Validating Hot-Spot Residues by Alanine Scanning: A Guide for Drug Discovery Scientists
This article provides a comprehensive resource for researchers and drug development professionals on the validation of hot-spot residues through alanine scanning mutagenesis.
Validating Hot-Spot Residues by Alanine Scanning: A Guide for Drug Discovery Scientists
Abstract
This article provides a comprehensive resource for researchers and drug development professionals on the validation of hot-spot residues through alanine scanning mutagenesis. We cover the foundational principles of protein-protein interactions and hot spots, detail state-of-the-art methodological approaches from experimental to high-throughput computational techniques, address common troubleshooting and optimization challenges, and present a framework for rigorous validation through comparative analysis with other biophysical methods. The integration of these strategies is crucial for accurately identifying key residues that drive binding affinity, thereby informing the rational design of targeted therapeutics.
Protein-Protein Interactions and Hot Spots: The Energetic Foundation of Binding
In protein-protein interactions (PPIs), hot spots are defined as residues that contribute significantly to the binding free energy. Experimental identification via alanine scanning mutagenesis is the gold standard, but it is costly and time-consuming. This has spurred the development of diverse computational methods to predict these critical residues. This guide provides an objective comparison of the performance, methodologies, and applicability of major computational hot spot prediction tools, contextualized within experimental validation frameworks crucial for researchers and drug development professionals.
Most cellular processes are governed by protein-protein interactions, and understanding their precise mechanisms is vital for drug discovery. Within the large surface of a protein-protein interface, the binding energy is not distributed uniformly. Instead, a small subset of residues, known as hot spots, accounts for the majority of the binding free energy [1] [2].
The canonical experimental method for identifying hot spots is alanine scanning mutagenesis. This technique involves systematically mutating individual interface residues to alanine and measuring the resulting change in binding free energy (ΔΔG) [1] [3]. A residue is typically defined as a hot spot if its mutation to alanine causes a ΔΔG ≥ 2.0 kcal/mol [1] [4]. This experimental definition, established by Clackson and Wells, forms the basis for validating computational predictions [1].
Despite its reliability, experimental alanine scanning is low-throughput. Each mutant must be expressed, purified, and analyzed separately, making it prohibitively expensive and slow for large-scale studies [1] [3]. Consequently, computational methods have been developed to predict hot spots from protein structure and sequence data, offering a rapid and scalable alternative.
Computational hot spot prediction methods can be broadly categorized into several types based on their underlying algorithms. The table below summarizes the key features of prominent methods.
Table 1: Overview of Major Computational Hot Spot Prediction Methods
| Method Name | Category | Required Input | Key Features/Algorithm | Availability |
|---|---|---|---|---|
| FoldX [1] [3] | Energy-Based / Empirical Force Field | Protein Complex Structure | Computational alanine scanning using an empirical force field. | Standalone Tool & Server |
| Robetta [1] [3] | Energy-Based / Physical Force Field | Protein Complex Structure | Computational alanine scanning using the Rosetta force field and conformational sampling. | Server |
| MM/GBSA/IE [5] | Molecular Dynamics (MD) Simulation | Protein Complex Structure | Binds free energy calculation from MD trajectories using Molecular Mechanics/Generalized Born Surface Area with Interaction Entropy. | Research Code |
| BudeAlaScan [3] | Energy-Based / Empirical Force Field | Protein Complex Structure or Ensemble | Adapted from a small-molecule docking algorithm (BUDE); can process structural ensembles from NMR or MD. | Command-Line Tool |
| HotSprint [6] | Knowledge-Based / Conservation | Protein Complex Structure | Combines evolutionary conservation from Rate4Site and solvent accessibility (ASA). | Database & Web Server |
| PredHS2 [7] | Machine Learning | Protein Complex Structure | Extreme Gradient Boosting (XGBoost) model with 26 optimal features including novel solvent exposure and disorder scores. | Research Code |
| PPI-hotspotID [8] | Machine Learning | Free Protein Structure | Ensemble classifier using only 4 residue features (conservation, aa type, SASA, and gas-phase energy). | Web Server |
| Min-SDS [4] | Graph Theory | Protein Complex Structure | Finds high-density subgraphs in residue interaction networks to identify potential hot spot clusters. | Research Code |
Performance Comparison of Prediction Tools
The predictive performance of these tools is typically benchmarked against experimental data from databases like ASEdb, BID, and SKEMPI. The following table summarizes quantitative performance metrics from independent studies.
Table 2: Comparative Performance of Select Prediction Methods
| Method | Reported Accuracy | Reported Sensitivity/Recall | Reported Precision | Reported F1-Score | Test Dataset |
|---|---|---|---|---|---|
| HotSprint [6] | 76.8% | 60.1% | 63.1% | 65.7% | ASEdb |
| PredHS2 [7] | Not Specified | Not Specified | Not Specified | 0.689 (10-fold CV) | Author's Dataset (313 residues) |
| PPI-hotspotID [8] | Not Specified | 0.67 | Not Specified | 0.71 | PPI-Hotspot+PDBBM (414 hot spots) |
| FTMap (PPI mode) [8] | Not Specified | 0.07 | Not Specified | 0.13 | PPI-Hotspot+PDBBM (414 hot spots) |
| Min-SDS [4] | Not Specified | 0.665 | Not Specified | F2-score: 0.364 | SKEMPI (67 complexes) |
| Mincut [4] | Not Specified | <0.400 | High (Best) | F2-score: <0.224 | SKEMPI (67 complexes) |
A comparative analysis of five computational alanine scanning (CAS) methods (FoldX, mCSM, BeAtMuSiC, Rosetta Flex_ddG, and BudeAlaScan) on the SKEMPI database revealed that while individual methods showed variable Pearson correlation coefficients with experimental ΔΔG, averaging the predictions from all five methods led to more accurate identification of hot spots than any single method alone [3].
Detailed Experimental Protocols for Validation
Experimental Alanine Scanning Mutagenesis
This protocol is the foundational experimental method for hot spot validation [1] [3].
- Site-Directed Mutagenesis: Create a series of plasmid constructs, each encoding a variant of the protein of interest where a single interfacial residue is mutated to alanine.
- Protein Expression and Purification: Express and purify the wild-type and each alanine mutant protein using standard systems (e.g., E. coli, mammalian cell culture).
- Binding Affinity Measurement: Determine the binding affinity (often reported as the dissociation constant, Kd) of each protein variant for its binding partner using a technique such as surface plasmon resonance (SPR), isothermal titration calorimetry (ITC), or similar.
- Energy Calculation: Calculate the change in binding free energy using the formula: ΔΔG = ΔGmut - ΔGwt = RT ln(Kdmut / Kdwt), where R is the gas constant and T is the temperature.
- Classification: A residue is classified as a hot spot if ΔΔG ≥ 2.0 kcal/mol.
Computational Alanine Scanning (CAS) with MM/GBSA
This MD-based protocol offers a quantitative theoretical approach [5].
- System Preparation: Obtain the 3D structure of the protein complex from the PDB. Add hydrogen atoms, solvate the complex in a water box, and add ions to neutralize the system.
- Molecular Dynamics Simulation: Perform energy minimization and equilibration, followed by a production MD run to generate a stable trajectory of the complex.
- Trajectory Generation for Mutants: Using the "single-trajectory approach," generate alanine mutant trajectories by computationally truncating the side chain of the target residue to alanine in each frame of the wild-type trajectory.
- Binding Free Energy Calculation: Use the MM/GBSA method on both wild-type and mutant trajectories to calculate the binding free energy. The difference, ΔΔG, is computed for each residue.
- Hot Spot Prediction: Residues with a predicted ΔΔG ≥ 2.0 kcal/mol are identified as computational hot spots.
Machine Learning-Based Prediction with PredHS2
This protocol outlines a modern machine-learning workflow [7].
- Dataset Curation: Compile a training dataset of known hot spots and non-hot spots from databases like ASEdb, BID, and SKEMPI.
- Feature Extraction: For each residue, compute a wide array of ~600 features, including:
- Sequence features: Amino acid type, evolutionary conservation.
- Structural features: Solvent Accessible Surface Area (SASA), secondary structure, atom packing density.
- Energetic features: Estimated energy terms.
- Neighborhood properties: Features from spatially adjacent residues (Euclidean and Voronoi neighborhoods).
- Feature Selection: Employ a two-step feature selection (e.g., mRMR and sequential forward selection) to identify an optimal, non-redundant feature set (e.g., 26 features for PredHS2).
- Model Training and Validation: Train a classifier (e.g., XGBoost for PredHS2) using the selected features and validate its performance via cross-validation and on independent test sets.
Successful hot spot analysis relies on a combination of experimental reagents and computational databases.
Table 3: Key Research Reagent Solutions for Hot Spot Analysis
| Item / Resource | Function / Description | Example / Source |
|---|---|---|
| Site-Directed Mutagenesis Kit | Enables creation of alanine point mutations in plasmid DNA for protein expression. | Commercial kits from Agilent, NEB, etc. |
| Protein Expression System | Produces the wild-type and mutant proteins for binding assays. | E. coli, insect cell (baculovirus), or mammalian cell systems. |
| Binding Affinity Instrument | Measures the strength of protein-protein binding for wild-type and mutants. | Surface Plasmon Resonance (SPR), Isothermal Titration Calorimetry (ITC). |
| Protein Data Bank (PDB) | Primary repository for 3D structural data of proteins and complexes, essential for computational methods. | https://www.rcsb.org/ [5] |
| SKEMPI Database | A curated database of binding free energy changes upon mutation, used for training and testing predictors. | SKEMPI 2.0 [3] [4] |
| ASEdb / BID | Legacy databases collecting experimental hot spot data from alanine scanning mutagenesis. | Alanine Scanning Energetics Database, Binding Interface Database [1] [6] |
The accurate prediction of hot spot residues is a critical step in understanding PPIs and for designing therapeutic agents that modulate these interactions. While experimental alanine scanning remains the validation gold standard, computational methods provide powerful and complementary high-throughput tools.
Energy-based methods like FoldX and MD-based MM/GBSA offer direct, physics-based interpretations but can be computationally demanding. Machine learning methods like PredHS2 and PPI-hotspotID often achieve high accuracy by integrating diverse features and are efficient for large-scale screening. Emerging graph theory approaches like Min-SDS show great promise in achieving high recall, identifying potential hot spots that other methods might miss.
For the most reliable results, a consensus approach—averaging predictions from multiple methods or using machine learning models that integrate various data types—is recommended. The choice of tool ultimately depends on the available input data (e.g., complex structure vs. free structure), required throughput, and the specific balance of precision and recall needed for the research objective.
Protein-protein interactions (PPIs) are fundamental to virtually all biological processes, from cell signaling to immune response [1] [3] [9]. The thermodynamic driving force for these interactions is not distributed uniformly across the large binding interfaces. Instead, it is frequently dominated by a small subset of residues known as "hot spots" [3]. These are residues that, when mutated to alanine, cause a significant reduction in binding free energy (typically ≥ 2.0 kcal/mol) [1]. Understanding the unique physicochemical properties of these hot spots is therefore crucial for deciphering the molecular logic of PPIs and for rational drug design aimed at modulating these interactions for therapeutic purposes [1] [10]. A consistent and striking finding from numerous experimental and computational studies is the marked prevalence of three aromatic and charged amino acids—tryptophan, arginine, and tyrosine—within these functionally critical regions [1] [11] [10]. This guide provides a comparative analysis of the roles of these amino acids in hot spot formation, supported by experimental data and the methodologies used to uncover them.
Quantitative Analysis of Amino Acid Prevalence in Hot Spots
Statistical analyses of known hot spot residues reveal a dramatically non-random distribution of amino acids. The enrichment of tryptophan, arginine, and tyrosine is particularly remarkable when compared to other residue types.
Table 1: Amino Acid Prevalence in Protein-Protein Interaction Hot Spots
| Amino Acid | Prevalence in Hot Spots (%) | Key Physicochemical Properties |
|---|---|---|
| Tryptophan (W) | 21.0% | Large aromatic side chain, hydrophobic surface, π-interactions, indole nitrogen for H-bonding [1] [11] |
| Arginine (R) | 13.3% | Positively charged guanidinium group, forms multiple H-bonds and salt bridges, long flexible side chain [1] [11] |
| Tyrosine (Y) | 12.3% | Aromatic hydroxyl group, capable of both hydrophobic interactions and H-bonding, planar structure [1] [11] [12] |
| Other Residues | <10% each | |
| Leucine, Valine, Serine, Threonine, Methionine | <3% each | [11] |
This data, consolidated from multiple studies including the seminal work by Bogan and Thorn (1998), underscores that a few specific residues provide a disproportionate contribution to binding energy [11]. The functional implication is clear: the distinct physicochemical properties of Trp, Arg, and Tyr make them exceptionally suited for forming strong, specific interactions at protein interfaces.
Experimental Validation Through Alanine Scanning
The primary experimental protocol for identifying and validating hot spot residues is alanine scanning mutagenesis [1] [3]. This method provides the foundational data against which all computational predictions are benchmarked.
Core Protocol Workflow
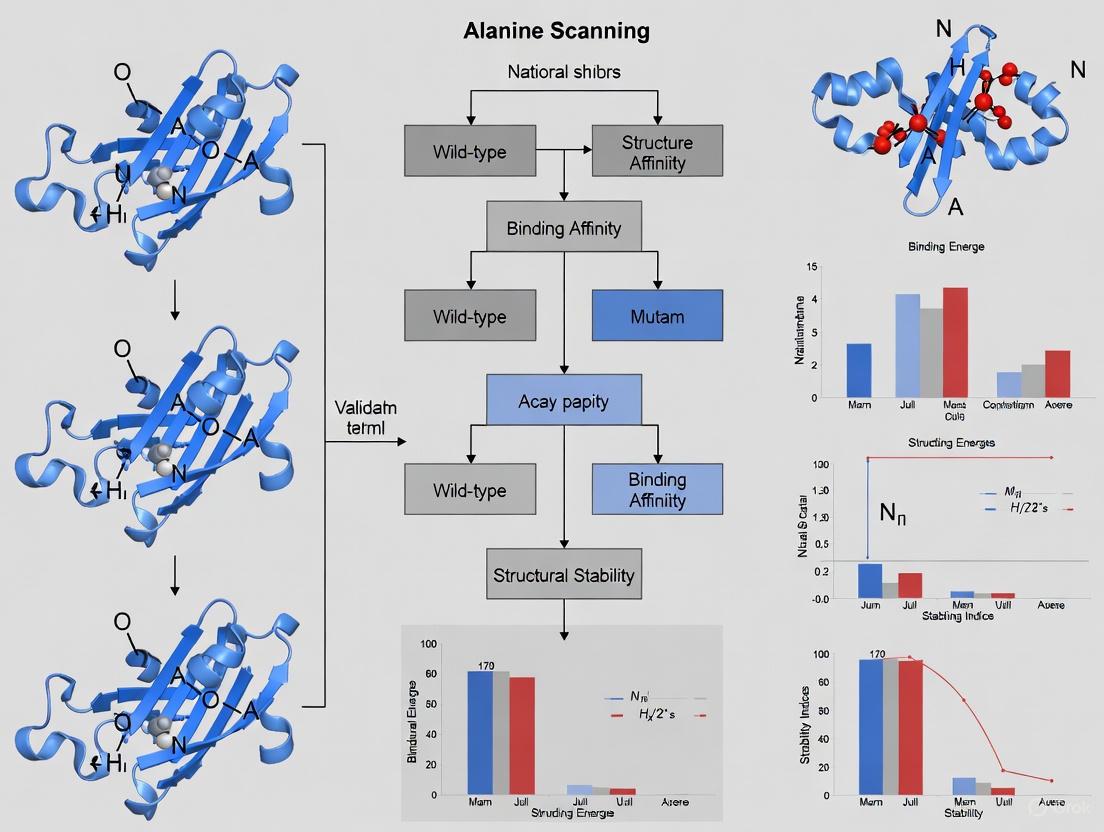
The following diagram outlines the standard workflow for experimental alanine scanning:
Methodology and Data Interpretation
The rationale for substituting residues with alanine lies in its inert methyl side chain, which removes all side-chain atoms beyond the β-carbon without introducing excessive conformational flexibility into the protein backbone, a problem associated with glycine mutations [1]. A measured binding free energy change (ΔΔG) of ≥ 2.0 kcal/mol upon mutation is the standard threshold for designating a residue as a hot spot, as this typically corresponds to a tenfold or greater decrease in binding affinity [1] [3].
The power of this protocol is illustrated by its application in seminal studies. For instance, alanine scanning of the human growth hormone (hGH) and its receptor identified key hot spot residues, two of which were tryptophans, establishing the very concept of hot spots [1]. Similarly, a study on insulin revealed TyrA19 as a critical hot spot, with an alanine mutation causing a 1,000-fold decrease in receptor binding affinity [13].
Table 2: Experimental Alanine Scanning Data for Selected Systems
| Protein Complex | Residue Mutated | ΔΔG (kcal/mol) | Classification | Citation/Context |
|---|---|---|---|---|
| Human Growth Hormone / Receptor | Tryptophan (W) | > 4.5 | Hot Spot | [1] |
| Insulin / Insulin Receptor | TyrA19 | ~4.1 (est. from 1000x loss) | Hot Spot | [13] |
| Insulin / Insulin Receptor | GlyB20 | ~ -0.6 (est. from increased affinity) | Non-Hot Spot (Affinity enhancing) | [13] |
| Model System | Any residue (e.g., Leu, Val, Ser) | < 2.0 | Non-Hot Spot | [11] |
Computational Prediction of Hot Spots
While accurate, experimental alanine scanning is time-consuming, expensive, and not scalable for proteome-wide studies [1] [3] [7]. This has driven the development of numerous computational methods for hot spot prediction, which can be broadly categorized into three groups.
Categories of Prediction Methods
- Energy-Based Methods: These tools use physical force fields or empirical scoring functions to calculate the difference in binding free energy upon alanine mutation. Examples include FoldX [1] [3], Robetta [1], and Rosetta Flex_ddG [3]. They effectively perform in silico alanine scanning.
- Machine Learning (ML) and AI-Based Methods: These approaches train classifiers on known hot spot data using a wide variety of features (sequence, structure, evolutionary conservation, etc.). Examples include KFC, HotPoint [3], PredHS2 [7], and mCSM [3]. Newer deep learning methods are also emerging [10].
- Hybrid and Novel Approaches: Newer methods like BudeAlaScan [3] and HotspotPred [9] combine empirical energy functions with the ability to analyze structural ensembles from NMR or MD simulations, or use structural templates to predict energetic contributions.
Performance Comparison of Computational Tools
Table 3: Comparison of Key Computational Hot Spot Prediction Tools
| Tool Name | Methodology Category | Key Inputs | Key Features | Considerations |
|---|---|---|---|---|
| FoldX | Energy-Based | Protein Structure | Fast, user-friendly [3] | Accuracy can be variable; less accurate than some modern tools [3] |
| Rosetta Flex_ddG | Energy-Based | Protein Structure | High accuracy, sophisticated sampling [3] | Computationally intensive (hours per mutation) [3] |
| BudeAlaScan | Energy-Based | Protein Structure or Ensembles (NMR, MD) | Fast, can process multiple mutations and ensembles [3] | Newer method, command-line interface [3] |
| PredHS2 | Machine Learning | Protein Structure & Sequence | High accuracy, uses novel features (solvent exposure, disorder) [7] | Performance depends on training data quality [7] |
| mCSM/BeAtMuSiC | Machine Learning / Statistical | Protein Structure | Uses statistical potentials and machine learning [3] | Trained on specific databases (e.g., SKEMPI) [3] |
Studies have shown that combining predictions from multiple methods (consensus approaches) often yields more accurate and reliable identification of hot spots than relying on a single tool [3].
