Unlocking Precision: A Guide to Droplet Digital PCR for Rare Allele Detection in Biomedical Research
Droplet Digital PCR (ddPCR) is a third-generation PCR technology that enables absolute quantification of nucleic acids with exceptional sensitivity, making it uniquely suited for detecting rare allele variants.
Unlocking Precision: A Guide to Droplet Digital PCR for Rare Allele Detection in Biomedical Research
Abstract
Droplet Digital PCR (ddPCR) is a third-generation PCR technology that enables absolute quantification of nucleic acids with exceptional sensitivity, making it uniquely suited for detecting rare allele variants. This article provides a comprehensive overview for researchers and drug development professionals, covering the foundational principles of ddPCR partitioning and Poisson statistics. It explores methodological workflows for applications in oncology, infectious disease, and blood screening, and offers practical troubleshooting guidance for complex samples. Finally, it presents validation strategies and comparative performance data against qPCR and NGS, highlighting ddPCR's superior sensitivity for low-frequency mutations, its tolerance to inhibitors, and its critical role in advancing liquid biopsy and non-invasive diagnostics.
The Digital Revolution: Core Principles of ddPCR and Why It Excels at Rare Event Detection
The accurate quantification of nucleic acids is a cornerstone of modern molecular research, driving discoveries in fields from oncology to infectious disease monitoring. For decades, quantitative PCR (qPCR) has served as the established workhorse for nucleic acid detection, providing relative quantification based on standard curves. However, the emergence of droplet digital PCR (ddPCR) represents a fundamental shift in quantification technology, offering unprecedented precision through absolute target molecule counting at the single-molecule level [1] [2]. This transition from analog to digital quantification is particularly transformative for applications requiring the detection of rare genetic events, such as somatic mutations in liquid biopsies or minimal residual disease in oncology patients [3] [2].
The core distinction between these technologies lies in their fundamental approach to quantification. While qPCR relies on monitoring amplification kinetics during PCR cycles relative to known standards, ddPCR employs a partitioning strategy that physically separates individual template molecules into thousands of nanoliter-sized droplets, effectively converting an analog measurement problem into a digital counting process [1] [4]. This methodological shift provides significant advantages in sensitivity, precision, and resistance to inhibitors, making ddPCR particularly suitable for detecting rare alleles present at frequencies below 0.1% in a wild-type background [3]. This technical guide explores the principles, applications, and practical implementation of ddPCR technology, with a specific focus on its transformative role in rare allele detection research.
Technological Comparison: qPCR vs. ddPCR
Fundamental Principles and Mechanisms
Quantitative PCR (qPCR) operates on the principle of monitoring DNA amplification in real-time using fluorescent chemistry. The cycle threshold (Ct), at which the fluorescence signal crosses a predetermined threshold, is proportional to the starting quantity of the target nucleic acid [5] [6]. This relative quantification requires construction of standard curves from samples with known concentrations, introducing potential variability and standardization challenges across experiments and laboratories. A significant limitation of qPCR is its susceptibility to amplification efficiency variations, where even a 5% difference in efficiency can result in greater than 2-fold difference in results after 30 cycles [6]. Furthermore, the presence of PCR inhibitors in complex biological samples can significantly affect Ct values, compromising quantification accuracy.
In contrast, droplet digital PCR (ddPCR) utilizes a water-oil emulsion droplet system to partition a single PCR reaction into approximately 20,000 nanoliter-sized droplets [1]. This massive partitioning statistically distributes target molecules such that each droplet contains zero, one, or a few template molecules. Following end-point PCR amplification, each droplet is analyzed individually for fluorescence using a droplet reader [1]. The ratio of positive to negative droplets, analyzed through Poisson statistics, enables absolute quantification of the target nucleic acid without the need for standard curves [1] [2]. This partitioning approach makes ddPCR less susceptible to PCR inhibitors and amplification efficiency variations, as the endpoint detection focuses on the presence or absence of amplification rather than its kinetics [6].
Comparative Performance Characteristics
dPCR demonstrates superior accuracy, particularly for high viral loads of influenza A, influenza B, and SARS-CoV-2, according to a 2025 study comparing both technologies during the 2023-2024 tripledemic [7]. The study found dPCR showed greater consistency and precision than Real-Time RT-PCR, especially in quantifying intermediate viral levels [7].
Table 1: Performance Characteristics of qPCR vs. ddPCR
| Parameter | qPCR | ddPCR |
|---|---|---|
| Quantification Method | Relative (requires standard curve) | Absolute (no standard curve) |
| Dynamic Range | 6-8 orders of magnitude [6] | ~4 orders of magnitude [6] |
| Precision & Sensitivity | Detects differences ≥20% between samples [6] | Detects differences as low as 10% between samples [6] |
| Tolerance to Inhibitors | Moderate to low susceptibility | High resistance [6] |
| Throughput | High (384-well formats) [6] | Moderate (16-96 sample formats) [6] |
| Cost Considerations | Lower reagent costs, interchangeable master mixes [6] | Higher costs, proprietary master mixes [6] |
| Rare Mutation Detection | Limited sensitivity for variants <1% [3] | High sensitivity (detection down to 0.1%) [3] |
The reduced dynamic range of ddPCR (approximately 4 orders of magnitude) compared to qPCR (6-8 orders of magnitude) represents a notable limitation for applications requiring quantification across extremely variable concentration ranges [6]. However, for most rare allele detection applications where target concentrations fall within a more limited range, this constraint rarely presents practical limitations. The higher sensitivity of ddPCR enables detection of rare sequences present at frequencies as low as 0.1% in a wild-type background, a critical capability for liquid biopsy applications in oncology [3] [2].
Core Principles of Droplet Digital PCR
Partitioning and Poisson Statistics
The foundational principle of ddPCR technology is massive partitioning, where each sample is divided into thousands to millions of discrete reactions. In droplet-based systems, this is achieved through microfluidic technology that generates a water-in-oil emulsion, typically creating approximately 20,000 nanoliter-sized droplets per sample [1]. The random distribution of target DNA molecules into these partitions follows a Poisson distribution, which accounts for the probability that any individual partition will contain zero, one, or multiple target molecules [1] [2].
The application of Poisson statistics is essential for accurate quantification in ddPCR. The formula for calculating the initial target concentration is:
[ \text{Target Concentration (copies/μL)} = \frac{-\ln(1 - p)}{V} ]
Where "p" represents the proportion of positive partitions, and "V" is the volume of each partition [1]. This mathematical approach enables absolute quantification without external standards, as the concentration is directly calculated from the binary readout of positive versus negative partitions [1] [2]. The accuracy of this quantification depends on having sufficient partitions to ensure statistical robustness, with higher partition counts improving detection limits and precision for rare targets [3].
Workflow and Instrumentation
The ddPCR workflow consists of several standardized steps that distinguish it from traditional qPCR approaches. First, the sample is prepared with primers, probes, and a specialized master mix designed for droplet formation. This mixture is then loaded into a droplet generator that uses microfluidics to create the water-in-oil emulsion [1]. After droplet generation, the sample undergoes standard PCR amplification to endpoint (typically 40 cycles) in a thermal cycler [1]. Following amplification, droplets are transferred to a reader that flows them in a single file through a detection system, classifying each droplet as positive or negative based on fluorescence thresholds [1]. Finally, specialized software applies Poisson statistics to calculate the absolute target concentration based on the ratio of positive to negative droplets [1].
ddPCR Workflow: From Sample to Results
Commercial ddPCR platforms have evolved significantly, offering researchers various options with different partitioning technologies and capabilities. The QX200 Droplet Digital PCR System from Bio-Rad utilizes droplet-based partitioning with an in-line detection system [8] [9]. The QIAcuity from QIAGEN employs a nanoplate-based system with planar imaging for signal detection [7] [8]. The Naica System from Stilla Technologies uses a crystal digital PCR approach with endpoint imaging [3] [9]. Recent comparative studies indicate that while these platforms differ in their partitioning mechanisms (droplets vs. nanowells) and detection methods, they demonstrate comparable performance in sensitivity and precision for most applications [8] [9].
Table 2: Comparison of Commercial Digital PCR Platforms
| Platform | Partitioning Technology | Detection Method | Typical Partitions | Throughput | Key Applications Cited |
|---|---|---|---|---|---|
| QX200 (Bio-Rad) | Droplet-based | In-line detection | ~20,000 droplets [1] | 96 samples [6] | Viral quantification, environmental monitoring [8] [9] |
| QIAcuity (QIAGEN) | Nanoplate-based | Planar imaging | 26,000 nanowells [7] | 96 samples [7] | Respiratory virus detection, copy number variation [7] [8] |
| Naica (Stilla Technologies) | Droplet-based | Endpoint imaging | ~30,000 droplets [3] | 16-96 samples [3] | Rare mutation detection, liquid biopsy [3] |
Experimental Design for Rare Allele Detection
Assay Design Considerations
The detection of rare alleles presents unique challenges that require careful experimental design. For rare mutation detection, the recommended approach uses two different hydrolysis probes (TaqMan chemistry) with a single set of primers that amplify the region of interest [3]. One probe targets the wild-type sequence, while the other targets the mutant allele, with each labeled with distinct fluorophores that must be compatible with the detection system's excitation and emission spectra [3]. This duplex approach enables simultaneous quantification of both wild-type and mutant sequences in the same reaction, improving accuracy by controlling for sample-to-sample variation.
Critical to successful rare allele detection is DNA input optimization, which directly determines assay sensitivity. The required DNA input can be calculated using the formula:
[ \text{Number of copies} = \frac{\text{mass of DNA (ng)}}{\text{haploid genome mass (ng)}} ]
For human genomic DNA, the haploid genome mass is approximately 3 pg (0.003 ng) [3]. The theoretical limit of detection with 95% confidence is approximately 0.2 copies/μL for systems like the Naica platform [3]. To calculate the minimum detectable mutant allele fraction:
[ \text{Sensitivity} = \frac{\text{Theoretical LOD}}{\text{Total target concentration}} ]
For example, with 10 ng of human genomic DNA input (approximately 3,333 genome copies) and a system LOD of 0.2 copies/μL, the theoretical detection sensitivity would be approximately 0.15% for the mutant allele [3]. This calculation highlights the importance of sufficient DNA input for detecting extremely rare variants.
Optimization and Validation Strategies
Fluorescence spillover compensation represents a critical optimization step in multiplexed ddPCR assays. When using multiple fluorophores, cross-talk between detection channels can create aberrant clusters that compromise accurate classification [3]. Establishing a compensation matrix requires running monocolor controls (samples containing only a single fluorophore) to quantify and correct for this spillover [3]. Proper compensation ensures clear separation between positive and negative populations, which is essential for accurate quantification, particularly for rare mutants present at very low frequencies.
Restriction enzyme selection can significantly impact assay precision, particularly for targets with potential secondary structure or tandem repeats. A 2025 study comparing ddPCR platforms found that choice of restriction enzyme affected precision, with HaeIII demonstrating superior performance compared to EcoRI, particularly for the QX200 system [8]. For the QIAcuity system, restriction enzyme choice had less impact on overall precision [8]. These findings highlight the importance of enzyme selection during assay development, particularly for challenging targets.
Validation of rare allele detection assays should include several quality control measures. Non-template controls (NTCs) must show minimal positive partitions to rule out contamination [3]. The total number of analyzable partitions should be maximized, as higher partition counts improve the statistical power for detecting rare events and reduce uncertainty in concentration measurements [3]. For the Naica system, typical analyzable partitions range between 19,000-22,000 [3]. Sample-specific factors such as DNA quality and the presence of inhibitors should also be assessed, though ddPCR demonstrates greater resilience to inhibitors compared to qPCR [6].
Research Reagent Solutions for ddPCR
Successful implementation of ddPCR workflows requires specific reagents optimized for partitioning and amplification. The following table outlines essential materials and their functions for rare allele detection experiments.
Table 3: Essential Research Reagents for ddPCR Rare Allele Detection
| Reagent/Material | Function | Specifications | Application Notes |
|---|---|---|---|
| ddPCR Master Mix | Provides essential components for amplification | Proprietary formulations specific to each platform [6] | Contains DNA polymerase, dNTPs, buffer; optimized for droplet stability |
| Hydrolysis Probes | Sequence-specific detection | FAM-labeled for wild-type, Cy3-labeled for mutant [3] | Must be compatible with instrument excitation/emission spectra |
| Primer Sets | Target sequence amplification | ~20 bp oligonucleotides, optimized concentrations [3] | Typically used at 500 nM final concentration [3] |
| Restriction Enzymes | Enhance target accessibility | HaeIII or EcoRI depending on application [8] | Improves precision, especially for tandem repeats [8] |
| Reference Dye | Normalization control | ROX/Atto590, instrument-specific [3] | Quality control for partition identification |
| Droplet Generation Oil | Creates stable water-in-oil emulsion | Surfactant-stabilized, thermostable [2] | Prevents droplet coalescence during thermal cycling |
| Digital PCR Plates/Cartridges | Partitioning vessels | Platform-specific (96-well, microfluidic chips) [3] | Varies by system: droplets vs. nanowells |
Application in Rare Allele Detection: Case Study
EGFR T790M Mutation Detection in NSCLC
A compelling application of ddPCR for rare allele detection is the identification of the EGFR T790M mutation in patients with non-small cell lung cancer (NSCLC). This mutation confers resistance to first- and second-generation tyrosine kinase inhibitors (TKIs) and often emerges during treatment [3]. Early detection of T790M is clinically significant as it directs therapeutic strategy toward third-generation TKIs [3]. The mutation is rarely detected during initial tumor characterization but becomes detectable during treatment, typically at low variant allele frequencies that challenge conventional qPCR methods.
The experimental protocol for EGFR T790M detection utilizes a single primer set amplifying the EGFR T790 locus, combined with two hydrolysis probes: a FAM-labeled probe targeting the wild-type sequence and a Cy3-labeled probe targeting the T790M mutation [3]. The PCR mix preparation follows a specific formulation with final primer concentrations of 500 nM and probe concentrations of 250 nM each [3]. Thermal cycling conditions typically involve an initial denaturation at 95°C for 10 minutes, followed by 45 cycles of 95°C for 30 seconds and 62°C for 15 seconds [3]. This optimized protocol enables specific detection of the T790M mutation at allele frequencies as low as 0.15% with 95% confidence using 10 ng of input DNA [3].
Rare Allele Detection Principle
Data Analysis and Interpretation
In ddPCR rare mutation analysis, results are typically visualized using 2D scatter plots showing fluorescence amplitudes for each probe channel. Distinct clusters emerge representing wild-type-only droplets (FAM-positive), mutant-only droplets (Cy3-positive), double-negative droplets, and occasionally double-positive droplets that may indicate heterozygotes or technical artifacts [3]. For rare mutant detection, the critical cluster is the mutant-only population, which should be clearly separated from the wild-type population and background noise.
The concentration of mutant alleles is calculated using Poisson statistics based on the number of mutant-positive partitions relative to the total number of partitions. The confidence in detection is influenced by both the number of mutant-positive partitions and the total number of partitions analyzed [3]. With systems generating over 20,000 partitions, detection limits below 0.1% variant allele frequency are achievable with appropriate DNA input [3]. This sensitivity surpasses conventional qPCR, which typically detects variants down to only 1-5% allele frequency, making ddPCR particularly valuable for monitoring emerging resistance mutations during cancer treatment.
The field of digital PCR continues to evolve with several emerging trends shaping its future applications. Increased automation represents a significant direction, with fully automatic ddPCR systems projected to reach a market value of $1,500 million by 2025, growing at a CAGR of 18.5% through 2033 [10]. This automation trend addresses current limitations in throughput and hands-on time, making the technology more accessible for clinical laboratories. Multiplexing capabilities are also expanding, with developments in three-color detection systems and advanced chemistries enabling simultaneous quantification of multiple targets [3] [10]. These advancements enhance the efficiency and information content of ddPCR experiments, particularly for complex applications like comprehensive mutation profiling.
The transition from qPCR to ddPCR represents more than a simple technological upgrade—it constitutes a fundamental shift in quantification philosophy. While qPCR will maintain its role as a high-throughput workhorse for applications where relative quantification suffices, ddPCR establishes a new standard for precision bioanalysis where absolute quantification and rare variant detection are paramount [6]. The technology's demonstrated superiority in detecting rare mutations, quantifying viral loads with precision, and analyzing complex samples positions it as an essential tool for advancing personalized medicine, liquid biopsy applications, and minimal residual disease monitoring [3] [2].
For research and drug development professionals, the decision to implement ddPCR workflows should be guided by specific application requirements. When the research question demands absolute quantification, exceptional sensitivity for rare variants, or analysis of complex samples with potential inhibitors, ddPCR offers compelling advantages that justify its implementation [6]. As the technology continues to mature with improvements in automation, multiplexing, and cost-effectiveness, its integration into research and clinical workflows will undoubtedly expand, further solidifying its role as a transformative technology in molecular analysis.
Droplet Digital PCR (ddPCR) represents a paradigm shift in molecular quantification, enabling unparalleled precision in rare allele detection and absolute nucleic acid quantification. By partitioning a sample into thousands of nano-droplets, this technology transforms the limitations of traditional bulk-phase PCR, allowing for single-molecule sensitivity. This technical guide explores the core principles of ddPCR, detailing how statistical Poisson distribution analysis of end-point PCR amplification across numerous partitions facilitates absolute target quantification without standard curves. Within the context of rare allele research, we examine how this partitioning power provides the foundation for detecting mutant frequencies as low as 0.1% and copy number variations with 95% concordance to gold-standard methods. We present comprehensive experimental protocols, quantitative validation data, and specialized reagent solutions that empower researchers to leverage this technology for advanced molecular diagnostics and drug development applications.
The fundamental principle underlying droplet digital PCR (ddPCR) is sample partitioning—a process that divides a PCR reaction into thousands to millions of discrete, nanoliter-volume water-in-oil droplets [11] [12]. This physical segregation of template molecules serves as the critical innovation that enables single-molecule detection by transforming a quantitative analog measurement into a simple digital counting exercise. Each droplet functions as an independent micro-reactor where PCR amplification occurs in isolation. Following thermocycling, droplets are analyzed for fluorescence, and the fraction of positive droplets is used to calculate the absolute initial copy number of the target nucleic acid sequence using Poisson statistics [13].
This partitioning approach directly addresses a significant limitation of traditional quantitative PCR (qPCR), where the quantification of rare alleles or precise copy number variations (CNVs) becomes increasingly unreliable at high copy numbers due to compounding errors from PCR inefficiency and pipetting variations [11]. In contrast, ddPCR provides absolute quantification without requiring a standard curve, demonstrating particular strength in applications requiring high precision, such as detecting low-abundance mutations in circulating free DNA (cfDNA) or resolving complex CNV patterns [11] [14]. The statistical power of ddPCR emerges from the large number of partitions created; modern systems routinely generate over 20,000 droplets per sample, providing thousands of data points that increase measurement accuracy and enable detection of rare variants present at frequencies as low as 0.1% [11] [14].
Core Principles of Single-Molecule Detection
The Partitioning Principle and Poisson Statistics
The analytical power of ddPCR stems from the combination of sample partitioning and Poisson distribution statistics. When a sample is partitioned into thousands of nanodroplets, template molecules are randomly distributed across these compartments. According to Poisson statistics, the probability of any droplet containing k template molecules is given by P(k) = (λ^k e^(-λ))/k!, where λ represents the average number of target molecules per droplet [12]. Critically, for single-molecule detection to be possible, most positive droplets must contain exactly one template molecule (k=1), which requires that λ is sufficiently low (typically λ < 0.3) to ensure that multiple templates in a single droplet are rare [12].
The fraction of negative droplets (P(0) = e^(-λ)) provides the most direct path to calculating the target concentration. By measuring the proportion of droplets without amplification, the initial template concentration in the original sample can be precisely determined using the equation: concentration = -ln(1 - p) × (total droplets/reaction volume), where p represents the positive droplet fraction [11]. This mathematical foundation allows ddPCR to achieve absolute quantification without standard curves, a significant advantage over qPCR methods that rely on relative quantification based on amplification curves [11].
Overcoming Background and Specificity Challenges
A significant challenge in single-molecule detection is distinguishing true positive signals from nonspecific amplification, which becomes particularly critical when detecting rare variants in a high background of wild-type sequences. Traditional digital PCR processes require several hours to complete, during which nonspecific amplification can accumulate and generate false-positive signals [12]. Recent advancements in ultrafast ddPCR have demonstrated that reducing PCR annealing time from minutes to seconds significantly mitigates nonspecific amplification [12]. This approach, implemented through flexible thin tube-based systems with high heat conduction efficiency, can complete the entire digital PCR process in just 5 minutes instead of 2 hours, virtually eliminating background signals in negative controls and enabling true single-molecule detection [12].
The specificity of ddPCR can be further enhanced through the use of allele-specific fluorescent oligonucleotide probes containing locked nucleic acids (LNA), which improve discrimination between wild-type and mutant sequences by increasing the thermal stability difference between perfectly matched and mismatched hybrids [13]. This approach enables detection sensitivity of up to 1 in 50,000 DNA copies, making it particularly valuable for identifying rare cancer mutations in liquid biopsies or monitoring minimal residual disease [13].
Quantitative Data and Performance Metrics
Copy Number Variation Analysis
Digital PCR provides exceptional accuracy in copy number variation (CNV) determination, as demonstrated by a 2025 study comparing ddPCR to pulsed field gel electrophoresis (PFGE), considered a gold standard in CNV identification [11]. The research focused on the human alpha defensin 1-3 (DEFA1A3) gene, a multiallelic gene with copy numbers ranging from 2 to 12 copies per diploid genome [11].
Table 1: Comparison of CNV Enumeration Methods for DEFA1A3 Gene
| Method | Concordance with PFGE | Spearman Correlation | Average Difference from PFGE | Key Limitations |
|---|---|---|---|---|
| ddPCR | 95% (38/40 samples) | r = 0.90 (p < 0.0001) | 5% | Minimal limitations observed |
| qPCR | 60% (24/40 samples) | r = 0.57 (p < 0.0001) | 22% | Underestimation at high copy numbers; compound error propagation |
| PFGE | Gold Standard | N/A | N/A | Low-throughput; time-intensive; requires special equipment |
The regression equation for ddPCR versus PFGE resulted in Y = 0.9953 × (95% CI [0.9607,1.030]), demonstrating nearly perfect 1:1 agreement between the methods [11]. In contrast, qPCR showed systematic underestimation with a regression equation of Y = 0.8889 × (95% CI [0.8114,0.9664]) [11]. This superior performance establishes ddPCR as a low-cost, high-throughput alternative for clinical CNV testing with accurate resolution across both low and high copy number ranges [11].
Rare Variant Detection Sensitivity
The partitioning power of ddPCR enables exceptional sensitivity in rare variant detection, as demonstrated in studies analyzing mutant alleles in urinary cfDNA and genomic DNA.
Table 2: Rare Variant Detection Performance in ddPCR Applications
| Application | Variant Target | Detection Sensitivity | Key Performance Metrics |
|---|---|---|---|
| Urinary cfDNA Analysis | NRAS (A59T) and EGFR (L858R) variants | 0.1% allelic frequency | Concordance between observed and expected frequencies across all tested allelic frequencies (5%, 1%, 0.1%) [14] |
| Genomic DNA Mutation Detection | Various rare mutations | 1 in 50,000 DNA copies | Precise quantitation using allele-specific LNA probes [13] |
| Ultrafast ddPCR (utPCR) | E. coli O157 in high background of E. coli K12 | Single-molecule detection | 10 CFU/mL blood detection limit; 5-minute workflow; elimination of nonspecific amplification [12] |
The precision of rare allele detection is further enhanced by recent advances in interpretable ddPCR assays that combine specialized neural networks for droplet segmentation and classification with multimodal large language models for context-aware explanations. This I2ddPCR framework achieves 99.05% accuracy in processing complex ddPCR images and can detect low-abundance targets as low as 90.32 copies/μL [15].
Experimental Protocols and Methodologies
Core ddPCR Workflow for Rare Allele Detection
The following workflow describes a standardized approach for rare variant detection using droplet digital PCR, incorporating best practices from recent applications.
Sample Preparation and cfDNA Extraction
- Urine Sample Preservation: Collect urine samples and preserve immediately using Colli-Pee UAS preservative at a 2.3:1 urine-to-preservative ratio [14].
- DNA Extraction: Extract cfDNA using magnetic bead-based kits (e.g., Mag-Bind cfDNA Kit). Input 4 mL of preserved urine and elute in 50 μL volume [14].
- Quality Assessment: Evaluate cfDNA yield and quality using the Cell-Free DNA ScreenTape Assay on the Agilent 4150 TapeStation system. Ideal samples show dark bands concentrated between 150-200 base pairs with minimal genomic DNA contamination [14].
Droplet Digital PCR Setup
- Reaction Composition: Combine 15 ng of cfDNA with ddPCR Supermix for Probes (No dUTP). Add allele-specific FAM-labeled probes for mutant alleles and SUN-labeled probes for wild-type sequences [14].
- Droplet Generation: Assemble reaction mixtures in a total volume of 300 μL and generate droplets using a QX200 Droplet Generator according to manufacturer's instructions [14].
- Thermal Cycling: Amplify using a C1000 Touch Thermal Cycler with optimized annealing temperatures for each probe set. Standard protocols typically involve 40 cycles of amplification [14].
Droplet Reading and Data Analysis
- Droplet Reading: Process droplets using a QX200 Droplet Reader [14].
- Threshold Setting: Manually set fluorescence thresholds based on 0% allelic frequency (100% wildtype) controls to ensure clear separation between positive and negative populations [14].
- Analysis: Use analysis software (e.g., QuantaSoft Analysis Pro) to determine mutant and wild-type counts and calculate variant allele frequencies [14].
Advanced Ultrafast ddPCR (utPCR) Protocol
For applications requiring maximum speed and elimination of nonspecific amplification, the utPCR protocol offers significant advantages:
Microfluidic Device Fabrication
- Design: Create microfluidic channel architectures using AutoCAD [12].
- Fabrication: Use soft lithographic techniques with SU-8 reagents on silicon wafers to create structured microfluidic substrates [12].
- Assembly: Bond polydimethylsiloxane (PDMS) casts to clean glass slides through plasma bonding [12].
- Hydrophobic Treatment: Treat microfluidic channels with aquapel and dry with nitrogen gas to render them hydrophobic [12].
Droplet Generation and Ultrafast Amplification
- Droplet Generation: Introduce oil phase (HFE-7500 with 5% w/w surfactant) and water phase into the PDMS device at constant flow rates (e.g., 800 μL/h oil, 400 μL/h water) [12].
- Droplet Collection: Collect formed droplets at the outlet [12].
- Ultrafast PCR: Transfer droplets into a flexible thin tube (0.4 mm inner diameter, polypropylene material) for ultrafast thermal cycling [12].
- Thermal Parameters: Reduce primer annealing/extension time to 5 seconds, completing the entire digital PCR process in 5 minutes instead of 2 hours [12].
Detection and Analysis
- Detection: Observe droplets under a confocal microscope or read with a droplet reader [12].
- Single-Molecule Confirmation: Verify complete absence of fluorescence in negative control droplets, indicating elimination of nonspecific amplification [12].
Essential Research Reagent Solutions
Successful implementation of ddPCR for single-molecule detection requires specific reagent systems optimized for partitioning and detection.
Table 3: Essential Research Reagent Solutions for ddPCR
| Reagent Category | Specific Product Examples | Function and Application |
|---|---|---|
| Sample Preservation | Colli-Pee UAS preservative [14] | Preserves urinary cfDNA integrity post-collection; maintains sample quality for rare variant detection |
| cfDNA Extraction | Mag-Bind cfDNA Kit (M3298) [14] | Maximizes cfDNA yield from urine samples with minimal genomic DNA contamination |
| ddPCR Master Mix | ddPCR Supermix for Probes (No dUTP) [14] | Provides optimal reaction environment for partitioned amplification; compatible with probe-based detection |
| Probe Chemistry | TaqMan-style allele-specific probes with LNA modifications [13] | Enhances discrimination between wild-type and mutant sequences; enables rare variant detection at 0.1% frequency |
| Droplet Generation Oil | HFE-7500 3M Novec engineered fluid with 5% surfactant [12] | Creates stable water-in-oil emulsions; enables formation of monodisperse droplets for partitioning |
| Reference Standards | Mimix Multiplex I cfDNA Set Reference Standard (HD780) [14] | Provides validated controls with known allelic frequencies (5%, 1%, 0.1%, 0%) for assay validation |
The partitioning power of droplet digital PCR through the creation of thousands of nano-droplets has fundamentally transformed single-molecule detection capabilities in molecular biology and diagnostic applications. By combining the physical separation of template molecules with precise Poisson statistical analysis, this technology enables absolute quantification of nucleic acids with unparalleled sensitivity and accuracy. The methodologies and data presented in this technical guide demonstrate how ddPCR outperforms traditional qPCR in both copy number variation analysis—achieving 95% concordance with gold-standard methods—and rare variant detection, reliably identifying mutant alleles at frequencies as low as 0.1%. As research continues to refine this technology through innovations such as ultrafast utPCR and AI-enhanced interpretable assays, the applications in cancer diagnostics, infectious disease monitoring, and genetic disorder screening will continue to expand. The experimental protocols and reagent solutions detailed herein provide researchers with a comprehensive toolkit for implementing this powerful technology in their own rare allele detection research, driving forward the capabilities of precision medicine and molecular diagnostics.
The Role of Poisson Statistics in Achieving Absolute Quantification
Digital PCR (dPCR) represents a transformative advancement in nucleic acid quantification, enabling absolute target measurement without standard curves. This technical guide explores the mathematical foundation of dPCR, focusing on the critical role of Poisson statistics in correcting for molecular distribution bias among partitions. We detail how this statistical framework facilitates rare allele detection at frequencies as low as 0.1% and provide comprehensive experimental protocols for implementing these principles in oncology research and drug development. The integration of microfluidic partitioning with Poisson modeling allows researchers to achieve unprecedented precision in quantifying genetic variants, making dPCR particularly valuable for liquid biopsy applications and minimal residual disease monitoring.
The Evolution of PCR Technologies
Digital PCR (dPCR) represents the third generation of PCR technology, following conventional PCR and real-time quantitative PCR (qPCR) [4]. Unlike its predecessors, dPCR enables absolute quantification of nucleic acid targets without requiring standard curves or reference materials [16]. This methodological advancement has proven particularly valuable for applications demanding high sensitivity and precision, including rare mutation detection, copy number variation analysis, and liquid biopsy applications in oncology [14] [17]. The core innovation of dPCR lies in its partitioning approach, where a single PCR reaction is divided into thousands to millions of parallel microreactions, allowing individual template molecules to be amplified in isolation [4].
