tools and cross-reference with any available experimental data.
For multi-chain complexes where accuracy declines with increasing chain numbers, integration of additional experimental data such as cross-linking mass spectrometry or NMR data becomes essential for validating predicted assemblies [72].
Table 2: Key Resources for Addressing Structural Data Gaps
| Resource | Type | Primary Function | Access |
|---|---|---|---|
| wwPDB Validation Server | Validation Tool | Pre-deposition structure validation | http://validate.wwpdb.org |
| PEPBI Database | Specialized Database | Protein-peptide complexes with thermodynamic data | https://www.nature.com/articles/s41597-025-05754-7 |
| MolProbity | Validation Tool | Stereochemical quality analysis | Integrated in wwPDB validation |
| UCSF Chimera | Visualization & Modeling | Molecular visualization and manual model building | https://www.cgl.ucsf.edu/chimera/ |
| RoseTTAFold | Prediction Software | Protein structure prediction for missing regions | https://robetta.bakerlab.org/ |
| Modeller | Modeling Software | Homology modeling of missing regions | Integrated in UCSF Chimera |
| DeepUrfold | Analysis Framework | Detecting distant structural relationships | Publication-based |
Best Practices for Structural Data Interpretation
Visualization Strategies for Quality Assessment
Effective visualization of structural quality requires careful color application:
- Color Palettes: Use sequential palettes with a single color in varying saturations to represent continuous data like resolution or confidence measures. Employ contrasting colors to highlight comparisons, such as well-resolved versus poorly-resolved regions [75].
- Accessibility: Ensure sufficient color contrast and avoid combinations problematic for color vision deficiencies. Use perceptual uniform color spaces like CIE Luv and CIE Lab that align with human vision perception [28].
- Context Awareness: Be mindful of color conventions in structural biology, such as B-factor putty representation or pLDDT coloring schemes in AlphaFold models [28] [75].
The RCSB PDB implements these principles in their visualization tools, mapping quality metrics directly onto 3D structures in the NGL viewer, with coloring schemes for "Geometry Quality" and "Density Fit" [76].
Context-Dependent Structure Interpretation
Different research questions require different approaches to handling structural gaps:
- Drug Binding Site Analysis: Prioritize structures with well-resolved binding pockets (high RSCC values for ligand and binding site residues) and minimal clashes. Use ligand quality plots to select the best ligand instances [24].
- Protein Engineering: Consider conformational flexibility in low-resolution regions, using computational sampling to explore alternative conformations.
- Evolutionary Analysis: Tools like DeepUrfold can detect distant structural relationships beyond traditional fold classification, potentially revealing functional insights even in lower-quality regions [77].
For intrinsically disordered regions (IDRs), recent research indicates that low-complexity regions (LCRs) within IDRs can induce local structure. PolyE and polyK regions frequently induce helical conformations, while other common LCRs tend to form coil structures [78]. This structural propensity should be considered when analyzing missing or disordered regions.
Structural biology remains an evolving field where data gaps and limitations are inherent to both experimental and computational approaches. By understanding the available quality metrics, implementing robust methodologies for addressing missing regions, and applying appropriate visualization and interpretation strategies, researchers can navigate these uncertainties effectively. The ongoing development of validation resources, computational tools, and specialized databases continues to enhance our ability to identify and address structural gaps, ultimately supporting more reliable biological insights and drug development efforts.
As the field advances with new AI-based approaches and integrated experimental-computational workflows, the fundamental principle remains unchanged: critical assessment of structural quality should precede any detailed analysis, with particular attention to regions directly relevant to the research question.
Within the Protein Data Bank (PDB), a fundamental challenge for researchers is the existence of dual labeling systems for chain and residue identifiers: one assigned by the depositing author and another by the PDB curation staff. These identifier conflicts can complicate tasks such as comparing structures, mapping mutations, and analyzing ligand-binding sites if not properly reconciled. This technical guide delineates the origins and implications of these discrepancies and provides a definitive protocol for their resolution, forming a critical component of a broader thesis on foundational concepts in structural bioinformatics. A clear understanding of these hierarchies is essential for accurate data retrieval, visualization, and computational analysis in structural biology and drug development.
The Protein Data Bank organizes structural data using a precise hierarchical framework: Entry > Entity > Instance > Assembly [10]. An Entry (denoted by a PDB ID, e.g., 2hbs) encompasses all data for a single deposited structure. An Entity describes a chemically unique molecule, such as a specific protein chain. An Instance refers to a specific copy of that entity within the entry, and an Assembly represents the biologically functional unit formed by one or more instances [10].
Identifiers are assigned at every level of this hierarchy to uniquely locate any atom. This guide focuses on identifiers at the instance level—specifically, chain IDs and residue numbers—where conflicts most frequently arise. The PDB format specification includes distinct record types (ATOM for standard residues and HETATM for non-standard residues, ligands, and solvents) that house these identifiers in defined columns [18]. Two parallel systems exist for labeling chains and residues:
- Author-Assigned Identifiers (
auth_*): Provided by the scientists who determined the structure, often to maintain consistency with related literature or sequence database numbering. - PDB-Assigned Identifiers (
label_*): Systematically assigned by the wwPDB biocuration team during processing to ensure internal consistency and compliance with archive-wide standards [11] [79].
The Core of the Conflict: Chain and Residue Identifiers
Chain Identifier Conflicts
Chain IDs uniquely identify each polymer chain instance within a structure. The assignment rules differ between the two systems, leading to potential mismatches.
| Aspect | Author-Assigned (auth_asym_id) |
PDB-Assigned (label_asym_id) |
|---|---|---|
| Origin | Provided by depositing scientist [11] | Assigned by wwPDB biocurators during processing [11] |
| Rationale | May use descriptive labels (e.g., 'L' for light antibody chain, 'R' for receptor) [11] | Often follows systematic order (e.g., A, B, C...) [10] |
| Flexibility | Can be any alphanumeric string [11] | Governed by wwPDB processing procedures [79] |
| Example (PDB: 2or1) | Author chains: 'L' and 'R' [11] | PDB chains: 'C' and 'D' [11] |
A common scenario occurs in structures of antibodies or protein complexes, where authors might assign chain IDs 'H' and 'L'. During curation, these may be reassigned to 'A' and 'B' [11]. Furthermore, ligands and solvent molecules are assigned the chain ID of their spatially closest macromolecular instance [11] [10].
Residue Numbering Conflicts
Residue numbers specify the position of an amino acid or nucleotide within a polymer chain. Discrepancies between numbering schemes are a frequent source of confusion.
| Aspect | Author-Assigned (auth_seq_id) |
PDB-Assigned (label_seq_id) |
|---|---|---|
| Origin | Provided by depositing scientist [11] | Assigned by wwPDB biocurators [11] |
| Numbering Scheme | Often matches related publications or UniProt sequence numbering [11] | Typically sequential, starting from 1 for the first residue in the chain [11] |
| Handling Gaps | May include gaps to align with reference sequences [11] | Usually a continuous numerical sequence [11] |
| Example (PDB: 6kr6) | Author numbering: 34-843 [11] | PDB numbering: 1-810 [11] |
The example of PDB entry 6kr6 illustrates a typical conflict: the author-defined residue numbers (34-843) align with the corresponding UniProt entry, while the PDB-assigned numbers are a sequential count from 1 to 810 [11]. This discrepancy must be accounted for when referencing specific residue positions from the literature.
Consequences of Unresolved Identifier Conflicts
Ignoring the duality of identifier systems can lead to significant errors in research:
- Incorrect Residue Selection: Specifying a residue by the wrong identifier can lead to analyzing the wrong location in the 3D structure.
- Failed Cross-Referencing: Mapping a mutation from literature (e.g., "Y20K" using author numbering) directly to a PDB file using its sequential numbering will target an incorrect residue if the numbering schemes differ [79].
- Visualization and Analysis Errors: Loading a structure and selecting residues by one numbering scheme while the visualization tool uses the other can produce misleading results.
- Hindered Reproducibility: Computational scripts and analysis pipelines that do not explicitly account for the identifier type may fail or produce inconsistent results across different PDB entries.
Experimental Protocols for Resolving Conflicts
A systematic approach is required to correctly identify and use the appropriate numbering system for a given PDB entry.
Protocol 1: Identifying the Conflict in a Given PDB Entry
Objective: To determine whether author-assigned and PDB-assigned chain IDs and residue numbers differ for a specific PDB entry.
- Access the Entry on RCSB PDB: Navigate to the RCSB PDB website (https://www.rcsb.org) and enter the PDB ID.
- Locate the "Sequence" Tab: View the protein sequence. The display often shows both numbering schemes.
- Inspect the "Structure" Tab in 3D View: Select a residue. The information panel that appears typically lists both the author-provided and PDB-assigned residue numbers and chain IDs.
- Download and Examine the PDB File:
Protocol 2: A Workflow for Consistent Residue Referencing
Objective: To establish a reproducible method for accurately selecting a specific residue across multiple PDB entries, suitable for computational drug development pipelines.
The following workflow outlines the critical steps for robustly handling identifier conflicts, from data acquisition to final selection.
This table lists key resources for researchers working with PDB identifiers.
| Resource Name | Type | Primary Function in Conflict Resolution |
|---|---|---|
| RCSB PDB Website [11] | Web Portal | Provides a user-friendly interface to inspect and compare both author and PDB-assigned identifiers directly in the sequence viewer and 3D structure viewer. |
| mmCIF Format File [11] [79] | Data File | The standard data file for the PDB archive, containing both auth_* and label_* identifiers, allowing for unambiguous programmatic access. |
| PDBx/mmCIF Dictionary [79] | Data Standard | The definitive documentation for the mmCIF format, specifying the definitions and relationships of all data items, including identifiers. |
| ChimeraX [18] [80] | Visualization Software | Molecular visualization tool that can import PDB and mmCIF files and allows users to select and display residues using different numbering schemes. |
The duality of author and PDB-assigned chain and residue identifiers is an inherent feature of the PDB archive, stemming from the need to balance depositor intent with data standardization. For researchers in structural biology and drug development, failing to recognize and resolve these identifier conflicts can compromise the integrity of their analyses, from basic visualization to advanced computational modeling. By understanding the PDB's organizational hierarchy, systematically employing the reconciliation protocols outlined herein, and leveraging the appropriate tools, scientists can transform this potential source of error into a manageable aspect of robust structural data analysis. This competence is a foundational skill for ensuring reproducibility and accuracy in all research that leverages the rich structural data within the Protein Data Bank.
Interpreting Alternate Atom Locations and Multi-Model NMR Ensembles
Within structural biology, the static depictions of proteins often belie their dynamic nature. This technical guide elucidates two fundamental concepts within Protein Data Bank (PDB) entries that capture molecular flexibility: alternate atom locations in X-ray crystallography and multi-model ensembles in Nuclear Magnetic Resonance (NMR) spectroscopy. Framed within the broader thesis that understanding conformational diversity is crucial for accurate biological interpretation and drug development, this document provides in-depth methodologies for identifying, visualizing, and analyzing these features. We summarize quantitative data for easy comparison, detail experimental protocols, and visualize workflows to equip researchers with the tools to move beyond single, static models and embrace the dynamic reality of macromolecular structures.
Proteins are inherently dynamic molecules, transitioning between ensembles of conformations to perform their biological functions [81]. The Protein Data Bank (PDB), the single worldwide archive of structural data of biological macromolecules, has evolved to capture this complexity beyond a one-sequence-one-structure framework [81] [82]. Two primary mechanisms within PDB entries encode information about structural flexibility and heterogeneity:
- Alternate Locations (Altlocs): In X-ray crystal structures, atoms are occasionally modelled in two or more discrete locations, termed "alternate locations" or "altlocs" [81]. This occurs when the experimental electron density provides evidence for multiple well-defined conformations of a residue or ligand, often indicating structural ambiguity or genuine flexibility.