Advancing research in this field relies on a suite of key reagents, databases, and software tools.
Table 4: Key Research Reagent Solutions for Hot Spot Analysis
| Resource Name | Type | Primary Function | Relevance to Hot Spot Research |
|---|---|---|---|
| SKEMPI Database | Database | Curated database of binding free energy changes for protein mutations [3] | Essential benchmark dataset for training and validating computational prediction tools [3] |
| ASEdb / BID | Database | Databases of experimental hot spots from alanine scanning mutagenesis [1] [7] | Provide ground-truth experimental data for analysis and method development [1] |
| Phage/Yeast Display | Experimental Platform | In vitro selection of high-affinity binding proteins [12] | Used to engineer synthetic binding proteins (e.g., nanobodies) that often target hot spots [9] [12] |
| QresFEP-2 | Computational Protocol | Hybrid-topology Free Energy Perturbation (FEP) simulation [14] | Physics-based method for predicting mutational effects on stability and binding with high accuracy [14] |
| Stable Protein Complexes | Experimental Reagent | Purified protein pairs for in vitro assays | Required for experimental binding affinity measurements (SPR, ITC) after mutagenesis. |
The empirical and computational data consistently affirm that tryptophan, arginine, and tyrosine are the quintessential components of protein-protein interaction hot spots. Their unique physicochemical properties—large hydrophobic surfaces, capabilities for π-interactions, and versatile hydrogen bonding—make them uniquely suited to form high-affinity interaction nodes. The validation of these principles rests on the foundation of alanine scanning mutagenesis, a protocol that has definitively linked atomic-level composition to binding energy. While experimental methods remain the gold standard, the growing suite of computational tools provides powerful and scalable alternatives for hot spot prediction. The integration of these experimental and computational approaches, guided by a deep understanding of the special roles of Trp, Arg, and Tyr, continues to drive progress in structural biology and the rational design of therapeutics aimed at modulating the human interactome.
Protein-protein interactions (PPIs) are fundamental to most biological processes, and their dysregulation is a cornerstone of many diseases. While PPI interfaces can be large, it has been established that their binding energy is not distributed uniformly. Instead, a small subset of residues, known as "hot spots," contributes the majority of the binding free energy. This article explores the critical role of hot spots, their validation through alanine scanning, and their growing implications for drug discovery, providing a comparative guide to the methods used to identify these pivotal regions.
What Are Hot Spot Residues?
In the context of PPIs, a hot spot is typically defined as a residue whose mutation to alanine causes a significant decrease in binding free energy (ΔΔG ≥ 2.0 kcal/mol) [1]. These residues are not randomly distributed; they are often clustered and structurally conserved within the protein interface [1]. Their composition is also distinctive, with tryptophan (W), arginine (R), and tyrosine (Y) being the most frequently occurring hot spot residues [1].
The seminal work that identified hot spots involved the study of human growth hormone binding to its receptor, revealing that only a small fraction of the interface residues were energetically critical for the interaction [1]. This discovery underscored that PPI interfaces are not monolithic; they contain specific, targetable regions of high functional importance.
The O-Ring Theory and Hot Regions
A key model for understanding hot spot function is the "O-Ring" theory. It proposes that hot spot residues are often surrounded by a ring of less critical residues that shield them from solvent water, thereby protecting the high-energy interactions [15]. Furthermore, hot spots tend not to act in isolation but are organized into densely packed modules called "hot regions," which are critical for binding affinity and specificity [15].
Experimental Validation: Alanine Scanning Mutagenesis
The gold standard for experimental identification of hot spot residues is alanine scanning mutagenesis [1] [15].
Detailed Experimental Protocol
- Site-Directed Mutagenesis: A residue of interest within the PPI interface is mutated to alanine using molecular biology techniques. Alanine is chosen because it removes all side-chain atoms past the β-carbon without introducing excessive flexibility or conformational strain to the protein backbone [1].
- Protein Expression and Purification: The wild-type and each alanine mutant protein are expressed in a suitable system (e.g., E. coli or mammalian cells) and subsequently purified [1].
- Binding Affinity Measurement: The binding constant of the mutant protein complex is measured and compared to that of the wild-type complex. Techniques such as isothermal titration calorimetry (ITC) or surface plasmon resonance (SPR) can be used to determine the binding affinity.
- Energy Calculation: The change in binding free energy (ΔΔG) is calculated as ΔGmut – ΔGwt. A residue is designated a hot spot if the ΔΔG is ≥ 2.0 kcal/mol, which typically corresponds to at least a tenfold drop in binding affinity [1] [15].
Workflow of Alanine Scanning
The diagram below illustrates the multi-step process of experimentally validating a hot spot residue through alanine scanning.
Challenges of Experimental Methods
While highly informative, experimental alanine scanning is costly, time-consuming, and low-throughput [1] [16]. Each mutant must be individually constructed, expressed, purified, and analyzed, making it impractical for large-scale studies. Data from these experiments are deposited in databases like the Alanine Scanning Energetics Database (ASEdb) and the Binding Interface Database (BID), but the available information remains limited to a relatively small number of complexes [1].
Computational Prediction of Hot Spots
To overcome the limitations of experimental methods, a variety of computational tools have been developed. These can be broadly categorized into methods that require the bound complex structure and those that can work with a single unbound protein structure or even just the protein sequence.
The following table summarizes the properties and performance of several key computational methods.
| Method Name | Input Requirement | Core Methodology | Reported Performance (F1-Score/Other) | Key Features |
|---|---|---|---|---|
| PPI-hotspotID [16] [8] | Free Protein Structure | Machine Learning (Ensemble Classifier) | F1: 0.71 (on largest benchmark to date) | Uses conservation, aa type, SASA, and gas-phase energy. Integrated with AlphaFold-Multimer. |
| Embed-1dCNN [17] | Protein Sequence | Pre-trained protein embedding + 1D CNN | F1: 0.82 (on its test set) | Avoids manual feature engineering; uses deep learning on sequence data. |
| FTMap (PPI Mode) [18] [16] | Free Protein Structure | Computational solvent mapping / probe docking | Low Recall (0.07) vs. PPI-hotspotID [8] | Identifies consensus sites for small molecule fragment binding. |
| SPOTONE [8] | Protein Sequence | Machine Learning (Extremely Randomized Trees) | F1: 0.17 (vs. 0.71 for PPI-hotspotID) [8] | Predicts from sequence using residue-specific features. |
| FoldX [1] | Protein Structure (Bound) | Energy-based computational alanine scanning | N/A (Widely used energy function) | Empirical force field; calculates energy changes upon mutation. |
| Robetta [1] | Protein Structure (Bound) | Energy-based computational alanine scanning | N/A (Widely used server) | Uses a physical energy function and backbone flexibility. |
Performance Comparison of Prediction Methods
A 2024 study compared modern methods on the largest collection of experimentally confirmed hot spots to date (414 hot spots across 158 proteins) [16] [8]. The results clearly show the advancement of new machine-learning approaches:
- PPI-hotspotID significantly outperformed both FTMap and SPOTONE, achieving a much higher recall (0.67 vs. 0.07 and 0.10, respectively) and F1-score (0.71 vs. 0.13 and 0.17) [8].
- The study also found that combining PPI-hotspotID with interface residues predicted by AlphaFold-Multimer yielded better performance than either method alone [16].
The Critical Link: Hot Spots in Drug Discovery
Targeting PPIs with small-molecule drugs was once considered impossible because their interfaces are often large, flat, and lack deep pockets. The discovery of hot spots has fundamentally changed this perception [1] [15].
Why Hot Spots Are Druggable
Hot spots create localized regions of high energy density that are amenable to targeting. Key principles include:
- Energetic Contribution: A small molecule that effectively displaces a hot spot residue can disrupt the entire PPI by eliminating a major source of binding energy [1].
- Structural Pre-organization: Hot spots often exist in somewhat pre-organized states in the unbound protein, making them more accessible and "druggable" [15].
- Ligand Binding "Hot Spots": There is a strong correlation between residues identified as hot spots by alanine scanning and regions on the protein surface that have a high propensity to bind small molecule fragments (identified by methods like FTMap or SAR by NMR) [18] [19]. This overlap provides a strategic starting point for drug design.
From Hot Spot Prediction to Drug Discovery
The following diagram outlines how hot spot identification integrates into the drug discovery pipeline.
This approach has proven to be a valid strategy for disrupting unwanted PPIs, and several potential drugs targeting hot spots show great promise [1].
The Scientist's Toolkit: Essential Research Reagents and Solutions
The following table catalogs key reagents and computational tools essential for hot spot research.
| Reagent / Tool | Category | Primary Function in Hot Spot Research |
|---|---|---|
| Alanine Scanning Mutagenesis Kits | Experimental Reagent | Streamline the process of creating site-directed alanine mutations for functional testing. |
| Sensitivity Labeled Nucleotides | Experimental Reagent | Used in sequencing to verify the correctness of introduced mutations. |
| Isothermal Titration Calorimetry (ITC) | Instrumentation / Assay | Gold-standard method for directly measuring binding affinity (Kd) and thermodynamics (ΔG) of wild-type vs. mutant complexes. |
| Surface Plasmon Resonance (SPR) | Instrumentation / Assay | Label-free technique for measuring binding kinetics (kon, koff) and affinity. |
| Fragment Libraries | Chemical Reagent | Collections of small, simple molecules used in X-ray crystallography or NMR to experimentally map protein surface hot spots. |
| PPI-hotspotID Web Server | Computational Tool | Predicts hot spot residues from the free protein structure using a machine-learning model [16]. |
| FTMap Server | Computational Tool | Computationally maps protein surfaces to identify regions with high propensity for binding small molecules [18]. |
| AlphaFold-Multimer | Computational Tool | Predicts the structure of a protein complex, which can be used to identify interface residues for subsequent hot spot analysis [16]. |
Hot spot residues are central to understanding and modulating protein-protein interactions. The rigorous validation of these residues through alanine scanning provides the foundational evidence of their energetic importance. While experimental methods remain the benchmark, advanced computational tools like PPI-hotspotID and Embed-1dCNN are now achieving high predictive performance, enabling large-scale analysis.
The convergence of two definitions of "hot spots"—energetic contributions from alanine scanning and small molecule fragment binding propensity—has created a powerful paradigm for drug discovery. By focusing therapeutic design efforts on these critical regions, researchers can develop targeted strategies to disrupt pathological PPIs, turning a fundamental biological insight into tangible clinical potential.
In the field of molecular biology and drug discovery, understanding the precise interactions that govern protein-protein and protein-ligand binding is paramount. Among the techniques available to researchers, the alanine scanning experiment has established itself as the gold standard for thermodynamic measurement of functional residues in protein interfaces. This methodology systematically quantifies the contribution of individual amino acid side chains to binding free energy through point mutation to alanine, providing a robust experimental approach for identifying "hot spots"—critical residues that account for the majority of binding energy in molecular interactions [1] [20]. The technique's preeminence stems from its elegant simplicity and profound thermodynamic basis: by replacing a side chain with alanine's inert methyl group, researchers can precisely determine the energetic consequences of removing specific chemical functionalities while minimizing structural perturbations [1] [20]. This review objectively compares alanine scanning with emerging computational alternatives, providing researchers with a comprehensive analysis of methodological performance in the critical context of hot-spot validation for drug development.
Fundamental Principles and Thermodynamic Basis
Core Mechanism and Energetic Interpretation
Alanine scanning operates on a fundamental thermodynamic principle: the change in binding free energy (ΔΔG) resulting from substituting a specific residue with alanine directly quantifies that residue's contribution to molecular recognition and binding. The experiment measures the discrepancy between the wild-type and mutant binding free energies (ΔGwt and ΔGmut), where ΔΔGbinding = ΔGmut – ΔGwt [1]. A residue is formally classified as a "hot spot" when its mutation to alanine causes a significant decrease in binding affinity, typically quantified as ΔΔG ≥ 2.0 kcal/mol [1]. This threshold identifies residues that contribute substantially to complex formation, with tryptophan, arginine, and tyrosine statistically overrepresented at these critical positions [1].
The choice of alanine as the substitution standard is deliberate and scientifically grounded. Alanine possesses a non-bulky, chemically inert methyl functional group that mimics the secondary structure preferences of many other amino acids without introducing extreme conformational flexibility or steric clashes [1] [20]. Notably, glycine is avoided despite its small size because its lack of a β-carbon can introduce unwanted backbone flexibility, potentially confounding results with structural artifacts rather than pure side-chain energetic contributions [1].
The Alanine-World Model and Structural Preservation
The theoretical foundation of alanine scanning is supported by the Alanine-World model, which predicts that most canonical amino acids can be exchanged with alanine while maintaining protein secondary structure integrity [20]. This preservation of structural framework is crucial for valid thermodynamic interpretation, as it ensures that measured energy differences primarily reflect the loss of specific side-chain interactions rather than global structural rearrangements. The model leverages alanine's unique ability to mimic the conformational preferences of diverse residue types, establishing it as an ideal neutral reference point for comparative thermodynamic analysis [20].
Experimental Methodologies and Protocols
Standard Experimental Workflow
The implementation of alanine scanning follows a systematic workflow that integrates molecular biology, protein engineering, and precise biophysical measurement. The following diagram illustrates the core experimental process:
Alanine Scanning Experimental Workflow
The process begins with site-directed mutagenesis of selected interface residues to alanine, creating a series of mutant proteins [20]. Each mutant undergoes recombinant expression and purification before precise measurement of binding affinity using biophysical techniques. Isothermal titration calorimetry (ITC) provides direct measurement of thermodynamic parameters including binding constant (K~a~), enthalpy (ΔH), and entropy (ΔS), offering comprehensive insight into the energetic drivers of molecular recognition [21] [22]. Alternative methods include surface plasmon resonance (SPR) for kinetic analysis and biological activity assays measuring second messenger production (e.g., IP1 accumulation for GPCRs) [23].
Advanced Implementation with Calorimetric Detection
Sophisticated implementations combine alanine scanning with ITC for enhanced mechanistic insight. In studying the CD4/gp120 interaction critical to HIV-1 entry, researchers employed thermodynamic guided alanine scanning to distinguish between binding hotspots and allosteric hotspots [22]. This approach revealed that not all residues contributing to binding affinity trigger the conformational changes associated with signaling, enabling design of inhibitors that block interaction without initiating unwanted signaling events [22]. For each alanine mutant, structural integrity must be verified through methods like differential scanning calorimetry (DSC) to ensure melting temperatures (T~m~) remain comparable to wild-type, confirming that observed effects stem from side-chain removal rather than global destabilization [22].
Performance Comparison: Experimental vs. Computational Methods
Direct Methodological Comparison
While alanine scanning provides experimental gold standard data, computational approaches offer complementary advantages in throughput and cost-efficiency. The table below summarizes the objective performance characteristics of major methodological categories:
| Method | Key Principle | Throughput | Cost | Accuracy/ Precision | Best Application Context |
|---|---|---|---|---|---|
| Experimental Alanine Scanning [1] [24] [21] | Direct measurement of ΔΔG via mutagenesis and biophysical measurement | Low to Moderate (individual mutants) | High (specialized equipment, reagents) | High (experimental precision) | Validation studies, drug optimization, fundamental mechanistic studies |
| Computational Alanine Scanning [1] [24] | In silico estimation of ΔΔG using molecular dynamics and energy functions | High (parallel processing) | Low (computational resources) | Moderate (correlation ~0.7-0.8 with experimental) [24] | Preliminary screening, large-scale interface analysis |
| Machine Learning Prediction [25] | Feature-based classification using trained models (e.g., SVM, random forests) | Very High (instant prediction) | Very Low | Variable (F1-score ~0.70 on benchmark sets) [25] | Proteome-wide analysis, initial target identification |
Empirical Accuracy Assessment
A comparative study examining the trypsin-synthetic peptide complex provides direct evidence of performance differences between methodological approaches. The research demonstrated that a 'post-process alanine scanning' computational protocol, which analyzes a single native complex trajectory, achieved better accuracy than running separate molecular dynamics simulations for individual mutants [24]. Notably, results from post-process alanine scanning were also more precise across 10 independent simulations and were obtained over five times faster than the full molecular dynamics protocol [24]. However, the same study reaffirmed that computational methods ultimately serve as approximations to experimental measurements, with even the most efficient algorithms requiring experimental validation for definitive hot spot identification.
Application Case Studies in Research and Development
Diverse Biological Systems Illustrate Methodological Versatility
Alanine scanning has delivered critical insights across diverse biological contexts, from neurotransmission to immunology. The following diagram illustrates the relationships between key application areas and their specific research outcomes:
Key Application Areas and Outcomes
The table below summarizes quantitative findings from these representative studies:
| Biological System | Key Residues Identified | Measured ΔΔG (kcal/mol) | Functional Impact |
|---|---|---|---|
| Bombesin Receptor (GPCR) [23] | Tryptophan, Histidine | Not specified | Critical for receptor binding and activation; guided SERS/SEIRA studies |
| Ca~V~α1-Ca~V~β Interaction [21] | Four conserved hotspot residues | ~2.0-4.5 (significant decrease) | Essential for channel trafficking and functional modulation |
| TCR-pMHC Interaction [26] | CDR3β residues | Not specified (significant reduction) | Enabled engineering of higher avidity TCRs for immunotherapy |
| CD4/gp120 (HIV-1) [22] | Distinct binding vs. allosteric hotspots | Variable by mutant | Enabled design of non-activating competitive inhibitors |
| Aβ(1-40) Amyloid Fibrils [27] | Core hydrophobic residues | Not specified (increased critical concentration) | Revealed determinants of fibril stability and elongation |
Industrial Research and Therapeutic Development
In pharmaceutical development, alanine scanning has proven invaluable for optimizing therapeutic agents. For instance, in the development of HIV-1 cell entry inhibitors, thermodynamic guided alanine scanning identified that not all binding hotspots are allosteric hotspots, enabling rational design of inhibitors that block the CD4/gp120 interaction without triggering the conformational changes that lead to viral entry [22]. Similarly, in T cell receptor engineering for cancer immunotherapy, alanine scanning rapidly mapped key interacting residues in TCR CDR3 regions, facilitating the design of focused mutant libraries and selection of TCRs with higher binding avidity for improved tumor recognition [26].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of alanine scanning requires specialized reagents and instrumentation. The following table details core components of the experimental toolkit:
| Reagent/Instrument | Specification/Function | Application Notes |
|---|---|---|
| Site-Directed Mutagenesis Kit | Commercial kits (e.g., Agilent QuikChange) for introducing point mutations | Critical for creating alanine substitutions; requires sequence verification [22] |
| Expression Systems | Mammalian (e.g., HEK-293), bacterial, or yeast expression systems | Choice affects proper folding and post-translational modifications [23] [22] |
| Purification Systems | Affinity chromatography (His-tag, antibody), FPLC | Required for obtaining pure protein for biophysical studies [22] |
| Isothermal Titration Calorimetry (ITC) | Direct measurement of binding thermodynamics (K~d~, ΔH, ΔS) | Gold standard for label-free binding measurements [21] [22] |
| Surface Plasmon Resonance (SPR) | Kinetic analysis of binding interactions (on/off rates) | Alternative to ITC when kinetics are of primary interest |
| Differential Scanning Calorimetry (DSC) | Assesses protein stability and folding integrity | Verifies mutations don't destabilize protein structure [22] |
Integrated Workflow for Hot-Spot Validation
The most effective contemporary research employs an integrated strategy that combines computational prediction with experimental validation. The optimal workflow begins with machine learning prediction (e.g., HEP, KFC2, or APIS methods) to prioritize target residues, followed by computational alanine scanning to refine predictions, and culminates in experimental alanine scanning for definitive validation [25]. This tiered approach maximizes efficiency while maintaining methodological rigor, with computational methods screening large interfaces and experimental verification providing thermodynamically precise data for critical residues.