The historical development of dPCR began with seminal work in 1992 when Morley and Sykes combined limiting dilution PCR with Poisson statistics to isolate, detect, and quantify single nucleic acid molecules [4]. This foundational approach was further refined in 1999 when Bert Vogelstein and colleagues formally coined the term "digital PCR" while developing methods to detect RAS oncogene mutations in patients with colorectal cancer [4]. The technology has since evolved through significant improvements in microfluidics and partitioning methodologies, leading to the commercial systems available today [16].
Fundamental Principles of dPCR
The digital PCR workflow consists of four key steps: (1) partitioning the PCR mixture containing the sample into thousands to millions of compartments; (2) amplifying individual target-containing partitions through endpoint PCR; (3) performing fluorescence analysis of the partitions; and (4) computing target concentration using Poisson statistics based on the fraction of positive and negative partitions [4]. This partitioning approach effectively dilutes the template molecules across many reaction chambers, with each partition ideally containing zero, one, or a few target molecules according to a Poisson distribution [4].
The binary nature of dPCR detection – where partitions are scored simply as positive or negative for amplification – provides the technique with its name and fundamental advantage [16]. This "digital" readout simplifies signal interpretation compared to analog measurement systems, as the instrument must only distinguish between two states rather than a full range of possibilities [16]. The resulting data robustness, combined with appropriate statistical modeling, enables dPCR to achieve precise absolute quantification even for rare targets present in complex biological backgrounds.
Poisson Statistics: The Mathematical Framework of dPCR
Theoretical Foundation
The Poisson distribution is a discrete probability distribution that expresses the probability of a given number of events occurring in a fixed interval of time or space if these events occur with a known constant mean rate and independently of the time since the last event [18]. In dPCR, this mathematical framework describes the random distribution of template molecules across partitions when the number of partitions greatly exceeds the number of target molecules [4] [16].
The Poisson model applies to dPCR because the partitioning process meets its core assumptions: (1) the probability of a molecule being in any partition is small and constant; (2) the presence of one molecule in a partition does not influence the presence of others; and (3) two molecules cannot occupy the same partition simultaneously [18]. When these conditions are met, the probability of a partition containing k template molecules follows the Poisson probability mass function:
Table 1: Core Variables in the Poisson Equation for dPCR
| Variable | Description | Application in dPCR |
|---|---|---|
| λ | Average number of target molecules per partition | Fundamental measurement unit in dPCR quantification |
| k | Actual number of target molecules in a specific partition | Not directly observed, statistically inferred |
| e | Base of the natural logarithm (~2.71828) | Mathematical constant |
| P(k) | Probability of a partition containing exactly k molecules | Determined from fraction of positive/negative partitions |
Practical Application in dPCR
In practice, dPCR quantification relies on measuring the proportion of partitions that contain no target molecules (k=0), as this value directly determines λ through the relationship:
P(k=0) = e^(-λ)
Rearranging this equation provides the fundamental calculation for dPCR quantification:
λ = -ln(1 - p)
Where p represents the ratio of positive partitions to the total number of partitions [19]. The copies per partition (λ) can then be converted to concentration (copies per microliter) based on the known partition volume [16]. This elegant mathematical relationship enables absolute quantification without external standards by simply counting positive and negative partitions [16] [19].
The following diagram illustrates the logical relationship between dPCR workflow and Poisson statistics:
The accuracy of Poisson-based quantification depends on several factors, including the number of partitions analyzed and the uniformity of partition volumes [20]. As partition count increases, the statistical power improves, particularly for rare variant detection where large partition numbers increase the likelihood of capturing low-frequency targets [3]. Modern dPCR systems typically generate 20,000 or more partitions, providing robust data for Poisson analysis [21] [19].
Experimental Protocols for Rare Allele Detection
Assay Design Considerations
Effective rare allele detection requires careful assay design to maximize specificity and sensitivity. The fundamental approach involves using two different hydrolysis probes (TaqMan) with a single set of primers that amplify the region of interest [3]. One probe targets the wild-type sequence while the other targets the mutant allele, with each labeled with distinct fluorophores (e.g., FAM and Cy3) that must be compatible with the dPCR system's detection capabilities [3].
For optimal performance, TaqMan PCR assays should amplify 60 to 150 bp fragments within the target region, with smaller products generally amplifying more efficiently [19]. Primers should be designed with a melting temperature (Tm) of approximately 60°C, while the internal hybridization probe should have a Tm 8-10°C higher than the primers [19]. To reduce secondary structure, avoid homopolymer runs of greater than 3 bases in the probe sequence, and avoid designing probes with a 5' guanine, which may partially quench fluorescence [19].
When detecting rare mutations in clinical samples, such as the EGFR T790M mutation in non-small cell lung cancer, the assay must distinguish mutant alleles present at frequencies below 1% against a background of wild-type sequences [3]. This requires not only well-designed probes but also appropriate DNA input calculations to ensure sufficient template is analyzed to detect the rare variant with statistical confidence.
DNA Preparation and Input Optimization
For rare variant detection, DNA quantity and quality critically impact assay sensitivity. The following calculation determines the minimum input required to detect a mutant allelic fraction with 95% confidence:
Number of copies in reaction volume = mass of DNA in reaction volume (in ng) / 0.003
The denominator (0.003) represents the approximate mass in nanograms of one haploid human genome (3 pg), applicable when working with human genomic DNA and detecting single-copy genes [3]. For example, with an input of 10ng of human genomic DNA in a 25μL reaction, the total copies of a single-copy gene would be 10/0.003 = 3,333 copies, or approximately 133 copies/μL [3].
The theoretical limit of detection (LOD) can then be calculated by dividing the system's theoretical LOD (e.g., 0.2 copies/μL) by the total target concentration. In this example, the sensitivity would be 0.2/133 = 0.15%, meaning the system could detect a mutated allelic fraction down to 0.15% with 95% confidence [3]. This calculation highlights the critical relationship between DNA input and rare variant detection capability.
Step-by-Step dPCR Protocol for EGFR T790M Detection
This protocol, optimized for the Naica System and Sapphire chip, demonstrates rare mutation detection in the context of non-small cell lung cancer research [3]:
Table 2: PCR Reaction Setup for EGFR T790M Detection
| Reagent | Final Concentration | Function |
|---|---|---|
| PCR Mastermix (2X or 5X) | 1X | Provides essential PCR components including DNA polymerase, dNTPs, and reaction buffer |
| Reference dye | As manufacturer's instructions | Internal control for partition identification |
| EGFR T790 Reverse and Forward primers | 500 nM | Amplify the target region containing the mutation site |
| EGFR T790WT probe | 250 nM | Detects wild-type EGFR sequence with specific fluorophore |
| EGFR T790M probe | 250 nM | Detects T790M mutant allele with distinct fluorophore |
| Human genomic DNA | Calculated based on desired sensitivity | Template containing wild-type and potential mutant sequences |
| Nuclease-free water | Volume adjust to 25 μL | Reaction volume adjustment |
PCR Mix Preparation: Assemble components in a clean area to prevent contamination. For n samples, prepare a master mix for n+1 reactions to account for pipetting error. Include monocolor controls for each probe to correct for fluorescence spillover [3].
Partitioning: Load the PCR mix into the appropriate dPCR system consumable according to manufacturer instructions. For droplet-based systems, this involves generating water-in-oil emulsion droplets; for chip-based systems, the sample is loaded into microchambers [3] [19].
Thermal Cycling: Amplify using the following conditions optimized for the EGFR T790M assay:
- Initial denaturation: 95°C for 10 minutes
- 45 cycles of:
- Denaturation: 95°C for 30 seconds
- Annealing/Extension: 62°C for 15 seconds [3]
Data Acquisition: Read partitions using appropriate instrumentation (imaging for chip-based systems, flow-based detection for droplet systems) [3].
Data Analysis: Apply fluorescence spillover compensation if necessary, then calculate mutant allele frequency using Poisson-corrected counts from positive partitions [3].
The following workflow diagram illustrates the complete dPCR process for rare allele detection:
Advanced Considerations in Poisson Modeling
Limitations of Standard Poisson Model
While the standard Poisson model provides robust quantification for most dPCR applications, its accuracy depends on the assumption of identical partition sizing [20]. In practice, partition volumes often vary, particularly in droplet-based dPCR systems where emulsion droplets may show size heterogeneity. This variation becomes particularly problematic at higher target concentrations, leading to underestimation of true target quantity when using standard Poisson modeling [20].
The impact of partition size variation increases with target concentration, as larger partitions are more likely to contain multiple target molecules while smaller partitions may contain none, distorting the relationship between positive partitions and actual target concentration [20]. Monte Carlo simulations have demonstrated that precision, defined as the absolute value of the maximum deviation from the true rate divided by the true rate, decreases significantly with increasing partition size variation, particularly at higher concentrations [20].
Poisson-Plus Model for Volume Variation
To address limitations of the standard Poisson model, the Poisson-Plus model incorporates partition volume variation into the quantification algorithm [20]. This advanced approach assumes that the mean number of molecules per partition (λ) is proportional to partition volume (v):
λ(v) = C·v
Where C represents the average number of molecules per unit volume (concentration) [20]. The model then integrates the probability distribution of partition volumes to derive a more accurate estimate of target concentration.
For a normally distributed partition volume with mean v₀ and standard deviation σ, the probability of a partition being negative is given by:
P(neg) = e^(½σ²C² - C·v₀)
This equation can be solved for C, providing a correction for partition volume variation [20]. The Poisson-Plus model demonstrates particular value in systems with significant partition size heterogeneity, enabling more accurate quantification across the entire dynamic range of dPCR applications [20].
Research Reagent Solutions for dPCR
Successful implementation of dPCR for rare allele detection requires specific reagents optimized for partitioning and amplification. The following table details essential materials and their functions:
Table 3: Essential Research Reagents for dPCR Rare Allele Detection
| Reagent/Category | Specific Examples | Function in dPCR Workflow |
|---|---|---|
| dPCR Mastermix | Bio-Rad's ddPCR Supermix for Probes | Provides optimized buffer, DNA polymerase, and dNTPs specifically formulated for stable droplet generation and efficient amplification [14] [19] |
| Hydrolysis Probes | TaqMan probes (FAM, HEX/VIC, Cy3 labeled) | Sequence-specific fluorescent probes that generate signal upon amplification; different fluorophores enable multiplex detection of wild-type and mutant alleles [3] [19] |
| Partitioning Consumables | DG8 droplet generator cartridges (Bio-Rad), microfluidic chips | Microfluidic devices that create uniform water-in-oil droplets or microchambers for sample partitioning [19] |
| Reference Dyes | ROX, Atto590 | Passive internal controls that identify partitions independent of amplification, correcting for partition volume variations [20] |
| Nucleic Acid Purification Kits | Mag-Bind cfDNA Kit | Specialized kits for extracting high-quality cell-free DNA from clinical samples like urine or plasma, maximizing yield and minimizing contamination [14] |
| Restriction Enzymes | AluI (4-cutter enzyme) | Digest genomic DNA to reduce viscosity and separate linked duplications that might affect partitioning efficiency [19] |
Poisson statistics provides the essential mathematical foundation that enables digital PCR to achieve absolute quantification of nucleic acid targets. By accounting for the random distribution of molecules across partitions, Poisson modeling transforms simple binary readouts (positive/negative partitions) into precise concentration measurements, particularly valuable for detecting rare genetic variants present at frequencies below 1%. As dPCR technology continues to evolve with improved partitioning methods and refined statistical models, its applications in rare allele detection—from liquid biopsies in oncology to pathogen detection and non-invasive prenatal testing—will continue to expand. The ongoing development of advanced modeling approaches, such as the Poisson-Plus model that accounts for partition volume variation, promises to further enhance the accuracy and reliability of this powerful quantification platform, solidifying its role as an indispensable tool in both basic research and clinical diagnostics.
Droplet Digital PCR (ddPCR) represents a paradigm shift in nucleic acid quantification, offering transformative capabilities for rare allele detection. This whitepaper details the core principles of ddPCR technology, highlighting its exceptional sensitivity, precision, and ability to provide absolute quantification without standard curves. We present quantitative performance data from recent studies, detailed experimental protocols for mutation detection, and essential reagent solutions to guide researchers in deploying this powerful technology for advanced research and drug development applications.
Droplet Digital PCR (ddPCR) is a third-generation PCR technology that enables absolute quantification of nucleic acid molecules with single-molecule sensitivity [2] [4]. The fundamental principle involves partitioning a PCR reaction into thousands to millions of nanoliter-sized water-in-oil droplets, effectively creating a massive array of independent reactions [2]. Following end-point PCR amplification, each droplet is analyzed individually to determine if it contains the target sequence (positive) or not (negative). The absolute concentration of the target nucleic acid is then calculated using Poisson statistics based on the ratio of positive to negative droplets, eliminating the requirement for standard curves [4] [19].
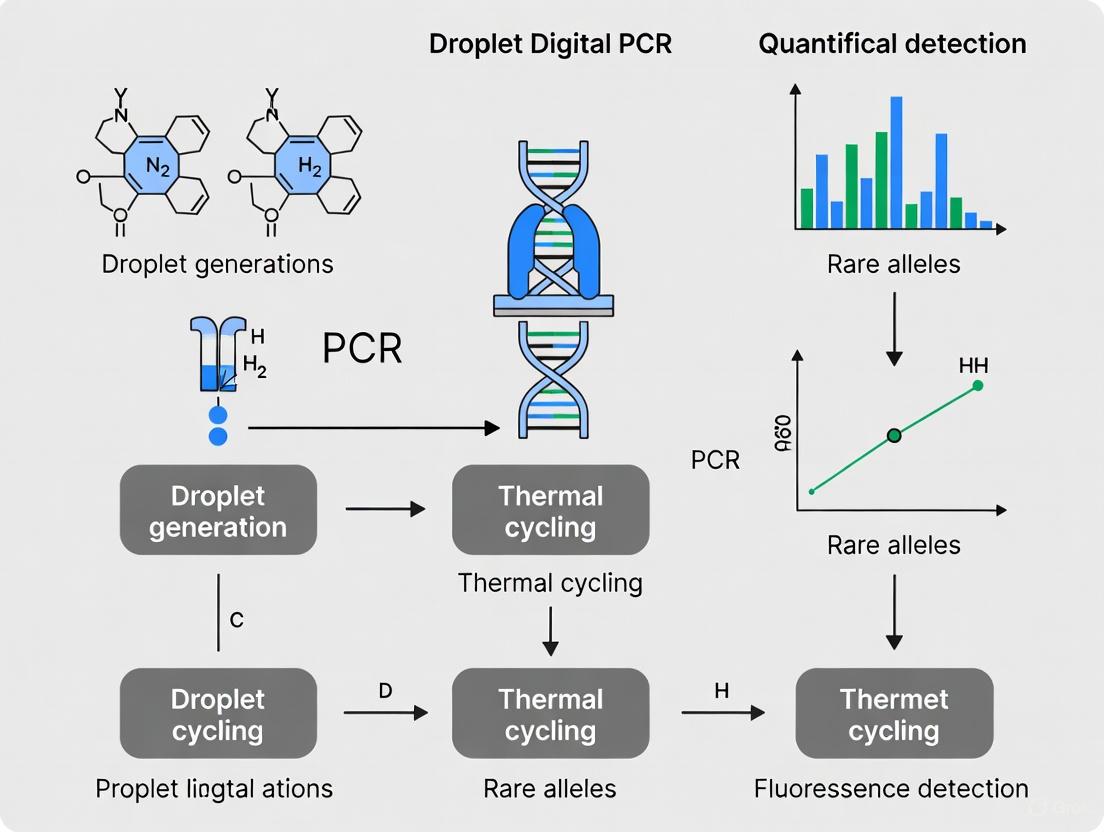
The ddPCR workflow consists of four critical stages: (1) partitioning the sample-reactio mixture into a monodisperse droplet emulsion; (2) performing PCR amplification on the droplet array; (3) analyzing each droplet's fluorescence; and (4) applying Poisson statistics to calculate absolute target concentration [2]. This partitioning approach provides ddPCR with significant advantages over quantitative PCR (qPCR), particularly for applications requiring detection of rare mutations, precise quantification of small fold-changes, or analysis of complex samples where PCR inhibitors may be present [22] [14].
Diagram 1: Core ddPCR workflow.
Unmatched Sensitivity in Rare Allele Detection
The partitioning technology underlying ddPCR enables exceptional sensitivity for detecting rare mutations against a background of wild-type sequences. This sensitivity makes it particularly valuable for liquid biopsy applications, monitoring minimal residual disease, and detecting emerging treatment-resistant clones in oncology [2] [23].
Quantitative Performance Data
Table 1: Sensitivity comparison between ddPCR and qPCR for low-abundance targets
| Application Context | Target Detected | qPCR Performance | ddPCR Performance | Reference |
|---|---|---|---|---|
| Hepatitis D Viral Load | HDV RNA | LOD: 14 IU/mL (clinical assay) | LOD: 0.56 IU/mL (10 copies/mL) | [22] |
| HDV Clinical Samples (Negative by qPCR) | HDV RNA | 0% detected (definition) | 31% positive by ddPCR | [22] |
| Urinary cfDNA Mutation Detection | NRAS & EGFR variants | Not reported | Reliable detection at 0.1% allelic frequency | [14] |
| Early dPCR Foundation | IgH mutated gene | Not applicable | 2 targets in 160,000 wild-type sequences | [2] [4] |
Experimental Protocol: Rare Variant Detection in Urinary cfDNA
A recent study demonstrated a complete workflow for detecting rare oncogenic mutations in urinary circulating free DNA (cfDNA) using ddPCR [14]:
Sample Preparation and Preservation
- Collect urine samples using Colli-Pee UAS preservative at a 2.3:1 urine-to-preservative ratio
- Spike with reference standards (e.g., Mimix Multiplex I cfDNA Set) at varying allelic frequencies (5%, 1%, 0.1%, 0% wildtype)
- Centrifuge at 4,000 × g for 15 minutes before extraction
cfDNA Extraction Methodology
- Use magnetic bead-based cfDNA extraction kits (e.g., Mag-Bind cfDNA Kit)
- Process 4 mL input volume of preserved urine
- Elute in 50 μL elution buffer
- Assess yield and quality using TapeStation analysis with Cell-Free DNA ScreenTape Assay
ddPCR Assay Configuration
- Utilize 15 ng of extracted cfDNA per reaction
- Prepare 300 μL total reaction volume with ddPCR Supermix for Probes
- Design mutant-specific FAM-labeled probes and wildtype-specific SUN-labeled probes
- Generate droplets using QX200 Droplet Generator
- Amplify with optimized annealing temperatures for each probe
- Analyze droplets using QX200 Droplet Reader
- Process data with QuantaSoft Analysis Pro Software
This protocol successfully detected NRAS (A59T) and EGFR (L858R) mutations at allelic frequencies as low as 0.1% with excellent concordance between observed and expected values [14].
Precision and Absolute Quantification
The digital nature of ddPCR provides exceptional precision and accuracy in nucleic acid quantification by counting individual molecules rather than relying on amplification curves relative to standards [19]. This absolute quantification capability eliminates inter-laboratory variability associated with standard curve construction in qPCR [24].
Calibration-Free Quantification
Unlike qPCR, which requires a standard curve of known concentrations for relative quantification, ddPCR provides absolute quantification by directly counting the number of positive partitions [2] [4]. The fundamental calculation is based on Poisson statistics:
λ = -ln(1 - p)
Where λ represents the average number of target molecules per partition and p is the ratio of positive partitions to total partitions [19]. This calculation is automatically performed by ddPCR analysis software, providing direct concentration measurements in copies per microliter without reference to external standards [19].
Enhanced Precision in Complex Matrices
Table 2: Precision assessment across sample types and targets
| Sample Matrix | Target | Precision Metric | Performance Outcome | Reference |
|---|---|---|---|---|
| Food Microbiology | Lacticaseibacillus casei | Limit of Detection | 10-fold higher sensitivity vs qPCR | [24] |
| Milk Samples | Lacticaseibacillus casei | Detection in inhibitors | Superior resistance to PCR inhibitors | [24] |
| Probiotic Products | Microbial content | Quantification accuracy | Absolute count without standards | [24] |
| Clinical HDV Samples | HDV RNA | Correlation with qPCR | ±1.12 log IU/mL standard deviation | [22] |
Essential Research Reagent Solutions
Successful implementation of ddPCR assays requires carefully selected reagents and materials optimized for droplet-based applications.
Table 3: Key research reagent solutions for ddPCR workflows
| Reagent/Material | Function | Application Notes | Example Products |
|---|---|---|---|
| ddPCR Supermix | Reaction buffer | Provides optimal chemistry for droplet generation and amplification | Bio-Rad ddPCR Supermix for Probes |
| Probe-Based Assays | Target detection | Sequence-specific detection with fluorophore-quencher pairs | TaqMan probes (FAM, HEX/VIC, TAMRA/Atto550, Cy5) |
| Droplet Generation Oil | Emulsion stabilization | Creates stable water-in-oil emulsion for partitioning | DG Oil for EvaGreen, Droplet Generation Oil |
| cfDNA Extraction Kits | Nucleic acid isolation | Maximizes yield of fragmented DNA while removing inhibitors | Mag-Bind cfDNA Kit, QIAamp Viral RNA Mini Kit |
| RNA Extraction Kits | RNA isolation | Maintains RNA integrity for reverse transcription ddPCR | INSTANT virus RNA/DNA kit, QIAamp Viral RNA Mini Kit |
| Preservation Solutions | Sample stabilization | Prevents nucleic acid degradation in liquid biopsies | Colli-Pee UAS preservative |
| Restriction Enzymes | DNA digestion | Reduces viscosity for improved partitioning; not always required | AluI (4-cutter) |
Advanced Applications in Research and Drug Development
The unique advantages of ddPCR have enabled its application across diverse research fields, particularly where sensitivity and precision are paramount.
Oncology and Liquid Biopsy
In liquid biopsy applications, ddPCR enables non-invasive monitoring of cancer dynamics through detection of circulating tumor DNA (ctDNA) [23]. Specific applications include:
- Measurable Residual Disease (MRD) Monitoring: Detection of minimal residual disease in acute myeloid leukemia (AML) with high sensitivity [23]
- Treatment Resistance Detection: Identification of emerging resistance mutations (e.g., ESR1 variants in breast cancer) during targeted therapy [23]
- Endometrial Cancer Management: Improved risk stratification and disease follow-up through ctDNA analysis [23]
Infectious Disease Quantification
ddPCR provides superior quantification of viral pathogens, particularly at low concentrations where qPCR assays show high variability [22]:
- Hepatitis D Virus (HDV) Monitoring: Accurate quantification for treatment initiation and response assessment [22]
- Pathogen Surveillance: Wastewater-based epidemiology for community-level pathogen tracking [23]
- Antibiotic Resistance Gene Detection: Monitoring dissemination of microbial antibiotic resistance genes [23]
Cell and Gene Therapy Development
In advanced therapy medicinal products (ATMPs), ddPCR provides critical quality control metrics [21]:
- Vector Copy Number (VCN) Quantification: Absolute quantification of transgene copies in gene-modified cells [21]
- Residual Plasmid DNA Detection: Sensitive detection of residual plasmid DNA post-transfection [21]
- Genome Edit Detection: Absolute quantification of genome editing events (e.g., CRISPR-Cas9) [21]
Diagram 2: Key application areas.
Droplet Digital PCR technology represents a significant advancement in molecular quantification, providing researchers with an unparalleled tool for detecting rare genetic events, precisely quantifying nucleic acids, and eliminating standardization variability. The core advantages of unmatched sensitivity, exceptional precision, and calibration-free quantification make ddPCR particularly valuable for challenging applications in oncology, infectious disease, and advanced therapy development. As research continues to push the boundaries of detection limits and quantification accuracy, ddPCR stands as an essential technology platform for both basic research and translational drug development.
From Theory to Bench: Designing and Executing a Rare Allele ddPCR Assay
Droplet Digital PCR (ddPCR) represents a third-generation polymerase chain reaction technology that has revolutionized the quantification of nucleic acids, particularly for the detection of rare genetic mutations. This technique enables the precise measurement of mutant alleles present at frequencies as low as 0.1% or even less, making it invaluable for applications in oncology, prenatal diagnostics, and infectious disease monitoring [14] [13] [4]. The core innovation of ddPCR lies in its partitioning process, where a single PCR reaction is divided into thousands of nanoliter-sized water-in-oil droplets, effectively creating individual microreactors [25]. This partitioning allows for the detection and absolute quantification of rare mutant alleles against a overwhelming background of wild-type DNA through a digital, binary readout of each droplet's fluorescence after amplification [4] [2].
The significance of ddPCR is particularly evident in liquid biopsy applications, where researchers must detect minute amounts of circulating tumor DNA (ctDNA) that often constitute less than 0.1% of the total circulating cell-free DNA in blood plasma or other biofluids like urine [14] [25]. Unlike quantitative PCR (qPCR), which relies on standard curves and relative quantification, ddPCR provides absolute quantification without calibration curves by applying Poisson statistics to the ratio of positive to negative droplets [25]. This technical deep dive explores the complete ddPCR workflow, from sample preparation through data analysis, with a specific focus on optimizing each step for rare allele detection in research and drug development settings.
Core Workflow of Droplet Digital PCR
The ddPCR process transforms analog molecular quantification into a digital counting exercise through a structured workflow consisting of four critical phases: sample preparation, partitioning, amplification, and droplet reading. Each phase must be meticulously optimized to achieve the sensitivity and specificity required for reliable rare variant detection [14] [26].
Sample Preparation and Preservation
The initial sample preparation phase is crucial for preserving the integrity of target nucleic acids, particularly when working with low-abundance targets like rare alleles. For urine-based circulating free DNA (cfDNA) analysis, preservation immediately after collection prevents degradation. Studies demonstrate that using preservatives such as Colli-Pee UAS at a 2.3:1 urine-to-preservative ratio effectively stabilizes samples for subsequent analysis [14]. The cfDNA extraction process must maximize yield while minimizing genomic DNA contamination; kits like Omega Bio-tek's Mag-Bind cfDNA Kit have shown effectiveness when processing 4 mL input volumes, with elution in 50 μL yielding sufficient material for downstream analysis [14].
The quality and quantity of extracted DNA should be verified using sensitive methods such as the Agilent Cell-Free DNA ScreenTape Assay on a 4150 TapeStation system [14]. This quality control step confirms the presence of the characteristic cfDNA fragment peak between 150-200 base pairs and assesses the degree of genomic DNA contamination, which appears as higher molecular weight bands. For rare allele detection, input DNA quantity must be optimized to ensure sufficient target molecules are present for reliable detection while avoiding overloading that can compromise partitioning efficiency. In research settings, using reference standards with known allelic frequencies (e.g., Horizon Discovery's Mimix Multiplex I cfDNA Set) helps validate assay performance and extraction efficiency through spike-in experiments [14].
Table 1: Sample Quality Assessment from Urinary cfDNA Analysis
| Experimental Group | Average Estimated Yield (ng) | cfDNA Fraction |
|---|---|---|
| Urine + UAS (No spike-in control) | 58.2 | 83% |
| Spike-in + Urine + UAS | 226.5 | ~46-53% recovery of spike-in |
| Spike-in + Elution Buffer (Control) | 161.9 | ~46% recovery |
| Spike-in + UAS (No urine control) | 186.1 | ~53% recovery |
Partitioning and Microfluidics
The partitioning phase represents the fundamental innovation that differentiates ddPCR from other PCR formats. In this step, the PCR reaction mixture—containing template DNA, primers, probes, and master mix—is partitioned into thousands to millions of nanoliter-sized droplets using sophisticated microfluidic systems [4] [2]. Commercially available platforms like the Bio-Rad QX200 system typically generate approximately 20,000 droplets per sample, with each droplet serving as an individual reaction vessel [14] [26].
The partitioning process follows a random distribution pattern described by Poisson statistics, where most droplets contain either zero or one target molecule, with a smaller fraction containing multiple copies [25]. This distribution is critical for accurate quantification, as it allows for the mathematical correction of droplets containing more than one target. The microfluidic technology employed must create monodisperse droplets (uniform in size) to ensure consistent amplification efficiency across all partitions [4]. Properly stabilized water-in-oil emulsions with appropriate surfactants prevent droplet coalescence during thermal cycling, maintaining partition integrity throughout the amplification process [4] [2].
Diagram 1: Complete ddPCR workflow from sample preparation to results, highlighting the partitioning process that separates the PCR mixture into thousands of individual reactions.
Amplification and Thermal Cycling
Following partitioning, the droplets undergo thermal cycling to amplify the target sequences. The amplification process occurs within each individual droplet, creating independent microreactors that physically separate rare mutant alleles from the abundant wild-type background [27]. This physical separation is what enables the exceptional sensitivity of ddPCR for rare variant detection, as it eliminates the competition for reagents that would occur in a bulk PCR reaction [25].
The thermal cycling parameters must be optimized for each specific assay, with particular attention to primer annealing temperatures to ensure specific amplification [14] [26]. Unlike qPCR, which monitors fluorescence in real-time during the exponential amplification phase, ddPCR utilizes end-point detection, measuring fluorescence after amplification is complete [25]. This approach makes ddPCR less susceptible to variations in amplification efficiency, as the final fluorescence signal depends only on whether the target sequence was present, not on how efficiently it was amplified [25]. The use of allele-specific fluorescent probes (such as TaqMan probes) with different fluorophores (e.g., FAM for mutant alleles, HEX/VIC for wild-type alleles) enables the discrimination between mutant and wild-type sequences within the same reaction [14] [27].
Droplet Reading and Analysis
After thermal cycling, the droplets are analyzed individually using a droplet reader that measures the fluorescence in each channel [26]. In systems like the Bio-Rad QX200, droplets flow sequentially through a detection channel where they are excited by a light source, and their fluorescence emissions are captured by detectors for each fluorophore [25] [26]. The analysis software, such as Bio-Rad's QuantaSoft, then classifies each droplet as positive (fluorescent) or negative (non-fluorescent) for each target based on predefined fluorescence thresholds [14].
