Protein Structure Determination by X-Ray Crystallography: A Comprehensive Guide from Principles to Practice
This article provides a comprehensive overview of the protein structure determination pipeline using X-ray crystallography, a cornerstone technique in structural biology.
Protein Structure Determination by X-Ray Crystallography: A Comprehensive Guide from Principles to Practice
Abstract
This article provides a comprehensive overview of the protein structure determination pipeline using X-ray crystallography, a cornerstone technique in structural biology. Tailored for researchers, scientists, and drug development professionals, it covers the foundational principles of diffraction, a step-by-step methodological walkthrough from crystallization to model refinement, and practical troubleshooting for common challenges. It further details critical structure validation protocols and offers a comparative analysis with other leading structural techniques like Cryo-EM and NMR, empowering readers to effectively apply and interpret crystallographic data in biomedical research and drug discovery.
The Fundamentals of Protein X-ray Crystallography: From Atoms to 3D Models
X-ray crystallography stands as a cornerstone technique for determining the three-dimensional atomic structure of matter, with its application to biological macromolecules like proteins revolutionizing our understanding of biology and empowering drug discovery efforts [1]. At the heart of this powerful method lies Bragg's Law, a fundamental physical principle that describes the condition for diffraction when X-rays interact with a crystalline lattice [2]. This law provides the essential link between the experimentally measured diffraction pattern and the atomic-scale structure of the crystal. For researchers, scientists, and drug development professionals, a deep understanding of Bragg's Law is not merely academic; it is critical for planning and interpreting crystallographic experiments, from obtaining the initial protein crystals to solving and validating a structural model [3]. This guide details the core physics, its practical application in protein structure determination, and the advanced methodologies that leverage this foundational principle.
Fundamental Physics of Bragg's Law
Historical Foundation and Theoretical Background
The phenomenon of X-ray diffraction was first demonstrated by Max von Laue in 1912, proving the wave-like nature of X-rays and the periodic arrangement of atoms in crystals [4]. Shortly thereafter, in 1913, Sir William Henry Bragg and his son, Sir William Lawrence Bragg, proposed a simpler explanation for the observed diffraction patterns [2] [5]. They modeled a crystal as a set of discrete, parallel planes of atoms separated by a constant distance, d. Lawrence Bragg proposed that the intense peaks of reflected radiation (now known as Bragg peaks) occurred when X-rays scattering off these different planes interfered constructively [2]. This seminal insight, for which the Braggs were jointly awarded the Nobel Prize in Physics in 1915, provided a powerful new tool for determining crystal structures and remains the most intuitive way to understand X-ray diffraction [2] [6].
Bragg's Law is a special case of the more general Laue diffraction and applies not only to X-rays but to all types of matter waves, including neutron and electron waves, provided the scattering object is a crystal with a large number of atoms [2].
The Bragg Condition and Mathematical Formulation
Bragg diffraction occurs when radiation, with a wavelength λ comparable to atomic spacings, is scattered in a specular (mirror-like) fashion by atomic planes and undergoes constructive interference [2]. The condition for this constructive interference is given by the well-known Bragg Equation:
Where:
nis a positive integer (1, 2, 3...) representing the order of the diffraction.λis the wavelength of the incident X-ray beam.dis the interplanar spacing between the atomic layers in the crystal.θis the glancing angle (or angle of incidence), measured between the incident ray and the scattering plane, not the surface normal [2].
This equation states that constructive interference, and hence an intense diffraction peak, will only be observed when the path difference between waves reflected from adjacent crystal planes is equal to an integer multiple of the X-ray wavelength [5] [6].
Figure 1: A schematic diagram illustrating the geometry of Bragg's Law. The path difference between waves reflecting from adjacent planes is AB + BC = 2d sinθ.
Derivation of Bragg's Law
The derivation of Bragg's Law stems from calculating the path difference between two parallel X-ray waves scattering off two adjacent atomic planes [2] [6].
- Consider two parallel X-ray waves, 1 and 2, incident on two crystal planes separated by a distance
dat a glancing angleθ. - Wave 2 travels an extra distance
ABto reach the second plane and an extra distanceBCafter scattering. The total path difference isAB + BC. - From geometry,
AB = BC = d sinθ. Therefore, the total path difference is2d sinθ. - For constructive interference to occur, this path difference must be an integer multiple of the wavelength
λ. - This leads directly to the Bragg equation:
nλ = 2d sinθ[2] [6].
It is crucial to note that while the phenomenon is described as "reflection," it is fundamentally a result of constructive interference from scattered waves. If this condition is not met, the waves will arrive out of phase and undergo destructive interference, resulting in no detectable signal [3].
Bragg's Law in Protein X-ray Crystallography
Protein X-ray crystallography is a multi-step process for determining the atomic structure of proteins, and Bragg's Law underpins the critical data collection and interpretation phases [3] [1].
The Central Role of Diffraction in Structure Determination
In protein crystallography, the protein is first purified and induced to form a highly ordered crystal [1]. When a crystal is placed in an intense X-ray beam, typically generated by a synchrotron radiation source, the electrons within the crystal scatter the X-rays [3]. Each atom acts as a source of scattered waves, and the regular, repeating arrangement of atoms in the crystal causes these scattered waves to interact and produce a diffraction pattern composed of discrete spots on a detector [3]. The position and intensity of these spots are the primary experimental data.
According to Bragg's model, each spot in the diffraction pattern corresponds to a specific set of atomic lattice planes within the crystal, defined by their Miller indices (hkl) [3]. The crystal is rotated in the X-ray beam (using a goniometer) to bring different lattice planes into their Bragg condition, thereby collecting a complete set of diffraction intensities [3]. Modern synchrotrons, with their high-intensity beams and advanced robotic sample handling, can collect an entire dataset in less than a minute [3].
Figure 2: A high-level workflow of protein structure determination by X-ray crystallography, highlighting where Bragg's Law is applied.
From Diffraction Spots to Electron Density
The final goal is to compute an electron density map into which the atomic model of the protein is built. The connection between the observed diffraction pattern and this map is made via a mathematical operation called a Fourier transform [3]. The electron density ρ(xyz) is calculated using the equation:
ρ(xyz) = 1/V Σ_h Σ_k Σ_l |F(hkl)| exp[-2πi(hx + ky + lz) + iϕ(hkl)]
Where:
Vis the volume of the unit cell.|F(hkl)|is the structure factor amplitude, derived from the measured intensity of the diffraction spot with Miller indiceshkl.ϕ(hkl)is the phase of the structure factor [3].
While the intensity of a diffraction spot (which gives |F(hkl)|) can be measured directly, the phase information ϕ(hkl) is lost during data collection. This is known as the "phase problem" in crystallography. Determining the phases requires additional experimental methods, such as heavy atom replacement (e.g., MIR or MAD), or molecular replacement if a related structure is known [3]. Bragg's Law is essential in the initial processing of diffraction images to correctly index and assign Miller indices to each spot, which is the first step in determining the unit cell parameters and preparing the data for phasing [3].
Advanced Quantitative Analysis and Methodologies
Quantitative X-ray Diffraction (XRD) Methods
Beyond biological crystallography, Bragg's Law is the foundation for quantitative phase analysis in materials science and geology. Several sophisticated software-based methods have been developed, each with specific strengths and limitations [7].
Table 1: Comparison of Quantitative X-ray Diffraction Mineral Analysis Methods
| Method | Principle | Typical Software | Advantages | Limitations |
|---|---|---|---|---|
| Reference Intensity Ratio (RIR) | Uses the intensity of a single peak and a known reference ratio to quantify phase abundance [7]. | JADE | A handy and simple approach [7]. | Lower analytical accuracy, especially in complex mixtures [7]. |
| Rietveld Refinement | A whole-pattern fitting method that refines a calculated pattern (based on crystal structure models) to match the observed pattern [7]. | HighScore, TOPAS, GSAS, BGMN, Maud | High accuracy for non-clay samples; can refine structural parameters (atom positions, cell parameters) [7]. | Struggles with phases with disordered or unknown crystal structures [7]. |
| Full Pattern Summation (FPS) | The observed pattern is modeled as the sum of reference patterns from pure phases [7]. | FULLPAT, ROCKJOCK | Wide applicability, considered most appropriate for sediments and clay-containing samples [7]. | Requires a comprehensive library of reference patterns [7]. |
Resolution and Data Quality in Protein Crystallography
In protein crystallography, the quality of the final atomic model is directly governed by the resolution of the diffraction data [3]. Resolution refers to the finest detail discernible in the electron density map and is determined by the highest angle diffraction spots collected. It is inversely related to the smallest d-spacing measured, as per Bragg's Law. The resolution dictates what structural features can be reliably interpreted.
Table 2: Interpretation of Resolution Ranges in Protein X-ray Crystallography
| Resolution Range | Structural Features Discernible |
|---|---|
| Low Resolution (> 5 Å) | The overall shape and envelope of the protein molecule; α-helices appear as rods. Individual amino acids cannot be distinguished [3]. |
| Medium Resolution (3.5 - 2.5 Å) | The protein backbone can be traced; side chains become distinguishable, allowing the sequence to be built into the density. Solvent (water) molecules can start to be identified [3]. |
| High/Atomic Resolution (2.4 Å or better) | Individual atoms become resolved; fine structural details are clear. The model-building process is more straightforward, and a large number of solvent molecules can be identified and modeled [3]. |
Modern Advances: Pushing the Limits of Bragg's Law
The field of X-ray diffraction continues to evolve, driven by advancements in source technology and data processing. The recent development of extremely brilliant synchrotron sources, such as the ESRF's Extremely Brilliant Source (EBS), has increased the available coherent flux by a factor of 100 [4]. This brilliance enables new techniques:
- Nanoscale Mapping: Advances in X-ray focusing optics now allow for diffraction mapping with a spatial resolution of 50–100 nm, enabling the quantification and localization of elastic strains and defects in crystalline materials, which is crucial for understanding material properties [4].
- Coherent Diffraction Imaging (CDI): This lensless technique can achieve a spatial resolution of 5–10 nm. It uses the coherent diffraction pattern and phase retrieval algorithms to reconstruct an image of the sample, bypassing the need for crystals large enough for traditional crystallography [4].
- Serial Crystallography: This method, often used with X-ray free-electron lasers (XFELs), involves collecting diffraction data from a stream of tiny, randomly oriented microcrystals. It has opened new avenues for studying proteins that are difficult to crystallize into large single crystals [1].
The Scientist's Toolkit: Essential Reagents and Materials
Successful X-ray crystallography relies on a suite of specialized reagents, materials, and instruments.
Table 3: Key Research Reagent Solutions and Materials for Protein X-ray Crystallography
| Item | Function / Purpose |
|---|---|
| Purified Protein Sample | The target macromolecule, typically produced via recombinant expression and purified to homogeneity, is the fundamental starting material [1]. |
| Crystallization Screening Kits | Commercial kits containing a wide array of chemical conditions (precipitants, buffers, salts, additives) to empirically identify initial conditions for protein crystal growth [3]. |
| Synchrotron Beamline | A large-scale facility that generates high-intensity, focused X-ray beams necessary for collecting high-quality diffraction data from protein crystals [3]. |
| Cryo-Protectant | A chemical (e.g., glycerol, ethylene glycol) used to soak crystals before flash-cooling in liquid nitrogen. This prevents ice formation and protects the crystal from radiation damage during data collection [3]. |
| Heavy Atom Compounds | Reagents containing atoms with high atomic numbers (e.g., mercury, platinum, selenium) used for experimental phasing. They are soaked into crystals or incorporated via expression (e.g., selenomethionine) to solve the "phase problem" [3]. |
| X-ray Detector | A two-dimensional hybrid pixel detector that captures the diffraction pattern. Modern detectors offer high dynamic range, fast readout speeds, and low noise, which are critical for efficient data collection [4]. |
Proteins are the fundamental workhorses of biology, orchestrating a vast array of cellular processes, from catalyzing chemical reactions and supporting immune responses to facilitating cellular communication [1]. The specific function of a protein is an direct consequence of its unique three-dimensional (3D) structure. The precise arrangement of atoms within a protein dictates its ability to bind other molecules, form complexes, and perform its biological role. Understanding protein structure is therefore paramount for deciphering the molecular mechanisms of life and disease. In drug discovery, this understanding empowers researchers to design targeted therapies that precisely modulate protein function, offering high efficacy and reduced side effects. This whitepaper explores how elucidating 3D protein architecture, primarily through the powerful technique of X-ray crystallography, provides critical insights into biological function and drives modern drug development.
The Principle of Structure Determination by X-ray Crystallography
X-ray crystallography is a premier method for visualizing the atomic structure of crystallized proteins, providing a detailed snapshot of their 3D architecture [1]. The technique relies on a fundamental principle of physics: when a beam of X-rays strikes a crystalline lattice, it scatters in a phenomenon known as diffraction. The resulting diffraction pattern, a collection of discrete spots captured on a detector, encodes information about the electron density within the crystal.
The birth of this field is credited to Max von Laue, who discovered the diffraction of X-rays by crystals, for which he was awarded the Nobel Prize in Physics in 1914 [8]. This was later formalized by William Lawrence Bragg, who formulated the seminal Bragg's Law:
This equation describes the condition for constructive interference, where n is an integer, λ is the wavelength of the X-rays, d is the distance between atomic lattice planes in the crystal, and θ is the angle of incidence. By measuring the angles and intensities of the diffracted beams, it is possible to compute a 3D electron density map and build an atomic model of the protein [3].
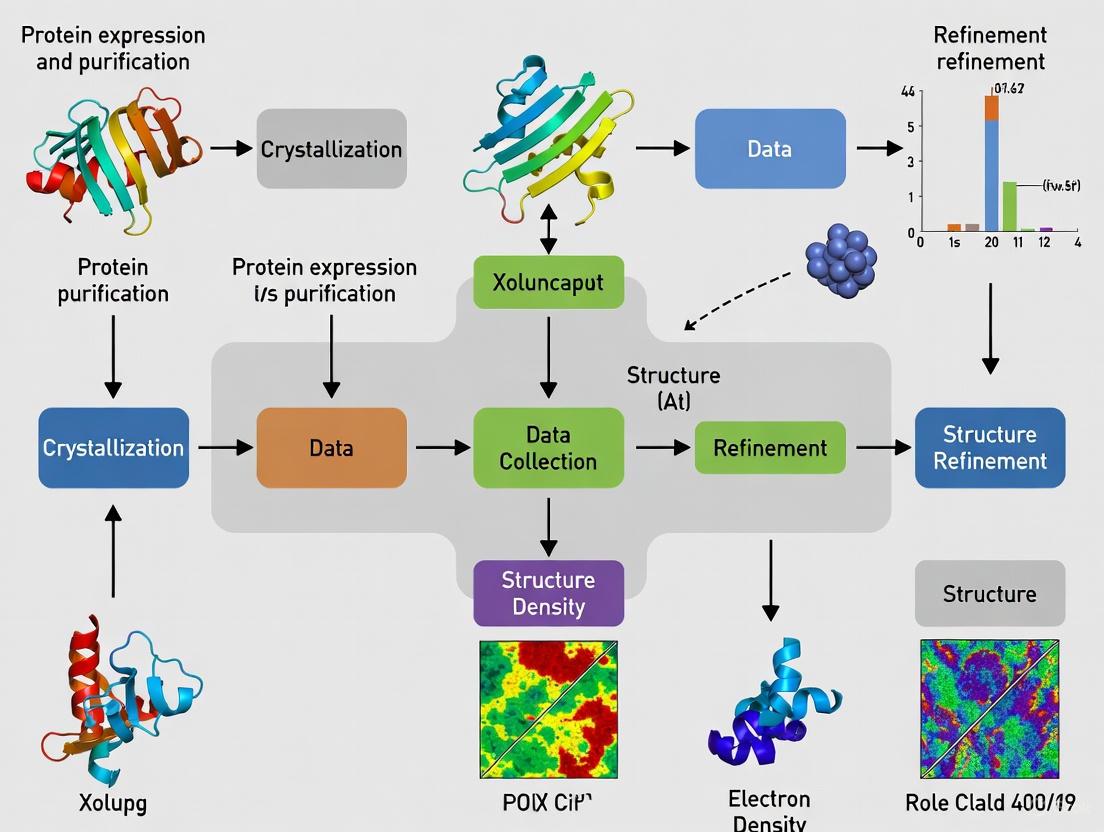
A Step-by-Step Technical Workflow
The process of determining a protein structure via X-ray crystallography is methodical and involves several critical stages, each with its own technical challenges and requirements.
Protein Purification and Crystallization
The journey begins with protein isolation to obtain a pure, homogeneous, and conformationally uniform sample [1] [9]. This typically involves recombinant protein expression followed by chromatographic purification. The next and often most critical hurdle is crystal formation. The purified protein solution is encouraged to form ordered, single crystals through careful manipulation of conditions like pH, temperature, and precipitant concentration [3]. This step is a major bottleneck, as not all proteins crystallize readily. Automation, using robots like the Mosquito crystallization robot which dispenses nanoliter volumes with high precision, has greatly improved the efficiency of screening thousands of crystallization conditions [9].
Data Collection and Diffraction Analysis
Once a suitable crystal is obtained, it is harvested and flash-frozen in liquid nitrogen to protect it from radiation damage in the X-ray beam [3]. The crystal is then mounted on a goniometer, which precisely rotates it in the path of an X-ray source. While laboratory X-ray sources exist, modern crystallography predominantly uses synchrotron radiation sources, which provide high-intensity, focused X-ray beams that enable rapid data collection—sometimes in less than a minute [3]. As the crystal diffracts the X-rays, a detector captures the resulting pattern of spots. A complete dataset requires collecting thousands of such diffraction spots from all possible orientations of the crystal.
Phase Determination and Model Building
The intensities of the diffraction spots are used to derive structure factors. However, to calculate an electron density map, the phases of the diffracted waves are required, and this phase information is lost during data collection. This is known as the "phase problem," and solving it is a central challenge in crystallography [3]. Common methods include heavy atom replacement, where heavy atoms (e.g., selenium) are introduced into the protein crystal, and the differences in diffraction are used to deduce phase information [3].
Once phases are estimated, an electron density map is calculated via a mathematical operation called Fourier summation. Researchers then build an atomic model of the protein by fitting its known amino acid sequence into this electron density map using specialized software [3] [1].
Model Refinement and Validation
The initial structural model is iteratively refined to achieve the best fit to the experimental electron density data. This process adjusts the atomic coordinates to minimize a value called the R-factor, which gauges the agreement between the observed data and the model [3]. The final step is validation, where the model's quality and stereochemical accuracy are thoroughly checked before it is deposited in the public Protein Data Bank (PDB) [3] [1]. The PDB itself performs validation checks before releasing the structure to the scientific community.
The workflow is summarized in the diagram below:
Interpreting Structural Data: Resolution and Quality
The quality of a crystallographic model is primarily judged by its resolution, a key parameter derived from the diffraction data [3]. Resolution reflects the level of detail visible in the electron density map and is a major determinant of the model's accuracy. The following table summarizes the interpretation of different resolution ranges:
Table: Interpretation of Resolution in X-ray Crystallography
| Resolution Range | Classification | Structural Details Observable |
|---|---|---|
| ≤ 5.0 Å | Low Resolution | The overall shape of the protein molecule is distinguishable; alpha-helices are visible as rods [3]. |
| 3.5 - 2.5 Å | Medium Resolution | Side chains begin to be distinguishable, allowing the model to be built; water molecules may be visible at better than 2.8 Å [3]. |
| ≥ 2.4 Å | Atomic Resolution | Detailed atomic modeling is possible; many solvent molecules can be identified and built into the density map [3]. |
Successful structure determination relies on a suite of specialized reagents, equipment, and software. The table below details key resources used in a typical crystallography pipeline.
Table: Essential Research Reagents and Resources for X-ray Crystallography
| Item Name | Type/Category | Function & Application |
|---|---|---|
| Homogeneous Protein Sample | Biological Reagent | A pure, conformationally uniform sample is the foundational starting material for successful crystallization [9]. |
| Crystallization Screen Solutions | Chemical Reagent | Pre-formulated solutions varying precipitants, salts, pH, etc., to empirically identify initial crystal growth conditions [9]. |
| Mosquito Robot | Laboratory Equipment | Automates the setup of crystallization trials by dispensing nanoliter-volume droplets with high precision, increasing throughput and reproducibility [9]. |
| Synchrotron Beamline | Large-Scale Facility | Provides a high-intensity, tunable X-ray source for rapid and high-resolution data collection, essential for modern crystallography [3]. |
| Cryoprotectant | Chemical Reagent | A compound (e.g., glycerol) used to protect the protein crystal from ice formation during flash-cooling in liquid nitrogen [3]. |
| Software for Data Processing | Computational Tool | Specialized packages for processing raw diffraction data ("data reduction"), solving structures, and refining models (e.g., PHENIX, CCP4) [3]. |
Application to Drug Discovery: From Structure to Therapy
The impact of protein structures on drug discovery is profound. Knowing the precise 3D structure of a therapeutic target, such as a enzyme or receptor, allows for rational drug design. Researchers can design small molecules that fit snugly into active sites or allosteric pockets to inhibit or activate the protein's function [10].
Analysis of ligands bound to proteins in the PDB reveals that most therapeutic molecules tend toward linear and planar geometries, with few having highly 3D conformations [10]. This "flatness" is partly due to synthetic challenges and adherence to rules for oral bioavailability. There is a growing recognition of the potential utility of libraries with greater 3D topological diversity to explore a wider range of biological targets and improve the success of drug discovery campaigns [10]. By studying protein-ligand complexes, scientists can optimize interactions like hydrogen bonding and hydrophobic contacts, leading to more potent and selective drug candidates.
While X-ray crystallography remains a cornerstone of structural biology, the field is continuously evolving. Techniques like serial crystallography allow data collection from microcrystals, opening avenues for studying challenging proteins [1]. Furthermore, cryo-electron microscopy (cryo-EM) has emerged as a powerful complementary technique, enabling the determination of high-resolution structures for proteins that are difficult to crystallize [1].
In conclusion, the determination of protein structure is indispensable for linking 3D architecture to biological function. X-ray crystallography provides a detailed, atomic-level view that is critical for understanding disease mechanisms and designing the next generation of targeted therapeutics. As technologies advance, our ability to visualize and interpret the molecular machinery of life will continue to deepen, fueling ongoing innovation in biomedicine and drug discovery.
X-ray crystallography stands as one of the most transformative techniques in the history of biology, enabling scientists to decipher the three-dimensional atomic structure of biological macromolecules. This breakthrough methodology has fundamentally advanced our understanding of life processes at the molecular level, from enzyme catalysis and immune recognition to genetic inheritance and disease mechanisms. The ability to visualize protein structures has revolutionized fields ranging from molecular biology and biochemistry to pharmaceutical development and biotechnology. This comprehensive review traces the key historical milestones of crystallography in biology, detailing the experimental protocols that enabled these discoveries and examining the technique's profound impact on modern biological research and drug development. By understanding this historical trajectory and the underlying methodologies, researchers can better appreciate both the current capabilities and future directions of structural biology.
Historical Timeline of Key Milestones
The application of X-ray crystallography to biological problems has unfolded over more than a century of innovation, with each breakthrough building upon previous technical and conceptual advances. The table below summarizes the pivotal moments in this scientific journey.
Table 1: Key Historical Milestones in Biological X-ray Crystallography
| Year | Milestone Achievement | Key Researchers/Group | Biological Significance |
|---|---|---|---|
| 1912 | First X-ray diffraction pattern from a crystal (copper sulfate) | Max von Laue, Walter Friedrich, Paul Knipping [11] | Established crystals as diffraction gratings for X-rays, founding the field of X-ray crystallography |
| 1913 | Formulation of Bragg's Law | William Lawrence Bragg & William Henry Bragg [11] [12] | Provided the fundamental mathematical relationship explaining X-ray diffraction by crystal planes |
| 1915 | Nobel Prize in Physics for X-ray crystal structure analysis | William Henry Bragg & William Lawrence Bragg [11] | Recognized the profound importance of X-ray crystallography for scientific discovery |
| 1934 | First X-ray diffraction data from a protein (pepsin) | J.D. Bernal & Dorothy Crowfoot Hodgkin [13] | Demonstrated that proteins, despite their complexity, could form crystals suitable for structural analysis |
| 1958 | First protein structure (myoglobin at 6 Å resolution) | John Kendrew [13] | Provided the first glimpse of a protein's three-dimensional structure, revealing its complex folding |
| 1960 | Structure of hemoglobin | Max Perutz [13] | Elucidated the structural basis of oxygen transport and cooperative binding in this complex protein |
| 1965 | First enzyme structure (lysozyme) | David Phillips [13] | Revealed the structural basis of enzymatic catalysis, identifying the active site and mechanism |
| 1971 | Foundation of the Protein Data Bank (PDB) | Brookhaven National Laboratory [14] | Established a central repository for structural data, enabling global sharing and collaboration |
| 1984 | Structure of the first virus | Extended structural determination to massive macromolecular complexes | |
| 2000 | Structural Genomics Initiatives launch | International consortiums | Systematized structure determination to cover entire protein fold spaces |
| 2020s | SARS-CoV-2 protein structures | Global research community [15] | Accelerated vaccine and therapeutic development during the COVID-19 pandemic |
| 2023+ | ALS beamlines deposit >10,000 protein structures | Advanced Light Source [14] | Demonstrated the high-throughput capabilities of modern synchrotron-based crystallography |
The progression from simple salt crystals to complex biological macromolecules illustrates how methodological advances have continually expanded the boundaries of what can be studied structurally. The early protein structures, while low resolution by today's standards, provided the first direct evidence that proteins had defined three-dimensional structures, confirming the thermodynamic hypothesis of protein folding. The visualization of enzyme active sites represented another transformative moment, moving biochemistry from kinetic inferences to mechanistic understanding based on atomic positioning. Contemporary structural biology continues to build upon this foundation, with recent advances including the determination of G-protein coupled receptors (GPCRs) and other membrane proteins that represent important drug targets, and the application of time-resolved crystallography to capture reaction intermediates [15].