Alanine scanning remains the unchallenged gold standard for experimental determination of residue-specific thermodynamic contributions to protein interactions. While computational methods continue to advance in predictive accuracy and throughput, their performance must still be evaluated against experimental alanine scanning data for validation [24] [25]. For research requiring definitive identification of functional hot spots—particularly in drug development contexts where precise energetic measurements inform optimization campaigns—experimental alanine scanning provides irreplaceable thermodynamic precision. The methodology's continued relevance is assured through integration with emerging structural techniques and computational approaches, maintaining its position as the definitive reference point for thermodynamic measurement of protein interaction interfaces.
From Bench to Code: Executing Experimental and Computational Alanine Scanning
Alanine scanning mutagenesis stands as a cornerstone experimental methodology for mapping protein-protein and protein-ligand interaction interfaces by systematically identifying "hot spot" residues that contribute significantly to binding energy. This guide provides a comprehensive comparison of experimental approaches, detailing the complete workflow from library design and mutagenesis through binding affinity measurement and data interpretation. We objectively evaluate the protocol's performance against alternative methodologies and present quantitative experimental data validating its crucial role in characterizing energetic landscapes at biological interfaces.
The systematic identification of functional epitopes represents a fundamental challenge in understanding molecular recognition, with profound implications for therapeutic antibody development, drug discovery, and protein engineering. Within this context, alanine scanning mutagenesis has emerged as a powerful experimental paradigm for quantitatively mapping binding energy contributions at amino acid resolution [28]. The technique operates on a straightforward biochemical principle: by substituting individual residues with alanine—thereby removing side-chain atoms beyond the β-carbon—researchers can probe the energetic contribution of specific side chains to binding interactions [29].
The conceptual foundation of this approach rests on the "hot spot" hypothesis, which proposes that binding energy is not uniformly distributed across interaction interfaces but is instead concentrated at a relatively small subset of residues [28]. Experimental validation of these hot spots through alanine scanning has revealed that protein interfaces display remarkable diversity in their energetic organization, with no simple patterns of hydrophobicity, shape, or charge reliably predicting which residues will prove functionally critical [28]. This methodology has been successfully applied to diverse systems including antibody-antigen complexes [30], insulin-receptor interactions [13], and G-protein coupled receptor signaling [31], establishing it as an indispensable tool for dissecting the thermodynamic determinants of molecular recognition.
Experimental Workflow: From Mutagenesis to Binding Measurement
The implementation of a comprehensive alanine scanning study requires the execution of a multi-stage experimental pipeline, each component of which must be carefully optimized to ensure reliable results.
Stage 1: Target Selection and Mutagenesis Strategy
The initial phase involves strategic selection of residues for mutagenesis based on structural or homology data to define the interface region. As exemplified in studies of antibody variable domains, researchers first identify permissive sites in complementarity-determining regions (CDRs) that can be mutated without complete loss of antigen binding [30]. This preliminary assessment may involve computational alanine scanning to prioritize residues for experimental investigation [3] [32]. The mutagenesis strategy must balance comprehensiveness against practical constraints, with typical studies examining 15-30 interface residues [13].
Stage 2: Mutant Generation and Protein Production
Following target selection, researcher employ site-directed mutagenesis to systematically substitute each selected residue with alanine. The canonical approach involves:
- Generating 21+ individual mutant constructs for comprehensive interface mapping [13]
- Expressing mutant proteins in suitable systems (typically bacterial or mammalian)
- Purifying variants using chromatography methods (e.g., metal-affinity chromatography)
- Verifying structural integrity through methods like SDS-PAGE analysis [30]
This phase represents one of the most resource-intensive aspects of the protocol, requiring production and purification of numerous individual protein variants [29].
Stage 3: Binding Affinity Measurement
With purified mutant proteins in hand, researchers quantitatively assess binding interactions using appropriate biophysical methods. Common approaches include:
- Fluorescence polarization to measure changes in binding affinity at multiple protein concentrations [30]
- Surface display technologies (yeast, phage) for higher-throughput screening [30]
- ELISA or Western blot assays as accessible alternatives for binding assessment [29]
Critical to this phase is measuring the change in free energy of binding (ΔΔG) relative to wild-type, calculated from changes in binding constants (Kd or Ki values) [13] [31].
Stage 4: Data Interpretation and Hot Spot Validation
The final stage involves classifying residues based on their energetic contributions, with hot spots typically defined as those where alanine substitution causes a ≥10-fold reduction in binding affinity (ΔΔG ≥ 1.36 kcal/mol) [28]. Researchers must exercise caution in interpretation, as mutations may indirectly affect binding through structural perturbations rather than direct involvement in the interface [28].
Table 1: Key Experimental Parameters in Alanine Scanning Studies
| Parameter | Typical Range | Considerations |
|---|---|---|
| Residues scanned | 15-30 positions | Focus on structural epitope; balance between comprehensiveness and practicality |
| Expression system | Bacterial (E. coli) or mammalian | Impacts folding, post-translational modifications |
| Binding assay | Fluorescence polarization, ELISA, surface display | Throughput, precision, and equipment requirements vary |
| ΔΔG threshold for hot spots | ≥1.36 kcal/mol (10-fold affinity loss) | Classification sensitivity/specificity trade-offs |
| Replicates | 2-3 independent experiments | Essential for statistical significance |
The following diagram illustrates the complete experimental workflow:
Quantitative Experimental Data: Representative Case Studies
Insulin Receptor Binding Interface
A comprehensive alanine scanning study of insulin revealed distinct energetic contributions across its receptor binding interface, with dramatic variations in mutational effects [13]. The data demonstrate how alanine scanning quantitatively identifies critical hot spots while also revealing potential affinity-enhancing mutations:
Table 2: Alanine Scanning Results for Insulin-Receptor Binding [13]
| Mutation Position | Fold Change in Affinity | ΔΔG (kcal/mol) | Classification |
|---|---|---|---|
| TyrA19 | 1,000-fold decrease | ~4.1 | Hot spot |
| GlyB8 | 33-fold decrease | ~2.1 | Hot spot |
| LeuB11 | 14-fold decrease | ~1.6 | Hot spot |
| GluB13 | 8-fold decrease | ~1.2 | Significant |
| GlyB20 | Increase | ~ -0.6 | Affinity enhancer |
| ArgB22 | Increase | ~ -0.4 | Affinity enhancer |
| SerA9 | Increase | ~ -0.3 | Affinity enhancer |
This dataset illustrates several key principles: (1) hot spot residues can produce dramatically different energetic penalties (from 8-fold to 1,000-fold reductions), (2) even conserved residues may not always be critical for binding (GlyB20), and (3) some mutations can paradoxically enhance affinity, providing insights for protein engineering.
Antibody Affinity Maturation Applications
In antibody engineering, alanine scanning guides affinity maturation strategies by identifying permissive sites for mutagenesis. One study of a single-domain antibody (VHH) specific for α-synuclein combined computational and experimental alanine scanning to identify CDR positions tolerant to mutagenesis [30]. The research team first identified 11 permissive sites that retained >50% wild-type binding when mutated to alanine, then designed focused libraries that yielded variants with >5-fold affinity improvements. This demonstrates how alanine scanning serves as a critical preliminary step in rational library design for antibody optimization.
Comparative Method Assessment: Performance Versus Alternatives
Direct Comparison with HDX-MS Epitope Mapping
Hydrogen-deuterium exchange mass spectrometry (HDX-MS) represents a leading alternative for epitope mapping that measures protection from exchange upon binding:
Table 3: Alanine Scanning vs. HDX-MS for Epitope Mapping [29]
| Parameter | Alanine Scanning | HDX-MS |
|---|---|---|
| Resolution | Single amino acid | Peptide-level (1-5 amino acids) |
| Throughput | Low (weeks-months) | Medium (days-weeks) |
| Sample consumption | High (each mutant individually) | Low (single complex analysis) |
| Energetic information | Direct ΔΔG measurement | Indirect (protection correlates with binding) |
| Structural perturbations | Possible conformational effects | Minimal (native conditions) |
| Equipment requirements | Standard molecular biology | Specialized mass spectrometry |
| Data interpretation | Straightforward (binding measurements) | Complex (exchange kinetics analysis) |
Computational Alanine Scanning Methods
Computational approaches provide complementary strategies for hot spot prediction with distinct performance characteristics:
Table 4: Computational Alanine Scanning Method Comparison [3]
| Method | Approach | Accuracy | Throughput | Requirements |
|---|---|---|---|---|
| Free Energy Perturbation (FEP) | Physics-based, molecular dynamics | High (R=0.7-0.8) | Low (hours-days/mutation) | Significant computational resources |
| FoldX | Empirical force field | Medium (R=0.6) | High (minutes/mutation) | Single structure |
| Robetta (Flex_ddG) | Physical energy function | Medium-High | Medium | Homology models acceptable |
| BudeAlaScan | Empirical free energy | Medium | High | Single structure or ensembles |
| Machine Learning (mCSM) | Statistical potentials | Medium | Very High | Structural features |
The performance metrics reveal inherent trade-offs between computational efficiency and predictive accuracy, with more rigorous physics-based methods requiring substantially greater resources but generally providing superior correlation with experimental data [3] [14].
Research Reagent Solutions: Essential Materials for Implementation
Successful execution of alanine scanning studies requires access to specialized reagents and instrumentation. The following table details key solutions employed in experimental protocols:
Table 5: Essential Research Reagents and Instruments
| Reagent/Instrument | Function | Examples/Alternatives |
|---|---|---|
| Site-directed mutagenesis kit | Introduction of alanine substitutions | Commercial kits (QuickChange) |
| Expression vector | Recombinant protein production | pET, pcDNA systems |
| Expression host | Protein synthesis | E. coli, mammalian cells |
| Purification system | Protein isolation | Metal-affinity, FPLC |
| Binding assay platform | Affinity measurement | Fluorescence polarization, SPR, ELISA |
| Structural modeling software | Interface analysis | Modeller, Rosetta |
Alanine scanning mutagenesis remains an indispensable tool for quantitatively mapping functional epitopes and validating hot spot residues, despite the emergence of complementary methodologies. The technique's unique strength lies in its direct measurement of side-chain energetic contributions through rigorous binding assays, providing a thermodynamic foundation for understanding molecular recognition. While the resource-intensive nature of comprehensive scanning studies presents practical limitations, the strategic integration of computational pre-screening with focused experimental validation creates an optimized approach for contemporary research. As protein therapeutics and targeted drug discovery continue to advance, the precise energetic mapping enabled by alanine scanning will maintain its critical role in rational protein design and interaction interface characterization.
Protein-protein interactions (PPIs) are vital to all biological processes, and identifying the key residues that drive these interactions—known as hot-spot residues—is crucial for understanding cellular function and advancing drug design [3]. Computational alanine scanning (CAS) has emerged as a rapid, in silico method to predict these residues by calculating the change in binding free energy (ΔΔG) when a residue is mutated to alanine [3]. This guide provides an objective comparison of five prominent CAS tools: FoldX, Robetta (Flex_ddG), mCSM, BeAtMuSiC, and BUDE Alanine Scanning (BudeAlaScan). Framed within a broader thesis on validating hot-spot residues, this article compares their methodologies, performance metrics, and experimental validation, supplying researchers and drug development professionals with data to inform their tool selection.
Methodology of Computational Alanine Scanning
Fundamental Principles
Computational alanine scanning is based on the thermodynamic principle that the binding free energy change (ΔΔG) upon mutating a residue to alanine quantifies its contribution to the protein-protein interaction [3]. A hot-spot residue is typically defined as one whose mutation to alanine causes a ≥2.0 kcal/mol drop in binding free energy [33]. These methods generally fall into two categories: physics-based/empirical energy functions (FoldX, Robetta Flex_ddG, BUDE Alanine Scanning) and statistical potentials/machine learning approaches (mCSM, BeAtMuSiC) [3].
Experimental Validation Workflow
The standard workflow for validating computational predictions involves experimental alanine scanning, which is time-consuming and costly [3] [33]. The following diagram illustrates the integrated computational-experimental workflow for hot-spot validation.
Tool Comparison: Mechanisms and Methodologies
The five tools employ distinct approaches, offering different trade-offs between speed, accuracy, and consideration of protein dynamics [3].
Table 1: Core Methodologies of the Five CAS Tools
| Tool | Underlying Method | Input Requirements | Key Features |
|---|---|---|---|
| FoldX | Empirical force field [3] | Single structure (e.g., from X-ray crystallography) [3] | Rapid calculations; widely used; empirical potentials [3] [34] |
| Robetta (Flex_ddG) | Physical energy function (Rosetta Ref2015/Talaris2014) [3] | Single structure [3] | Sophisticated Monte Carlo sampling & minimization; high accuracy [3] |
| mCSM | Machine learning (graph-based signatures) [3] | Single structure [3] | Uses signature patterns of the protein environment; trained on SKEMPI [3] |
| BeAtMuSiC | Statistical potentials [3] | Single structure [3] | Coarse-grained predictor; statistical potentials derived from known structures [3] |
| BUDE Alanine Scanning (BudeAlaScan) | Empirical free-energy (BUDE force field) [3] | Single structures or ensembles (NMR, MD) [3] | Handles structural ensembles; scans multiple mutations simultaneously [3] |
Performance and Benchmarking
A comparative analysis using the SKEMPI database—a comprehensive collection of binding free energy changes upon mutation—reveals variations in predictive accuracy and computational speed [3].
Table 2: Performance Metrics on the SKEMPI Database
| Tool | Pearson Correlation (ΔΔG) | Computational Speed | Strengths and Limitations |
|---|---|---|---|
| FoldX | Data available in source [3] | ~8 minutes (single core) [3] | Fast but can suffer from lower accuracy, especially in antibody-antigen systems [34] |
| Robetta (Flex_ddG) | Data available in source [3] | ~1-2 hours per mutation (single core) [3] | High accuracy but computationally intensive; not ideal for high-throughput screening [3] |
| mCSM | Data available in source [3] | Not specified | Good performance; machine learning approach trained on structural data [3] |
| BeAtMuSiC | Data available in source [3] | Not specified | Uses statistical potentials; performance benchmarked on SKEMPI [3] |
| BUDE Alanine Scanning | Data available in source [3] | ~5 minutes (single core) [3] | Fast; unique capability to process structural ensembles from NMR or MD [3] |
A notable finding is that a consensus approach—averaging the ΔΔG values for each residue across the five methods—often leads to more accurate prediction than any single method alone [3].
Experimental Validation Case Studies
Validation on Diverse Protein Complexes
The comparative predictive capability of these tools was tested through detailed experimental analyses on three diverse PPI targets [3]:
- NOXA-B/MCL-1: An α-helix-mediated PPI, relevant in oncology [3].
- SIMS/SUMO: A β-strand-mediated interaction involved in SUMOylation regulation [3].
- GKAP/SHANK-PDZ: A β-strand-mediated scaffolding interaction at synaptic junctions [3].
For these targets, the computational predictions were followed by experimental alanine scanning to measure the actual ΔΔG values, validating the in silico predictions [3]. The consensus approach proved particularly effective across these diverse interfaces [3].
Workflow for Target Validation
The process for validating predictions on a target like the NOXA-B/MCL-1 complex involves a multi-stage workflow, integrating computational predictions with experimental assays.
Research Reagent Solutions
The following table details key databases, software, and experimental reagents essential for conducting and validating computational alanine scanning studies.
Table 3: Essential Research Reagents and Resources
| Resource Name | Type | Function in CAS Research |
|---|---|---|
| SKEMPI/SKEMPIv2.0 | Database [3] [34] | Curated database of binding free energy changes upon mutation; used for training and benchmarking CAS tools [3] [34] |
| ProTherm | Database [3] [34] | Database of thermodynamic data for protein stability and mutations; used for folding stability benchmarks [3] |
| PDB Fixer | Software Tool [34] | Pre-processes 3D crystal structures for CAS by adding missing residues and heavy atoms, and correcting errors [34] |
| MOE (Molecular Operating Environment) | Software Suite [35] | Commercial software that can perform site-directed mutagenesis computations, including alanine scanning [35] |
| Alanine Scanning Mutagenesis Kit | Experimental Reagent | Commercial kits for performing site-directed mutagenesis to create alanine variants for experimental validation |
| Surface Plasmon Resonance (SPR) | Experimental Instrument | Measures real-time binding affinity (KD) of wild-type and mutant proteins to determine experimental ΔΔG [34] |
Emerging Trends and Future Outlook
The field of computational alanine scanning continues to evolve. Recent efforts focus on integrating machine learning to correct and improve existing force fields. For instance, a neural network framework applied to FoldX output significantly improved its correlation with experimental data, especially for higher-order mutations [34]. Another trend involves predicting hot spots from free protein structures (without the bound complex). Tools like PPI-hotspotID, which uses machine learning with features like conservation, amino acid type, and solvent accessibility, show promise in this area [33]. The integration of AlphaFold-predicted structures with these methods further expands the potential for probing PPIs where experimental structures are unavailable [33]. Finally, the ability to handle structural ensembles from NMR or molecular dynamics simulations, as seen in BUDE Alanine Scanning, provides a crucial avenue for accounting for protein dynamics and disordered regions in PPI analysis [3].
The identification of hot spot residues—a small subset of amino acids that contribute disproportionately to binding free energy—is crucial for understanding protein interactions and guiding drug discovery. While traditionally applied to protein-protein interactions (PPIs), alanine scanning mutagenesis is now being extended to map protein-lipid interactions, a frontier in membrane protein biology. This guide compares experimental and computational approaches for hot spot validation, focusing on their application in characterizing lipid binding sites on membrane proteins. We provide objective performance comparisons and detailed methodologies to help researchers select appropriate techniques for studying these critical interactions.
Most cellular processes involve complex protein interactions, with a small fraction of interfacial residues termed "hot spots" contributing the majority of binding free energy [1]. A residue is defined as a hot spot when its mutation to alanine causes a decrease in binding free energy (ΔΔG) of ≥ 2.0 kcal/mol [1]. Alanine scanning mutagenesis, the experimental gold standard for identifying these residues, systematically substitutes individual amino acids with alanine, removing side-chain atoms past the β-carbon without introducing conformational flexibility or steric effects [1] [36].
The composition of hot spots is distinctive, with tryptophan (21%), arginine (13.3%), and tyrosine (12.3%) occurring with high frequency due to their unique physicochemical properties [1]. These residues often form cooperative, structurally conserved networks that make attractive targets for therapeutic intervention [1].