The quantification of target molecules relies on Poisson statistics to calculate the original concentration from the fraction of positive droplets [25]. The formula for this calculation is:
Concentration (copies/μL) = -ln(1 - p) / V
Where p is the fraction of positive droplets and V is the volume of each droplet [25]. This mathematical correction accounts for the Poisson distribution of targets among droplets, including those that contained multiple target molecules. For rare allele detection, the variant allele frequency (VAF) is calculated as the ratio of mutant-positive droplets to the total target-positive droplets (mutant + wild-type) [14] [27]. Research has demonstrated excellent concordance between observed and expected allelic frequencies down to 0.1% using this approach [14].
Table 2: Example Concordance Analysis of Observed vs. Expected Allelic Frequencies
| Gene Target | Expected AF | Observed AF | Concordance |
|---|---|---|---|
| NRAS | 5% | 5% | Excellent |
| NRAS | 1% | 1% | Excellent |
| NRAS | 0.1% | 0.1% | Excellent |
| EGFR | 5% | 2.5% | Outside acceptance criteria |
| EGFR | 1% | 1% | Excellent |
| EGFR | 0.1% | 0.1% | Excellent |
Technical Considerations for Rare Allele Detection
Assay Design and Optimization
Effective rare allele detection requires meticulous assay design and validation. Allele-specific probes must discriminate effectively between wild-type and mutant sequences, often incorporating modified nucleotides like locked nucleic acids (LNA) to enhance specificity by increasing the melting temperature difference between matched and mismatched probes [13] [27]. Assays should be validated using reference standards with known allelic frequencies to establish sensitivity, specificity, and the limit of detection [14] [27].
Multiplexing capabilities represent another advantage of ddPCR, allowing simultaneous detection of multiple mutations in a single reaction [27]. For example, research has demonstrated successful development of three- and four-plex panels for EGFR mutations (L858R, T790M, L861Q, and Del19) in non-small cell lung cancer [27]. However, multiplex assays require careful optimization to ensure that amplification efficiency remains consistent across all targets and that fluorescence signals can be clearly distinguished [27].
Handling PCR Inhibitors and Complex Samples
A significant advantage of ddPCR for rare allele detection is its enhanced resistance to PCR inhibitors compared to qPCR [25]. In traditional bulk PCR, inhibitors present in the sample matrix (such as hemoglobin, heparin, or bile salts) affect the entire reaction, potentially reducing amplification efficiency and compromising quantification [25]. In ddPCR, these inhibitors are distributed across thousands of partitions, effectively diluting them to sub-inhibitory concentrations in most droplets [25]. Additionally, because ddPCR relies on endpoint detection rather than amplification kinetics, slight delays in amplification caused by residual inhibitors have minimal impact on quantification as long as amplification reaches the detection threshold [25].
This robustness makes ddPCR particularly valuable for analyzing challenging sample types such as urine, stool, and formalin-fixed paraffin-embedded (FFPE) tissues, which often contain various PCR inhibitors [14] [2]. Nevertheless, appropriate sample purification remains important, as high levels of inhibitors can still affect droplet generation and amplification efficiency [14].
The Scientist's Toolkit: Essential Reagents and Equipment
Successful implementation of ddPCR for rare allele detection requires specific reagents, equipment, and consumables. The following table summarizes key components of the ddPCR workflow and their functions based on current protocols and commercial systems.
Table 3: Essential Research Reagent Solutions for ddPCR Rare Allele Detection
| Category | Specific Product Examples | Function in Workflow |
|---|---|---|
| Sample Preservation | Colli-Pee UAS Preservative (DNA Genotek) | Stabilizes urine samples at collection to prevent cfDNA degradation [14] |
| Nucleic Acid Extraction | Mag-Bind cfDNA Kit (Omega Bio-tek) | Extracts high-quality cfDNA from biofluids with minimal gDNA contamination [14] |
| ddPCR Master Mix | ddPCR Supermix for Probes (No dUTP) (Bio-Rad) | Optimized reaction buffer for droplet generation and amplification [14] [26] |
| Partitioning Oil | Droplet Generation Oil (Bio-Rad) | Creates stable water-in-oil emulsion for droplet formation [26] |
| Reference Standards | Mimix Multiplex I cfDNA Set (Horizon Discovery) | Validates assay performance with known allelic frequencies (5%, 1%, 0.1%, 0%) [14] |
| Allele-Specific Probes | TaqMan Probes, ZEN Probes, castPCR Assays | Detect wild-type and mutant alleles with high specificity using different fluorophores [27] |
| Droplet Generator | QX200 Droplet Generator (Bio-Rad) | Partitions samples into ~20,000 nanoliter-sized droplets [14] [26] |
| Thermal Cycler | C1000 Touch Thermal Cycler (Bio-Rad) | Executes precise temperature cycling for PCR amplification [14] |
| Droplet Reader | QX200 Droplet Reader (Bio-Rad) | Measures end-point fluorescence in individual droplets [14] [26] |
| Analysis Software | QuantaSoft Analysis Pro Software (Bio-Rad) | Analyzes droplet fluorescence and calculates target concentration using Poisson statistics [14] |
Droplet Digital PCR represents a transformative technology for rare allele detection, combining exceptional sensitivity with absolute quantification. The complete workflow—from careful sample preservation through partitioning, amplification, and droplet reading—enables researchers to detect mutant alleles at frequencies as low as 0.1% with high precision and accuracy [14]. The technology's independence from standard curves, resistance to PCR inhibitors, and ability to provide direct copy number quantification make it particularly valuable for applications in liquid biopsy, minimal residual disease monitoring, and tumor heterogeneity studies [25] [27].
As molecular diagnostics continue to advance toward non-invasive approaches and personalized medicine, ddPCR offers researchers and drug development professionals a robust platform for detecting and quantifying rare genetic variants in complex biological samples. Following optimized protocols for each step of the workflow and utilizing appropriate controls and reference standards ensures reliable, reproducible results that can inform clinical decision-making and therapeutic development [14] [27]. The ongoing development of multiplex assays and automated platforms will further expand the utility of ddPCR in both research and clinical settings [4] [2].
Circulating tumor DNA (ctDNA) refers to fragmented DNA molecules that are shed into the bloodstream by tumor cells through processes such as apoptosis, necrosis, and active secretion [28] [29]. These fragments, typically 20-50 base pairs in length, carry tumor-specific genetic alterations and represent a subset of circulating cell-free DNA (cfDNA), which is predominantly derived from normal leukocytes and stromal cells [28]. In cancer patients, ctDNA typically constitutes approximately 0.1-1.0% of total cfDNA, though this proportion can increase with tumor burden [28]. Liquid biopsy describes the minimally invasive collection and analysis of these tumor-derived components from various bodily fluids, most commonly blood, but also urine, cerebrospinal fluid, and pleural effusions [30] [29].
The clinical significance of ctDNA stems from its ability to provide a real-time snapshot of the tumor's molecular landscape, capturing its genetic heterogeneity without the limitations of traditional tissue biopsy [29] [31]. Compared to tissue biopsy, liquid biopsy offers several distinct advantages: it is minimally invasive, can be performed repeatedly to monitor disease dynamics, captures heterogeneity from multiple tumor sites, and may detect molecular changes earlier than radiographic imaging [28] [31]. These characteristics make ctDNA analysis particularly valuable for cancer monitoring, including detection of minimal residual disease (MRD), assessment of treatment response, and identification of emerging resistance mechanisms [29] [31].
Principles of Droplet Digital PCR for Rare Allele Detection
Droplet Digital PCR (ddPCR) represents a transformative approach in nucleic acid quantification that enables absolute measurement of target sequences without reliance on standard curves [17] [32]. The fundamental principle involves partitioning a single PCR reaction into thousands to millions of nanoliter-sized water-in-oil droplets, effectively creating a multi-well plate where each droplet functions as an individual PCR microreactor [33] [17]. Following endpoint amplification, each droplet is analyzed fluorometrically to determine whether it contains the target sequence (positive) or not (negative) [17]. The absolute concentration of the target molecule in the original sample is then calculated using Poisson statistics based on the ratio of positive to negative droplets [17].
For rare mutant allele detection in ctDNA, ddPCR offers distinct advantages over traditional quantitative PCR (qPCR). Its partitioning nature significantly enhances sensitivity by effectively concentrating rare targets against a background of wild-type DNA [3] [32]. This technology demonstrates substantially higher resistance to various PCR inhibitors that may be present in biological samples, reducing artifacts caused by variable amplification efficiency [17] [32]. Furthermore, ddPCR provides absolute quantification without requiring standard curves, resulting in improved reproducibility and precision, particularly for low-abundance targets [17] [32]. The direct counting approach of ddPCR enables reliable detection of rare mutations at frequencies as low as 0.1% or even 0.001% in some applications, a sensitivity level difficult to achieve with conventional qPCR [3] [17].
Table 1: Comparison of Digital PCR Platforms
| Feature | Chip-Based dPCR (cdPCR) | Droplet Digital PCR (ddPCR) |
|---|---|---|
| Partitioning Method | Microfluidic chambers | Water-in-oil emulsion droplets |
| Typical Partition Count | 20,000-40,000 [17] | 20,000-10,000,000 [17] |
| Partition Volume | Nanoliter range [17] | Picoliter range [17] |
| Commercial Systems | BioMark, QuantStudio3D [17] | QX100/200, RainDrop [17] |
| Key Advantage | Even partition volume, minimal evaporation [17] | Higher throughput, lower cost per reaction [17] |
Technical Workflow for ctDNA Detection via ddPCR
Sample Collection and Processing
The pre-analytical phase is critical for reliable ctDNA analysis. Blood samples should be collected in specialized tubes that stabilize nucleated blood cells and prevent genomic DNA contamination, such as Cell-Free DNA BCT tubes [31]. Plasma separation through centrifugation typically follows a two-step protocol: an initial centrifugation at 1,600-2,000 × g for 10-20 minutes to separate plasma from blood cells, followed by a second centrifugation at 16,000 × g for 10 minutes to remove remaining cellular debris [29]. ctDNA is then extracted from the plasma using commercial circulating nucleic acid extraction kits, with quantification performed via fluorometric methods to assess quality and yield [3].
Assay Design for Rare Mutation Detection
Effective ddPCR assay design requires careful consideration of several factors. For rare mutation detection, the typical approach utilizes two differently labeled hydrolysis probes (TaqMan) with a single primer set that amplifies the region of interest [3]. One probe targets the wild-type sequence while the other targets the mutant allele, each labeled with distinct fluorophores compatible with the ddPCR system's excitation and emission spectra [3]. The DNA input must be optimized to achieve the desired sensitivity; for human genomic DNA, the number of copies can be calculated using the formula: Number of copies = mass of DNA in nanograms / 0.003 (since the mass of one haploid human genome is approximately 3 pg) [3]. This calculation is essential for determining the theoretical limit of detection for rare alleles.
Droplet Generation and PCR Amplification
The PCR reaction mixture is prepared according to the manufacturer's specifications for the chosen ddPCR system [3]. A typical 25μL reaction volume might contain 1X PCR mastermix, 500nM forward and reverse primers, 250nM of each probe, and the extracted ctDNA sample [3]. This mixture is then partitioned into thousands of nanoliter-sized droplets using appropriate droplet generation technology [17]. The thermal cycling protocol must be optimized for the specific assay; for example, an EGFR T790M assay might utilize an initial denaturation at 95°C for 10 minutes, followed by 45 cycles of 95°C for 30 seconds and 62°C for 15 seconds [3]. Following amplification, droplets are read using a droplet reader that measures fluorescence in each droplet [17].
Diagram 1: ddPCR Workflow for ctDNA Analysis
Data Analysis and Interpretation
Following droplet reading, data analysis involves several critical steps. Quality control assessment should include verification of the non-template control (NTC), which should display only negative partitions, and evaluation of the total number of analyzable partitions, with higher numbers increasing detection sensitivity for rare events [3]. For multiplexed assays, fluorescence spillover compensation may be necessary to correct for spectral overlap between different fluorophores [3]. The absolute concentration of the target molecule is calculated using Poisson statistics based on the ratio of positive to negative droplets, with the formula: Concentration = -ln(1 - p) / V, where p is the fraction of positive droplets and V is the droplet volume [17].
Data visualization typically employs two-dimensional scatter plots where each dot represents an individual droplet, with fluorescence amplitudes plotted for each channel [3]. Distinct clusters should form for negative droplets (double-negative), wild-type alleles (positive in one channel), mutant alleles (positive in the other channel), and potentially double-positive droplets [3]. For rare mutation detection, the critical parameter is the fractional abundance, calculated as the ratio of mutant alleles to total (mutant + wild-type) alleles [3]. The theoretical limit of detection with 95% confidence is approximately 0.2 copies/μL for some systems, though this varies by platform and application [3].
Diagram 2: Data Analysis Pipeline
Clinical Applications in Oncology
Treatment Response Monitoring
ctDNA monitoring has demonstrated significant utility in tracking treatment response across various cancer types. The ctDNA to Monitor Treatment Response (ctMoniTR) Project, which pooled patient-level data from eight clinical studies, found that advanced non-small cell lung cancer (NSCLC) patients treated with tyrosine kinase inhibitors (TKIs) who achieved undetectable ctDNA levels within 10 weeks had significantly better overall survival and progression-free survival [31]. Similar findings have been reported in colorectal cancer, where changes in ctDNA mutation rates for genes such as APC, KRAS, TP53, and PIK3CA correlated with tumor burden and therapeutic response during treatment [28]. These dynamic changes in ctDNA levels often precede radiographic evidence of response or progression by several weeks to months, providing an opportunity for earlier treatment modification [31].
Minimal Residual Disease (MRD) Detection
The exceptional sensitivity of ddPCR makes it particularly valuable for detecting MRD following curative-intent surgery or treatment [31]. Studies have demonstrated that the presence of ctDNA after complete resection of solid tumors is strongly predictive of future recurrence, while its absence correlates with prolonged remission [29] [31]. In bladder, breast, prostate, and head and neck squamous cell carcinomas, ctDNA has shown promise for MRD detection and early relapse identification [31]. This capability enables potential intervention while disease burden is still low, potentially improving patient outcomes through earlier initiation of additional therapy.
Resistance Mechanism Identification
ddPCR facilitates monitoring for emerging resistance mutations during targeted therapy. A prime example is the detection of the EGFR T790M mutation in NSCLC patients undergoing treatment with first- and second-generation EGFR TKIs [3]. This mutation, which rarely appears during initial tumor characterization but emerges during TKI treatment, confers resistance to these agents but creates sensitivity to third-generation TKIs [3]. Early detection of T790M and other resistance mechanisms such as mutations in KRAS, ESR1, BRAF, and AKT genes enables timely adjustment of treatment strategies, optimizing therapeutic sequencing and improving long-term disease control [31].
Table 2: Clinical Applications of ctDNA Monitoring in Different Cancers
| Cancer Type | Key Applications | Common Genetic Targets | Clinical Utility |
|---|---|---|---|
| Non-Small Cell Lung Cancer | EGFR TKI response monitoring, resistance detection [3] [31] | EGFR (T790M, L858R), KRAS [3] [31] | Guides TKI selection and switching; predicts survival [31] |
| Colorectal Cancer | Treatment response monitoring, MRD detection [28] [30] | APC, KRAS, TP53, PIK3CA [28] | Correlates with tumor burden and CEA concentration [28] |
| Breast Cancer | Prognostic assessment, therapy selection [28] [29] | ESR1, HER2, PIK3CA [31] | Included in CSCO and AJCC guidelines for prognostic assessment [28] |
| Multiple Solid Tumors | MRD detection, early relapse identification [31] | Tumor-specific mutations | Earlier detection of recurrence than imaging [31] |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents and Materials for ddPCR-based ctDNA Analysis
| Reagent/Material | Function | Specification Considerations |
|---|---|---|
| Blood Collection Tubes | Stabilize cellular and cell-free components during storage/transport | Cell-free DNA BCT tubes or similar specialized collection systems [31] |
| Nucleic Acid Extraction Kit | Isolate ctDNA from plasma | Optimized for low-abundance circulating nucleic acids [3] |
| ddPCR Master Mix | Provide essential components for PCR amplification | Should include DNA polymerase, dNTPs, reaction buffer, MgCl₂ [3] |
| Sequence-Specific Primers | Amplify target genomic region | Typically used at 500nM final concentration; must flank mutation site [3] |
| Hydrolysis Probes (TaqMan) | Detect wild-type and mutant sequences | FAM-labeled for wild-type, Cy3/HEX-labeled for mutant; 250nM final concentration [3] |
| Droplet Generation Oil | Create water-in-oil emulsion for partitioning | Specific to ddPCR system; ensures stable droplet formation [17] |
| Reference DNA | Positive and negative controls | Wild-type and mutant sequences to validate assay performance [3] |
Current Challenges and Future Directions
Despite significant advances, several challenges remain in the implementation of ctDNA analysis via ddPCR. Pre-analytical variables including blood collection timing, tube types, and processing protocols require standardization to ensure reproducible results across laboratories [31]. Biological confounding factors such as clonal hematopoiesis of indeterminate potential (CHIP) can lead to false-positive calls, necessitating careful interpretation of results [31]. Additionally, the limited number of targets that can be simultaneously analyzed with ddPCR (compared to next-generation sequencing) constrains its utility for comprehensive tumor genotyping [31].
Future developments are likely to focus on several key areas. Technological innovations including enhanced assay sensitivity, integration of DNA methylation analysis, and fragmentomics (analysis of ctDNA fragmentation patterns) may improve early detection capabilities [31]. Collaborative efforts led by organizations such as Friends of Cancer Research, the Foundation for the National Institutes of Health, and the Blood Profiling Atlas in Cancer are working to establish analytical standards and build consensus around clinical validation [31]. Furthermore, expanding clinical applications in areas such immunotherapy response prediction and ultra-early cancer detection continue to be active areas of investigation [29] [31]. As evidence accumulates, ctDNA monitoring is progressively being incorporated into clinical guidelines, with organizations including the National Comprehensive Cancer Network, American Society of Clinical Oncology, and European Society for Medical Oncology already recommending ctDNA testing for specific clinical scenarios in certain cancer types [31].
Droplet Digital PCR has emerged as a powerful methodology for ctDNA analysis in oncology, offering the sensitivity, precision, and reproducibility required for detecting rare mutant alleles in circulation. Its application to cancer monitoring provides a minimally invasive approach for assessing treatment response, detecting minimal residual disease, and identifying emerging resistance mechanisms. While challenges remain in standardization and interpretation, ongoing technological improvements and collaborative efforts to establish best practices are accelerating the integration of ctDNA analysis into routine cancer management. As validation studies continue to demonstrate its clinical utility across diverse cancer types and stages, liquid biopsy using ddPCR is poised to become an increasingly essential tool in precision oncology, enabling more dynamic and personalized treatment approaches for cancer patients.
Droplet Digital PCR (ddPCR) represents a transformative approach in molecular diagnostics, particularly for the detection and quantification of low-abundance pathogens and resistance genes in infectious diseases. As a third-generation PCR technology, ddPCR enables absolute quantification of nucleic acids without the need for standard curves by partitioning a single PCR reaction into thousands of nanoliter-sized droplets, each functioning as an individual micro-reactor [17]. This digital partitioning allows for precise detection of rare targets—including viral reservoirs, emerging resistant strains, and low-level pathogens—that often evade conventional diagnostic methods like quantitative PCR (qPCR) [32] [17].
The fundamental principle of ddPCR lies in its binary endpoint detection. After PCR amplification, each droplet is analyzed for fluorescence signals, categorizing it as positive (containing the target sequence) or negative (without the target). The absolute copy number of the target molecule in the original sample is then calculated using Poisson statistics based on the ratio of positive to negative droplets [17] [24]. This approach provides exceptional sensitivity for detecting targets present at frequencies as low as 0.001%–0.1%, making it uniquely suited for identifying minimal residual infection, early treatment response monitoring, and detecting emerging resistance mutations before clinical manifestation [3] [17].
Comparative Advantages of ddPCR for Pathogen Detection
Technical Superiority Over qPCR
When compared to traditional quantitative PCR (qPCR), ddPCR demonstrates significant advantages for detecting low-abundance targets in complex clinical samples. While qPCR measures amplification fluorescence at the quantification cycle (Cq) and relies on standard curves for relative quantification, ddPCR utilizes end-point detection and absolute quantification through binary counting of partitioned reactions [32] [17]. This fundamental difference makes ddPCR particularly valuable for infectious disease applications where target concentrations are minimal or sample inhibitors are present.
Research has demonstrated that for sample/target combinations with low nucleic acid levels (Cq ≥ 29) and variable amounts of chemical and protein contaminants, ddPCR technology produces more precise, reproducible, and statistically significant results [32]. The partitioning process in ddPCR effectively dilutes inhibitors that would typically compromise PCR efficiency in qPCR assays, allowing for accurate detection even in partially inhibited reactions [32]. This characteristic is particularly valuable for direct pathogen detection from complex clinical matrices such as stool, blood, and sputum, where inhibitory substances are frequently co-extracted with nucleic acids.
Table 1: Performance Comparison Between ddPCR and qPCR for Pathogen Detection
| Parameter | ddPCR | qPCR |
|---|---|---|
| Quantification Method | Absolute counting without standard curves | Relative quantification requiring standard curves |
| Sensitivity | Can detect rare targets at 0.001%-0.1% frequency [17] | Typically limited to 1-10% frequency for rare variants [17] |
| Tolerance to Inhibitors | High - partitioning dilutes inhibitors [32] | Low - requires adequate dilution of inhibitors [32] |
| Precision at Low Target Concentration | High precision (CV < 10%) even at single-copy levels [32] | Variable precision, highly dependent on reaction efficiency [32] |
| Dynamic Range | Linear over wide range but limited by partition count [17] | Wider dynamic range but compromised at extremes [17] |
| Data Output | Absolute copy number/μL | Cq values (relative quantification) |
Detection of Gastrointestinal Pathogens: A Case Study
The enhanced sensitivity of ddPCR is particularly evident in detecting SARS-CoV-2 in non-respiratory samples. A 2023 study evaluating droplet digital qRT-PCR for detecting SARS-CoV-2 RNA in stool and urine samples from COVID-19 patients demonstrated remarkable differences in detection efficiency between ddPCR and traditional qRT-PCR [34]. While urine samples showed only 27.1% positivity by ddPCR, stool samples demonstrated 100% detection rate, highlighting both the technology's sensitivity and the viral tropism for gastrointestinal tissue [34].
In this study, the average cycle threshold (Ct) of qRT-PCR strongly correlated with the average copy number of 327.10 copies/μL analyzed in ddPCR, validating ddPCR as a quantitative reference method [34]. The researchers concluded that ddPCR's ability to identify positive samples with low concentrations of viral targets helps resolve cases of inconclusive diagnosis, particularly important for discharge criteria and transmission prevention [34]. This application demonstrates ddPCR's clinical utility for detecting pathogens in non-conventional sample types where target concentrations are minimal.
Experimental Design and Protocols
Core ddPCR Workflow for Pathogen Detection
The following diagram illustrates the fundamental workflow for ddPCR-based detection of pathogens and resistance genes in infectious disease applications:
Detailed Protocol for Absolute Quantification of Bacterial Pathogens
The detection and absolute quantification of specific bacterial pathogens using ddPCR requires careful assay design and optimization. Based on established methodology for detecting Lacticaseibacillus casei as a model system, the following protocol can be adapted for various bacterial targets in infectious disease diagnostics [24]:
Sample Processing and DNA Extraction:
- Centrifuge liquid samples (e.g., stool suspensions, urine, broth cultures) at 4000 rpm for 10 minutes to remove debris [34]
- For bacterial cultures, centrifuge at 11,000×g for 10 minutes, remove supernatant [24]
- Extract DNA using commercial kits (e.g., Qiagen Viral RNA extraction Kit for viruses, specialized bacterial DNA extraction kits) following manufacturer's protocols [34] [24]
- Quantify DNA concentration using fluorometric analysis (e.g., Qubit system) [24]
Primer and Probe Design:
- Identify unique genetic markers through comparative genomic analysis to ensure species-specific detection [24]
- Design primers and TaqMan hydrolysis probes with appropriate fluorophore labels (FAM/HEX) compatible with ddPCR systems [3] [24]
- Validate specificity against closely related species to minimize cross-reactivity [24]
ddPCR Reaction Setup:
- Prepare reaction mixture containing: ddPCR supermix, primers (900 nM final concentration), probe (250 nM final concentration), and DNA template (optimized concentration) [24]
- Adjust total volume to accommodate droplet generation system requirements (typically 20-25 μL) [3] [24]
Droplet Generation and Thermal Cycling:
- Generate droplets using appropriate droplet generator according to manufacturer's instructions [24]
- Transfer droplets to PCR plate and seal properly
- Perform PCR amplification with optimized thermal cycling conditions:
Droplet Reading and Data Analysis:
- Read droplets using droplet reader system
- Set fluorescence amplitude thresholds based on positive and negative controls
- Apply Poisson correction to calculate absolute copy number concentration (copies/μL) [24]
Table 2: Essential Research Reagents for Pathogen Detection ddPCR Assays
| Reagent Category | Specific Examples | Function/Purpose |
|---|---|---|
| Nucleic Acid Extraction | Qiagen Viral RNA Kit, specialized bacterial DNA kits [34] [24] | Isolation of pathogen nucleic acids from clinical samples |
| ddPCR Mastermix | ddPCR Supermix, PerfeCTa Multiplex mastermix [3] [24] | Provides essential components for amplification in droplet format |
| Primer/Probe Sets | Species-specific primers, FAM/HEX-labeled TaqMan probes [3] [24] | Target-specific amplification and detection |
| Reference Materials | Synthetic DNA standards, reference strains [34] [3] | Assay validation and quantification standards |
| Droplet Generation Oil | DG Oil, Droplet Generation Oil [24] | Creates stable water-in-oil emulsion for partitioning |
| Quality Controls | No-template controls, positive controls, inhibition controls [3] | Ensures assay specificity and detects contamination |
Advanced Applications in Resistance Gene Detection
Rare Mutation Detection for Antimicrobial Resistance
The detection of rare mutations associated with antimicrobial resistance represents one of the most impactful applications of ddPCR in infectious disease diagnostics. The technology's ability to detect variant alleles present at frequencies below 0.1% enables early identification of emerging resistance before conventional phenotypic methods can detect it [3]. This capability is particularly valuable for monitoring treatment response in chronic infections (e.g., tuberculosis, HIV) where resistant subpopulations may initially represent only a minor fraction of the total pathogen population.
The experimental approach for rare resistance mutation detection adapts the general ddPCR workflow with specific modifications:
Assay Design for Mutation Detection:
- Design two hydrolysis probes with different fluorophores: one targeting the wild-type sequence and another targeting the mutant allele [3]
- Use a single primer set to amplify the region containing the mutation site
- Validate probe specificity using control samples with known genotype [3]
Determining Sample Input and Sensitivity:
- Calculate DNA input based on desired sensitivity using the formula: Number of copies = mass of DNA (ng)/0.003 (for human genomic DNA) [3]
- For 10ng human genomic DNA input: 10/0.003 = 3,333 haploid genomes
- Theoretical detection limit with 95% confidence: 0.2 copies/μL [3]
- Sensitivity = Theoretical LOD / Total target concentration = 0.2/133 = 0.15% [3]
Data Analysis and Interpretation:
- Analyze using 2D scatter plots (FAM vs. HEX fluorescence)
- Apply fluorescence spillover compensation when using multiple fluorophores [3]
- Calculate mutant allele frequency as: (Mutant-positive droplets / Total positive droplets) × 100 [3]
Implementation in Clinical Settings
The translation of ddPCR from research to clinical applications requires careful consideration of several factors. A 2023 study highlighted ddPCR's utility for resolving inconclusive diagnoses, particularly when low viral loads create challenges for standard qPCR methods [34]. In this application, ddPCR detected SARS-CoV-2 in 100% of stool samples from confirmed COVID-19 patients, compared to significantly lower detection rates in other sample types [34].
For clinical implementation, ddPCR assays must demonstrate:
- Robust performance across different sample matrices (blood, stool, urine, respiratory samples)
- Consistency between different operators and instruments
- Stability of results across multiple reagent lots
- Clear interpretation guidelines for clinical decision-making
The establishment of such validated protocols has been demonstrated for various pathogens, including the absolute quantitative detection of Lacticaseibacillus casei in food matrices, which provides a template for adapting similar approaches for clinical bacterial pathogens [24].
Data Analysis and Quality Control
Analysis Algorithms and Software Tools
The analysis of ddPCR data requires specialized approaches to accurately classify droplets and calculate target concentrations. The ddpcr R package provides an open-source solution for analyzing two-channel ddPCR experiments, addressing limitations of proprietary software [35] [36]. The package implements a multi-step analysis algorithm:
- Identify failed wells - exclude wells with failed ddPCR reactions based on predefined quality metrics [35] [36]
- Identify outlier droplets - remove droplets with aberrant fluorescence signals potentially caused by reader errors [35] [36]
- Identify empty droplets - exclude droplets with very low fluorescence amplitudes to reduce data size and noise [35] [36]
- Gating droplets - assign droplets to clusters using kernel density estimation and Gaussian mixture models [36]
- Calculate concentration - determine absolute template concentration (copies/μL) using Poisson statistics [35] [36]
The analysis approach is particularly effective for PNPP (Positive-Negative/Positive-Positive) type experiments, which are common in infectious disease applications for detecting wild-type and mutant variants [35]. The software generates two-dimensional scatter plots visualizing droplet clusters based on their fluorescence amplitudes in both channels, enabling visual verification of automated gating results [36].
Quality Control Measures
Implementing robust quality control measures is essential for generating reliable ddPCR data in clinical diagnostics:
Experimental Controls:
- Include no-template controls (NTC) to detect contamination [3]
- Use positive controls with known target concentration to verify assay performance
- Implement reference gene assays for sample quality assessment
Data Quality Assessment:
- Monitor the total number of analyzed partitions (typically >10,000 valid droplets) [3]
- Check for expected cluster patterns in two-dimensional fluorescence plots
- Verify low droplet counts in NTC wells (<10 positive droplets) [3]
- Assess separation between positive and negative droplet populations
Quantification Standards:
- Use standardized reference materials for absolute quantification
- Apply Poisson correction to account for multiple targets per partition
- Report confidence intervals based on partition count and positive fraction
Droplet Digital PCR has established itself as an indispensable technology for detecting low-abundance pathogens and resistance genes in infectious disease diagnostics. Its superior sensitivity, precision, and tolerance to inhibitors provide significant advantages over qPCR for challenging diagnostic scenarios where target concentrations are minimal or sample quality is compromised [34] [32] [17]. As the technology continues to evolve, several emerging trends are likely to shape its future applications in infectious disease diagnostics.