Fundamental Principles and Experimental Protocols
Core Physical Principles
The theoretical foundation of X-ray crystallography rests on the wave nature of X-rays and the periodic arrangement of atoms within crystals. When X-rays encounter a crystalline lattice, they are scattered by the electrons surrounding the atoms. The scattered waves interfere with each other, producing a diffraction pattern when the conditions for constructive interference are met according to Bragg's Law: nλ = 2d sinθ, where n is an integer, λ is the wavelength of the incident X-ray beam, d is the spacing between atomic planes in the crystal, and θ is the angle of incidence [3] [12]. This relationship allows researchers to calculate the distances between atomic planes from measured diffraction angles.
The diffraction pattern captured on a detector represents the amplitudes of the structure factors, but the phase information is lost during measurement—this constitutes the fundamental "phase problem" in crystallography [3]. Solving this problem requires specialized methods such as molecular replacement (using a known homologous structure), multiple isomorphous replacement (using heavy atom derivatives), or anomalous dispersion (using the anomalous scattering of atoms at specific wavelengths) [3].
Protein Crystallography Workflow
The determination of a protein structure via X-ray crystallography follows a multi-step workflow with specific technical requirements at each stage. The following diagram illustrates this process:
Diagram 1: Protein X-ray Crystallography Workflow
Protein Purification and Crystallization
The process begins with the purification of the target protein to homogeneity, typically using chromatographic methods such as affinity, ion-exchange, and size-exclusion chromatography [16] [17]. High protein purity (>95%) and conformational homogeneity are essential prerequisites for obtaining diffraction-quality crystals. The purified protein is concentrated to high levels (typically 5-20 mg/mL, depending on the protein) in an appropriate buffer [16].
Crystallization represents a critical bottleneck in structural determination and is typically achieved through vapor diffusion methods (hanging or sitting drops) [16]. In these setups, a small volume of protein solution is mixed with a precipitant solution and equilibrated against a larger reservoir containing a higher concentration of the same precipitant. Water vapor diffuses from the protein drop to the reservoir, slowly increasing the concentration of both protein and precipitant until supersaturation is achieved, promoting crystal nucleation and growth [16]. Commercial sparse matrix screens systematically explore combinations of precipitants (e.g., polyethylene glycols, salts), buffers, and additives to identify initial crystallization conditions [16].
Data Collection and Structure Solution
Once suitable crystals are obtained (typically >0.1 mm in smallest dimension), they are harvested and cryo-cooled in liquid nitrogen to mitigate radiation damage during data collection [3] [16]. Modern data collection occurs predominantly at synchrotron facilities, which provide high-intensity, tunable X-ray beams [3]. The crystal is mounted on a goniometer and exposed to the X-ray beam while being rotated, with diffraction patterns collected at small angular increments (typically 0.1-1.0°) [3] [16].
The resulting diffraction patterns are processed through computational "data reduction" to correct for experimental artifacts and extract structure factor amplitudes [3]. As mentioned previously, the phase problem is then solved using molecular replacement, multiple isomorphous replacement, or anomalous dispersion methods. With both amplitudes and phases determined, an electron density map is calculated through Fourier transformation, into which an atomic model is built and iteratively refined against the experimental data [3]. The quality of the structural model is assessed using validation metrics including the R-factor and R-free, with final structures typically deposited in the Protein Data Bank for public access [3].
Table 2: Key Reagents and Materials in Protein Crystallography
| Reagent/Material Category | Specific Examples | Function and Application |
|---|---|---|
| Precipitants | Polyethylene glycol (PEG) of various molecular weights, ammonium sulfate, sodium chloride, MPD | Promote protein crystallization by reducing solubility and inducing supersaturation |
| Buffers | HEPES, Tris, phosphate, citrate buffers across pH range (3-10) | Maintain protein stability and consistent protonation states during crystallization |
| Additives | Various salts, divalent cations, detergents, small organics | Modulate crystallization by affecting protein interactions, particularly for challenging targets |
| Cryoprotectants | Glycerol, ethylene glycol, sucrose, low-molecular-weight PEG | Prevent ice formation during cryo-cooling by replacing water molecules in crystal lattice |
| Crystallization Plates | 24-well, 96-well format plates for sitting or hanging drops | Enable high-throughput screening of crystallization conditions with minimal sample consumption |
| Synchrotron Beamlines | Advanced Light Source (ALS), MAX IV, other international facilities | Provide high-intensity X-ray sources with advanced optics and detectors for data collection |
Impact on Biological Understanding and Drug Development
Elucidation of Biological Mechanisms
X-ray crystallography has provided unparalleled insights into fundamental biological processes by visualizing their molecular components. The technique revealed the structural basis of enzyme catalysis through the first enzyme structure (lysozyme), which showed how enzymes position substrates for reaction and stabilize transition states [13]. Subsequent structures of numerous enzymes have illuminated the chemical mechanisms underlying virtually every metabolic pathway.
In immunology, crystallography has elucidated how the immune system recognizes pathogens. Structures of major histocompatibility complex (MHC) molecules revealed their peptide-binding grooves, explaining how the immune system displays foreign and self-peptides for T-cell recognition [13]. The structures of antibodies and their complexes with antigens have illuminated the molecular basis of immunological specificity and cross-reactivity [13].
Perhaps most famously, X-ray crystallography played a crucial role in determining the structure of DNA, with Rosalind Franklin's diffraction data from DNA fibers providing key measurements that informed the Watson-Crick model of the double helix [12]. Her "Photo 51" revealed the 3.4 Å spacing between base pairs, the 34 Å helical repeat, and the 20 Å helix diameter [12]. This breakthrough launched the era of molecular biology and our modern understanding of genetics.
Applications in Structure-Based Drug Design
The pharmaceutical industry has leveraged crystallography to transform drug discovery from a largely empirical process to a rational, structure-based endeavor. The approach involves determining the three-dimensional structure of a drug target, typically an enzyme or receptor, and using this information to design small molecules that modulate its activity. The iterative process of structure-based drug design is illustrated below:
Diagram 2: Structure-Based Drug Design Cycle
The impact of this approach is exemplified by the development of HIV protease inhibitors, where crystallographic structures of inhibitor-enzyme complexes guided the design of compounds that effectively treated AIDS [13]. Similarly, the determination of kinase structures has enabled the development of targeted cancer therapies such as imatinib (Gleevec), designed to fit specifically into the ATP-binding pocket of aberrant signaling proteins [13]. More recently, structural studies of the SARS-CoV-2 spike protein and other viral components accelerated the development of therapeutics and vaccines during the COVID-19 pandemic [15].
Crystallography continues to drive drug discovery for challenging target classes, including G-protein coupled receptors (GPCRs) and other membrane proteins. The ability to visualize how drugs bind to their targets at atomic resolution enables more precise optimization of potency, selectivity, and physicochemical properties, ultimately leading to improved clinical candidates.
Modern Advancements and Future Perspectives
Technical Innovations in Crystallography
The field of X-ray crystallography has undergone revolutionary technical advances that have dramatically expanded its capabilities and applications. Synchrotron radiation sources have largely replaced laboratory X-ray generators, providing beams that are orders of magnitude more intense and enabling the use of smaller crystals and faster data collection [3] [14]. The development of cryo-crystallography (flash-cooling crystals to cryogenic temperatures) has mitigated radiation damage, allowing for more extensive data collection from single crystals [3].
More recently, serial crystallography approaches, particularly at X-ray free-electron lasers (XFELs), have enabled structure determination from microcrystals that are too small for conventional methods [15]. These techniques use the "diffraction before destruction" principle, where ultrashort, extremely bright X-ray pulses collect diffraction patterns before the crystal is vaporized by the beam [15]. Serial crystallography has opened new possibilities for studying radiation-sensitive materials and for time-resolved studies of enzymatic reactions and other dynamic processes [15].
Advances in sample delivery methods have been crucial for enabling these approaches. Liquid injectors stream crystal suspensions across the X-ray beam, while fixed-target devices present crystals on solid supports [15]. These technical innovations have progressively reduced sample requirements—from gram quantities in early serial crystallography experiments to microgram amounts today—making structural studies feasible for more challenging biological targets [15].
Integration with Complementary Methods
Modern structural biology increasingly integrates crystallography with complementary techniques to address complex biological questions. Cryo-electron microscopy (cryo-EM) has emerged as a powerful alternative for determining structures of large macromolecular complexes that may be difficult to crystallize [13] [17]. Nuclear magnetic resonance (NMR) spectroscopy provides information about protein dynamics and solution-state conformations that complements the static snapshots from crystallography [17].
Computational methods, particularly artificial intelligence-based structure prediction as exemplified by AlphaFold2, now provide accurate models for many proteins without experimental determination [17]. These predicted structures can facilitate molecular replacement in crystallographic analyses and guide experimental design [17]. The integration of these diverse approaches represents the future of structural biology, where hybrid methodologies provide comprehensive understanding of biological macromolecules in both static and dynamic contexts.
From its origins in physics laboratories to its current status as an indispensable biological tool, X-ray crystallography has fundamentally transformed our understanding of life at the molecular level. The historical milestones outlined in this review—from the first diffraction patterns to the current era of synchrotron-based high-throughput structural biology—demonstrate how technical innovations have continuously expanded the frontiers of biological knowledge. The experimental protocols developed over decades now enable researchers to visualize biological macromolecules with atomic precision, providing insights into mechanisms of disease and facilitating rational drug design. As crystallography continues to evolve alongside complementary techniques like cryo-EM and computational prediction, its capacity to illuminate biological structure and function will undoubtedly yield further breakthroughs in basic science and therapeutic development. For researchers pursuing protein structure determination, understanding this historical context and methodological foundation provides both practical guidance and inspiration for future investigations.
The determination of three-dimensional protein structures is fundamental to modern biology and drug discovery. X-ray crystallography has been the predominant technique for elucidating atomic-level structures for over a century [18]. This guide details three interconnected concepts that form the theoretical foundation of X-ray crystallography: resolution, which defines the level of detail obtainable from an experiment; electron density, which provides the map for model building; and the phase problem, the central challenge in converting raw diffraction data into meaningful structural information.
Understanding these concepts is critical for researchers interpreting structural data and for drug development professionals relying on accurate protein models for rational drug design. Recent advances, particularly in deep learning, are transforming how we approach these fundamental problems [19] [20].
Core Concept 1: Resolution
Definition and Physical Basis
In X-ray crystallography, resolution describes the finest level of detail discernible in an experimental electron density map. It is quantitatively defined by the smallest interplanar spacing (d-spacing) for which diffraction spots can be measured, typically reported in Ångströms (Å) [16]. The relationship between the diffraction angle and resolution is governed by Bragg's Law: ( nλ = 2d \sin(θ) ), where ( d ) represents the lattice spacing, ( λ ) is the X-ray wavelength, and ( θ ) is the diffraction angle [18].
Higher resolution (corresponding to a smaller numerical value in Å) results from measuring diffraction data to wider angles and provides greater atomic detail. The quality of the crystal primarily determines the achievable resolution; well-ordered crystals with perfectly repeating unit cells produce diffraction to higher angles [16].
Resolution Ranges and Structural Interpretability
The table below summarizes how different resolution ranges affect the interpretability of electron density maps.
Table 1: Interpretation of Electron Density Maps at Various Resolution Ranges
| Resolution Range (Å) | Structural Features Resolvable | Common Applications |
|---|---|---|
| < 1.2 Å | Individual atoms clearly resolved; alternative conformations discernible. | Small molecule crystallography; ultra-high-resolution protein studies. |
| 1.2 - 1.8 Å | Well-resolved backbone and side chains; water molecules and ions can be placed. | High-accuracy ligand binding studies; detailed mechanism analysis. |
| 1.8 - 2.5 Å | Polypeptide chain trace is clear; bulky side chains are distinguishable. | Standard for protein-ligand complex determination and drug design. |
| 2.5 - 3.2 Å | Secondary structures (α-helices, β-sheets) are visible. | Large complexes or membrane proteins where high resolution is challenging. |
| > 3.2 Å | Course molecular outline and protein domains may be visible. | Low-resolution phasing; often combined with other data for large assemblies. |
Low-resolution data (e.g., >2.5 Å) presents a significant challenge because the resulting electron density map lacks clearly defined atomic features, making the subsequent building of an accurate atomic model subjective, time-consuming, and often intractable [19]. This creates a critical bottleneck in structure determination.
Core Concept 2: Electron Density
Theoretical Foundation
Electron density, denoted as ( ρ(\mathbf{r}) ), is a three-dimensional function that describes the distribution of electrons within the crystal's unit cell [21]. The fundamental goal of X-ray crystallography is to determine this function. The structure factors, ( F(\mathbf{h}) ), obtained from the diffraction experiment are the Fourier components of the electron density.
The mathematical relationship is given by the inverse Fourier transform: [ ρ(\mathbf{r}) = \frac{1}{V} \sum_{\mathbf{h}} e^{-2\pi i \mathbf{h} \cdot \mathbf{r}} F(\mathbf{h}) ] where ( V ) is the volume of the unit cell, ( \mathbf{r} ) is a position vector in real space, and ( \mathbf{h} ) represents the Miller indices (h, k, l) [19] [20].
From Map to Atomic Model
The electron density map is calculated using both the measured amplitudes and the estimated phases of the structure factors. A crystallographer then interprets this map to build an atomic model that fits the observed density, taking into account prior knowledge of protein chemistry, such as known amino acid sequences and standard bond lengths and angles [22]. The quality of the final atomic model is therefore directly dependent on the quality and resolution of the electron density map.
Core Concept 3: The Phase Problem
The Fundamental Challenge
The phase problem is the central obstacle in X-ray crystallography. In a diffraction experiment, the detector records only the intensity of each diffracted beam, which is proportional to the square of the structure factor amplitude, ( |F(\mathbf{h})| ) [23]. However, the structure factor is a complex number characterized by both an amplitude and a phase, ( ϕ(\mathbf{h}) ): [ F(\mathbf{h}) = |F(\mathbf{h})| e^{i ϕ(\mathbf{h})} ] The loss of phase information upon measurement is critical because both amplitude and phase are required to compute the electron density map via Fourier synthesis [23] [20]. This is often described as a holistic relationship: every detail of the real-space structure depends on the totality of information (both amplitudes and phases) in reciprocal space, and vice versa [21].
Historical and Modern Methods for Phase Retrieval
Several experimental and computational methods have been developed to solve the phase problem.
Table 2: Methods for Solving the Crystallographic Phase Problem
| Method | Underlying Principle | Key Requirements / Limitations |
|---|---|---|
| Direct Methods [23] | Uses probabilistic relationships between phases of strong reflections. | Requires atomic-resolution data (typically better than 1.2 Å). Works for small molecules, rarely for proteins. |
| Molecular Replacement (MR) [22] [24] | Uses phases from a known, homologous structure as an initial model. | Requires a previously solved structure with significant sequence or structural similarity. |
| Heavy-Atom Methods (MIR) [16] [24] | Involves comparing native data with data from crystals containing incorporated heavy atoms (e.g., Hg, Pt). | Requires derivatives that are isomorphous with the native crystal. Labor-intensive. |
| Anomalous Dispersion (MAD/SAD) [23] [24] | Uses differences in diffraction intensity near the absorption edge of an atom (e.g., Se in selenomethionine). | Requires tunable X-ray source (synchrotron) and incorporation of anomalous scatterers. |
| Patterson Methods [20] | Analyzes a Fourier map calculated with squared amplitudes and zero phases to find heavy atom positions. | Becomes uninterpretable for large structures due to peak overlap (n² peaks for n atoms). |
| Deep Learning (e.g., XDXD) [19] | An end-to-end generative model that predicts a complete atomic model directly from low-resolution diffraction data. | Bypasses traditional phasing and map interpretation; shows 70.4% match rate at 2.0 Å resolution. |
The following workflow diagram illustrates how these methods integrate into the overall structure determination process.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful structure determination relies on a suite of specialized reagents and materials. The following table details key components used in the process.
Table 3: Essential Research Reagents and Materials for Protein Crystallography
| Reagent / Material | Function / Purpose | Application Notes |
|---|---|---|
| Pure Protein Sample (>95% purity) [16] [24] | The target molecule for crystallization. High purity and homogeneity are critical for forming well-ordered crystals. | Assessed by SDS-PAGE and Dynamic Light Scattering (DLS). |
| Precipitant Solutions [16] [24] | Agents (e.g., PEGs, salts) that reduce protein solubility, encouraging precipitation into an ordered crystal lattice. | A "sparse matrix" of ~50 conditions is typically screened. |
| Cryoprotectants (e.g., glycerol) [16] [24] | Protect crystals from radiation damage by forming a glassy state during flash-cooling in liquid nitrogen. | Replaces water in and around the crystal to prevent ice formation. |
| Heavy Atom Derivatives [16] [24] | Used in MIR phasing. Heavy atoms (e.g., Hg, Pt, Au) are incorporated into crystals to provide phasing information. | Soaked into pre-grown crystals or incorporated via protein expression (e.g., selenomethionine). |
| Detergents / Lipids [24] | Solubilize and stabilize membrane proteins for crystallization, which is a major challenge in the field. | Used in lipidic cubic phase crystallization for membrane proteins. |
Advanced Topics and Future Directions
The Low-Resolution Bottleneck and AI Solutions
A significant frontier in crystallography is overcoming the limitations of low-resolution data. While methods like molecular replacement can provide initial phases, the resulting electron density maps at resolutions like 2.0 Å are often ambiguous and lack clear atomic features, making model building difficult [19].
Recent breakthroughs in deep learning are offering end-to-end solutions. For instance, the XDXD framework is a diffusion-based generative model that predicts a complete atomic model directly from low-resolution single-crystal X-ray diffraction data, bypassing the need for manual map interpretation [19]. This model has demonstrated a 70.4% match rate for structures with data limited to 2.0 Å resolution, showing robust performance on a benchmark of 24,000 experimental structures [19]. Other approaches involve using convolutional neural networks to interpret Patterson maps, which are computed directly from diffraction intensities without phase information, to produce initial electron-density estimates [20].
Emerging Experimental Modalities
Technologies like X-ray Free Electron Lasers (XFEL) are revolutionizing data collection. In serial femtosecond crystallography, a stream of microcrystals is passed through an extremely bright, pulsed XFEL beam, allowing diffraction data to be collected before the crystals are destroyed by radiation damage [22]. This enables the study of challenging samples and the capture of molecular movies of dynamic processes, such as enzyme catalysis, on femtosecond timescales [22] [24].
The following diagram illustrates the core logical relationship between the three essential concepts discussed in this guide.
The concepts of resolution, electron density, and the phase problem are not merely sequential steps but are deeply intertwined pillars supporting the edifice of X-ray crystallography. The quality of the final atomic model is contingent upon the resolution of the data and the accuracy of the phases obtained to compute the electron density map. For researchers and drug development professionals, a firm grasp of these principles is indispensable for critically evaluating structural models deposited in databases and for designing effective experiments. The field is dynamic, with emerging computational methods, particularly deep learning, poised to automate structure determination and overcome long-standing challenges like the low-resolution bottleneck, thereby paving the way for new discoveries in structural biology.
A Step-by-Step Workflow: From Protein Purification to Refined Model
Within the framework of protein structure determination via X-ray crystallography, the initial steps of protein purification and crystallization constitute the most significant bottleneck, often determining the success or failure of entire structural initiatives. This whitepaper provides an in-depth technical analysis of this critical phase, detailing the foundational principles of crystallization, comprehensive purification methodologies, and contemporary experimental protocols. Aimed at researchers and drug development professionals, this guide synthesizes current practices with emerging trends—such as AI integration and microfluidics—that are poised to alleviate these longstanding challenges. The content is contextualized within the growing protein crystallization market, which is projected to expand from $1.62 billion in 2024 to $2.8 billion by 2029, driven largely by demands in biopharmaceutical development [25] [26].
The Premise of Protein Crystallization
Protein crystallization is the process of inducing proteins to form a highly ordered, three-dimensional lattice. The fundamental principle governing this process is supersaturation [27] [28]. A protein solution becomes supersaturated when the concentration of the protein exceeds its solubility limit under specific chemical and physical conditions. This non-equilibrium state drives the first-order phase transition of nucleation, where spontaneous clusters of protein molecules (nuclei) form and become stable enough to serve as templates for crystal growth [27].
The path from an undersaturated solution to a viable crystal is typically visualized using a phase diagram, which plots protein concentration against precipitant concentration. This diagram reveals several key zones:
- Undersaturated Zone (Stable): The protein remains fully soluble. No crystallization occurs.
- Metastable Zone: Nuclei may form but are metastable. Crystal growth can occur if seeds are present, but new nucleation is unlikely.
- Labile Zone (Nucleation): The region of highest supersaturation where spontaneous nucleation and crystal growth can occur.
- Precipitation Zone: Excessive supersaturation leads to disordered, amorphous aggregation instead of ordered crystals [27] [29].
The objective of any crystallization experiment is to navigate the solution from the undersaturated zone into the labile zone to initiate nucleation, and then to maintain conditions in the metastable zone to allow for slow, ordered crystal growth [27]. The success of this process is highly dependent on the precise control of numerous variables, including pH, temperature, ionic strength, and the nature of the precipitating agent.
The Imperative of High-Quality Protein Purification
The journey to a high-resolution structure begins with the production of a pure, monodisperse, and stable protein sample. The homogeneity of the preparation is arguably the most critical factor in obtaining crystals that diffract to high resolution [30] [28].
Core Purification Methodologies
A combination of chromatographic techniques is typically employed to achieve the requisite purity (>95-99%) [30] [31].
Table 1: Key Chromatographic Techniques for Protein Purification
| Technique | Principle of Separation | Primary Application in Crystallography |
|---|---|---|
| Affinity Chromatography | Utilizes highly specific biological interactions (e.g., His-tag/Ni-NTA, antibody/Protein A) [30]. | Initial capture and significant purification in a single step. |
| Ion-Exchange Chromatography | Separates proteins based on their net surface charge. | Polishing step to remove impurities and isoforms with minor charge differences. |
| Size-Exclusion Chromatography (SEC) | Separates molecules based on their hydrodynamic radius or size [30]. | Final polishing step to isolate monodisperse populations and remove aggregates. |
Analysis of Purity and Monodispersity
Beyond chromatographic purity, the conformational homogeneity of a protein sample is vital. Techniques used for assessment include:
- SDS-PAGE: Confirms denaturing purity and approximate molecular weight.
- Dynamic Light Scattering (DLS): Measures the hydrodynamic radius of particles in solution, providing a critical assessment of monodispersity. A single, narrow peak is indicative of a homogeneous sample suitable for crystallization trials [30].
- Mass Spectrometry: Verifies protein identity and can analyze post-translational modifications (e.g., glycosylation) that can hinder crystallization [30].
Experimental Crystallization Protocols
Once a pure protein sample is concentrated to a suitable level (typically 5-50 mg/mL), systematic crystallization trials begin [28]. Several established methods are used to achieve supersaturation in a controlled manner.
Vapor Diffusion (Hanging and Sitting Drop)
Vapor diffusion is the most widely used technique in high-throughput crystallization screens [27] [28]. The following protocol is typical for a 24-well tray setup:
Materials: Protein sample (≥ 5 mg/mL), 24-well hanging or sitting drop tray, reservoir solutions (precipitants, buffers, salts), silicon grease, siliconized cover slides, micropipette with low-retention tips [28].
Procedure:
- Reservoir Preparation: Fill each well of the tray with 500 μL of a precipitant solution [28].
- Sealing: For hanging drop, place a greased ring around the rim of each well. For sitting drop, optically clear tape is used later [28].
- Drop Setup:
- On a clean cover slide (hanging) or on the shelf (sitting), pipette 1-2 μL of concentrated protein.
- Add an equal volume of reservoir solution to the protein drop, mixing carefully to avoid bubbles [28].
- Sealing and Incubation: For hanging drop, invert the cover slide and carefully place it over the well, pressing down to form a seal. For sitting drop, seal the entire tray row with transparent tape. Place the tray in a stable, vibration-free incubator at a controlled temperature (commonly 4°C or 20°C) [28].
- Monitoring: Check trays daily for crystal growth, handling with extreme care to prevent disturbances. Crystals can appear within hours or take several months [28].
Principle: The drop containing protein and precipitant is initially at a lower concentration than the reservoir. Water vapor diffuses from the drop to the reservoir, slowly concentrating both the protein and the precipitant in the drop until equilibrium is reached. This gradual increase in concentration ideally drives the solution into the labile zone for nucleation and then into the metastable zone for crystal growth [27] [28].
Batch Crystallization under Oil
Batch crystallization is a method where the protein is immediately mixed into a supersaturated state [27] [28].
Materials: Protein sample, 96-well microbatch tray, paraffin/mineral oil mixture, micropipette [28].
Procedure:
- Fill the wells of the microbatch tray with oil to a depth of about 3 mm [28].
- Pipette 1 μL of protein solution directly to the bottom of an oil-filled well.
- Add 1 μL of precipitant solution to the same well, ensuring it fuses with the protein droplet [28].
- Seal the tray and incubate under stable conditions.
Principle: The protein and precipitant are mixed at their final concentrations, immediately establishing supersaturation. The layer of inert oil prevents evaporation of water from the drop, allowing for very small volumes and protecting the sample from airborne contamination [27] [28]. This method is particularly useful for producing microcrystals for serial crystallography experiments [27].
The following workflow diagram illustrates the key steps and decision points in the purification and crystallization process:
Navigating the Crystallization Bottleneck: Challenges and Optimization
Despite standardized protocols, crystallization remains the primary unpredictable element in structure determination. Key challenges and optimization strategies include:
Intrinsic Protein Challenges
- Membrane Proteins: Their hydrophobic nature requires detergents or lipidic cubic phases (e.g., monoolein) for solubilization, which drastically complicates the phase diagram and crystal lattice formation [27] [29]. As of 2010, membrane proteins represented less than 0.5% of non-redundant entries in the PDB [29].
- Flexible Regions: Dynamic loops and termini prevent the formation of a repeating lattice. Construct optimization via limited proteolysis, sequence alignment, and truncation of flexible regions is a common and often essential strategy to rigidify the protein [27]. For example, truncations in S. typhimurium aspartate receptor improved crystal diffraction from 3.0 Å to 1.85 Å [27].
Screening and Additives
- Sparse Matrix Screening: Initial screens use incomplete factorial design to test a wide range of conditions (pH, precipitants, salts) with minimal experiments [28]. Commercial screens are widely available.
- Rational Additives: Small molecules, cofactors, ligands, or specific ions can enhance crystal quality by stabilizing a particular protein conformation or mediating crystal contacts [27] [30]. For instance, introducing antibody fragments or using PDZ domains as scaffolding modules can facilitate nucleation for problematic targets [27].