While classical alanine scanning has revolutionized the study of soluble protein complexes, extending this methodology to membrane protein-lipid interactions presents unique challenges and opportunities. Membrane proteins, which constitute over 30% of the human proteome and represent a major class of drug targets, rely on specific lipid interactions for their structure, function, and stability [37] [38]. This guide compares established and emerging methods for hot spot validation in the context of protein-lipid interactions.
Computational Prediction of Hot Spots
Computational approaches provide valuable alternatives to experimental alanine scanning, offering greater throughput and lower cost. These methods predict binding free energy changes (ΔΔG) upon alanine mutation using various algorithms and force fields.
Table 1: Comparison of Computational Hot Spot Prediction Methods
| Method | Approach | Features | Performance (Pearson Correlation) | Throughput |
|---|---|---|---|---|
| FoldX | Empirical force field | Physical energy terms | 0.55-0.65 | High (minutes) |
| Rosetta Flex_ddG | Physical energy function with sampling | Talaris2014/Ref2015 force fields | 0.60-0.70 | Low (hours per mutation) |
| mCSM | Machine learning | Signature-based patterns | 0.60-0.65 | High |
| BeAtMuSiC | Statistical potentials | Coarse-grained potentials | 0.55-0.62 | High |
| BudeAlaScan | Empirical free energy | BUDE force field, ensemble processing | 0.58-0.63 | Medium (minutes) |
Table 2: Practical Considerations for Method Selection
| Method | Best For | Limitations | Availability |
|---|---|---|---|
| FoldX | Quick assessments, large interfaces | Limited conformational sampling | Standalone tool & server |
| Rosetta Flex_ddG | High-accuracy predictions, flexible interfaces | Computationally intensive, requires expertise | Standalone package |
| mCSM | Rapid screening, non-experts | Dependent on training data coverage | Web server |
| BeAtMuSiC | Conservative estimates, initial screening | May miss subtle effects | Web server |
| BudeAlaScan | Structural ensembles, multiple mutations | Newer method, limited validation | Command-line tool |
Comparative analyses reveal that consensus approaches, which average predictions across multiple methods, often outperform individual tools [3]. For membrane protein-lipid interactions, computational methods must account for the unique membrane environment, with tools like FoldX and Rosetta requiring adaptation for lipid bilayers.
Experimental Alanine Scanning for Protein-Lipid Interactions
Traditional alanine scanning faces challenges when applied to membrane proteins, particularly due to difficulties in protein solubilization, purification, and stability in detergent micelles [38]. Recent advances have adapted this methodology specifically for mapping protein-lipid interactions.
Native Mass Spectrometry Approach
Jayasekera et al. developed an innovative native mass spectrometry (MS) method to profile lipid binding sites on Aquaporin Z (AqpZ), a bacterial water channel [37] [39] [40]. This approach quantifies the thermodynamic contributions of specific residues to lipid binding.
Table 3: Key Research Reagents for Native MS Alanine Scanning
| Reagent/Solution | Function/Application |
|---|---|
| AqpZ mutants | Target membrane protein with systematic alanine substitutions |
| Tetraethylene glycol monooctyl ether (C8E4) | Mild detergent for protein solubilization and stabilization |
| Ammonium acetate buffer (0.2 M) | Volatile buffer compatible with native MS |
| Cardiolipin (POCL, TOCL) | Anionic lipid species for binding studies |
| Phosphatidylglycerol (POPG) | Comparison anionic lipid |
| Phosphatidylethanolamine (POPE) | Zwitterionic phospholipid control |
| Q-Exactive HF UHMR Orbitrap | High-resolution mass spectrometer for native MS |
Experimental Workflow:
- Mutant Selection and Design: Residues are selected based on: (1) prior known lipid-interacting residues (e.g., W14), (2) hydrophobic residues likely interacting with lipid tails (e.g., F10, F13, F196), and (3) cationic residues at the lipid interface that could interact with anionic headgroups (e.g., R3, R75, R224) [37].
- Site-Directed Mutagenesis: Selected residues are systematically mutated to alanine using primer-based mutagenesis.
- Protein Expression and Purification: Mutant proteins are expressed in E. coli C43 cells and purified in C8E4 detergent to maintain native structure.
- Sample Preparation for Native MS: Wild-type and mutant proteins are mixed at approximately 1:1 molar ratio with added lipids at specific protein:lipid ratios (1:1:50 for cardiolipins, 1:1:100 for weaker-binding lipids).
- Data Acquisition: Mass spectra are acquired using temperature ramping from 15-35°C with key settings including spray voltage of 1.2 kV and collision voltage of 75-85 V.
- Data Analysis: Peak areas for lipid-bound and unbound states are quantified using UniDec and custom Python scripts. The dissociation constant ratio (K) and free energy difference (ΔΔG) are calculated using the equations:
- K = [W] × [ML] / [WL] × [M]
- ΔΔG = -RTlnK where W and M represent wild-type and mutant proteins, and WL and ML represent their lipid-bound states [37].
This native MS approach revealed that AqpZ is selective toward cardiolipins at specific sites, with CL orienting with its headgroup facing the cytoplasmic side and its acyl chains interacting with a hydrophobic pocket at the monomeric interface [37] [40].
Integrated Workflow: Combining Native MS with Molecular Dynamics
The most powerful applications combine experimental alanine scanning with computational simulations:
Diagram: Integrated Workflow for Mapping Protein-Lipid Interactions
This integrative approach provides unique insights into lipid binding sites and selectivity, enabling researchers to map protein structure based on lipid affinity [37] [40]. For AqpZ, this revealed that cardiolipin orients with its headgroup facing the cytoplasmic side, with acyl chains interacting specifically with a hydrophobic pocket at the monomeric interface within the lipid bilayer [37].
Performance Comparison and Validation
Accuracy of Computational Predictions
When benchmarked against experimental data from the SKEMPI database (containing 3,047 binding free energy changes upon mutation), computational methods show varying performance levels:
Prediction Accuracy Metrics:
- FoldX: Moderate accuracy (Pearson correlation ~0.55-0.65), fast execution (~8 minutes for typical interface) [3]
- Rosetta Flex_ddG: Higher accuracy (Pearson correlation ~0.60-0.70), but computationally intensive (1-2 hours per mutation) [3]
- BudeAlaScan: Balanced approach (Pearson correlation ~0.58-0.63), medium throughput (~5 minutes), capable of processing structural ensembles [3]
- Consensus approaches: Typically outperform individual methods by averaging predictions across multiple tools [3]
For membrane protein-lipid interactions, accuracy may be reduced due to limited structural data and challenges in modeling the membrane environment.
Experimental Validation
Computational predictions require experimental validation. For the AqpZ study, native MS alanine scanning identified W14 as contributing to the highest affinity CL binding site, with R224 contributing to a secondary site [37]. These findings were validated through complementary molecular dynamics simulations showing high lipid occupancy and residence times at these residues.
The combination of native MS with MD simulations creates a powerful validation cycle: computational predictions guide experimental targets, while experimental results refine computational models. This approach confirmed CL selectivity at specific AqpZ sites and elucidated the molecular orientation of bound lipids [37] [40].
Extending alanine scanning to protein-lipid interactions represents a significant advancement in membrane protein biology. While computational methods offer rapid screening capabilities, integrated approaches that combine computational predictions with experimental validation provide the most robust strategy for identifying lipid interaction hot spots.
For researchers studying membrane protein-lipid interactions, we recommend:
- Initial Screening: Use consensus computational approaches (e.g., FoldX + mCSM + BudeAlaScan) to identify potential hot spot residues.
- Experimental Validation: Apply native MS-based alanine scanning to quantify binding contributions of selected residues.
- Mechanistic Insight: Complement with molecular dynamics simulations to visualize lipid interactions and understand molecular orientation.
- Functional Assessment: Corrogate identified hot spots with functional assays to establish physiological relevance.
This multifaceted approach enables comprehensive mapping of protein-lipid interactions, providing critical insights for drug discovery and understanding membrane protein function in health and disease. As methods continue to advance, particularly in cryo-EM and computational modeling, our ability to precisely characterize these interactions will further improve, opening new avenues for therapeutic intervention.
Best Practices for Selecting Residues to Mutate
A central goal in molecular biology and drug development is understanding the relationship between protein structure and function. A powerful approach to probing this relationship is alanine scanning mutagenesis, a method designed to identify "hot spot" residues—those where mutation significantly disrupts protein function or binding affinity [41] [42]. This technique systematically substitutes target residues with alanine, effectively removing side-chain atoms past the beta-carbon, thereby testing the functional contribution of the native side chain without introducing major conformational distortions [42]. Selecting the right residues to mutate is critical for efficient experimental design. This guide compares the performance of computational prediction tools with experimental alanine scanning and provides best-practice protocols for validating hot-spot residues.
Performance Comparison: Computational Predictions vs. Experimental Alanine Scanning
Selecting residues for mutagenesis often begins with in silico predictions before moving to costly experimental validation. The table below objectively compares the performance of different methodological approaches.
Table 1: Performance Comparison of Residue Selection and Mutagenesis Methods
| Method | Key Principle | Typical Throughput | Key Performance Metrics | Primary Advantages | Primary Limitations |
|---|---|---|---|---|---|
| Computational Alanine Scanning (e.g., SNAP) | Predicts functional effects of mutations using neural networks [41]. | Very High (exhaustive in silico mutagenesis feasible) | ~70% sensitivity for hot spots (ΔΔG ≥1 kcal/mol); higher accuracy for severe changes [41]. | Fast, low-cost; can probe all residues and all possible amino acid substitutions [41]. | Predictions require experimental validation; accuracy can vary. |
| Experimental Alanine Scanning | Measures binding energy change (ΔΔG) after substituting a residue with alanine [41] [42]. | Low (each mutant expressed and assayed separately) [42]. | Identifies hot spots based on empirical energy thresholds (e.g., ΔΔG ≥1 kcal/mol) [41]. | Gold standard for defining functional epitopes and energetic contributions [42]. | Laborious, costly, and low-throughput [42]. |
| Shotgun Scanning | Phage-displayed combinatorial library with binomial substitution (wild-type or alanine) at multiple positions [42]. | High (large libraries >10^10 clones screened via binding selection) [42]. | Identifies hot spots via enrichment ratios (Ala/WT) from sequencing, correlating with ΔΔG [42]. | Rapidly maps functional epitopes; combines throughput of libraries with functional insight of alanine scanning [42]. | Requires phage display expertise; indirect measurement of energy change. |
| Base Editing (BE) Screens | Uses CRISPR base editors to make endogenous transitions (C>T or A>G) guided by sgRNA pools [43]. | High (surrogate genotyping via sgRNA sequencing) [43]. | Correlation with gold-standard DMS data; quality depends on filters for single-edit sgRNAs [43]. | Endogenous genomic context; scalable across cell lines [43]. | Limited to transition mutations; bystander edits; PAM sequence requirements [43]. |
| Deep Mutational Scanning (DMS) | Heterologous expression of saturated cDNA mutant libraries [43]. | High | Comprehensive measurement of all amino acid substitutions [43]. | Broad mutational repertoire; well-established analysis [43]. | Non-endogenous expression; potential scaling challenges with large genes [43]. |
The performance data in Table 1 highlights that computational tools like SNAP achieve a good balance of speed and reasonable accuracy, serving as an excellent filter before experimental work. A study validating SNAP against the ASEdb database of alanine scans found it identified 70% of true hot spots, with performance improving for residues causing more severe functional disruptions [41].
Experimental Protocols for Key Methodologies
Protocol: Computational Prediction with SNAP
SNAP is a neural network-based tool that predicts the functional effect of single amino acid substitutions, providing a score from -100 (neutral) to +100 (deleterious effect) [41].
- Input Preparation: Provide the protein sequence or structure for analysis.
- In Silico Mutagenesis: The tool performs an exhaustive all-against-all mutagenesis in silico.
- Score Analysis: The output is a SNAP score for each possible substitution. Higher positive scores predict a greater functional effect. Residues with high average SNAP scores across multiple mutations, or specifically for the alanine substitution, are prioritized as candidate hot spots [41].
- Accessibility Consideration: Predictions can be further refined by considering solvent accessibility, as functional residues are often found at the protein surface [41].
Protocol: Shotgun Scanning Alanine Mutagenesis
This combinatorial method maps functional epitopes by creating and screening a library of protein variants where multiple positions can be either wild-type or alanine [42].
- Library Design:
- Identify target residues at a protein-protein interface.
- Design degenerate oligonucleotides that encode for a binomial mixture (wild-type or alanine) at each targeted codon position using split-pool synthesis [42].
- Phage Display Library Construction:
- Clone the mutagenic oligonucleotide pool into a phagemid vector (e.g., pW1205a) for display on the surface of M13 bacteriophage as a fusion to a coat protein.
- Use the Kunkel mutagenesis method with mutagenic oligonucleotides to construct the library [42].
- Selection and Screening:
- Subject the phage library to successive rounds of binding selection against the immobilized target receptor (e.g., hGHbp).
- Amplify bound phage in an E. coli host after each round to enrich functional clones [42].
- Data Analysis:
- Isolate individual binding phage and sequence the mutated region.
- For each varied position, calculate the ratio of wild-type to alanine occurrences in the selected pool.
- A high wild-type to alanine ratio indicates a position where alanine substitution is detrimental to binding, marking a potential hot spot. This ratio can be correlated with the change in free energy of binding (ΔΔG) [42].
Protocol: Base Editor Screening for Endogenous Mutagenesis
This protocol uses a pooled CRISPR base editor library to mutate residues in their endogenous genomic context [43].
- sgRNA Library Design: Design a library of sgRNAs targeting genomic regions encoding the residues of interest, considering the base editor's editing window [43].
- Library Delivery and Cell Sorting: Transduce the sgRNA library into cells expressing the base editor and apply the relevant phenotypic selection (e.g., growth factor independence in Ba/F3 cells) [43].
- Next-Generation Sequencing (NGS): Isolate genomic DNA from pre- and post-selection populations. Amplify and sequence the sgRNA region to quantify sgRNA abundance.
- Variant Effect Calculation: The functional effect of a variant is inferred from the depletion or enrichment of its corresponding sgRNA(s) after selection.
- Data Filtering for High-Quality Results: Apply a critical filter to retain only sgRNAs that are predicted to produce a single amino acid edit within the editing window. This filter significantly enhances the agreement between base editor data and gold-standard DMS datasets [43].
Workflow Visualization for Residue Selection and Validation
The following diagram illustrates the logical workflow for selecting and validating hot-spot residues, integrating both computational and experimental approaches.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful mutagenesis studies rely on specific reagents and tools. The following table details key materials required for the experiments discussed in this guide.
Table 2: Essential Research Reagents and Solutions for Mutagenesis Studies
| Reagent / Tool | Function in Experiment | Key Characteristics |
|---|---|---|
| SNAP Software | Provides in silico predictions of functional residue importance [41]. | Neural network-based; output score from -100 to +100; trained on PMD database [41]. |
| Phagemid Vector (e.g., pW1205a) | Serves as the backbone for constructing phage-displayed protein libraries in shotgun scanning [42]. | Allows fusion of protein variant to M13 bacteriophage coat protein for surface display [42]. |
| Degenerate Oligonucleotides | Encodes the binomial (wild-type/alanine) mutagenesis for combinatorial libraries [42]. | Synthesized with mixed bases at target codons; used in Kunkel mutagenesis [42]. |
| Base Editor System | Enables targeted, single-nucleotide changes in the endogenous genomic context [43]. | Typically consists of a cytosine (CBE) or adenosine (ABE) deaminase fused to nCas9 [43]. |
| sgRNA Library | Guides the base editor to specific genomic loci for mutagenesis [43]. | Pooled library of sgRNAs targeting codons of interest; sequenced to surrogate genotype cells [43]. |
| Next-Generation Sequencing (NGS) Platform | Essential for quantifying sgRNA depletion/enrichment in BE screens and sequencing selected clones in shotgun scanning [43] [42]. | Enables high-throughput, parallel sequencing of pooled samples. |
Overcoming Challenges: Optimizing Alanine Scanning for Accuracy and Efficiency
Alanine scanning mutagenesis stands as a cornerstone technique in the validation of hot-spot residues at protein-protein interfaces, providing critical insights into the energetic contributions of individual amino acids to binding affinity. This method systematically substitutes residues with alanine to measure changes in binding free energy (ΔΔG), identifying hot spots as those whose mutation disrupts binding by ≥ 2.0 kcal/mol [1]. Despite its foundational role, traditional alanine scanning faces significant challenges that can constrain its application and interpretation in modern drug discovery research. This guide objectively compares these limitations against emerging computational and experimental alternatives, providing researchers with a framework for selecting optimal validation strategies.
Core Limitations of Traditional Alanine Scanning
The standard alanine scanning approach presents three primary constraints that impact its efficiency and the breadth of information it can deliver.
Labor Intensity and Low Throughput: Experimental alanine scanning requires constructing, expressing, and purifying individual mutant proteins, with each variant analyzed separately through binding assays. This process is noted to be "time-consuming, costly, and labor-intensive," making systematic analysis of large interfaces a substantial undertaking [1] [8]. The low-throughput nature inherently limits the scale at which hot spots can be experimentally validated.
Challenge of Conformational Changes: Mutations can induce local or global structural perturbations that indirectly disrupt binding, making it difficult to distinguish between direct energetic contributions and indirect structural effects [29]. Alanine scanning primarily reveals the energetic consequences of side-chain removal but does not directly report on the protein's dynamic conformational states, potentially leading to misinterpretation of residues that are critical for structural integrity rather than direct binding.
Single-Residue Focus: By investigating one residue at a time, the method may overlook cooperative effects between multiple residues [29]. The energetic contribution of a hot spot often depends on its structural and chemical context, including shielding from solvent by surrounding residues (the O-ring effect) [44]. This limitation can underestimate the complexity of binding interfaces.
Comparative Methodologies and Performance Data
Innovative computational and hybrid approaches have been developed to address the constraints of traditional alanine scanning. The following table summarizes their key characteristics and performance metrics.
Table 1: Comparison of Hot Spot Prediction and Validation Methods
| Method | Key Features | Throughput | Handling of Conformational Changes | Resolution | Reported Performance |
|---|---|---|---|---|---|
| Traditional Alanine Scanning | Experimental measurement of ΔΔG from side-chain removal [1] | Low (individual mutant analysis) [1] | Limited; mutations may induce confounding conformational changes [29] | Single residue | Experimental benchmark; identifies residues with ΔΔG ≥ 2.0 kcal/mol [1] |
| Computational Alanine Scanning (e.g., FoldX, Robetta) | Energy-based calculations on protein structures [1] [44] | High | Models static structures; limited explicit dynamics | Single residue | Varies; Robetta is a well-established standard for comparison [44] |
| Machine Learning & Energy-Based Hybrids (e.g., TSVM) | Combines energy terms as features in machine learning models [44] | High | Implicitly accounts for local environment via energy terms [44] | Single residue | Precision: 56%, Recall: 65% (using Transductive SVM) [44] |
| PPI-HotspotID | Machine learning using free protein structures; integrates conservation, SASA, etc. [8] | High | Uses features from free (unbound) structures | Single residue | Recall: 0.67, Precision: 0.75, F1-score: 0.71 [8] |
| Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS) | Measures deuterium uptake changes in bound vs. unbound states [29] | Medium-High | Detects binding-induced conformational changes and dynamics [29] | Peptide (1-5 amino acids) | Provides structural context and dynamics information [29] |
| BUDE Alanine Scanning | Computational method applicable to structural ensembles from NMR or MD [45] [46] | High | Can account for conformational flexibility through ensembles | Single residue | Validated on diverse targets like NOXA-B/MCL-1 and SIMS/SUMO [45] |
The data demonstrates a trade-off between experimental throughput and mechanistic insight. Computational methods offer high throughput but differ in their ability to capture protein dynamics and conformational changes.