The integration of ddPCR with next-generation sequencing (NGS) represents a promising direction for comprehensive pathogen characterization. ddPCR can serve as a quality control tool for NGS libraries and provide orthogonal validation for sequencing results [17]. Additionally, the development of multiplex ddPCR assays with increased target capacity will enable simultaneous detection of multiple pathogens or resistance markers from limited clinical samples.
The clinical adoption of ddPCR is expected to expand as standardized protocols emerge and cost-effectiveness improves. Current applications in non-invasive prenatal testing, oncology, and virology demonstrate the technology's potential to transform diagnostic paradigms [17]. For infectious diseases, ddPCR's ability to detect minimal residual disease and emerging resistance will likely play an increasingly important role in personalized antimicrobial therapy and infection control.
In conclusion, ddPCR technology provides the sensitivity, precision, and robustness required for modern infectious disease diagnostics. Its ability to absolutely quantify low-abundance targets enables earlier detection of pathogens and resistance mechanisms, ultimately supporting improved patient outcomes through timely intervention and tailored therapeutic approaches.
Non-Invasive Prenatal Testing and Rare Blood Donor Screening Applications
Digital PCR (dPCR) represents a transformative approach in molecular diagnostics, enabling absolute quantification of nucleic acids without requiring standard curves. This technology operates by partitioning a PCR reaction into thousands of nanoscale reactions, each functioning as an individual microreactor. Following amplification, the proportion of positive reactions is counted and applied to Poisson statistics to determine the absolute target concentration [37]. For rare allele detection, this partitioning strategy provides exceptional sensitivity by effectively concentrating rare targets within isolated partitions while diluting abundant wild-type sequences across the majority of partitions [3]. This physical enrichment allows dPCR to detect mutant alleles present at frequencies as low as 0.001% in some applications, far surpassing the capabilities of conventional quantitative PCR (qPCR) [17].
The fundamental difference between dPCR and qPCR lies in their quantification strategies. While qPCR monitors amplification kinetics throughout the reaction and relies on standard curves for relative quantification, dPCR utilizes end-point detection and binary counting of positive partitions to achieve absolute quantification [37]. This methodological distinction makes dPCR particularly suitable for applications requiring precise measurement of low-abundance targets, including circulating fetal DNA in maternal plasma, rare mutations in oncology, and minor blood group antigens in transfusion medicine [38] [17]. The technology's independence from amplification efficiency and its superior tolerance to PCR inhibitors further enhance its reliability for complex clinical samples [17].
Principles of Droplet Digital PCR for Rare Allele Detection
Statistical Foundation
The absolute quantification capability of dPCR hinges on Poisson statistics, which describe the random distribution of molecules across many partitions. According to Poisson distribution, the probability of a partition containing at least one target molecule is expressed as p = 1 - e^(-λ), where λ represents the average number of target molecules per partition [37]. The proportion of positive partitions (k/n) enables calculation of the initial target concentration using the formula λ = -ln(1 - k/n) [37]. This statistical framework mathematically defines the accuracy and performance metrics of dPCR, with precision increasing alongside the number of partitions analyzed [37].
The confidence in target concentration estimation depends significantly on the number of empty partitions. Optimal precision occurs at λ = 1.6, corresponding to approximately 20% empty partitions, where the pattern of positive and negative partitions provides the most statistical information [37]. This relationship has practical implications for experimental design, as sample dilution may be necessary to achieve optimal target concentration per partition. For 10,000 partitions, the optimal precision at λ = 1.6 scales as the inverse square root of the partition count, demonstrating how increased partitioning enhances measurement accuracy [37].
Platform Technologies
Two primary dPCR platforms dominate current applications: chip-based dPCR (cdPCR) and droplet-based dPCR (ddPCR). Chip-based systems utilize microfluidic technology to partition reactions into nanoliter chambers arrayed on integrated fluidic circuits. Commercial systems like BioMark and QuantStudio3D contain 20,000-40,000 microchambers, providing even volume distribution and stable reaction environments [17]. Droplet-based systems employ water-in-oil emulsion technology to generate millions of picoliter-sized droplets, dramatically increasing partitioning density. Systems such as QX200/200 create approximately 20,000 droplets per reaction, while platforms like RainDrop can generate 1-10 million droplets [17]. The exceptional partitioning capacity of ddPCR makes it particularly suitable for rare allele detection applications requiring maximum sensitivity.
Assay Design Considerations
Effective rare mutation detection requires careful assay design, typically utilizing dual-labeled hydrolysis probes (TaqMan) with a single primer set. The primers amplify the target region, while two different fluorescent probes distinguish wild-type and mutant sequences [3]. For EGFR T790M mutation detection, for example, a FAM-labeled probe targets the wild-type sequence while a Cy3-labeled probe targets the mutant allele [3]. This approach enables clear discrimination between homozygous wild-type, heterozygous, and homozygous mutant genotypes based on distinct fluorescence clustering patterns.
DNA input optimization represents another critical consideration for rare allele detection. The theoretical limit of detection with 95% confidence is approximately 0.2 copies/μL for systems like the Naica platform [3]. Sensitivity for rare alleles can be calculated using the formula: Sensitivity = Theoretical LOD / Total target concentration. For example, with 10ng of human genomic DNA (containing approximately 133 copies/μL of a single-copy gene), the detectable mutant allelic fraction would be 0.15% [3]. This mathematical relationship guides experimental design to ensure sufficient DNA input for detecting desired mutation frequencies.
Applications in Non-Invasive Prenatal Testing
Fetal Blood Group Antigen Genotyping
Non-invasive prenatal testing for blood group antigens enables targeted management of pregnancies at risk for hemolytic disease of the fetus and newborn (HDFN). dPCR assays have been developed for RHD, KEL1, HPA-1a, and HPA-5b genotyping from cell-free fetal DNA in maternal plasma [38]. These assays demonstrate exceptional sensitivity, with the RHD exon 5 assay detecting 0.05% RHD target in an RhD-negative background, while the HPA-1a assay achieves detection limits as low as 0.05% HPA-1a [38]. This sensitivity enables reliable fetal genotyping even in early pregnancy when fetal DNA fraction is low.
The dPCR workflow for blood group antigen testing involves cell-free DNA extraction from maternal plasma, followed by target preamplification to increase evaluable signals. Multiplex preamplification protocols enable simultaneous analysis of multiple targets: Multiplex I covers RHD exons 3, 5, and 7 with GAPDH and AMEL; Multiplex II includes KEL, HPA-1, HPA-5, and AMEL; while Multiplex III addresses control SNPs [38]. Following purification, the preamplified products undergo chip-based dPCR using two-color TaqMan probe chemistry for allelic discrimination [38]. This methodology provides unambiguous fetal genotyping results that guide clinical decisions regarding anti-D prophylaxis administration.
Table 1: Sensitivity of dPCR Assays for Fetal Blood Group Antigen Detection
| Target | Limit of Detection | Clinical Utility |
|---|---|---|
| RHD Exon 5 | 0.05% RHD in RhD-negative background | HDFN prevention in RhD-negative pregnancies |
| RHD Exon 7 | 0.25% RHD in RhD-negative background | Complementary target for RHD typing |
| RHD Exon 3 | 2.5% RHD in RhD-negative background | Lower specificity limits utility |
| KEL1/2 | 0.5% target allele | HDFN prevention in Kell-sensitized pregnancies |
| HPA-1a | 0.05% target allele | FNAIT risk assessment |
| HPA-5b | 0.5% target allele | FNAIT risk assessment |
Chromosomal Aneuploidy Screening
dPCR applications in NIPT extend to screening for common chromosomal aneuploidies, particularly trisomy 21 (Down syndrome), trisomy 18 (Edwards syndrome), and trisomy 13 (Patau syndrome). The technology enables precise quantification of fetal DNA and chromosome ratios through counting of individual DNA molecules [39] [17]. dPCR demonstrates particular utility for trisomy 21 detection, with studies showing accurate diagnosis through quantification of chromosome 21 representation in maternal plasma [17]. The digital counting approach provides inherent quantitative precision that benefits aneuploidy detection.
The development of single nucleotide polymorphism (SNP)-based NIPT methodologies, such as the Panorama test, represents a technological advancement in the field. This approach evaluates approximately 13,000 SNPs distributed across the genome, leveraging the 1% of DNA that differs between individuals to screen for common trisomies, microdeletions, and triploidy [40]. The SNP-based methodology provides additional capabilities including determination of zygosity and fetal fraction, while accounting for ethnic and population variability [40]. Validation studies involving over 20,000 pregnancies demonstrate combined sensitivity exceeding 99% for trisomies 21, 18, and 13, with a positive predictive value surpassing 95% for trisomy 21 [40].
Table 2: dPCR Performance in Prenatal Applications
| Application | Detection Sensitivity | Specificity | Notable Advantages |
|---|---|---|---|
| Trisomy 21 Screening | >99% [40] [41] | >99% [40] | Lower false positive rate than traditional screening |
| Fetal RHD Genotyping | 99.9% [38] | 99.1% [38] | Enables targeted anti-D prophylaxis |
| Fetal HPA-1a Detection | 0.05% target [38] | High [38] | Superior to qPCR for SNP detection |
| 22q11.2 Deletion | 83% [40] | >99% [40] | Non-invasive microdeletion screening |
| Fetal Sex Determination | ~99% [42] | ~99% [42] | Guides management of X-linked disorders |
Applications in Rare Blood Donor Screening
High-Throughput Antigen Typing
dPCR technology enables highly sensitive large-scale screening for rare blood group phenotypes in donor populations. The technology's precision and sensitivity facilitate identification of donors with uncommon antigen combinations, particularly valuable for patients requiring chronic transfusion support who develop multiple alloantibodies. The digital methodology provides absolute quantification without standard curves, making it suitable for automated high-throughput applications [17]. This capability enhances rare donor registry development and supports antigen-negative blood product provision.
The exceptional sensitivity of dPCR for single nucleotide polymorphism detection benefits blood group genotyping where conventional serological methods face limitations. For HPA-1a/b system typing, dPCR reliably distinguishes heterozygous and homozygous genotypes based on differential probe fluorescence, detecting minor allele frequencies below 0.1% [38]. This precision exceeds conventional qPCR, which typically requires at least 1% rare alleles for stable detection [38]. The technology's partitioning strategy reduces template competition, enhancing detection of minor population variants in pooled samples.
Rare Mutation Detection in Transfusion Medicine
Beyond blood group antigen screening, dPCR applications extend to detecting rare mutations associated with transfusion-transmitted diseases and platelet function disorders. The technology's partitioning approach provides concentration effects that enhance detection of low-frequency variants in large donor pools [3]. This capability supports blood safety initiatives through highly sensitive pathogen detection and identification of carriers for rare bleeding disorders.
The procedural workflow for rare mutation detection in blood donor screening mirrors established dPCR methodologies, incorporating careful assay design, optimal DNA input calculation, and appropriate fluorescence compensation controls. For rare mutation detection, the use of monocolor controls (non-template control, target-specific controls) enables correction of fluorescence spillover between channels, ensuring accurate cluster identification and variant calling [3]. This rigorous approach maintains assay specificity when screening for very low-frequency mutations in diverse donor populations.
Comparative Performance Analysis
dPCR Versus qPCR for Rare Allele Detection
Direct comparison between dPCR and qPCR reveals distinct advantages for rare allele detection applications. dPCR demonstrates significantly enhanced sensitivity, detecting rare alleles at frequencies of 0.1% or lower compared to 1% for qPCR [38] [17]. This order-of-magnitude improvement stems from dPCR's partitioning strategy, which reduces background signal and enables detection of rare mutants against abundant wild-type sequences [3]. The technology also shows superior tolerance to PCR inhibitors, as partitioning dilutes inhibitors across multiple reactions, preventing comprehensive reaction failure [37].
The absolute quantification capability of dPCR represents another key advantage, eliminating requirement for standard curves and reducing inter-laboratory variability [17]. Studies demonstrate highly reproducible dPCR results with error rates below 5%, surpassing qPCR reproducibility [17]. For non-invasive prenatal testing and rare blood donor screening, this precision provides reliable quantitative data for clinical decision-making without additional reference materials. However, qPCR maintains advantages in dynamic range and established implementation in clinical laboratories, ensuring both technologies will continue complementary roles in molecular diagnostics.
Table 3: Method Comparison for Nucleic Acid Quantification
| Parameter | Digital PCR | Quantitative PCR |
|---|---|---|
| Quantification Method | Absolute counting via Poisson statistics | Relative to standard curve |
| Sensitivity for Rare Alleles | 0.001%-0.1% [17] | ~1% [38] |
| Precision | High (CV <5%) [17] | Moderate (CV 10-25%) [43] |
| Tolerance to Inhibitors | High [37] [17] | Moderate to low |
| Dynamic Range | Narrow (limited by partition count) | Wide (up to 10-log range) |
| Throughput | Moderate to high | High |
| Cost per Sample | Higher [17] | Lower [17] |
dPCR Versus Next-Generation Sequencing
dPCR complements next-generation sequencing (NGS) in molecular diagnostics, providing orthogonal validation for NGS findings through highly precise quantification [17]. While NGS offers comprehensive mutation screening across multiple genomic regions, dPCR delivers exceptional sensitivity for specific known variants. This synergistic relationship positions dPCR as both a validation tool for NGS results and a primary detection method for targeted applications requiring utmost sensitivity [17].
In non-invasive prenatal testing, dPCR and NGS represent complementary approaches with distinct strengths. NGS methodologies like massively parallel shotgun sequencing (MPSS) and chromosome selective sequencing (CSS) provide genome-wide aneuploidy assessment but face limitations including average failure rates of 1.58-3.56% and higher computational requirements [39]. dPCR offers focused analysis of specific targets with minimal technical failure rates, making it suitable for high-priority applications including fetal RHD genotyping and single-gene disorder assessment [39] [38]. The technologies' combined use provides comprehensive prenatal genetic assessment balancing breadth and precision.
Experimental Protocols
dPCR Protocol for Fetal RHD Genotyping
The established dPCR protocol for non-invasive fetal RHD genotyping begins with blood sample collection and processing. Blood samples should be stored at room temperature and processed within three days, with plasma obtained through sequential centrifugation: first at 2,000 × g for 10 minutes, then at 16,000 × g for 10 minutes at 4°C [38]. Cell-free DNA is extracted from 1mL plasma using commercial kits (e.g., QIAamp Circulating Nucleic Acid Kit), with elution in 100μL sterile water yielding typical concentrations of 2.1-19.5ng/μL [38].
Target preamplification precedes dPCR analysis to increase evaluable signals. Multiplex PCR reactions (25μL) contain Multiplex PCR Master Mix, 0.1μM of each primer, and 2-9ng cfDNA [38]. Cycling conditions include: 15 minutes at 95°C; 30 cycles of 30 seconds at 94°C, 90 seconds at 63°C, 90 seconds at 72°C; final extension for 10 minutes at 72°C [38]. Purified PCR products undergo chip-based dPCR using two-color TaqMan probes with commercial systems, with data analysis software enumerating positive partitions for target quantification.
Rare Mutation Detection Protocol
The procedural workflow for rare mutation detection (exemplified by EGFR T790M detection) incorporates careful assay design and optimization. PCR reactions should include 1X PCR mastermix, reference dye (if required), 500nM primers, 250nM each wild-type and mutant probe, and optimized DNA input in 25μL total volume [3]. Human genomic DNA input should be calculated based on copy number requirements, using the conversion: number of copies = mass of DNA (ng) / 0.003 (for human genomic DNA) [3].
Thermal cycling conditions must be optimized for specific mastermixes and detection systems. For the EGFR T790M assay using PerfeCTa Multiplex mastermix, recommended conditions include: initial denaturation at 95°C for 10 minutes; 45 cycles of 95°C for 30 seconds and 62°C for 15 seconds [3]. Following amplification, data acquisition occurs via chip imaging or droplet reading per manufacturer instructions, with subsequent analysis using specialized software applying fluorescence compensation and Poisson statistics for absolute quantification [3].
Essential Research Reagent Solutions
Table 4: Essential Research Reagents for dPCR Applications
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Nucleic Acid Extraction | QIAamp Circulating Nucleic Acid Kit [38] | Cell-free DNA isolation from plasma samples |
| Preamplification | Multiplex PCR Master Mix [38] | Target enrichment prior to dPCR analysis |
| dPCR Mastermix | LightCycler 480 SYBR Green I Master [43] | Provides essential PCR components with optimized chemistry |
| Hydrolysis Probes | TaqMan probes (FAM, VIC labels) [38] [3] | Sequence-specific detection with fluorescent reporting |
| Reference Dyes | Instrument-specific reference dyes [3] | Normalization for partition volume variation |
| Partitioning Oil | QX200 Droplet Generation Oil [37] | Creates stable water-in-oil emulsions for ddPCR |
| Positive Controls | Synthetic DNA targets [3] | Assay validation and performance monitoring |
| Negative Controls | Non-template controls [3] | Contamination monitoring and background assessment |
Digital PCR technology has revolutionized non-invasive prenatal testing and rare blood donor screening through exceptional sensitivity and absolute quantification capabilities. The partitioning strategy underlying dPCR enables rare allele detection at frequencies approaching 0.001%, supporting critical clinical applications including fetal blood group genotyping, aneuploidy screening, and rare donor identification [38] [17]. As the technology continues evolving, emerging applications in liquid biopsy, infectious disease monitoring, and complex genetic analysis will further expand its diagnostic utility.
Future dPCR development will likely focus on increasing partitioning density, enhancing multiplexing capabilities, and streamlining workflows for clinical implementation. Integration with microfluidic technologies promises improved automation and reduced costs, while novel detection chemistries may expand multiplexing capacity [37] [17]. For non-invasive prenatal testing, dPCR may enable comprehensive single-gene disorder screening through highly multiplexed assays. In transfusion medicine, the technology could support extensive antigen profiling for rare donor registry development. These advancements will solidify dPCR's role as an essential tool for molecular diagnostics requiring ultimate sensitivity and precision.
Circulating free DNA (cfDNA) derived from urine has emerged as a promising, non-invasive biomarker in oncology, nephrology, and other medical research fields [44]. Urinary cfDNA (ucfDNA) reflects both local and systemic genetic changes, offering insights into the underlying pathophysiology of diseases not only in the urinary tract but also in distant organs [44]. This capacity to capture both localized and systemic alterations makes ucfDNA an exciting tool for personalized medicine, where patient-specific molecular profiles can guide treatment decisions and improve outcomes [44]. The analysis of ucfDNA holds the potential to revolutionize diagnostics, enabling earlier detection, better risk stratification, and more precise monitoring of therapeutic efficacy with minimal patient burden [44].
However, ucfDNA analysis presents significant challenges that must be addressed to harness its full potential. The harsh environment within urine, characterized by variable and acidic pH (ranging from 5.0 to 7.0) and high activity of nucleic acid hydrolyzing enzymes (100-fold more active than in plasma), rapidly degrades cfDNA [45]. This degradation, if unchecked, compromises DNA integrity and complicates the extraction process [45]. Furthermore, ucfDNA is typically more degraded, shorter, and variably sized compared to plasma cfDNA [45]. Therefore, a successful workflow must incorporate three critical elements: (1) pre-analytical preservation to minimize cfDNA degradation post-collection, (2) reliable cfDNA extraction methodology to maximize yield with minimal genomic DNA contamination, and (3) appropriate downstream technology with high sensitivity to draw relevant conclusions [44]. This case study details a complete workflow solution that addresses these challenges for the detection of rare NRAS and EGFR mutations.
Experimental Workflow for Urinary cfDNA Analysis
The complete workflow from urine sample collection to mutation detection involves several critical stages, each optimized to preserve and analyze the labile ucfDNA. The following diagram illustrates this integrated process.
Sample Collection and Preservation
The first and most critical pre-analytical step is the immediate preservation of urine upon collection.
- Urine Collection and Preservation: Urine sample was collected and added to DNA Genotek's Colli-Pee UAS preservative at a ratio of 2.3 to 1 for sample preservation [44]. This stabilization is crucial because, without it, ucfDNA degrades rapidly. Studies show that in native (unstabilized) urine, an increase in smaller DNA fragments (<150 bp) occurs after just 6 hours, and after 72 hours, very few fragments remain, with cfDNA concentration decreasing significantly [45]. In contrast, stabilized urine preserves ucfDNA quantity and integrity for several days [45].
- Reference Standard Spike-In: To mimic a clinical sample and validate the assay's sensitivity, Mimix Multiplex I cfDNA Set Reference Standard (HD780) from Horizon Discovery was spiked into the preserved urine sample [44]. This standard consists of 8 onco-relevant mutations at different allelic frequencies: 5%, 1%, 0.1%, and 0% (100% Wildtype) [44].
cfDNA Extraction and Quality Control
After preservation, the next step involves isolating the cfDNA while maximizing yield and purity.
- Extraction Protocol: The preserved urine samples were centrifuged at 4,000 × g for 15 minutes before the start of the protocol [44]. cfDNA was then extracted from a 4 mL input volume using Omega Bio-tek’s Mag-Bind cfDNA Kit, following a modified protocol for urine samples, and eluted in a 50 µL volume [44].
- Yield and Quality Assessment: The yield and quality of the extracted cfDNA were evaluated using the Cell-Free DNA ScreenTape Assay on the Agilent 4150 TapeStation system [44]. This analysis assesses the fragment size profile and the fraction of cfDNA relative to the total DNA extracted. A successful extraction shows dark bands concentrated between 150 and 200 base pairs, indicating high-yielding, high-quality cfDNA [44]. The presence of a higher molecular weight band indicates genomic DNA contamination, which was observed in samples containing actual urine [44].
Table 1: Average cfDNA Yield from Different Experimental Conditions
| Experimental Group | Average Estimated Yield (ng) |
|---|---|
| Urine + UAS (No spike-in control) | 58.2 ng |
| Spike-in + Urine + UAS | 226.5 ng |
| Spike-in + Elution Buffer (No urine, no preservative control) | 161.9 ng |
| Spike-in + UAS (No urine control) | 186.1 ng |
Data sourced from [44].
Droplet Digital PCR for Rare Mutation Detection
Droplet Digital PCR (ddPCR) was selected as the downstream detection method due to its superior sensitivity and ability to absolutely quantify rare alleles without the need for a standard curve [17].
- Principle of ddPCR: In ddPCR, the PCR reaction mixture is partitioned into thousands of nanoliter-sized water-in-oil droplets, effectively creating a massive number of individual PCR reactions [17]. After end-point amplification, each droplet is analyzed for fluorescence. The fraction of positive droplets is then counted, and the original concentration of the target molecule in the sample is calculated using Poisson statistics [17]. This partitioning allows for the precise detection and absolute quantification of rare targets, such as somatic mutations, present at very low frequencies in a background of wild-type DNA [3].
- ddPCR Reaction Setup: For this study, 15 ng of the extracted cfDNA was mixed with Bio-Rad’s ddPCR Supermix for Probes (No dUTP) in a total reaction volume of 300 µL according to the manufacturer’s protocol [44]. Two assays were developed to detect:
- NRAS variant A59T
- EGFR variant L858R
- The assays used a FAM-labeled probe for the mutant allele and a SUN-labeled probe for the wild-type allele [44]. The droplets were generated using a Bio-Rad QX200 Droplet Generator, amplified on a C1000 Touch Thermal Cycler, and subsequently scanned using a QX200 Droplet Reader [44]. Data were analyzed using Bio-Rad’s QuantaSoft Analysis Pro software [44].
Key Findings and Data Analysis
Sensitivity and Concordance of Mutation Detection
The ddPCR assays successfully detected mutant alleles for both NRAS and EGFR genes at all tested allelic frequencies down to 0.1%, demonstrating the high sensitivity of the integrated workflow [44]. The following table summarizes the concordance between the expected and observed allelic frequencies.
Table 2: Concordance Analysis of Observed vs. Expected Allelic Frequencies
| Gene | Spiked Allelic Frequency | Observed Allelic Frequency (Spike-in + Urine + UAS) | Within Expected Range? |
|---|---|---|---|
| NRAS | 5% | ~5% | Yes |
| NRAS | 1% | ~1% | Yes |
| NRAS | 0.1% | ~0.1% | Yes |
| EGFR | 5% | 2.5%* | No (Acceptance: 3.5-6.5%) |
| EGFR | 1% | ~1% | Yes |
| EGFR | 0.1% | ~0.1% | Yes |
Data synthesized from [44]. *The outlier for EGFR at 5% was attributed to a likely experimental error, as all other data points showed excellent concordance.
The data show that the observed allelic frequencies for the NRAS gene were in excellent concordance with the expected frequencies for all experimental groups and were well within the manufacturer's acceptance criteria [44]. A similar trend was observed for the EGFR assay, with the exception of the 5% allelic frequency spike-in in the urine sample, where the observed frequency was lower; this was considered a likely experimental error rather than a systematic failure [44]. Crucially, mutant alleles at frequencies as low as 0.1% were reliably detected, underscoring the workflow's capability for rare variant detection [44].
The Critical Role of Sample Stabilization
The results underscore the non-negotiable requirement for urine stabilization. Research has demonstrated that in unstabilized urine, ucfDNA degrades rapidly, with a substantial increase of smaller fragments (<150 bp) occurring after 6 hours and minimal quantities remaining after 72 hours [45]. One study found that native urine retained just 16% of the initial cfDNA after 3 days, indicating more than 80% degradation [45]. In contrast, when using stabilized urine, the number of amplifiable gene copies (e.g., PIK3CA wild type) remained relatively constant over time, preserving the original genetic information necessary for accurate mutation detection [45].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Urinary cfDNA Workflow
| Product Name | Function / Application |
|---|---|
| Colli-Pee UAS Preservative (DNA Genotek) | Stabilizes urine samples at collection to prevent degradation of cfDNA during storage and transport. |
| Mag-Bind cfDNA Kit (Omega Bio-tek) | Solid-phase magnetic bead-based extraction of high-quality cfDNA from preserved urine and other liquid biopsy samples. |
| Mimix cfDNA Reference Standards (Horizon Discovery) | Multiplexed, quantitative reference materials containing defined oncogenic mutations at specific allelic frequencies for assay validation and quality control. |
| ddPCR Supermix for Probes (Bio-Rad) | Optimized PCR mastermix containing DNA polymerase, dNTPs, and buffer, formulated for droplet digital PCR applications. |
| Cell-Free DNA ScreenTape Assay (Agilent) | Automated electrophoresis assay for sizing, quantifying, and assessing the quality of cell-free DNA extracts. |
Discussion: Principles of ddPCR in Rare Allele Research
The success of this case study is fundamentally rooted in the principles of ddPCR, which make it uniquely suited for rare allele detection in complex backgrounds like ucfDNA. The core advantage of ddPCR over traditional qPCR is its partitioning step, which enhances sensitivity and enables absolute quantification.
- Superior Sensitivity and Precision for Low-Abundance Targets: ddPCR's partitioning effectively dilutes PCR inhibitors present in biological samples like urine across thousands of droplets, reducing their local concentration and mitigating their impact on amplification [32] [17]. This allows for the analysis of samples that would otherwise yield variable and unreliable results with qPCR [32]. Furthermore, by partitioning a sample into thousands of reactions, ddPCR enriches rare targets, allowing for the detection of mutations with an allelic frequency as low as 0.001% in some configurations, far surpassing the 1-5% typical limit of qPCR [17].
- Absolute Quantification without Standard Curves: Unlike qPCR, which relies on Cq values and a standard curve for relative or absolute quantification, ddPCR uses end-point fluorescence detection and binomial Poisson statistics for direct, absolute quantification of target DNA [32] [17]. This eliminates variability associated with standard curve construction and makes the data more reproducible and reliable, a critical feature for clinical applications [17].
- Application in Liquid Biopsies and Beyond: The principles demonstrated in this case study extend to various research areas. In oncology, ddPCR is widely used for monitoring treatment response and resistance via plasma cfDNA analysis [46] [17]. It also finds applications in non-invasive prenatal testing (NIPT) for detecting fetal alleles in maternal plasma [17] and in infectious disease diagnostics for precise viral load quantification [17]. The method's robustness and sensitivity make it an indispensable tool in the modern molecular laboratory.
This case study demonstrates a complete and robust workflow for the detection of rare NRAS and EGFR mutations from urinary cfDNA. The integration of immediate sample preservation with Colli-Pee UAS preservative, efficient extraction using the Mag-Bind cfDNA kit, and highly sensitive detection via droplet digital PCR provides a reliable solution for non-invasive molecular diagnostics. The data confirm that this workflow can detect mutant alleles at frequencies as low as 0.1%, a level critical for identifying emerging therapy-resistant clones in cancer patients.
The success of this approach underscores the transformative potential of urinary cfDNA when paired with appropriate pre-analytical and analytical methods. For researchers and drug development professionals, this workflow offers a validated template for implementing urinary cfDNA analysis in studies of urological cancers, non-urological cancers via trans-renal DNA, and other pathological conditions. As the field of liquid biopsy continues to evolve, the combination of a truly non-invasive sample like urine with the precision of ddPCR is poised to play an increasingly important role in personalized medicine, enabling frequent monitoring, early detection of relapse, and guiding targeted therapeutic strategies with minimal patient burden.
Navigating Challenges: A Practical Guide to Optimizing ddPCR for Complex Samples
Droplet Digital PCR (ddPCR) represents a significant advancement in molecular diagnostics, enabling the absolute quantification of nucleic acid targets by partitioning a sample into thousands to millions of individual droplets [2]. This technology is particularly valuable for detecting rare genetic mutations, such as in circulating tumor DNA (ctDNA) analysis, where it can identify mutant alleles amidst a vast background of wild-type sequences [47]. However, a persistent technical challenge known as the "rain" phenomenon can compromise data interpretation and assay sensitivity. In ddPCR, "rain" refers to the population of droplets that fall between the clearly defined negative and positive clusters on the fluorescence amplitude plot [48]. These intermediate-signal droplets create a nebulous background, obscuring the clear separation needed for accurate binary quantification.
The presence of rain is particularly problematic in rare event detection, such as monitoring minimal residual disease in oncology, where the false classification of a small number of droplets can significantly impact clinical interpretation [48]. For instance, in BCR-ABL monitoring for chronic myelogenous leukemia, optimized ddPCR must reliably detect transcript levels as low as 1/100,000 with minimal background false-positive signals [48]. Understanding the origins of rain and implementing strategies to minimize it are therefore critical for maximizing the analytical performance of ddPCR assays in research and clinical settings. This phenomenon stems from multiple factors in the experimental workflow, which this guide will explore in detail while providing actionable optimization strategies.