Table 2: Common Precipitants and Additives in Crystallization Screens
| Category | Examples | Mode of Action | Frequency of Use |
|---|---|---|---|
| Salts | Ammonium Sulfate, Sodium Chloride | Reduces protein solubility by competing for water molecules (salting out). | High (~30% of conditions) [28] |
| Polymers | Polyethylene Glycol (PEG) 3350, PEG 1000 | Excludes volume, increasing effective protein concentration. | Very High (~30% of conditions) [27] [28] |
| Organic Solvents | MPD, Ethanol, Isopropanol | Reduces dielectric constant of solution, promoting association. | Moderate |
| Additives | Ions (e.g., Zn²⁺), Ligands, Detergents | Stabilizes specific conformations or mediates crystal contacts. | Condition-dependent [27] |
The Scientist's Toolkit: Key Research Reagent Solutions
A successful crystallization pipeline relies on a suite of specialized reagents and instruments.
Table 3: Essential Materials and Reagents for Protein Crystallization
| Item | Function | Example Vendors/Products |
|---|---|---|
| Crystallization Plates | Platforms for setting up nanoliter- to microliter-scale trials. | 24-well hanging/sitting drop trays; 96-well microbatch trays [28]. |
| Sparse Matrix Screens | Pre-formulated solutions to efficiently sample chemical space. | Hampton Research (Crystal Screen), Molecular Dimensions (JCSG+), Rigaku (Morpheus) [32] [28]. |
| Precipitant Reagents | Chemicals to induce supersaturation. | PEGs, Ammonium Sulfate, MPD [27] [28]. |
| Cryoprotectants | Agents like glycerol to protect crystals during flash-cooling in liquid nitrogen. | Glycerol, Ethylene Glycol, Paratone-N Oil [25] [30]. |
| Liquid Handling Robots | Automation for high-throughput, reproducible screen setup. | Tecan Group, Formulatrix Inc., SPT Labtech's mosquito crystal [25] [26]. |
Future Perspectives and Market Context
The persistent challenge of the crystallization bottleneck is driving innovation and significant market growth. The global protein crystallization market is projected to grow from $1.62 billion in 2024 to $2.8 billion by 2029, at a Compound Annual Growth Rate (CAGR) of 11.5% [25]. Key trends shaping the future include:
- Automation and AI: Integration of artificial intelligence for predicting crystallization conditions and automated imaging systems for crystal detection is becoming standard, improving success rates and throughput [25] [26].
- Microfluidics: These platforms reduce sample volume requirements to nanoliters, enabling thousands of conditions to be screened with minimal protein material [29] [26].
- Advanced Light Sources: Techniques like serial femtosecond crystallography (SFX) at X-ray free electron lasers (XFELs) allow the use of microcrystals, thereby lowering the barrier for proteins that only form small crystals [30] [22].
- Hybrid Methods: The rise of cryo-electron microscopy (cryo-EM) provides an alternative for structures that are persistently recalcitrant to crystallization, especially large complexes and membrane proteins [30] [22].
Protein purification and crystallization represent the critical, rate-limiting step in de novo structure determination via X-ray crystallography. Mastering this phase requires a deep understanding of biophysical principles, meticulous execution of purification and screening protocols, and strategic optimization. While it remains a significant bottleneck, ongoing technological advancements in automation, microfluidics, and computational prediction are steadily increasing the throughput and success rates. For researchers in structural biology and drug development, a rigorous and systematic approach to this first step is the indispensable foundation upon which all subsequent atomic insights are built.
Modern macromolecular X-ray crystallography predominantly relies on synchrotron radiation sources, which have revolutionized the field by providing X-ray beams of unparalleled intensity and quality. These facilities have become indispensable for structural biology, enabling the determination of over 70% of all macromolecular structures in the Protein Data Bank [33]. Synchrotrons generate X-rays through the acceleration of charged particles in magnetic fields, producing beams that are orders of magnitude more brilliant than traditional laboratory X-ray sources [33]. This high brilliance enables researchers to work with smaller crystals, collect data at higher resolutions, and employ advanced experimental techniques such as anomalous dispersion for solving the phase problem in crystallography.
The evolution of synchrotron sources has progressed through distinct generations, each offering significant improvements in beam characteristics and experimental capabilities. Fourth-generation synchrotrons, such as those featuring multi-bend achromat lattice designs, provide dramatically increased coherent flux, enabling novel imaging techniques like coherent X-ray diffraction imaging (CXDI) and expanding the possibilities for studying non-crystalline biological specimens [34]. These advanced sources offer unprecedented opportunities for structural biology, particularly when combined with the latest detector technologies and experimental methodologies.
Key Components of a Synchrotron Beamline
A synchrotron beamline consists of several critical components that work together to deliver optimized X-ray beams for protein crystallography experiments:
Front End and Optics: The beamline begins with insertion devices (undulators or wigglers) that generate specific X-ray characteristics. This is followed by sophisticated optical systems including monochromators for selecting X-ray wavelengths and mirrors for focusing and harmonic rejection. Modern beamlines often feature micro-focusing optics that can produce beam sizes below 10 microns, enabling data collection from microcrystals [15].
Experimental Station: The core of the beamline contains a goniometer for precise crystal positioning and rotation, sample visualization systems, and a detector positioned at optimal distance from the sample. Cryogenic cooling systems are standard for conventional data collection, while specialized environmental chambers are available for room-temperature studies [35].
Robotic Sample Handling: Automated sample changers, such as systems capable of storing 460 samples in liquid nitrogen, allow for high-throughput data collection by automatically mounting and centering crystals in the X-ray beam [3]. This automation has dramatically increased the efficiency of synchrotron facilities, enabling the collection of multiple complete datasets per hour.
Beam Conditioning: Advanced beamlines incorporate adaptive optics and collimation systems to optimize beam characteristics for specific experiments, such as serial crystallography or time-resolved studies.
Modern X-ray Detectors: Technology and Performance
The development of modern X-ray detectors represents a critical advancement in macromolecular crystallography. Current detectors predominantly use hybrid photon counting technology, which provides noise-free detection with high dynamic range and fast readout capabilities [36]. These detectors directly convert X-ray photons into electrical charge, enabling precise counting of individual photons without readout noise.
Table 1: Modern X-ray Detectors for Synchrotron Crystallography
| Detector Model | Technology | Pixel Size | Frame Rate | Key Features | Applications |
|---|---|---|---|---|---|
| EIGER2 [36] | Photon-counting | 75 µm | High (kHz range) | High dynamic range, no readout noise | Still/multi-series collection, SX |
| PILATUS4 [36] | Photon-counting | 150 µm | Moderate | Renewed version of popular PILATUS | Standard rotation data collection |
| SELUN [36] | Hybrid Photon Counting | Not specified | 120,000 fps | Sustained frame rate at high count rates | 4th-gen synchrotrons, high-speed SX |
| MYTHEN2 [36] | Microstrip photon-counting | Strip detector | Continuous | Compact, modular design | Specialized applications |
The performance characteristics of modern detectors have enabled new experimental modalities in crystallography. High frame rates are essential for serial crystallography, where thousands of diffraction patterns must be collected in rapid succession [15]. The absence of readout noise ensures accurate measurement of weak diffraction signals, which is particularly important for detecting high-resolution information and for working with small crystals that produce weak diffraction.
Data Collection Strategies and Methodologies
Conventional Rotation Data Collection
Traditional macromolecular crystallography employs the rotation method, where a single crystal is continuously rotated in the X-ray beam while collecting diffraction images at small angular intervals (typically 0.1-1°). This method requires well-diffracting crystals of sufficient size (typically >10-50 μm) and remains the workhorse for most structural biology projects [3]. The crystal is maintained at cryogenic temperatures (approximately 100 K) to mitigate radiation damage, allowing complete datasets to be collected from individual crystals [35].
The optimal data collection strategy depends on crystal characteristics including symmetry, unit cell dimensions, and diffraction quality. Modern beamline control software automatically determines optimal rotation ranges, exposure times, and beam parameters to maximize data quality while minimizing radiation damage. Complete datasets can typically be collected within minutes at modern synchrotron beamlines, a dramatic improvement over the hours or days required with earlier generations of synchrotron sources or laboratory X-ray generators [3].
Serial Crystallography Methods
Serial crystallography (SX) has emerged as a powerful alternative approach, particularly for challenging systems that only produce microcrystals. This method involves collecting diffraction "still" images from thousands of microcrystals, with each crystal typically exposed to a single X-ray pulse before being replaced [15]. The individual diffraction patterns are then indexed and merged to create a complete dataset.
Table 2: Serial Crystallography Delivery Methods and Sample Consumption
| Delivery Method | Principle | Sample Consumption | Advantages | Limitations |
|---|---|---|---|---|
| Liquid Injection [15] | Crystal slurry injected as liquid stream | ~μL to mL range | High speed, compatibility with time-resolved studies | High sample waste, jet stability issues |
| Fixed-Target [35] | Crystals mounted on solid support | <1 μL | Minimal sample waste, compatibility with slow data collection | Lower throughput, potential background scattering |
| High-Viscosity Extrusion [15] | Crystal suspension in viscous matrix | Reduced waste compared to liquid injection | Reduced flow rates, lower background | Potential damage to crystals, complex operation |
Serial crystallography can be performed at both X-ray free-electron lasers (XFELs), where it is termed serial femtosecond crystallography (SFX), and at synchrotrons, where it is known as serial millisecond crystallography (SMX) [15]. The "diffraction before destruction" approach at XFELs uses ultrashort femtosecond pulses to outrun radiation damage, while SMX at synchrotrons distributes dose across many crystals to minimize damage [15].
Experimental Workflow and Protocol
The following diagram illustrates the core decision-making process and workflow for X-ray data collection at synchrotrons:
Sample Preparation and Mounting
For conventional crystallography, crystals are harvested from crystallization drops and cryo-cooled in liquid nitrogen to prevent radiation damage. This process requires adding cryoprotectants to prevent ice formation during freezing [3]. Crystals are then mounted on standardized pins and stored in automated sample changers until data collection.
For fixed-target serial crystallography, advanced approaches involve growing crystals directly on microporous sample holders containing multiple compartments for different protein/ligand complexes [35]. After crystal growth, crystallization solution is removed by blotting through the porous membrane, and ligand solutions are added by pipetting. This approach minimizes sample handling and enables high-throughput screening.
Data Collection Parameters
Critical parameters for data collection include:
Beam Energy: Typically selected between 5-15 keV (0.8-2.5 Å wavelength), with specific energies chosen for anomalous diffraction experiments near elemental absorption edges [33].
Detector Distance: Optimized based on desired resolution, with larger distances providing higher resolution but requiring longer exposure times.
Exposure Time: Balanced between achieving sufficient signal-to-noise and minimizing radiation damage, typically ranging from milliseconds to seconds per image.
Rotation Range: Complete datasets typically require 180-360° of total rotation, depending on crystal symmetry.
Beam Size: Matched to crystal dimensions to minimize background scattering, with micro-focus beams (<10 μm) used for microcrystals [15].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Synchrotron Data Collection
| Item | Function | Application Notes |
|---|---|---|
| Cryoprotectants [3] | Prevent ice formation during cryo-cooling | Glycerol, ethylene glycol, or commercial solutions; concentration optimized empirically |
| Crystal Mounting Loops [3] | Secure crystal during data collection | Various sizes matched to crystal dimensions; nylon or micro-meshed |
| Microporous Sample Holders [35] | Fixed-target serial crystallography | Enable on-chip crystallization and ligand soaking; 12 compartments for high-throughput |
| Crystallization Screens [37] | Initial crystal condition screening | Commercial 96-well format screens; require ~100-250 μL of purified protein |
| Liquid Handling Robots [37] | Automated crystallization setup | Mosquito robot for nanoliter-volume dispensing; improves reproducibility |
| Fragment Libraries [35] | Ligand screening | F2X entry library (95 molecules) for identifying protein-ligand interactions |
Advanced Applications: Room-Temperature and Time-Resolved Studies
Recent advancements have enabled more physiologically relevant data collection through room-temperature serial crystallography. This approach captures protein conformations closer to native states and can reveal previously unobserved conformational states [35]. Fixed-target serial crystallography has been advanced to enable high-throughput fragment screening at room temperature, achieving resolutions comparable to cryogenic methods while providing insights into biologically relevant protein dynamics [35].
Time-resolved serial crystallography (TR-SX) represents another frontier, enabling the visualization of reaction intermediates in biological processes. Two primary approaches have been developed: light-activated studies using pump-probe lasers for photosensitive proteins, and mix-and-inject serial crystallography (MISC) for enzyme-substrate interactions [15]. These "molecular movie" techniques allow researchers to observe structural changes in real-time, providing unprecedented insights into biochemical mechanisms.
Data Quality Assessment and Optimization
The quality of X-ray diffraction data fundamentally determines the achievable resolution and accuracy of the final atomic model. Key quality metrics include:
Resolution: Determined by the smallest Bragg spacing (dmin) measurable from the diffraction pattern. Higher resolution (lower dmin values) provides greater atomic detail:
Completeness and Redundancy: Essential for accurate intensity measurement and reduction of random errors. Modern detectors and collection strategies typically achieve >95% completeness and high redundancy.
Signal-to-Noise Ratio: Determined by crystal quality, beam intensity, and detector performance. Photon-counting detectors provide essentially noise-free detection [36].
Radiation Damage Management: Particularly critical for room-temperature data collection, where radiation sensitivity is more than 100 times higher than at cryogenic temperatures [35]. Serial crystallography approaches mitigate this by distributing dose across many crystals.
The continuing evolution of synchrotron sources, detector technologies, and experimental methodologies ensures that X-ray crystallography will remain a cornerstone technique for understanding biological mechanisms and advancing drug discovery initiatives. Fourth-generation synchrotrons and the ongoing development of even more advanced detectors promise to further expand the capabilities of this powerful structural biology tool.
In the field of X-ray crystallography, determining the three-dimensional structure of a protein relies on measuring the intensities of X-rays diffracted by a crystal. These intensities provide the amplitudes of the structure factors but crucially lack information about their phases [38] [23]. This fundamental limitation is known as the "phase problem."
To reconstruct an electron density map and ultimately determine the atomic structure, both amplitude and phase information are required [38]. This article explores the primary experimental methods used to overcome this challenge: Molecular Replacement (MR), Single-wavelength Anomalous Dispersion (SAD), and Multi-wavelength Anomalous Dispersion (MAD).
Core Phasing Methods
Molecular Replacement (MR)
Molecular Replacement is a phasing method used when a structurally similar model is already available.
- Principle: MR grafts the known phases from a homologous structure onto the experimentally determined amplitudes of the target molecule [23]. The process involves positioning the search model within the unit cell of the unknown crystal.
- Requirements: Successful MR typically requires a search model with >25% sequence identity and a root-mean-square deviation (RMSD) of less than 2.0 Å between the Cα atoms of the model and the target structure [38].
- Process: The method employs the Patterson function, a map calculated using squared structure factors (intensities) that does not require phases. This map reveals peaks at interatomic vectors. The search model is rotated and translated to match the Patterson map of the target crystal [38].
Experimental Phasing: SAD and MAD
Experimental phasing methods solve the phase problem by using measurable differences in diffraction intensities, introduced by specific atoms, to derive phase information.
- Single-wavelength Anomalous Dispersion (SAD): Uses a single dataset collected at one wavelength, typically near the absorption edge of an anomalous scatterer present in the crystal [39] [40]. Native-SAD utilizes light atoms naturally present in proteins, such as sulfur in cysteine and methionine residues, eliminating the need for derivatization [40].
- Multi-wavelength Anomalous Dispersion (MAD): Requires data collection at multiple wavelengths near the absorption edge of an anomalous scatterer. The anomalous dispersion effect causes a wavelength-dependent phase shift in the reflections, which can be analyzed to solve the structure [23].
The following table summarizes the characteristics of SAD and MAD phasing.
Table 1: Comparison of SAD and MAD Phasing Methods
| Feature | SAD (Single-wavelength Anomalous Dispersion) | MAD (Multi-wavelength Anomalous Dispersion) |
|---|---|---|
| Data Requirement | Single dataset | Multiple datasets at different wavelengths |
| Anomalous Scatterers | Selenium (incorporated), Sulfur (native), or other heavy atoms | Selenium, mercury, or other atoms with strong edges |
| Key Advantage | Simpler data collection; suitable for native phasing (e.g., S-SAD) | Provides multiple independent measurements for more robust phasing |
| Experimental Consideration | Requires highly accurate data; benefits from high multiplicity | Requires tunable X-ray source (synchrotron); all data from one crystal ensures isomorphism |
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful experimental phasing relies on several key reagents and materials.
Table 2: Key Research Reagents and Materials for Experimental Phasing
| Reagent/Material | Function in Phasing |
|---|---|
| Selenomethionine | Biosynthetically incorporated into proteins to provide strong anomalous scatterers (selenium atoms) for SAD/MAD phasing [38] [40]. |
| Heavy Atom Soaks | Compounds containing atoms like mercury, platinum, or gold used to derivatize crystals via soaking, creating isomorphous heavy-atom derivatives for MIR/SIR phasing [38]. |
| Native Protein Crystals | Crystals of the wild-type protein containing endogenous light atoms (e.g., sulfur, phosphorus, chlorine, calcium) for native-SAD phasing [40]. |
| Cryoprotectants | Chemicals like glycerol or ethylene glycol used to protect crystals from ice formation during flash-cooling in liquid nitrogen for data collection [16]. |
Technical Workflows and Modern Implementations
Integrated MR-SAD Workflow
For challenging structures with low-resolution data, a combined MR-SAD approach can be powerful. The following diagram illustrates a modern computational pipeline for this method, which simultaneously uses information from a partial model and anomalous scattering to overcome model bias and completeness issues [41].
Workflow for MR-SAD Structure Solution
Practical SAD Phasing Protocol
The following is a detailed methodology for determining a macromolecular structure using the SAD method, as implemented in modern software suites like Phenix [39]:
- Data Collection and Preparation: Collect a single X-ray diffraction dataset from a single crystal at a wavelength optimized for the anomalous scatterer (e.g., selenium, sulfur, or a heavy atom). The data should be indexed, integrated, and scaled.
- Substructure Determination: Use a program like
phenix.hyssto locate the positions of the anomalous scatterers in the unit cell. This step is critical and relies on a significant anomalous signal. - Initial Phasing: Calculate initial experimental phases based on the located anomalous substructure.
- Density Modification: Apply algorithms such as solvent flattening to improve the quality of the initial electron density map. This step leverages the fact that a large portion of the crystal is disordered solvent.
- Model Building: An automated model-building program (e.g.,
phenix.autosol) interprets the improved electron density map and builds an initial atomic model. If the map is of sufficient quality, this model can be quite complete. - Model Refinement: The initial model is iteratively refined against the original diffraction data to improve its fit to the electron density and its stereochemical quality.
Advanced Applications and Future Directions
Native-SAD at Long Wavelengths
A significant advancement in SAD phasing is the use of long-wavelength X-rays (e.g., λ = 2.75 Å to 5.9 Å) for native-SAD experiments. The anomalous scattering factor (f") of light atoms like sulfur increases dramatically at wavelengths approaching their absorption edge (λ = 5.02 Å for sulfur) [40]. This makes it possible to solve structures using only the weak anomalous signal from native sulfurs, even in proteins with a sulfur content as low as 0.25% [40]. Dedicated beamlines operating in a vacuum environment overcome the technical challenges of air absorption and increased scattering at these long wavelengths, making native-SAD a more routine and powerful phasing method.
The Role of AI and Automation
Artificial intelligence is beginning to impact how the phase problem is approached. While tools like AlphaFold provide highly accurate models that can serve as search models for Molecular Replacement, experimental phasing remains essential for ~20% of novel structures where predictions are insufficient for MR [40]. Furthermore, new deep learning models are being developed to tackle the phase problem directly. For instance, the XDXD framework uses a diffusion-based generative model to predict a complete atomic model directly from low-resolution single-crystal X-ray diffraction data, potentially bypassing traditional map interpretation challenges [19].
In protein X-ray crystallography, the construction and refinement of an atomic model is the crucial step that transforms an experimental electron density map into a detailed, three-dimensional structure. This process is inherently iterative, cycling between manual model building and computational refinement to achieve the best possible agreement between the atomic model and the observed X-ray diffraction data [42] [43]. The primary metric for assessing this agreement is the R-factor (also called the residual factor or R-value), a single number that quantifies the global fit of the model to the experimental data [44] [45]. This step is foundational for all subsequent analyses, such as understanding enzyme mechanisms or structure-based drug design, making its rigorous execution paramount for researchers and drug development professionals [16].
This guide provides an in-depth technical overview of the model building and refinement workflow, the mathematical and practical interpretation of R-factors, and the essential validation procedures required to produce a reliable protein structure.
The Mathematical Foundation: Understanding the R-Factor
The R-factor provides a measure of the disagreement between the experimental X-ray diffraction data and the data calculated from the proposed atomic model [44].
The R-Factor Equation
The conventional crystallographic R-factor is defined by the following equation:
[ R = \frac{\sum ||F{\text{obs}}| - |F{\text{calc}}||}{\sum |F_{\text{obs}}|} ]
where:
- ( F_{\text{obs}} ) is the observed structure factor amplitude (derived from the measured intensity of a diffraction spot, ( I \propto |F|^2 )) [44].
- ( F_{\text{calc}} ) is the structure factor amplitude calculated from the current atomic model [44] [43].
- The sum extends over all measured X-ray reflections [44].
The minimum possible R-factor is zero, indicating perfect agreement, while a totally random set of atoms will give an R-value of about 0.63 [42] [45].
The Free R-Factor: A Guard Against Overfitting
A significant risk in refinement is overfitting, where the model becomes too tailored to the specific noise and features of the dataset used for refinement, compromising its predictive power [44]. To mitigate this, the Free R-factor (( R_{\text{free}} )) was introduced [44].
- Methodology: Before refinement begins, approximately 5-10% of the experimental observations are randomly selected and set aside [44] [42].
- Calculation: The ( R{\text{free}} ) is calculated using *only* this unused subset of data, while the standard R-factor (( R{\text{work}} )) is calculated using the remaining 90-95% of the data that was used in refinement [42].
- Interpretation: For an ideal model that is not over-refined, the ( R{\text{free}} ) value will be similar to, though typically slightly higher than, the ( R{\text{work}} ) value [42]. A large discrepancy between ( R{\text{free}} ) and ( R{\text{work}} ) is a strong indicator of overfitting or other problems in the refinement process [44].
The Model Building and Refinement Workflow
The process of transforming an initial electron density map into a final, refined model is a cycle of manual and computational steps. The following diagram illustrates this iterative workflow and the relationship between the key R-factors.
Diagram 1: The iterative cycle of protein model building, refinement, and validation. The process is complete when validation checks confirm the model is both accurate and geometrically sound.
Initial Model Building
The initial model is built into an electron density map by placing atoms—specifically, the protein's known amino acid sequence—to fit the map's contours [42] [22]. This is typically done using molecular graphics software.
- Interpreting Electron Density: The clarity of the electron density map is directly tied to the resolution of the diffraction data [42].
- High-Resolution (e.g., 1.0 Å): Allows for clear visualization of individual atoms and the placement of accurate side-chain and main-chain conformations [42] [46].
- Low-Resolution (e.g., 3.0 Å): Only reveals the basic contours of the protein chain, making the atomic structure harder to infer and more prone to error [42] [46].
- Handling Disorder: Flexible regions on the protein surface may have weak or smeared electron density. These are often modeled with high B-factors or as multiple alternate conformations [43].
Computational Refinement
Refinement is the process of optimizing the parameters of the atomic model to improve its agreement with the ( F_{\text{obs}} ) data, thereby lowering the R-factor [43]. This is performed using least-squares or maximum-likelihood algorithms in refinement software [43].
The parameters refined for each atom are:
- Atomic Coordinates (x, y, z): The position of the atom in 3D space.
- Atomic Displacement Parameter (B-factor): A measure of the vibrational motion or static disorder of an atom, usually expressed in Ų [43]. Typical values range from ~2 for very rigid, well-ordered atoms to ~100 for highly flexible surface loops [43].
Refinement is conducted under stereochemical restraints [43]. These restraints incorporate prior knowledge of chemical geometry—such as standard bond lengths, bond angles, and planarity of groups—preventing the model from moving into chemically unrealistic conformations while minimizing the R-factor [43]. For larger systems, TLS (Translational, Librational, Screw) refinement is often used, which models the movement of entire groups of atoms (e.g., domains) as rigid bodies, reducing the number of refined parameters [43].
Validation and Quality Assessment
A low R-factor alone is not a guarantee of a correct structure [45]. Comprehensive validation is essential.
Quantitative Metrics and Benchmarks
The table below summarizes the key quantitative indicators used to assess a refined model.
Table 1: Key quantitative metrics for assessing refined protein structures [42] [46] [45].
| Metric | Definition | Typical Target Values for a Good Quality Structure |
|---|---|---|
| Resolution | The level of detail present in the diffraction data [42]. | High: < 2.0 Å Medium: ~2.5 Å Low: > 3.0 Å |
| R-work (( R )) | Agreement between the model and the data used in refinement [44]. | ~0.20 (or 20%) for 2.5 Å resolution. Generally decreases with higher resolution. |
| R-free (( R_{\text{free}} )) | Agreement between the model and the data not used in refinement [44] [42]. | Slightly higher than R-work (e.g., ~0.24 for 2.5 Å resolution). A difference of >0.05 can indicate problems [45]. |
| Ramachandran Outliers | Percentage of amino acid residues in energetically disallowed regions of the Ramachandran plot [46]. | < 1% for a well-refined structure. |
| Clashscore | A measure of the number of steric overlaps (atoms too close) per thousand atoms [46]. | As low as possible. A value of 5 is excellent for a high-resolution structure [46]. |
Beyond the R-Factor: Holistic Validation
- Real-Space Fit: The real-space correlation coefficient (RSCC) or real-space residual (RSR) measures how well the model fits the experimental electron density within a local region (e.g., a single residue) [45].
- Stereochemical Quality: The geometry of the model—including bond lengths, bond angles, and torsion angles—should be close to ideal values derived from high-resolution small-molecule structures [43]. The Ramachandran plot is a critical check of the protein's main-chain conformation [46].