Experimental Protocols for Key Methodologies
Detailed Workflow: Integrated Native MS Alanine Scanning
Recent advancements illustrate how alanine scanning can be adapted to address its traditional limitations. A study on membrane protein-lipid interactions employed a native Mass Spectrometry (MS) approach to enhance throughput and provide complementary data [37].
- Mutant Design and Selection: Residues are selected based on criteria such as prior experimental data, hydrophobic character, or charge. Neutral alanine substitutions are designed via site-directed mutagenesis [37].
- Sample Preparation for Native MS: Wild-type and a single mutant protein are purified and mixed at an approximately 1:1 molar ratio. Lipids or binding partners are added from detergent-solubilized stocks at defined molar ratios [37].
- Data Acquisition: Native MS is performed using instruments like a Q-Exactive HF UHMR Orbitrap mass spectrometer. Key settings include positive ion mode, a mass range of 4,000-15,000 m/z, and controlled collision voltages. Data is often collected across a temperature gradient (e.g., 15-35°C) [37].
- Data Analysis: Peak areas for lipid-bound and unbound states of both wild-type and mutant proteins are deconvoluted using software like UniDec. The difference in free energy (ΔΔG) is calculated using the formula: ΔΔG = -RT ln(K), where the equilibrium constant K = [W] × [ML] / [WL] × [M] [37]. (W = wild-type protein, M = mutant protein, L = ligand, WL/WL = ligand-bound complexes)
- Validation with MD Simulations: Results are complemented with coarse-grain Molecular Dynamics (MD) simulations of the wild-type protein in a model membrane to visualize lipid interactions and residence times at identified sites, confirming the structural context of binding hot spots [37].
Workflow: HDX-MS for Epitope Mapping
HDX-MS offers an orthogonal approach that directly probes binding interfaces and conformational changes [29].
- Deuterium Labeling: The antigen protein in its unbound state is diluted into a deuterated buffer and incubated for various time points.
- Binding Complex Analysis: The same process is repeated for the antigen pre-incubated with its antibody binding partner.
- Quenching and Digestion: The labeling reaction is quenched at low pH and temperature. The protein sample is digested using an immobilized pepsin column.
- LC-MS Analysis: Peptides are separated by liquid chromatography and analyzed by a high-resolution mass spectrometer.
- Data Processing: Deuteration levels of peptide fragments from the bound and unbound antigen are compared. Regions showing significant reduction in deuterium uptake in the bound state constitute the binding epitope, as they become protected from solvent exchange.
The following diagram illustrates the logical decision-making process for selecting the appropriate methodology based on research goals and constraints.
Method Selection Workflow for Hot-Spot Validation
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful experimental validation of hot spots relies on a suite of specialized reagents and tools. The following table details key materials and their applications.
Table 2: Essential Research Reagents and Solutions for Hot-Spot Analysis
| Reagent / Material | Function in Hot-Spot Analysis | Specific Application Example |
|---|---|---|
| Site-Directed Mutagenesis Kits | Creates plasmid DNA encoding specific alanine mutations [37] | Generation of mutant libraries for traditional or native MS scanning |
| Expression Systems (e.g., C43 E. coli) | Produces recombinant wild-type and mutant proteins [37] | High-yield expression of membrane proteins like AqpZ for binding studies |
| Detergent Solubilization Buffers | Extracts and solubilizes membrane proteins while preserving native structure [37] | Purification of AqpZ in tetraethylene glycol monooctyl ether (C8E4) [37] |
| Native MS Buffer (Ammonium Acetate) | Volatile salt compatible with mass spectrometry, preserves non-covalent interactions [37] | Analysis of protein-lipid complexes in native MS alanine scanning [37] |
| Deuterium Oxide (D₂O) | labeling solvent for HDX-MS experiments [29] | Identifying protein regions protected from exchange upon binding |
| Stable Lipid Stocks (e.g., POCL, POPG) | Provides defined binding partners for interaction studies [37] | Probing lipid binding specificity to AqpZ hot spots in native MS assays |
| Molecular Dynamics Software (e.g., CHARMM-GUI) | Simulates protein dynamics and ligand interactions in silico [37] | Visualizing lipid residence times and validating predicted hot spots |
The validation of hot-spot residues through alanine scanning remains a vital component of understanding protein-protein interactions. While the classical experimental method provides direct energetic measurements, researchers now have a powerful array of complementary and alternative strategies. Computational alanine scanning and machine learning predictors like PPI-HotspotID offer high-throughput screening capabilities. Integrated approaches, such as native MS applied to mutant analysis, enhance throughput and provide quantitative data while mitigating the risk of misinterpreting conformational effects. Orthogonal techniques like HDX-MS uniquely capture binding-induced dynamics and conformational changes. The optimal research strategy often involves a combination of these methods, leveraging their respective strengths to achieve a comprehensive and validated map of critical binding residues, thereby accelerating drug discovery and fundamental biological research.
In the field of computational biology, the accurate prediction of protein-protein interaction (PPI) hot spots—residues critical for binding affinity—is a cornerstone for understanding cellular mechanisms and advancing structure-based drug discovery [1]. Experimental validation of these residues through alanine scanning mutagenesis is the gold standard, but it is notoriously time-consuming, costly, and labor-intensive, as each mutant must be purified and analyzed separately [1] [8]. This experimental bottleneck has catalyzed the development of computational methods designed to prioritize residues for laboratory validation. Among the most powerful of these computational strategies are ensemble learning and consensus approaches, which combine multiple models or predictions to achieve a level of accuracy and robustness that is difficult to attain with any single model [47] [48] [49]. This guide objectively compares the performance of various predictive methodologies, focusing on their integration with the experimental framework of alanine scanning research.
Ensemble and Consensus Methods: A Primer
Ensemble learning is a machine learning paradigm that operates on a simple but powerful principle: combining the outputs of multiple models, or "base learners," to produce a single, superior prediction [50]. The core idea is that by leveraging the strengths and compensating for the weaknesses of individual models, the ensemble can make more robust and accurate predictions than any single model alone [49].
Key Mechanisms of Ensemble Learning
- Reducing Variance (Addressing Overfitting): Methods like Bagging (Bootstrap Aggregating) train multiple models on different bootstrapped samples of the dataset. By averaging their predictions, they smooth out the overfitting inherent in individual, complex models. A prime example is the Random Forest algorithm [51] [50].
- Reducing Bias (Addressing Underfitting): Methods like Boosting train models sequentially, with each new model focusing on correcting the errors of its predecessors. This iterative refinement allows the ensemble to capture complex patterns that simple models might miss [51] [50].
- Leveraging Diversity: Techniques such as Stacking combine models with different inductive biases (e.g., decision trees, support vector machines) and use a meta-model to learn how best to integrate their predictions. This leverages the unique strengths of each algorithm [50].
The Consensus Approach
A specific and powerful form of ensemble modeling is the consensus approach. In this framework, predictions from multiple, distinct models are collected, and a final decision is made only when there is agreement, or consensus, among them [48] [49]. For instance, if separate linear regression, support vector machine, and decision tree models all independently predict a particular residue to be a hot spot, the confidence in that prediction is significantly higher [49]. This method effectively filters out false positives caused by the quirks of any single model, thereby increasing precision, albeit potentially at the cost of fewer overall predictions [49].
The following diagram illustrates a generic workflow for a consensus-based prediction system.
Quantitative Comparison of Hot Spot Prediction Methods
Computational methods for predicting hot spots range from energy-based calculations to machine learning classifiers that leverage sequence and structural features [1] [8] [25]. The performance of these methods is typically evaluated on benchmark datasets derived from experimental databases like ASEdb and BID [25]. Key metrics include sensitivity (recall), which measures the ability to identify true hot spots; precision, which reflects the correctness of the predictions; and the F1-score, which harmonizes the balance between sensitivity and precision [8].
The table below summarizes the reported performance of several established prediction methods.
Table 1: Performance Comparison of Hot Spot Prediction Methods on Benchmark Datasets
| Method | Underlying Principle | Key Features Used | Sensitivity (Recall) | Precision | F1-Score |
|---|---|---|---|---|---|
| PPI-hotspotID [8] | Ensemble Machine Learning | Conservation, amino acid type, solvent-accessible surface area (SASA), gas-phase energy (ΔGgas) | 0.67 | N/R | 0.71 |
| FTMap [8] | Probe-based molecular docking | Consensus binding sites on protein surface | 0.07 | N/R | 0.13 |
| SPOTONE [8] | Ensemble of Extremely Randomized Trees | Sequence-based residue-specific features | 0.10 | N/R | 0.17 |
| HEP [25] | Support Vector Machine (SVM) | Electron-ion interaction pseudopotential, pseudo hydrophobicity, protrusion index | N/R | N/R | 0.70 |
N/R: Not explicitly reported in the reviewed literature from the provided search results.
The data reveals that modern machine learning methods, particularly those employing ensemble strategies like PPI-hotspotID, achieve significantly higher sensitivity and F1-scores than older approaches. The performance of HEP also underscores the importance of effective feature selection for building accurate predictive models [25].
Experimental Protocols for Validation
The development and validation of computational predictors are intrinsically linked to experimental techniques. The following protocols detail the key methodologies.
Experimental Protocol: Alanine Scanning Mutagenesis
This protocol is used to experimentally identify and validate hot spot residues [1].
- Site-Directed Mutagenesis: A residue of interest within the protein-protein interface is mutated to alanine using molecular biology techniques. Alanine is chosen because its substitution removes all side-chain atoms past the β-carbon without introducing excessive flexibility to the protein backbone [1].
- Protein Purification: The wild-type and alanine-mutated proteins are expressed and purified separately.
- Binding Affinity Measurement: The binding constant (Kd) between the mutated protein and its binding partner is measured using techniques such as isothermal titration calorimetry (ITC) or surface plasmon resonance (SPR).
- Energetic Analysis: The change in binding free energy (ΔΔG) is calculated using the formula: ΔΔG = -RT ln(ΔKd,mut/ΔKd,wt), where ΔKd,mut and ΔKd,wt are the dissociation constants for the mutant and wild-type complexes, respectively.
- Classification: A residue is definitively classified as a hot spot if its mutation to alanine results in a ΔΔG ≥ 2.0 kcal/mol [1].
Computational Protocol: Building an Ensemble Predictor
This protocol outlines the steps for creating a machine learning-based ensemble predictor for hot spots, reflecting methodologies used in tools like PPI-hotspotID and HEP [8] [25].
- Feature Extraction: For each residue in a dataset of known hot spots and non-hot spots, compute a wide array of features. These can include:
- Evolutionary: Sequence conservation scores.
- Structural: Solvent-accessible surface area (SASA), protrusion index (PI), depth index.
- Energetic: Gas-phase energy, van der Waals interactions.
- Physicochemical: Amino acid type, hydrophobicity, electron-ion interaction pseudopotential (EIIP) [25].
- Feature Selection: Employ feature selection algorithms (e.g., minimum Redundancy Maximum Relevance, or mRMR) to identify the most discriminative subset of features, reducing model complexity and enhancing generalizability [25].
- Model Training and Ensemble Construction: Train multiple base classifiers (e.g., Support Vector Machines, Random Forests) on the selected features. The ensemble can be created via:
- Validation: The ensemble model's performance is rigorously evaluated on a held-out test set or via cross-validation using metrics from Table 1.
The workflow for this protocol is visualized below.
The Scientist's Toolkit: Research Reagent Solutions
This section details key computational and experimental resources used in hot spot research.
Table 2: Essential Research Reagents and Resources for Hot Spot Analysis
| Item Name | Type/Function | Brief Description of Role |
|---|---|---|
| Alanine Scanning Kits | Experimental Reagent | Commercial kits that streamline the process of site-directed mutagenesis for alanine substitution. |
| ASEdb / BID [1] [25] | Data Repository | Public databases (Alanine Scanning Energetics Database; Binding Interface Database) that curate experimentally determined hot spots for training and benchmarking. |
| PPI-HotspotDB [8] | Data Repository | An expanded database that includes hot spots from UniProtKB manually curated as significantly disrupting PPIs, providing a larger benchmark. |
| Rosetta / FoldX [1] | Software Suite | Energy-based modeling suites capable of performing computational alanine scanning to estimate ΔΔG changes in silico. |
| FTMap [8] | Web Server | A tool that identifies hot spots on protein surfaces by finding consensus sites that bind multiple small molecular probes. |
| PPI-hotspotID [8] | Web Server | An ensemble machine learning-based webserver for predicting hot spots from free protein structures. |
| SPOTONE [8] | Web Server | A sequence-based predictor using ensemble learning to identify hot spots. |
Integrated Workflow: From Computation to Experimental Validation
For researchers in drug development, the most effective strategy is a synergistic cycle that integrates computational prediction with experimental validation. The following diagram outlines this holistic workflow.
This workflow begins with a protein structure, runs multiple prediction tools (e.g., PPI-hotspotID, FTMap), and applies a consensus filter to generate a high-confidence list of putative hot spots. These prioritized residues are then validated experimentally via alanine scanning. Crucially, the experimental results feed back into the computational models, allowing for retraining and continuous improvement of the predictive framework [48] [8]. This closed-loop process maximizes efficiency by ensuring that costly experimental resources are focused on the most promising candidates identified by robust computational consensus.
The relentless pursuit of novel therapeutics demands technologies that can accelerate the identification and validation of drug targets. Within this landscape, protein-protein interactions (PPIs) represent particularly attractive yet challenging targets, as their modulation often depends on identifying key residues known as "hot spots"—areas where mutations disproportionately disrupt binding energy [9]. Traditional alanine scanning mutagenesis, while powerful for hotspot identification, has been hampered by its low throughput, high costs, and labor-intensive nature [8]. This comparison guide examines the integrated workflow of high-throughput mutagenesis and native mass spectrometry (native MS)—two complementary technologies that together create a powerful platform for rapid hotspot validation and drug discovery.
High-throughput mutagenesis enables the systematic generation of hundreds to millions of protein variants through methods such as site-saturation mutagenesis and two-fragment PCR assembly [52] [53]. When coupled with native MS—a technique that preserves non-covalent protein-ligand interactions during analysis—researchers gain an unparalleled ability to rapidly screen these variants for binding characteristics [54]. This guide objectively compares this integrated approach against traditional methods, providing experimental data and protocols to illustrate its transformative potential for researchers, scientists, and drug development professionals focused on hotspot validation.
High-Throughput Mutagenesis Methodologies
Modern high-throughput mutagenesis has evolved significantly beyond single-point mutation strategies. Current approaches leverage combinatorial library generation to create diverse variant populations for screening:
Two-Fragment PCR Mutagenesis: This method utilizes mutagenic primers in two separate PCR reactions to generate overlapping vector fragments, which are subsequently assembled using Gibson assembly. This approach significantly reduces PCR artefacts such as misannealing and tandem primer repeats that commonly plague traditional methods. A complete alanine scanning library of 400 single-point mutations can be systematically generated in approximately 6 weeks with this pipeline [53].
Saturation Mutagenesis with Degenerate Primers: By incorporating degeneracy in targeted regions through overlap extension PCR, libraries with diversity ranging from 10⁴ to 10⁷ variants can be created. This approach is particularly valuable for promoter engineering and targeted randomization of specific protein domains [52].
Fluorescence-Activated Cell Sorting (FACS) Integration: When combined with suitably engineered fluorescent reporters, these mutagenesis libraries can be rapidly screened through multiple rounds of positive and negative sorting, rapidly converging to optimal variants with desired phenotypes [52].
Native Mass Spectrometry Fundamentals
Native MS has emerged as a powerful biophysical technique for studying intact proteins and their non-covalent complexes in a native-like folded state. Key advantages include:
Label-Free Detection: Unlike fluorescence-based methods that may suffer from autofluorescence and false positives, native MS provides direct detection without labeling requirements [55] [54].
Minimal Sample Consumption: The technique requires only picomoles of material while providing information on binding thermodynamics, stoichiometry, and ternary and quaternary protein structure [54].
Preservation of Non-Covalent Interactions: Under controlled electrospray ionization (ESI) conditions, native MS maintains non-covalent protein-ligand interactions during transfer from solution to gas phase, allowing researchers to study binding events directly [54].
Table 1: Comparison of Binding Characterization Techniques
| Technique | Sample Consumption | Throughput | Kd Range | Label-Free | Information Obtained |
|---|---|---|---|---|---|
| Native MS | Picomoles | Medium-High | nM-μM | Yes | Stoichiometry, binding affinity, complex integrity |
| ITC | Nanomoles | Low | nM-μM | Yes | Binding affinity, stoichiometry, thermodynamics |
| SPR | Low nanomoles | Medium | pM-mM | No (typically) | Binding affinity, kinetics, specificity |
| Fluorescence | Variable | High | Variable | No | Binding activity, high-throughput compatible |
Integrated Workflow: From Mutagenesis to Hotspot Validation
The synergy between high-throughput mutagenesis and native MS creates a robust pipeline for systematic hotspot identification and validation. The following diagram illustrates this integrated approach:
Experimental Protocols for Integrated Hotspot Validation
Protocol 1: High-Throughput Two-Fragment PCR Mutagenesis [53]
Primer Design: Design mutagenic primers containing desired mutations using high-throughput programs like AAscan. Include ColE1 primers annealing to the vector's bacterial origin of replication.
Fragment Amplification: Perform two separate PCR reactions using:
- Reaction 1: Mutagenic forward primer + ColE1 reverse primer
- Reaction 2: ColE1 forward primer + Mutagenic reverse primer
- Use step-down PCR protocol with annealing temperature decrease of 0.5°C per cycle
Template Digestion: Combine PCR fragments and digest methylated template DNA with DpnI at 37°C for 18 hours
Fragment Assembly: Clean up DNA to remove primers and enzymes, then perform Gibson assembly at 50°C for 10 minutes followed by 37°C for 1 hour
Transformation and Screening: Transform with 2μL assembly product into chemically competent E. coli cells, plate on selective media, and sequence confirm colonies
Protocol 2: Native MS Analysis of Protein-Ligand Complexes [54]
Sample Preparation: Buffer exchange protein variants into volatile ammonium acetate solution (150-200mM) using size exclusion chromatography
Instrument Parameters:
- Nano-electrospray ionization source voltage: 1.0-1.5 kV
- Capillary temperature: 150-200°C
- Collision energies: 50-200 eV (optimized for each complex)
- Backing pressure: 5-10 mbar
Data Acquisition:
- Acquire spectra under non-denaturing conditions
- Monitor charge state distribution for native conformation preservation
- Perform tandem MS to determine complex stability
Data Analysis:
- Deconvolute mass spectra to determine intact masses
- Calculate binding stoichiometry from mass shifts
- Determine relative binding affinities from complex abundances
Performance Comparison: Integrated Approach vs. Traditional Methods
Throughput and Efficiency Metrics
The integration of high-throughput mutagenesis with native MS demonstrates clear advantages over traditional approaches for hotspot identification:
Table 2: Throughput Comparison of Hotspot Identification Methods
| Method | Time per 100 Mutants | Success Rate | Cost per Mutant | Data Quality | Automation Potential |
|---|---|---|---|---|---|
| Integrated Mutagenesis-Native MS | 1-2 weeks | 83% [53] | Medium | High (direct binding data) | High |
| Traditional Cloning + ITC | 8-12 weeks | 60-70% | High | High (thermodynamic parameters) | Low |
| Computational Prediction Only | 1-2 days | 63-73% [9] | Low | Variable (requires validation) | High |
| Yeast Two-Hybrid Screening | 4-6 weeks | 50-60% | Medium | Medium (indirect measurement) | Medium |
Data Quality and Informational Value
Beyond throughput, the integrated approach provides superior data quality and richness:
Direct Binding Measurements: Native MS directly quantifies product formation based on mass-to-charge (m/z) differences between substrates and products, providing unambiguous evidence of binding events [55].