Fundamental Causes of Rain in ddPCR
The rain phenomenon in ddPCR arises from technical imperfections that prevent complete binary separation between positive and negative droplets. A primary cause is suboptimal PCR amplification efficiency within individual partitions. When amplification is inefficient, droplets containing the target molecule may generate insufficient fluorescence, causing them to appear in the intermediate signal region rather than the definitive positive cluster [48]. This inefficiency can result from several factors, including imperfect primer/probe design, inhibition of the polymerase enzyme, or suboptimal thermal cycling conditions.
Another significant contributor is probe chemistry issues. Hydrolysis probes, the most common detection method in ddPCR, rely on complete cleavage during amplification to generate a robust fluorescence signal. Incomplete cleavage, premature probe degradation, or inappropriate probe concentrations can all lead to intermediate fluorescence levels [49]. The design of the probes themselves is also crucial; probes with imperfect specificity may hybridize to non-target sequences, generating background signal that elevates the negative droplet cluster and narrows the separation from positive droplets [48].
Template quality and quantity further influence rain formation. Degraded or fragmented DNA, common in samples like formalin-fixed paraffin-embedded (FFPE) tissues or cell-free DNA, may amplify less efficiently [49]. Additionally, overloading the reaction with too much template DNA can lead to the formation of double-positive droplets (containing both wild-type and mutant alleles) in mutation detection assays, creating an intermediate signal population that complicates clear cluster identification [49]. This is particularly relevant in drop-off ddPCR assays, where optimal separation depends on most droplets containing no more than one copy of the targeted gene [49].
Table 1: Primary Causes of Rain in ddPCR and Their Effects on Data Quality
| Cause Category | Specific Factor | Impact on Signal Separation |
|---|---|---|
| PCR Amplification | Inefficient primer design | Reduced fluorescence amplitude in positive droplets |
| Suboptimal thermal cycling | Incomplete target amplification | |
| Polymerase inhibition | Diminished signal generation | |
| Probe Chemistry | Incomplete hydrolysis | Intermediate fluorescence levels |
| Non-specific binding | Elevated background in negative droplets | |
| Suboptimal concentration | Weak signal development | |
| Template Quality | DNA degradation/fragmentation | Reduced amplification efficiency |
| Excessive template loading | Increased double-positive events | |
| Impurities/inhibitors | Interference with amplification |
Experimental Optimization Strategies
Primer and Probe Design
The foundation of a robust ddPCR assay with minimal rain lies in optimal primer and probe design. For primer design, specific parameters dramatically impact performance. Primers should target an amplicon smaller than 150 base pairs, especially when analyzing fragmented DNA sources like cfDNA [49]. The melting temperature (Tm) should be between 55-70°C, calculated using the Santa Lucia 1998 thermodynamic parameters with 50 mM salt concentration and 300 nM oligonucleotide concentration [49]. Select sequences with a GC content of 50-60%, avoid repeats of Gs and Cs longer than 3 bases, and prioritize primers with Gs and Cs at their 3' end when possible [49]. Always verify primer specificity using tools such as PrimerBlast to avoid non-target amplification.
Probe design requires equally meticulous attention. Hydrolysis probes should be labeled with a fluorescent reporter at the 5' end and a quencher at the 3' end [49]. In drop-off ddPCR assays, which can detect multiple mutations within a target region using a single pair of wild-type hydrolysis probes, the positioning of the two probes is critical [49]. Probe 1 should cover a non-variable sequence adjacent to the target region, while Probe 2 must be complementary to the wild-type sequence where mutations are expected [49]. This design enables Probe 2 to discriminate wild-type and mutant alleles through suboptimal hybridization to mutant sequences. For duplex assays detecting two targets simultaneously, ensure that the fluorophores selected have non-overlapping emission spectra and are compatible with the detection channels of your ddPCR instrument.
Reaction Optimization
Systematic optimization of the ddPCR reaction conditions can significantly reduce rain by enhancing amplification efficiency and signal-to-noise ratio. Thermal cycling parameters must be empirically optimized for each assay. While standard cycling conditions provide a starting point, adjustments to annealing temperature and extension time can dramatically improve performance. A gradient thermal cycler can help identify the optimal annealing temperature that maximizes fluorescence separation between positive and negative droplets.
Reaction composition also plays a crucial role. As demonstrated in the development of an optimized duplex ddPCR for BCR-ABL, careful titration of primer and probe concentrations can increase target signals by two- to five-fold, significantly enhancing resolution between positive and negative droplets [48]. Magnesium concentration, often suboptimal in standard buffers, should be optimized as it directly affects polymerase processivity and fidelity. Additionally, the inclusion of additives such as betaine or DMSO can help amplify difficult templates by reducing secondary structures, thereby minimizing rain formation.
Table 2: Key Research Reagent Solutions for ddPCR Assay Development
| Reagent Category | Specific Examples | Function in Assay |
|---|---|---|
| Polymerase Systems | Thermostable DNA polymerases | Enzymatic amplification of target sequences |
| dNTP mixes | Nucleotide substrates for DNA synthesis | |
| Probe Chemistry | Hydrolysis probes (FAM, HEX/VIC) | Sequence-specific detection with fluorescent reporter |
| Quenchers (BHQ, TAMRA) | Fluorescence quenching in uncleaved probes | |
| Sample Preparation | Cell-free DNA extraction kits | Isolation of circulating nucleic acids |
| Streck Cell-Free DNA BCT tubes | Blood collection tube for cfDNA stabilization | |
| Partitioning | Droplet generation oil | Immiscible phase for creating water-in-oil emulsions |
| Surfactants | Stabilization of droplets during thermal cycling |
Droplet Generation and Reading
The physical processes of droplet generation and analysis represent additional critical points for rain minimization. During droplet generation, ensure consistent and monodisperse droplet formation by using fresh, properly formulated oil and surfactant combinations. Different ddPCR systems require specific surfactant compositions to maintain droplet integrity throughout thermal cycling; using incompatible surfactants can lead to droplet coalescence or degradation, contributing to rain [2].
The endpoint fluorescence reading process also requires optimization. Adjust the detection threshold carefully to distinguish between true positive and negative droplets while accounting for the rain population. Some ddPCR instruments offer amplitude threshold settings that can be fine-tuned based on the specific assay characteristics. When setting these thresholds, it's advisable to include multiple negative controls to establish the baseline fluorescence of genuine negative droplets and better identify the rain population.
For challenging assays, consider technical replication to improve detection reliability. As demonstrated in BCR-ABL detection, analyzing quadruplicates instead of duplicates increased the detection rate of MR4.5 (a 4.5-log reduction in transcript level) from 92% to 100% [48]. This approach helps account for stochastic effects in rare target detection and provides more robust data despite the presence of some rain.
Workflow and Troubleshooting
The following workflow diagram illustrates a comprehensive approach to addressing rain in ddPCR assays, integrating the optimization strategies discussed in previous sections:
Step-by-Step Troubleshooting Protocol
When confronting significant rain in a ddPCR assay, follow this systematic troubleshooting protocol to identify and address the root cause:
Initial Assessment: Begin by examining the raw fluorescence data to characterize the rain pattern. Determine whether the intermediate signals appear closer to the negative cluster, positive cluster, or as a continuous spread between them. This pattern offers initial clues about potential causes.
Primer and Probe Verification: Check the specificity of your primer and probe sequences using in silico tools. Verify that amplicon size is appropriate for your template material (especially important for fragmented DNA). For mutation detection assays using the drop-off approach, confirm that Probe 2 is positioned correctly to detect the expected mutations through suboptimal hybridization [49].
Thermal Cycling Optimization: Perform a thermal gradient experiment to identify the optimal annealing temperature. The ideal temperature will maximize the separation between positive and negative clusters while minimizing intermediate populations. Also ensure that extension times are sufficient for complete amplification, particularly for longer targets.
Reaction Composition Titration: Systematically titrate primer and probe concentrations. A typical starting point is 900 nM for primers and 250 nM for probes, but optimal concentrations may vary. As demonstrated in BCR-ABL assay development, proper optimization can increase target signals by several-fold, significantly improving resolution [48].
Template Quality Control: Assess template quality using appropriate methods. For cfDNA, use a fluorometer with high-sensitivity assays to accurately quantify the typically low concentrations [49]. Ensure the DNA is not degraded and is free of inhibitors that could cause incomplete amplification. Avoid excessive template input, which increases the probability of double-positive droplets.
Partitioning and Detection Verification: Confirm that droplet generation is producing a consistent, monodisperse population. Check that the oil and surfactant are fresh and properly formulated for your ddPCR system. Verify instrument calibration and fluorescence detection settings according to manufacturer recommendations.
Advanced Rain Reduction Strategies
For particularly challenging applications, consider these advanced strategies:
Digital Annealing Algorithm: Following data acquisition, computational approaches can help distinguish true positive signals from rain. These methods analyze the distribution pattern of droplets and apply statistical models to reclassify intermediate signals based on their proximity to established clusters.
Background Limitation Techniques: In rare allele detection, limit the background false-positive rate by establishing strict thresholds based on extensive negative controls. In the optimized BCR-ABL assay, researchers achieved a background false-positive rate of just 5% through systematic optimization [48].
Multiplexing Considerations: When designing multiplex assays, ensure sufficient spectral separation between fluorophores to prevent channel bleed-through, which can create rain-like effects. Proper compensation using single-color controls can minimize this issue.
The rain phenomenon presents a significant challenge in ddPCR, particularly for applications requiring high sensitivity and precision such as rare allele detection in oncology research [47]. However, through systematic investigation of potential causes and implementation of targeted optimization strategies, researchers can significantly improve signal separation and data reliability. The key lies in a methodical approach addressing primer and probe design, reaction composition, thermal cycling conditions, template quality, and instrumental factors.
As ddPCR technology continues to evolve with applications expanding in clinical diagnostics [2], drug development, and biomedical research, mastering these optimization principles becomes increasingly important. The strategies outlined in this guide provide a framework for developing robust, rain-minimized assays capable of detecting rare mutations with the sensitivity and precision required for advanced research and clinical applications. By implementing these practices, researchers can maximize the analytical performance of their ddPCR assays, ensuring reliable results even in the most demanding detection scenarios.
Digital PCR (dPCR), particularly droplet digital PCR (ddPCR), demonstrates superior robustness against PCR inhibitors compared to quantitative PCR (qPCR) through the core principle of sample partitioning. This technical guide explores the mechanisms by which partitioning mitigates inhibition in complex matrices, enabling precise detection of rare alleles and low-abundance targets. By examining specific inhibitor classes, their modes of action, and experimental validation data, this review provides researchers with methodologies to optimize ddPCR assays for challenging samples in drug development and clinical diagnostics.
The analysis of nucleic acids from complex sample matrices represents a significant challenge in molecular diagnostics and biomedical research. Samples such as wastewater, blood, soil, and processed tissues contain various substances that interfere with polymerase chain reaction (PCR) amplification, collectively known as PCR inhibitors [50]. These compounds can originate from the sample matrix itself, from target cells or tissues, or from reagents added during sample processing. In standard quantitative PCR (qPCR), these inhibitors affect amplification efficiency and fluorescent signaling, leading to skewed quantification, false negatives, and inaccurate results [50] [51]. The fundamental strength of PCR—amplifying specific sequences from minimal target molecules—becomes compromised when inhibitors are present, particularly for applications requiring high sensitivity such as rare allele detection, circulating tumor DNA analysis, and pathogen surveillance [50] [52].
The emergence of digital PCR (dPCR) and its droplet-based implementation (ddPCR) has provided a powerful alternative for nucleic acid quantification in inhibitor-rich environments. The core innovation lies in sample partitioning, where a single PCR reaction is divided into thousands to millions of nanoliter-scale reactions [53] [16]. This partitioning confers inherent robustness against PCR inhibitors through both dilution and statistical effects. Understanding these mechanisms is crucial for researchers and drug development professionals working with challenging sample types where inhibitor removal through purification is impractical or would result in unacceptable target loss [50].
Mechanisms of PCR Inhibition
PCR inhibitors interfere with amplification through diverse biochemical mechanisms, primarily targeting DNA polymerase activity, nucleic acid integrity, or fluorescent detection.
Biochemical Targets of Common Inhibitors
- DNA Polymerase Interference: Many inhibitors directly affect DNA polymerase activity. Humic substances from soil and plant materials, immunoglobulin G from blood, and complex polysaccharides from feces can bind to or denature DNA polymerases, reducing their enzymatic efficiency [50] [51].
- Nucleic Acid Interaction: Some inhibitors interact directly with nucleic acids. Humic acids share structural similarities with DNA and can co-purify with nucleic acids, while certain metal ions may promote nucleic acid degradation or compromise integrity [50].
- Fluorescence Quenching: Substances that quench fluorescence, such as colored compounds from plant materials or heme from blood, interfere with detection in probe-based assays by absorbing the emitted fluorescence, leading to underestimation of target concentration [50]. This occurs through collisional quenching (where the quenching molecule contacts the excited-state fluorophore) or static quenching (where the quencher forms a non-fluorescent complex with the fluorophore) [50].
- Cofactor Chelation: Certain inhibitors, including EDTA and heparin (common anticoagulants in blood collection), chelate magnesium ions that are essential cofactors for DNA polymerase activity [50] [51].
Table 1: Common PCR Inhibitors and Their Mechanisms of Action
| Inhibitor Category | Example Sources | Primary Mechanism | Impact on PCR |
|---|---|---|---|
| Humic Substances | Soil, sediment, plants | DNA polymerase binding, fluorescence quenching | Reduced amplification efficiency, false negatives |
| Blood Components | Blood, serum, plasma | Hemoglobin: fluorescence quenching; IgG: polymerase binding; Heparin: Mg²⁺ chelation | Reduced fluorescence signal, inhibited polymerization |
| Complex Polysaccharides | Feces, plant tissues | Polymerase interaction, increased viscosity | Delayed amplification, reduced efficiency |
| Metal Ions | Industrial effluents, wastewater | Nucleic acid degradation, enzyme denaturation | Template degradation, reduced polymerase activity |
| Detergents | Sample processing reagents | Enzyme denaturation, membrane disruption | Complete reaction failure at high concentrations |
Impact on qPCR versus dPCR
In qPCR, inhibitors directly affect the amplification efficiency, which is reflected in the quantification cycle (Cq) values. Since quantification relies on comparing Cq values to a standard curve, any inhibition-induced efficiency reduction skews quantification accuracy [50]. In contrast, dPCR utilizes end-point detection and absolute quantification based on Poisson statistics, making it less dependent on amplification kinetics [50] [53]. While inhibitors may still prevent amplification in some partitions, the remaining unaffected partitions provide accurate quantification of the target molecules that were successfully amplified [50] [16].
How Partitioning Confers Robustness
The partitioning of a PCR reaction into thousands of individual compartments provides dPCR with inherent resistance to inhibitors through several interconnected mechanisms.
Dilution Effect and Limited Inhibitor-Polymerase Interaction
When a sample containing PCR inhibitors is partitioned into numerous nanoliter-sized reactions, inhibitor molecules are distributed across all partitions. This dramatically reduces the local concentration of inhibitors in each individual reaction chamber [54]. Since the probability of inhibitor-polymerase interaction depends on concentration, this dilution effect decreases the likelihood that an inhibitor molecule will encounter and interfere with the DNA polymerase in any given partition [50]. Partitions that receive no or minimal inhibitor molecules will amplify normally, providing accurate binary data (positive/negative) for quantification.
Partitioning Dilutes Inhibitors Across Reactions
End-point Detection and Poisson Statistics
Unlike qPCR, which relies on amplification kinetics and the critical quantification cycle (Cq), dPCR uses end-point detection [50] [53] [16]. After amplification is complete, each partition is simply scored as positive or negative based on fluorescence intensity. This binary readout is then analyzed using Poisson statistics to calculate the absolute concentration of target molecules in the original sample [16]. Since this approach does not depend on the rate or efficiency of amplification, it is inherently less affected by inhibitors that merely slow amplification rather than prevent it entirely [50]. Partitions with sufficient target and minimal inhibitor will still amplify to detectable levels, even if amplification kinetics are slower, preserving quantification accuracy.
Experimental Evidence of Enhanced Robustness
Multiple studies have demonstrated dPCR's superior performance in inhibitor-rich environments. Research on wastewater samples for SARS-CoV-2 surveillance found that ddPCR provided more reliable viral load measurements compared to qPCR in the presence of inhibitors [51]. Similarly, in forensic science, dPCR has shown better quantification accuracy for samples containing humic acids, with complete inhibition occurring at significantly higher humic acid concentrations compared to qPCR [50]. This enhanced tolerance extends to various sample types, including blood, feces, and soil extracts [50] [54].
Table 2: Comparative Performance of qPCR vs. dPCR in Inhibitor-Rich Matrices
| Sample Matrix | Primary Inhibitors | qPCR Impact | dPCR Impact | Experimental Findings |
|---|---|---|---|---|
| Wastewater | Humic substances, metals, polysaccharides | Significant Cq delays, underestimation | Minimal impact on quantification | ddPCR detected SARS-CoV-2 in undiluted samples where qPCR failed [51] |
| Blood/Serum | Hemoglobin, immunoglobulin G, heparin | Reduced efficiency, fluorescence quenching | Maintained accuracy in quantification | dPCR enabled direct detection without extensive purification [50] [55] |
| Soil/Sediment | Humic and fulvic acids | Complete inhibition at low concentrations | Partial inhibition only at high concentrations | dPCR provided accurate quantification at 10x higher humic acid concentrations [50] |
| Feces | Complex polysaccharides, bilirubin | False negatives, variable efficiency | Consistent detection across samples | Partitioning reduced matrix effects, improving reproducibility [54] |
| Plant Tissues | Polyphenols, polysaccharides | Inhibited amplification, delayed Cq | Reliable quantification with minor efficiency loss | dPCR successfully quantified targets in complex plant extracts [56] |
Methodologies for Enhanced Inhibition Resistance
Beyond the inherent benefits of partitioning, several methodological approaches can further improve dPCR performance in complex matrices.
Sample Preparation and Inhibitor Removal
Effective sample preparation remains crucial for handling severely inhibited samples:
- Silica-based purification: Effectively removes many inhibitors but may result in DNA loss (10-80% recovery) [50].
- Magnetic bead-based extraction: Provides efficient purification suitable for automation, with better inhibitor removal than simple extraction methods [50].
- Dilution approach: Simple dilution of nucleic acid extracts reduces inhibitor concentration but also decreases target concentration, potentially affecting sensitivity [51]. A 10-fold dilution is commonly used but may be insufficient for highly inhibited samples [51].
- Inhibitor-tolerant polymerases: Specialized enzyme blends containing inhibitor-resistant DNA polymerases (e.g., those derived from extreme thermophiles) can significantly improve amplification in challenging samples [50].
PCR Enhancers and Additives
Various chemical additives can counteract specific inhibition mechanisms when added to the PCR reaction mix:
Table 3: Research Reagent Solutions for Combating PCR Inhibition
| Reagent Solution | Concentration Range | Mechanism of Action | Target Inhibitors |
|---|---|---|---|
| Bovine Serum Albumin (BSA) | 0.1-0.5 μg/μL | Binds to inhibitors, prevents polymerase interaction | Humic substances, polyphenols, blood components |
| T4 Gene 32 Protein (gp32) | 0.1-0.5 nM | Stabilizes single-stranded DNA, binds humic acids | Humic acids, tannins |
| Dimethyl Sulfoxide (DMSO) | 1-5% | Lowers DNA melting temperature, destabilizes secondary structures | Complex templates, some polysaccharides |
| Tween-20 | 0.1-1% | Non-ionic detergent, counteracts inhibitory effects on Taq | Fecal contaminants, some tissue extracts |
| Glycerol | 1-5% | Enzyme stabilizer, protects against degradation | Multiple inhibitor classes |
| Formamide | 1-3% | Destabilizes DNA helix, reduces melting temperature | Secondary structures, GC-rich targets |
Evaluations of these enhancers in wastewater samples found that BSA and gp32 provided the most significant improvements in detection, sometimes enabling detection in samples that were completely inhibited without enhancers [51]. The effectiveness of specific enhancers varies by sample type and inhibitor profile, requiring empirical optimization.
Digital PCR Protocol Optimization
Optimized dPCR Workflow for Complex Matrices
Applications in Rare Allele Detection
The robustness of partitioned dPCR systems is particularly valuable for detecting rare genetic variants in complex backgrounds, a crucial requirement in cancer diagnostics and disease monitoring.
Circulating Tumor DNA and Cancer Mutations
Detection of rare somatic mutations in circulating tumor DNA (ctDNA) represents one of the most demanding applications for dPCR. Studies have demonstrated dPCR's ability to detect mutant alleles at frequencies as low as 0.01%-0.1% in a background of wild-type DNA [52] [13] [57]. This sensitivity is crucial for early cancer detection, treatment monitoring, and resistance mutation screening. For example, multiplex dPCR assays have been developed to detect resistance mutations in BTK inhibitors with superior sensitivity compared to next-generation sequencing, enabling identification of emerging resistant clones earlier during treatment [57].
Enhanced Specificity with Specialized Primers
The combination of partitioning with specialized primer designs further enhances specificity for rare allele detection. SuperSelective primers employ a unique design with a long 5' "anchor" sequence for strong hybridization and a very short 3' "foot" sequence containing the interrogated nucleotide, enabling highly selective amplification of single-nucleotide variants (SNVs) even in vast excess of wild-type sequences [52]. When coupled with ddPCR, this approach has demonstrated precise quantification of SNVs at frequencies below 0.5% with linear correlation between expected and measured variant allele frequencies [52].
Partitioning confers exceptional robustness against PCR inhibitors through fundamental physical and statistical principles. By diluting inhibitors across thousands of partitions and utilizing end-point detection with Poisson statistics, dPCR maintains quantification accuracy in complex matrices where qPCR fails. Combined with optimized sample preparation, inhibitor-resistant polymerases, and chemical enhancers, partitioning technologies enable reliable detection of rare alleles and low-abundance targets in clinically and environmentally relevant samples. For researchers in drug development and clinical diagnostics working with challenging sample types, dPCR provides a powerful tool that transforms inhibitor management from a technical obstacle into a manageable parameter through its partitioned architecture.
Optimizing Thermal Cycling Conditions and Primer/Probe Concentrations
In the field of molecular diagnostics, droplet digital PCR (ddPCR) has emerged as a powerful third-generation technology capable of absolute quantification of nucleic acids with exceptional sensitivity. This calibration-free technology partitions a PCR reaction into thousands of nanoliter-sized droplets, enabling single-molecule detection through end-point fluorescence measurement and Poisson statistics [2]. For rare allele detection—a application crucial in oncology for identifying circulating tumor DNA (ctDNA) in liquid biopsies, monitoring minimal residual disease, and detecting emerging treatment-resistant mutations—optimal assay performance is paramount [58] [3]. The ability to reliably detect mutations present at frequencies as low as 0.1% requires meticulous optimization of thermal cycling conditions and primer/probe concentrations [58] [44]. Even minor deviations in these parameters can significantly impact amplification efficiency, specificity, and ultimately, the detection limit of rare mutant alleles against a background of abundant wild-type sequences.
This technical guide provides a comprehensive framework for optimizing these critical parameters, with a specific focus on applications in rare allele detection research. The principles and methodologies outlined herein are designed to help researchers achieve the precision, sensitivity, and reproducibility required for confident detection of low-frequency genetic variants, thereby enabling advancements in cancer research, liquid biopsy development, and personalized medicine approaches.
Core Principles of ddPCR in Rare Mutation Detection
Droplet digital PCR's exceptional sensitivity for rare mutation detection stems from its fundamental working principle: sample partitioning. By dividing a PCR reaction into thousands to millions of individual compartments, ddPCR effectively enriches low-level targets by isolating them from the dominant wild-type background [2] [58]. Each partition acts as an independent micro-reactor, with target-containing droplets generating a fluorescent signal while those without target remain dark. This binary readout (positive or negative) enables absolute quantification of target molecules without the need for standard curves, a significant advantage over quantitative PCR (qPCR) [2].
For rare allele detection, researchers typically employ a duplex assay design using two differently labeled hydrolysis probes (e.g., TaqMan probes)—one specific for the wild-type sequence and another for the mutant allele [3]. The probe targeting the mutant allele is typically labeled with a fluorophore such as FAM, while the wild-type probe is labeled with HEX, VIC, or another spectrally distinct dye [44]. This design allows simultaneous detection of both alleles in a single reaction, with the rare mutant fraction calculated based on the ratio of mutant-positive to total positive partitions [3]. The extreme sensitivity of this approach enables detection of mutant allele frequencies as low as 0.1% under optimized conditions [58], making it invaluable for detecting minimal residual disease, characterizing tumor heterogeneity, and monitoring treatment response through liquid biopsies [2] [58].
Optimizing Thermal Cycling Conditions
Thermal cycling parameters directly impact amplification efficiency, specificity, and the robustness of ddPCR assays. Suboptimal conditions can lead to reduced fluorescence amplitude, non-specific amplification, and impaired partition classification, particularly problematic when detecting rare mutations against a high wild-type background.
Key Thermal Cycling Parameters
Initial Denaturation: A prolonged initial denaturation step (typically 10 minutes at 95°C) is critical for ensuring complete separation of DNA strands and activating hot-start DNA polymerases [59] [60]. This step enhances reaction specificity and improves the reproducibility of amplification kinetics across all partitions.
Denaturation: Standard denaturation conditions in ddPCR protocols typically involve temperatures of 94–95°C for 30 seconds [59] [60]. This brief, high-temperature step ensures DNA strand separation without significantly compromising polymerase activity over many cycles.
Annealing Temperature: The annealing temperature is arguably the most critical parameter requiring optimization, as it determines primer-binding specificity. The optimal temperature must be empirically determined for each primer-probe set and typically ranges from 58°C to 62°C [59] [60]. Higher annealing temperatures generally enhance specificity but may reduce overall signal intensity if set too high. Research demonstrates that efficient primer-probe sets maintain stable amplification efficiency even at higher annealing temperatures (e.g., 62°C) [60].
Annealing/Eextension Time: A common annealing and extension time is 1 minute [59], though this can sometimes be reduced for shorter amplicons. Sufficient time must be allocated for complete primer extension, especially for amplicons exceeding 150 base pairs.
Cycle Number: Digital PCR assays typically employ 40-50 amplification cycles [59] [60] to ensure sufficient signal generation from partitions containing single DNA molecules. This higher cycle number compared to many qPCR applications is necessary because the endpoint fluorescence measurement must distinguish positive from negative partitions with high confidence.
Optimization Strategy and Experimental Approach
A systematic approach to thermal cycling optimization should begin with a temperature gradient experiment across the theoretical annealing temperature range of your primers. Evaluate performance using both positive controls (samples with known mutations) and negative controls (wild-type only samples). The optimal conditions will maximize the separation between positive and negative droplet clusters while minimizing non-specific amplification [60].
Recent advances include the development of Ultra-Rapid ddPCR (UR-ddPCR), which utilizes innovative thermal cycling methods such as preheated water baths and thin stainless-steel capillaries for heat transfer to reduce overall processing time to approximately 15 minutes while maintaining analytical performance comparable to conventional ddPCR (R² = 0.995) [61].
Table 1: Standard vs. Optimized Thermal Cycling Conditions for Rare Allele Detection
| Parameter | Standard Conditions | Optimized Range | Considerations for Rare Alleles |
|---|---|---|---|
| Initial Denaturation | 95°C for 10 min [59] [60] | 95°C for 5–10 min | Critical for complex samples; do not reduce time with inhibitor-prone matrices |
| Denaturation | 94°C for 30 sec [60] | 94–95°C for 20–30 sec | Shorter times may be possible with rapid-cycling instruments |
| Annealing Temperature | 58°C [59] | 58–62°C [60] | Must be empirically determined; higher temperatures improve specificity for rare mutants |
| Annealing/Extension Time | 60 sec [59] | 45–60 sec | Sufficient for amplicons <200 bp; increase for longer targets |
| Cycle Number | 45 cycles [59] | 40–50 cycles [60] | Higher cycles (50) enhance sensitivity for very low-abundance targets |
| Final Extension | 98°C for 10 min [59] | 98°C for 5–10 min | Ensures complete extension; may inactivate enzymes |
Optimizing Primer and Probe Concentrations
Optimal primer and probe concentrations are fundamental to achieving maximum amplification efficiency while maintaining assay specificity. Improper concentrations can lead to reduced fluorescence amplitude, non-specific amplification, and impaired ability to distinguish genuine rare mutations from background noise.
Recommended Concentration Ranges
Based on established ddPCR protocols, the following concentration ranges typically serve as a starting point for optimization:
- Primers: Final concentration of 500 nM for each forward and reverse primer [59] [3]. This concentration is generally sufficient to drive efficient amplification without promoting non-specific binding or primer-dimer formation.
- Probes: Final concentration of 250 nM for each hydrolysis probe [59] [3]. This concentration typically provides strong fluorescent signals without exacerbating background fluorescence or incurring unnecessary costs.
These values align with concentrations commonly used in qPCR, though they must be validated specifically for ddPCR applications due to differences in reaction dynamics and detection methods [3].
Optimization Experimental Protocol
Prepare a Master Mix containing all reaction components except primers and probes, including ddPCR supermix, DNA template, and nuclease-free water.
Design a Matrix Experiment testing multiple combinations of primer and probe concentrations:
- Primers: Test 400 nM, 500 nM, and 600 nM
- Probes: Test 200 nM, 250 nM, and 300 nM
Generate Droplets and perform thermal cycling using a standardized protocol with your target annealing temperature.
Analyze Results using the following criteria:
- Fluorescence Amplitude: The difference in fluorescence between positive and negative droplets should be maximized.
- Cluster Separation: Clear distinction between positive and negative droplet populations for both mutant and wild-type targets.
- Rain: Minimal intermediate fluorescence droplets ("rain") between clear positive and negative clusters.
- Specificity: No false-positive signals in non-template controls and wild-type-only samples.
Table 2: Optimization Guidelines for Primer and Probe Concentrations
| Component | Recommended Concentration | Effect of Too Low | Effect of Too High |
|---|---|---|---|
| Primers | 500 nM [59] [3] | Reduced amplification efficiency, higher Cq, failed detection of rare mutants | Increased non-specific amplification, primer-dimer formation, increased background |
| Probes | 250 nM [59] [3] | Low fluorescence amplitude, poor cluster separation, uncertain classification | Increased background fluorescence, higher assay cost, potential inhibition |
| Template DNA | 1–20 ng/μL (sample-dependent) [3] | Insufficient mutant copies for reliable detection | Inhibition of amplification, droplet overcrowding violating Poisson assumption |
For rare mutation detection, special attention must be paid to the DNA input amount. The required sensitivity dictates the necessary DNA input, which can be calculated using the formula: Number of copies = mass of DNA (ng) / 0.003 (for human genomic DNA) [3]. This ensures sufficient mutant copies are present for reliable detection at the desired allele frequency.