- B-Factor Analysis: The B-factors should be consistent with the protein's structural biology; for example, atoms in a stable hydrophobic core should have lower B-factors than those in a flexible loop [43].
The Scientist's Toolkit: Essential Software for Refinement
A range of specialized software is used for the tasks of model building, refinement, and validation.
Table 2: Essential software tools for model building, refinement, and validation.
| Software Tool | Primary Function | Application in the Workflow |
|---|---|---|
| Coot | Model building and manipulation | Interactive manual model building into electron density maps, fitting ligands, and correcting errors [43]. |
| ShelXL / ShelXle | Structure refinement | The industry-standard program (ShelXL) for computational refinement, often used via a graphical interface (ShelXle) [47] [48]. |
| Phenix | Comprehensive crystallography suite | Integrates tools for phasing, refinement, and validation in a single software package. |
| MolProbity / wwPDB Validation Server | Structure validation | Provides detailed analysis of stereochemistry, Ramachandran plot, clashscore, and other quality indicators [46]. |
| Mercury / VESTA | Structure visualization | For visualizing and analyzing the final refined model and electron density [48]. |
| PLATON | Crystallography toolbox | A multi-purpose tool for checking for missed symmetry, validation, and creating graphics [48]. |
Advanced Topics and Future Directions
Quantum Crystallography
Emerging methods, collectively known as quantum crystallography, are pushing the boundaries of accuracy. Techniques like Hirshfeld Atom Refinement (HAR) use quantum-mechanically derived electron densities instead of independent spherical atoms, allowing for more accurate determination of hydrogen atom positions and bond lengths, even from data collected at conventional X-ray wavelengths [47].
Serial Femtosecond Crystallography (SFX)
Using X-ray Free Electron Lasers (XFELs), SFX involves collecting diffraction patterns from a stream of microcrystals before they are destroyed by the powerful X-ray pulse [22]. This allows the study of radiation-sensitive samples and the capture of molecular movies of reaction intermediates [22].
Model building, refinement, and the critical assessment of the R-factor represent the culmination of the protein crystallography process. Success hinges on understanding that the R-factor, while vital, is just one part of a larger validation picture. A high-quality structure is the product of an iterative cycle of building and refinement, rigorously cross-validated by ( R_{\text{free}} ), and scrutinized for its stereochemical and real-space fit. By adhering to this rigorous protocol and leveraging modern software tools, researchers can produce atomic models of the highest reliability, providing a solid foundation for scientific discovery and rational drug design.
Structure-Based Drug Design (SBDD) and Fragment-Based Drug Discovery (FBDD) represent paradigm shifts in modern pharmaceutical development, leveraging atomic-level insights into protein targets to rationally design therapeutic agents. These approaches have evolved from specialized techniques into mainstream methodologies integral to most industrial drug discovery programs, contributing to numerous approved therapies and over 70 clinical-stage candidates [49] [50] [51]. This technical guide examines the core principles, workflows, and cutting-edge technological advances enabling SBDD/FBDD, with particular focus on the pivotal role of protein structure determination through X-ray crystallography and emerging complementary methods. The integration of sophisticated computational approaches, enhanced biophysical screening technologies, and innovative structural biology methods continues to expand the druggable proteome, allowing researchers to tackle increasingly challenging targets with unprecedented efficiency and precision.
Core Principles and Strategic Value
Conceptual Foundations of SBDD and FBDD
Structure-Based Drug Design operates on the fundamental principle of utilizing three-dimensional structural information about biological targets to guide the design and optimization of small molecule therapeutics [52]. Unlike traditional methods that rely on indirect inference from known active compounds, SBDD provides direct blueprints of molecular interactions, enabling researchers to engineer compounds with precise steric and electronic complementarity to their targets [53]. This approach has become "an integral part of most industrial drug discovery programs" according to industry assessments [52].
Fragment-Based Drug Discovery represents a specialized implementation of SBDD principles, beginning with very small chemical fragments (typically <300 Da) that bind weakly to target proteins [49]. These fragments offer high ligand efficiency and access to cryptic binding pockets, serving as ideal starting points for rational optimization into potent leads [49] [51]. Compared to high-throughput screening (HTS), FBDD libraries are smaller but provide broader coverage of chemical space with higher hit rates and more favorable physicochemical properties for downstream development [49] [51].
Demonstrated Impact and Clinical Success
The translational impact of these approaches is evidenced by their growing contribution to the pharmaceutical landscape. A 2025 bibliometric analysis confirmed that FBDD alone has contributed to eight FDA-approved drugs and more than 50 clinical-stage candidates [51]. Notable successes include:
- Venetoclax (Bcl-2 inhibitor) and Sotorasib (KRAS-G12C inhibitor), which demonstrate FBDD's capability to tackle previously "undruggable" targets like protein-protein interactions and oncogenic mutants [51].
- Asciminib (allosteric BCR-ABL1 inhibitor), showcasing the ability to develop compounds with novel mechanisms that overcome resistance to conventional therapies [51].
Table 1: FDA-Approved Drugs Originating from FBDD Campaigns
| Drug Name | Primary Target | Indication | Approval Year |
|---|---|---|---|
| Vemurafenib | BRAF | Melanoma | 2011 |
| Pexidartinib | CSF-1R | Tenosynovial giant cell tumor | 2015 |
| Venetoclax | Bcl-2 | Chronic lymphocytic leukemia | 2016 |
| Erdafitinib | FGFR | Urothelial carcinoma | 2019 |
| Berotralstat | Plasma kallikrein | Hereditary angioedema | 2020 |
| Sotorasib | KRAS-G12C | Non-small cell lung cancer | 2021 |
| Asciminib | BCR-ABL1 | Chronic myeloid leukemia | 2021 |
| Capivasertib | AKT kinase | Breast cancer | 2023 |
The FBDD Workflow: From Fragments to Leads
The fragment-based drug discovery process follows a systematic, iterative workflow that transforms weak fragment hits into potent lead compounds through cycles of design, synthesis, and testing [49].
Rational Fragment Library Design
Success in FBDD hinges on the quality of the fragment library. These libraries are meticulously curated, typically containing hundreds to a few thousand compounds (compared to millions in HTS), selected for maximum diversity and optimal physicochemical properties [49]. Key design principles include:
- Rule of 3 Compliance: Molecular weight <300 Da, cLogP <3, hydrogen bond donors and acceptors <3, rotatable bonds <3 to ensure favorable solubility and synthetic tractability [49].
- Broad Chemical Functionality: Inclusion of diverse hydrogen bond donors/acceptors, hydrophobic centers, aromatic rings, and ionizable groups to probe various interaction types [49].
- Shape and Property Diversity: Ensuring coverage of different molecular geometries and physicochemical profiles to sample diverse binding pocket contours [49].
- Growth Vectors: Incorporation of synthetically tractable sites for straightforward chemical elaboration during optimization [49].
High-Throughput Biophysical Screening
Due to fragments' inherently weak binding affinities (micromolar to millimolar range), highly sensitive biophysical techniques are required for detection [49] [51]. These methods provide direct, label-free detection of binding events:
- Surface Plasmon Resonance (SPR): Real-time optical technique monitoring refractive index changes as fragments bind immobilized targets; provides comprehensive kinetic data (KD, kon, koff) [49]. Next-generation systems enable high-throughput screening across target panels, revealing selectivity patterns in days rather than years [50].
- MicroScale Thermophoresis (MST): Measures directed movement of molecules in temperature gradients; highly sensitive with minimal sample consumption in solution [49].
- Isothermal Titration Calorimetry (ITC): Gold standard for thermodynamic characterization, measuring heat changes during binding to provide complete KD, ΔH, and ΔS profiles [49].
- Nuclear Magnetic Resonance (NMR) Spectroscopy: Powerful for identifying binders in complex mixtures and mapping binding sites through ligand-observed or protein-observed techniques [49] [51].
- Thermal Shift Assays (TSA): Rapid, cost-effective measurement of thermal stability changes upon ligand binding [49].
Structural Elucidation of Fragment Hits
Following hit identification, atomic-level structural characterization becomes paramount for rational optimization [49]. This critical phase employs several complementary techniques:
- X-ray Crystallography: Remains the gold standard, providing unambiguous 3D maps of fragment-protein interactions through co-crystallization [49] [54]. Reveals specific interactions (hydrogen bonds, hydrophobic contacts, π-stacking) and identifies unoccupied pockets for growth [49]. Recent advances in room-temperature crystallography capture proteins in states closer to physiological conditions, revealing conformations missed by traditional cryo-crystallography [55].
- Cryo-Electron Microscopy (Cryo-EM): Increasingly viable for structural determination of challenging targets that resist crystallization, particularly membrane proteins [49] [52].
- Protein-Observed NMR: Provides solution-state insights into dynamic interactions, conformational changes, and multiple binding poses not captured in static crystal structures [49].
Fragment-to-Lead Optimization Strategies
With precise structural information, initial fragment hits undergo systematic optimization into drug-like leads through several strategic approaches [49]:
- Fragment Growing: Systematic addition of chemical moieties to the initial fragment core, extending into adjacent unoccupied pockets identified through structural analysis to improve affinity and selectivity [49] [51].
- Fragment Linking: Covalent connection of two or more distinct fragments binding to separate but adjacent sites, often resulting in synergistic affinity increases through additive interactions [49] [51].
- Fragment Merging: Combination of overlapping features from multiple fragments binding to the same region into a single, optimized scaffold incorporating key interactions from all parent fragments [49] [51].
This optimization occurs through iterative Design-Make-Test-Analyze (DMTA) cycles, where compounds are designed, synthesized, biologically evaluated, and structurally characterized to inform subsequent design iterations [53].
Technological Advances Driving Innovation
Revolutionary Structural Biology Methods
Recent advances in structural biology methods have significantly enhanced the resolution, speed, and biological relevance of structures obtained for SBDD/FBDD:
Room-Temperature X-ray Crystallography: Traditional cryocooling (–170°C) can distort molecular structures and mask dynamic states. New serial crystallography methods at room temperature (e.g., the HiPhaX instrument at DESY's PETRA III) visualize proteins under near-physiological conditions, revealing previously unknown conformations relevant to drug binding [55]. This approach recently uncovered a novel conformation in an antibiotic resistance enzyme that was invisible in cryo-crystallography and unpredicted by AlphaFold 3 [55].
Serial Crystallography with XFELs/Synchrotrons: Serial femtosecond crystallography (SFX) with X-ray free electron lasers (XFELs) uses ultrashort pulses in a "diffraction-before-destruction" approach, enabling studies of microcrystals and time-resolved "molecular movies" of dynamic processes [56]. These methods have been adapted for synchrotron sources (serial millisecond crystallography, SMX), making the technology more accessible [56].
Advanced Sample Delivery Systems: Diverse delivery methods have been developed to support serial crystallography, including high-viscosity extrusion (HVE) injectors for membrane protein crystals in lipidic cubic phase (LCP), fixed target approaches, and microfluidic chips that minimize sample consumption while maximizing data quality [56].
Computational and AI-Enabled Approaches
Computational methods play increasingly vital roles throughout SBDD/FBDD workflows [49] [52] [53]:
- Machine Learning-Powered Structure Prediction: Models like AlphaFold3, HelixFold3, and Chai enable protein-ligand co-folding, predicting binding modes directly from sequence even for targets without experimental structures [52] [53].
- Molecular Dynamics (MD) Simulations: Provide dynamic insights into protein-ligand complex behavior over time, revealing transient interactions, conformational flexibility, and the role of water molecules in binding [49] [52].
- Free Energy Perturbation (FEP): Advanced alchemical methods that accurately predict relative binding affinities of small chemical modifications, significantly accelerating lead optimization by prioritizing synthesis [49].
- Generative AI for Molecular Design: Deep learning models that incorporate 3D structural information to generate novel molecular structures optimized for specific target binding, exploring chemical space beyond human intuition [53].
Table 2: Key Computational Methods in Modern SBDD/FBDD
| Method | Primary Application | Key Advantages | Current Limitations |
|---|---|---|---|
| Molecular Docking | Binding pose prediction, virtual screening | Fast screening of large compound libraries | Accuracy dependent on scoring functions |
| Molecular Dynamics (MD) | Understanding dynamic binding processes | Captures flexibility and solvation effects | Computationally intensive for large systems |
| Free Energy Perturbation (FEP) | Relative binding affinity prediction | High accuracy for congeneric series | Requires significant computational resources |
| Machine Learning Structure Prediction | Protein-ligand complex modeling | No experimental structure required | Accuracy varies with target complexity |
| Generative AI Models | De novo molecule design | Explores novel chemical space | Chemical synthesizability challenges |
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful SBDD/FBDD campaigns rely on specialized reagents, materials, and instrumentation throughout the workflow.
Table 3: Essential Research Reagents and Solutions for SBDD/FBDD
| Category | Specific Examples | Function/Application |
|---|---|---|
| Protein Production | Expression vectors, cell lines, purification resins, detergents | Production of pure, stable, functional target proteins |
| Crystallization | Sparse matrix screens, precipitants, additives, LCP materials | Formation of diffraction-quality crystals |
| Fragment Libraries | Rule-of-3 compliant compounds with growth vectors | Source of initial hits with optimized properties |
| Biophysical Screening | Sensor chips, capillaries, fluorescent dyes | Detection and characterization of binding events |
| Structural Biology | Cryoprotectants, crystal loops, sample supports | Sample preparation for structural analysis |
| Computational Resources | Molecular visualization software, simulation packages, AI platforms | Data analysis, modeling, and design |
Structure-Based and Fragment-Based Drug Design have matured into indispensable approaches in modern pharmaceutical research, driven by continuous technological innovation. The integration of advanced structural methods like room-temperature crystallography with computational approaches such as AI-powered prediction and generative molecular design represents the cutting edge of the field [53] [55]. Future developments will likely focus on several key areas:
- Enhanced Time-Resolved Studies: Serial crystallography methods will enable more detailed "molecular movies" of drug binding events and protein dynamics, providing unprecedented insights into mechanisms of action [56].
- Tackling Challenging Target Classes: Improved methods for membrane proteins, protein-protein interactions, and intrinsically disordered regions will expand the druggable proteome [50] [53].
- Data Integration and Management: The treatment of structural data as a strategic product rather than a byproduct will accelerate discovery through better organization, accessibility, and integration with AI systems [57].
- Covalent and Targeted Protein Degradation: Emerging applications of FBDD principles to covalent inhibitors and proteolysis-targeting chimeras (PROTACs) open new therapeutic modalities [50].
As these technologies converge, the drug discovery process will continue to accelerate, reducing late-stage failures and delivering innovative therapeutics for challenging diseases with greater precision and efficiency.
Solving Common Crystallography Challenges: From Poor Crystals to Radiation Damage
In the realm of protein structure determination via X-ray crystallography, the quality of the crystalline sample serves as the fundamental determinant of success. This technical guide examines advanced strategies for optimizing crystal growth with a specific focus on three interconnected pillars: purity, monodispersity, and the specialized case of membrane proteins. The path to a high-resolution structure begins long before X-rays interact with a crystal; it originates with the meticulous preparation of protein samples whose integrity directly dictates their crystallographic potential [58]. Recent advances in serial crystallography have further intensified the demand for high-quality microcrystals, where sample consumption considerations make optimization protocols even more critical [15].
The challenges are particularly pronounced for membrane proteins, which represent biologically significant targets for drug development but have historically resisted crystallization efforts due to their hydrophobic nature and complex stabilization requirements [59] [60]. This guide synthesizes current methodologies and experimental protocols to provide researchers with a comprehensive framework for navigating the intricate process of crystal optimization, ultimately enabling more reliable structure determination of challenging targets.
Foundational Principles of Protein Crystallization
The Crystallography Pipeline and Crystal Quality
Protein X-ray crystallography follows a defined pipeline wherein each step profoundly influences the final structural outcome. The process encompasses protein purification, crystallization, X-ray diffraction, data collection, and model building [37]. The initial step of protein purification establishes the foundation for successful crystallization, as contaminants, heterogeneity, and aggregation directly compromise crystal formation and diffraction quality [58]. Subsequently, the crystallization step itself represents a major bottleneck, particularly because difficulties associated with crystallization constitute the primary limitation in X-ray crystallographic structure determination [3].
The quality of the final crystallographic structural model is primarily determined by the resolution of the collected X-ray data. Resolution depends on the number of diffraction spots collected during data acquisition, with more spots providing finer details in the calculated electron density map [3]. The relationship between resolution and structural detail follows these general parameters:
- Low resolution (5 Å and below): The overall shape of the protein molecule is distinguishable; α-helices are visible as long rods, but detailed building of amino acids is not possible.
- Medium resolution (3.5-2.5 Å): Researchers can distinguish side chains and build the model; beyond 2.8 Å, water molecules become clearly distinguishable.
- Atomic resolution (2.4 Å or better): The model-building process becomes significantly more accurate, and many solvent molecules can be identified and built into the density map [3].
The Link Between Sample Quality and Diffraction
The physical basis of X-ray crystallography rests on Bragg's Law (nλ = 2d sinθ), which describes how X-rays diffract when they encounter the regular atomic lattice of a crystal [3]. This diffraction occurs due to the scattering of electromagnetic waves by electrons within the crystal lattice. Each electron acts as a miniature X-ray source when struck by the X-ray beam, with scattered waves from all electrons combining through interference [3].
A protein crystal represents a periodic arrangement of molecules, and the quality of this periodicity directly impacts the sharpness and intensity of diffraction spots. imperfections in the crystal lattice, whether from structural heterogeneity, impurities, or irregular packing, manifest as poor diffraction quality, ultimately limiting the resolution achievable in the final structure [3].
Strategies for Optimizing Crystal Purity
Protein Engineering and Construct Design
The optimization process begins with intelligent protein construct design, which can dramatically enhance expression, solubility, and stability—all prerequisites for successful crystallization. Several strategic considerations guide this process:
Truncation of Flexible Regions: Disordered regions, long loops, and flexible termini often interfere with crystallization by introducing heterogeneity and structural instability. Bioinformatics tools such as DISOPRED and IUPred can predict disordered regions, while experimental techniques like limited proteolysis followed by mass spectrometry can identify stable fragments for crystallization [58]. A case study demonstrated that a kinase domain with a disordered N-terminal region failed to crystallize, but after truncating the first 50 residues, researchers obtained high-quality crystals [58].
Stabilizing Mutations: Introducing rational mutations can enhance protein stability and promote crystallization without disrupting function. Surface entropy reduction (SER) mutations reduce conformational flexibility, while replacing glycine or serine residues in loop regions with proline can enhance thermostability. For G-protein coupled receptors (GPCRs), introducing thermostabilizing mutations enabled crystallization of receptors that previously failed to form ordered crystals [58].
Fusion Tags: Fusion tags such as His-tag, maltose-binding protein (MBP), SUMO, and GB1 can enhance expression levels and solubility, preventing aggregation. However, they should be used judiciously as they might interfere with crystallization. Best practice involves designing constructs with cleavable linkers (e.g., TEV or PreScission protease sites) so the tag can be removed before crystallization trials [58].
Advanced Purification Methodologies
Achieving high purity requires a multi-step purification strategy tailored to the specific protein target. The following table summarizes the primary chromatographic techniques employed for crystallization-grade protein purification:
Table 1: Protein Purification Techniques for Crystallography
| Technique | Basis of Separation | Advantages | Optimization Tips |
|---|---|---|---|
| Affinity Chromatography | Specific binding to tags | High selectivity; rapid purification; minimal sample loss | Use competitive elution; test different resin types; enzymatic tag cleavage |
| Ion Exchange Chromatography (IEX) | Net charge at given pH | High resolution purification; removes protein variants | Gradient elution; adjust buffer pH; test ionic strengths |
| Size Exclusion Chromatography (SEC) | Hydrodynamic radius | Removes aggregates and oligomers; buffer exchange | Choose appropriate pore size; use low-concentration detergents for membrane proteins |
Rigorous quality control assessment must follow purification to ensure sample suitability for crystallization. Key analytical techniques include SDS-PAGE & Western Blot (confirming purity and molecular weight), Mass Spectrometry (verifying protein identity and modifications), Dynamic Light Scattering (detecting aggregation and measuring monodispersity), Circular Dichroism Spectroscopy (evaluating secondary structure and folding integrity), and Thermal Shift Assay (assessing stability under varying buffer conditions) [58].
The Purity-Morphology Relationship in Crystallization
The relationship between crystal morphology and purity extends beyond protein crystallography to small molecules, offering insights into general principles of crystal optimization. Research on potassium chloride (KCl) crystallization demonstrates how different crystal morphologies exhibit varying impurity levels and purification behaviors [61].
In KCl systems containing octadecylamine hydrochloride (ODA-H) as an impurity, three distinct crystal morphologies emerge under different crystallization conditions: cubic, spherical, and ellipsoidal. Each morphology demonstrates characteristic purity profiles [61]:
- Spherical crystals exhibit superior flowability but the lowest purity due to extensive agglomeration during formation, which entraps impurities.
- Ellipsoidal crystals show better flowability than cubic crystals but inferior to spherical ones; their purity exceeds cubic crystals before washing but falls below after washing.
- Cubic crystals, with poorer flowability, achieve the highest purity following washing treatment because their well-defined faces and reduced agglomeration minimize impurity incorporation [61].
This morphology-purity relationship underscores how crystallization conditions can be manipulated to favor crystal habits that inherently exclude or facilitate impurity removal, a principle applicable to protein crystallization.
Achieving Monodisperse Protein Samples
The Critical Importance of Monodispersity
Monodispersity—the state of a protein existing as a uniform population of well-folded, non-aggregated molecules—represents a non-negotiable prerequisite for successful crystallization. Aggregated or heterogeneous protein samples introduce disorder that prevents the formation of a regular crystal lattice, leading to poor diffraction quality or complete crystallization failure [58]. The empirical criteria for crystallizable membrane proteins illustrate this requirement: >98% pure, >95% homogeneous, and >95% stable when stored unconcentrated at 4°C for two weeks [60].
Controlling Agglomeration in Crystallization Systems
Agglomeration, the process where primary crystals adhere to form larger clusters, represents a major obstacle to achieving monodispersity. The fundamental mechanism driving agglomeration can be quantified through the Lifshitz-van der Waals acid-base theory, which describes the adhesive force (Fadh) between particles as Fadh = πd²/2 × ΔGSLS, where d is the particle diameter and ΔGSLS is the adhesion free energy [61].
In practical crystallization systems, controlling agglomeration requires manipulating experimental parameters to disrupt these adhesive forces. For ammonium paratungstate pentahydrate, studies have demonstrated that understanding the agglomeration mechanism enables controllable preparation of pure monodisperse crystals [62]. Similar principles apply to protein systems, where optimization of crystallization conditions can prevent undesirable particle interactions.
Several parameters influence agglomeration behavior during crystallization:
- Cooling Rate: affects supersaturation levels and crystal growth kinetics
- Stirring Rate: influences shear forces that can break apart agglomerates
- Additive Concentration: specific additives can modify crystal surface properties
- Supersaturation Control: precise control prevents rapid, disordered crystal growth [61]
The shear force (Fimp) provided by stirring can be calculated as Fimp = ρsπd³NaπDl²/12la, where ρs is the crystal density, Na is the stirring rate, Dl is the diameter of the stirring paddle, and la is the acceleration distance (approximately equal to particle diameter d) [61]. By balancing adhesive forces with disruptive shear forces, researchers can achieve the optimal equilibrium for monodisperse crystal growth.
Novel Approaches to Monodisperse Crystal Production
Emerging technologies offer innovative pathways to monodisperse crystal production. Researchers at Michigan State University have developed a method to "draw" crystals using laser pulses focused on gold nanoparticles, enabling unprecedented control over crystallization timing and location [63]. This approach allows scientists to grow crystals at precise locations and times, essentially providing "a front-row seat to watch the very first moments of a crystal's life under a microscope" while steering its development [63].
For high-throughput crystallography applications, serial crystallography methods have driven advances in monodisperse microcrystal production. These approaches require vast numbers of uniform microcrystals, placing a premium on monodispersity [15].
Specialized Strategies for Membrane Proteins
Unique Challenges in Membrane Protein Crystallization
Membrane proteins represent particularly challenging targets for crystallography due to their inherent structural characteristics. Analysis of the protein fold space reveals that 1,075 membrane proteins exhibit unique topologies not found in the soluble proteome, with only 189 folds present in both soluble and membrane environments [59]. This structural segregation underscores the specialized approach required for their crystallization.
The amphipathic nature of membrane proteins necessitates extraction from lipid membranes using mild detergents and purification to a stable, homogeneous population before crystallization attempts can begin [60]. The hydrophobic surfaces normally embedded in the lipid bilayer must be stabilized during purification, often requiring detergents or lipid systems that maintain native structure while allowing protein-protein contacts necessary for crystal lattice formation.
Expression and Solubilization Strategies
Successful membrane protein crystallography begins with appropriate expression system selection. While Escherichia coli remains popular for its simplicity and low cost, alternative systems including Pichia pastoris yeast, Saccharomyces cerevisiae yeast, and Sf9 insect cells may be necessary for proteins requiring eukaryotic folding machinery or post-translational modifications [60].
Detergent screening represents a critical step in membrane protein preparation. A systematic protocol involves:
- Aliquotting resuspended membrane fractions into multiple tubes
- Adding different detergents (e.g., OG, DDM, LDAO, CHAPS, FC-12)
- Gentle mixing by pipetting (avoiding vortexing to prevent foam)
- Agitation for 12-18 hours at 4°C
- Centrifugation to pellet unsolubilized material
- Analysis of supernatant by SDS-PAGE and Western blot to identify optimal detergents [60]
Table 2: Key Reagents for Membrane Protein Crystallography
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Detergents | DDM, OG, LDAO, CHAPS, FC-12 | Solubilize and stabilize membrane proteins while maintaining structure |
| Affinity Tags | His-tag, GST-tag, MBP-tag | Facilitate purification; enhance solubility |
| Protease Cleavage Sites | TEV, thrombin, Factor Xa | Remove affinity tags after purification |
| Lipid Systems | Bicelles, lipidic cubic phases | Mimic native membrane environment for crystallization |
Computational Design of Soluble Membrane Protein Analogues
Recent breakthroughs in computational protein design have opened new avenues for membrane protein structural studies. Researchers have developed a deep learning pipeline that designs soluble analogues of integral membrane proteins, effectively recapitulating complex membrane topologies such as GPCRs in solution [59].