- Multi-Model NMR Ensembles: Structures determined by solution-state NMR spectroscopy are typically represented as an ensemble of multiple models [83] [84]. These ensembles reflect the conformational space sampled by the protein in solution and are consistent with the averaged geometric and dynamic parameters measured by NMR.
Interpreting these features is foundational for research areas where dynamics are linked to function, such as enzyme catalysis, allosteric regulation, and drug binding. Overlooking them can lead to an underappreciation of protein flexibility and its functional consequences [81].
Core Concepts and Quantitative Data
Alternate Atom Locations in X-ray Crystallography
Where experimental electron density evidence exists for multiple conformations, atoms are modelled in alternate locations [81]. The PDB file format uses a single-letter code (e.g., 'A', 'B') on ATOM or HETATM records to distinguish these locations [84]. Programs reading PDB files often ignore these by default, which has limited the accessibility of this high-resolution data representing structural ensembles [81].
Table 1: Key Characteristics of Alternate Locations
| Feature | Description | Data Source |
|---|---|---|
| Prevalence | Found in a significant number of X-ray structures; can involve side chains and backbone segments. | PDB-wide surveys [81] |
| Identification in PDB | Labeled with a single-character code (e.g., label_alt_id in mmCIF; column 17 in legacy PDB format). |
PDBx/mmCIF specification [84] |
| Structural Impact | Can show variations in dihedral angles, side-chain rotamers, and backbone displacements. | Dataset of alternately located segments [81] |
| Thermal Parameter | Each altloc has its own B-factor, representing the positional uncertainty for that specific conformation. | PDB entry data [81] |
Multi-Model Ensembles in NMR Spectroscopy
NMR structures are deposited as ensembles of models because the experimental observables are time and ensemble averages over dynamically fluctuating molecules [83]. A key conceptual shift is that NMR parameters must be interpreted as properties of the ensemble rather than of any single conformer [83].
Table 2: Characteristics of Multi-Model NMR Ensembles
| Feature | Description | Typical Values/Examples |
|---|---|---|
| Ensemble Size | Number of models representing the conformational diversity. | Often 10-50 models per entry [84] |
| Representative Model | A single model from the ensemble often designated as the "best representative." | PDB entry 2n3q [85] |
| Restraints Used | Experimental data (e.g., NOEs, J-couplings, chemical shifts, residual dipolar couplings) used to generate the ensemble. | Recommendations of the wwPDB NMR Validation Task Force [86] |
| Validation | Assessed using metrics like the Random Coil Index, which reports on protein flexibility. | wwPDB Validation Report [85] [86] |
Methodologies and Experimental Protocols
Protocol for Interpreting Alternate Locations
The following methodology is adapted from analyses of alternately located backbone segments [81].
- Data Retrieval: Query the PDB for structures of interest, filtering by method (X-Ray Diffraction), resolution (e.g., ≤ 3.5 Å), and R-free value (e.g., ≤ 0.33) to ensure data quality.
- File Parsing: Use a structural bioinformatics library (e.g., BioPython [81] or ProDy [87]) to parse the PDB or mmCIF file and extract all atoms with alternate location indicators.
- Coordinate Extraction: For each residue with alternate locations, extract the Cartesian coordinates (x, y, z) for each altloc for all backbone atoms (N, Cα, C, O).
- Distance Calculation: Calculate the distance between equivalent atoms in different alternate conformations. For the alpha-carbon (Cα), the distance between altlocs X and Y is calculated as: dX,Y = ‖pCA,X - pCA,Y‖, where pCA,X is the coordinate vector.
- Dihedral Angle Analysis: Calculate the backbone dihedral angles (φ, ψ) for each alternate conformation. This reveals the impact of the alternate location on the protein's backbone conformation.
- Contact Analysis: Identify all atoms within a defined radius (e.g., 5 Å) of the residue in each of its alternate conformations. Classify contacts as intra-chain, inter-chain, or ligand contacts.
Protocol for Generating and Validating NMR Ensembles
This protocol is based on ensemble-based interpretations of NMR data and wwPDB recommendations [83] [86].
- Data Collection: Acquire a full set of NMR experimental data, including chemical shifts, J-couplings, NOE-derived distances, and residual dipolar couplings (RDCs).
- Restraint Derivation: Convert raw experimental data into geometric restraints for structure calculation (e.g., distance bounds from NOEs, torsion angle restraints from J-couplings).
- Ensemble Calculation: Use computational methods to generate an ensemble of structures that satisfies the experimental restraints. Two primary approaches are:
- Ensemble Restraining: A number of parallel replicas are simulated, and the applied restraints are treated as an ensemble property, ensuring the collective ensemble matches the data [83].
- Pool-and-Select: A large pool of candidate conformations is generated, and a selection algorithm identifies a subset that best fulfills the experimental parameters [83].
- Validation: The final ensemble is rigorously validated against the experimental data and geometric quality standards. The wwPDB validation report includes checks for restraint violations, stereochemical quality, and agreement with measured chemical shifts (e.g., via the Random Coil Index) [86].
The following workflow diagram illustrates the parallel processes for interpreting these two types of conformational data.
The Scientist's Toolkit: Essential Research Reagents and Materials
This table details key resources and tools required for working with alternate locations and NMR ensembles.
Table 3: Essential Research Reagents and Tools
| Item Name | Function/Application | Example/Source |
|---|---|---|
| RCSB PDB Website | Primary portal for searching, retrieving, and analyzing PDB entries, including access to validation reports. | https://www.rcsb.org/ [35] |
| Mol* Viewer | Web-based and standalone 3D structure viewer for simultaneously visualizing alternate locations and NMR ensembles. | Integrated into RCSB PDB [62] [85] |
| BioPython | A library for computational biology; the Bio.PDB module can parse PDB files and handle alternate locations. |
https://biopython.org [81] |
| ProDy | A Python package for protein dynamics analysis; can fetch, parse, and write PDB files, handling models and altlocs. | http://prody.org [87] |
| wwPDB Validation Report | Provides an assessment of the quality and reliability of a PDB entry, including key metrics for NMR ensembles. | Available for each entry on the RCSB PDB site [86] |
| PDBx/mmCIF Data Format | The standard format for PDB entries, which robustly handles complex data like multiple models and altlocs. | PDB Data Distribution [84] |
| Alternate Location Dataset | Curated datasets for surveying the landscape of alternate conformations across the PDB. | Harvard Dataverse [81] |
Visualization and Analysis in Mol*
The Mol* viewer, integrated into the RCSB PDB website, is an indispensable tool for visualizing these complex structural features [62] [85].
Visualizing Alternate Locations
In Mol*, atoms with alternate locations are typically represented by switching between different conformations. The Components Panel allows users to select and display specific alternate locations. By creating separate components for each altloc, one can compare conformations, measure distances, and analyze interactions specific to each state [62].
Visualizing NMR Ensembles
For an NMR entry (e.g., PDB ID 2n3q), the Structure Panel provides options to view the representative model, all models in the ensemble, or individual models using a slider [85]. The Preset menu includes an "Annotation" view colored by the Random Coil Index, a validation metric that highlights flexible protein regions based on NMR chemical shifts [85]. This directly links the ensemble's appearance to experimental data quality.
The following diagram outlines the logic for managing the display of these features within Mol*.
Alternate atom locations and multi-model NMR ensembles are not mere technical footnotes but are central to a modern, dynamic understanding of protein structure and function. By applying the methodologies outlined in this guide—leveraging the quantitative data, adhering to the analytical protocols, and utilizing the powerful visualization tools available—researchers and drug developers can extract profound insights into conformational heterogeneity. Mastering the interpretation of these features is a foundational skill for anyone leveraging the PDB, enabling a more accurate and biologically relevant analysis that can inform everything from basic mechanistic studies to targeted drug design.
Best Practices for Selecting the Right Structure for Your Research Goal
The Protein Data Bank (PDB) is one of the richest open-source repositories in biology, housing over 242,000 macromolecular structural models alongside much of the experimental data that underpins these models [88]. For researchers in drug discovery and basic science, selecting the most appropriate structure from this vast archive is a critical first step that underpins the validity of all subsequent analyses. The PDB is maintained as a single, global archive through the Worldwide Protein Data Bank (wwPDB) consortium, which coordinates deposition, validation, and dissemination of macromolecular structures [88]. Leveraging this wealth of data, structural bioinformatics has uncovered patterns—such as conserved protein folds, binding-site features, or subtle conformational shifts among related proteins—that would be impossible to detect from any single structure [88].
However, good structural bioinformatics requires understanding the nuances of the underlying experimental data, data encoding conventions, and quality control metrics that can affect a model's precision, fit-to-data, and comparability [88]. This guide provides a comprehensive framework for selecting optimal protein structures tailored to specific research objectives, ensuring reliable and biologically relevant conclusions.
Foundational Concepts: Understanding PDB Organization
The Structural Hierarchy of the PDB
Biomolecules in the PDB archive are organized and represented using a hierarchical structure to simplify searching and exploration [10]. Understanding this hierarchy is essential for meaningful structural selection and analysis.
Table: Levels of Structural Organization in the PDB
| Level | Definition | Example |
|---|---|---|
| Entry | All data pertaining to a particular structure deposited in the PDB | PDB ID 2hbs (sickle cell hemoglobin) |
| Entity | A chemically unique molecule (polymeric or non-polymeric) | Alpha chain protein, beta chain protein, heme group |
| Instance | A particular occurrence of an entity | Two instances of alpha chain in hemoglobin tetramer |
| Assembly | Biologically relevant group of instances forming a stable complex | Hemoglobin tetramer (functional oxygen-binding unit) |
The entry is designated with a 4-character alphanumeric identifier called the PDB ID [10]. Since there can be multiple instances of a given entity in an entry, each instance of a polymer or branched entity is given a unique chain identifier (e.g., A, AA, ...) [10]. Critically, chain IDs assigned to an entity in two different entries of the same protein may be different, as there is no specific rationale for their assignment [10].
Accessing and Visualizing Structures
The RCSB PDB provides powerful tools for structural exploration. The default visualization tool is Mol, a web-based tool that can be used without downloading or installing any software or apps [26]. Each PDB entry has a Structure tab that uploads coordinate files and displays them for interactive analysis [26]. The Mol interface simultaneously displays molecules in 3D, sequences of polymers, and ligands, ions, and water molecules [26]. The tool enables researchers to selectively display parts of a structure, change molecular representations, color components meaningfully, and analyze interactions throughout the structure or in the neighborhood of a single residue or ligand [26].
A Systematic Framework for Structure Selection
Selecting the optimal structure requires a methodical approach that aligns with specific research goals. The following workflow provides a robust framework for this process.
Define Your Biological Selection Criteria
When starting a structural bioinformatics project, the first step is to define the biological criteria for your study [88]. Consider the structures you need to answer your research question, whether it involves all lysozymes, a specific tyrosine kinase, or all enzymes [88]. Key considerations include:
- Protein Identity and Source: Specify the exact protein, organism, and potentially specific isoforms or variants relevant to your research.
- Ligand and Cofactor Requirements: Determine if you need structures with specific bound molecules (substrates, inhibitors, cofactors, or drug candidates). Small molecules such as glycerol or DMSO are often crystallographic additives, while other molecules may be native or synthetic ligands [88].
- Complex Composition: Assess whether your protein is part of large complexes by examining the identities of other chains. Identical chains typically reflect symmetry-related protomers, whereas distinct macromolecules reveal a multi-protein complex [88].
- Redundancy Reduction: A significant fraction of PDB entries corresponds to homologous proteins or multiple structures of the same protein [88]. Depending on your question, you may want to filter based on sequence or structure using tools like MMseq or CDHit for sequence clustering, or TM-score and CATH for structural clustering [88].