Integrated Experimental Workflow
The following workflow diagram illustrates the comprehensive process for optimizing ddPCR assays for rare allele detection, from initial setup through validation:
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful rare allele detection by ddPCR requires careful selection of reagents and components. The following table outlines key solutions and their functions in the experimental workflow:
Table 3: Essential Research Reagent Solutions for ddPCR Rare Mutation Detection
| Reagent Solution | Function | Example Implementation |
|---|---|---|
| ddPCR Mastermix | Provides core PCR components: DNA polymerase, dNTPs, buffer, MgCl₂ [59] [3] | Bio-Rad's ddPCR Supermix for Probes; 2× concentration used at 1× final [59] [44] |
| Sequence-Specific Primers | Amplify the target genomic region containing the mutation site [3] | One set of forward/reverse primers (e.g., 500 nM final) to amplify both wild-type and mutant alleles [3] |
| Hydrolysis Probes (TaqMan) | Specifically detect and differentiate wild-type vs. mutant alleles [3] | FAM-labeled mutant probe and HEX/VIC-labeled wild-type probe (e.g., 250 nM each) [3] [44] |
| Reference Dye | Normalizes fluorescence signals and corrects for well-to-well variation | ROX/Atto590, included in mastermix or added separately per manufacturer's instructions [3] |
| Digital PCR System | Partitions samples, performs thermal cycling, and reads endpoint fluorescence | QX200 Droplet Digital PCR System (Bio-Rad) [59] [44] or QuantStudio Absolute Q [58] |
| cfDNA Extraction Kits | Isolate and purify cell-free DNA from liquid biopsy samples (plasma, urine) | Mag-Bind cfDNA Kit with optimized protocols for 4 mL urine input volume [44] |
The exquisite sensitivity of droplet digital PCR for rare allele detection is critically dependent on meticulously optimized thermal cycling conditions and primer/probe concentrations. Through systematic evaluation of annealing temperatures (typically 58–62°C) and confirmation of appropriate reagent concentrations (500 nM for primers, 250 nM for probes), researchers can achieve robust assays capable of detecting mutant alleles at frequencies as low as 0.1% [58] [60] [44]. This level of sensitivity enables groundbreaking applications in liquid biopsy analysis, cancer monitoring, and personalized medicine. The optimization framework presented in this guide provides a structured pathway for researchers to validate their ddPCR assays, ensuring the precision and accuracy required to detect genetic needles in genomic haystacks.
The Impact of Restriction Enzymes on Assay Precision and Gene Copy Number Quantification
Digital PCR (dPCR) has emerged as a powerful tool for absolute nucleic acid quantification, enabling highly precise gene copy number analysis essential for applications ranging from cancer diagnostics to environmental monitoring. A critical, yet often underexplored, factor influencing the performance of these assays is the use of restriction enzymes. This technical guide examines the profound impact of restriction enzyme selection on assay precision and accuracy within the context of droplet digital PCR (ddPCR). Evidence from comparative platform studies demonstrates that the strategic choice of restriction enzyme can significantly reduce technical variation, thereby enhancing the reliability of copy number quantification—a cornerstone of robust rare allele detection research.
Digital PCR (dPCR) represents a transformative advancement in nucleic acid quantification, enabling the absolute measurement of target DNA without the need for standard curves [17]. The core principle involves partitioning a PCR reaction into thousands of nanoliter-sized reactions, performing end-point amplification, and applying Poisson statistics to calculate the absolute copy number of the target sequence based on the ratio of positive to negative partitions [8] [17]. This technology offers exceptional sensitivity and precision, particularly for applications requiring the detection of rare alleles, copy number variations (CNVs), and subtle changes in gene expression [3] [17] [27].
Within the dPCR workflow, sample preparation is a critical step, and for copy number variation (CNV) analysis, this frequently involves the use of restriction enzymes. The primary function of restriction enzymes in this context is to fragment high-molecular-weight genomic DNA (gDNA). This fragmentation is not merely procedural; it is essential for achieving confluent sample disbursement across the thousands of partitions and ensuring the reliability of subsequent results [62]. By cleaving DNA at specific recognition sites, restriction enzymes mitigate the risk of multiple target copies residing on a single DNA fragment, which could lead to their co-compartmentalization in the same partition and consequent underestimation of the true copy number. The considerations for restriction enzyme selection are multifaceted, encompassing target amplicon compatibility to ensure the intended amplification region remains intact, and methylation sensitivity, which can influence digestion efficiency depending on the epigenetic status of the DNA sample [62].
The Critical Impact of Restriction Enzyme Selection on Assay Precision
The choice of restriction enzyme is not a neutral experimental parameter; it directly and measurably influences the precision and reproducibility of dPCR assays. A landmark 2025 study provides compelling quantitative evidence for this effect, having systematically compared the performance of the QX200 droplet digital PCR (ddPCR) system from Bio-Rad and the QIAcuity One nanoplate digital PCR (ndPCR) system from QIAGEN using DNA from the ciliate Paramecium tetraurelia [8].
The study evaluated two different restriction enzymes, HaeIII and EcoRI, across varying cell numbers. The precision of the assays was assessed using the coefficient of variation (CV), a statistical measure of relative variability, with lower CV values indicating higher precision and reproducibility. The results, summarized in the table below, reveal a striking enzyme-dependent effect, particularly for the ddPCR platform.
Table 1: Impact of Restriction Enzyme Choice on Assay Precision (Coefficient of Variation, %CV)
| Platform | Cell Number | CV with EcoRI (%) | CV with HaeIII (%) |
|---|---|---|---|
| QX200 ddPCR | 10 | 13.6 | 2.8 |
| 50 | 62.1 | 4.9 | |
| 100 | 18.9 | 1.7 | |
| QIAcuity ndPCR | 10 | 27.7 | 14.6 |
| 50 | 15.8 | 2.8 | |
| 100 | 0.6 | 1.6 |
Data adapted from [8].
The data demonstrates a general tendency for higher precision when using HaeIII instead of EcoRI, with this effect being especially pronounced for the QX200 ddPCR system [8]. For instance, in the 50-cell sample, the CV for ddPCR plummeted from 62.1% with EcoRI to just 4.9% with HaeIII. This suggests that HaeIII may provide more consistent and efficient digestion for this specific experimental setup, leading to a more uniform distribution of target molecules across partitions. While the ndPCR platform also benefited from HaeIII, its performance was generally less variable with enzyme choice compared to the ddPCR system. These findings underscore that restriction enzyme selection is a critical variable that must be optimized for each specific dPCR application and platform to achieve the highest possible data quality.
Experimental Protocols for Restriction Enzyme Digestion in dPCR
Integrating restriction enzyme digestion into the dPCR workflow can be accomplished through different methodological approaches. Below are two validated protocols from the literature.
Traditional Restriction Enzyme Digestion Protocol
This method involves a separate digestion step prior to setting up the dPCR reaction, as employed in the comparative study of the QX200 and QIAcuity platforms [8].
- Digestion Reaction Setup: Combine 1 µg of genomic DNA with the selected restriction enzyme (e.g., HaeIII or EcoRI) and its corresponding manufacturer-recommended buffer in a total volume of 20-50 µL.
- Incubation: Incubate the reaction mixture at the optimal temperature for the enzyme (typically 37°C) for a defined period, often 1-2 hours, to ensure complete digestion.
- Enzyme Inactivation: Heat-inactivate the restriction enzyme (e.g., 20 minutes at 65°C or as per manufacturer's instructions). Alternatively, the digested DNA can be purified.
- dPCR Reaction: Use an aliquot of the digested (and potentially purified) DNA as the template in the dPCR reaction mix.
Streamlined "One-pot" Digestion Protocol
A more efficient, single-tube protocol was validated for CYP2D6 copy number determination on the Absolute Q dPCR system, simplifying the workflow and reducing hands-on time [62].
- Master Mix Preparation: Combine the dPCR supermix, forward and reverse primers, fluorescent probes, and the selected restriction enzyme directly in the reaction tube or plate.
- Template Addition: Add the undigested genomic DNA template to the master mix.
- Integrated Digestion and Amplification: Place the reaction plate directly into the thermocycler. The thermal cycler program is initiated with an extended hold at the restriction enzyme's optimal temperature (e.g., 37°C for 30-60 minutes) to allow for DNA fragmentation within the same reaction mix.
- dPCR Amplification: Following the digestion hold, the thermocycling program proceeds directly to the standard PCR amplification steps (e.g., initial denaturation at 95°C, followed by 40-50 cycles of denaturation and annealing/extension).
This "One-pot" method has been demonstrated to yield results consistent with the traditional, separate digestion method while offering significant gains in workflow efficiency [62].
The Scientist's Toolkit: Essential Reagents for dPCR with Restriction Enzymes
Table 2: Key Research Reagent Solutions for Restriction Enzyme dPCR
| Reagent / Solution | Function / Explanation |
|---|---|
| Restriction Enzymes (e.g., HaeIII, EcoRI) | Enzymes that fragment genomic DNA at specific recognition sequences to ensure unbiased distribution of target molecules across partitions, crucial for accurate copy number determination [8] [62]. |
| dPCR Supermix | A specialized master mix containing DNA polymerase, dNTPs, buffer, and MgCl₂, optimized for the partitioning and endpoint amplification required in dPCR [3] [63]. |
| Sequence-Specific Probes & Primers | Hydrolysis probes (e.g., TaqMan) and primer sets designed to bind and amplify the wild-type and/or mutant allele of interest, enabling target-specific detection and quantification [3] [63] [27]. |
| Microfluidic Partitioning Oil/Consumables | Surfactant chemistries and cartridges (for ddPCR) or nanoplate-based chips (for ndPCR) that generate the thousands of individual reaction partitions essential for digital quantification [8] [3]. |
The integration of restriction enzyme digestion is a fundamental component of a robust dPCR workflow for gene copy number quantification. Empirical evidence clearly demonstrates that the selection of the specific enzyme is not merely a technical detail but a decisive factor influencing assay precision. As shown in direct platform comparisons, enzymes like HaeIII can drastically reduce coefficients of variation compared to alternatives like EcoRI, thereby enhancing the reliability of data used for critical applications in rare allele detection, pharmacogenetics, and cancer diagnostics [8]. The advent of streamlined protocols, such as the "One-pot" digestion method, further integrates this crucial step efficiently into the dPCR process [62]. For researchers, the mandatory optimization of restriction enzyme selection and digestion conditions is a prerequisite for achieving the full potential of digital PCR's precision in quantifying gene copies and detecting rare genetic events.
Visual Appendix: Workflows and Logical Relationships
dPCR Workflow with Restriction Enzyme Digestion
Enzyme Selection Logic for Assay Optimization
Droplet Digital PCR (ddPCR) has emerged as a powerful tool for the precise quantification of nucleic acids, enabling the detection of rare alleles with unparalleled sensitivity. This technical guide details the establishment of a rigorous ddPCR protocol, framing the critical importance of environmental and organismal controls within the broader principles of rare allele detection research. We provide an in-depth examination of core methodologies, supported by structured data and workflow visualizations, to empower researchers and drug development professionals in implementing robust, reproducible ddPCR assays.
Digital PCR (dPCR), and specifically Droplet Digital PCR (ddPCR), represents the third generation of PCR technology. Its principle involves partitioning a PCR reaction mixture into thousands to millions of nanoliter-sized droplets, effectively creating a massive array of individual reactions. Following end-point PCR amplification, the fraction of positive (fluorescent) and negative droplets is counted, allowing for the absolute quantification of the target nucleic acid sequence using Poisson statistics, without the need for a standard curve [4] [64].
This calibration-free technology presents powerful advantages for rare allele detection, including high sensitivity, absolute quantification, high accuracy, and reproducibility [4]. The ability to detect rare genetic mutations within a background of wild-type genes was among the first clinically relevant applications of dPCR, paving the way for liquid biopsy applications and tumor heterogeneity analysis in oncology [4]. The sensitivity of ddPCR allows for the detection of mutations with a variant allele frequency as low as 0.1% to 0.002% (1 in 50,000 copies), a critical capability for monitoring minimal residual disease or early cancer signals [13] [44].
Core Principles and Advantages of ddPCR
The fundamental workflow of ddPCR involves four key steps: i) partitioning of the sample, ii) PCR amplification to end-point, iii) fluorescence reading of each partition, and iv) data analysis based on Poisson statistics [4]. This section breaks down the core principles that make ddPCR exceptionally suitable for sensitive detection applications.
Absolute Quantification via Poisson Statistics
In ddPCR, the sample is randomly distributed across thousands of partitions. A partition containing one or more target molecules will generate a positive fluorescence signal, while those with no target will remain negative. The ratio of positive to total partitions allows for the calculation of the absolute concentration of the target molecule in the original sample, based on the Poisson distribution model. This eliminates the reliance on external standard curves, a significant source of variability and error in quantitative PCR (qPCR) [64] [65].
Superior Sensitivity and Tolerance to Inhibitors
Partitioning the sample effectively concentrates the target molecules and dilutes PCR inhibitors present in the reaction mix. This makes ddPCR notably more resistant to the effects of inhibitors commonly found in complex biological samples (e.g., sputum, urine) compared to qPCR [64] [65]. Studies have shown that ddPCR can successfully detect pathogens and rare alleles in samples where qPCR fails or provides suboptimal quantification due to inhibition or low target abundance [65].
Table 1: Key Comparative Advantages of ddPCR over qPCR for Rare Allele Detection
| Feature | Droplet Digital PCR (ddPCR) | Quantitative PCR (qPCR) |
|---|---|---|
| Quantification | Absolute, without a standard curve | Relative, requires a standard curve |
| Precision & Sensitivity | High; capable of detecting rare alleles (<0.1% VAF) | Lower; limited by amplification efficiency and background noise |
| Tolerance to Inhibitors | High; sample partitioning dilutes inhibitors | Moderate to Low; inhibitors can skew Cq values |
| Data Output | Direct count of target molecules | Cycle threshold (Cq) value relative to standards |
| Dynamic Range | Linear over a wide range due to partition counting | Limited by the efficiency and quality of the standard curve |
Establishing a Rigorous ddPCR Protocol
The reliability of ddPCR data, especially for low-abundance targets, is critically dependent on a rigorous protocol that incorporates comprehensive controls at every stage. These controls are essential for validating the assay's performance, ensuring specificity, and verifying the integrity of the sample and reaction components.
The Critical Role of Environmental Controls
Environmental controls are designed to detect contamination introduced from reagents, laboratory surfaces, or aerosol during sample and reaction preparation.
- No-Template Control (NTC): This is the most crucial environmental control. It consists of all PCR reagents (master mix, primers, probes, water) but no sample DNA. The NTC is partitioned and amplified alongside experimental samples. An acceptable result is the generation of zero or a very low, predetermined number of positive droplets, confirming the absence of reagent contamination or amplicon carryover [44]. The presence of significant positive droplets in the NTC invalidates the entire run.
The Critical Role of Organismal and Sample Controls
Organismal and sample controls verify the quality of the sample and the specificity of the assay for its intended target.
- No Spike-in Control (Endogenous Control): When working with complex sample matrices like urine or plasma, a sample processed without any spiked-in target (e.g., a reference standard) should be analyzed. This control assesses the background level of the endogenous target or potential non-specific amplification from the sample's native nucleic acids [44].
- Wild-type (Negative) Control: A sample known to contain only the wild-type allele is essential for rare mutation detection. This control is used to set the fluorescence amplitude threshold that robustly distinguishes mutant-positive from wild-type-only droplets, ensuring the assay does not generate false positives from non-specific amplification [44].
- Positive Controls: Samples with known concentrations of the mutant allele, ideally at different variant allele frequencies (e.g., 5%, 1%, 0.1%), should be included. These controls validate the assay's sensitivity, dynamic range, and quantification accuracy by comparing observed versus expected allele frequencies [13] [44].
The following workflow diagram integrates these essential controls into the standard ddPCR procedure:
Experimental Protocol: A Case Study in Rare Variant Detection
The following detailed protocol is adapted from a study that successfully detected NRAS and EGFR mutant alleles from urinary cfDNA at frequencies as low as 0.1% [44].
Sample Preparation and DNA Extraction
- Sample Preservation: Urine samples were collected and immediately preserved with a commercial preservative (e.g., Colli-Pee UAS) at a 2.3:1 ratio to prevent cfDNA degradation.
- Spike-in Controls: To mimic clinical samples and validate the workflow, a multiplex cfDNA reference standard (e.g., Horizon Discovery's Mimix Set) containing oncogenic mutations at known allelic frequencies (5%, 1%, 0.1%, 0% [wild-type]) was spiked into the preserved urine.
- cfDNA Extraction: cfDNA was extracted from a 4 mL input volume using a specialized kit (e.g., Mag-Bind cfDNA Kit). The extraction included the following control groups:
- Reference standard spiked into preserved urine.
- Preserved urine alone (No spike-in control).
- Reference standard spiked into preservative alone (No urine control).
- Reference standard spiked into elution buffer (No urine, no preservative control).
- Quality Assessment: The yield and quality of the extracted cfDNA were evaluated using a high-sensitivity automated electrophoresis system (e.g., Agilent TapeStation) to confirm the presence of a dominant ~150-200 bp cfDNA fragment and assess potential genomic DNA contamination [44].
Droplet Digital PCR Setup and Workflow
- Reaction Assembly: A 300 µL PCR reaction was assembled using 15 ng of cfDNA, ddPCR Supermix for Probes, and allele-specific TaqMan assays. For example, a duplex reaction can be set up with a FAM-labeled probe for the mutant allele (e.g., NRAS A59T or EGFR L858R) and a HEX- or SUN-labeled probe for the wild-type allele [44].
- Droplet Generation: The reaction mixture was loaded into a droplet generator (e.g., Bio-Rad QX200 Droplet Generator) to create an emulsion of approximately 20,000 nanoliter-sized droplets per sample.
- PCR Amplification: The droplet emulsion was transferred to a 96-well plate and amplified on a conventional thermal cycler (e.g., C1000 Touch Thermal Cycler) using a optimized protocol with adjusted annealing temperatures for the specific probes.
- Droplet Reading and Analysis: Post-amplification, the plate was loaded into a droplet reader (e.g., QX200 Droplet Reader) which aspirated each sample and measured the fluorescence in each droplet. Data was analyzed using dedicated software (e.g., QuantaSoft Analysis Pro).
Data Analysis and Validation
- Threshold Setting: Fluorescence amplitude thresholds for positive droplets were set manually based on the droplet cloud patterns of the wild-type (0% AF) control and the No Template Control (NTC) to ensure clear separation between negative and positive populations [44].
- Concordance Analysis: For the positive controls, the observed mutant allele frequency was calculated by the software (Positive Droplets / Total Droplets) and compared to the expected frequency from the reference standard's certificate of analysis. The results should fall within the manufacturer's stated acceptance criteria [44].
- Result Interpretation: A valid run requires the NTC to have zero positive droplets for the mutant channel, and the positive controls to show concordance. Only then can the experimental sample results be trusted.
Table 2: Essential Research Reagent Solutions for ddPCR Rare Allele Detection
| Reagent / Solution | Function / Purpose | Example Product / Note |
|---|---|---|
| Nucleic Acid Preservation Buffer | Stabilizes cfDNA in biofluids post-collection to prevent degradation. | Colli-Pee UAS Preservative [44] |
| cfDNA Extraction Kit | Purifies short, fragmented cfDNA from large-volume biofluids; minimizes gDNA contamination. | Mag-Bind cfDNA Kit [44] |
| ddPCR Supermix for Probes | Optimized buffer, enzymes, and dNTPs for probe-based amplification in droplet formats. | Bio-Rad ddPCR Supermix for Probes (No dUTP) [44] |
| Allele-Specific TaqMan Assays | FAM-labeled probes for mutant alleles and HEX/VIC-labeled probes for wild-type alleles. | Custom or commercially available LNA-enhanced probes [13] |
| Reference Standard | Provides genetically defined, quantitated targets for assay validation and run controls. | Horizon Discovery Mimix Multiplex cfDNA Set [44] |
The power of ddPCR for rare allele detection is fully realized only through a rigorously controlled experimental protocol. The strategic implementation of environmental controls like the NTC, and organismal controls including wild-type, positive, and endogenous sample controls, forms the bedrock of reliable and interpretable data. The detailed workflow and reagent solutions outlined in this guide provide a framework for researchers in oncology, infectious disease, and drug development to establish robust ddPCR assays. By adhering to these principles, scientists can confidently push the boundaries of sensitivity, enabling discoveries and diagnostics in the challenging realm of rare genetic targets.
Proving Performance: Validation, Platform Comparisons, and Diagnostic Accuracy
In the field of molecular diagnostics, particularly in droplet digital PCR (ddPCR) for rare allele detection, understanding the fundamental capabilities of an analytical method is paramount. Robust assay validation ensures that data generated for research and drug development is reliable, accurate, and fit for purpose. Limit of Blank (LoB), Limit of Detection (LoD), and Limit of Quantitation (LoQ) are three critical performance metrics that describe the smallest concentration of an analyte that can be reliably measured by an analytical procedure [66] [67]. These parameters are especially crucial in applications like detecting rare cancer mutations in cell-free DNA (cfDNA), where the target concentration is minimal and the potential for false positives or negatives must be rigorously controlled [68] [69]. This guide provides an in-depth technical framework for establishing these metrics within the context of ddPCR, forming the foundation for credible and reproducible research.
Core Definitions and Theoretical Foundations
The terms LoB, LoD, and LoQ describe a hierarchy of an assay's sensitivity, each with a distinct statistical and practical meaning.
Limit of Blank (LoB) is the highest apparent analyte concentration expected to be found when replicates of a blank sample containing no analyte are tested. It essentially defines the background "noise" of the assay. Statistically, the LoB is calculated as the mean of blank measurements plus 1.645 times their standard deviation (SD), capturing the 95th percentile of the blank signal distribution [66]. In ddPCR, this translates to the false-positive signal that can arise even from a sample with zero mutant alleles.
Limit of Detection (LoD) is the lowest analyte concentration that can be reliably distinguished from the LoB. It is a concentration at which detection is feasible, but not necessarily with acceptable precision or accuracy for exact quantification. The LoD is positioned above the LoB and accounts for the variability of both blank and low-concentration samples. Its standard calculation is LoB + 1.645(SD of a low-concentration sample) [66]. For a ddPCR assay detecting EGFR mutations, an LoD of one mutant in 180,000 wild-type molecules has been demonstrated [68].
Limit of Quantitation (LoQ) is the lowest concentration at which the analyte can not only be reliably detected but also measured with predefined goals for bias and imprecision [66]. The LoQ cannot be lower than the LoD and is often found at a much higher concentration. It is sometimes defined as the concentration that yields a specific coefficient of variation (CV), such as 20%, a metric also known as "functional sensitivity" [66].
Table 1: Summary of LoB, LoD, and LoQ Characteristics
| Parameter | Definition | Sample Type | Typical Replicates (Verification) | Key Equation |
|---|---|---|---|---|
| LoB | Highest result likely from a blank sample | Sample containing no analyte | 20 [66] | LoB = Mean~blank~ + 1.645(SD~blank~) [66] |
| LoD | Lowest concentration distinguished from LoB | Sample with low concentration of analyte | 20 [66] | LoD = LoB + 1.645(SD~low concentration sample~) [66] |
| LoQ | Lowest concentration meeting bias and imprecision goals | Sample at or above the LoD | 20 [66] | LoQ ≥ LoD [66] |
Calculation Methods and Statistical Approaches
Multiple analytical approaches can be employed to determine these limits, and the choice of method should be matched to the specific technology and data characteristics.
Standard Deviation-Based Methods
This is the most referenced approach for immunoassays and PCR methods and is outlined in guidelines like CLSI EP17 [66] [67].
- For LoB: Multiple replicates (n ≥ 20) of a blank sample are measured. The LoB is calculated as the mean of the blank results + 1.645 * SD of the blank results (assuming a normal distribution) [66]. This one-sided confidence interval ensures 95% of blank measurements fall below this limit.
- For LoD: Replicates (n ≥ 20) of a sample containing a low concentration of analyte near the expected LoD are tested. The LoD is derived using the previously determined LoB and the standard deviation of this low-level sample: LoD = LoB + 1.645(SD~low concentration sample~) [66]. This ensures that 95% of measurements at the LoD will exceed the LoB, minimizing false negatives.
Alternative Calculation Methods
Other common methods include:
- Signal-to-Noise Ratio: This approach sets the LoD at a signal-to-noise ratio of 2:1 or 3:1, where the noise is the measured signal of the blank control. This method is practical for assays with observable background noise [67].
- Standard Deviation of the Response and Slope: Defined in ICH Q2 guidelines, this method uses the standard error of the regression line (often the standard deviation of the y-intercept) and the slope of the calibration curve. The formulas are LOD = 3.3σ/Slope and LOQ = 10σ/Slope, where σ is the standard deviation of the response [67].
Experimental Protocols for ddPCR Assay Validation
Establishing LoB, LoD, and LoQ for a ddPCR assay requires a meticulous experimental design. The following protocol uses the development of a KRAS exon 2 drop-off ddPCR assay as a model [69].
Sample Preparation and Experimental Replicates
- Blank Samples: To establish the LoB, analyze a minimum of 20 replicates of a blank sample. This should be a biological matrix known to not contain the target analyte, such as wild-type genomic DNA or cfDNA from healthy donors, processed identically to test samples [66] [69].
- Low-Concentration Samples: To determine the LoD and LoQ, prepare a dilution series of the target analyte in the same blank matrix. A recommended practice is to test at least 20 replicates at each of several low concentrations around the expected LoD [66]. For the KRAS assay, this involved testing samples with known low variant allele frequencies.
Data Collection and Analysis Workflow
The process for determining the metrics follows a logical sequence, where each step informs the next.
Protocol: KRAS Drop-off ddPCR Assay Example
In a 2025 study, researchers developed a novel KRAS drop-off ddPCR assay for detecting mutations in cfDNA [69]. The experimental procedure for determining its limits was as follows:
- Assay Design: A wild-type (drop-off) probe, labeled with HEX, was designed to span the KRAS codon 12/13 hotspot. A reference probe, labeled with FAM, targeted a stable upstream region within the same amplicon.
- LoB Determination: The LoB was established by repeatedly testing a wild-type cfDNA sample, which should contain zero mutant alleles. The number of false-positive "mutant" droplets was recorded across many replicates. The LoB was calculated to be 0.13 copies/µL [69].
- LoD Determination: Serial dilutions of synthetic mutant DNA in a background of wild-type DNA were created to simulate low variant allele frequencies. The LoD, defined as the lowest concentration detectable with 95% confidence, was determined to be 0.57 copies/µL [69]. This process also confirmed the assay's high sensitivity for detecting rare mutants.
- LoQ Considerations: While not explicitly stated, the high inter-assay precision (r² = 0.9096) suggests that the LoD and LoQ were likely very close for this specific application, meaning the LoD concentration was also sufficient for quantification in this context [69].
The Scientist's Toolkit: Research Reagent Solutions
Successful assay validation relies on specific, high-quality reagents. The following table details essential materials and their functions based on the featured experiments.
Table 2: Essential Research Reagents for ddPCR Assay Validation
| Reagent / Material | Function in Validation | Example from Literature |
|---|---|---|
| Blank Matrix | Provides the analyte-free background for determining LoB and for making dilutions for LoD/LoQ. | Wild-type genomic DNA or cfDNA from healthy donors [69]. |
| Synthetic Target Analytes | Used to spike the blank matrix at precisely known low concentrations to establish LoD and LoQ. | Synthetic oligonucleotides with specific KRAS mutations [69]. |
| LNA-modified Probes | Enhance hybridization specificity and discrimination between wild-type and mutant sequences, crucial for low LoD. | LNA-based TaqMan probes used in the KRAS drop-off assay [69]. |
| Reference/Internal Control Probe | Quantifies the total number of molecules present, serving as an internal control for reaction efficiency and DNA quality. | FAM-labeled reference probe binding outside the KRAS mutation hotspot [69]. |
| cfDNA Extraction Kit | Isolves cell-free DNA from plasma samples with high efficiency and minimal contamination, preserving low-abundance targets. | PME-free circulating DNA extraction kit (Analytik Jena) [69]. |
| Fluorometric Quantifier | Accurately measures the concentration of extracted DNA to ensure consistent input amounts across validation experiments. | Qubit 4 fluorometer [69]. |
Advanced Concepts: Multiplexing and Data Analysis in ddPCR
The principles of LoB, LoD, and LoQ also apply to more complex, multiplexed ddPCR assays, which are valuable for screening multiple mutations simultaneously.
- Multicolor Combinatorial Probe Coding (MCPC): This strategy uses a limited number of fluorophores in various combinations to label probes, dramatically increasing the number of targets detectable in a single reaction. With n fluorophores, up to 2^n - 1 targets can be uniquely identified [70]. For example, a system with 7 colors can theoretically detect up to 127 targets [71].
- Impact on Limits: While multiplexing increases throughput, careful validation is required. The use of multiple probes can potentially increase background noise or cause fluorescence spillover, which must be characterized and compensated for as it can impact the effective LoB and LoD of each individual channel [72] [70].
- Data Analysis and Thresholding: In ddPCR, the clear separation between positive and negative droplet populations is fundamental. Software tools with population editors allow for precise thresholding, which is critical for accurately determining the number of mutant and wild-type molecules, and thus for calculating the metrics like LoB and LoD [71]. The relationship between the statistical definitions and the raw data is visualized below.
The rigorous determination of Limit of Blank, Limit of Detection, and Limit of Quantitation is a non-negotiable component of assay development, especially in the demanding field of rare allele detection using droplet digital PCR. These metrics provide a standardized, statistical framework for understanding the true sensitivity and quantitative capability of an assay, from distinguishing signal from noise (LoB) to achieving reliable detection (LoD) and finally, to generating precise and accurate quantitative data (LoQ). By adhering to detailed experimental protocols and leveraging advanced reagent solutions, researchers can ensure their ddPCR assays are thoroughly validated, generating data that is robust, reproducible, and ultimately, fit for the critical purpose of advancing research and drug development.