This approach utilizes an AF2-based design method (AF2seq) combined with ProteinMPNN sequence optimization to generate stable, soluble proteins that adopt membrane protein folds naturally found only in lipid environments [59]. The pipeline involves:
- Using AF2seq to generate sequences adopting a desired target membrane protein fold
- Applying ProteinMPNN sequence optimization to AF2seq-generated starting topologies
- Repredicting structures of all resulting sequences with AlphaFold2
- Filtering based on structural similarity to target topology, confidence scores, and sequence novelty [59]
This computational approach has successfully designed soluble analogues of previously challenging membrane protein folds, including claudin, rhomboid protease, and GPCRs, without need for experimental optimization [59]. The method demonstrates remarkable accuracy, with experimental structures showing high design precision and thermal stability, effectively expanding the functional soluble fold space and potentially enabling new approaches in drug discovery [59].
Emerging Techniques and Future Directions
Serial Crystallography and Sample Consumption
Serial crystallography (SX), conducted at synchrotrons and X-ray free-electron lasers (XFELs), has revolutionized structural biology by enabling studies of biomolecular reaction mechanisms [15]. However, these techniques present significant sample consumption challenges, as crystals must be continuously replenished in the X-ray beam path to acquire complete datasets—typically requiring around ten thousand diffraction patterns to resolve an electron density map [15].
The theoretical minimum sample consumption for SX can be calculated based on specific parameters. Assuming 10,000 indexed patterns are sufficient for a full dataset, with microcrystal dimensions of 4×4×4 μm and a protein concentration in the crystal of ~700 mg/mL (for a 31 kDa protein), approximately 450 ng of protein would be ideally required for a complete SX experiment [15]. This calculation establishes a benchmark for evaluating the efficiency of sample delivery methods.
Advanced Sample Delivery Methods
To address sample consumption challenges, three primary sample delivery systems have emerged:
- Fixed-Target Systems: Microcrystals are deposited on a solid support and scanned through the X-ray beam, minimizing sample waste
- Liquid Injection Systems: Crystal slurries are continuously injected as a liquid stream in the path of the X-ray beam
- Hybrid Methods: Combine elements of both fixed-target and liquid injection approaches [15]
Recent advances have dramatically reduced sample requirements from gram quantities in early SX experiments to microgram amounts in current studies, significantly expanding the range of biologically relevant targets accessible to structural studies [15].
Workflow Integration for Optimal Crystal Growth
The complex interplay between various optimization strategies necessitates an integrated workflow. The following diagram illustrates the comprehensive pathway from protein engineering to final crystal optimization, highlighting critical decision points and quality control checkpoints:
This integrated approach emphasizes the iterative nature of crystal optimization, where feedback from each stage informs adjustments to previous steps, gradually converging on conditions that yield diffraction-quality crystals.
The pursuit of optimized crystal growth through enhanced purity, monodispersity, and specialized membrane protein strategies remains fundamental to advancing structural biology and drug discovery. As techniques continue to evolve—from computational design of soluble membrane protein analogues to increasingly efficient serial crystallography methods—the field moves closer to overcoming traditional barriers in structure determination. By systematically applying the principles and protocols outlined in this guide, researchers can significantly improve their success rates in generating high-quality crystals, particularly for challenging targets that have historically resisted structural characterization. The continued integration of innovative computational and experimental approaches promises to further expand the structural landscape, opening new frontiers in our understanding of protein function and therapeutic intervention.
The determination of three-dimensional protein structures is fundamental to advancing our comprehension of biological processes, enabling rational drug design, and elucidating disease mechanisms. X-ray crystallography remains a predominant technique for this purpose, with over 200,000 macromolecular structures deposited in the Protein Data Bank (PDB) [64]. However, a central challenge in this field is the "phase problem," a fundamental hurdle that arises because a diffraction experiment records the intensities (amplitudes) of X-rays scattered by a crystal but loses the phase information, which is essential for reconstructing the electron density map [65] [66]. Solving this problem is a prerequisite for building an accurate atomic model of the protein.
This whitepaper provides an in-depth examination of both established and cutting-edge methods for conquering the phase problem. It covers traditional experimental phasing techniques, explores the transformative impact of artificial intelligence (AI) in protein structure prediction and phasing, and details the latest AI-driven methodologies that are pushing the boundaries of crystallography. Aimed at researchers, scientists, and drug development professionals, this guide also presents quantitative comparisons of these methods, detailed experimental protocols, and essential resource toolkits to facilitate advanced structural biology research.
The Core Challenge: Understanding the Phase Problem
In a single-crystal X-ray diffraction experiment, a crystal is exposed to an X-ray beam, producing a diffraction pattern of regularly spaced spots known as reflections [16] [66]. The position of each spot provides information about the crystal lattice and symmetry, while its intensity is proportional to the square of the structure factor amplitude, |F(hkl)| [66]. To compute an electron density map and determine the positions of atoms within the crystal, both the amplitude and the phase of the structure factor, F(hkl), are required. The relationship is defined by the inverse Fourier transform:
ρ(x, y, z) = 1/V ∑∑∑ |F(hkl)| e^(-iα(hkl)) e^(-2πi(hx+ky+lz))
Where:
ρ(x, y, z)is the electron density at point (x, y, z).Vis the volume of the unit cell.|F(hkl)|is the amplitude of the structure factor.α(hkl)is the phase angle for reflection (hkl).
The core of the phase problem is that the phase angle α(hkl) cannot be directly measured in a standard diffraction experiment, creating a fundamental gap between the collected data and the desired atomic structure [65] [66]. The following diagram illustrates the pivotal position of the phase problem within the overall X-ray crystallography workflow.
Traditional Phasing Methodologies
Traditional methods for solving the phase problem can be broadly categorized into experimental phasing and molecular replacement. These techniques have been the backbone of structural biology for decades.
Experimental Phasing
Experimental phasing relies on introducing heavy atoms into the protein crystal and collecting additional diffraction data to extract phase information.
- Single/Multiple Isomorphous Replacement (SIR/MIR): This method involves introducing heavy atoms (e.g., mercury, platinum) into the crystal lattice without disturbing its structure (isomorphous replacement). The differences in diffraction intensities between the native and derivative crystals are used to deduce initial phase estimates [65].
- Single/Multiple Anomalous Dispersion (SAD/MAD): This is now the most commonly used experimental phasing technique [65]. It exploits the anomalous scattering that occurs when X-rays interact with specific atoms (such as selenium in selenomethionine, or heavy atoms like mercury) at a wavelength near their absorption edge. The anomalous signal, which is present in a single dataset, provides both amplitude and phase information, allowing the phase problem to be solved [65].
Table 1: Comparison of Traditional Experimental Phasing Methods
| Method | Key Principle | Requirements | Advantages | Limitations |
|---|---|---|---|---|
| SIR/MIR | Intensity differences from heavy-atom derivatives | Multiple isomorphous crystals with heavy atoms | Well-established, can work with larger proteins | Requires isomorphism; challenging to prepare multiple derivatives |
| SAD/MAD | Anomalous scattering from specific atoms | Tunable X-ray source (e.g., synchrotron); incorporation of anomalous scatters | Requires only a single crystal; highly effective | Needs a strong anomalous signal; radiation damage can be an issue |
Molecular Replacement
Molecular Replacement (MR) is an alternative approach used when a structurally similar protein is already known. The known structure is used as a search model to determine the initial phases for the unknown structure [64]. This method avoids the need for heavy-atom derivatization but is entirely dependent on the availability and similarity of a known model. The rise of AI-predicted structures has dramatically expanded the scope and success of molecular replacement, as discussed in Section 4 [64].
The AI Revolution in Protein Structure Prediction and Phasing
Artificial intelligence has revolutionized structural biology by providing highly accurate protein structure predictions, which in turn offer powerful new solutions to the phase problem.
AI-Generated Models as Molecular Replacement Search Models
Tools like AlphaFold2 and RoseTTAFold can predict protein structures directly from their amino acid sequences with an estimated precision often better than 1 Å for backbone and sidechain positions [64]. These AI-generated models are now widely used as search models in molecular replacement, enabling the determination of structures for proteins that lack close homologs of known structure [64]. Furthermore, AI models can be used to correct and improve existing, imperfect experimental models by fitting them into ambiguous electron density maps, thereby enhancing the precision of structures already in the PDB [64].
AI for Direct Phasing
Recent research has focused on using AI to directly attack the phase problem, moving beyond the need for a search model.
- The PhAI Network: A pioneering deep learning architecture was designed to solve crystal structures ab initio using only experimental amplitude data [67]. Initially, this network was successful for small structures (unit-cell volumes up to 1000 ų) in centrosymmetric space groups [67].
- AI-Based Phase-Seeding (AI-PhaSeed): This method combines the PhAI network with a phase-seeding algorithm to extend the applicability of AI phasing to larger structures (unit-cell volumes up to 3500 ų) [67]. The workflow, detailed in the diagram below, involves using the AI to generate reliable seed phases for a small subset of reflections, which are then extended and refined to produce a complete electron density map [67].
End-to-End AI Structure Determination
The latest innovation seeks to bypass the interpretation of electron density maps entirely.
- The XDXD Framework: This is an end-to-end deep learning model that predicts a complete atomic crystal structure directly from low-resolution (e.g., 2.0 Å) single-crystal X-ray diffraction data [19]. Using a diffusion-based generative model conditioned on the experimental diffraction amplitudes, XDXD generates chemically plausible crystal structures without the intermediate step of producing and interpreting an electron density map [19]. This approach has demonstrated a 70.4% match rate for structures with data limited to 2.0 Å resolution, successfully handling unit cells with up to 200 non-hydrogen atoms [19].
Table 2: Performance of the XDXD Model on Experimental Data from the COD [19]
| Number of Non-Hydrogen Atoms in Unit Cell | Match Rate (%) | Typical RMSE |
|---|---|---|
| 0 - 40 | High (Baseline) | < 0.05 |
| 40 - 80 | ~80% | ~0.05 |
| 80 - 120 | ~70% | ~0.06 |
| 120 - 160 | ~55% | ~0.07 |
| 160 - 200 | ~40% | > 0.07 |
Advanced Applications and Future Directions
AI-driven phasing methods are enabling new scientific frontiers, particularly in the study of dynamic biological processes.
Time-Resolved Serial Crystallography
Serial crystallography (SX), conducted at X-ray free-electron lasers (XFELs) and synchrotrons, uses microcrystals to study protein structures and dynamics [15]. Time-resolved serial femtosecond crystallography (TR-SFX) allows for the capture of molecular movies, visualizing reaction intermediates at atomic resolution on timescales from femtoseconds to milliseconds [15]. This is achieved by initiating reactions in protein crystals with light (for light-sensitive proteins) or via rapid mixing (Mix-and-Inject Serial Crystallography, MISC) [15]. A significant challenge in these experiments is the massive sample consumption, which has driven the development of low-volume sample delivery systems such as fixed-target chips and high-viscosity extruders [15].
Limitations and Complementary Strategies
Despite their power, AI-based predictions have limitations. They may not fully capture protein dynamics, flexible regions, or conformations influenced by specific environmental conditions [68]. Therefore, experimental structure determination techniques like X-ray crystallography remain essential for validating AI predictions, discovering novel protein folds, and studying proteins in complex with drugs, nucleic acids, or other partners. The future lies in a synergistic approach, where AI models provide initial phases or structures that are then rigorously refined and validated against experimental diffraction data.
The Scientist's Toolkit: Essential Reagents and Materials
Successful protein structure determination, especially with advanced phasing methods, relies on a suite of specialized reagents and materials.
Table 3: Key Research Reagent Solutions for Protein Crystallography
| Item | Function/Description |
|---|---|
| Crystal Screen Kits | Sparse matrix solutions (e.g., 50+ conditions) varying in precipitant, buffer, pH, and salt to identify initial crystallization conditions via trial and error [16]. |
| Selenomethionine | An amino acid used in SAD phasing; incorporated into recombinant proteins to provide a strong anomalous scattering signal from selenium atoms [65]. |
| Heavy-Atom Derivatives | Compounds containing atoms like Hg, Pt, or Au used for experimental phasing via MIR or SAD to solve the phase problem [65]. |
| Liquid Injection Systems | Devices (e.g., GDVN, high-viscosity extruders) that deliver a stream of microcrystals suspended in mother liquor for serial crystallography at XFELs and synchrotrons [15]. |
| Fixed-Target Sample Supports | Microfluidic chips (e.g., silicon, polymer) with micro-wells or apertures used to array microcrystals for low-sample-consumption serial crystallography [15]. |
| Cryoprotectants | Chemicals (e.g., glycerol, ethylene glycol) used to prepare crystals for cryo-cooling in a stream of liquid nitrogen, reducing radiation damage during data collection [16]. |
The conquest of the phase problem has entered a new era. Traditional experimental phasing and molecular replacement remain vital tools for the structural biologist. However, the integration of artificial intelligence is fundamentally transforming the field. From providing superior search models for molecular replacement to enabling direct phasing through networks like PhAI and AI-PhaSeed, and even generating complete atomic structures end-to-end with frameworks like XDXD, AI is overcoming long-standing limitations. These advancements are expanding the scope of structural biology to include more challenging targets and enabling the visualization of protein dynamics at unprecedented temporal and spatial resolutions. For researchers in drug development and biotechnology, these tools provide faster, more accurate, and more detailed structural insights, thereby accelerating the pace of discovery and innovation.
Radiation damage is a fundamental and resolution-limiting factor in macromolecular X-ray crystallography and cryo-electron microscopy (cryo-EM) [69]. When biological samples are exposed to ionizing radiation during structural studies, the deposited energy invariably destroys the delicate native structures of biological specimens through both primary and secondary damage mechanisms [69] [70]. This phenomenon has necessitated the development of sophisticated mitigation strategies, with cryo-cooling and serial crystallography emerging as two powerful approaches that have revolutionized the field of structural biology.
The dose, expressed as energy deposited per mass unit (measured in Gray, where 1 Gy = 1 J kg⁻¹), provides a crucial standardized metric for quantifying radiation damage [69] [70]. In protein crystallography, the Henderson limit (also known as D₀.₅) defines the dose at which diffraction power drops by half, originally set at 20 MGy and later revised to 30 MGy [70]. However, local chemical changes, including alterations to oxidation states and bond lengths, begin occurring at much lower doses—as small as 0.2-0.4 MGy for some high-valent metal centers [70]. This review examines how cryo-cooling and serial crystallography techniques address these challenges, enabling researchers to push the boundaries of structural biology while minimizing radiation-induced artifacts.
Fundamental Mechanisms of Radiation Damage
Primary and Secondary Damage Processes
Radiation damage to biological samples occurs through distinct primary and secondary mechanisms. Primary damage results from direct inelastic interactions between ionizing radiation and sample components. In X-ray crystallography, the most dominant primary interaction is photoelectric absorption, where atoms eject photoelectrons upon absorbing incident X-rays [69]. At transmission electron microscopy energies (100-300 keV), inelastic scattering is approximately three times more likely than elastic scattering, with plasmon scattering, K- and L-shell ionization, Bremsstrahlung, and secondary electron emission being the most significant processes [69].
Secondary damage arises from the diffusion and chemical activity of species generated during primary damage events. The emitted photoelectrons cause numerous ionization events (approximately 500 per photoelectron at 12 keV), creating electron-loss and electron-gain centers that damage protein structures directly or indirectly through diffusion in the vitrified buffer [69]. In aqueous environments, X-ray irradiation of water generates hydrogen (H) and hydroxyl (OH) radicals, electrons (e⁻), and hydrated electrons (e⁻ₐq) [69]. These diffusible radicals can break chemical bonds, reduce redox-active metal centers, and cause irreversible local structural changes that ultimately compromise diffraction quality [70].
Table 1: Radiation Damage Mechanisms in Structural Biology
| Damage Type | Time Scale | Primary Effects | Consequences |
|---|---|---|---|
| Primary Damage | Femtoseconds | Photoelectric absorption, electron ejection | Core-hole creation, initial ionization |
| Secondary Damage | Femtoseconds to milliseconds | Radical diffusion, chemical reactions | Bond rupture, metal center reduction, structural rearrangements |
| Global Damage | Seconds to minutes | Loss of long-range order | Reduced diffraction intensity and resolution |
Quantitative Dose Limits in Structural Biology
The radiation sensitivity of biological samples varies significantly depending on composition, temperature, and the specific metric being measured. The well-established Henderson limit of 30 MGy for global damage to protein crystals was determined based on the decay of diffraction intensity [70]. However, more sensitive spectroscopic techniques have revealed that local chemical damage occurs at substantially lower doses. For example, high-valent metal centers in metalloproteins can undergo reduction at doses between 0.1 and 10 MGy, with lower D₀.₅ values observed at room temperature compared to cryogenic conditions [70].
In cryo-electron microscopy, studies have documented linear increases in image brightness with dose (approximately 0.1% per 10 MGy) prior to gas bubble formation, with complete sample sublimation occurring at extreme doses up to 5500 MGy [69]. These quantitative relationships between dose and damage provide essential guidelines for designing experimental protocols that maximize information recovery while minimizing structural degradation.
Cryo-Cooling: Principles and Implementation
Theoretical Foundation of Cryo-Cooling
Cryo-cooling mitigates radiation damage primarily by reducing the diffusion rates of radical species generated during irradiation. At cryogenic temperatures (typically around 100 K), the mobility of destructive radicals is significantly restricted, thereby slowing secondary damage processes [69] [70]. The theoretical basis for this approach stems from the temperature dependence of radical diffusion in amorphous ice—protons become mobile at approximately 115 K, while OH radicals gain mobility above 130 K [69]. Positive holes rapidly trap at 77 K, forming amido radicals on protein backbone chains, whereas electrons remain sufficiently mobile to encounter and damage disulfide bonds [69].
The effectiveness of cryo-cooling depends on achieving rapid vitrification that prevents crystalline ice formation, which can damage protein crystals and create scattering artifacts. Proper cryo-cooling produces vitreous ice—an amorphous state that preserves the native hydration shell around macromolecules while immobilizing potentially damaging radical species [69].
Practical Implementation and Protocols
Standard cryo-crystallography protocols involve several critical steps to ensure optimal sample preservation:
Cryoprotectant Selection and Optimization: Samples must be transferred to solutions containing cryoprotectants (e.g., glycerol, ethylene glycol, or various salts) at appropriate concentrations (typically 15-25%) to prevent ice formation during cooling [71].
Snap-Cooling Process: Samples are rapidly plunged into cryogenic liquids (typically liquid nitrogen at 77 K or liquid propane at higher cooling rates) to achieve vitreous ice formation [71]. The cooling rate must exceed the critical velocity for ice crystallization (approximately 10⁴ K/s) to ensure proper vitrification.
Storage and Transfer: Cryo-cooled samples are maintained under continuous cryogenic conditions during storage, transport, and data collection to prevent devitrification and temperature cycling effects [71].
Recent advancements have introduced sophisticated variations such as the Cryo2RT method, which enables high-throughput room-temperature data collection from cryo-cooled crystals by thawing them on the goniometer immediately before X-ray exposure [71]. This approach leverages the practical advantages of cryo-shipping while enabling room-temperature structural studies that may better represent physiological conformations.
Table 2: Temperature-Dependent Radiation Damage Effects
| Temperature | Radical Mobility | Radiation Dose Limit (D₀.₅) | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Room Temperature (296 K) | High mobility for all radical species | Lower doses (e.g., 0.1-1 MGy for metal centers) | Physiological relevance, no cryo-artifacts | Rapid damage progression, limited data quality |
| Standard Cryo (100 K) | Restricted mobility for most radicals | ~30 MGy for global damage | Significant damage reduction, practical handling | Potential conformational trapping |
| Ultra-Low Cryo (30 K) | Greatly restricted radical diffusion | Extended dose limits | Further reduction in secondary damage | Technical complexity, limited availability |
Serial Crystallography: Emerging Paradigms
The "Diffraction Before Destruction" Principle
Serial crystallography (SX) represents a paradigm shift in radiation damage mitigation through the "diffraction before destruction" principle [56] [72]. This approach leverages the extraordinary brightness and ultrashort pulse duration of X-ray free-electron lasers (XFELs) to collect diffraction patterns from micrometre-sized crystals in a "one crystal, one shot" mode [56] [72]. Each crystal is exposed to a single femtosecond X-ray pulse (typically 10-50 fs) that provides sufficient photons for measurable diffraction while depositing devastating radiation damage—but the damage manifestations occur after the diffraction event due to the temporal separation between electronic and nuclear dynamics [72].
The theoretical foundation for this approach was established through simulations suggesting that usable diffraction could be captured before nuclear motion leads to structural disintegration [72]. This was subsequently demonstrated experimentally at XFEL facilities worldwide, enabling high-resolution structure determination from crystals that would be unusable at synchrotron sources due to radiation sensitivity [56] [72].
Serial Methods at Different Source Types
Serial crystallography methodologies have been adapted for various X-ray sources with distinct technical implementations:
Serial Femtosecond Crystallography (SFX) at XFELs: Utilizing the extreme peak brilliance of XFEL sources (approximately ten orders of magnitude higher than third-generation synchrotrons), SFX enables damage-free data collection from micrometre- and sub-micrometre-sized crystals at room temperature [56] [72]. The short pulse duration (femtoseconds) essentially decouples the diffraction measurement from radiation damage processes.
Serial Millisecond Crystallography (SMX) at Synchrotrons: With the development of higher-flux synchrotron beamlines, serial methods have been adapted to synchrotron radiation sources [56] [15]. Although the longer exposure times (milliseconds) do not completely avoid radiation damage, SMX enables data collection with significantly reduced damage by spreading the total dose across many crystals and outrunning most secondary radiation damage processes [56].
Time-Resolved Serial Crystallography (TR-SX): Serial approaches naturally enable time-resolved studies of biomolecular dynamics through pump-probe methodologies, where a reaction is initiated by light or rapid mixing followed by delayed X-ray probing [56] [15]. This has enabled the creation of "molecular movies" that capture functional structural transitions at atomic resolution [56].
Experimental Protocols and Workflows
Cryo-Cooling Experimental Protocol
The following protocol outlines the standard methodology for cryo-cooling protein crystals, based on established procedures with recent enhancements from the Cryo2RT approach [71]:
Sample Preparation:
- Grow crystals using standard vapor diffusion or batch methods.
- Prepare cryoprotectant solution by adding 20-25% glycerol, ethylene glycol, or similar cryoprotectant to the mother liquor.
- Transfer crystals through cryoprotectant solutions in stepwise or direct manner, depending on crystal sensitivity.
Mounting and Cooling:
- Mount individual crystals using MicroLoops or specialized chips (e.g., IMISX plates).
- Wrap crystals in a protective oil layer (Paratone-N or paraffin oil) to prevent dehydration.
- Plunge mounted crystals directly into liquid nitrogen for snap-cooling.
- Store and transport cooled crystals in specialized containers (UniPucks) under continuous liquid nitrogen conditions.
Data Collection Strategies:
- For conventional cryo-crystallography: Maintain samples at 100 K using cryostream during entire data collection.
- For Cryo2RT approach: Shield and retract cryostream to thaw crystals on goniometer, collect room temperature data, then recool for additional cycles if needed [71].
Serial Crystallography Experimental Protocol
Serial crystallography requires specialized instrumentation and methodologies that differ significantly from conventional approaches:
Sample Delivery Systems:
- Liquid Injectors: Gas Dynamic Virtual Nozzle (GDVN) injectors create focused liquid streams containing crystal suspensions (flow rates: 10-40 μL/min) [72]. Recent modifications have reduced sample consumption up to eightfold [72].
- High-Viscosity Extruders: Specifically designed for lipidic cubic phase (LCP) samples, these injectors enable data collection from membrane protein crystals with controlled flow rates of 0.001-0.3 μL/min [56] [72].
- Fixed-Target Systems: Crystals are deposited on solid supports (silicon chips, nylon loops, or microfluidic devices) that are raster-scanned through the beam, significantly reducing sample consumption [56] [15].
Data Collection Workflow:
- Deliver crystal suspension or fixed target to interaction point with X-ray beam.
- Synchronize X-ray pulses with sample delivery for optimal hit rates.
- Collect diffraction patterns using specialized detectors (e.g., CSPAD, ePix10k, JUNGFRAU, AGIPD) capable of handling high repetition rates and large dynamic ranges [72].
- Monitor hit rates (images containing discernible diffraction patterns) to ensure sufficient data quality.
Data Processing:
- Identify diffraction patterns with indexable Bragg peaks from the vast datasets (often millions of images).
- Index, integrate, and merge partial reflections from multiple crystals using specialized software (e.g., CrystFEL).
- Reconstruct complete diffraction data through Monte Carlo integration or similar approaches.
Workflow Comparison: Cryo-Cooling vs. Serial Crystallography
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Radiation Damage Mitigation
| Item Name | Function/Purpose | Technical Specifications | Application Context |
|---|---|---|---|
| Cryoprotectants | Prevent ice formation during cooling | Glycerol (15-25%), ethylene glycol, low-molecular weight PEG | Cryo-cooling protocols |
| High-Viscosity Carriers | Matrix for crystal embedding and delivery | Lipid cubic phase (LCP), hydroxyethyl cellulose, agarose | Serial crystallography with viscous injectors |
| Microfabricated Chips | Fixed-target sample supports | Silicon nitride membranes, polymer-based devices | Fixed-target serial crystallography |
| Gas Dynamic Virtual Nozzle (GDVN) | Liquid jet crystal delivery | Flow rates: 10-40 μL/min, focused stream diameter: few μm | SFX at XFEL facilities |
| High-Viscosity Extruder | Delivery of viscous samples | Flow rates: 0.001-0.3 μL/min, precise extrusion control | LCP-SFX for membrane proteins |
| Specialized Detectors | High-speed X-ray detection | CSPAD, JUNGFRAU, AGIPD; high dynamic range, fast readout | Data collection at XFELs and synchrotrons |
Comparative Analysis and Future Directions
Quantitative Comparison of Damage Mitigation Efficacy
Table 4: Radiation Damage Mitigation Technique Comparison
| Technique | Typical Dose per Dataset | Sample Consumption | Resolution Limit | Temperature Regime |
|---|---|---|---|---|
| Traditional RT Crystallography | Limited by global damage | Single crystal | Limited by radiation damage | Room temperature |
| Cryo-Crystallography | Up to 30 MGy per crystal | Single crystal | Near atomic (<1.5 Å) | 100 K (cryogenic) |
| Serial Femtosecond Crystallography | ~0 MGy (per diffraction pattern) | 0.1-10 mg protein | Atomic (1.5-3.0 Å) | Room temperature preferred |
| Serial Synchrotron Crystallography | Distributed across many crystals | 0.01-1 mg protein | Atomic (1.5-3.0 Å) | Room temperature or cryogenic |
Integrated Approaches and Future Developments
The future of radiation damage mitigation lies in integrated approaches that combine the strengths of multiple techniques. The Cryo2RT method exemplifies this trend by bridging cryo-cooling practicalities with room-temperature structural insights [71]. Similarly, hybrid methods that employ serial approaches with cryo-cooled samples are expanding the experimental parameter space available to structural biologists.