Multi-Parameter Assessment: Unlike single-parameter methods, native MS simultaneously determines stoichiometry, binding affinity, and complex integrity from a single experiment [54].
False Positive Reduction: The label-free nature of native MS eliminates optical artifacts common in fluorescence-based assays, significantly reducing false positives [55].
Case Studies: Experimental Validation of Hotspot Residues
USP1 Inhibitor Development Using Affinity Selection-MS
An optimized Affinity Selection Mass Spectrometry (AS-MS) workflow was developed for efficient identification of potent USP1 inhibitors. In this study:
- USP1 was immobilized on agarose beads with low small molecule retention and efficient protein capture
- The binding affinity of 49 compounds was evaluated using AS-MS with calculation of binding index (BI) values
- Biochemical inhibition assays validated AS-MS results, revealing correlation between higher BI values and lower IC50 values
- The optimized workflow enabled rapid identification of high-quality USP1 inhibitor hits, facilitating structure-activity relationship studies [56]
High-Throughput Mutagenesis for GPCR Studies
Complete alanine scanning libraries for two human GPCRs—cannabinoid CB2 receptor (360 residues) and vasopressin V2 receptor (371 residues)—were generated using high-throughput mutagenesis:
- For V2R, 467 out of 565 sequencing reactions confirmed designed mutations (83% success rate)
- For CB2, 390 out of 537 colonies showed correct sequences (73% success rate)
- The entire process for 400 single-point mutations was completed within 6 weeks
- This systematic approach enabled comprehensive mapping of functional residues in these pharmaceutically important membrane proteins [53]
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagent Solutions for Integrated Mutagenesis-Native MS Workflows
| Reagent/Equipment | Function | Example Applications | Key Characteristics |
|---|---|---|---|
| Gibson Assembly Master Mix | Enzymatic assembly of DNA fragments | High-throughput mutagenesis fragment assembly | Isothermal, simultaneous exonuclease, polymerase, and ligase activities |
| Ammonium Acetate Buffer | Volatile buffer for native MS | Protein complex stabilization during ESI | Volatile, MS-compatible, maintains native protein structure |
| Degenerate Primers | Saturation mutagenesis | Library generation for targeted regions | Incorporates NNK/NNN codons for maximal diversity |
| Echo MS+ System | Acoustic ejection mass spectrometry | High-throughput compound screening | Enables analysis of one sample per second from DMSO stocks |
| ZenoTOF 8600 System | High-sensitivity native MS detection | Intact protein complex analysis | Electron Activated Dissociation for structural characterization |
Comparative Analysis: Advantages and Limitations
Advantages of the Integrated Approach
Speed: Acoustic ejection MS (AEMS) can achieve analysis speeds of one sample per second, dramatically accelerating screening workflows [55].
Versatility: The integrated approach handles various sample types including membrane proteins, which represent the majority of pharmacological targets [54].
Synergistic Data Generation: High-throughput mutagenesis generates variant libraries while native MS provides rapid functional characterization, creating a virtuous cycle of hypothesis generation and testing.
Limitations and Considerations
Specialized Expertise Required: Both techniques require specialized training not typically part of standard molecular biology curricula.
Data Management Challenges: The high throughput of these technologies generates vast datasets requiring sophisticated bioinformatics support and AI-assisted analysis tools [55].
Initial Investment: Implementation requires significant capital investment in specialized instrumentation such as acoustic liquid handlers and high-performance mass spectrometers.
The integration of high-throughput mutagenesis with native mass spectrometry represents a paradigm shift in hotspot identification and validation. This comparison demonstrates clear advantages in throughput, data quality, and informational value compared to traditional approaches. As drug discovery increasingly targets complex protein-protein interactions, this integrated workflow provides researchers with a powerful tool for rapidly identifying and validating critical binding residues, ultimately accelerating the development of novel therapeutics targeting previously undruggable pathways.
For research teams equipped with the necessary expertise and instrumentation, this integrated approach offers an unprecedented capacity to systematically map protein interaction interfaces and identify hotspot residues with efficiency and precision that dramatically outperforms traditional methodologies.
In alanine scanning research, the definitive interpretation of experimental results hinges on a critical distinction: determining whether a measured loss in binding affinity stems from the direct disruption of a key interaction at a protein-protein interface or from an indirect, structural perturbation that alters the protein's conformation. This distinction is fundamental to the accurate validation of true hot-spot residues, which are defined as residues that contribute significantly (typically ≥ 2.0 kcal/mol) to the binding free energy of a complex [1].
The core challenge lies in the nature of the alanine mutation itself. Substituting a residue with alanine removes all side-chain atoms past the β-carbon, which can affect binding through two primary mechanisms:
- Direct Effect: The mutated side chain was directly involved in favorable interactions (e.g., hydrogen bonds, van der Waals contacts) with the binding partner. Its removal directly eliminates these interactions.
- Indirect Effect: The mutation causes a local or even global conformational change in the protein. This structural perturbation can destabilize the binding interface even if the mutated residue itself was not directly involved in binding, leading to false positives in hot-spot identification [29]. As one analysis notes, distinguishing between these direct and indirect effects can be particularly challenging when mutations introduce structural artifacts [29].
Experimental Approaches for Distinguishing Direct from Indirect Effects
No single experimental method can unequivocally resolve this dilemma. Instead, researchers rely on a combination of techniques to triangulate the true nature of a hot spot. The following table summarizes the capabilities of key methodologies in this process.
Table 1: Comparison of Methodologies for Interpreting Alanine Scanning Results
| Method | Key Principle | Ability to Detect Direct Effects | Ability to Detect Indirect Perturbations | Key Limitations |
|---|---|---|---|---|
| Alanine Scanning Mutagenesis | Measures binding affinity change (ΔΔG) upon mutation to alanine [1]. | Direct measurement of energetic contribution. | Limited. Infers indirect effects from context but cannot directly detect structural changes [29]. | Cannot distinguish mechanism without complementary data [29]. |
| Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS) | Probes protein dynamics by measuring hydrogen-deuterium exchange rates [29]. | Indirect. Identifies regions protected from exchange upon binding. | Strong. Can reveal conformational changes and dynamics in both bound and unbound states [29]. | Limited resolution (typically 1-5 amino acids); requires specialized equipment/expertise [29]. |
| X-ray Crystallography | Provides a high-resolution, static 3D structure of the protein or complex [57]. | Direct. Visualizes atomic-level interactions at the interface. | Strong. Can reveal structural changes by comparing mutant and wild-type structures [57]. | Technically challenging; provides static picture, may miss dynamic changes [57]. |
| Computational Alanine Scanning | Uses physics-based or machine learning models to calculate ΔΔG [1] [58]. | Direct prediction of energetic contribution. | Varies by method. Advanced molecular dynamics can model flexibility. | Accuracy depends on model and input data; requires experimental validation [14]. |
Workflow for Integrated Analysis
A robust strategy for interpreting alanine scanning data involves integrating multiple techniques. The following diagram illustrates a synergistic workflow designed to differentiate direct binding effects from indirect structural perturbations.
Detailed Experimental Protocols
To implement the workflow above, researchers can follow these detailed protocols for key experiments.
Alanine Scanning Mutagenesis
Objective: To systematically identify residues critical for binding by measuring the change in binding free energy (ΔΔG) upon mutation to alanine.
- Library Generation: Utilize site-directed mutagenesis to create a library of mutant antigens where each residue in the suspected interface is individually substituted with alanine [29] [59]. For high-throughput studies, employ custom peptide libraries or shotgun scanning methodologies [30] [20].
- Protein Expression and Purification: Express and purify each mutant protein. This step is often the most labor-intensive and time-consuming part of the process [29].
- Binding Affinity Measurement: Determine the binding affinity (e.g., KD, IC50) of each alanine mutant for its partner using a robust quantitative assay. Common techniques include:
- Surface Plasmon Resonance (SPR)
- Isothermal Titration Calorimetry (ITC)
- Enzyme-Linked Immunosorbent Assay (ELISA) [29]
- Energetic Analysis: Calculate the change in binding free energy using the formula: ΔΔG = -RT ln(KD(mutant) / KD(wild-type)), where R is the gas constant and T is the temperature [1]. A residue is typically classified as a hot spot if ΔΔG ≥ 2.0 kcal/mol [1].
Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS)
Objective: To detect changes in protein dynamics and solvent accessibility that occur upon mutation or binding, revealing allosteric or structural perturbations.
- Sample Preparation: Prepare wild-type and alanine mutant proteins (in both bound and unbound states) in a suitable buffer. The buffer must be compatible with MS and the deuterium exchange reaction (typically pH 7-8) [29].
- Deuterium Labeling: Dilute the protein sample into a deuterated buffer (D₂O) and allow the hydrogen-deuterium exchange to proceed for a defined set of time points (e.g., from 10 seconds to several hours) [29].
- Quenching and Digestion: Quench the exchange reaction by lowering the pH and temperature. Pass the quenched sample through an immobilized pepsin column to generate peptide fragments [29].
- Mass Spectrometry Analysis: Inject the digested peptides into a high-resolution mass spectrometer to measure the mass increase due to deuterium uptake [29].
- Data Interpretation: Compare the deuterium uptake rates and levels for the wild-type vs. mutant proteins. A region showing altered dynamics in the unbound state of the mutant suggests an indirect structural perturbation. A region that becomes protected from exchange only in the bound state of the wild-type, but not the mutant, indicates a direct binding site [29].
The Scientist's Toolkit: Key Research Reagent Solutions
Successful execution of these protocols relies on specific reagents and tools. The following table catalogues essential solutions for hot-spot validation studies.
Table 2: Research Reagent Solutions for Hot-Spot Validation
| Reagent / Solution | Function in Research | Key Features & Considerations |
|---|---|---|
| Custom Alanine Scanning Peptide Libraries [59] | Systematically test the role of each residue in a linear epitope or peptide-protein interaction. | High-purity peptides; available in 96-well plates or individual tubes; modifiable with probes or tags. |
| Site-Directed Mutagenesis Kits | Create plasmid DNA encoding for alanine mutations in full-length proteins for structural studies. | High mutation efficiency; variety of vector compatibilities. |
| Stable Cell Lines for Protein Expression | Express and purify large quantities of wild-type and mutant proteins for SPR, ITC, or crystallography. | Consistent protein yield and quality; essential for reproducible results. |
| Biosensor Chips (e.g., SPR) | Immobilize binding partners for real-time, label-free kinetic analysis (e.g., determination of KD, kon, koff). | High sensitivity; requires specialized instrumentation. |
| HDX-MS Automation & Analysis Software [29] | Automate sample handling for HDX-MS and process complex mass spectrometry data. | Reduces manual error; improves reproducibility and throughput of HDX experiments. |
| Robetta Server or FoldX Software [1] | Perform computational alanine scanning to predict ΔΔG values and prioritize residues for experimental testing. | Fast and inexpensive initial screen; provides structural context for mutations. |
Validating a true hot-spot residue requires moving beyond a simple observation of binding energy loss. It demands a multi-faceted approach that dissects whether that energetic penalty originates from the removal of a direct binding interaction or from a secondary structural disruption. By strategically integrating quantitative alanine scanning with techniques like HDX-MS and X-ray crystallography, as outlined in this guide, researchers can confidently assign the mechanistic role of interfacial residues. This rigorous interpretation is paramount for leveraging alanine scanning data in foundational research and for guiding the rational design of therapeutic molecules that target protein-protein interactions with high precision.
Beyond Alanine: Corroborating Findings with Complementary Techniques
Epitope mapping, the process of identifying the precise binding sites of antibodies on their target antigens, is a cornerstone of therapeutic antibody development, vaccine design, and diagnostic innovation [60]. The exact epitope dictates the mechanism of action, efficacy, and potential immunogenicity of a biologic, making its identification a critical step in the research and development pipeline [61]. Among the numerous techniques available, alanine scanning mutagenesis (Ala Scan) and hydrogen-deuterium exchange mass spectrometry (HDX-MS) have emerged as two powerful yet fundamentally distinct methods.
This guide provides an objective comparison of these two techniques, with a specific focus on their utility in validating hot-spot residues—the subset of amino acids that contribute the most to binding free energy [7]. Understanding their respective strengths, limitations, and performance data is essential for researchers to select the optimal strategy for their specific project stage and goals.
Alanine Scanning Mutagenesis
Alanine scanning is a hypothesis-driven, site-directed mutagenesis approach. Its core principle involves systematically substituting individual amino acid residues in the antigen with alanine and measuring the consequent change in binding affinity to the antibody [29].
- Mechanism: Alanine is chosen because its small, non-reactive side chain (a methyl group) effectively removes atoms past the β-carbon of the original residue's side chain. This mutation eliminates the side chain's specific chemical interactions (e.g., hydrogen bonds, van der Waals forces, electrostatic interactions) without dramatically altering the protein's backbone conformation or introducing new chemical properties [29].
- Readout: The binding affinity of each mutant antigen is tested against the antibody using techniques like surface plasmon resonance (SPR) or ELISA. A significant reduction in binding affinity (typically quantified as a change in binding free energy, ΔΔG ≥ 2.0 kcal/mol) identifies that residue as a potential "hot spot" critical for the interaction [29] [7].
Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS)
HDX-MS is a solution-based, biophysical technique that probes protein structure and dynamics by measuring the exchange rate of amide hydrogen atoms with deuterium from the solvent [29] [62].
- Mechanism: In a typical HDX-MS experiment, the antigen (free state) and the antigen-antibody complex (bound state) are exposed to a deuterated buffer for varying time periods. Amide hydrogens that are solvent-accessible and not involved in stable hydrogen bonding will exchange with deuterium. When an antibody binds, the epitope region on the antigen becomes shielded from the solvent, resulting in reduced deuterium uptake compared to the free state [29] [62].
- Readout: After a quench step, the protein is digested into peptides, and the mass increase due to deuterium incorporation is measured by mass spectrometry. By comparing the deuterium uptake levels of peptic peptides between the free and bound states, the regions protected by antibody binding—and thus constituting the epitope—can be identified [29] [63].
Comparative Workflow and Methodologies
The experimental pathways for Ala Scan and HDX-MS are fundamentally different, with direct implications for project timelines, resource allocation, and the type of information obtained. The following diagrams illustrate these core workflows.
Alanine Scanning Mutagenesis Workflow
HDX-MS Epitope Mapping Workflow
Direct Performance Comparison and Experimental Data
A comprehensive 2023 study by Dang et al. directly compared multiple epitope mapping techniques, including Ala Scan and HDX-MS, across five different antibody-antigen pairs, using X-ray crystallography as the structural benchmark [61]. This provides robust, head-to-head experimental data on their performance.
Table 1: Experimental Performance Comparison Across Antibody-Antigen Pairs [61]
| Antibody-Antigen Pair | Ala Scan Performance | HDX-MS Performance |
|---|---|---|
| Pembrolizumab + PD-1 | Identified 10 residues; only 1 matched X-ray; 6 were distant false positives | Not specified in the provided excerpt |
| Nivolumab + PD-1 | Identified residues distantly removed from the true epitope | Not specified in the provided excerpt |
| Ipilimumab + CTLA-4 | No partial epitope identified; many mutations disrupted native structure | Not specified in the provided excerpt |
| Tremelimumab + CTLA-4 | No partial epitope identified; many mutations disrupted native structure | Not specified in the provided excerpt |
| MK-5890 + CD27 | All 4 identified residues matched the X-ray epitope | Not specified in the provided excerpt |
The data highlights a critical finding: the performance of alanine scanning is highly dependent on the structural rigidity of the antigen. For the well-structured CD27 antigen with 8 intra-disulfide bonds, Ala Scan was highly accurate. However, for other targets, it identified false positives or failed altogether due to mutation-induced structural disruption [61].
Table 2: Overall Technical and Operational Comparison [29] [62] [64]
| Parameter | Alanine Scanning Mutagenesis | HDX-MS |
|---|---|---|
| Epitope Type | Best for conformational/discontinuous; can map linear components | Best for conformational/discontinuous |
| Information Level | Single amino acid residue | Peptide-level (typically 5-10 amino acids) |
| Structural Context | Indirect inference; can be confounded by long-range structural effects | Direct analysis of solvent accessibility in native state |
| Dynamic Information | No (provides a static, energetic snapshot) | Yes (can probe protein dynamics and conformational changes) |
| Throughput | Low (labor-intensive, requires production of hundreds of mutants) | Medium-High (fewer samples, automated handling possible) |
| Resource Intensity | High (time, cost, labor) | Medium (requires specialized, high-cost equipment) |
| Key Strength | Pinpoints critical "hot spot" residues and quantifies their energetic contribution | Maps binding footprint under native conditions without mutagenesis |
| Key Limitation | May cause conformational changes, leading to false positives/negatives | Cannot achieve single-residue resolution; complex data analysis |
Strategic Application in Research and Development
Validation of Hot-Spot Residues
The validation of hot-spot residues is a primary application of alanine scanning. By quantifying the change in binding energy (ΔΔG) for each mutation, it provides a direct, functional readout of a residue's energetic contribution to the interaction [29] [7]. This is invaluable for confirming computational predictions of hot spots [65] and for understanding the structure-activity relationship of an antibody. However, as the performance data shows, this requires careful control to ensure mutations do not disrupt the global protein fold [61].
HDX-MS complements this by providing the broader structural context. It can confirm that the proposed hot-spot residues are indeed part of the binding interface and can reveal if a mutation that affects binding did so through allosteric effects rather than direct contact [29] [62].
Use Cases in Drug Development
- Therapeutic Antibody Characterization: HDX-MS is excellent for initial, rapid epitope binning and to understand the mechanism of action (e.g., blocking a ligand-receptor interaction) [29] [62]. Ala Scan is then deployed for fine-mapping to identify the critical residues for patent protection and to guide engineering for higher affinity [61] [66].
- Biosimilar Development: Demonstrating epitope similarity to the originator product is a regulatory requirement. HDX-MS can quickly show that the biosimilar binds to the same region, while Ala Scan can provide residue-level validation [60].
- Engineering Enhanced Therapeutics: HDX-MS has been used to elucidate the mechanism by which destabilizing Fc-region mutations (e.g., YTE, JAWA) enhance antibody function, such as extending serum half-life or promoting agonism, by revealing changes in dynamics and free energy [63] [67].
Essential Research Reagent Solutions
Successful execution of either technique relies on specialized reagents and tools.