The evolution of Polymerase Chain Reaction (PCR) technology from quantitative PCR (qPCR) to digital PCR (dPCR), and specifically droplet digital PCR (ddPCR), represents a paradigm shift in molecular diagnostics and research. While qPCR has long been the gold standard for nucleic acid detection and quantification, the emergence of ddPCR offers distinct advantages for applications requiring absolute quantification and high sensitivity, particularly in rare allele detection research. This technical guide provides an in-depth comparison of these two powerful technologies, focusing on their performance in sensitivity, accuracy, and tolerance to inhibitors—critical parameters for researchers and drug development professionals working with challenging samples. Understanding these differences is essential for selecting the optimal method for specific research objectives, from clinical diagnostics to environmental surveillance [5].
Fundamental Principles and Workflows
Quantitative PCR (qPCR)
qPCR, also known as real-time PCR, monitors the amplification of a targeted DNA molecule during each PCR cycle (in "real time") using fluorescent reporters. The principle of quantification relies on the cycle threshold (Ct), the number of cycles required for the fluorescent signal to cross a predetermined threshold. The Ct value is inversely proportional to the starting quantity of the target nucleic acid. Quantification is achieved by comparing the Ct values of unknown samples to a standard curve generated from samples with known concentrations. This method is highly effective for relative quantification but introduces potential variability due to its dependence on external standards and amplification efficiency [5] [73].
Droplet Digital PCR (ddPCR)
ddPCR takes a different approach by partitioning a PCR reaction into thousands of nanoliter-sized droplets. Each droplet functions as an individual PCR reactor. After endpoint PCR amplification, each droplet is analyzed for fluorescence. Critically, quantification is absolute and does not require a standard curve. The count of positive (fluorescent) and negative (non-fluorescent) droplets is used in conjunction with Poisson statistics to calculate the absolute copy number of the target molecule in the original sample. This partitioning is the source of ddPCR's enhanced precision and resistance to inhibitors [7] [74].
Comparative Workflow Visualization
The diagram below illustrates the core procedural differences between the two techniques.
Head-to-Head Performance Comparison
Sensitivity and Limit of Detection
Sensitivity, often defined by the Limit of Detection (LOD), is a key differentiator between the two technologies. Multiple studies consistently demonstrate ddPCR's superior sensitivity, especially at very low target concentrations.
- Viral Detection in Wastewater: A 2025 study comparing RT-qPCR and RT-ddPCR for detecting model viruses found that while both methods performed similarly for high and medium viral loads, RT-ddPCR showed significantly greater sensitivity for low viral loads. It reliably detected trace levels of viral particles where RT-qPCR struggled [75].
- Respiratory Virus Diagnostics: A comparative study from 2025 on respiratory viruses (Influenza A/B, RSV, SARS-CoV-2) found that dPCR demonstrated "superior accuracy, particularly for high viral loads of influenza A, influenza B, and SARS-CoV-2, and for medium loads of RSV." Its precision was especially notable for quantifying intermediate viral levels [7].
- Probiotic Detection in Complex Matrices: Research comparing qRT-PCR and ddPCR for detecting multi-strain probiotics in human fecal samples reported that ddPCR demonstrated a 10–100 fold lower limit of detection than qRT-PCR, highlighting its power in challenging, inhibitor-rich environments [74].
Accuracy and Precision
Accuracy refers to the closeness of a measurement to the true value, while precision relates to the reproducibility of repeated measurements.
- Absolute vs. Relative Quantification: The core advantage of ddPCR is its ability for absolute quantification without a standard curve, eliminating one major source of variability and potential inaccuracy inherent in qPCR [7] [5].
- Precision in Environmental Monitoring: A 2025 cross-platform dPCR study, while comparing two dPCR systems, highlighted that dPCR, in general, is "considered to have great potential as it enables the absolute quantification of nucleic acids" and is "the better choice for applications that require a higher precision" compared to qPCR [8].
- Precision in Copy Number Analysis: The same study found that both the QX200 ddPCR and QIAcuity ndPCR platforms yielded "high precision across most analyses," with coefficients of variation (CV) often below 5-10% for a wide range of target concentrations, underscoring the reliability of the digital approach [8].
Tolerance to PCR Inhibitors
Complex sample matrices, such as wastewater, feces, and blood, often contain substances that can inhibit the PCR reaction, leading to underestimation of target concentration or false negatives.
- Wastewater Analysis: The wastewater virus study concluded that "RT-ddPCR measurement on extracted wastewater samples demonstrated improved performance against inhibitors" compared to RT-qPCR. This resilience is attributed to the partitioning step, which dilutes inhibitors across thousands of droplets, thereby preserving amplification efficiency in a majority of partitions [75].
- Inherent Robustness: The partitioning step in ddPCR effectively dilutes out PCR inhibitors present in the sample. This means that even if an inhibitor suppresses the fluorescence in some droplets, the vast number of other droplets can still be amplified and counted accurately, providing a more robust result in complex matrices [74].
The following table synthesizes key performance metrics from recent comparative studies.
Table 1: Comparative Performance Metrics of qPCR and ddPCR
| Performance Metric | qPCR / RT-qPCR | ddPCR / RT-ddPCR | Supporting Evidence |
|---|---|---|---|
| Limit of Detection (LOD) | Higher LOD, struggles with trace targets | 10-100x lower LOD; reliably detects low copies | Probiotic detection [74], Wastewater viruses [75] |
| Quantification Method | Relative (via standard curve) | Absolute (via Poisson statistics) | Respiratory virus study [7] |
| Precision (at low copies) | Lower precision due to Ct variability | High precision (CV often <10%) | Platform comparison [8] |
| Inhibitor Tolerance | Susceptible to suppression of amplification | High tolerance; partitioning dilutes inhibitors | Wastewater [75], Probiotic [74] |
| Multiplexing Capacity | Well-established | Advanced high-plex assays (e.g., 9-plex) | 9-plex viral assay [76] |
| Throughput & Cost | High throughput, lower cost per sample | Lower throughput, higher cost per sample | General comparison [5] |
Experimental Protocols for Key Applications
Protocol: Comparing Viral Load Quantification in Respiratory Samples
This protocol is adapted from a 2025 study comparing dPCR and Real-Time RT-PCR for respiratory virus detection [7].
Sample Collection and Nucleic Acid Extraction:
- Collect nasopharyngeal swabs or other respiratory samples in appropriate transport media.
- Extract total nucleic acids using an automated system (e.g., KingFisher Flex system with MagMax Viral/Pathogen kit or STARlet platform with STARMag kit). Include an internal control to monitor extraction efficiency.
Real-Time RT-PCR Workflow:
- Use commercial multiplex respiratory panel kits (e.g., Allplex Respiratory Panel).
- Perform amplification on a standard thermocycler (e.g., CFX96 from Bio-Rad).
- Record Ct values for each target. Stratify samples based on Ct values: high (Ct ≤ 25), medium (Ct 25.1–30), and low viral load (Ct > 30).
Digital PCR Workflow:
- Use a dPCR system (e.g., QIAcuity from QIAGEN or QX200 from Bio-Rad).
- Prepare reactions using a multiplex assay for the same respiratory targets (e.g., Influenza A, Influenza B, RSV, SARS-CoV-2).
- Load samples into nanoplates or droplet generators. Perform endpoint PCR.
- Analyze partitions and calculate absolute copy numbers (copies/μL) using the instrument's software (e.g., QIAcuity Suite or QuantaSoft).
Data Analysis:
- Compare the quantitative results from both methods across the different viral load strata.
- Perform statistical analysis (e.g., Kruskal-Wallis test) to assess differences in quantification and precision.
Protocol: Assessing Inhibitor Tolerance in Wastewater Matrices
This protocol is based on a 2025 study evaluating model viruses in wastewater [75].
Sample Preparation and Spiking:
- Collect wastewater from various treatment stages (e.g., influent, effluent) to obtain matrices with different chemical characteristics.
- Spike a known quantity of a model virus (e.g., Pseudomonas phage Φ6) into both real wastewater and synthetic wastewater samples.
RNA Extraction and Omission:
- Split each sample into two portions.
- Perform RNA extraction on one portion using a commercial kit.
- Use the second portion directly in the PCR reaction without RNA extraction to test resilience to inhibitors.
Parallel PCR Analysis:
- Analyze all sample portions (extracted and non-extracted) using both RT-qPCR and RT-ddPCR.
- Use identical primer/probe sets for the model virus in both platforms to ensure a direct comparison.
Data Analysis:
- Calculate the percentage recovery of the spiked virus for each method and sample type.
- Compare the measured concentrations between RT-qPCR and RT-ddPCR, particularly in non-extracted samples, to evaluate the impact of inhibitors. Statistical analysis will show ddPCR's superior performance in inhibitor-rich, non-extracted samples [75].
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of ddPCR and qPCR, especially for demanding applications like rare allele detection, relies on a carefully selected toolkit. The following table details key reagents and their functions, as evidenced by the reviewed studies.
Table 2: Key Research Reagent Solutions for ddPCR and qPCR Workflows
| Reagent / Kit | Function | Application Example |
|---|---|---|
| Mag-Bind cfDNA Kit | Extracts circulating free DNA (cfDNA) with high yield and minimal gDNA contamination, critical for rare variant detection. | Extraction of cfDNA from urine for oncology research [44]. |
| Colli-Pee UAS Preservative | Preserves urine samples at the point of collection to prevent degradation of labile cfDNA biomarkers. | Stabilization of urine samples for downstream ddPCR analysis of NRAS/EGFR mutations [44]. |
| One-step RT-ddPCR Advanced Kit | Integrates reverse transcription and PCR in a single tube, reducing hands-on time and pipetting errors. | Development of a multiplex 9-plex assay for respiratory and hepatitis viruses [76]. |
| Enviro Wastewater TNA Kit | Purifies total nucleic acids from complex and inhibitor-rich environmental samples like wastewater. | Concentration and extraction of viral RNA from wastewater for public health surveillance [76]. |
| QX200 Droplet Generator/Oil | Creates the nanoliter-sized droplet partitions essential for the ddPCR reaction. | All ddPCR applications using the Bio-Rad QX200 system [74] [44]. |
| Taqman Fast Advanced Mastermix | A robust enzyme mix for fast, highly specific qPCR assays using hydrolysis probes. | qRT-PCR detection of probiotic strains in fecal samples [74]. |
Application in Rare Allele Detection Research
The principles of ddPCR make it uniquely suited for rare allele detection, a critical area in cancer research and liquid biopsy development.
- Detection of Oncogenic Mutations: A 2025 application note demonstrated the detection of NRAS and EGFR gene variants from urinary cfDNA at allelic frequencies as low as 0.1%. This workflow, combining sample preservation, optimized cfDNA extraction, and ddPCR, highlights the technology's power for non-invasive cancer monitoring and early detection [44].
- Inherent Sensitivity for Rare Events: The ability to partition a sample allows ddPCR to detect a single mutant molecule among a background of 10,000 wild-type molecules. This sensitivity is fundamentally superior to qPCR for such applications, as the rare event is physically separated and concentrated in individual droplets, making its amplification and detection unambiguous [13].
The choice between ddPCR and qPCR is not a matter of one being universally better than the other, but rather of selecting the right tool for the specific research question. qPCR remains the workhorse for high-throughput, routine diagnostics and applications where relative quantification is sufficient, largely due to its established protocols, lower cost, and high throughput.
In contrast, ddPCR is the unequivocal choice for applications demanding the highest levels of sensitivity, precision, and robustness. Its capabilities in absolute quantification without standard curves, superior performance with low-abundance targets, and resilience to inhibitors make it indispensable for advanced research, including rare allele detection in oncology, precise viral load monitoring, and analysis of complex sample matrices like wastewater and stool. As molecular diagnostics continue to evolve towards more precise and liquid biopsy-based applications, the role of ddPCR is poised to expand significantly.
The analysis of circulating tumor DNA (ctDNA) has emerged as a cornerstone of liquid biopsy, enabling non-invasive cancer detection, monitoring of minimal residual disease (MRD), and real-time assessment of treatment response. ctDNA represents a tiny fraction (0.01% to <10%) of the total cell-free DNA (cfDNA) in circulation, presenting a significant technological challenge for reliable detection [47]. Two principal methodologies have dominated this field: droplet digital PCR (ddPCR) and next-generation sequencing (NGS). Each technology offers distinct advantages and limitations, creating a fundamental trade-off between ultra-sensitive detection of known variants and the ability to simultaneously screen multiple genomic regions for novel alterations.
ddPCR achieves exceptional sensitivity for detecting predefined mutations by partitioning samples into thousands of nanoliter-sized droplets, effectively creating individual PCR reactions that are read digitally to quantify target sequences with precision [77]. In contrast, NGS enables comprehensive profiling of multiple genomic regions in a single assay, making it ideal for discovery applications where the exact mutational landscape is unknown [47]. This technical guide examines the core principles, performance characteristics, and practical implementation of both platforms within the context of ctDNA analysis, providing researchers and drug development professionals with the framework to select appropriate methodologies for specific experimental and clinical questions.
Fundamental Technical Principles
Droplet Digital PCR (ddPCR) Core Mechanics
ddPCR operates on the principle of sample partitioning and digital counting, providing absolute quantification of nucleic acid targets without the need for standard curves. The workflow begins with partitioning of the PCR reaction mixture into approximately 20,000 water-in-oil droplets, where each droplet functions as an individual micro-reactor [77]. Following thermal cycling, each droplet is analyzed sequentially through a droplet reader that detects fluorescence signals. The fundamental advantage of this system lies in its binary endpoint measurement—droplets are classified as positive or negative based on fluorescence amplitude, enabling precise calculation of target concentration based on the ratio of positive to total droplets according to Poisson statistics [77].
The partitioning process is particularly powerful for rare allele detection because it effectively enriches the mutant signal by separating it from the abundant wild-type background. In a conventional PCR, a sample containing one mutant copy and 99 wild-type copies (1% variant allele frequency or VAF) would yield an undetectable mutant signal due to exponential amplification dominance by wild-type sequences [77]. However, in ddPCR, partitioning generates one droplet containing the mutant copy and 99 droplets containing wild-type copies, allowing independent amplification and detection of the rare mutant allele [77]. This technical approach enables ddPCR to achieve remarkable sensitivity down to 0.008% VAF under optimal conditions, making it particularly suitable for detecting low-frequency somatic variants in ctDNA analysis [77].
Next-Generation Sequencing (NGS) Core Mechanics
NGS employs a fundamentally different approach, simultaneously sequencing millions of DNA fragments in parallel to provide a comprehensive view of the genomic landscape. The core process begins with library preparation, where DNA fragments are ligated with adapter sequences and often amplified to create sequencing templates. During sequencing, bases are incorporated and detected through various detection methods (e.g., fluorescence, pH change, or luminescence), depending on the specific platform technology. The resulting data undergoes sophisticated bioinformatic processing including alignment to reference genomes, variant calling, and annotation to identify somatic alterations.
The key advantage of NGS in ctDNA analysis is its ability to interrogate multiple genomic regions simultaneously without prior knowledge of specific mutations. For example, targeted panels like the Ion AmpliSeq Cancer Hotspot Panel v2 can interrogate >2800 COSMIC variants across 50 oncogene and tumor suppressor gene hotspot regions, providing theoretical coverage of 99% in rectal cancer patients [47]. However, this comprehensive approach typically comes with reduced sensitivity compared to ddPCR, especially at lower sequencing depths. Standard NGS panels optimized for ctDNA analysis typically achieve detection thresholds around 0.1% VAF or higher, though this can be improved with specialized error-suppression methods and ultra-deep sequencing at the cost of significantly increased expense and computational requirements [47].
Performance Comparison in ctDNA Analysis
Direct Comparison of Sensitivity and Detection Rates
Multiple studies have directly compared the performance of ddPCR and NGS for ctDNA detection in clinical settings. A 2025 study focusing on localized rectal cancer provides compelling quantitative data, demonstrating that ddPCR detected ctDNA in 24 of 41 patients (58.5%) from baseline plasma samples, while NGS panels identified ctDNA in only 15 of the same 41 patients (36.6%)—a statistically significant difference (p = 0.00075) [47] [78]. This substantial disparity in detection rates highlights ddPCR's superior sensitivity for identifying low-abundance ctDNA, particularly in non-metastatic cancers where tumor DNA shedding into circulation may be limited.
The same study further validated these findings in an independent cohort, where 21 of 26 patients (80.8%) showed detectable ctDNA in pre-therapy plasma using the optimized detection methods [47]. Importantly, the researchers noted that positive ctDNA results correlated with established clinical indicators of disease severity, including higher clinical tumor stage and lymph node positivity identified by MRI [47]. These findings suggest that while both technologies can identify ctDNA, ddPCR offers enhanced capability for detecting the minimal disease burden that might be missed by standard NGS approaches.
Table 1: Direct Performance Comparison of ddPCR vs. NGS in Rectal Cancer ctDNA Detection
| Parameter | ddPCR Performance | NGS Performance | Study Details |
|---|---|---|---|
| Detection Rate | 24/41 patients (58.5%) | 15/41 patients (36.6%) | Development group, baseline plasma [47] |
| Statistical Significance | p = 0.00075 | p = 0.00075 | Comparison between methods [47] |
| Validation Cohort Detection | 21/26 patients (80.8%) | Not reported | Independent validation group [47] |
| Theoretical Coverage | Limited to pre-defined mutations | 99% in rectal patients (hotspot panel) | Targeted NGS panel covering 50 genes [47] |
| Limit of Detection (LOD) | As low as 0.008% VAF [77] | Approximately 0.1% VAF (standard panels) [47] | Dependent on sequencing depth and error correction |
Practical Considerations: Cost, Turnaround Time, and Workflow
Beyond pure performance metrics, practical considerations significantly influence technology selection for ctDNA analysis. Operational costs present a substantial differentiator, with studies indicating that ddPCR maintains a 5–8.5-fold cost advantage over NGS for ctDNA detection [47]. This economic efficiency stems from ddPCR's streamlined workflow, which requires minimal bioinformatic processing and utilizes standardized reagent kits. The simplified analytical process also translates to faster turnaround times, making ddPCR particularly suitable for clinical applications where rapid results are valuable for time-sensitive treatment decisions.
NGS, while more resource-intensive, offers unparalleled breadth of genomic interrogation. The technology's multiplexing capabilities enable simultaneous assessment of hundreds of genomic regions, providing a comprehensive mutational profile from limited specimen material. However, this advantage comes with operational complexities including extended library preparation protocols, lengthy sequencing runs, and computationally intensive bioinformatic analyses for base calling, alignment, and variant annotation [77]. These factors collectively extend turnaround times and increase personnel requirements compared to targeted ddPCR approaches.
Table 2: Practical Implementation Comparison for ctDNA Analysis
| Characteristic | ddPCR | NGS |
|---|---|---|
| Operational Cost | 5–8.5-fold lower than NGS [47] | High (reagents, sequencing, bioinformatics) |
| Turnaround Time | Rapid (hours to 1-2 days) | Extended (several days to weeks) |
| Multiplexing Capacity | Limited (typically 2-6 targets per reaction) | Extensive (hundreds to thousands of targets) |
| Input DNA Requirements | Low (as little as 1 ng gDNA demonstrated) [77] | Moderate to high (typically ≥50 ng) [77] |
| Bioinformatic Complexity | Minimal (binary classification of droplets) | Extensive (alignment, variant calling, filtering) |
| Ideal Application | Tracking known mutations, MRD monitoring | Discovery, comprehensive profiling, unknown targets |
Advanced Methodologies and Applications
Innovative Approaches Enhancing ddPCR Sensitivity
Recent methodological innovations have further expanded ddPCR's capabilities for rare variant detection. The SP-ddPCR approach utilizes SuperSelective primers that incorporate a long 5'-"anchor" sequence for strong hybridization and a very short 3'-"foot" sequence containing the interrogated nucleotide [52]. A "bridge" sequence between these regions creates a single-stranded bubble that enhances selective amplification of single-nucleotide variants (SNVs), enabling precise quantification of mutations present at frequencies below 0.1% [52]. This methodology has successfully confirmed the presence of potentially pathogenic variants in normal colonic mucosa with allelic frequencies ≤5%, demonstrating utility for detecting early molecular alterations in field carcinogenesis [52].
Methylation-specific ddPCR represents another advanced application, leveraging cancer-specific epigenetic alterations as detection markers. A 2025 study developed a multiplex ddPCR assay targeting five tumor-specific methylation markers for lung cancer detection, demonstrating ctDNA-positive rates of 38.7–46.8% in non-metastatic disease and 70.2–83.0% in metastatic cases [79]. This epigenetic approach capitalizes on the fact that methylation changes often occur early in carcinogenesis and are highly recurrent across tumors, providing an alternative to mutation-based detection that may be particularly valuable for cancer types with heterogeneous mutational profiles [79].
Clinical Applications Across Cancer Types
The complementary strengths of ddPCR and NGS have enabled diverse applications across oncology. In HPV-associated oropharyngeal squamous cell carcinoma (OPSCC), ddPCR assays targeting E7 genes of high-risk HPV genotypes (16, 18, 33, 35, 56, and 59) demonstrated 91.6% sensitivity for detecting circulating tumor HPV DNA (ctHPV-DNA), with levels correlating with TNM stage and tumor viral load [80]. Notably, 72.5% of post-treatment samples achieved undetectable ctHPV-DNA levels, highlighting the utility for treatment response monitoring [80].
In colorectal cancer, ddPCR has proven valuable for detecting mutations in genes such as NRAS and EGFR at allelic frequencies as low as 0.1%, even in challenging sample types like urinary cfDNA [44]. The technology's robustness with minimal input material (as little as 1 ng of formalin-fixed paraffin-embedded tissue) further enhances its applicability to real-world clinical specimens where sample quantity may be limited [77].
Experimental Protocols for ctDNA Analysis
Tumor-Informed ddPCR Protocol for MRD Detection
The tumor-informed approach represents a methodologically rigorous strategy for ctDNA detection, wherein mutations identified in tumor tissue are subsequently tracked in plasma using patient-specific ddPCR assays. The protocol begins with tumor tissue collection from surgical resection specimens or biopsies, with DNA extraction performed using validated kits such as the Maxwell FFPE Plus DNA Kit [79]. Primary tumor DNA sequencing follows, typically using targeted NGS panels (e.g., Ion AmpliSeq Cancer Hotspot Panel v2) to identify somatic mutations with high variant allele frequency [47].
For plasma collection and processing, blood samples are collected in specialized cfDNA preservation tubes (e.g., Streck Cell-Free DNA BCT) before any therapeutic intervention [47] [80]. Plasma is separated through two consecutive centrifugations (1600g for 10 minutes followed by 3000g for 10 minutes) to remove cellular components [80]. cfDNA extraction employs kits specifically designed for low-abundance nucleic acids, such as the QIAamp Circulating Nucleic Acid Kit or DSP Circulating DNA Kit, with elution volumes typically ranging from 50-60 μL [80] [79].
The ddPCR assay design phase involves selecting 1-2 mutations with the highest variant allele frequencies identified in tumor tissue and designing custom probes or utilizing SuperSelective primers for these specific targets [47] [52]. The ddPCR reaction assembly includes 15-20 ng of cfDNA mixed with ddPCR Supermix and target-specific assays in a total reaction volume that is partitioned into 20,000 droplets using automated droplet generators [47] [44]. Following PCR amplification with optimized thermal cycling conditions, droplets are analyzed on droplet readers, and data analysis is performed using companion software (e.g., QuantaSoft) with manual threshold setting based on control samples to distinguish positive and negative droplet populations [80] [44].
Tumor-Uninformed NGS Protocol for ctDNA Profiling
The tumor-uninformed NGS approach enables comprehensive mutation profiling without prior knowledge of tumor genetics, making it suitable for initial molecular characterization. The protocol begins with blood collection and plasma separation using identical methods to the ddPCR protocol to ensure sample quality [47]. Following cfDNA extraction, library preparation is performed using kits optimized for fragmented DNA, with incorporation of unique molecular identifiers (UMIs) to mitigate amplification biases and PCR errors [47].
For targeted sequencing, hybridization capture or amplicon-based panels (e.g., cancer hotspot panels) are used to enrich relevant genomic regions, with adjustments to potentially lower variant calling thresholds to 0.01% VAF to enhance sensitivity for ctDNA detection [47]. Sequencing is typically performed at high depth (>300× coverage) to ensure sufficient sampling of low-frequency variants, followed by bioinformatic analysis including alignment to reference genomes, UMI-based error suppression, and variant calling with statistical filtering to distinguish true somatic variants from technical artifacts [47].
Research Reagent Solutions
Table 3: Essential Research Reagents for ctDNA Analysis
| Reagent Category | Specific Products | Application Notes |
|---|---|---|
| Blood Collection Tubes | Streck Cell-Free DNA BCT [47] [80] | Preserves cfDNA integrity by preventing leukocyte lysis and nuclease degradation |
| cfDNA Extraction Kits | QIAamp Circulating Nucleic Acid Kit [80], DSP Circulating DNA Kit [79], Mag-Bind cfDNA Kit [44] | Optimized for low-concentration, fragmented DNA; critical for yield and purity |
| DNA Quantification | Cell-Free DNA ScreenTape Assay (Agilent 4150 TapeStation) [44] | Assesses cfDNA size distribution (peak ~160-200 bp) and quantifies yield |
| ddPCR Master Mix | ddPCR Multiplex Supermix [80], ddPCR Supermix for Probes [44] | Provides optimized reagents for partitioned PCR reactions with fluorescence detection |
| Bisulfite Conversion | EZ DNA Methylation-Lightning Kit [79] | Essential for methylation-based ddPCR assays; converts unmethylated cytosines to uracils |
| Control Materials | Mimix Multiplex I cfDNA Reference Standard [44], gBlocks [80] | Contains predefined mutations at specific allelic frequencies for assay validation |
Technology Selection Framework and Future Directions
The decision between ddPCR and NGS for ctDNA analysis depends primarily on the experimental objectives, with each technology occupying a distinct niche. ddPCR excels in scenarios requiring maximum sensitivity for tracking known mutations, particularly in MRD detection, treatment response monitoring, and surveillance for recurrence. Its cost-effectiveness, rapid turnaround, and technical simplicity make it ideal for focused applications in both research and clinical settings. Conversely, NGS remains indispensable for discovery-phase research, comprehensive genomic profiling, and situations where tumor tissue is unavailable for informed assay design.
Emerging technological developments suggest increasing convergence between these platforms. Novel approaches like the SP-ddPCR method are expanding the multiplexing capabilities of digital PCR while maintaining its exceptional sensitivity [52]. Simultaneously, advances in error-corrected NGS methodologies are progressively lowering the detection limits of sequencing-based approaches. The optimal ctDNA analysis strategy frequently involves a complementary approach, using NGS for initial molecular characterization and ddPCR for longitudinal monitoring of validated biomarkers. This integrated paradigm leverages the respective strengths of both platforms to provide comprehensive molecular insights into cancer dynamics, supporting both biological discovery and clinical translation in precision oncology.
Digital PCR (dPCR) represents a transformative evolution in nucleic acid quantification, enabling the absolute counting of target DNA molecules without the need for a standard curve. This third-generation PCR technology operates by partitioning a single PCR reaction into thousands to millions of individual reactions, allowing for the detection and quantification of target sequences with exceptional precision and sensitivity [2]. The principle of limiting dilution ensures that each partition contains zero, one, or a few target molecules according to a Poisson distribution. Following end-point amplification, the fraction of positive partitions is counted, and the absolute concentration of the target sequence is calculated using Poisson statistics [2] [81]. This partitioning creates an artificial enrichment of low-abundance sequences, making dPCR particularly powerful for detecting rare genetic mutations within a background of wild-type DNA—a critical capability in oncology and liquid biopsy applications [2] [81].
The emergence of dPCR has been particularly significant for rare allele detection in circulating tumor DNA (ctDNA), where it can identify mutant alleles present at frequencies as low as 0.1% amidst a vast excess of wild-type DNA [14]. This exquisite sensitivity enables non-invasive cancer detection, monitoring of minimal residual disease, and assessment of treatment response through liquid biopsy approaches [82] [2]. As commercial dPCR platforms have diversified, two dominant partitioning methodologies have emerged: droplet-based dPCR (ddPCR) and nanoplate-based dPCR (ndPCR), each with distinct technological implementations and performance characteristics that merit detailed comparative analysis for researchers engaged in rare variant detection.
Droplet-Based Digital PCR (ddPCR)
Droplet-based dPCR systems, exemplified by the Bio-Rad QX200 and QX600 platforms, employ water-in-oil emulsion technology to partition PCR reactions into thousands of nanoliter-sized droplets. These systems utilize microfluidic circuits and specialized surfactant chemistry to generate monodispersed droplets at high speeds (typically 1-100 kHz) [2]. The resulting droplets function as independent reaction vessels, with each droplet potentially containing a single target molecule. Following PCR amplification, the droplets are streamed through a microfluidic channel in single file and interrogated individually using a laser detection system and fluorescence reader [2] [8]. This in-line detection method allows for the analysis of a large number of partitions but requires precise control of droplet flow. The QX200 system typically generates approximately 20,000 droplets per reaction, though newer systems can produce significantly more partitions to enhance dynamic range and detection sensitivity [11].
Nanoplate-Based Digital PCR (ndPCR)
Nanoplate-based systems, such as the QIAGEN QIAcuity, utilize integrated microfluidic chips containing fixed arrays of nanoscale wells. The QIAcuity platform represents an all-in-one system that performs partitioning, thermal cycling, and imaging on a single instrument without the need for transfer between separate devices [83]. The system employs proprietary nanoplates that automatically partition the PCR mixture into either 8,000, 24,000, or 26,000 individual reactions depending on the specific plate configuration [83] [8]. Following partitioning and amplification, the entire nanoplate is imaged using a fluorescence microscope or scanner, providing a static snapshot of all partitions simultaneously [2]. This approach offers simplified workflow integration and eliminates potential issues with droplet stability or transfer, though it is constrained by the fixed number of partitions available on each plate [83].
The following diagram illustrates the core workflow differences between these two partitioning technologies:
Direct Performance Comparison: Sensitivity, Precision, and Accuracy
Limits of Detection and Quantification
Recent comparative studies provide critical insights into the performance characteristics of droplet-based versus nanoplate-based dPCR systems. In a 2025 study comparing the Bio-Rad QX200 ddPCR system with the QIAGEN QIAcuity ndPCR system for gene copy number analysis in protists, researchers established that the Limit of Detection (LOD) for ddPCR was approximately 0.17 copies/µL input (3.31 copies/reaction), while ndPCR demonstrated a slightly higher LOD of 0.39 copies/µL input (15.60 copies/reaction) [8]. Conversely, the Limit of Quantification (LOQ) showed an opposite trend, with ddPCR requiring 4.26 copies/µL input (85.2 copies/reaction) compared to ndPCR's 1.35 copies/µL input (54 copies/reaction) [8]. This suggests that while droplet-based systems may offer superior initial detection capabilities for extremely low-abundance targets, nanoplate-based systems might provide more reliable quantification at slightly higher concentrations.