Emerging directions include:
- Ultra-low temperature crystallography (10-20 K) to further suppress radical diffusion [70]
- Advanced sample delivery systems that minimize consumption while maximizing hit rates [15]
- Computational damage correction algorithms that account for radiation-induced structural perturbations
- Multi-modal integration with spectroscopic techniques to monitor and correct for radiation-induced changes during data collection
These developments continue to push the boundaries of structural biology, enabling researchers to extract increasingly precise structural information from radiation-sensitive biological systems while minimizing the confounding effects of radiation damage.
Radiation damage remains an inherent challenge in structural biology, but the development and refinement of cryo-cooling and serial crystallography techniques have fundamentally transformed our ability to overcome this limitation. Cryo-cooling provides a practical and widely accessible method for significantly extending sample lifetime through temperature-controlled suppression of radical diffusion. Serial crystallography, particularly at XFEL facilities, represents a paradigm shift that essentially eliminates radiation damage through the "diffraction before destruction" principle. Together, these approaches have enabled unprecedented insights into the structure and dynamics of biological macromolecules, pushing the boundaries of resolution, reducing sample requirements, and opening new possibilities for time-resolved studies. As these technologies continue to evolve and converge, they will undoubtedly unlock new frontiers in our understanding of biological structure and function.
Protein X-ray crystallography remains the gold standard for determining high-resolution three-dimensional structures of biological macromolecules, providing indispensable insights into function and guiding drug discovery [73] [74]. However, obtaining high-quality crystals suitable for diffraction analysis consistently presents a major bottleneck. The challenging "crystallization step" often stalls projects for months or even years, particularly for membrane proteins, flexible complexes, and proteins with dynamic surfaces [75] [76]. This technical guide details three innovative approaches—lipid cubic phase (LCP) crystallization, microseeding, and surface entropy reduction (SER)—developed to overcome these hurdles. When integrated strategically into the protein structure determination pipeline, these methods significantly increase the success rate of obtaining well-diffracting crystals from challenging targets [77] [78] [76].
Lipid Cubic Phase (LCP) Crystallization for Membrane Proteins
Principle and Rationale
The lipid cubic phase (LCP) is a highly ordered, three-dimensional lipid matrix that mimics the native membrane environment. It is formed by specific lipids, such as monoolein (MO), upon mixing with water in a specific ratio, typically around 60:40 to 70:30 (v/v) MO:water [77] [79]. This structure consists of a single continuous lipid bilayer that curves through space, forming two interpenetrating but non-contacting water channels. For integral membrane proteins, this in meso (within the mesh) method provides a stabilizing, membrane-like milieu that supports proper folding and function, which is often lost in detergent-based crystallization [79] [80]. The LCP system has been successfully used to solve the structures of numerous G protein-coupled receptors (GPCRs) and other membrane proteins [77].
Detailed Experimental Protocol
Materials:
- Purified membrane protein in a suitable detergent.
- Monoolein (or other suitable monoacylglycerol).
- Coupled syringe mixing device or a mechanical mixer.
- Precipitant screening kits.
- Glass sandwich plates or syringes for crystal observation.
- Humidified environment chamber (>80% humidity) to prevent LCP dehydration [79].
Procedure:
- LCP Preparation: Pre-load a syringe with liquid monoolein. Using a coupled syringe system, mix the monoolein with your purified protein solution in an approximate 60:40 (v/v) ratio (lipid:protein solution). Vigorously mix by passing the material back and forth between the syringes hundreds of times until a transparent, viscous LCP is formed. The quality of the LCP can be checked by its optical birefringence when viewed between crossed polarizers [79] [80].
- Crystallization Setup: Dispense 20-50 nL boluses of the protein-laden LCP onto a glass plate or into wells. Overlay each bolus with 0.8-1.0 µL of precipitant solution using standard vapor-diffusion or micro-batch techniques [77].
- Incubation and Monitoring: Seal the plates and incubate at a constant temperature. Monitor for crystal growth using a microscope. Crystals grown in LCP often appear as bright, birefringent needles or plates.
- Harvesting and Data Collection: Due to the high viscosity of LCP, crystals are often harvested directly using a special MicroMesh or loops designed for viscous samples. For serial crystallography at XFELs or synchrotrons, the entire LCP sample, with embedded microcrystals, can be extruded directly using a high-viscosity extruder (HVE) injector [79] [15].
Table 1: Viscosity Profile of Monoolein-based LCP and Modifications
| LCP Composition (MO:Water, v/v) | Additive | Zero-Shear Viscosity (η₀) | Phase State | Suitability for HVE |
|---|---|---|---|---|
| 70:30 | None | 6.2 ± 0.13 kPa·s | Cubic-Pn3m | Excellent |
| 60:40 | 6% DDM | ~1 order of magnitude lower | Cubic-Pn3m | Good |
| 50:50 | 10% DDM | ~1 order of magnitude lower | Cubic-Pn3m | Metastable |
| 40:60 | 14% DDM | ~1 order of magnitude lower | Cubic-Pn3m | Unstable |
Workflow Visualization
Microseeding Strategies for Crystal Optimization
Principle and Rationale
Microseeding is a technique that uses very small, often microscopic, crystal fragments ("seeds") to initiate and promote growth in new crystallization experiments. This approach bypasses the stochastic nucleation phase, which is a major source of failure and irreproducibility. By providing pre-formed nucleation sites, microseeding can induce crystallization in conditions that would otherwise not form crystals and dramatically improve crystal size, uniformity, and diffraction quality [78]. A specialized variant, hetero-micro-seeding, uses crystals from a closely related protein variant (e.g., a point mutant) to nucleate growth of a target protein that is otherwise recalcitrant to crystallization [78].
Detailed Experimental Protocol
Materials:
- Source of seeds (e.g., crystals from a related protein, crushed crystals, or microcrystals from a previous experiment).
- Seed bead homogenizer (optional).
- Crystallization solutions for the target protein.
Procedure:
- Seed Stock Preparation: Harvest a few source crystals (even poorly diffracting ones are suitable) into a microtube containing 20-50 µL of a stabilizing solution (e.g., mother liquor or a compatible reservoir solution). Gently crush the crystals using a seed bead or by vigorous pipetting to create a heterogeneous mixture of microcrystals and crystal fragments.
- Seed Serial Dilution: Prepare a serial dilution of the seed stock (e.g., 1:10, 1:100, 1:1000, 1:10,000) in the stabilizing solution. The optimal dilution is empirical and must be determined experimentally.
- Setting Up Seeded Trials: Prepare crystallization trials for the target protein using conditions that are close to, but below, the nucleation threshold. Introduce a small volume (e.g., 0.1-0.2 µL) of the diluted seed stock into the protein-drop before setting up the crystallization experiment (e.g., sitting drop or hanging drop).
- Incubation and Evaluation: Incubate the trays and monitor for crystal growth. The ideal dilution will yield a small number of single, well-formed crystals per drop. A high seed concentration may lead to showers of small crystals, while a very low concentration may yield no growth.
The hetero-micro-seeding strategy was successfully used to determine the structures of Bovine Pancreatic Trypsin Inhibitor (BPTI) variants. Micro-crystal seeds from BPTI variants with Gly, Ile, or Leu at position 38 were used to nucleate the crystallization of a target BPTI variant, leading to successful structure determination where conventional methods failed [78].
Surface Entropy Reduction (SER)
Principle and Rationale
Surface Entropy Reduction (SER) is a rational mutagenesis approach that aims to reduce the conformational flexibility of surface residues to promote crystal contact formation. Protein surfaces are often rich in flexible, high-entropy residues like lysine and glutamate, which can hinder the formation of ordered crystal lattices. SER involves mutating these residues to smaller, less flexible amino acids like alanine or serine. This strategy reduces the entropic penalty of immobilizing these residues during crystal contact formation, thereby increasing the probability of obtaining well-diffracting crystals [75] [76].
Detailed Experimental Protocol
Materials:
- Protein sequence and structural model (if available).
- Site-directed mutagenesis kit.
- SER prediction server (e.g., SERp: http://services.mbi.ucla.edu/SER/).
Procedure:
- In Silico Prediction: Submit your protein's amino acid sequence to the SER prediction server. The server identifies surface-exposed clusters of high-entropy residues (Lys, Glu, Gln) that are not part of conserved regions or predicted secondary structure elements [76].
- Mutant Design: Based on the server output, design one or more mutants. Common strategies include:
- Protein Production: Use site-directed mutagenesis to create the SER construct. Express and purify the mutant protein using standard protocols.
- Crystallization Screening: Subject the SER mutant to crystallization screening. It is advisable to test the mutant in parallel with the wild-type protein to directly assess the impact of the mutation.
A related chemical biology approach is Surface Lysine Methylation (SLM), which chemically modifies lysine ε-amines to N,N-dimethyl-lysine (dmLys). This modification reduces surface entropy without the need for mutagenesis and has been shown to increase the frequency of crystal contacts, particularly with glutamate residues, through the formation of new C–H···O interactions [76]. Statistical analysis of the PDB shows that dmLys-Glu contacts occur more frequently than Lys-Glu contacts, explaining the success of this method [76].
Table 2: Impact of Surface Modifications on Intermolecular Contact Rates
| Residue Type | Contact Partner | Relative Contact Rate | Proposed Mechanism |
|---|---|---|---|
| Lysine (Lys) | Glutamate (Glu) | Baseline | Ionic/H-bond |
| Arginine (Arg) | Glutamate (Glu) | Higher than Lys | Bidentate H-bonds |
| dimethyl-Lys (dmLys) | Glutamate (Glu) | Higher than Lys [76] | H-bonds & C–H···O interactions |
| dmLys | Isoleucine (Ile) | Higher than Lys [76] | Increased hydrophobic contact |
Integrated Applications and Advanced Techniques
The true power of these methods is revealed when they are combined or applied within cutting-edge crystallographic workflows.
Integration with MicroED: For samples that yield only microcrystals or nanocrystals (a common outcome with LCP or seeding), Microcrystal Electron Diffraction (MicroED) can be a powerful solution. MicroED allows for high-resolution structure determination from crystals billions of times smaller than those used in conventional X-ray crystallography [81] [77]. A notable example is the determination of a structure from Proteinase K microcrystals embedded in LCP using MicroED [77].
Serial Crystallography at XFELs/Synchrotrons: LCP is the carrier medium of choice for high-viscosity extrusion (HVE) injectors in serial femtosecond (SFX) and serial millisecond crystallography (SMX) at X-ray free-electron lasers (XFELs) and synchrotrons [79] [15]. The precise characterization of LCP's viscoelastic properties is critical for stabilizing the jet and minimizing sample consumption, which has been reduced to theoretically as low as 450 ng of protein for a complete dataset in optimal conditions [15].
Combining SER with Seeding: The development of the Top7sm2-I68R mutant demonstrates how SER can be combined with strategic interface engineering. After initial SER mutations (Top7sm2) failed to produce a refinable model due to persistent poor crystal packing, a subsequent I68R mutation was introduced to disrupt a specific, continuous intermolecular β-sheet. This yielded a new crystal form that diffracted to 1.4 Å resolution with excellent refinement statistics (R/Rfree = 0.20/0.24) [75].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Advanced Crystallization Techniques
| Reagent / Material | Function / Application |
|---|---|
| Monoolein (1-Oleoyl-rac-glycerol) | Primary lipid for forming the Lipid Cubic Phase (LCP) matrix for membrane protein crystallization [77] [79]. |
| Dimethylamine-trifluoroborane (ABC) | Reducing agent used in the reductive methylation protocol for Surface Lysine Methylation (SLM) [76]. |
| Formaldehyde | Methylating agent used in Surface Lysine Methylation (SLM) to convert Lys to dimethyl-Lys [76]. |
| Pluronic F-127 Polymer | Stabilizing additive used to optimize the viscosity and jetting stability of LCP for HVE injection [79]. |
| SER Prediction Server (SERp) | Bioinformatics tool to identify surface residue clusters suitable for mutagenesis in Surface Entropy Reduction [76]. |
| High-Viscosity Extrusion (HVE) Injector | Device for delivering LCP-embedded microcrystals in a stable jet for serial crystallography at XFELs/synchrotrons [15]. |
| Coupled Syringe Mixing Device | Tool for homogenously reconstituting membrane proteins into LCP by mechanical mixing [80]. |
The integration of Lipid Cubic Phase crystallization, Microseeding, and Surface Entropy Reduction into the structural biologist's toolkit has fundamentally expanded the frontier of protein structure determination. These methods address the core challenge of crystallization from complementary angles: LCP provides a biomimetic environment for membrane proteins, microseeding controls and amplifies nucleation, and SER rationally engineers crystal contacts. As these approaches continue to mature and converge with revolutionary techniques like MicroED and serial crystallography, they will undoubtedly play a pivotal role in elucidating the structures of ever more challenging targets, from dynamic enzyme complexes to integral membrane receptors, thereby accelerating the pace of discovery in basic science and rational drug design.
Ensuring Accuracy and Choosing the Right Tool: Validation, Cryo-EM, and NMR
In protein structure determination via X-ray crystallography, the final atomic model is derived from a combination of experimental X-ray diffraction data and knowledge-based modeling [22]. Structure validation is therefore a critical step that assesses the reliability and quality of the determined structure, ensuring it is both experimentally faithful and stereochemically reasonable [82]. This process uses a suite of metrics to identify potential errors in the model, which can arise from misinterpretation of ambiguous electron density data, particularly at lower resolutions [82]. For researchers in structural biology and drug development, a rigorously validated structure is a prerequisite for meaningful biological interpretation, such as understanding enzyme mechanisms or performing structure-based drug design [16]. This guide provides an in-depth technical examination of three cornerstone validation techniques: R-factors, Ramachandran plots, and the all-atom contact analysis implemented in MolProbity.
The R-Factor: Quantifying Model-to-Data Agreement
Definition and Calculation
The R-factor, also known as the residual factor or R-work, is a primary quantitative measure of how well the refined atomic model explains the observed experimental X-ray diffraction data [44]. It is calculated using the formula:
where F_obs is the observed structure factor amplitude and F_calc is the structure factor amplitude calculated from the model [44]. The structure factor is intrinsically related to the intensity of each reflection in the diffraction pattern (I_hkl ∝ |F(hkl)|²) [44]. The R-factor sums the absolute differences between the observed and calculated amplitudes across all measured reflections, normalized by the sum of the observed amplitudes. A value of zero indicates perfect agreement, while higher values indicate greater discrepancy. In practice, even for poor models, values are typically less than one due to the inclusion of a scaling factor [44].
The Free R-Factor and Data Splitting
A significant advancement in crystallographic validation was the introduction of the Free R-factor (R_free) [44]. This metric is calculated in an identical manner to the conventional R-factor, but it uses a subset of reflections (typically 5-10%) that were excluded from the refinement process [44]. This reserved test set serves as an unbiased control to detect overfitting—a scenario where a model becomes overly tailored to the refinement data, including its noise, rather than reflecting the true underlying structure. Consequently, a large discrepancy between R and R_free can indicate overfitting or other model errors [44].
Table 1: Key R-factor Metrics in Crystallographic Validation
| Metric | Calculation | Interpretation | Purpose |
|---|---|---|---|
| R-factor (R-work) | Σ | |F_obs| - |F_calc| | / Σ |F_obs| |
Measures agreement between model and data used in refinement. Lower is better. | Primary indicator of model fit during refinement. |
| Free R-factor (R-free) | Same as R-work, but calculated on a reflection subset excluded from refinement. | Unbiased measure of model quality; guards against overfitting. | Key validation metric; should track R-work closely. |
The Ramachandran Plot: Validating Protein Backbone Geometry
Principles and Methodology
The Ramachandran plot is a fundamental tool for validating the conformational geometry of a protein's backbone [83]. It is a two-dimensional plot that visualizes the distribution of the phi (φ) and psi (ψ) torsion angles for each amino acid residue in the structure (except proline) [84]. These angles describe the rotations around the N-Cα (φ) and Cα-C (ψ) bonds, defining the polypeptide chain's conformation [84]. The plot reveals allowed and disallowed regions based on steric hindrance between atoms in the backbone and side chains [84].
The analysis typically categorizes residues into four regions:
- Favored (Core) regions: The most sterically favorable combinations of φ and ψ angles, commonly associated with secondary structures like alpha-helices and beta-sheets [84].
- Allowed regions: Less favorable but still permitted conformations.
- Generously allowed regions: Conformations that are possible but less likely.
- Outlier (Disallowed) regions: Conformations that are sterically impossible and almost always indicate an error in the model [84].
Advanced Metrics: The Ramachandran Z-Score
While reporting the number or percentage of residues in the "outlier," "allowed," and "favored" regions is standard practice, this can be misleading [85]. A model might have zero outliers and a high percentage of favored residues, yet the overall distribution of (φ, ψ) angles might not match the expected statistical distribution observed in high-quality structures [85].
To address this, the Ramachandran Z-score (Rama-Z) provides a single, global numerical metric [85]. It quantifies how "normal" the entire (φ, ψ) distribution of a model is compared to a reference set of high-resolution, high-quality structures [85]. A Rama-Z score near zero indicates a typical distribution, while strongly negative scores suggest an improbable backbone conformation, even in the absence of dramatic outliers [85]. This makes the Rama-Z score a more sensitive and robust metric for backbone validation, especially for structures determined at lower resolutions [85].
Table 2: Interpreting Ramachandran Plot Statistics
| Region | Typical Color | Structural Meaning | Implication for Model Quality |
|---|---|---|---|
| Favored (Core) | Red/Dark Blue | Most favorable, low-energy conformations (e.g., α-helices, β-sheets). | High percentage (>98% in good structures) expected. |
| Allowed | Yellow/Light Blue | Less favorable but sterically permitted conformations. | A small percentage is acceptable. |
| Generously Allowed | Green/Light Green | Conformations that are possible but with some strain. | Should be minimal. |
| Outlier | White/Grey | Sterically impossible conformations. | Almost always indicates a local error; requires investigation. |
MolProbity: All-Atom Contact Analysis and Comprehensive Validation
MolProbity is a powerful, general-purpose web server and software suite that provides expert-system consultation on the accuracy of macromolecular structure models [82] [86]. It integrates several validation analyses into a single workflow, with a unique emphasis on all-atom contact analysis [82]. Its typical usage involves a clear sequence of steps, as visualized below.
Core Methodologies and Diagnostics
MolProbity's strength lies in its combination of multiple high-accuracy diagnostics.
All-Atom Contact Analysis and Clashscore: Unlike methods that use a "united-atom" approach, MolProbity adds and optimizes the positions of all hydrogen atoms using the program Reduce [82]. It then uses the program Probe to calculate all-atom contacts, identifying steric overlaps or clashes [82]. These clashes are visualized as red spikes, representing physically impossible atomic overlaps [82]. The results are integrated into a Clashscore, defined as the number of serious clashes (overlaps > 0.4 Å) per 1,000 atoms [82]. A lower Clashscore indicates a better, more chemically reasonable model.
Sidechain and Backbone Conformation Validation: MolProbity uses up-to-date, quality-filtered empirical distributions to identify outliers in sidechain rotamer conformations [82]. Furthermore, it checks for a specific type of fitting error: the 180-degree misorientation of the amide groups of Asn and Gln and the imidazole rings of His, which is common due to symmetric electron density [82]. The Reduce program automatically tests and corrects these flips during hydrogen optimization, which often improves the model's hydrogen-bonding network and reduces steric clashes [82].
The MolProbity Score: To provide a single, overall quality metric, MolProbity calculates a composite MolProbity Score that combines the Clashscore, the percentage of Ramachandran outliers, and the percentage of rotamer outliers [86]. A lower MolProbity Score indicates a higher-quality model, helping researchers quickly assess and compare structures.
Table 3: Essential Research Reagent Solutions for Structure Validation
| Tool or Resource | Type | Primary Function in Validation |
|---|---|---|
| MolProbity Server | Web Server / Software Suite | Integrated all-atom contact analysis, Ramachandran/rotamer checks, and flip validation [82] [86]. |
| PHENIX Software Suite | Software Platform | Integrates MolProbity validation tools directly into the crystallographic refinement and model-building workflow [86]. |
| Coot | Molecular Graphics Software | Used for interactive model building and rebuilding; can integrate MolProbity analysis to generate a "to-do" list for corrections [86]. |
| PDB Validation Server | Web Server | Provides automated validation reports upon deposition to the Protein Data Bank, including key quality indicators [22]. |
| PROCHECK | Software | An earlier but widely used program for stereochemical quality analysis, including Ramachandran plots [84]. |
An Integrated Validation Protocol
For robust validation, these tools should be used in concert and iteratively throughout the model building and refinement process, not just as a final step before deposition [86]. A recommended protocol is:
- After initial model building, run a MolProbity analysis to get a baseline assessment of R-work, R-free, Clashscore, Ramachandran outliers, and rotamer outliers.
- Use the 3D graphics output in KiNG or Coot to visually inspect and locate specific problems flagged by MolProbity, such as steric clashes, Ramachandran outliers, and questionable rotamers [82] [86].
- Prioritize corrections starting with the most severe issues, like large steric clashes and Ramachandran outliers, as these can indicate significant local errors.
- Rebuild and refine the model to address these issues, taking advantage of automated correction suggestions for Asn/Gln/His flips and rotamer outliers where appropriate.
- Re-validate the model and monitor the improvement in all quality metrics. Ensure that R-free improves or remains stable and does not diverge significantly from R-work.
By systematically applying this integrated approach, researchers can produce highly reliable protein structures, ensuring the integrity of subsequent biological conclusions and drug development efforts based on these models.
The Protein Data Bank (PDB) serves as the global repository for three-dimensional structural data of biological macromolecules, enabling advancements in fields ranging from basic biology to drug discovery. However, the quality of structures within the PDB is not uniform. Assessment of structure quality is therefore a critical step before any subsequent analysis, as conclusions drawn from unreliable models can be misleading [87] [88]. This guide provides researchers, scientists, and drug development professionals with a comprehensive framework for evaluating and selecting the most reliable protein structures, with a particular emphasis on those determined by X-ray crystallography.
A structural model is an interpretation of experimental data and is inherently imperfect. Limitations can arise from various sources, including mismatches between the model and experimental data, regions of local disorder, distortions in atomic geometry, or errors in model building and refinement [87]. The process of validation ensures that the atomic model not only fits the experimental data but also conforms to known stereochemical principles [46].
Key Quality Metrics for X-ray Crystallographic Structures
The evaluation of an X-ray crystallography structure relies on a set of well-established metrics that assess both the agreement with experimental data and the geometric plausibility of the model. These metrics can be broadly categorized into global measures, which describe the overall structure, and local measures, which assess specific regions.
Global Quality Measures
Global metrics provide a quick overview of the entire structure's quality.
- Resolution: This is one of the most critical indicators of structure quality. It measures the level of detail visible in the electron density map and indicates how well two adjacent atoms can be distinguished. Lower values represent better resolution (e.g., 1.5 Å is better than 3.0 Å). High-resolution structures (typically < 2.0 Å) allow for more precise atomic placement and better definition of side chains and solvent molecules [87] [46].
- R-factor and R-free: The R-factor quantifies the agreement between the experimental diffraction data and the data simulated from the atomic model. A perfect, but unattainable, agreement would yield an R-factor of zero. For protein structures, R-factors typically range from 14% to 25% [87] [46]. R-free is calculated using a small subset of the experimental data that was excluded from the refinement process, making it an unbiased validation metric. The R-free value is usually slightly higher (by ~0.05 or 5%) than the R-factor. A large discrepancy between the R-factor and R-free may indicate over-fitting or errors in the model [87].
- Clashscore: Generated by MolProbity, the clashscore identifies steric overlaps where non-bonded atoms are positioned too close to each other. A lower clashscore is indicative of a more favorable and realistic model geometry [46].
Table 1: Interpretation of Global Quality Metrics for X-ray Structures
| Metric | Excellent | Good | Acceptable | Poor | Interpretation |
|---|---|---|---|---|---|
| Resolution (Å) | ≤ 1.5 | 1.5 - 2.5 | 2.5 - 3.2 | > 3.2 | Lower values allow for more atomic detail [87] [46]. |
| R-free (%) | ≤ 20 | 20 - 25 | 25 - 30 | > 30 | Should be close to, but higher than, the R-factor [87]. |
| Clashscore | ≤ 2 | 2 - 5 | 5 - 10 | > 10 | Lower scores indicate fewer atom-atom clashes [46]. |
Local Quality and Model Geometry
While global metrics are informative, the quality of a structure is not uniform. Certain regions, like flexible loops or surface residues, may be less well-defined.
- Real Space Correlation Coefficient (RSCC): This is a local measure of how well the atomic coordinates of a specific residue agree with the experimental electron density. The RSCC ranges from 0 to 1, with higher values indicating better agreement. Atomic coordinates for residues with an RSCC in the lowest 1% should not be trusted, while those in the lowest 1-5% should be considered with caution [87].
- Ramachandran Plot: This plot visualizes the torsion angles (Phi and Psi) of each amino acid residue in the protein backbone. A high-quality structure will have most of its residues in the "favored" regions of the plot. The percentage of residues in "allowed" regions should be small, and "outliers" should be minimal (ideally < 0.5%) and require justification. A high percentage of outliers can indicate backbone geometry problems [46].
- Rotamer Outliers: This metric assesses the conformations of amino acid side chains. The percentage of side chains in unlikely, or "outlier," rotameric states should be low. A high percentage suggests poor side-chain placement, potentially due to weak electron density [46].
- Hydrogen-Bonding Geometry: A newly developed validation method analyzes the geometry of hydrogen bonds within a structure. Systematic analysis of high-resolution models shows that hydrogen-bond parameters have a distinct and conserved distribution. Deviations from these expected distributions can reveal subtle geometry issues that may be missed by other validation tools, making it a powerful independent validator [89].