Determine How You Will Quality Control Your Data
Beyond determining the biological selection criteria, it is crucial to consider the experimental data underlying structures to ensure a quality dataset [88]. The table below summarizes key quality metrics across different structure determination methods.
Table: Quality Assessment Metrics for Different Structure Determination Methods
| Method | Key Quality Metric | High-Quality Range | Additional Metrics |
|---|---|---|---|
| X-ray Crystallography | Resolution | <2.5 Å for side chains; <3.5 Å for backbone | R-factor, R-free, Clashscore |
| Cryo-EM | Resolution (FSC 0.143) | <3.5 Å for atomic detail | Map-model correlation, Q-score |
| NMR | Not applicable | Not applicable | Clashscore, Ramachandran outliers |
| All Methods | Stereochemical accuracy | Within expected ranges | Rotamer outliers, Ramachandran plot |
High resolution is essential for accurate side chain positioning, whereas lower resolution models can still yield valuable insights into overall fold and backbone conformation [88]. In cryo-EM, resolution is estimated differently than in crystallography: it is typically calculated using the Fourier Shell Correlation (FSC) between two independently reconstructed half-maps [88].
The Structure Summary page on RCSB PDB provides a quick assessment of structure quality through the wwPDB Validation slider, where each row denotes a measure of structure quality [27]. The location of percentile bars indicates quality, with blue/right indicating better and red/left indicating worse metrics [27].
Table: Key Resources for Structural Selection and Analysis
| Resource | Type | Function and Application |
|---|---|---|
| RCSB PDB | Database | Primary portal for accessing and searching structural data [35] |
| Mol* | Visualization Tool | Web-based 3D structure visualization and analysis [26] |
| ProteinTools | Analysis Toolkit | Web server for identifying hydrophobic clusters, hydrogen bonds, salt bridges [89] |
| PISCES Server | Curation Tool | Removes sequence redundancy and selects highest-quality structures [88] |
| SIFTS Database | Mapping Resource | Maps PDB entries to UniProt, CATH, SCOP, and other databases [88] |
| AlphaFold DB | Prediction Database | Access to AI-predicted protein structures for comparison [90] |
Experimental Protocols for Structure Validation
Protocol for Assessing Structure Quality Using RCSB PDB Tools
Access the Structure Summary Page: Enter the PDB ID in the search bar on RCSB.org to access the dedicated page for your structure of interest [27].
Review the Header Section: Examine the experimental method, resolution (for X-ray and cryo-EM), and source organisms [27].
Analyze Validation Sliders: For experimental structures, check the wwPDB Validation slider in the Header section. The solid bar represents the structure's quality percentile relative to all structures, while the hollow bar represents quality relative to structures solved by the same method [27].
Examine Ligand Quality: For X-ray structures with ligands, check the Ligand Structure Quality Assessment slider. The closer the bar is to the blue end, the better the goodness of fit to experimental data [27].
Download Validation Report: Click the "Download Full Validation Report" button for comprehensive quality metrics including Ramachandran outliers, rotamer outliers, and clash scores [27].
Visualize in 3D: Click "Validate in 3D" to open the structure in Mol* with validation data mapped directly onto the 3D model [27].
Protocol for Selecting Biologically Relevant Assemblies
Identify Potential Assemblies: On the Structure Summary page, view the "Snapshot of the Structure" section. Click the arrows in the gray heading bar to view different biological assemblies [27].
Evaluate Assembly Symmetry: Underneath each biological assembly snapshot, examine the local, global, and pseudo symmetries of the structure [27].
Compare Assembly Contents: Assess the number and arrangement of chains in each assembly to determine which represents the functional biological unit [10].
Use "Find Similar Assemblies": Click this hyperlink below the symmetry information to search for structures with similar quaternary organization [27].
Validate with External Resources: For membrane proteins, check links to membrane protein-specific databases (OPM, PDBTM) for additional validation of assembly organization [27].
Advanced Applications and Special Cases
Working with Computed Structure Models
For Computed Structure Models (CSMs) from sources like AlphaFold DB, the Structure Summary page provides specific information including model confidence metrics [27]. CSMs are colored by a model confidence score (pLDDT) where regions of high confidence are colored dark blue and regions of lower confidence are colored yellow or orange [27]. The Model Confidence section lists a pLDDT global score and a histogram showing residue-level confidence [27].
Utilizing External Databases and Tools
The RCSB PDB integrates with numerous specialized resources to enhance structural analysis:
- SIFTS Database: Provides comprehensive mapping between PDB entries and external databases including UniProt, CATH, SCOP, and Pfam [88].
- Structural Alignment Tools: FATCAT, TM-align, CE, or Smith-Waterman 3D alignment provide insights into sequence and structural relationships, powerful for identifying similar protein shapes despite sequence differences [88].
- Specialized Analysis Servers: Tools like ProteinTools offer dedicated analysis of hydrophobic clusters, hydrogen bond networks, salt bridges, and contact maps through an interactive web interface [89].
Structure Prediction as a Complementary Approach
When experimental structures are unavailable or incomplete, prediction tools can provide valuable structural insights. Key resources include:
- AlphaFold DB: Database of pre-computed models from DeepMind's AlphaFold system [90].
- ColabFold: Web server combining fast homology search with AlphaFold2 for rapid model generation [90].
- ESMFold: Language model-based approach that can predict structures without multiple sequence alignments [90].
These tools are particularly valuable for designing mutants, understanding alternative splicing variants, and preliminary screening of ligands [91]. However, they have limitations with antibodies, intrinsically disordered regions, and allosteric mechanisms [91].
Selecting the right protein structure is a foundational step in structural bioinformatics that requires careful consideration of biological relevance, experimental quality, and functional context. By following the systematic framework outlined in this guide—defining precise biological criteria, applying rigorous quality controls, identifying biologically relevant assemblies, and leveraging integrated bioinformatics resources—researchers can ensure their structural analyses yield reliable, biologically meaningful insights. As the PDB continues to grow and evolve, these best practices will remain essential for leveraging structural data to advance scientific discovery and drug development.
Ensuring Reliability: Model Validation and Cross-Method Comparisons
Understanding the wwPDB Validation Report and Key Metrics
Within the framework of foundational Protein Data Bank (PDB) research, the validation of three-dimensional structural models is a critical pillar for ensuring data quality, reliability, and reproducibility. The worldwide PDB (wwPDB) validation report provides a standardized, comprehensive assessment of structural models and their associated experimental data. These reports are integral to the scientific process, serving as a crucial checkpoint for depositors, reviewers, and journal editors, and are a required component of manuscript submission for many leading scientific journals [92]. This guide provides an in-depth technical examination of the wwPDB validation report, its core metrics, and its practical application for researchers and drug development professionals.
The wwPDB Validation Service and Report Generation
The wwPDB Validation Service (https://validate.wwpdb.org) is a standalone web server that allows researchers to upload their structural models and experimental data to generate a validation report identical to the one produced during the official deposition process [93]. This pre-deposition check is highly recommended to identify and correct potential issues prior to formal submission.
To use the service, users must create a validation account. The process involves uploading coordinate files (e.g., for X-ray crystallography, NMR, or 3D Electron Microscopy) and, optionally, the corresponding experimental data files. The server performs automated checks and sends an email notification upon completion, typically within 5-10 minutes for most structures, though NMR ensembles or large models may take longer [93]. It is crucial to note that the report generated by this standalone service is preliminary and should not be submitted to journals. The official, confidential validation report is provided by wwPDB biocurators only after the structure has been formally deposited via the OneDep system [93].
The following diagram illustrates the two-stage process of validation and deposition:
Global Quality Assessment Metrics
A central feature of the wwPDB validation report is the "Overall quality at a glance" section, which provides percentile-based sliders for key global quality indicators. These sliders position the deposited structure relative to all other structures in the PDB archive and to a resolution-similar subset, offering an immediate, contextualized quality assessment [94] [92].
Core Global Quality Metrics
Table 1: Core Global Quality Metrics in the wwPDB Validation Report
| Metric | Description | Interpretation | Method Relevance |
|---|---|---|---|
| Clashscore | Number of severe atomic overlaps per 1000 atoms. | Lower values indicate better steric quality. A high Clashscore suggests problematic van der Waals contacts. | Primarily X-ray, also 3DEM |
| Ramachandran outliers | Percentage of residues in disallowed regions of the Ramachandran plot. | Lower percentages indicate more plausible protein backbone conformations. | X-ray, NMR, 3DEM |
| Sidechain outliers | Percentage of residues with unlikely rotamer conformations. | Lower percentages indicate more accurate sidechain placement. | X-ray, NMR, 3DEM |
| RSRZ outliers | Real-Space R Z-score for poor model-to-density fit (X-ray/3DEM). | Identifies residues where the atomic model does not fit the experimental density well. | X-ray, 3DEM |
| Q-score | Average per-atom quality index measuring resolvability in 3DEM maps. | Ranges from 0 (unresolved) to 1 (well-resolved). Higher values indicate better model-map fit [94]. | 3DEM |
Method-Specific Enhancements
The wwPDB continuously refines its validation offerings. A significant recent development for 3DEM structures is the introduction of a Q-score percentile slider in the validation report. This slider, added in October 2025, compares an entry's average Q-score against the entire EMDB/PDB archive and a resolution-similar subset [94]. Because Q-score correlates strongly with resolution between 1–10 Å, an unusually low percentile can flag issues with model-map fit or map quality, providing a powerful at-a-glance assessment for depositors, reviewers, and users [94].
For NMR structures, validation includes an assessment of the Random Coil Index (RCI), which predicts protein flexibility using secondary chemical shifts. The validation report can display this information, coloring the structure by the RCI to help identify regions of intrinsic disorder [86] [85].
Ligand and Small Molecule Validation
The accurate representation of small molecule ligands, inhibitors, cofactors, and drugs is paramount, especially in structural biology-driven drug discovery. The wwPDB validation report provides a detailed analysis of ligand geometry and fit.
During deposition, ligands in the uploaded coordinate file are compared against the wwPDB Chemical Component Dictionary (CCD) [95]. The validation report includes:
- Geometry Checks: Bond lengths, bond angles, and chiral centers are compared to ideal values from small-molecule crystallographic data.
- Fit to Density: The Real Space R (RSR) and Real Space Correlation Coefficient (RSCC) quantify how well the ligand's atomic model explains the experimental electron density (X-ray) or map (3DEM).
- Steric Clashes: The report identifies severe atomic overlaps between the ligand and the surrounding macromolecular environment.
Ongoing efforts to improve ligand validation include the PDBe's updated pipeline using pdbeccdutils and PDBe Arpeggio software. These tools systematically identify covalently linked ligands and calculate interatomic contacts between ligands and proteins, respectively, standardizing and enriching interaction data across the PDB archive [96].
Method-Specific Validation Protocols
The wwPDB validation pipeline tailors its checks based on the experimental method used for structure determination. The following sections outline the core methodologies and validation criteria for the three primary techniques.
X-ray Crystallography
Table 2: Key Validation Metrics for X-ray Crystallography Structures
| Category | Specific Metrics | Data Requirements |
|---|---|---|
| Data Quality | Resolution, Rmerge, Rmeas, I/σ(I), CC1/2, Completeness, Multiplicity | Structure factor file (e.g., .mtz, .cif) |
| Model Quality | R-work, R-free, Clashscore, Ramachandran outliers, Sidechain outliers, RSRZ outliers | Coordinate file (.pdb, .cif) |
| Ligand Fit | Real Space Correlation Coefficient (RSCC), Real Space R (RSR) | Coordinates and structure factors |
Nuclear Magnetic Resonance (NMR) Spectroscopy
For NMR structures, the validation process assesses both the coordinates of the structural ensemble and the underlying experimental restraints [95]. The key components and checks include:
- Restraint Validation: The report analyzes the completeness of chemical shift assignments and the agreement between the deposited coordinates and the experimental restraint data (e.g., distance, dihedral angle, and residual dipolar coupling restraints) [86].