Table 3: Key Research Reagents and Materials
| Reagent / Material | Function in Ala Scan | Function in HDX-MS |
|---|---|---|
| Site-Directed Mutagenesis Kits | Generates plasmid DNA for each alanine mutant | Not directly applicable |
| Recombinant Protein Expression Systems | Produces and purifies each mutant antigen | Produces and purifies the antigen and antibody |
| Biosensors (e.g., for SPR or BLI) | Measures binding kinetics/affinity of mutants | Not typically used in core HDX-MS workflow |
| Deuterium Oxide (D₂O) | Not applicable | The labeling agent for hydrogen-deuterium exchange |
| Acid Labile Proteases (e.g., Pepsin) | Not applicable | Digests the labeled protein into peptides for MS analysis |
| Ultra-Performance LC-MS System | Not applicable | Separates and analyzes deuterated peptides (high resolution required) |
| HDX-MS Data Analysis Software | Not applicable | Processes complex data to calculate deuterium uptake and identify protected regions |
Alanine scanning mutagenesis and HDX-MS are not mutually exclusive but are powerful complementary techniques in the epitope mapping toolkit.
- Choose Alanine Scanning when your goal is the quantitative validation of hot-spot residues at single-amino-acid resolution, particularly for patent applications, or for guiding affinity maturation campaigns, especially when working with structurally robust antigens.
- Choose HDX-MS for a rapid, medium-resolution mapping of the epitope on the natively folded antigen, to understand binding-induced conformational changes, or to study antigens that are structurally sensitive to point mutations.
For the most comprehensive and reliable understanding of an antibody-antigen interaction, an integrated approach is often optimal. Using HDX-MS to first define the binding footprint, followed by targeted alanine scanning of the identified region, provides both the structural context and the energetic validation required to confidently characterize therapeutic antibodies.
Protein-protein interactions (PPIs) serve as the foundation for nearly all biological processes, from signal transduction to cellular regulation [68] [1]. Within the extensive interfaces of these interactions, research has revealed that binding energy is not distributed uniformly; instead, a small subset of residues contributes disproportionately to the binding affinity. These critical residues, known as "hot spots," are typically defined as residues whose mutation to alanine causes a decrease in binding free energy (ΔΔG) of ≥2.0 kcal/mol [1]. The identification and characterization of these hot spots has become a pivotal strategy in drug discovery, particularly for designing small molecules that can disrupt pathological PPIs—a challenging yet increasingly viable therapeutic approach [69] [1].
Hot spots exhibit distinctive compositional preferences, with tryptophan (21%), arginine (13.3%), and tyrosine (12.3%) occurring with particularly high frequency [1]. These residues often form cooperative networks within structurally conserved regions of the protein interface [1]. The ability of small molecules to target these hot spots despite their relatively small footprint compared to typical PPI interfaces is attributed to this energetic clustering, making hot spots attractive targets for therapeutic intervention [68] [69].
Theoretical Foundation: The Overlap Between Natural Binding Sites
Bi-Functional Binding Sites
The conceptual bridge between energetic hot spots and small-molecule binding sites is supported by systematic structural analyses across protein families. A comprehensive study of known protein structures revealed that ligand and protein binding sites significantly overlap in many protein families [69]. This analysis identified 197 protein families with statistically significant (p<0.01) overlap between small-molecule and protein-binding positions—termed "bi-functional positions" [69].
These bi-functional positions share remarkable similarities with traditional hot spots: they are particularly enriched in tyrosine and tryptophan residues and are significantly less conserved than mono-functional or solvent-exposed positions [69]. This relationship suggests an evolutionary and functional link between regions that bind small molecules and those that mediate critical protein-protein interactions.
Structural and Energetic Relationships
The relationship between hot spots and small-molecule binding sites extends beyond mere spatial overlap. Hot spots often create favorable binding environments that can be exploited by small molecules [65]. Computational analyses using methods like FTMap have demonstrated that consensus sites—regions on protein surfaces that bind multiple small-molecule probe clusters—often correspond to hot spot regions important for PPIs [65] [33]. These regions are characterized by specific physicochemical properties that make them amenable to small-molecule binding, including structural complementarity, hydrophobicity, and the presence of polar interactions [69] [1].
Experimental Validation: Methodologies and Technologies
Alanine Scanning Energetics
The gold standard for experimental identification of hot spot residues is alanine scanning mutagenesis [1]. This method involves systematically mutating interface residues to alanine, which removes side-chain atoms past the β-carbon while minimizing effects on backbone flexibility [1]. Each mutant protein is then purified and analyzed for binding affinity changes, with residues causing ≥2.0 kcal/mol reduction in binding energy designated as hot spots [1].
Table 1: Key Experimental Methods for Hot Spot Identification
| Method | Principle | Applications | Advantages | Limitations |
|---|---|---|---|---|
| Alanine Scanning | Measures binding energy changes after mutation to alanine | Identification of energetic hot spots; Validation of computational predictions | Direct energetic measurement; High accuracy | Time-consuming; Low throughput; Costly |
| Native Mass Spectrometry | Measures thermodynamic contributions of mutations to lipid binding under non-denaturing conditions | Mapping lipid binding sites and selectivity; Membrane protein interactions | Preserves non-covalent interactions; Can test multiple mutants simultaneously | Requires specialized instrumentation; Limited to certain protein types |
| X-ray Crystallography | Determines atomic-level structure of protein-ligand complexes | Structural characterization of binding modes; Identification of key interactions | High resolution; Direct visualization of binding | Technically challenging; May not capture dynamics |
Recent adaptations have improved throughput, such as "shotgun scanning" and the use of reflectometric interference spectroscopy [1]. Additionally, native mass spectrometry has emerged as a valuable tool for mapping interactions, particularly for membrane proteins, allowing researchers to measure the thermodynamic contributions of specific mutations to ligand binding [37].
Structural Biology Approaches
X-ray crystallography of protein complexes with small molecules provides critical structural insights into how hot spots are engaged. For example, the crystal structure of urokinase receptor (uPAR) bound to a pyrrolinone small-molecule inhibitor revealed a π-cation interaction with Arg-53, a residue not previously considered a traditional hot spot [68]. This finding illustrates that small molecules can engage not only classic hot spots but also peripheral residues that interact cooperatively with hot spots to enhance binding [68].
Computational Prediction: From Structures to Hot Spots
Emerging Computational Tools
Computational methods for hot spot prediction have advanced significantly, addressing the time and cost limitations of experimental approaches. These tools generally fall into two categories: energy-based methods that compute binding energy differences, and machine learning approaches that use various structural and evolutionary features [33].
Table 2: Computational Methods for Hot Spot Prediction
| Tool/Method | Input | Underlying Approach | Key Features | Performance Notes |
|---|---|---|---|---|
| PPI-hotspotID | Free protein structure | Automated machine-learning ensemble | Conservation, aa type, SASA, ΔGgas | Better performance than FTMap and SPOTONE in validation [33] |
| FTMap | Protein structure | Probe-based rigid body docking | Identifies consensus binding sites | Can predict hot spots from free structures [65] [33] |
| FoldX | Protein structure | Energy-based computational alanine scanning | Force field-based energy calculations | Widely used for protein engineering [1] |
| Robetta | Protein structure | Energy-based computational alanine scanning | Physical energy functions | Web server available [1] |
| SPOTONE | Protein sequence | Extremely randomized trees | Sequence-based features | Accessible via webserver [33] |
PPI-hotspotID represents a recent advancement that uses an automated machine-learning framework incorporating only four residue features: conservation, amino acid type, solvent-accessible surface area (SASA), and gas-phase energy (ΔGgas) [33]. This method can identify hot spots from free protein structures (without requiring complex structures) and has demonstrated the ability to detect hot spots not obvious from complex structures, including those in indirect contact with binding partners [33].
Molecular Dynamics Simulations
Explicit-solvent molecular dynamics (MD) simulations have emerged as powerful tools for understanding the dynamic behavior of hot spots and small-molecule binding. MD simulations of uPAR bound to various small molecules revealed that different inhibitors engage hot spots through distinct patterns of correlated motion [68]. Free energy calculations combined with energy decomposition can quantify the contribution of individual residues to protein-protein and protein-compound interactions, providing insights into the cooperative nature of hot spot engagement [68].
Case Studies: Successful Targeting of Hot Spots
uPAR·uPA Interaction Inhibition
The interaction between urokinase receptor (uPAR) and urokinase-type plasminogen activator (uPA) represents a compelling case study. This high-affinity (K_D = 1 nM) interaction occurs over a large interface (>1000 Ų), making it a challenging target for small-molecule inhibition [68]. Through a combination of computational screening and structure-based design, researchers discovered small-molecule inhibitors that engage key residues at this interface.
The crystal structure of uPAR bound to a pyrrolinone-based inhibitor (compound 12) revealed a critical π-cation interaction with Arg-53, which was not initially identified as a hot spot through traditional alanine scanning [68]. This engagement altered the contributions of traditional hot spots to binding affinity, demonstrating that small molecules can leverage cooperative interactions with peripheral residues to achieve potent inhibition [68].
Bcl-xL and IL-2 Inhibition
Other successful examples of hot spot targeting include small-molecule inhibitors of Bcl-xL and IL-2 receptor, which represent some of the few known examples of small molecules that disrupt tight PPIs (K_d = 1–100 nM) [68]. These successes typically involve small molecules that bind to hot spot regions with higher effective affinity than the surrounding interface, effectively competing out the natural protein partner despite the small molecule's smaller footprint [68].
Key Research Reagents and Databases
Table 3: Essential Research Resources for Hot Spot Studies
| Resource | Type | Function/Application | Key Features |
|---|---|---|---|
| ASEdb | Database | Repository of alanine scanning energetics data | Curated experimental ΔΔG values [1] |
| BID | Database | Binding Interface Database | Verified experimental hot spots [1] |
| SKEMPI 2.0 | Database | Structural Kinetic and Energetic database of Mutant Protein Interactions | Includes single-point mutation data [33] |
| PPI-HotspotDB | Database | Expanded collection of experimentally determined PPI-hot spots | Includes UniProtKB curated mutations [33] |
| Wild-type and Mutant Proteins | Biological Reagents | Alanine scanning experiments | Requires protein expression and purification systems [1] [37] |
| Crystallization Reagents | Chemical Reagents | Structure determination of protein-ligand complexes | Enable atomic-level structural insights [68] |
Experimental Workflows
The following diagram illustrates a typical integrated workflow for identifying and validating hot spots and their relationship to small-molecule binding:
The strategic targeting of hot spot residues represents a promising approach for modulating PPIs with small molecules. The relationship between energetic hot spots and small-molecule binding sites is well-established structurally and energetically, with both shared physicochemical properties and overlapping spatial distributions. As computational methods like PPI-hotspotID continue to improve, and experimental techniques like native mass spectrometry advance, the systematic identification and targeting of hot spots will become increasingly efficient.
The integration of computational prediction with experimental validation—particularly through alanine scanning and structural biology—provides a powerful framework for drug discovery. The ability of small molecules to engage not only traditional hot spots but also cooperatively interacting peripheral residues [68] expands the targetable landscape of PPIs. As these approaches mature, targeting hot spots with small molecules will likely yield new therapeutic options for diseases driven by pathological protein-protein interactions.
Protein-protein interactions (PPIs) are fundamental to virtually all cellular processes, including signal transduction, DNA replication, and immune responses [3] [70]. The dysregulation of these interactions is implicated in numerous diseases, making PPIs attractive targets for therapeutic intervention [71] [72]. A pivotal concept in PPI research is the "hot spot"—a small subset of interface residues that accounts for the majority of the binding free energy [3] [25]. Conventionally, hot spots are defined as residues whose mutation to alanine causes a significant decrease (≥ 2.0 kcal/mol) in binding affinity [71] [7].
Experimental identification of hot spots through techniques like alanine scanning mutagenesis is resource-intensive, time-consuming, and difficult to implement on a large scale [3] [71]. This has motivated the development of computational methods to predict hot spot residues, offering a rapid and cost-effective alternative. However, the ultimate value of these predictions hinges on their experimental validation. This guide objectively compares the performance of various computational methods and presents the experimental data that validates them, providing researchers with a clear framework for assessing tool selection in hot spot identification.
Computational Methods for Hot Spot Prediction
Computational methods for predicting PPI hot spots generally fall into three main categories: energy-based methods, machine learning (ML)-based methods, and hybrid methods that combine multiple approaches [71] [7].
Table 1: Categories of Computational Hot Spot Prediction Methods
| Method Category | Underlying Principle | Representative Tools |
|---|---|---|
| Energy-Based | Calculate the change in binding free energy (ΔΔG) upon mutation using force fields or empirical scoring functions. | Robetta [7], FOLDEF [7], BUDE Alanine Scanning (BudeAlaScan) [3], FoldX [3] |
| Machine Learning (ML)-Based | Use classifiers trained on sequence and structural features of known hot spots to discriminate new ones. | PredHS2 [7], PPI-hotspotID [71], HEP [25] |
| Consensus/Ensemble | Combine multiple prediction methods or models to improve accuracy and reliability. | Averaging results from FoldX, mCSM, BeAtMuSiC, Rosetta Flex_ddG, and BudeAlaScan [3]; PPI-hotspotID with AlphaFold-Multimer [71] |
Energy-based methods, such as computational alanine scanning (CAS), operate by calculating the difference in binding free energy between the wild-type complex and the alanine-mutated variant (ΔΔG) [3]. Tools like BudeAlaScan offer the advantage of being applicable not only to single structures from crystallography but also to structural ensembles from NMR or molecular dynamics simulations, thereby accounting for protein dynamics [3]. Machine learning methods have gained prominence for their ability to handle complex, high-dimensional data. These methods, including PredHS2 and PPI-hotspotID, typically employ a wide array of features—such as solvent accessibility, conservation scores, protrusion index, and physicochemical properties—to train classifiers like Support Vector Machines (SVM) or Extreme Gradient Boosting (XGBoost) [71] [25] [7]. A significant innovation is the use of protein language models (e.g., ESM-2), which learn evolutionary information from millions of protein sequences and can be used to predict hot spots from sequence data alone, even with limited training examples [73].
Performance Comparison of Prediction Methods
The accuracy of computational methods is benchmarked against experimental databases like ASEdb, BID, and SKEMPI. Key performance metrics include Sensitivity (Recall), Precision, F1-score (which balances precision and recall), and Matthews Correlation Coefficient (MCC) [7].
Table 2: Quantitative Performance Comparison of Selected Prediction Methods
| Method | Sensitivity/Recall | Precision | F1-Score | MCC | Key Supporting Experimental Data |
|---|---|---|---|---|---|
| Consensus of 5 Methods (FoldX, etc.) | N/A | N/A | (More accurate than any single method) [3] | N/A | Validated on NOXA-B/MCL-1, SIMS/SUMO, GKAP/SHANK-PDZ [3] |
| PPI-hotspotID | 0.67 | N/A | 0.71 | N/A | Validated on eukaryotic elongation factor 2 (eEF2) [71] |
| PredHS2 | 0.81 | 0.73 | 0.77 | 0.54 | Tested on independent BID dataset [7] |
| HEP | N/A | N/A | 0.70 | 0.46 | Tested on ASEdb and BID datasets [25] |
| FTMap (PPI mode) | 0.07 | N/A | 0.13 | N/A | Benchmark on PPI-Hotspot+PDBBM dataset [71] |
The data reveals that modern ML-based methods consistently outperform older techniques. For instance, PPI-hotspotID demonstrated a dramatically higher F1-score (0.71) compared to FTMap (0.13) on the same benchmark dataset [71]. Similarly, PredHS2 showed superior performance over other state-of-the-art methods on an independent test set [7]. Furthermore, evidence suggests that a consensus approach, which averages the results of multiple independent methods (e.g., FoldX, mCSM, BeAtMuSiC, Rosetta Flex_ddG, and BudeAlaScan), can achieve higher accuracy than relying on any single method alone [3]. The integration of interface residues predicted by AlphaFold-Multimer with methods like PPI-hotspotID also yields better performance than using either method in isolation [71].
Experimental Validation: Methodologies and Case Studies
Computational predictions are only as valuable as their experimental confirmation. The following case studies illustrate the rigorous process of validation across diverse protein systems.
Case Study 1: α-Helix and β-Strand Mediated PPIs (BudeAlaScan Validation)
Experimental System and Protocol: A comparative analysis of five CAS methods (FoldX, mCSM, BeAtMuSiC, Rosetta Flex_ddG, and BudeAlaScan) was conducted, and their consensus predictions were experimentally validated using alanine scanning mutagenesis on three distinct PPI targets [3]:
- NOXA-B/MCL-1: An α-helix-mediated interaction involved in apoptosis regulation.
- SIMS/SUMO and GKAP/SHANK-PDZ: Both are β-strand-mediated interactions, with the latter functioning as a scaffold at synaptic junctions [3].
Workflow Overview: The general process for such validation is systematic, as shown in the diagram below.
Key Findings and Validation: The experimental results confirmed that the consensus prediction approach successfully identified key hot-spot residues across all three topographically diverse interfaces [3]. This study demonstrated that leveraging multiple computational methods provides a robust strategy for accurate hot-spot prediction, which was corroborated by quantitative binding measurements of the alanine mutants.
Case Study 2: Eukaryotic Elongation Factor 2 (PPI-hotspotID Validation)
Experimental System and Protocol: The novel method PPI-hotspotID, which uses an ensemble of classifiers and only four residue features (conservation, amino acid type, solvent-accessible surface area, and gas-phase energy), was developed and tested [71]. Its predictions were experimentally verified on eukaryotic elongation factor 2 (eEF2), a translation factor essential for peptide elongation [71] [8].
Key Findings and Validation: Experimental validation confirmed several PPI-hot spots predicted by PPI-hotspotID in eEF2 [71]. A notable strength of PPI-hotspotID is its ability to identify hot spots from the free protein structure (i.e., the unbound state), and it can reveal hot spots that are in indirect contact with binding partners, which might be overlooked by analyzing static complex structures alone [71] [8].
Case Study 3: A Topographically Novel Affimer/BCL-xL Interface
Experimental System and Protocol: The consensus prediction approach was further applied to a novel Affimer/BCL-xL protein-protein interface, where two protein loops project into a hydrophobic cleft on BCL-xL, presenting a unique topological challenge [3] [46].
Key Findings and Validation: Based on the computational predictions, a minimal number of residues were selected for experimental testing. Alanine scanning experiments confirmed these residues as hot spots, validating the predictive approach even for a non-canonical, topographically novel interface [3]. This case underscores the utility of computational guidance in prioritizing residues for mutation in complex systems.
Successful experimental validation relies on a suite of reliable reagents and databases.
Table 3: Key Research Reagent Solutions for Hot Spot Validation
| Reagent/Resource | Function and Application in Validation | Example/Source |
|---|---|---|
| Site-Directed Mutagenesis Kits | Systematically create alanine point mutations in the gene of interest for functional testing. | Commercial kits (e.g., from Agilent, NEB) [3] |
| Protein Expression & Purification Systems | Produce and purify large quantities of wild-type and mutant proteins for biophysical assays. | E. coli, insect, or mammalian cell expression systems [3] |
| Biophysical Binding Affinity Assays | Quantitatively measure the change in binding free energy (ΔΔG) upon alanine mutation. | Surface Plasmon Resonance (SPR), Isothermal Titration Calorimetry (ITC) [70] |
| Alanine Scanning Energetics Database (ASEdb) | Public database of experimental hot spots for method benchmarking and training. | https://www.asedb.org [25] [7] |
| SKEMPI Database | A comprehensive database of binding free energy changes for mutant protein interactions. | SKEMPI 2.0 [3] [73] |
| PPI-HotspotDB | An expanded database incorporating hot spots from UniProtKB manual curation. | Contains 4,039 experimentally determined hot spots [71] |
The experimental workflow from prediction to validation involves multiple stages, each with its own critical resources, as summarized below.