Table 2: Key Model Geometry Metrics from a PDB Validation Report
| Metric | Target Value | Caution Flag | Description |
|---|---|---|---|
| Ramachandran Favored (%) | > 98% | < 90% | Percentage of residues in most favorable phi/psi angles [46]. |
| Ramachandran Outliers (%) | < 0.5% | > 2% | Percentage of residues in disallowed regions of the plot [46]. |
| Rotamer Outliers (%) | < 2% | > 5% | Percentage of side chains in unlikely conformations [46] [89]. |
| Cβ Deviations | 0% | > 0.1% | Indicates errors in backbone chirality [89]. |
| RSCC Z-score | ~ 0 | < -2.5 | Measures local fit to density; low Z-score indicates poor fit [87]. |
A Practical Workflow for Structure Assessment
The following diagram and protocol outline a systematic approach for assessing any PDB entry, helping you to quickly identify the most reliable structures for your research.
Assessment Workflow
Step-by-Step Protocol
- Check Global Metrics: Begin by reviewing the overall resolution, R-value, and R-free from the PDB entry's summary page. Compare these values to the benchmarks in Table 1. A structure with a resolution of 2.5 Å or better and an R-free below 25% is generally a good candidate [87] [46].
- Review the Validation Report: Every PDB entry has a validation report. Access this report and examine the Ramachandran plot statistics, clashscore, and rotamer outliers. Prioritize structures with >98% of residues in the favored regions and <0.5% outliers [46].
- Inspect Local Fit for Regions of Interest: If your research focuses on a specific active site, binding pocket, or mutation site, check the local quality metrics. Use the Real Space Correlation Coefficient (RSCC) to ensure the electron density strongly supports the modeled conformation in that region. Residues with an RSCC below the 5th percentile should be treated with caution [87].
- Examine the Electron Density: For the most critical assessments, visually inspect the electron density maps. The
2mFo-DFcmap (typically contoured at 1.0 σ) should clearly show density for the main chain and side chains of your region of interest. ThemFo-DFcdifference map (typically contoured at +3.0 σ and -3.0 σ) should show no major positive (indicating missing atoms) or negative (indicating misplaced atoms) peaks [46] [90]. - Final Assessment: Synthesize the information from all previous steps. A structure that passes all global and local checks can be considered high-quality. If a structure has global or local weaknesses, consider whether they impact your intended use. If they do, search for a better structure or interpret the unreliable regions with skepticism.
The following tools and databases are indispensable for accessing, evaluating, and analyzing protein structures.
Table 3: Key Research Reagent Solutions for Structure Validation
| Tool / Resource | Primary Function | Key Features |
|---|---|---|
| RCSB PDB | Primary Database | Provides access to experimental structures, validation reports, and summary metrics [87]. |
| PDB Validation Report | Quality Assessment | Offers a detailed analysis of model geometry and fit to experimental data for each entry [87] [46]. |
| MolProbity | Structure Validation | Generates clashscores, Ramachandran plots, and rotamer analyses; integrated into PDB validation [46] [89]. |
| Phenix Software Suite | Structure Determination & Validation | Includes tools for refinement and validation, such as phenix.hbond for analyzing hydrogen-bonding geometry [89]. |
| COOT | Model Building & Visualization | Used for manual model building and inspection and for visualizing electron density maps [90]. |
Selecting a high-quality protein structure is a fundamental step in ensuring the integrity of structural analysis. By applying the systematic workflow outlined in this guide—evaluating global metrics, scrutinizing the validation report, and critically examining local regions of interest—researchers can make informed decisions about which PDB entries to use. The integration of traditional metrics like resolution and R-free with modern validation tools, including hydrogen-bonding analysis, provides a robust framework for identifying reliable structural models. As structural biology continues to evolve, with an increasing number of structures determined by cryo-EM and computed by AI, these core principles of validation and critical assessment will remain essential for extracting meaningful biological insights.
For decades, the precise determination of protein structures has been a cornerstone of modern biology and drug discovery, providing invaluable insights into molecular function and mechanisms of disease. Among the experimental techniques available, X-ray crystallography has long served as the workhorse of structural biology, responsible for determining approximately 84% of structures in the Protein Data Bank [91]. However, the recent "resolution revolution" in cryo-electron microscopy (cryo-EM) has transformed the landscape, offering a powerful complementary approach for visualizing biological macromolecules [92] [93]. This paradigm shift, acknowledged by the 2017 Nobel Prize in Chemistry, has enabled researchers to tackle increasingly complex biological questions that were previously inaccessible.
Understanding the relative strengths and limitations of these techniques is crucial for structural biologists, researchers, and drug development professionals seeking to determine the three-dimensional architecture of proteins and their complexes. This comparative analysis examines the fundamental principles, technical requirements, and applications of both methods, providing a framework for selecting the appropriate technique based on specific research objectives, sample characteristics, and available resources.
Fundamental Principles and Methodologies
X-ray Crystallography: A Crystalline Approach
X-ray crystallography determines atomic structures by analyzing how X-rays diffract when passed through a crystallized sample. The technique relies on the ordered, repeating arrangement of molecules in a crystal to amplify the diffraction signal [94] [11]. When X-rays interact with the electron clouds of atoms in a crystal, they produce a characteristic diffraction pattern of spots. The intensities and angles of these spots are measured and, combined with phase information (often derived through molecular replacement or experimental methods), used to calculate an electron density map [91] [95]. Researchers then build and refine an atomic model that fits this electron density, resulting in a high-resolution structure.
The requirement for high-quality crystals represents both a strength and limitation of this method. Well-ordered crystals can yield extraordinary resolution, often surpassing 1.0 Å, but many biological molecules resist crystallization due to flexibility, complexity, or inherent instability [91] [94].
Cryo-Electron Microscopy: The Vitrification Revolution
Cryo-EM bypasses the crystallization requirement by preserving samples in a near-native state through vitrification - rapid freezing that transforms aqueous solutions into amorphous ice without crystal formation [96]. This process immobilizes biological molecules in a thin layer of glass-like ice, preserving their native structure. When a beam of electrons passes through these vitrified samples, multiple two-dimensional projection images are captured from different angles [96] [93].
Advanced computational algorithms then process these images to reconstruct a three-dimensional density map. In single-particle analysis (SPA), one of the most powerful cryo-EM approaches, images of thousands to millions of individual particles are classified, aligned, and averaged to generate high-resolution structures [97]. This ability to analyze molecules without crystallization has made cryo-EM particularly valuable for studying large complexes, membrane proteins, and dynamic systems.
Technical Comparison: Workflows and Requirements
Comparative Workflow Diagrams
The following diagrams illustrate the distinct workflows for X-ray crystallography and cryo-EM, highlighting key differences in their methodological approaches.
X-ray Crystallography Workflow: The process begins with protein purification and crystallization, followed by crystal harvesting, X-ray diffraction, data processing, phase determination, model building, and final refinement [91] [94].
Cryo-EM Single Particle Analysis Workflow: The process involves sample purification, vitrification, EM imaging with a cryo-electron microscope, motion correction, particle picking, 2D classification, 3D reconstruction, and final model building and refinement [96] [97].
Sample Requirements and Technical Specifications
Table 1: Sample Requirements and Technical Specifications Comparison
| Parameter | X-ray Crystallography | Cryo-EM |
|---|---|---|
| Sample Purity | High homogeneity required (>95%) [97] | Moderate heterogeneity acceptable [94] |
| Sample Amount | >2 mg typically [94] | 0.1-0.2 mg [94] |
| Sample Concentration | ~10 mg/ml for soluble proteins [97] | ≥ 2 mg/mL [97] |
| Molecular Size | Optimal <100 kDa [94] | Optimal >100 kDa [94] |
| Structural Stability | Requires rigid structure [94] | Flexible/Dynamic acceptable [94] |
| Buffer Conditions | Low phosphate buffers preferred [91] | Low organic solvents, salt ≤300 mM [97] |
Table 2: Operational Considerations and Output Parameters
| Consideration | X-ray Crystallography | Cryo-EM |
|---|---|---|
| Typical Timeline | Weeks to months [94] | Weeks typically [94] |
| Maximum Resolution | Sub-1.0 Å possible [94] | 2-3 Å [94] |
| Typical Resolution | 1.5-2.5 Å [94] | 3-4 Å [94] |
| Data Collection Time | Minutes to hours per dataset [94] | Hours to days per dataset [94] |
| Data Volume | Gigabytes [94] | Terabytes [94] |
| Equipment Access | Synchrotron facilities needed [91] [94] | High-end microscope needed [92] [94] |
| Cost Considerations | Synchrotron access needed [94] | High microscope costs (~$10M for high-end) [92] [94] |
Strengths and Limitations Analysis
Advantages of X-ray Crystallography
X-ray crystallography remains a powerful technique for structural determination with several distinct advantages:
- Atomic Resolution: When high-quality crystals are obtained, X-ray crystallography routinely achieves resolutions finer than 2 Å, providing extraordinary detail of molecular architecture, including precise atomic positions and chemical bond characteristics [94].
- High Precision for Small Molecules: The technique offers exceptional precision for studying small molecules, fragment binding, and detailed ligand interactions, making it invaluable for structure-based drug design [94].
- Well-Established Pipelines: Decades of development have resulted in robust, standardized data processing pipelines, validation methods, and software ecosystems that streamline structure determination [94].
- High-Throughput Capability: For well-behaved targets that crystallize readily, X-ray approaches can be scaled for high-throughput analysis, enabling rapid screening of multiple ligand complexes or mutant variants [91].
Limitations of X-ray Crystallography
Despite its strengths, the technique faces several significant challenges:
- Crystallization Requirement: The necessity for high-quality crystals represents the most significant bottleneck, particularly for membrane proteins, flexible complexes, and large assemblies that are difficult to crystallize [91] [94].
- Crystal Packing Artifacts: The crystal environment may stabilize specific conformations that don't represent physiological states or introduce packing constraints that distort native structures [94].
- Radiation Damage: Despite cryo-cooling techniques, X-ray exposure can cause radiation damage during data collection, potentially altering sensitive structural features [96].
- Limited Dynamic Information: Traditional crystallography typically provides static snapshots, making it challenging to capture conformational dynamics and transitions without specialized time-resolved approaches [94].
Advantages of Cryo-EM
The rise of cryo-EM has addressed many limitations of crystallography, offering several compelling advantages:
- No Crystallization Required: By eliminating the crystallization requirement, cryo-EM enables the study of targets that have resisted crystallographic approaches, including large complexes, membrane proteins, and flexible assemblies [92] [94].
- Native State Preservation: Vitrification preserves samples in a near-native state, allowing structural studies in conditions that closely mimic the physiological environment [96] [92].
- Conformational Heterogeneity: Single-particle analysis can often resolve multiple conformational states from a single sample, providing insights into molecular dynamics and functional mechanisms [94].
- Minimal Sample Consumption: The technique requires significantly less sample material compared to crystallography (0.1-0.2 mg vs. >2 mg), advantageous for scarce or difficult-to-express targets [94].
Limitations of Cryo-EM
Despite its transformative impact, cryo-EM faces several important limitations:
- Resolution Limitations: While improving rapidly, cryo-EM typically achieves lower resolution than crystallography (typically 2.5-4.0 Å vs. often better than 2.0 Å), potentially missing atomic-level details [94].
- Sample Preparation Challenges: Optimizing vitrification conditions, ice thickness, and particle distribution requires significant expertise and can be challenging for some targets [96] [97].
- Equipment Cost and Access: High-end cryo-EM microscopes carry price tags approaching $10 million, creating accessibility barriers despite efforts to develop more affordable options [92].
- Computational Demands: Processing cryo-EM data requires substantial computational resources and expertise in specialized image processing software, creating steep learning curves [94].
Research Reagent Solutions and Essential Materials
Table 3: Key Research Reagents and Materials for Structural Biology
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Specialized EM Grids | Support film for vitrified samples | Graphene or graphene oxide grids (e.g., GraFuture) reduce background noise and address preferred orientation [97] |
| Cryogenic Fluids | Sample vitrification | Liquid ethane or propane for rapid freezing to form amorphous ice [96] [93] |
| Crystallization Screens | Crystal formation | Sparse matrix screens with various precipitants, salts, and pH conditions to identify initial crystallization conditions [91] |
| Detergents/Membrane Mimetics | Solubilize membrane proteins | Used for both techniques; nanodiscs, amphipols, or detergents for cryo-EM; detergents for cubic phase crystallography [91] [94] |
| Direct Electron Detectors | Electron detection in cryo-EM | Critical hardware advancement enabling the "resolution revolution" with improved signal-to-noise [92] [95] |
| Synchrotron Access | X-ray source for crystallography | Essential for high-resolution data collection; requires beamtime allocation at facilities [91] [94] |
| Lipidic Cubic Phase (LCP) Materials | Membrane protein crystallization | Monoolein-based matrices for creating membrane-like environments for crystallizing membrane proteins [91] [95] |
Application-Based Method Selection
Target-Specific Considerations
Choosing between crystallography and cryo-EM depends heavily on the specific research target and objectives:
- Membrane Proteins: Cryo-EM has demonstrated remarkable success with membrane proteins, particularly G protein-coupled receptors (GPCRs), ion channels, and transporters, by preserving them in near-native lipid environments [92] [94]. Crystallography remains valuable for stable membrane protein constructs, especially using lipidic cubic phase methods that mimic membrane environments [91] [95].
- Large Complexes and Assemblies: For complexes exceeding 100-200 kDa, cryo-EM generally excels, enabling structural analysis without disruption of quaternary structure [94]. The technique has proven particularly powerful for ribosomes, viral capsids, and transcriptional machinery.
- Dynamic Systems and Multiple Conformations: Cryo-EM's ability to resolve conformational heterogeneity from a single sample makes it ideal for studying allosteric mechanisms, enzyme catalysis, and structural transitions [94]. Advanced crystallographic approaches like time-resolved serial crystallography can capture dynamics but require specialized facilities.
- Drug Discovery Applications: X-ray crystallography remains the gold standard for fragment-based drug design and high-resolution ligand binding studies due to its exceptional resolution for well-behaved targets [91] [94]. Cryo-EM is increasingly valuable for validating drug binding to complex targets like membrane proteins and large assemblies [94].
Integrated Approaches and Future Directions
The most powerful structural biology research often integrates multiple techniques, leveraging their complementary strengths:
- Hybrid Methodologies: Combining crystallography of individual domains with cryo-EM of full complexes provides comprehensive structural understanding across spatial scales [98].
- AI and Computational Integration: Artificial intelligence tools like AlphaFold are increasingly integrated with both experimental techniques, with predicted models facilitating molecular replacement in crystallography and aiding map interpretation in cryo-EM [95].
- Time-Resolved Studies: Both techniques are advancing toward dynamic structural biology, with microcrystallography at XFELs and time-resolved cryo-EM capturing transient states and mechanistic intermediates [95].
- Accessibility Improvements: Efforts to develop cheaper, simpler cryo-EM instruments using 100 keV electrons rather than 300 keV aim to democratize access to this transformative technology [92].
X-ray crystallography and cryo-EM represent complementary, rather than competing, approaches to structural biology. X-ray crystallography remains unparalleled for obtaining atomic-resolution structures of proteins that form high-quality crystals, providing exquisite detail for small molecules, ligands, and well-behaved soluble proteins. In contrast, cryo-EM has dramatically expanded the scope of structural biology to encompass large complexes, membrane proteins, and dynamic systems that have long resisted crystallization.
The choice between these techniques depends critically on research objectives, sample characteristics, and available resources. For high-resolution studies of stable targets that crystallize readily, crystallography remains optimal. For structurally challenging targets, particularly large complexes and membrane proteins in near-native states, cryo-EM offers a powerful alternative. The most successful structural biology pipelines increasingly integrate both approaches, leveraging their complementary strengths to tackle increasingly complex biological questions and accelerate drug discovery efforts.
As both technologies continue to advance—with crystallography pushing toward more challenging targets and faster time-resolved studies, and cryo-EM achieving higher resolutions and greater accessibility—the future of structural biology lies in their synergistic application, combined with emerging computational methods, to illuminate the molecular mechanisms of life and disease.
The determination of three-dimensional protein structures is fundamental to understanding biological function at a molecular level. Among the experimental techniques available, X-ray crystallography and Nuclear Magnetic Resonance (NMR) spectroscopy have emerged as the principal methods for atomic-resolution structure determination. Together, these two techniques account for the vast majority of structures deposited in the Protein Data Bank (PDB) [99]. While both methods aim to elucidate atomic structures, they differ profoundly in their physical principles, sample requirements, and the nature of the structural information they provide.
The choice between X-ray crystallography and NMR spectroscopy is not merely a matter of convenience but a strategic decision that can significantly impact the quality and biological relevance of the structural data obtained. This technical guide provides an in-depth comparison of these two foundational techniques, offering researchers, scientists, and drug development professionals a framework for selecting the appropriate method based on their specific research objectives, sample characteristics, and desired structural information.
Fundamental Principles and Historical Context
X-ray Crystallography
X-ray crystallography determines atomic structures by measuring how X-rays are diffracted by the electron clouds of atoms arranged in a crystalline lattice. The technique is based on Bragg's Law (nλ = 2d sinθ), which describes the condition for constructive interference when X-rays interact with parallel planes of atoms in a crystal [100] [101] [102]. When a crystal is exposed to an X-ray beam, the resulting diffraction pattern provides information about the electron density distribution within the crystal. Through computational methods, this electron density map is used to determine the positions of atoms and build a three-dimensional molecular model [11] [16].
The science of X-ray crystallography began with Max von Laue's 1912 discovery that crystals could diffract X-rays, confirming that X-rays are electromagnetic waves and that crystals possess a regular, periodic structure [11]. This was rapidly followed by William Henry Bragg and William Lawrence Bragg developing the foundational principles of crystal structure analysis, earning them the 1915 Nobel Prize in Physics [101]. The first atomic-resolution structure (table salt) was solved in 1914, followed by the structure of diamond that same year [11].
NMR Spectroscopy
Protein NMR spectroscopy exploits the quantum-mechanical properties of atomic nuclei, typically hydrogen, carbon, and nitrogen, when placed in a strong magnetic field [103]. Unlike crystallography, which directly visualizes electron density, NMR detects the absorption of radio frequency signals by atomic nuclei. The precise absorption frequency (chemical shift) of each nucleus depends on its local molecular environment, providing information about the atom's chemical identity and spatial position [104] [103].
NMR for protein structure determination developed significantly later than crystallography, emerging as a viable method in the 1980s [105]. The technique relies on measuring through-space interactions between nuclei (Nuclear Overhauser Effect) to determine interatomic distances, which are then used as constraints to calculate three-dimensional structures that satisfy all experimental observations [103].
Technical Comparison: Advantages and Limitations
Table 1: Comprehensive comparison of X-ray crystallography and NMR spectroscopy
| Parameter | X-ray Crystallography | NMR Spectroscopy |
|---|---|---|
| Sample State | Solid crystalline state | Solution (primarily) or solid state |
| Sample Requirement | High-quality single crystals (typically >0.1 mm) [16] [66] | Concentrated solution (0.1-3 mM) in aqueous buffer [103] |
| Typical Sample Volume | Single crystal | 300-600 μL [103] |
| Molecular Size Limit | Essentially none; structures of viral capsids determined [104] [16] | Limited for solution NMR; challenging above ~50 kDa [104] [103] |
| Resolution/Precision | High atomic resolution (often 1-3 Å) [104] [16] | Lower resolution; global RMSD 1.5-2.5 Å for backbone atoms [99] |
| Time Requirements | Days to years for crystallization; hours for data collection | Minutes to days for data collection [103] |
| Key Advantage | Unmatched resolution for large structures | Studies dynamics and native state in solution |
| Major Limitation | Requires crystallization; static picture | Limited for large molecules; complex interpretation |
| Structural Output | Single, precise model | Ensemble of models satisfying distance constraints |
Key Advantages and Disadvantages
X-ray crystallography advantages include its ability to handle very large molecular complexes without size limitations, provide high atomic resolution structures, and directly visualize ordered water molecules and ligands in binding sites [104] [102]. Its disadvantages center on the crystallization requirement, which can be insurmountable for some proteins (particularly membrane proteins), and the static nature of the structures obtained from the crystalline environment [104] [16].
NMR spectroscopy advantages include the ability to study proteins in near-native solution conditions, probe molecular dynamics and flexibility, identify conformational changes, and monitor interactions in real time [104] [103]. Its disadvantages include limitations on protein size, the need for large amounts of pure sample, lower effective resolution, and complex data analysis requiring specialized expertise [104] [103].
Experimental Workflows
X-ray Crystallography Workflow
The following diagram illustrates the multi-step process of structure determination by X-ray crystallography:
Protein Purification and Crystallization: The target protein must be purified to homogeneity and concentrated to 5-20 mg/mL [16]. Crystallization is typically achieved through vapor diffusion methods (hanging or sitting drop) where a drop containing protein and precipitant is equilibrated against a reservoir with higher precipitant concentration [16] [66]. This slowly increases the precipitant concentration in the drop, encouraging ordered crystal formation rather than amorphous precipitation.
Data Collection: A single crystal is mounted on a goniometer and exposed to an X-ray beam [16] [102]. The crystal is rotated to record diffraction patterns from multiple orientations. Modern detectors capture these patterns with exposure times ranging from seconds at synchrotron sources to hours with in-house generators [16].
Data Processing: The diffraction images are processed to determine the unit cell parameters, space group symmetry, and to measure the intensities of all diffraction spots [16] [66]. The quality of diffraction is assessed by the resolution limit, with better crystals diffracting to higher resolution (lower Ångström values).
Phasing: The "phase problem" is the major computational challenge in crystallography - while diffraction intensities can be measured directly, phase information is lost and must be determined indirectly [16] [66]. Common phasing methods include molecular replacement (using a homologous structure), multiple-wavelength anomalous dispersion (MAD), or single-wavelength anomalous dispersion (SAD) [101] [66].
Model Building and Refinement: An atomic model is built into the experimental electron density map and iteratively refined to improve the fit to the diffraction data while maintaining realistic geometric parameters [16] [102]. The quality of the final model is assessed by R-factor and R-free values [66].
NMR Spectroscopy Workflow
The following diagram illustrates the process of protein structure determination by NMR spectroscopy:
Sample Preparation: Protein samples for NMR are typically isotopically labeled with ¹⁵N and/or ¹³C to facilitate the assignment process and enable multidimensional experiments [103]. The protein is dissolved in an aqueous buffer at concentrations of 0.1-3 mM in a volume of 300-600 μL [103].
Data Collection: A suite of multidimensional NMR experiments is performed to correlate the signals of nuclei connected through chemical bonds or through space [103]. Key experiments include HSQC (heteronuclear single quantum coherence), TOCSY (total correlation spectroscopy), and NOESY (nuclear Overhauser effect spectroscopy). Data collection times range from hours to days depending on the experiment dimensionality and sample concentration [103].
Resonance Assignment: The first major interpretive step involves assigning each resonance in the NMR spectrum to specific atoms in the protein [103]. For isotopically labeled proteins, this is achieved primarily through triple-resonance experiments that connect nuclei through chemical bonds along the protein backbone and side chains.
Distance Restraints: NOESY experiments provide information about protons that are close in space (typically <5-6 Å), which are converted into distance restraints for structure calculation [103]. The number and quality of these distance restraints directly determine the accuracy and precision of the final structure.
Structure Calculation and Refinement: Structures are calculated using computational methods that generate three-dimensional models satisfying all experimental distance and angle constraints [103]. The final output is an ensemble of structures that collectively represent the protein's conformation in solution, with regions of greater flexibility showing higher structural variability.
Research Reagent Solutions and Essential Materials
Table 2: Essential research reagents and materials for structural biology techniques
| Item | Function/Purpose | Used in Technique |
|---|---|---|
| Crystallization Screens | Sparse matrix of conditions to identify initial crystallization hits [16] | X-ray Crystallography |
| Cryoprotectants | Protect crystals from radiation damage during flash-cooling in liquid N₂ [16] | X-ray Crystallography |
| Isotope-Labeled Media | Production of ¹⁵N, ¹³C-labeled proteins for multidimensional NMR [103] | NMR Spectroscopy |
| Deuterated Solvents | Reduces background ¹H signals in NMR experiments [103] | NMR Spectroscopy |
| Size Exclusion Chromatography | Final purification step to ensure sample homogeneity [16] | Both Techniques |
| Synchrotron Access | High-intensity X-ray source for challenging crystals [16] | X-ray Crystallography |
| High-Field NMR Spectrometer | High-sensitivity detection for biomolecular NMR [103] | NMR Spectroscopy |
Strategic Selection Guide
When to Prefer X-ray Crystallography
Choose X-ray crystallography when:
Working with large proteins or complexes - The technique has essentially no upper size limit, with structures determined for viral capsids and ribosomes [104] [16].
Atomic resolution is critical - For visualizing precise ligand binding, catalytic mechanisms, or ion coordination, the high resolution of crystallography is essential [104] [102].
Membrane proteins that form stable crystals - While crystallization is challenging, successful cases provide the most detailed structural insights into membrane protein function [104].
Time-resolved studies of reversible processes - Using Laue diffraction or serial crystallography, kinetic processes can be studied at atomic resolution [101].
Industrial drug discovery applications - The ability to rapidly determine structures of protein-ligand complexes makes crystallography ideal for structure-based drug design [11] [102].
When to Prefer NMR Spectroscopy
Choose NMR spectroscopy when:
Studying protein dynamics and flexibility - NMR uniquely provides information on molecular motions across multiple timescales [104] [103].
The protein does not crystallize - For proteins refractory to crystallization, NMR may be the only method for atomic-resolution structure determination [104].
Investigating weak interactions and binding - Chemical shift perturbations can identify interaction surfaces and measure binding affinities [103].
Studying intrinsically disordered proteins - These proteins lack fixed structure and cannot be crystallized, but can be characterized by NMR [103].
When solution-state behavior is critical - For understanding protein function under physiological conditions without crystal packing artifacts [104] [99].
Complementary Use of Both Techniques
In many research scenarios, X-ray crystallography and NMR spectroscopy provide complementary information that together give a more complete understanding of protein structure and function. Crystallography can provide high-resolution structural frameworks, while NMR offers insights into dynamics and solution behavior [105] [99]. A systematic comparison of structures determined by both methods revealed that while the overall folds are highly similar (backbone RMSD 1.5-2.5 Å), local differences often reflect genuine biological flexibility rather than methodological artifacts [99].