- Ensemble Quality: Checks for the structural integrity of each model in the ensemble and the overall compactness of the ensemble.
- Chemical Shift Analysis: Validation includes an assessment of the chemical shift data itself, checking for values outside expected ranges and potential atom nomenclature issues during file upload [95].
- Random Coil Index (RCI): As mentioned, the RCI is used to predict protein flexibility and is integrated into the validation report's annotation presets [86] [85].
3D Electron Microscopy (3DEM)
3DEM validation has seen significant advances, particularly with the formalization of Q-score as a standard metric. The validation report for a 3DEM structure with an atomic model includes both model and map quality assessments [94] [97].
- Map Quality: The global resolution is reported based on the Fourier Shell Correlation (FSC) threshold criterion (typically FSC=0.143). The deposition of half-maps used for the FSC calculation is mandatory [95].
- Model-Map Fit: The Q-score is a central metric, measuring the resolvability of individual atoms in the map. The recently introduced Qrelativeall and Qrelativeresolution percentiles allow for a statistical assessment of the model-map fit quality compared to the entire archive and to structures of similar resolution [94].
- Model Quality: Standard geometry checks (Clashscore, Ramachandran, etc.) are performed, and the fit of the model to the map is quantified by per-residue RSRZ scores.
The workflow for processing and validating a 3DEM entry is summarized below:
Table 3: Essential Tools and Resources for PDB Deposition and Validation
| Tool/Resource | Function | Access/URL |
|---|---|---|
| wwPDB OneDep System | Unified system for depositing structures to the PDB. | http://deposit.wwpdb.org [95] |
| Standalone Validation Server | Pre-deposition validation service for generating preliminary reports. | https://validate.wwpdb.org [93] |
| MolProbity | All-atom contact geometry validation tool integrated into the wwPDB pipeline. | http://molprobity.biochem.duke.edu [86] [97] |
| Mol* Viewer | 3D structure viewer used in wwPDB sites; allows visualization of validation annotations. | Integrated at RCSB PDB, PDBe, PDBj [85] |
| MolViewSpec | A Mol* extension for creating, sharing, and reproducing molecular scenes and figures. | molstar.org [94] |
PDBe pdbeccdutils |
Software library for processing small molecule ligands in the PDB. | https://pdbeurope.github.io/ccdutils/ [96] |
| Chemical Component Dictionary (CCD) | Reference dictionary of all approved small molecule components in PDB entries. | Accessible via wwPDB sites |
Future Directions and Ongoing Remediation
The wwPDB is a dynamic resource, with continuous efforts to enhance validation and maintain archive-wide data quality. Two significant ongoing initiatives are:
- Metalloprotein Remediation: Planned for Q3 2026, this project will update approximately 13,000 PDB entries and 900 metal-containing chemical components. The goal is to ensure FAIR data practices for metalloproteins by standardizing calculated charge, improving ideal coordinate calculation, and adding detailed metal coordination and pi-bond annotations using community software like FindGeo and MetalCoord [94].
- Transition to PDBx/mmCIF and Extended PDB IDs: The wwPDB strongly encourages all users to adopt the PDBx/mmCIF file format and the new 12-character alphanumeric PDB IDs (e.g.,
pdb_00001abc). This transition is essential to support the continued growth of the archive, as the legacy four-character IDs are expected to be fully assigned before 2028 [94].
The wwPDB validation report is an indispensable tool in the structural biologist's arsenal, providing a standardized, authoritative, and comprehensive assessment of the quality of macromolecular structures. For researchers in academia and drug development, a deep understanding of its metrics—from global indicators like the Clashscore and Ramachandran plot to method-specific measures like the R-free and Q-score—is fundamental for critical data evaluation. As the field advances with higher-resolution structures and increasingly complex macromolecular machines, the wwPDB's ongoing development of validation methods, such as the recent Q-score percentiles for 3DEM, ensures that the foundation of structural biology remains robust, reliable, and fit for the demands of modern science.
For researchers, scientists, and drug development professionals, selecting and critically evaluating three-dimensional macromolecular structures from the Protein Data Bank (PDB) is a fundamental task. The reliability of any structural analysis, whether for understanding enzyme mechanisms, interpreting disease-associated mutations, or designing new therapeutics, hinges on the quality of the underlying model. This guide provides an in-depth examination of three core metrics—Resolution, R-factors, and Clash Scores—which serve as essential indicators of the confidence one can place in a PDB entry. These metrics are foundational for assessing the quality of experimentally determined structures, a crucial skill for effective research within the structural biology and drug discovery ecosystem [25].
Core Quality Metrics Explained
Resolution
In X-ray crystallography, resolution is a primary measure of overall structure quality, indicating the level of detail visible in the electron density map used to build the atomic model. It is reported in angstroms (Å), and it fundamentally describes how well two adjacent atoms in the structure can be distinguished [25]. The numerical value has an inverse relationship with quality; a lower resolution value corresponds to a higher-quality structure. For instance, a structure determined at 1.8 Å resolution provides more atomic detail and is more reliable than one determined at 3.0 Å. However, resolution is a global metric and does not, by itself, highlight regions of local disorder or inaccuracies within the model [25].
Table: Interpretation of Resolution Ranges for X-ray Crystal Structures
| Resolution (Å) | Quality Tier | Typical Information Level |
|---|---|---|
| < 1.5 | Very High | Fine details are clear, including individual atoms and some hydrogen atoms. Ideal for detailed mechanistic studies and drug design. |
| 1.5 - 2.0 | High | Clear tracing of the polypeptide chain; distinct side-chain densities. Suitable for most analyses, including ligand binding. |
| 2.0 - 2.5 | Medium | Overall fold is clear, but side-chain conformations may be ambiguous. Requires more caution in interpretation. |
| 2.5 - 3.0 | Low | The polypeptide chain path may be unclear in regions. Bulk side-chain positions can be modeled. |
| > 3.0 | Very Low | The model is less reliable; often only the coarse fold and main chain are visible. |
R-factors
R-factors are statistical measures that quantify the agreement between the atomic model and the experimental X-ray diffraction data collected during the structure determination process [25]. The most commonly reported R-factor is the R-work, which assesses the fit for the data used in refining the model. To prevent over-fitting, an independent validation metric called R-free is calculated using a small, withheld portion of the experimental data (a "test set") that was not used during refinement [25]. In a high-quality structure, the R-work and R-free values are typically low and relatively close, often differing by about 0.05 (or 5%). A large discrepancy between R-work and R-free may indicate over-interpretation of the data or errors in the model [25].
Clashscores and Stereochemical Checks
While resolution and R-factors assess the fit to experimental data, clashscores and other stereochemical checks evaluate the model's agreement with known physical and chemical constraints. The clashscore is a specific metric calculated by MolProbity and reported in wwPDB validation reports. It is defined as the number of serious, steric atom-atom overlaps per 1000 atoms [98]. A lower clashscore indicates a more favorable and physically realistic model, as it has fewer atomic clashes.
Beyond the clashscore, comprehensive validation includes checks for covalent bond distances and angles against standard values, correct stereochemistry at chiral centers, and proper atom nomenclature [82]. These checks are run as part of the PDB's integrated data processing system, and serious errors are corrected through annotation and correspondence with the depositing authors [82].
Table: Key Quality Assessment Metrics and Their Interpretation
| Metric | What It Measures | Ideal Values / Interpretation |
|---|---|---|
| Resolution | Level of detail in experimental data [25]. | Lower is better (< 2.0 Å is generally good). |
| R-work | Fit of model to refinement data [25]. | Lower is better; context-dependent on resolution. |
| R-free | Fit of model to validation data [25]. | Should be close to R-work (within ~0.05). |
| Clashscore | Steric hindrance in the model [98]. | Lower is better (fewer atomic clashes). |
| Ramachandran Outliers | Protein backbone torsion angle sanity [85]. | Lower percentage is better; indicates favored conformations. |
| Rotamer Outliers | Protein side-chain conformation sanity. | Lower percentage is better; indicates standard side-chain packing. |
| Real Space Correlation (RSCC) | Local fit of model to electron density [25]. | Ranges from 0 to 1; higher is better (>0.8 is generally acceptable). |
The Structure Validation Workflow
Understanding how these quality metrics are generated and integrated is key to interpreting them. The worldwide PDB (wwPDB) employs a rigorous, multi-stage validation pipeline for every deposited experimental structure. The following workflow diagram outlines this standardized process, from data deposition to the final report used by researchers.
Validation Pipeline
A Practical Protocol for Assessing Model Quality
This section provides a detailed, step-by-step methodology for researchers to systematically evaluate the quality of a PDB structure, integrating the core metrics discussed above.
Objective: To perform a comprehensive quality assessment of a PDB entry, focusing on its global quality, local fit, and stereochemical sanity, thereby determining its suitability for specific research applications such as mechanistic analysis or molecular docking.
Required Tools & Resources:
- A PDB entry identifier (e.g., 4HHB).
- Access to the RCSB PDB website (www.rcsb.org).
- The wwPDB validation report for the entry.
- Molecular graphics software (e.g., Mol*, PyMOL, UCSF Chimera).
Procedure:
Access the Structure Summary Page: Navigate to the RCSB PDB and enter the PDB ID in the search bar. The Structure Summary Page is the central hub for information about the entry.
Evaluate Global Quality Metrics:
- Locate the "Experimental Data & Validation" section on the page.
- Record the resolution (for X-ray structures). Compare the value to the tiers in Table 1 to establish an initial expectation of model quality.
- Record the R-work and R-free values. Confirm that the values are reasonably low for the given resolution and that the difference between them is not large (typically within ~0.05). A large discrepancy warrants caution.
Analyze the wwPDB Validation Report:
- Click the link to download or view the full validation report. This report provides a unified summary of all quality checks.
- Identify the Clashscore. This is typically found in a section summarizing "Geometry" or "Stereochemistry." Lower scores are better; the report often provides a percentile ranking compared to structures of similar resolution.
- Check the Ramachandran plot statistics. Note the percentage of residues in the "favored" and "outlier" regions. A high-quality model typically has >90% in favored regions and <1% outliers.
- Review the rotamer outlier statistics. This indicates the percentage of side chains with unusual conformations.
Inspect Local Quality and Ligand Fit:
- For regions of specific interest (e.g., an active site or a ligand-binding pocket), consult the per-residue validation data.
- Use the Real Space Correlation Coefficient (RSCC) to assess local fit. This value ranges from 0 (no correlation) to 1 (perfect correlation). Residues and ligands with RSCC values below 0.8 should be treated with caution, and those below 0.7 are considered poorly supported by the electron density [25].
- For structures containing small-molecule ligands, consult the dedicated ligand quality plots available on the "Ligands" tab of the RCSB PDB site. These 2D plots visually summarize the ligand's fit to the density (X-axis) and its geometric correctness (Y-axis) [24].
Visualize the Model and Data:
- Open the structure in a molecular graphics viewer like Mol*.
- Utilize "Validation Report" presets in the Mol* "Components" panel to color the structure by metrics like "Density Fit" or "Geometry Quality," which can visually highlight problematic regions [85].
- For critical regions, visually inspect the atomic model against the experimental electron density map (2mFo-DFc map) to verify the interpretation.
The following diagram summarizes the logical decision process a researcher should employ when evaluating a structure using these metrics.
Quality Decision Tree
This table details key resources and tools used in the validation and analysis of PDB structures.