The case studies presented herein demonstrate a powerful paradigm: computational predictions, particularly those from robust ML models and consensus approaches, can accurately identify hot spot residues across a wide range of protein interfaces. The experimental validation via alanine scanning provides the critical ground truth that confirms these predictions and builds confidence in the methods.
Future directions in the field are poised to be shaped by several key technologies. The integration of protein language models like ESM-2 shows promise in predicting residue properties from sequence alone, potentially reducing dependency on solved 3D structures [73]. Furthermore, the application of AlphaFold-Multimer for predicting complex structures and interface residues can be synergistically combined with hot spot predictors to create end-to-end pipelines [71] [74]. As these computational tools become more accurate and accessible, they will continue to accelerate the understanding of PPI mechanisms and provide a solid foundation for rational drug design, ultimately facilitating the discovery and optimization of PPI modulators for therapeutic applications [72].
The identification of hot-spot residues—amino acids that contribute significantly to the binding free energy of protein-protein interactions (PPIs)—is a critical step in understanding cellular physiology and developing targeted therapeutics [8]. Alanine scanning mutagenesis, the experimental gold standard, systematically replaces residues with alanine to measure their energetic contribution to binding [3]. However, this process is time-consuming, costly, and labor-intensive, creating bottlenecks in drug discovery pipelines [3] [8].
Integrative computational approaches have emerged to overcome these limitations by combining the precision of alanine scanning with the predictive power of molecular dynamics (MD) simulations and fragment screening [75] [76]. These methodologies enable researchers to rapidly identify and validate hot-spot residues with greater efficiency and reduced resource expenditure [3]. This guide objectively compares the performance of leading integrative techniques, providing experimental data and protocols to inform selection for drug discovery applications.
Performance Comparison of Computational Alanine Scanning Methods
Computational alanine scanning (CAS) methods predict hot-spot residues from protein structures by calculating the change in binding free energy (ΔΔG) upon mutation to alanine [3]. Their performance varies in accuracy, precision, and computational demand.
Table 1: Performance Comparison of Computational Alanine Scanning Methods
| Method | Approach Category | Reported Pearson Correlation (Experimental vs. Predicted ΔΔG) | Computational Speed | Key Advantage |
|---|---|---|---|---|
| Post-Process Alanine Scanning [24] | Molecular Dynamics (MD) Analysis | High (Precise vs. Experimental) [24] | >5x faster than full MD [24] | High precision from a single trajectory |
| BUDE Alanine Scanning [3] | Empirical Free Energy | Benchmark F1-Score: 0.71 [8] | ~5 minutes for a scan [3] | Handles multiple mutations & structural ensembles |
| Flex_ddG (Rosetta) [3] | Physicochemical Force Field | High (Accurate) [3] | 1-2 hours per mutation [3] | Sophisticated sampling & specialized force fields |
| FoldX [3] | Empirical Force Field | Benchmark F1-Score: N/A | ~8 minutes for a scan [3] | One of the first rapid CAS tools |
| FTMap (PPI Mode) [8] | Probe-Based Mapping | Low (F1-Score: 0.13) [8] | Rapid | Identifies consensus binding sites |
| SPOTONE [8] | Machine Learning (Sequence-Based) | Low (F1-Score: 0.17) [8] | Rapid | Requires only protein sequence |
The "post-process" protocol, which analyzes a single MD trajectory of the native complex, provides better accuracy and precision than running separate, extensive MD simulations for each mutant (the "full MD" protocol) [24]. This method achieved a five-fold increase in computational speed while delivering more reliable results [24].
For rapid, high-throughput screening, fast CAS tools like BUDE Alanine Scanning, FoldX, and FlexddG offer a practical balance between speed and accuracy [3]. Notably, a comparative analysis demonstrated that a consensus approach—averaging the ΔΔG predictions from multiple methods (e.g., BUDE Alanine Scanning, FoldX, FlexddG, mCSM, and BeAtMuSiC)—yielded more accurate identification of hot-spot residues than any single method alone [3].
Table 2: Experimental Validation of CAS Predictions Across Diverse PPI Targets
| Protein-Protein Interaction (PPI) Target | PPI Interface Type | Validation Outcome | Key Finding |
|---|---|---|---|
| NOXA-B / MCL-1 [3] | α-helix-mediated | Accurate prediction | Confirmed role in oncology target |
| SIMS / SUMO [3] | β-strand-mediated | Accurate prediction | Representative of SUMOylation regulation |
| GKAP / SHANK-PDZ [3] | β-strand-mediated | Accurate prediction | Scaffolding function at synaptic junctions |
| Affimer / BCL-xL [3] | Topographically novel | Accurate prediction | Validated hot-spots in a loop-projection interface |
| IAPP Octapeptide [77] | Amyloidogenic peptide | Insights corroborated | Identified Isoleucine-5 role in β-rich cluster formation |
Experimental Protocols for Key methodologies
Protocol: Post-Process Alanine Scanning with Molecular Dynamics
This protocol leverages a single MD simulation of the wild-type complex to efficiently estimate ΔΔG for alanine mutations [24].
- System Preparation: Obtain the protein-protein complex structure from the Protein Data Bank (PDB) [75]. Prepare the structure using standard molecular simulation tools (e.g., LEaP in AMBER) by adding hydrogen atoms and solvating the complex in an explicit water box, such as TIP3P, with over 30,000 water molecules to ensure a proper hydration shell [77]. Add counterions to neutralize the system's charge.
- Molecular Dynamics Simulation: Energy-minimize the system to remove steric clashes. Gradually heat the system to the target temperature (e.g., 310 K) and then perform a production MD simulation under constant temperature and pressure (NPT ensemble) using a software package like AMBER [77]. The simulation should be sufficiently long to capture relevant motions (e.g., 100 ns). A single, well-converged trajectory of the native complex is sufficient [24].
- Trajectory Analysis and Free Energy Calculation: Instead of running separate simulations for each mutant, analyze the saved trajectory frames from the wild-type simulation. For each residue of interest, use the trajectory to computationally mutate the side chain to alanine in silico for every frame, effectively creating a "virtual mutant" trajectory. The binding free energy change (ΔΔG) is then calculated using an end-state method such as the Molecular Mechanics Poisson-Boltzmann Surface Area (MM/PBSA) method [3]. The formula is: ΔΔG = ΔG(mutant) - ΔG(wild-type), where ΔG is the binding free energy calculated for each virtual structure from the trajectory. A residue is typically identified as a hot-spot if ΔΔG ≥ 2 kcal/mol [3] [8].
Protocol: Integrating Fragment Screening with Computational Predictions
Fragment-based screening identifies small, low molecular weight compounds that bind to key sites on a protein, which can validate predicted hot-spot regions [75] [3].
- Target Identification: Perform computational alanine scanning (using a method from Table 1) on the target protein to predict hot-spot residues and define a putative binding site [3].
- Fragment Library Screening: Use a biophysical technique such as surface plasmon resonance (SPR), isothermal titration calorimetry (ITC), or X-ray crystallography to screen a library of small fragment compounds (typically 150-300 Da) for binding to the target protein [75] [3]. The goal is to find fragments that bind to the predicted hot-spot region.
- Validation and Triangulation: If a fragment is found to bind, determine its binding pose, typically via X-ray crystallography [3]. Analyze whether the fragment makes key contacts with the computationally predicted hot-spot residues. This experimental confirmation validates the computational prediction. Furthermore, multiple fragments binding to adjacent sub-pockets within the hot-spot region can be used to guide the design of a larger, more potent lead compound by chemically linking these fragments [3].
Successful implementation of integrative approaches relies on specific computational and experimental resources.
Table 3: Essential Research Reagents and Resources
| Item Name | Function / Application | Relevant Context |
|---|---|---|
| Protein Data Bank (PDB) [75] | Repository for 3D structural data of proteins and nucleic acids. | Source of initial protein-complex structures for MD simulations and CAS. |
| SKEMPI Database [3] | Database of binding free energy changes upon mutation for PPIs. | Used for benchmarking and training computational alanine scanning methods. |
| AMBER Software Suite [77] | Package for molecular dynamics simulations of biomolecules. | Used for running all-atom MD simulations of protein complexes in explicit solvent. |
| Fragment Library [75] [3] | A collection of small, low molecular weight compounds. | Used in fragment-based screening to experimentally probe and validate predicted hot-spot regions. |
| Surface Plasmon Resonance (SPR) [75] [3] | Technique for real-time analysis of biomolecular interactions. | Used to measure binding affinities (KD) of fragments or mutant proteins. |
| PubChem/ChEMBL [75] | Databases of chemical molecules and their bioactivities. | Sources of chemical compounds and bioactivity data for lead discovery. |
| BUDE Alanine Scan [3] | Command-line tool for empirical free-energy based CAS. | Rapid scanning of single structures or ensembles from NMR/MD. |
| PPI-HotspotID [8] | Machine-learning method for predicting hot spots from free protein structures. | Useful when only the structure of a single protein (not the complex) is available. |
The integration of alanine scanning with MD simulations and fragment screening represents a powerful paradigm for accelerating the identification and validation of PPI hot-spot residues. While traditional experimental alanine scanning remains the validation cornerstone, computational methods like post-process MD analysis and BUDE Alanine Scanning offer significant gains in speed and cost-efficiency [24] [3]. The synergistic use of these tools, followed by experimental validation through fragment screening or direct mutagenesis, provides a robust framework for informing drug discovery efforts, ultimately facilitating the design of novel therapeutics that target critical protein-protein interactions [75] [3] [76].
Conclusion
The validation of hot-spot residues through alanine scanning remains a cornerstone technique for deciphering the energetic landscape of protein-protein interactions. A synergistic approach that combines robust computational predictions with targeted experimental validation, and supplements these findings with complementary techniques like HDX-MS, provides the most reliable identification of functionally critical residues. Future directions will be shaped by advances in high-throughput experimental methodologies, more accurate and dynamic computational models that account for protein flexibility, and the increasing integration of AI. These developments will further solidify the role of hot-spot validation in accelerating the discovery of novel therapeutics that target previously intractable PPIs, ultimately bridging fundamental biochemical insights to clinical applications.
References
- 1. Computational Prediction of Hot Spot Residues - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3807665/]
- 2. Hot Regions in Protein–Protein Interactions [https://www.sciencedirect.com/science/article/abs/pii/S0022283604013981]
- 3. Predicting and Experimentally Validating Hot-Spot Residues ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6804253/]
- 4. Densest subgraph-based methods for protein-protein ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04996-1]
- 5. Computational analysis of hot spots and binding mechanism ... [https://pubs.rsc.org/en/content/articlehtml/2019/ra/c9ra01369e]
- 6. database of computational hot spots in protein interfaces [https://pmc.ncbi.nlm.nih.gov/articles/PMC2238999/]
- 7. Enhanced Prediction of Hot Spots at Protein ... [https://www.nature.com/articles/s41598-018-32511-1]
- 8. PPI-hotspot ID : A Method for Detecting Protein ... [https://elifesciences.org/reviewed-preprints/96643v2]
- 9. Predicting interacting hotspots for nanobodies' binding using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12268110/]
- 10. Artificial intelligence based methods for hot spot prediction [https://www.sciencedirect.com/science/article/abs/pii/S0959440X21001469]
- 11. Anatomy of hot spots in protein interfaces [https://www.sciencedirect.com/science/article/abs/pii/S0022283698918435]
- 12. The Importance of Being Tyrosine: Lessons in Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2829252/]
- 13. Alanine Scanning Mutagenesis of Insulin [https://www.sciencedirect.com/science/article/pii/S002192581956882X]
- 14. Accurate predictions of protein mutational effects ... [https://www.nature.com/articles/s42004-025-01771-0]
- 15. Hot spots in protein–protein interfaces: Towards drug ... [https://www.sciencedirect.com/science/article/abs/pii/S0079610714000455]
- 16. PPI-hotspotID for detecting protein-protein interaction hot ... [https://pubmed.ncbi.nlm.nih.gov/39283314/]
- 17. Prediction of hot spots towards drug discovery by protein ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0290899]
- 18. Relationship between Hot Spot Residues and Ligand ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3623692/]
- 19. Relationship between hot spot residues and ligand binding ... [https://pubmed.ncbi.nlm.nih.gov/22770357/]
- 20. Alanine scanning [https://en.wikipedia.org/wiki/Alanine_scanning]
- 21. Alanine-scanning mutagenesis defines a conserved energetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3018278/]
- 22. Optimization of CD4/gp120 Inhibitors by Thermodynamic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3527665/]
- 23. Surface-Enhanced Raman/Infrared Spectroscopy versus ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8279479/]
- 24. Comparing experimental and computational alanine ... [https://pubmed.ncbi.nlm.nih.gov/20656696/]
- 25. Predicting hot spots in protein interfaces based on ... [https://www.oncotarget.com/article/7695/text/]
- 26. Engineering improved T cell receptors using an alanine-scan ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3668434/]
- 27. Alanine Scanning Mutagenesis of Aβ(1-40) Amyloid Fibril ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283606000696]
- 28. Unraveling hot spots in binding interfaces: progress and ... [https://www.sciencedirect.com/science/article/abs/pii/S0959440X0200283X]
- 29. Epitope Mapping: Alanine Scanning Mutagenesis vs. HDX- ... [https://www.rapidnovor.com/alanine-scanning-mutagenesis-vs-hdxms/]
- 30. Facile Affinity Maturation of Antibody Variable Domains ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5591402/]
- 31. Computational Prediction of Alanine Scanning and Ligand ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1003585]
- 32. Prediction of hot spot residues at protein-protein interfaces by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2777894/]
- 33. PPI-hotspotID for detecting protein–protein interaction hot ... [https://elifesciences.org/articles/96643]
- 34. Leveraging neural networks to correct FoldX free energy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11463479/]
- 35. 2.3. Alanine scanning (mutagenesis) [https://bio-protocol.org/exchange/minidetail?type=30&id=6987472]
- 36. Alanine Scanning Mutagenesis based CreMap™ Epitope ... [https://www.creative-biolabs.com/cremap-alanine-scanning-mutagenesis-mapping-service.html]
- 37. Alanine Scanning to Define Membrane Protein-Lipid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11527333/]
- 38. Stabilizing membrane proteins through protein engineering [https://www.sciencedirect.com/science/article/abs/pii/S136759311300063X]
- 39. Alanine Scanning to Define Membrane Protein-Lipid ... [https://pubmed.ncbi.nlm.nih.gov/39484449/]
- 40. Alanine Scanning to Define Membrane Protein-Lipid ... [https://experts.arizona.edu/en/publications/alanine-scanning-to-define-membrane-protein-lipid-interaction-sit]
- 41. Comprehensive in silico mutagenesis highlights ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2597370/]
- 42. Alanine Scanning | MyBioSource Learning Center [https://www.mybiosource.com/learn/testing-procedures/alanine-scanning/]
- 43. A side-by-side comparison of variant function ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12311789/]
- 44. Prediction of hot at protein-protein interfaces by... spot residues [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-10-365]
- 45. Predicting and Experimentally - Validating at... Hot Spot Residues [https://pubmed.ncbi.nlm.nih.gov/31525028/]
- 46. Predicting and Experimentally - Validating at... Hot Spot Residues [https://chemrxiv.org/engage/chemrxiv/article-details/60c742b19abda23d09f8c08f]
- 47. A New Approach to Efficient Ensemble Learning [https://arxiv.org/html/2506.20814v1]
- 48. A consensus approach for estimating the predictive ... [https://www.sciencedirect.com/science/article/abs/pii/S0169260715000243]
- 49. Increase the Accuracy of Machine Learning Models Using this ... [https://abouttrading.substack.com/p/increase-the-accuracy-of-machine]
- 50. How Do Ensemble Methods Improve Prediction Accuracy? [https://www.cohorte.co/blog/how-do-ensemble-methods-improve-prediction-accuracy]
- 51. Comparative analysis of algorithmic approaches in ... [https://www.nature.com/articles/s41598-025-15971-0]
- 52. Targeted mutagenesis and high-throughput screening of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10400447/]
- 53. High-throughput mutagenesis using a two-fragment PCR ... [https://www.nature.com/articles/s41598-017-07010-4]
- 54. High-Throughput Native Mass Spectrometry Screening in ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2022.837901/full]
- 55. How is mass spectrometry enhancing speed and ... [https://www.drugdiscoverynews.com/how-is-mass-spectrometry-enhancing-speed-and-throughput-in-drug-discovery-16563]
- 56. Optimizing the affinity selection mass spectrometry ... [https://www.sciencedirect.com/science/article/pii/S2472630324000566]
- 57. Structural perturbations of substrate binding and oxidation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9653361/]
- 58. Hot spot prediction in protein-protein interactions by an ... [https://bmcsystbiol.biomedcentral.com/articles/10.1186/s12918-018-0665-8]
- 59. Alanine Scanning Library Service [https://www.jpt.com/products-services/peptide-libraries/alanine-scanning-library/?srsltid=AfmBOooet5YyQSzJtWljz4uMjiURPma0vyq1mEpuCxvPU2diVWRUS-5Q]
- 60. Linear vs. conformational epitope mapping: Methods, ... [https://www.pepperprint.com/downloads-resources/blog/article/linear-vs-conformational-epitope-mapping-methods-differences-and-strategic-use-in-drug-development]
- 61. of monoclonal antibodies: a comprehensive... Epitope mapping [https://pmc.ncbi.nlm.nih.gov/articles/PMC10730160/]
- 62. Comparing Epitope Mapping Methods [https://www.rapidnovor.com/comparing-epitope-mapping-methods/]
- 63. Enhancement of Protein–Protein Interactions by ... [https://www.mdpi.com/2218-273X/15/8/1201]
- 64. Difference Between Linear and Conformational Epitope Mapping [https://www.creative-biostructure.com/resource-linear-vs-conformational-epitope-mapping.htm]
- 65. PPI-hotspotID: A Method for Detecting Protein- ... [https://elifesciences.org/reviewed-preprints/96643/reviews]
- 66. The rise of in silico epitope : faster insights, near X-ray... mapping [https://blog.biostrand.ai/the-rise-of-in-silico-epitope-mapping-faster-insights-near-x-ray-precision]
- 67. Enhancement of Protein-Protein Interactions by ... [https://pubmed.ncbi.nlm.nih.gov/40867645/]
- 68. Small Molecules Engage Hot Spots through Cooperative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5812370/]
- 69. The Overlap of Small Molecule and Protein Binding Sites ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1000668]
- 70. Computational Methods for Predicting Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6097941/]
- 71. PPI-hotspotID for detecting protein–protein interaction hot ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11405013/]
- 72. New insights into protein–protein interaction modulators in ... [https://www.nature.com/articles/s41392-024-02036-3]
- 73. Using protein language models for protein interaction hot spot ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05737-2]
- 74. Recent progress and future challenges in structure-based ... [https://www.sciencedirect.com/science/article/pii/S1525001625002771]
- 75. Integrative computational approaches for discovery and ... [https://www.frontiersin.org/journals/drug-discovery/articles/10.3389/fddsv.2024.1362456/full]
- 76. Integrative computational approaches in pharmaceuticals [https://pubmed.ncbi.nlm.nih.gov/40175049/]
- 77. Large-scale all-atom molecular dynamics alanine - scanning of IAPP... [https://www.nature.com/articles/s41598-018-38401-w?error=cookies_not_supported]