X-ray crystallography and NMR spectroscopy remain the cornerstone techniques for protein structure determination, each with distinct strengths and limitations. Crystallography excels at providing high-resolution static structures of large complexes, while NMR offers unique insights into dynamics and solution behavior. The strategic selection between these methods should be guided by the specific research question, protein characteristics, and desired structural information. As both techniques continue to evolve, their complementary application will continue to drive advances in our understanding of protein structure and function, enabling innovations across biochemistry, molecular biology, and drug discovery.
The field of structural biology is undergoing a profound transformation, moving from a paradigm dominated by individual experimental techniques to an integrative approach that combines computational predictions with experimental data. The determination of protein three-dimensional structures is fundamental to understanding biological function and advancing drug development. While X-ray crystallography has long been a cornerstone technique, responsible for approximately 89% of structures in the Protein Data Bank (PDB), it faces persistent challenges including the protein crystallization bottleneck, phase problem, and difficulties with membrane proteins and flexible complexes [106] [107]. The recent emergence of highly accurate artificial intelligence (AI)-based structure prediction tools, particularly AlphaFold, has revolutionized the field, not as a replacement for experimental methods but as a powerful complementary approach [108]. These hybrid methodologies leverage the strengths of both computational and experimental paradigms—harnessing the rapid, comprehensive nature of AI predictions while grounding results in experimental observation—to accelerate and enhance structure determination workflows for researchers and drug development professionals.
Theoretical Foundation: Complementary Strengths and Limitations
X-ray Crystallography: Established Gold Standard with Persistent Challenges
X-ray crystallography determines protein structures by analyzing the diffraction patterns produced when X-rays interact with protein crystals. The resulting electron density maps enable the construction of atomic models with high precision. The technique has been instrumental in numerous breakthroughs, from the first protein structures of myoglobin and hemoglobin to detailed enzymatic mechanisms [95] [107]. However, its limitations are significant: many proteins resist crystallization, particularly membrane proteins such as G protein-coupled receptors (GPCRs) and ion channels; crystal packing forces may distort native conformations; and the phase problem complicates map interpretation [106] [107]. Additionally, crystallography typically captures static snapshots, making it challenging to study dynamic processes and conformational heterogeneity.
AlphaFold Prediction: The AI Revolution with Contextual Gaps
AlphaFold and similar AI tools have demonstrated remarkable accuracy in predicting protein structures from amino acid sequences alone. These methods leverage deep learning algorithms trained on known structures in the PDB and evolutionary information from multiple sequence alignments. However, even high-confidence predictions (pLDDT > 90) have important limitations. They typically represent consensus conformations rather than ligand-bound or condition-specific states, often lacking structural nuances induced by environmental factors, post-translational modifications, or binding partners [108]. Comparative analyses reveal that while AlphaFold predictions often match experimental maps closely, they can display global distortions and domain orientation differences, with median Cα root-mean-square deviation (RMSD) values of 1.0 Å compared to experimental structures [108]. Critically, these predictions do not include ligands, covalent modifications, or the influence of environmental factors such as pH or solvent composition [108].
Table 1: Comparative Analysis of Structure Determination Methods
| Feature | X-ray Crystallography | AlphaFold Prediction | Hybrid Approaches |
|---|---|---|---|
| Atomic Resolution | Typically high (often <2.5 Å) | Variable accuracy (pLDDT dependent) | Enhanced by combining information sources |
| Ligand Binding Sites | Directly observable in electron density | Not natively predicted (except AlphaFold3) | Integrative modeling possible |
| Throughput | Slow (weeks to years) | Rapid (hours to days) | Intermediate |
| Sample Requirements | High-quality crystals needed | Amino acid sequence only | Crystals still required but methods more efficient |
| Conformational Flexibility | Typically single conformation | Consensus conformation | Can model multiple states |
| Key Limitations | Crystallization bottleneck, phase problem | No environmental context, limited complexes | Method integration challenges |
Methodological Framework: Integrated Workflows
The integration of AlphaFold predictions with crystallographic data occurs at multiple stages of the structure determination pipeline, from initial phasing to final refinement. Three principal integration paradigms have emerged: input-level fusion, output-level hybrid modeling, and AI-guided experimental refinement.
Input-Level Integration: Molecular Replacement with Predicted Models
AlphaFold predictions have dramatically transformed molecular replacement (MR), the most common method for solving the phase problem in crystallography. Traditional MR relies on homologous structures with sufficient sequence similarity, often failing for proteins without close relatives. AlphaFold-generated models now serve as high-quality search models, even in the absence of close homologs.
Protocol 1: Molecular Replacement with AlphaFold Models
- Model Generation: Predict the target structure using AlphaFold with the complete amino acid sequence.
- Model Preparation: Trim low-confidence regions (pLDDT < 70) to reduce noise in the initial phasing.
- MR Search: Use standard MR software (Phaser, Molrep) with the trimmed model.
- Iterative Building and Refinement: Cycles of manual and automated rebuilding (with Buccaneer, Phenix) followed by refinement.
This approach has significantly expanded the applicability of MR to previously intractable targets, reducing the need for experimental phasing methods like MAD/SAD that require specialized data collection and sample preparation [108].
Output-Level Integration: Hybrid Model Building
Output-level integration creates hybrid models that combine experimental density maps with computational predictions. The MICA framework (Multimodal Integration of Cryo-EM and AlphaFold) exemplifies this approach, using a deep learning architecture with a Feature Pyramid Network to simultaneously process cryo-EM density maps and AlphaFold3-predicted structures [109]. Although developed for cryo-EM, analogous approaches are being adapted for crystallography.
Protocol 2: Hybrid Model Construction
- Initial Backbone Tracing: Use experimental maps to trace protein backbone.
- Confidence-Based Replacement: Replace low-confidence regions in experimental maps with corresponding high-confidence AlphaFold predictions.
- Sequence-Guided Threading: Align sequence to backbone using structural information from AlphaFold predictions.
- Full-Atom Refinement: Generate all-atom models and refine against experimental maps [109].
This approach demonstrates robust performance across varying protein sizes and resolution qualities, achieving an average TM-score of 0.93 on high-resolution cryo-EM maps [109].
AI-Guided Ligand Building and Complex Refinement
For drug discovery applications, accurately modeling protein-ligand interactions is crucial. A recently developed pipeline integrates AlphaFold3-like models (Chai-1) with molecular dynamics simulations to fit ligands into experimental cryo-EM maps [110]. This approach is equally applicable to crystallographic electron density maps.
Protocol 3: Ligand Building Workflow
- Complex Prediction: Input protein sequence and ligand SMILES string into AlphaFold3-like model.
- Rigid Body Alignment: Align predicted complex to experimental density.
- Density-Guided Simulation: Perform molecular dynamics simulations with additional forces scaling with the gradient of similarity between simulated and experimental density.
- Validation: Assess model-to-map cross-correlation, protein-ligand interaction energy, and geometry scores [110].
This method has successfully modeled ligands for kinases, GPCRs, and transporters, achieving cross-correlation values of 82-95% with experimental maps [110].
The following diagram illustrates the core workflow for integrating AlphaFold predictions with crystallographic data, showing the key decision points and processes:
Quantitative Performance and Validation
Rigorous evaluation demonstrates the significant advantages of hybrid approaches over standalone methods. In comprehensive benchmarking using the Cryo2StructData test dataset (resolution range: 2.05Å-3.9Å), the MICA framework outperformed state-of-the-art methods ModelAngelo and EModelX(+AF) across multiple metrics [109].
Table 2: Performance Comparison of Structure Determination Methods
| Method | Average TM-score | Cα Match (%) | Cα Quality Score | Aligned Cα Length | Sequence Identity | Sequence Match |
|---|---|---|---|---|---|---|
| ModelAngelo | 0.87 | 84.5 | 0.79 | 1125 | 95.2 | 89.1 |
| EModelX(+AF) | 0.89 | 86.2 | 0.81 | 1187 | 95.4 | 90.3 |
| MICA (Hybrid) | 0.93 | 91.8 | 0.88 | 1256 | 95.5 | 88.7 |
The table above illustrates that MICA achieved superior performance in most structural accuracy metrics, particularly in TM-score (0.93), which measures global fold accuracy, and Cα match (91.8%), indicating more complete backbone tracing [109]. The method demonstrated robustness across protein sizes (384-4128 residues) and resolution ranges.
When comparing AlphaFold predictions directly with experimental crystallographic data, studies reveal important nuances. In an analysis of 102 high-quality crystal structures, the mean map-model correlation for AlphaFold predictions was 0.56 after superposition, substantially lower than the 0.86 for deposited models [108]. This indicates that while predictions capture the overall fold accurately, significant local deviations exist. Through "morphing" to minimize structural differences, the correlation improved to 0.67, suggesting that both domain-level distortions and local conformational variations contribute to the discrepancies [108].
Table 3: Key Research Reagents and Computational Tools for Hybrid Methods
| Tool/Resource | Type | Function | Application in Hybrid Methods |
|---|---|---|---|
| AlphaFold2/3 | Software | Protein structure prediction | Generate search models for MR; initial hybrid models |
| Phenix | Software | Crystallography structure solution | Molecular replacement, refinement, and validation |
| CCP4 | Software Suite | Crystallographic computation | Data processing, molecular replacement, model building |
| Chai-1 | Software | Protein-ligand complex prediction | Predict ligand binding poses for experimental refinement |
| GROMACS | Software | Molecular dynamics | Density-guided simulations for flexible fitting |
| PyMOL | Software | Molecular visualization | Structure analysis, comparison, and figure generation |
| DIALS | Software | Diffraction data processing | Data reduction and integration for synchrotron data |
| Crystallization Kits | Laboratory Reagents | Protein crystallization | Sparse matrix screens for initial crystal formation |
The integration of AlphaFold predictions with crystallographic data represents a fundamental shift in structural biology methodology. As these hybrid approaches mature, several emerging trends promise to further transform the field. The development of condition-specific predictors that incorporate environmental factors like pH, ligands, and post-translational modifications will enhance the biological relevance of predictions. Automated multi-state modeling will enable researchers to capture conformational ensembles from crystallographic data, particularly important for understanding allosteric regulation and drug mechanisms. Real-time experimental integration, where AI predictions guide data collection strategies at synchrotrons, will optimize the use of valuable beamtime and accelerate structure solution [111].
In conclusion, the emerging hybrid methods for integrating AlphaFold predictions with crystallographic data are transforming structural biology from a predominantly experimental endeavor to an integrated computational-experimental science. These approaches leverage the complementary strengths of both methodologies: the rapid, comprehensive nature of AI predictions with the empirical grounding of experimental observation. For researchers and drug development professionals, these advances translate to accelerated structure determination, particularly for challenging targets like membrane proteins and dynamic complexes. As the field continues to evolve, these integrated workflows will become increasingly central to extracting maximal biological insight from structural data, ultimately advancing our understanding of biological mechanisms and accelerating therapeutic development.
Conclusion
X-ray crystallography remains an indispensable and powerful technique for determining high-resolution protein structures, directly fueling advancements in understanding disease mechanisms and rational drug design. While the path from protein to model presents challenges in crystallization and phasing, robust methodologies and innovative solutions like serial crystallography continue to expand its capabilities. The critical importance of rigorous structure validation cannot be overstated, as it ensures the reliability of the structural data that underpins scientific discovery. Looking forward, the integration of crystallography with predictive AI models and its synergistic use with complementary techniques like Cryo-EM promise a future of even more dynamic and comprehensive molecular understanding, accelerating the development of novel therapeutics and deepening our insight into biological function at the atomic level.
References
- 1. Protein Structure Determination by X-ray Crystallography [https://www.molecularcloud.org/p/protein-structure-determination-by-x-ray-crystallography]
- 2. Bragg's law [https://en.wikipedia.org/wiki/Bragg%27s_law]
- 3. Protein X-ray Crystallography: Principles of the Technique [https://www.proteinstructures.com/protein-crystallography-basics/]
- 4. X-ray Diffraction Imaging of Deformations in Thin Films and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9024510/]
- 5. Bragg's Law [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Supplemental_Modules_(Analytical_Chemistry)/Instrumentation_and_Analysis/Diffraction_Scattering_Techniques/Bragg's_Law]
- 6. Bragg's Law [https://byjus.com/physics/braggs-law/]
- 7. Comparison of Quantitative X-ray Diffraction Mineral ... [https://www.mdpi.com/2075-163X/13/4/566]
- 8. 7.3: X-ray Crystallography [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Physical_Methods_in_Chemistry_and_Nano_Science_(Barron)/07%3A_Molecular_and_Solid_State_Structure/7.03%3A_X-ray_Crystallography]
- 9. X-ray crystallography - Structural Biology Core [https://www.mayo.edu/research/core-resources/structural-biology-core/services/x-ray-crystallography]
- 10. Evaluation of 3-Dimensionality in Approved and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7294711/]
- 11. X-ray crystallography [https://en.wikipedia.org/wiki/X-ray_crystallography]
- 12. XRD Explained: Principles, Analysis & Applications [https://www.technologynetworks.com/analysis/articles/x-ray-diffraction-xrd-xrd-principle-xrd-analysis-and-applications-404260]
- 13. 100 Years Later: Celebrating the Contributions of X-ray ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4502579/]
- 14. ALS Structural Biology Beamlines Surpass 10,000 Protein ... [https://als.lbl.gov/als-structural-biology-beamlines-surpass-10000-protein-structures-in-the-pdb/]
- 15. Sample delivery methods for protein X-ray crystallography ... [https://www.nature.com/articles/s41467-025-65173-5]
- 16. x Ray crystallography - PMC - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC1186895/]
- 17. Next-Generation Protein Sequencing: Nanopore and AI ... [https://premierscience.com/pjs-25-1205/]
- 18. X-ray Protein Crystallography [https://phys.libretexts.org/Courses/University_of_California_Davis/Biophysics_200A%3A_Current_Techniques_in_Biophysics/X-ray_Protein_Crystallography]
- 19. XDXD: End-to-end crystal structure determination with low ... [https://arxiv.org/html/2510.17936v1]
- 20. A deep learning solution for crystallographic structure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10324481/]
- 21. 1.7: Structural Resolution [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Crystallography_in_a_Nutshell_(Ripoll_and_Cano)/01%3A_Chapters/1.07%3A_New_Page]
- 22. Methods for Determining Atomic Structures - PDB-101 [https://pdb101.rcsb.org/learn/guide-to-understanding-pdb-data/methods-for-determining-structure]
- 23. Phase problem [https://en.wikipedia.org/wiki/Phase_problem]
- 24. A Beginner's Guide to Protein Crystallography [https://www.creative-biostructure.com/protein-crystallography-452.htm?srsltid=AfmBOopTjJWA-0aRpKuosKyxl0J4u0xx80mc_7hBJSXMudLj1K4HMNBY]
- 25. Protein Crystallization Market Report 2025: Projected $2.8 [https://www.globenewswire.com/news-release/2025/06/24/3104062/28124/en/Protein-Crystallization-Market-Report-2025-Projected-2-8-Billion-Growth-by-2029.html]
- 26. Protein Crystallization Market Size, Growth, Trends & ... [https://www.mordorintelligence.com/industry-reports/protein-crystallization-market]
- 27. Protein crystallization: Eluding the bottleneck of X-ray ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5645037/]
- 28. Protein Crystallization for X-ray Crystallography - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3182643/]
- 29. Challenges and Opportunities for New Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2992350/]
- 30. A Beginner's Guide to Protein Crystallography [https://www.creative-biostructure.com/protein-crystallography-452.htm?srsltid=AfmBOopweRXr77xbKPr7K7t8_O3f2kcRPJZ7EK7lR5bz5kaTR27h1Uuf]
- 31. 2.3: Protein Structural Determination [https://chem.libretexts.org/Courses/University_of_Arkansas_Little_Rock/CHEM_4320_5320%3A_Biochemistry_1/02%3A__Protein_Structure/2.3%3A_Protein_Structural_Determination]
- 32. Global Protein Crystallography Product Market Growth 2025-2031 [https://infinitymarketresearch.com/report/protein-crystallography-product-market/21123]
- 33. Synchrotron Radiation as a Tool for Macromolecular X-Ray ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7886262/]
- 34. Prospects for coherent X-ray diffraction imaging at fourth ... [https://journals.iucr.org/m/issues/2025/03/00/ro5044/]
- 35. Room-temperature X-ray fragment screening with serial ... [https://www.nature.com/articles/s41467-025-64918-6]
- 36. X-Ray Detectors [https://www.dectris.com/detectors/x-ray-detectors/]
- 37. Protein Crystallography | Discovery Research Platform for ... [https://biology.ed.ac.uk/drp/structural-biology-core/protein-crystallography]
- 38. Introduction to phasing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2852296/]
- 39. Experimental Phasing [https://www.phenix-online.org/version_docs/dev-2486/overviews/xray-experimental-phasing.html]
- 40. Experimental phasing opportunities for macromolecular ... [https://www.nature.com/articles/s42004-023-01014-0]
- 41. A new MR-SAD algorithm for the automatic building of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5947721/]
- 42. Learn: Guide to Understanding PDB Data: Crystallographic Data [https://pdb101.rcsb.org/learn/guide-to-understanding-pdb-data/crystallographic-data]
- 43. Protein crystallography for non-crystallographers, or how to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4465431/]
- 44. R-factor (crystallography) [https://en.wikipedia.org/wiki/R-factor_(crystallography)]
- 45. R Factor - an overview [https://www.sciencedirect.com/topics/neuroscience/r-factor]
- 46. Validation and Quality Assessment of X-ray Protein Structures [https://www.proteinstructures.com/protein-structure-validation/]
- 47. A quantum crystallographic protocol for general use [https://www.nature.com/articles/s41598-025-96400-0]
- 48. X-Ray Crystallography - Software - Purdue Chemistry [https://www.chem.purdue.edu/xray/software.html]
- 49. Fragment-based drug discovery: A unified workflow for ... [https://www.ddw-online.com/fragment-based-drug-discovery-a-unified-workflow-for-efficient-lead-generation-38290-202510/]
- 50. Fragment-Based Drug Discovery | April 15-16, 2025 [https://www.drugdiscoverychemistry.com/25/fragment-based-drug-discovery]
- 51. Bibliometrics Analysis and Knowledge Mapping of Fragment ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12106911/]
- 52. Structure-Based Drug Design - Rowan Scientific [https://www.rowansci.com/technologies/structure-based-drug-design]
- 53. Incorporating targeted protein structure in deep learning ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12560950/]
- 54. X-Ray Crystallography - UF Health Cancer Center [https://cancer.ufl.edu/research/supported-shared-resources/other-supported-resources/drug-design-core/x-ray-crystallography/]
- 55. DESY scientists advance drug development with room- ... [https://desy.de/desy_latest_news/2025/hiphax/index_eng.html]
- 56. Serial crystallography for structure-based drug discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC7572805/]
- 57. Data as a Product for Structure-Based Drug Design (SBDD) [https://www.desertsci.com/2025/04/17/data-as-a-product-for-structure-based-drug-design-sbdd/]
- 58. Optimizing Protein Production and Purification for ... [https://www.creative-biostructure.com/resource-optimizing-protein-production-purification-crystallography.htm?srsltid=AfmBOorvRkIGpSETfyszm5Ne321NCVAf73Q5t22YsblilR7Zq1aq8Mfk]
- 59. Computational design of soluble and functional membrane ... [https://www.nature.com/articles/s41586-024-07601-y]
- 60. A general protocol for the crystallization of membrane ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4075773/]
- 61. Preparation of High-Purity Potassium Chloride Crystal ... [https://www.mdpi.com/2073-4352/15/11/958]
- 62. Study on agglomeration mechanism of ammonium ... [https://www.sciencedirect.com/science/article/abs/pii/S0032591024002146]
- 63. Using crystals and light, scientists unlock new ways to grow ... [https://natsci.msu.edu/news/2025/25-10-scientists-unlock-new-ways-to-grow-materials-on-demand.aspx]
- 64. AI-based protein models enhance the accuracy of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10324573/]
- 65. Crystallography Technique: Experimental phasing [https://rigaku.com/products/crystallography/techniques/experimental-phasing-method]
- 66. X-ray Crystallography [https://www.creativebiomart.net/resource/principle-protocol-x-ray-crystallography-393.htm]
- 67. (IUCr) The AI-based phase-seeding (AI-PhaSeed) method [https://journals.iucr.org/j/issues/2025/06/00/po5167/]
- 68. Critical assessment of AI-based protein structure prediction [https://www.sciencedirect.com/science/article/pii/S2950363925000353]
- 69. Radiation damage in single-particle cryo-electron microscopy [https://pmc.ncbi.nlm.nih.gov/articles/PMC3083915/]
- 70. Probing soft X-ray induced photoreduction of a model Mn ... [https://journals.iucr.org/s/issues/2025/02/00/rv5186/]
- 71. Cryo2RT: a high-throughput method for room-temperature ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11301757/]
- 72. A bright future for serial femtosecond crystallography with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5600683/]
- 73. X Ray Crystallography [https://www.ijpsjournal.com/article/X+Ray+Crystallography]
- 74. X Ray Crystallography in the Real World: 5 Uses You'll ... [https://www.linkedin.com/pulse/x-ray-crystallography-real-world-5-uses-youll-actually-pl0mf/]
- 75. Surface Engineering of Top7 to Facilitate Structure ... [https://www.mdpi.com/1422-0067/23/2/701]
- 76. New surface contacts formed upon reductive lysine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2974831/]
- 77. Structure Determination from Lipidic Cubic Phase ... [https://www.sciencedirect.com/science/article/pii/S0969212620302392]
- 78. A hetero-micro-seeding strategy for readily crystallizing ... [https://www.sciencedirect.com/science/article/abs/pii/S0006291X17317357]
- 79. Viscoelastic characterization of the lipid cubic phase ... [https://www.nature.com/articles/s41598-025-25449-8]
- 80. Solid-state NMR of membrane peptides and proteins in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12256886/]
- 81. Standards for MicroED - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12231505/]
- 82. MolProbity: all-atom contacts and structure validation for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1933162/]
- 83. Ramachandran Plot - Proteopedia, life in 3D [https://proteopedia.org/wiki/index.php/Ramachandran_plots]
- 84. Structure Validation [http://binf.gmu.edu/nkengunt/struc_val.html]
- 85. A global Ramachandran score identifies protein structures ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7642142/]
- 86. MolProbity - An Active Validation Tool [https://www.phenix-online.org/documentation/reference/molprobity_tool.html]
- 87. Assessing the Quality of 3D Structures [https://www.rcsb.org/docs/general-help/assessing-the-quality-of-3d-structures]
- 88. The Quality and Validation of Structures from Structural Genomics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4081469/]
- 89. Overall protein structure quality assessment using hydrogen-bonding parameters - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10394671/]
- 90. User Guide - Data Collection and Processing [https://smb.slac.stanford.edu/users_guide/manual/Data_collection_processing.html]
- 91. Comparison Of Structural Techniques: X-ray, NMR, Cryo-EM [https://peakproteins.com/a-comparison-of-the-structural-techniques-used-at-sygnature-discovery-x-ray-crystallography-nmr-and-cryo-em/]
- 92. The powerful imaging technique revealing a hidden world ... [https://www.nature.com/articles/d42473-025-00158-y]
- 93. Microscopic Magnificence [https://www.utsouthwestern.edu/ctplus/stories/2025/cryo-em-research.html]
- 94. X-ray Crystallography vs Cryo-EM: Which is Best for Your ... [https://www.creative-biostructure.com/resource-cryo-em-vs-xray-crystallography-comparison.htm?srsltid=AfmBOopp-XGeZT9wc5broDrrtUwNb9jui5BCmj5_ILNV2sPLT76NK9lV]
- 95. High-resolution protein modeling through Cryo-EM and AI ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2025.1688455/full]
- 96. Cryo Electron Microscopy: Principle, Strengths, Limitations ... [https://www.technologynetworks.com/analysis/articles/cryo-electron-microscopy-principle-strengths-limitations-and-applications-377080]
- 97. Cryo-EM vs. X-Ray: Which Structural Method Is Best? [https://shuimubio.com/blogs/cryo-em-vs-x-ray-which-structural-method-is-best]
- 98. X-Ray Crystal and Cryo-Electron Microscopy Structure ... [https://www.mdpi.com/2077-0375/15/5/143]
- 99. Systematic Comparison of Crystal and NMR Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3032220/]
- 100. X-ray Crystallography [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Supplemental_Modules_(Analytical_Chemistry)/Instrumentation_and_Analysis/Diffraction_Scattering_Techniques/X-ray_Crystallography]
- 101. Comparison of X-ray Crystallography, NMR and EM [https://www.creative-biostructure.com/comparison-of-crystallography-nmr-and-em_6.htm?srsltid=AfmBOoqJKiQzjMNDcL-fTl6KgxvJyLDDqqbuOpzcguzr6GQwlxmZ42X0]
- 102. X-ray Crystallography [https://wiki.anton-paar.com/us-en/x-ray-crystallography/]
- 103. Nuclear magnetic resonance spectroscopy of proteins [https://en.wikipedia.org/wiki/Nuclear_magnetic_resonance_spectroscopy_of_proteins]
- 104. X-Ray Crystallography vs. NMR Spectroscopy [https://www.news-medical.net/life-sciences/X-Ray-Crystallography-vs-NMR-Spectroscopy.aspx]
- 105. Protein structure determination in solution by NMR ... [https://pubmed.ncbi.nlm.nih.gov/2266107/]
- 106. Hybrid methods for combined experimental and computational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7773420/]
- 107. Protein structure determination techniques : X - ray ... | Penprofile [https://penprofile.com/protein-structure-determination-techniques-x-ray-crystallography-vs-cryo-electron-microscopy/]
- 108. AlphaFold predictions are valuable hypotheses and ... [https://www.nature.com/articles/s41592-023-02087-4]
- 109. Multimodal deep learning integration of cryo-EM and ... [https://www.nature.com/articles/s42004-025-01718-5]
- 110. Cryo-EM ligand building using AlphaFold3-like model and ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1013367]
- 111. 2025 User Meeting Workshops & Tutorials [https://als.lbl.gov/2025-user-meeting-workshops-tutorials/]