Table: Essential Resources for Structure Validation and Analysis
| Tool / Resource | Type | Primary Function in Quality Assessment |
|---|---|---|
| wwPDB Validation Server | Web Service | Provides the official validation report for a PDB entry, integrating all major metrics (R-factors, clashscore, Ramachandran, etc.) into a single document [98]. |
| MolProbity | Software Suite | An all-atom contact analysis tool that calculates the clashscore and identifies steric outliers, rotamer outliers, and provides Ramachandran analysis [98]. |
| Mol* | Visualization Software | An integrated 3D structure viewer on RCSB.org that allows visualization of the model, electron density maps, and validation annotations like clashes and geometric outliers [85]. |
| Uppsala Electron Density Server (EDS) | Web Service | Calculates real-space fit measures (RSR and RSCC) for models against electron density, providing crucial local quality indicators [24]. |
| PDB Chemical Component Dictionary (CCD) | Data Dictionary | Defines the ideal chemical geometry for all small-molecule ligands found in the PDB, serving as the reference for ligand geometry validation [24]. |
Structural biology has been fundamentally transformed by the advent of high-accuracy computed structure models (CSMs), which complement the experimentally determined structures archived in the Protein Data Bank (PDB). For decades, the PDB has served as the single global archive for experimentally determined 3D structures of biological macromolecules, with more than 210,000 structures as of 2023 [99] [22]. The Worldwide PDB (wwPDB) partnership manages this archive, ensuring FAIR (Findability, Accessibility, Interoperability, and Reusability) principles for the global scientific community [22]. However, experimental structure determination remains time-consuming, expensive, and technically challenging, leaving billions of proteins in nature without structural characterization [63].
The revolutionary development of artificial intelligence and machine learning (AI/ML) systems, particularly AlphaFold2 and RoseTTAFold, has enabled accurate protein structure prediction from amino acid sequences alone [100] [63]. These advances have expanded the structural universe by three orders of magnitude, with resources like the AlphaFold Protein Structure Database now containing predictions for over 214 million sequences [99]. The integration of approximately one million CSMs with traditional PDB structures on the RCSB.org portal provides researchers with an unprecedented comprehensive view of structural proteomes [100] [99]. This integration is particularly valuable for drug development professionals who require structural insights for target identification and characterization.
Table 1: Fundamental Characteristics of Experimental Structures and CSMs
| Characteristic | Experimental Structures (PDB) | Computed Structure Models (CSMs) |
|---|---|---|
| Source | Experimental measurement (X-ray, NMR, EM) | Computational prediction from sequence |
| Data Foundation | Experimental diffraction patterns, magnetic resonance data, electron density maps | Protein sequences, multiple sequence alignments, existing PDB structures |
| Confidence Metrics | Resolution, R-factor, R-free, RSCC, Q-score | pLDDT (per-residue and global) |
| Coverage | ~200,000 structures (as of 2022) | ~1,000,000+ models available via RCSB.org |
| Environmental Factors | Include ligands, solvents, modifications | Generally apo forms without ligands |
| Dynamic Information | May capture multiple states/conformations; NMR provides ensembles | Typically single static conformation |
Methodological Foundations: Experimental Determination vs. Computational Prediction
Experimental Structure Determination Methods
Experimental methods for structure determination have evolved significantly over the past five decades, with technical innovations driving exponential growth in PDB archival holdings [22]. The three primary experimental methods each have distinct methodological approaches:
Macromolecular Crystallography (MX) represents approximately 87% of the PDB archive and involves protein crystallization followed by X-ray irradiation [22] [25]. The resulting diffraction patterns are used to solve the phase problem through molecular replacement (MR), multiple-wavelength anomalous dispersion (MAD), or other phasing methods [22]. The quality of MX structures is primarily assessed by resolution (lower values indicating better quality), with most structures determined at resolutions between 1.0-3.0 Å [22]. Additional validation metrics include R-factor and R-free values (lower values indicating better agreement with experimental data), with typical R-free values around 0.25 (25%) for high-quality structures [25].
Nuclear Magnetic Resonance (NMR) spectroscopy accounts for approximately 7% of PDB structures and provides solution-state structural information [22] [25]. NMR exploits the magnetic properties of atomic nuclei to measure interatomic distances and dihedral angles, which serve as restraints for calculating structural ensembles [25]. Key quality indicators include the number of restraints per residue and restraint violations, with the Random Coil Index (RCI) providing information on residue flexibility and disordered regions [25].
3D Electron Microscopy (3DEM) represents the fastest-growing experimental method, with archival holdings increasing approximately six-fold in just four years [22]. This method is particularly valuable for studying large macromolecular complexes and membrane proteins. 3DEM quality is assessed through Fourier Shell Correlation (FSC) resolution estimates and Q-scores that evaluate the fit between atomic models and EM maps [25]. Recent technical advances have pushed 3DEM resolution to near-atomic levels (e.g., 1.15 Å for apoferritin) [22].
Computed Structure Model Generation
The emergence of AI/ML approaches has revolutionized protein structure prediction, with AlphaFold2 and RoseTTAFold representing the current state-of-the-art [100] [63]. These methods employ sophisticated neural network architectures that leverage evolutionary information and physical constraints:
AlphaFold2 generates structures through an iterative process that starts with multiple sequence alignment generation [63]. The system then employs an Evoformer module to process related sequences and extract co-evolutionary signals, followed by a structure module that combines these signals with physical principles to generate atomic coordinates [100] [63]. The final output includes both the 3D coordinates and a per-residue confidence score (pLDDT) ranging from 0-100 [100] [63].
RoseTTAFold utilizes a three-track neural network that simultaneously processes sequence, distance, and coordinate information [100]. This architecture allows the system to efficiently integrate patterns at the sequence and structural levels, producing accurate models particularly for protein-protein complexes [100].
Both methods rely heavily on the growing wealth of experimental structures in the PDB, which serve as essential training data and structural templates, creating a synergistic relationship between experimental and computational approaches [63].
Diagram 1: Methodological Pathways for Structure Determination. Experimental and computational approaches represent distinct pathways for deriving 3D structural information from protein sequences.
Quality Assessment and Validation Frameworks
Experimental Structure Validation
The wwPDB has established comprehensive validation pipelines for each experimental method, developed by expert Validation Task Forces [25]. These pipelines assess both global and local structure quality using multiple orthogonal metrics:
X-ray crystallography structures are validated against the experimental structure factor data [25]. The Real-Space-Correlation-Coefficient (RSCC) has emerged as a particularly valuable local quality metric, measuring agreement between atomic coordinates and electron density for individual residues [25]. RSCC values range from 0-1, with higher values indicating better agreement. Statistical analysis of over 100 million amino acid residues in PDB structures has established that residues with RSCC in the lowest 1% should not be trusted, while those in the lowest 1-5% should be used with caution [25].
NMR structures undergo chemical shift validation and restraint analysis [25]. Unusual chemical shifts may indicate truly strained conformations or assignment errors, requiring careful interpretation. The number and magnitude of restraint violations provide crucial information about how well the structural ensemble satisfies the experimental data [25].
3DEM validation has advanced significantly with the development of quantitative metrics like the Q-score, which measures the resolvability of atoms in cryo-EM maps [25]. Q-scores can be calculated for individual atoms and averaged across residues or complete models, providing a standardized assessment of map-model fit [25].
Table 2: Quality Assessment Metrics for Experimental Structures and CSMs
| Method | Primary Global Metric | Primary Local Metric | Interpretation Guidelines |
|---|---|---|---|
| X-ray Crystallography | Resolution (Å); R-free | Real-Space-Correlation-Coefficient (RSCC) | Resolution < 2.0 Å = high quality; RSCC < 5th percentile = unreliable |
| NMR Spectroscopy | Restraint violations; RCI | Per-residue restraint violations | Few violations with small magnitudes = high quality; High RCI = disordered regions |
| 3D Electron Microscopy | FSC resolution (Å); Map-model fit | Q-score (per-residue/atom) | Resolution < 3.0 Å = high quality; Q-score > 0.8 = well-resolved |
| AlphaFold2/RoseTTAFold | Global pLDDT | Per-residue pLDDT | pLDDT > 90 = high confidence; pLDDT < 50 = very low confidence |
CSM Confidence Estimation
For CSMs, the predicted Local Distance Difference Test (pLDDT) serves as the primary confidence metric [100] [63] [25]. This score estimates the reliability of the predicted structure based on how well it agrees with the multiple sequence alignment data and reference structures used during prediction [25]. The pLDDT ranges from 0-100 and is interpreted as follows:
- Very high confidence (pLDDT > 90): Comparable to high-quality experimental structures
- Confident (70 < pLDDT < 90): Generally reliable for structural analysis
- Low confidence (50 < pLDDT < 70): Should be interpreted with caution
- Very low confidence (pLDDT < 50): Likely disordered or unreliable [100] [25]
Recent evaluations comparing AlphaFold predictions with experimental electron density maps have provided crucial insights into their real-world accuracy [101]. While many high-confidence predictions match experimental maps remarkably closely, some show significant deviations despite high pLDDT scores [101]. Systematic analysis reveals that AlphaFold predictions typically show median Cα RMSD of 1.0 Å from experimental structures, compared to 0.6 Å between different experimental determinations of the same protein [101]. This indicates that while highly accurate, CSMs generally exhibit greater deviations from experimental references than pairs of experimental structures determined under different conditions.
Access and Utilization in Research
Accessing Structures via RCSB.org
The RCSB.org portal provides unified access to both experimental structures and CSMs, with specific visual cues to distinguish between them [100] [27]. Experimental structures are marked with a dark blue flask icon, while CSMs are identified with a cyan computer icon [100] [27]. This distinction is maintained throughout search results, structure summary pages, and visualization tools.
The portal offers multiple search paradigms, including options to include or exclude CSMs in searches via a toggle switch [100]. Advanced search capabilities allow querying based on CSM-specific attributes such as source database and confidence levels [100]. Search results can be filtered to show only experimental structures or only CSMs, and can be ordered by relevance, pLDDT scores, or other criteria [100].
Structure Summary Pages for CSMs include several specialized sections not found in experimental structure pages [27]. The Model Confidence section displays global pLDDT scores and histograms showing the distribution of per-residue confidence scores, enabling rapid assessment of model reliability [27]. These pages also provide direct links to source databases and associated protein sequence information [27].
Practical Applications and Limitations
Both experimental structures and CSMs have distinct strengths and limitations that make them suitable for different research applications:
Experimental structures remain essential for understanding molecular interactions with ligands, drugs, cofactors, and nucleic acids [63] [101]. They capture the effects of post-translational modifications, crystallization conditions, and environmental factors [101]. Approximately 95% of high-resolution MX structures (better than 2.5 Å) provide more accurate atomic-level information than corresponding CSMs [63]. Experimental methods also excel at capturing conformational flexibility, multimeric assemblies, and transient states [63].
CSMs provide unprecedented coverage of proteomes and are particularly valuable for proteins that have resisted experimental structure determination [100] [63]. They serve as excellent starting models for molecular replacement in crystallography, guide hypothesis generation about protein function, and facilitate structure-based drug discovery for targets without experimental structures [63]. The case study of the Src oncoprotein illustrates both the power and limitations of current CSMs—while well-folded domains are accurately predicted, flexible regions and domain orientations may be less reliable [63].
Diagram 2: Decision Framework for Structure Selection and Validation. This workflow guides researchers in selecting appropriate structural models and applying rigorous quality assessment before analysis.
Research Reagent Solutions: Essential Tools for Structural Biology
Table 3: Key Research Resources for Structural Analysis
| Resource | Type | Primary Function | Access |
|---|---|---|---|
| RCSB.org PDB Portal | Database & Tools | Unified access to experimental structures and CSMs; search, visualization, analysis | https://www.rcsb.org/ |
| AlphaFold Protein Structure DB | Database | Repository for AlphaFold2 predictions; >214 million models | https://alphafold.ebi.ac.uk/ |
| ModelArchive | Database | Repository for computational models from various methods; >74,000 models | https://modelarchive.org/ |
| Mol* | Visualization Tool | Interactive 3D structure visualization for both experimental structures and CSMs | Integrated in RCSB.org |
| wwPDB Validation Server | Analysis Tool | Structure validation against experimental data | https://validate.wwpdb.org/ |
| UniProt | Database | Protein sequence and functional information used for cross-referencing | https://www.uniProt.org/ |
The complementary strengths of experimental structures and CSMs create a powerful synergy for structural biology research and drug development. Experimental structures provide higher accuracy, especially for atomic-level details, and capture environmental influences, ligands, and dynamic states [63] [101]. CSMs offer unprecedented coverage of protein sequence space and serve as excellent starting points for further investigation [100] [63].