Precision and Accuracy Metrics
The same study demonstrated that both platforms achieved high precision across most analyses, with coefficients of variation (CV) ranging between 6-13% for ddPCR and 7-11% for ndPCR when using synthetic oligonucleotides [8]. However, precision was significantly influenced by experimental conditions, particularly the choice of restriction enzymes when analyzing DNA from Paramecium tetraurelia. For ddPCR, CV values varied dramatically between 2.5% and 62.1% with EcoRI, but improved to less than 5% with HaeIII. In contrast, ndPCR showed less variability between enzymes, with CVs ranging from 0.6% to 27.7% for EcoRI and 1.6% to 14.6% for HaeIII [8]. This indicates that nanoplate-based systems may offer more consistent performance across varying experimental conditions—a valuable characteristic for standardized clinical applications.
For copy number variation (CNV) analysis, ddPCR has demonstrated remarkable accuracy when validated against gold-standard methods. In a 2025 study examining the DEFA1A3 gene, ddPCR showed 95% concordance with pulsed field gel electrophoresis (PFGE), with a strong Spearman correlation of r = 0.90 (p < 0.0001) and results differing by only 5% on average from the reference method [11]. This performance establishes ddPCR as a robust methodology for CNV applications requiring high accuracy.
Table 1: Comparative Performance Metrics of ddPCR vs. ndPCR Platforms
| Performance Parameter | Droplet-Based (QX200) | Nanoplate-Based (QIAcuity) | Experimental Context |
|---|---|---|---|
| Limit of Detection (LOD) | 0.17 copies/µL input [8] | 0.39 copies/µL input [8] | Synthetic oligonucleotides |
| Limit of Quantification (LOQ) | 4.26 copies/µL input [8] | 1.35 copies/µL input [8] | Synthetic oligonucleotides |
| Precision (CV Range) | 6-13% [8] | 7-11% [8] | Synthetic oligonucleotides |
| Restriction Enzyme Impact | High (CV: 2.5-62.1%) [8] | Moderate (CV: 0.6-27.7%) [8] | Paramecium DNA with EcoRI |
| Accuracy in CNV | 95% concordance with PFGE [11] | Not reported | DEFA1A3 gene copy number |
| Methylation Detection | Specificity: 100%, Sensitivity: 98.03% [84] | Specificity: 99.62%, Sensitivity: 99.08% [84] | CDH13 gene in breast cancer |
Method-Specific Performance Characteristics
In DNA methylation analysis, a 2025 comparative study of the CDH13 gene in breast cancer tissues revealed that both platforms performed exceptionally well, with ddPCR achieving 100% specificity and 98.03% sensitivity, while ndPCR reached 99.62% specificity and 99.08% sensitivity [84]. The methylation levels measured by both methods showed a strong correlation (r = 0.954), indicating that the primary differentiators between platforms may not be raw performance but rather workflow considerations [84].
For viral load quantification in respiratory infections, dPCR has demonstrated superior accuracy compared to Real-Time RT-PCR, particularly for high viral loads of influenza A, influenza B, and SARS-CoV-2, and for medium loads of RSV [7]. This enhanced performance is attributed to dPCR's resistance to PCR inhibitors and its ability to provide absolute quantification without standard curves, though the study noted that platform choice should consider throughput requirements and automation capabilities [7].
Experimental Design and Protocol Implementation
Workflow Comparison and Practical Implementation
The practical implementation of dPCR experiments reveals significant workflow differences between droplet-based and nanoplate-based systems. The QIAcuity ndPCR system integrates partitioning, thermal cycling, and imaging on a single instrument, with a total time from system start to results of less than two hours [83]. The system uses microfluidic nanoplate technology where preparation involves transferring mastermix, probes, primers, and samples directly to a nanoplate, which is then sealed and placed in the instrument for automated processing [83]. Data analysis occurs through the QIAcuity Software Suite, which provides concentration in copies per microliter and quality controls, with remote analysis capability within the same local area network.
In contrast, droplet-based systems typically involve a multi-step process where the PCR mixture is first converted to a water-in-oil emulsion using a droplet generator, transferred to a standard thermal cycler for amplification, and then moved to a droplet reader for fluorescence detection [2] [14]. This process requires more manual handling and transfer between instruments but offers greater flexibility in reaction setup and the ability to archive droplets for potential re-analysis.
The following workflow diagram illustrates the procedural differences between these platforms:
Rare Allele Detection Protocol for Liquid Biopsy
For rare allele detection in liquid biopsy applications, such as identifying oncogenic mutations in urinary cfDNA, a validated protocol involves several critical steps. First, sample preservation is essential to minimize cfDNA degradation—urine samples can be preserved using specialized preservatives like Colli-Pee UAS at a 2.3:1 ratio immediately after collection [14]. cfDNA extraction follows, with kits specifically designed for cell-free DNA (such as Mag-Bind cfDNA Kit) processing 4mL input volumes and eluting in 50μL to maximize concentration [14]. Extraction quality should be verified using fragment analysis systems like the Agilent 4150 TapeStation with Cell-Free DNA ScreenTape Assay, looking for the characteristic ~160-200bp banding pattern of cfDNA without significant genomic DNA contamination [14].
For ddPCR analysis, approximately 15ng of extracted cfDNA is mixed with ddPCR Supermix for Probes in a 300μL total reaction volume [14]. Mutant and wildtype alleles are detected using sequence-specific FAM and SUN probes, with droplets generated using a QX200 Droplet Generator. Thermal cycling employs optimized annealing temperatures for each probe set, followed by droplet reading on a QX200 Droplet Reader and analysis with QuantaSoft Analysis Pro Software [14]. This approach reliably detects mutant alleles at frequencies as low as 0.1% with appropriate validation controls, including no-template controls and wildtype-only controls to establish background signals and specificity thresholds [14].
Research Reagent Solutions for dPCR Experiments
Table 2: Essential Research Reagents for dPCR-Based Rare Allele Detection
| Reagent Category | Specific Examples | Function & Importance |
|---|---|---|
| Nucleic Acid Preservation | Colli-Pee UAS preservative [14] | Stabilizes cfDNA in urine samples immediately post-collection to prevent degradation |
| cfDNA Extraction Kits | Mag-Bind cfDNA Kit [14] | Isolves cell-free DNA from body fluids with minimal genomic DNA contamination |
| dPCR Master Mix | ddPCR Supermix for Probes (No dUTP) [14] | Optimized reaction chemistry for digital PCR with probe-based detection |
| Reference Standards | Mimix Multiplex I cfDNA Set Reference Standard [14] | Provides validated controls with known mutation allele frequencies for assay validation |
| Quality Assessment | Agilent Cell-Free DNA ScreenTape Assay [14] | Verifies cfDNA size distribution and quantifies yield without genomic DNA contamination |
| Probe-Based Detection | Target-specific FAM/SUN probe sets [14] | Enables multiplex detection of wildtype and mutant alleles with different fluorophores |
| Universal Probe Systems | USE-PCR color-coded tags [85] | Streamlines assay development through universal probe chemistry compatible with multiple platforms |
Emerging Technologies and Future Directions
The dPCR landscape continues to evolve with emerging technologies that enhance multiplexing capabilities and workflow efficiency. Universal Signal Encoding PCR (USE-PCR) represents a significant innovation that combines universal hydrolysis probes with amplitude modulation and multispectral encoding to enable highly multiplexed target detection [85]. This approach utilizes color-coded tags in allele-specific primers that generate distinct fluorescent signatures when amplified, allowing a single universal probe mixture to detect up to 32 different targets through ternary encoding [85]. This system decouples analyte detection from signal generation, significantly streamlining assay development and enabling standardized analysis across multiple dPCR platforms including the Absolute Q, QIAcuity, QX600, and Digital Light Cycler [85].
For rare variant detection specifically, methods like SPIDER-seq (sensitive genotyping based on peer-to-peer network-derived identifier for error reduction in amplicon sequencing) enable molecular identity tracking in PCR-derived libraries using overwritten barcodes [82]. This approach constructs cluster identifiers (CIDs) from peer-to-peer networks of daughter molecules derived from original templates, effectively reducing errors and detecting mutations at frequencies as low as 0.125% [82]. Such advancements address fundamental challenges in rare allele detection, particularly the discrimination of true mutations from polymerase errors introduced during early amplification cycles.
Platform Selection Guidelines for Research Applications
Selection between droplet-based and nanoplate-based dPCR systems depends heavily on specific research requirements and operational considerations. The following decision framework can guide researchers in selecting the most appropriate platform for their rare allele detection applications:
Choose Droplet-Based dPCR When:
- Maximum sensitivity for ultra-rare variants (<0.1% allele frequency) is the primary priority [8] [14]
- Reaction setup flexibility and the ability to archive samples (as stable droplets) are valuable
- Budget constraints favor lower consumable costs per sample
- Experimental protocols require customization beyond standardized workflows
Choose Nanoplate-Based dPCR When:
- Workflow integration and minimal manual handling are critical for throughput needs [83]
- Standardized operation across multiple users is essential for reproducibility
- Rapid turnaround time (<2 hours from start to results) benefits project timelines [83]
- Laboratory space optimization favors all-in-one instrumentation over multiple devices [83]
For research focused specifically on rare allele detection, both platforms demonstrate excellent capabilities with slight variations in performance characteristics. The emerging consensus from recent comparative studies indicates that the core differentiators increasingly involve workflow efficiency, operational simplicity, and integration with existing laboratory processes rather than fundamental performance disparities [8] [84] [7]. As dPCR technology continues to mature, platform selection should consider both current application needs and future requirements for assay development, multiplexing expansion, and potential clinical translation.
Droplet Digital PCR (ddPCR) is establishing a new paradigm for the detection of rare alleles in clinical diagnostics, particularly in the identification of bloodstream pathogens. In the context of sepsis and other bloodstream infections (BSIs), the rapid and accurate identification of causative agents is a critical determinant of patient outcomes. Blood culture, the long-standing gold standard, is hampered by a prolonged time-to-result (often 24-72 hours) and significantly reduced sensitivity in patients who have previously received antibiotics [86] [87]. This diagnostic delay necessitates the use of broad-spectrum antimicrobials, contributing to the global challenge of antimicrobial resistance. The core principle of ddPCR for rare allele detection lies in its ability to partition a sample into thousands of nanoliter-sized droplets, effectively isolating individual DNA molecules for amplification. This process allows for the absolute quantification of target pathogen DNA with a sensitivity and speed that culture-based methods cannot match [87]. This technical guide explores the clinical validation of ddPCR findings by correlating them with conventional blood culture and imaging results, providing a framework for researchers and clinicians to integrate this advanced molecular technique into diagnostic and therapeutic pipelines.
Performance Comparison: ddPCR vs. Blood Culture
Clinical studies consistently demonstrate that ddPCR offers superior sensitivity and a drastically reduced detection time compared to blood culture. The following table summarizes key quantitative findings from recent clinical investigations.
Table 1: Comparative Performance of ddPCR and Blood Culture in Clinical Studies
| Study Focus | Blood Culture Positivity | ddPCR Positivity | Key Pathogens Detected by ddPCR | Average Detection Time |
|---|---|---|---|---|
| BSI Detection (n=149) [86] [88] | 6/149 specimens (4.0%); 6 strains | 42/149 specimens (28.2%); 63 strains | Acinetobacter baumannii, Streptococcus spp., Cytomegalovirus, Klebsiella pneumoniae, Staphylococcus aureus | ddPCR: 4.8 ± 1.3 hoursBlood Culture: 94.7 ± 23.5 hours |
| CLABSI Detection (Central Line Samples) [89] | Reference Standard | Sensitivity: 91% (95% CI: 77-98)Specificity: 96% (95% CI: 85-99) | Not Specified | ddPCR: ~2.5 hours |
| Analytical Sensitivity (Spiked Blood) [90] | Reference Standard (CFU/mL) | Almost perfect correlation with culture (r ≥ 0.997, p ≤ 0.001) | Escherichia coli, Klebsiella pneumoniae, Staphylococcus aureus, Enterococcus spp. | ddPCR: 3.5 to 4 hours |
The data reveal ddPCR's enhanced capability to detect polymicrobial infections. One study identified 14 cases of polymicrobial infections via ddPCR, including double, triple, and even quadruple infections, which are often missed by blood culture [86]. Furthermore, ddPCR's quantitative output (in copies/mL) provides a potential metric for assessing pathogen load, which could correlate with disease severity and therapeutic response [86] [90].
Experimental Protocols for Clinical Validation
Validating ddPCR against blood culture requires a rigorous and standardized experimental workflow. The following protocol outlines the key steps for a correlative study.
Sample Collection and Preparation
- Patient Cohort: Enroll patients with suspected bloodstream infections based on clinical signs (e.g., fever ≥38.0°C, elevated white blood cell count, increased C-reactive protein or procalcitonin) [86] [88].
- Blood Collection: Draw venous blood using standard aseptic technique. Collect samples for both blood culture and ddPCR simultaneously.
- For Blood Culture: Collect two sets (aerobic and anaerobic) with a recommended volume of 10 mL per set [86] [88]. Incubate using an automated system like BacT/ALERT 3D.
- For ddPCR: Collect blood into tubes containing EDTA [86] [88]. Centrifuge at 1,600 × g for 10 minutes to separate plasma, which is then used for DNA extraction.
DNA Extraction and ddPCR Assay
- cfDNA Extraction: Extract circulating free DNA (cfDNA) from plasma using specialized kits (e.g., Mag-Bind cfDNA Kit) and automated systems to maximize yield and minimize contamination [86] [14].
- ddPCR Reaction Setup: Design primer-probe sets for target pathogens. Common targets for sepsis include:
- Partitioning and Amplification: Combine the extracted DNA with a PCR mastermix and primers/probes. Generate droplets (e.g., using the QX200 Droplet Generator, Bio-Rad). Perform PCR amplification with a optimized thermal cycling protocol [87] [3].
- Data Analysis: Read the droplets using a droplet reader (e.g., QX200 QuantaSoft Droplet Reader). Use analysis software (e.g., QuantaSoft Analysis Pro) to quantify the absolute number of target DNA copies per milliliter of blood [14] [87].
The following diagram illustrates the parallel workflow for comparing ddPCR with the gold standard.
Correlation with Imaging Results
While direct detection of pathogens is not the function of most clinical imaging, correlating ddPCR findings with imaging studies like computed tomography (CT) or magnetic resonance imaging (MRI) can provide a comprehensive clinical picture.
- Identify the Focus of Infection: A positive ddPCR result for a specific pathogen in a patient with pulmonary infiltrates on chest X-ray or CT adds microbiological confirmation to radiological findings [86].
- Assess Severity and Complication: In cases of suspected metastatic infection, ddPCR can help confirm the causative organism. For example, detecting Staphylococcus aureus DNA via ddPCR in a patient with bacteremia and a new vertebral lesion on MRI strongly supports a diagnosis of spondylodiscitis.
- Monitor Treatment Response: A decreasing pathogen DNA load in serial ddPCR tests, coupled with improved imaging findings (e.g., resolving abscesses or infiltrates), can serve as a robust indicator of successful antimicrobial therapy.
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of a ddPCR assay for pathogen detection relies on a suite of specialized reagents and instruments.
Table 2: Key Research Reagent Solutions for ddPCR-Based Pathogen Detection
| Item | Function | Example Products & Notes |
|---|---|---|
| Blood Collection Tubes | Preserves cell-free DNA and prevents degradation for accurate molecular results. | Streck Cell-Free DNA BCT tubes; EDTA tubes for plasma separation [86] [47]. |
| cfDNA Extraction Kit | Isolves high-purity circulating free DNA from plasma; critical for yield and sensitivity. | Mag-Bind cfDNA Kit; other kits from Pilot Gene Technology [86] [14]. |
| ddPCR System | Partitions samples, performs amplification, and reads results for absolute quantification. | QX200 Droplet Digital PCR System (Bio-Rad); Naica System (Stilla Technologies) [14] [87] [3]. |
| PCR Mastermix | Provides optimized reagents for efficient amplification in a droplet environment. | ddPCR Supermix for Probes (No dUTP); PerfeCTa Multiplex mastermix [14] [3]. |
| Species-Specific Primers/Probes | Binds to and detects unique genetic sequences of the target pathogen(s). | Custom-designed assays for genes like coa (S. aureus), uidA (E. coli), 16S rRNA [87] [90]. |
| Droplet Generator Oil | Creates the nanoliter-sized droplets that are the core of the digital PCR process. | DG Droplet Generation Oil for Probes (Bio-Rad); specific oils are system-dependent. |
| Positive Control DNA | Validates assay performance and serves as a quantitative standard. | Genomic DNA from ATCC strains; synthetic DNA controls [87]. |
Analysis and Interpretation of Correlative Data
Interpreting the results from ddPCR and blood culture requires a nuanced understanding of the strengths and limitations of each method.
- Concordant Positive Results: When both ddPCR and blood culture identify the same pathogen, the result provides high confidence, with ddPCR offering the added advantage of quantification [86] [90].
- ddPCR Positive / Culture Negative: This is a frequent and clinically significant finding. It may indicate:
- True Infection: The patient has a current infection, but the pathogen is non-culturable, slow-growing, or the patient has received prior antibiotic therapy [86] [87].
- Non-Viable Pathogen DNA: Detection of nucleic acids from non-viable organisms, which can occur during the effective clearance of an infection.
- Culture Positive / ddPCR Negative: This scenario may arise if:
- Quantifying Pathogen Load: The absolute quantification provided by ddPCR (copies/mL) can be correlated with clinical severity scores and inflammatory markers like CRP and PCT to assess disease burden and monitor response to treatment [86] [90].
The integration of Droplet Digital PCR into the diagnostic workflow for bloodstream infections represents a significant advancement. Clinical validation studies consistently confirm that ddPCR exhibits higher sensitivity and a dramatically faster time-to-result than traditional blood culture. Its ability to quantify pathogen DNA load and detect polymicrobial and pre-treated infections provides clinicians with a powerful tool for making informed, timely therapeutic decisions. Correlating ddPCR findings with blood culture and imaging results creates a robust, multi-modal diagnostic picture, enhancing patient care and supporting antimicrobial stewardship efforts. As the technology continues to evolve and standardize, its role in the precise diagnosis and management of infectious diseases is poised to expand further.
Conclusion
Droplet Digital PCR has firmly established itself as a transformative technology for rare allele detection, offering unparalleled sensitivity and absolute quantification that are critical for modern biomedical research and clinical diagnostics. Its proven success in liquid biopsy for oncology, non-invasive pathogen detection, and rare variant screening underscores its potential to personalize medicine and improve patient outcomes. Future directions will focus on standardizing protocols for clinical adoption, expanding multiplexing capabilities, and integrating ddPCR into large-scale therapeutic monitoring and early detection programs. As the technology continues to evolve, its role in unlocking the diagnostic information contained within rare nucleic acid populations will only become more profound, solidifying its position as an indispensable tool in the precision medicine arsenal.
References
- 1. How does ddPCR work? [https://mogene.com/how-does-ddpcr-work/]
- 2. Digital PCR: from early developments to its future application ... [https://pubs.rsc.org/en/content/articlehtml/2025/lc/d5lc00055f]
- 3. Rare Mutation Detection Digital PCR Tutorial [https://www.stillatechnologies.com/digital-pcr/primer-design/rare-mutation-detection/]
- 4. Digital PCR: from early developments to its future application ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12278255/]
- 5. Quantitative or digital PCR? A comparative analysis for ... [https://www.sciencedirect.com/science/article/abs/pii/S0165993624001584]
- 6. When to Choose Quantitative PCR vs Digital PCR [https://kcasbio.com/blogs/quantitative-pcr-vs-digital-pcr-choosing-the-right-platform-for-bioanalysis/]
- 7. Comparative Performance of Digital PCR and Real-Time RT ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12474457/]
- 8. Comparing the precision of two digital PCR applications for ... [https://www.nature.com/articles/s41598-025-13143-8]
- 9. Performance and characteristics of three digital PCR ... [https://pubmed.ncbi.nlm.nih.gov/41004253/]
- 10. Fully Automatic Droplet Digital PCR Systems 2025 Trends ... [https://www.archivemarketresearch.com/reports/fully-automatic-droplet-digital-pcr-systems-206536]
- 11. Digital droplet PCR is an accurate and precise method to ... [https://www.nature.com/articles/s41598-025-20944-4]
- 12. utPCR: A Strategy for the Highly Specific and Absolutely ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10605045/]
- 13. Variant Quantitation Using Rare Droplet Digital PCR [https://pubmed.ncbi.nlm.nih.gov/30350210/]
- 14. Variant Rare from Urinary cfDNA by Allele ... Detection Droplet Digital [https://omegabiotek.com/application-notes/rare-variant-allele-detection-from-urinary-cfdna-by-droplet-digital-pcr/]
- 15. [2501.09218] Interpretable Droplet Digital PCR Assay for ... [https://arxiv.org/abs/2501.09218]
- 16. Fundamentals of digital PCR [https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/pcr/digital-pcr/what-is-digital-pcr]
- 17. Principles of digital PCR and its applications in current ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6943456/]
- 18. Poisson distribution [https://en.wikipedia.org/wiki/Poisson_distribution]
- 19. Digital Droplet PCR: CNV Analysis and Other Applications [https://pmc.ncbi.nlm.nih.gov/articles/PMC4355013/]
- 20. Poisson Plus Quantification for Digital PCR Systems [https://www.nature.com/articles/s41598-017-09183-4]
- 21. dPCR vs. ddPCR: Why Precision Matters in Cell and Gene ... [https://www.roslinct.com/insights/dpcr-vs-ddpcr/]
- 22. Droplet Digital PCR: A Powerful Tool for Accurate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12080313/]
- 23. Droplet Digital PCR World 2025 [https://info.bio-rad.com/droplet-digital-pcr-world-2025?WT.mc_id=250401046234]
- 24. Droplet digital PCR method for the absolute quantitative ... [https://www.sciencedirect.com/science/article/pii/S0740002023000527]
- 25. : Advantages and Applications in Molecular... Droplet Digital PCR [https://www.labx.com/resources/droplet-digital-pcr-advantages-and-applications-in-molecular-diagnostics/5581]
- 26. dPCR Protocol [https://www.sigmaaldrich.com/US/en/technical-documents/protocol/genomics/qpcr/dpcr?srsltid=AfmBOoo82OLZ-DkqZVEOBih5Yp6IZYavyDynzacomXJyG_bALKJ2agdN]
- 27. Multiplex Detection of Rare Mutations by Picoliter Droplet ... | PLOS One [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0159094]
- 28. Liquid biopsy in cancer: current status, challenges and ... [https://www.nature.com/articles/s41392-024-02021-w]
- 29. Liquid biopsy for human cancer: cancer screening, monitoring ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11130638/]
- 30. Current and Emerging Applications of Droplet Digital PCR ... [https://pubmed.ncbi.nlm.nih.gov/34773243/]
- 31. Drug Discovery News — ctDNA monitoring is providing a ... [https://friendsofcancerresearch.org/news/drug-discovery-news-ctdna-monitoring-is-providing-a-smarter-way-to-track-and-treat-cancer/]
- 32. Droplet Digital PCR versus qPCR for gene expression ... [https://www.nature.com/articles/s41598-017-02217-x]
- 33. Droplet-Based Digital PCR: Application in Cancer Research [https://www.sciencedirect.com/science/article/abs/pii/S0065242316300956]
- 34. Evaluation of droplet digital qRT-PCR (dd ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10352106/]
- 35. ddpcr: Analysis and visualization of Droplet Digital PCR ... [https://cran.r-project.org/web/packages/ddpcr/readme/README.html]
- 36. ddpcr: an R package and web application for analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5031129/]
- 37. dPCR: A Technology Review - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC5948698/]
- 38. Introduction of Noninvasive Prenatal Testing for Blood ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7443669/]
- 39. Non-invasive prenatal testing: a revolutionary journey in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10666054/]
- 40. Panorama – Non-Invasive Prenatal Testing (NIPT) [https://www.natera.com/womens-health/panorama-nipt-prenatal-screening/]
- 41. NIPT Test (Noninvasive Prenatal Testing): What To Expect [https://my.clevelandclinic.org/health/diagnostics/21050-nipt-test]
- 42. A Comprehensive Guide to Non-Invasive Prenatal Testing [https://www.thebump.com/a/non-invasive-prenatal-testing]
- 43. Comparison of Analytic Methods for Quantitative Real-Time ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4642823/]
- 44. Rare Variant Allele Detection from Urinary cfDNA by ... [https://omegabiotek.com/rare-variant-allele-detection-from-urinary-cfdna-by-droplet-digital-pcr/]
- 45. Evaluation of urinary cfDNA workflows for the molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12547846/]
- 46. Diagnostic Accuracy of Droplet Digital PCR and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7063461/]
- 47. Performance Comparison of Droplet Digital PCR and Next ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12062871/]
- 48. Optimized Digital Droplet PCR for BCR-ABL [https://www.sciencedirect.com/science/article/pii/S1525157818300710]
- 49. Single Droplet Digital Polymerase Chain Reaction for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6235282/]
- 50. PCR inhibition in qPCR, dPCR and MPS—mechanisms and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7072044/]
- 51. Evaluation of PCR-enhancing approaches to reduce ... [https://www.sciencedirect.com/science/article/abs/pii/S0048969724069250]
- 52. Quantification of rare somatic single nucleotide variants by ... [https://www.nature.com/articles/s41598-023-39874-0]
- 53. Digital Assays Part I: Partitioning Statistics and Digital PCR [https://www.sciencedirect.com/science/article/pii/S2472630322013334]
- 54. Digital PCR | Protocols | Applications [https://www.oncodesign-services.com/cpt_ressource/five-reasons-why-you-should-switch-to-digital-pcr-dpcr/]
- 55. Improvement of digital PCR conditions for direct detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7439326/]
- 56. Effect of the Matrix and Target on the Accurate ... [https://www.mdpi.com/2077-0472/13/1/127]
- 57. Multiplex digital PCR enables sensitive detection of ... [https://pubmed.ncbi.nlm.nih.gov/39849159/]
- 58. Digital PCR for Rare Mutation Detection [https://www.thermofisher.com/us/en/home/life-science/pcr/digital-pcr/mutation-detection.html]
- 59. Development and validation of a droplet digital PCR assay ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1573949/full]
- 60. Optimization of TaqMan-based quantitative PCR diagnosis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12165414/]
- 61. Ultra-Rapid Droplet Digital PCR Expedites and Refines ... [https://www.insideprecisionmedicine.com/topics/oncology/ultra-rapid-droplet-digital-pcr-expedites-and-refines-intraoperative-brain-cancer-surgery/]
- 62. CYP2D6 copy number determination using digital PCR [https://pmc.ncbi.nlm.nih.gov/articles/PMC11349684/]
- 63. Droplet digital PCR-based analyses for robust, rapid, and ... [https://actaneurocomms.biomedcentral.com/articles/10.1186/s40478-022-01335-6]
- 64. Droplet Digital Polymerase Chain Reaction - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/droplet-digital-polymerase-chain-reaction]
- 65. Evaluation of Droplet Digital Polymerase Chain Reaction ... [https://www.mdpi.com/1422-0067/21/9/3043]
- 66. Limit of Blank, Limit of Detection and Limit of Quantitation [https://pmc.ncbi.nlm.nih.gov/articles/PMC2556583/]
- 67. Method Validation Essentials, Limit of Blank ... [https://www.biopharminternational.com/view/method-validation-essentials-limit-blank-limit-detection-and-limit-quantitation]
- 68. Determining lower limits of detection of digital PCR assays ... [https://www.sciencedirect.com/science/article/pii/S2214753514000047]
- 69. A novel KRAS exon 2 drop-off digital PCR assay for mutation ... [https://diagnosticpathology.biomedcentral.com/articles/10.1186/s13000-025-01637-y]
- 70. Multicolor Combinatorial Probe Coding for Real-Time PCR [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0016033]
- 71. How to multiplex with color combination in Crystal Digital ... [https://www.stillatechnologies.com/digital-pcr/naica-system-analysis/how-to-multiplex-with-color-combination-in-crystal-digital-pcr/]
- 72. Three-color crystal digital PCR [https://www.sciencedirect.com/science/article/pii/S2214753516300328]
- 73. Basic Principles of RT-qPCR [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/basic-principles-rt-qpcr.html]
- 74. Comparison of qRT-PCR and ddPCR for multi-strain ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1579797/full]
- 75. Comparison of RT-qPCR and RT-ddPCR on Assessing ... [https://pubmed.ncbi.nlm.nih.gov/40673468/]
- 76. One Assay, Nine Targets: Advancing Viral Surveillance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12529474/]
- 77. Utility of droplet digital polymerase chain reaction for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10360090/]
- 78. Performance Comparison of Droplet Digital PCR and Next- ... [https://pubmed.ncbi.nlm.nih.gov/40346007/]
- 79. Development and validation of a methylation-specific ... [https://www.nature.com/articles/s41598-025-15228-w]
- 80. Sensitive and Specific Droplet Digital PCR Assays for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11512825/]
- 81. Digital PCR Principle, techniques and applications [https://www.stillatechnologies.com/digital-pcr/principle/]
- 82. Correcting errors in PCR-derived libraries for rare allele ... [https://www.nature.com/articles/s42003-025-08537-3]
- 83. Digital PCR workflow and products [https://www.qiagen.com/us/applications/digital-pcr/workflow-and-products]
- 84. Comparative Analysis of Two Digital PCR Platforms for ... [https://pubmed.ncbi.nlm.nih.gov/40826981/]
- 85. Multiplexed, universal probe-based rare variant detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12227673/]
- 86. Comparative analysis between digital PCR and blood ... [https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2025.1615409/full]
- 87. Multiplex Droplet Digital Polymerase Chain Reaction ... [https://www.researchprotocols.org/2021/12/e33746]
- 88. Comparative analysis between digital PCR and blood ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12515817/]
- 89. ddPCR enables rapid detection of bloodstream infections ... [https://www.sciencedirect.com/science/article/pii/S1684118224000495]
- 90. Quantification of bacterial DNA in blood using droplet ... [https://www.sciencedirect.com/science/article/pii/S0732889323001840]