For researchers and drug development professionals, strategic integration of both approaches will be essential. Experimental structures should be preferred when available, especially for studying molecular interactions, ligand binding, and detailed mechanistic analysis [63]. CSMs provide invaluable insights for the millions of proteins without experimental characterization and can guide experimental design, hypothesis generation, and preliminary investigations [100] [63]. As the field advances, the continued refinement of both experimental and computational methods promises to further expand our understanding of protein structure and function, ultimately accelerating drug discovery and biomedical innovation.
When utilizing any 3D model—experimental or computational—researchers must consistently consider quality metrics and limitations, interpret findings within the context of these constraints, and prioritize experimental validation for definitive conclusions, particularly when exploring structural details involving interactions not included in predictions [101] [25].
Evaluating Ligand and Ion Placement with Electron Density and Map Validation
The Protein Data Bank (PDB) serves as the global repository for three-dimensional structural models of biological macromolecules. For structures determined using X-ray crystallography, which represent approximately 87% of the PDB archive, the atomic model is interpreted from experimental electron density data [25]. The agreement between the deposited atomic coordinates and the experimental electron density provides a critical measure of structural reliability, particularly for small-molecule ligands and metal ions that often play key functional roles in macromolecular function [24] [102].
Validation of these components has emerged as a crucial discipline in structural biology because incorrectly modeled ligands and ions can significantly impact downstream research, including drug discovery efforts and mechanistic interpretations [103] [104]. With over 70% of PDB structures containing one or more small-molecule ligands (excluding water molecules), establishing standardized validation metrics and protocols ensures that researchers can identify reliable structural models for their investigations [24].
This technical guide examines the foundational concepts, metrics, and methodologies for evaluating ligand and ion placement using electron density maps and validation tools, providing researchers with a framework for assessing structural quality within the context of broader PDB entry research.
Foundational Concepts of Electron Density Maps
Types of Electron Density Maps
For X-ray crystallography structures, the PDB archive contains both atomic coordinates and structure factor files representing the intensity and phase information derived from the diffraction pattern [105]. These data are combined to generate electron density maps, which visually represent the experimental data against which the atomic model is validated. Two primary map types are essential for validation:
2mFo-DFc Map (2Fo-Fc Map): This map uses observed structure factors (Fo) and calculated structure factors (Fc) to represent the overall fit of the model to the experimental data [105] [102]. It typically shows density contours surrounding all well-determined atoms in the model and is conventionally colored blue, grey, or white in molecular visualization software.
mFo-DFc Map (Fo-Fc Map): Known as a difference map, this representation highlights discrepancies between the model and experimental data [105] [102]. Positive difference density (conventionally colored green) indicates features present in the experimental data but not accounted for in the atomic model, potentially suggesting missing atoms or alternative conformations. Negative difference density (conventionally colored red) indicates features included in the model but lacking support in the experimental data, potentially representing over-interpretation or errors in model building.
Table 1: Electron Density Map Types and Their Applications in Validation
| Map Type | Calculation | Interpretation | Common Visualization Conventions |
|---|---|---|---|
| 2mFo-DFc | 2mFo-DFc | Overall fit of model to experimental data; should cover well-determined atoms | Blue, grey, or white surface |
| mFo-DFc | mFo-DFc | Differences between model and experimental data | Green (positive) and red (negative) surfaces |
| Anomalous | Based on anomalous scattering | Identification of elements with significant anomalous scattering | Varies by software; often magenta or yellow |
Accessing Electron Density Maps
Electron density maps can be accessed through several routes. The RCSB PDB provides coefficient files for 2Fo-Fc and Fo-Fc maps in PDBx/mmCIF format, available for download from the Structure Summary Page of individual entries [105]. These coefficient files can be converted to various formats compatible with molecular visualization programs:
- MTZ format for use with CCP4 programs via cif2mtz or GEMMI
- CCP4 format for use with Chimera, PyMOL, and other visualization software
- DSN6 format for use in Coot [105]
The PDBe website also offers integrated visualization of electron density maps through its online interface using the LiteMol viewer, providing accessibility for researchers who may not have specialized software installed [102].
Validation Metrics for Ligand Placement
Goodness of Fit to Experimental Data
The agreement between a ligand model and the experimental electron density data is quantified using two primary metrics:
Real Space Correlation Coefficient (RSCC): This measure evaluates the correlation between the electron density calculated from the atomic model and the observed experimental electron density in the region surrounding the ligand [24] [25]. RSCC values range from 0 to 1, with higher values indicating better agreement. For a well-fit ligand, RSCC values typically exceed 0.9, while values below 0.8 may indicate potential issues with the model [24].
Real Space R-factor (RSR): This metric measures the goodness of fit between the observed and calculated electron density [24]. Unlike RSCC, lower RSR values indicate better agreement, with values below 0.2 generally representing well-fit models.
The wwPDB validation reports provide both RSCC and RSR values for each ligand instance in a structure, allowing researchers to assess local fit to experimental data [24].
Geometric Quality Assessment
The chemical and geometric合理性 of ligand structures is assessed by comparing bond lengths and angles to established values from high-quality small-molecule crystal structures in the Cambridge Structural Database (CSD) [24] [103]. Key metrics include:
RMSD Z-scores for bond lengths and bond angles: These scores represent how much the observed geometry deviates from expected values in terms of standard deviations [24] [103]. Z-scores near zero indicate excellent agreement with expected geometry, while absolute values exceeding 2.0 are typically flagged as outliers.
Ligand geometry composite ranking scores: The RCSB PDB employs principal component analysis to aggregate correlated quality indicators into composite scores that rank ligand quality across the entire PDB archive [24]. These scores follow a uniform distribution from 0% (worst) to 100% (best), with 50% representing median quality.
Table 2: Key Validation Metrics for Ligand Quality Assessment
| Validation Category | Metric | Interpretation | Threshold Values |
|---|---|---|---|
| Experimental Data Fit | Real Space Correlation Coefficient (RSCC) | Correlation between calculated and observed electron density | >0.9 (Good), 0.8-0.9 (Acceptable), <0.8 (Poor) |
| Experimental Data Fit | Real Space R-factor (RSR) | Goodness of fit between observed and calculated electron density | <0.2 (Good), 0.2-0.3 (Acceptable), >0.3 (Poor) |
| Geometry Quality | RMSD Z-score (Bond Lengths) | Deviation of bond lengths from CSD expectations | <1.0 (Good), 1.0-2.0 (Acceptable), >2.0 (Outlier) |
| Geometry Quality | RMSD Z-score (Bond Angles) | Deviation of bond angles from CSD expectations | <1.0 (Good), 1.0-2.0 (Acceptable), >2.0 (Outlier) |
| Composite Metrics | PC1-fitting Score | Composite indicator for experimental data fit | Higher values indicate better fit |
| Composite Metrics | PC1-geometry Score | Composite indicator for geometric parameters | Higher values indicate better geometry |
Ligand Quality Assessment Workflow
The following diagram illustrates the logical workflow for assessing ligand quality using electron density maps and validation metrics:
Special Considerations for Metal Ion Validation
Challenges in Metal Ion Modeling
Metal ions present unique challenges in macromolecular structure determination due to their distinct coordination geometries and the potential for misidentification, particularly at lower resolutions [104]. Studies indicate that a substantial portion of metal ions in the PDB are either misidentified or poorly refined, highlighting the importance of specialized validation approaches [104].
The CheckMyMetal (CMM) server provides specialized validation for metal binding sites, assessing multiple parameters to identify potential issues with metal ion assignment and refinement [104].
Metal Ion Validation Metrics
CheckMyMetal evaluates metal binding sites using eight key parameters, classified into three categories (Acceptable, Borderline, and Dubious) based on established coordination chemistry principles [104]:
- Valence: Measures how well the observed bond lengths match the expected bond-valence sum for the proposed metal ion.
- Coordination geometry: Assesses whether the coordination number and geometry match known preferences for the proposed metal.
- Atomic contacts: Evaluates whether the types of atoms coordinating the metal are chemically reasonable donors for that metal.
- B-factor ratio: Compares the B-factors of the metal to those of its coordinating atoms.
- Occupancy: Checks whether the metal occupancy is consistent with the surrounding structure.
Additional parameters include nVECSUM (a measure of deviation from ideal geometry), gRMSD (geometry root-mean-square deviation), and vacancy (unoccupied coordination sites) [104].
Table 3: Metal Ion Validation Parameters from CheckMyMetal
| Parameter | Description | Acceptable Range | Borderline Range | Dubious Range |
|---|---|---|---|---|
| Valence (for Zn) | Agreement between observed and expected bond-valence | 1.7-2.3 | 1.3-1.7 or 2.3-2.7 | <1.3 or >2.7 |
| Coordination Geometry | Match to preferred coordination geometry | Preferred geometry | Other coordination numbers | Unusual geometry |
| Atomic Contacts | Chemical合理性 of coordinating atoms | Usual donor atoms | Occasionally found donors | Unusual donors |
| B-factor Ratio | Ratio of metal B-factor to ligand B-factors | 0.86-1.0 | 0.54-0.86 | <0.54 |
| Occupancy | Metal site occupancy | 0.9-1.0 | 0.1-0.9 | 0.0-0.1 |
| nVECSUM | Deviation from ideal geometry | 0-0.10 | 0.10-0.23 | 0.23-1.0 |
Practical Protocols for Validation
Accessing and Interpreting Validation Reports
The wwPDB provides comprehensive validation reports for all structures in the PDB archive. These reports are available in both human-readable PDF format and machine-readable XML format [106]. To access and interpret these reports:
- Access the validation report through the Structure Summary page on the RCSB PDB website for any entry of interest.
- Review the executive summary which provides key quality metrics at a glance, including percentile scores comparing the structure to others in the archive.
- Examine ligand-specific sections which include RSCC and RSR values for each ligand instance, along with geometric quality Z-scores.
- Consult the detailed outlier listings which identify specific residues, ligands, or geometric parameters that deviate from expected values.
For metal ions, the CheckMyMetal server (https://cmm.minorlab.org) provides specialized validation that can be used alongside the wwPDB reports [104].
Visual Inspection Workflow
While quantitative metrics are essential, visual inspection of electron density maps remains a critical component of validation:
- Generate appropriate maps: Ensure both 2mFo-DFc and mFo-DFc maps are generated at appropriate contour levels (typically 1.0σ for 2mFo-DFc and ±3.0σ for mFo-DFc maps).
- Inspect ligand density: Verify that the 2mFo-DFc map completely and continuously covers the ligand structure without significant breaks.
- Check difference density: Look for significant positive mFo-DFc density that might indicate missing atoms or alternative conformations, or negative density that might indicate over-modeling.
- Examine the binding site context: Ensure that the ligand interactions with the macromolecule are well-defined and chemically reasonable.
The following workflow illustrates the comprehensive process for validating ligand and ion placement:
Table 4: Essential Tools and Resources for Electron Density Validation
| Tool/Resource | Type | Primary Function | Access Method |
|---|---|---|---|
| wwPDB Validation Reports | Validation Service | Comprehensive quality assessment of PDB entries | https://www.rcsb.org/ or https://www.wwpdb.org/ |
| CheckMyMetal (CMM) | Specialized Validation Server | Metal binding site validation | https://cmm.minorlab.org |
| GEMMI | Software Library | Conversion of map coefficients and format manipulation | Standalone or through CCP4 |
| CCP4 Suite | Software Package | Crystallographic computation, including FFT for map generation | https://www.ccp4.ac.uk/ |
| Coot | Visualization Software | Model building and validation with electron density visualization | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PDBe LiteMol | Web-based Viewer | Integrated visualization of structures and electron density maps | https://www.ebi.ac.uk/pdbe/pdbj/ |
| UCSF Chimera | Visualization Software | Molecular visualization with electron density support | https://www.cgl.ucsf.edu/chimera/ |
| PyMOL | Visualization Software | Molecular graphics with electron density capabilities | https://pymol.org/ |
Rigorous validation of ligand and ion placement using electron density maps and quantitative metrics represents a fundamental practice in structural biology research. The integration of multiple complementary approaches—including quantitative metrics from wwPDB validation reports, specialized tools like CheckMyMetal for metal ions, and careful visual inspection of electron density maps—provides a comprehensive framework for assessing structural reliability.
As structural biology continues to play an essential role in drug discovery and mechanistic studies, proper evaluation of these critical components ensures that subsequent research, design, and development efforts build upon a foundation of reliable structural data. The protocols and metrics outlined in this guide provide researchers with a standardized approach to assess ligand and ion placement, facilitating more informed use of structural data from the PDB archive.
Within the foundational concepts of protein data bank entries research, the ability to seamlessly navigate and integrate information from specialized biological databases is paramount. The volume of data generated by modern scientific research necessitates robust data management systems. As of August 2025, GenBank release 268.0 contained 47.01 trillion bases and 5.90 billion sequence records [107]. Similarly, the UniProt Knowledgebase (UniProtKB), a central resource for protein sequence and functional information, provides expertly curated data on millions of proteins [108]. For researchers and drug development professionals, the true power of these resources is unlocked through strategic cross-referencing, creating a network of interconnected knowledge that supports complex queries and facilitates discoveries in areas such as drug target identification and enzyme function annotation. This technical guide provides a detailed methodology for navigating and integrating data from UniProt, GenBank, and chemical dictionaries like ChEBI, forming a core competency for modern bioinformatic research.
A clear understanding of the scope, content, and primary function of each database is the first step in effective cross-referencing. The following table summarizes the core characteristics of these key resources.
Table 1: Core Biological and Chemical Databases for Cross-Referencing
| Database Name | Primary Content Scope | Key Quantitative Metrics | Role in Protein Research |
|---|---|---|---|
| UniProt Knowledgebase (UniProtKB) | Protein sequences and functional annotations [108] | ~246 million sequence records (as of 2024_04 release); Contains reviewed (Swiss-Prot) and unreviewed (TrEMBL) sections [108] | Provides a centralized, curated resource for protein functional data, including enzymatic activity, pathways, and post-translational modifications. |
| GenBank | Nucleotide sequences (DNA/RNA) [107] | 47.01 trillion bases; 5.90 billion records (Release 268.0, Aug 2025) [107] | Serves as the foundational repository for genetically encoded information, enabling the link from gene to protein sequence. |
| ChEBI | Chemical entities of biological interest [109] | >195,000 manually curated entries [109] | An ontological dictionary for small molecules, critical for describing enzyme substrates, products, and drugs in structured annotations. |
| RCSB Protein Data Bank (PDB) | Experimentally-determined 3D macromolecular structures [35] | Contains over 200,000 structures (e.g., 9RC6, 9OHV) [35] | Provides the structural context for protein function, ligand binding, and rational drug design. |
Experimental Protocols for Cross-Referencing and Data Integration
Protocol 1: Establishing the Gene-Protein-Chemical Relationship
This protocol details the steps to trace a genetic sequence to its corresponding protein and associated small molecule ligands or substrates, a common workflow in target validation for drug discovery.
1. Objective: To identify the protein product of a gene of interest and characterize its interaction with relevant chemical entities.
2. Materials and Reagents:
- Computational Tools: NCBI Entrez/GenBank, UniProt BLAST, RCSB PDB Ligand Explorer.
- Biological Reagents: cDNA clone of the target gene, relevant cell line for functional expression.
- Chemical Reagents: Purified putative substrate or drug candidate (structure defined in ChEBI).
3. Methodology:
- Step 1: Gene Identification in GenBank. Initiate the search using a unique gene symbol, nucleotide accession number (e.g., NM_XXXXXX), or a sequence via BLAST [107]. The record provides the official gene name, taxonomic identifier, and the CDS (Coding Sequence) region which defines the protein product.
- Step 2: Transition to Protein Record in UniProtKB. Use the GenBank protein accession number (provided in the /translation qualifier of the CDS feature in GenBank) to query UniProtKB. Alternatively, perform a BLAST search of the nucleotide coding sequence against the UniProtKB database to retrieve the corresponding UniProt entry [108].
- Step 3: Functional Annotation in UniProtKB. In the retrieved UniProtKB/Swiss-Prot record, examine the "Function" section for enzyme classification (EC number), Gene Ontology (GO) terms, and annotated catalytic activity. These annotations often use the ChEBI ontology to describe reactions [108]. For example, the entry for human flavin reductase (P30043) describes its role in S-nitrosylation using ChEBI terms [108].
- Step 4: Chemical Entity Lookup in ChEBI. Note the ChEBI identifiers (e.g., CHEBI:XXXXX) from the UniProtKB annotation. Query the ChEBI database with these IDs to obtain the precise chemical structure, IUPAC name, and synonyms for the involved small molecules [109].
- Step 5: Structural Validation in RCSB PDB. Search the RCSB PDB for structures of the target protein, possibly in complex with its substrate or a drug. Use the Ligand Explorer tool to visualize the binding interactions. The chemical components in the structure will be annotated with links to ChEBI or similar dictionaries [35].
Diagram: Database Relationships for Gene-Protein-Chemical Workflow
Protocol 2: Retrospective Curation of Enzyme Function from Literature
This protocol leverages cross-referencing to update database entries with new functional information from recent publications, a key activity for biocuration and database enrichment.
1. Objective: To extract and formally annotate a newly discovered enzymatic activity for a protein using standardized ontologies.
2. Materials and Reagents:
- Literature Source: Peer-reviewed publication providing experimental evidence for the new function (e.g., identification of BLVRB as a nitrosylase [108]).
- Computational Tools: UniProt curation interface, ChEBI ontology browser, Rhea reaction database.
- Analytical Reagents: Assay kits to validate the purported function (e.g., S-nitrosylation detection assay).
3. Methodology:
- Step 1: Protein and Publication Identification. Identify the UniProtKB entry for the protein of interest (e.g., P30043 for BLVRB). Use tools like LitSuggest to identify key publications that report new functional data for this protein [108].
- Step 2: Data Extraction. From the publication, extract the precise details of the biochemical reaction: enzyme, substrates, cofactors, and products. Identify the specific protein residues involved in catalysis or post-translational modifications (e.g., the target cysteine residue for S-nitrosylation) [108].
- Step 3: Ontology Mapping. For each small molecule participant, find the corresponding ChEBI ID. For the overall reaction, query the Rhea database to find or request a new reaction ID. Rhea uses ChEBI for its participants, ensuring ontological consistency [108].
- Step 4: Record Annotation. In the UniProtKB/Swiss-Prot record, add the following:
- Catalytic activity annotation: Using the Rhea reaction ID.
- Gene Ontology (GO) terms: e.g., S-nitrosylation (GO:0017014).
- Active site/MOD residue annotation: Document the specific modified residue (e.g., Cys-X-X).
- Free-text comments: Summarize the finding in the "Function" section [108].
- Step 5: Causal Model Building (Advanced). Construct a GO-CAM model to describe the flow of the biochemical reaction and its role in a larger biological process, formally linking the protein, its molecular function, and the affected pathways [108].
Diagram: Workflow for Literature-Based Functional Annotation
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table details key reagents and computational tools essential for conducting research that relies on database cross-referencing, particularly for experimental validation.
Table 2: Key Research Reagents and Tools for Cross-Referencing Experiments
| Reagent / Tool Name | Category | Function in Cross-Referencing Workflow |
|---|---|---|
| BLAST (Basic Local Alignment Search Tool) | Computational Tool | Identifies homologous sequences across GenBank and UniProt, enabling the transfer of functional annotations from well-characterized proteins to novel sequences [108]. |
| cDNA Clone | Biological Reagent | Provides the exact protein-coding sequence for a gene, serving as the physical link between a GenBank record and a recombinantly expressed protein for functional study. |
| ChEBI Ontology | Computational Resource | Provides standardized, machine-readable identifiers for small molecules, enabling precise annotation of metabolites, drugs, and reaction participants in UniProtKB and RCSB PDB [109] [108]. |
| S-nitrosylation Detection Assay | Analytical Reagent Kit | Validates functional predictions (e.g., from UniProtKB annotations) by experimentally confirming the transfer of a nitrosyl group (ChEBI: CHEBI:16480) to a target protein [108]. |
| Rhea Reaction Database | Computational Resource | A curated resource of biochemical reactions that uses ChEBI identifiers; used by UniProt curators to annotate enzymatic activities in a computable form [108]. |
Advanced Applications in Drug Development
For drug development professionals, sophisticated cross-referencing is indispensable. A prime application is in the study of Antimicrobial Resistance (AMR). Researchers can identify proteins in ESKAPE pathogens (e.g., E. coli, S. aureus) that play direct roles in AMR, such as beta-lactamases and efflux pumps, through UniProtKB annotations [108]. By tracing these proteins back to their genes in GenBank, one can analyze sequence variation across clinical isolates. Furthermore, by querying the RCSB PDB, researchers can obtain 3D structures of these proteins, often co-crystallized with inhibitors [35]. The chemical components of these drugs and their targets are systematically described using the ChEBI ontology, enabling a unified view from genetic determinant to chemical inhibitor. This integrated approach facilitates the identification of resistance mechanisms and the rational design of new drugs to overcome them.
Conclusion
Mastering the foundational concepts of the Protein Data Bank is indispensable for modern biomedical research. By understanding the archive's organization, the strengths and limitations of different structure determination methods, and the principles of data validation, researchers can confidently extract meaningful biological insights. The ongoing expansion of the archive, the integration of high-quality computed models, and advancements in techniques like cryo-EM promise an even richer structural understanding of biological processes. This progress will continue to be a major catalyst for innovation in structure-guided drug discovery, the design of novel biologics, and the fundamental understanding of disease mechanisms, ultimately accelerating the translation of structural knowledge into clinical applications.
References
- 1. Supporting the PDB Archive [https://www.rcsb.org/news/6171a4a9ef055f03d1f22326]
- 2. wwPDB: PDB Archive Downloads [https://www.wwpdb.org/ftp/pdb-ftp-sites]
- 3. wwPDB: Remediation [https://www.wwpdb.org/documentation/remediation]
- 4. wwPDB Format version 3.3: Introduction [https://www.wwpdb.org/documentation/file-format-content/format33/sect1.html]
- 5. Remediation of the protein data bank archive - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2238854/]
- 6. wwPDB: PDB Versioned Repository [https://www.wwpdb.org/ftp/pdb-versioned-ftp-site]
- 7. Overall Growth of Released Structures Per Year [https://www.rcsb.org/stats/growth/growth-released-structures]
- 8. PDB Statistics: Data Storage Growth [https://www.rcsb.org/stats/data_storage_growth]
- 9. Worldwide Protein Data Bank: wwPDB [https://www.wwpdb.org/]
- 10. Organization of 3D Structures in the Protein Data Bank [https://www.rcsb.org/docs/general-help/organization-of-3d-structures-in-the-protein-data-bank]
- 11. Identifiers in PDB [https://www.rcsb.org/docs/general-help/identifiers-in-pdb]
- 12. Molecular Graphics Programs - PDB-101 [https://pdb101.rcsb.org/learn/guide-to-understanding-pdb-data/molecular-graphics-programs]
- 13. Getting Started [https://www.rcsb.org/docs/3d-viewers/mol*/getting-started]
- 14. RCSB PDB Newsletter: Fall 2025 [https://cdn.rcsb.org/rcsb-pdb/general_information/news_publications/newsletters/2025q4/home.html]
- 15. Announcing the New PDBx/mmCIF User Guide [https://www.rcsb.org/news/666a056d526f3281a10043bc]
- 16. Pairwise Structure Alignment [https://www.rcsb.org/docs/tools/pairwise-structure-alignment]
- 17. Residue.num — PDB-style residue number [https://salilab.org/modeller/9.24/manual/node352.html]
- 18. Introduction to Protein Data Bank Format [https://www.cgl.ucsf.edu/chimera/docs/UsersGuide/tutorials/pdbintro.html]
- 19. What is "chain identifier" in PDB? [https://biology.stackexchange.com/questions/37495/what-is-chain-identifier-in-pdb]
- 20. PDB residue serial inconsistency and PDB id&UniProt ID ... [https://www.biostars.org/p/9588718/]
- 21. Structures Without Legacy PDB Format Files [https://www.rcsb.org/docs/general-help/structures-without-legacy-pdb-format-files]
- 22. Protein Data Bank: A Comprehensive Review of 3D Structure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9599165/]
- 23. Protein-Nucleic Acid Complexes Released Per Year [https://www.rcsb.org/stats/growth/growth-protein-na-complex]
- 24. Ligand Structure Quality in PDB Structures [https://www.rcsb.org/docs/general-help/ligand-structure-quality-in-pdb-structures]
- 25. Assessing the Quality of 3D Structures [https://www.rcsb.org/docs/general-help/assessing-the-quality-of-3d-structures]
- 26. Structure [https://www.rcsb.org/docs/exploring-a-3d-structure/structure]
- 27. Structure Summary Page [https://www.rcsb.org/docs/exploring-a-3d-structure/structure-summary-page]
- 28. Ten simple rules to colorize biological data visualization - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7561171/]
- 29. Structure comparison tool [https://docs.gpcrdb.org/structure_comparison.html]
- 30. News - Protein Data Bank Japan [https://pdbj.org/news?year=2025&lang=en]
- 31. Data Format [https://pdbj.org/help/data-format?lang=en]
- 32. File Download Services [https://www.rcsb.org/docs/programmatic-access/file-download-services]
- 33. RCSB PDB Batch Download Service [https://www.rcsb.org/downloads]
- 34. Benchmarks for reading and interpreting mmCIF files [https://github.com/project-gemmi/mmcif-benchmark]
- 35. RCSB PDB: Homepage [https://www.rcsb.org/]
- 36. Comparison of X-ray Crystallography, NMR and EM [https://www.creative-biostructure.com/comparison-of-crystallography-nmr-and-em_6.htm?srsltid=AfmBOoo-gW7Bu-BjLod1-C0lmbaqS1qO2LyIWIEn9AO0gfz7Jhuyxxrq]
- 37. Comparison Of Structural Techniques: X-ray, NMR, Cryo-EM [https://peakproteins.com/a-comparison-of-the-structural-techniques-used-at-sygnature-discovery-x-ray-crystallography-nmr-and-cryo-em/]
- 38. The Applications & Principles of X-Ray Crystallography [https://www.azom.com/article.aspx?ArticleID=18684]
- 39. XRD Explained: Principles, Analysis & Applications [https://www.technologynetworks.com/analysis/articles/x-ray-diffraction-xrd-xrd-principle-xrd-analysis-and-applications-404260]
- 40. X-Ray Crystallography: Principles, Steps, and Applications ... [https://www.preachbio.com/2025/09/X-RAY-CRYSTALLOGRAPHY.html]
- 41. Comparing Analytical Techniques for Structural Biology [https://www.nanoimagingservices.com/about/blog/unraveling-the-mysteries-of-the-molecular-world-structural-biology-techniques]
- 42. High-resolution protein modeling through Cryo-EM and AI [https://pmc.ncbi.nlm.nih.gov/articles/PMC12590954/]
- 43. Methods of protein structure comparison - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4321859/]
- 44. Structural determination of small proteins by cryo-EM using ... [https://www.nature.com/articles/s41598-025-20608-3]
- 45. Ultra-fast spinning NMR: towards detailed analysis of ... [https://www.myscience.org/news/wire/ultra_fast_spinning_nmr_towards_detailed_analysis_of_complex_proteins-2025-ens-lyon]
- 46. Protein structure prediction via deep learning: an in-depth ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12003282/]
- 47. Highly accurate protein structure prediction with AlphaFold [https://www.nature.com/articles/s41586-021-03819-2]
- 48. AlphaFold Protein Structure Database [https://alphafold.ebi.ac.uk/]
- 49. Machine learning in protein structure prediction [https://www.sciencedirect.com/science/article/pii/S1367593121000508]
- 50. AlphaFold Protein Structure Database 2025: a redesigned ... [https://pubmed.ncbi.nlm.nih.gov/41273079/]
- 51. High-accuracy protein complex structure modeling based ... [https://www.nature.com/articles/s41467-025-65090-7]
- 52. Cutoff Scanning Matrix (CSM): structural classification and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3287581/]
- 53. RCSB Protein Data Bank: Enabling biomedical research and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6933845/]
- 54. What information does a PDB entry contain? | PDBe [https://www.ebi.ac.uk/training/online/courses/exploring-pdb-entry/what-information-does-a-pdb-entry-contain/]
- 55. Find Drugs in the PDB [https://www.rcsb.org/news/5764422199cccf72e74ca355]
- 56. Drug and target protein structures in the PDB [https://www.ebi.ac.uk/thornton-srv/databases/drugport/]
- 57. Experiment [https://www.rcsb.org/docs/exploring-a-3d-structure/experiment]
- 58. an interactive WebGL visualizer for protein-ligand complex [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-15-56]
- 59. Consistent two-dimensional visualization of protein-ligand ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3224603/]
- 60. A Concise Review of Biomolecule Visualization - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10887528/]
- 61. Notice: NGL Viewer Deprecation [https://www.rcsb.org/news/feature/65b42d3fc76ca3abcc925d15]
- 62. Maneuvering in Mol* [https://www.rcsb.org/docs/3d-viewers/mol*/maneuvering-in-mol*]
- 63. Guide to Understanding PDB Data: Computed Structure Models [https://pdb101.rcsb.org/learn/guide-to-understanding-pdb-data/computed-structure-models]
- 64. Considering best practices in color palettes for molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9377702/]
- 65. Integrative Structures on RCSB.org [https://www.rcsb.org/docs/general-help/integrative-structures-on-rcsborg]
- 66. wwPDB: Processing Procedures and Policies [https://www.wwpdb.org/documentation/policy]
- 67. Integrative protein assembly with LZerD and deep learning ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12353303/]
- 68. Introduction to Biological Assemblies and the PDB Archive [https://pdb101.rcsb.org/learn/guide-to-understanding-pdb-data/biological-assemblies]
- 69. Principles and characteristics of biological assemblies in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9169879/]
- 70. Learn: Guide to Understanding PDB Data: Missing Coordinates [https://pdb101.rcsb.org/learn/guide-to-understanding-pdb-data/missing-coordinates]
- 71. Protein Data Bank: Asymmetric Unit/Biological Assembly [https://chemistry.stackexchange.com/questions/47065/protein-data-bank-asymmetric-unit-biological-assembly]
- 72. Challenges in bridging the gap between protein structure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11623436/]
- 73. Multivariate Analyses of Quality Metrics for Crystal Structures ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7182075/]
- 74. A Paired Database of Predicted and Experimental Protein ... [https://www.nature.com/articles/s41597-025-05754-7]
- 75. Your Data Visualization Color Guide: 7 Best Practices | Sigma [https://www.sigmacomputing.com/blog/7-best-practices-for-using-color-in-data-visualizations]
- 76. Access Structure Quality Metrics [https://www.rcsb.org/news/5d7bb149ea7d0653b99c8815]
- 77. New Study from UVA Researchers Challenges Traditional ... [https://datascience.virginia.edu/news/new-study-uva-researchers-challenges-traditional-views-protein-structure]
- 78. Low Complexity Induces Structure in Protein Regions ... [https://www.mdpi.com/2218-273X/12/8/1098]
- 79. wwPDB: Processing Procedures and Policies [https://www.wwpdb.org/documentation/procedure]
- 80. using protein structures to inspire creative expression and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12094914/]
- 81. A dataset of alternately located segments in protein crystal ... [https://www.nature.com/articles/s41597-024-03595-4]
- 82. The Protein Data Bank - PMC - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC102472/]
- 83. Ensemble-Based Interpretations of NMR Structural Data to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6269897/]
- 84. Dealing with Coordinates - PDB-101 [https://pdb101.rcsb.org/learn/guide-to-understanding-pdb-data/dealing-with-coordinates]
- 85. Managing the Display [https://www.rcsb.org/docs/3d-viewers/mol*/managing-the-display]
- 86. User guide to the wwPDB NMR validation reports [http://www.wwpdb.org/validation/NMRValidationReportHelp]
- 87. PDB files — ProDy [http://www.bahargroup.org/prody/tutorials/structure_analysis/pdbfiles.html]
- 88. Ten rules for a structural bioinformatic analysis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12578330/]
- 89. ProteinTools: a toolkit to analyze protein structures - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8262690/]
- 90. Structure Prediction [https://www.rcsb.org/docs/additional-resources/structure-prediction]
- 91. Protein Structure Prediction: A Beginner's Guide to Free ... [https://bitesizebio.com/74900/protein-structure-prediction/]
- 92. wwPDB: Validation Reports [https://www.wwpdb.org/validation/validation-reports]
- 93. wwPDB: Validation Servers [https://www.wwpdb.org/validation/validation-servers]
- 94. wwPDB: 2025 News [https://www.wwpdb.org/news/news]
- 95. wwPDB: Tutorial [https://www.wwpdb.org/deposition/tutorial]
- 96. Updated pipeline for small molecule data processing at ... [http://www.ebi.ac.uk/pdbe/news/updated-pipeline-small-molecule-data-processing-pdbe]
- 97. User guide to the wwPDB EM validation reports [http://www.wwpdb.org/validation/2017/EMValidationReportHelp]
- 98. User guide to the wwPDB X-ray validation reports [https://www.wwpdb.org/validation/XrayValidationReportHelp]
- 99. RCSB Protein Data Bank: visualizing groups of ... [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2023.1311287/full]
- 100. Computed Structure Models and RCSB.org [https://www.rcsb.org/docs/general-help/computed-structure-models-and-rcsborg]
- 101. AlphaFold predictions are valuable hypotheses and ... [https://www.nature.com/articles/s41592-023-02087-4]
- 102. PDBe brings electron-density viewing to the masses [http://www.ebi.ac.uk/pdbe/news/pdbe-brings-electron-density-viewing-masses]
- 103. Validation of ligands in macromolecular structures ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5947763/]
- 104. CMM—An enhanced platform for interactive validation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9794025/]
- 105. Electron Density Maps and Coefficient Files [https://www.rcsb.org/docs/general-help/electron-density-maps-and-coefficient-files]
- 106. Validation of Structures in the Protein Data Bank - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5718880/]
- 107. GenBank Release 268.0 is Available! - NCBI Insights - NIH [https://ncbiinsights.ncbi.nlm.nih.gov/2025/08/26/genbank-release-268-0/]
- 108. UniProt: the Universal Protein Knowledgebase in 2025 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11701636/]
- 109. ChEBI - Chemical Entities of Biological Interest [https://www.ebi.ac.uk/chebi/]