Validating Ligand Binding in Electron Density Maps: A Guide to Methods, Challenges, and Best Practices
Accurately identifying and validating small-molecule ligands in experimental density maps from techniques like X-ray crystallography and cryo-electron microscopy (cryo-EM) is a critical yet challenging step in structure-guided drug design.
Validating Ligand Binding in Electron Density Maps: A Guide to Methods, Challenges, and Best Practices
Abstract
Accurately identifying and validating small-molecule ligands in experimental density maps from techniques like X-ray crystallography and cryo-electron microscopy (cryo-EM) is a critical yet challenging step in structure-guided drug design. This article provides a comprehensive overview for researchers and drug development professionals, covering the foundational principles of ligand validation, modern computational methods including deep learning, strategies for troubleshooting common issues in map interpretation, and a comparative analysis of validation metrics and benchmarks. By synthesizing recent advancements, such as those from the 2024 EMDataResource Ligand Challenge, this guide aims to equip scientists with the knowledge to improve the reliability of their structural models and avoid the pitfalls of misidentified ligands.
The Critical Foundation: Why Ligand Validation is Essential in Structural Biology
The Central Role of Ligands in Understanding Protein Function and Drug Mechanism
Protein-ligand interactions represent a fundamental molecular mechanism governing critical biological processes in living organisms. These interactions, which involve the formation of complexes between proteins and ligands (small molecules or other macromolecules), are indispensable for enzyme catalysis, signal transduction, gene regulation, and molecular recognition [1]. The precise binding of ligands to protein targets initiates conformational changes that modulate protein function, making this process a cornerstone for understanding cellular physiology and pathology. From a therapeutic perspective, the majority of pharmaceutical compounds function as ligands that selectively bind to protein targets to alter their activity, underscoring the pivotal role of ligand interactions in drug discovery and development [1] [2].
The study of these interactions has evolved significantly from Emil Fischer's 1894 "lock-and-key" principle to contemporary understanding that incorporates induced-fit mechanisms and conformational selection dynamics [1]. Modern research has revealed that protein-ligand interactions exhibit remarkable complexity, involving weak and transient binding, allosteric modulation, and multivalent interactions that enhance affinity and specificity [1]. The accurate characterization of these interactions provides invaluable insights for rational drug design, enabling researchers to develop therapeutic agents with optimized binding affinity, specificity, and pharmacokinetic properties.
Computational Methods for Predicting Ligand Binding
Computational approaches for predicting protein-ligand interactions have become indispensable tools in modern drug discovery, offering cost-effective and scalable strategies for exploring chemical and biological spaces [3]. These methods span various techniques, from traditional physics-based simulations to cutting-edge machine learning algorithms, each with distinct strengths and limitations for specific applications in structure-based drug design.
Comparative Analysis of Computational Prediction Methods
Table 1: Performance Comparison of Computational Methods for Ligand Binding Site Prediction
| Method Name | Method Type | Key Features | Performance Metrics | Limitations |
|---|---|---|---|---|
| LABind [2] | Structure-based (Multi-ligand) | Graph transformer with cross-attention mechanism; ligand-aware binding site prediction | AUPR: 0.72 (DS1), 0.70 (DS2); Effective for unseen ligands | Requires protein structure information |
| LigBind [2] | Structure-based (Single-ligand) | Pre-training on broad ligand sets; requires fine-tuning for specific ligands | Limited effectiveness without fine-tuning | Cannot predict sites for completely unseen ligands without retraining |
| P2Rank/DeepPocket [2] | Structure-based (Multi-ligand) | Relies on protein structure features (e.g., solvent accessible surface) | Moderate performance across diverse ligands | Ignores ligand-specific binding patterns |
| SEGSA_DTA [4] | Affinity Prediction | SuperEdge graph convolution; supervised attention for protein-ligand interactions | Outperforms state-of-the-art affinity prediction methods | Specifically designed for affinity prediction, not binding site identification |
| ENTess QSBR [5] | Affinity Prediction | Delaunay tessellation with electronegativity descriptors; k-Nearest Neighbor QSBR | Cross-validated R²: 0.83 (test set), 0.85 (validation set) | Traditional QSAR approach with limited applicability to diverse complexes |
Experimental Protocols for Binding Affinity Prediction
The ENTess QSBR (Quantitative Structure-Binding Relationship) method employs a well-defined protocol for binding affinity prediction [5]. The methodology begins with dataset preparation, involving the collection of high-resolution (below 3.0Å) X-ray crystal structures of protein-ligand complexes from the PDB. Hydrogen atoms and water molecules are removed, and ligands are extracted using molecular modeling software such as SYBYL. Delaunay tessellation is then applied to the protein-ligand interface, partitioning the 3D structure into space-filling, irregular tetrahedra (simplices) with atoms as vertices. Each atomic quadruplet composition is characterized by a single descriptor calculated as the sum of the Pauling electronegativity values for the four participating atom types. These ENTess descriptors serve as independent variables in k-Nearest Neighbor QSBR studies, with models validated through rigorous training/test set splits and leave-one-out cross-validation.
For SEGSA_DTA implementation, the protocol involves several key steps [4]. First, protein and ligand structures are converted into graph representations with comprehensive edge features. The SuperEdge graph convolution then fuses node and edge information to capture intricate structural relationships. A multi-supervised attention module learns attention distributions consistent with real protein-ligand interactions, with model interpretability enhanced through SHapley Additive exPlanations (SHAP) analysis. Training incorporates multi-task learning on binding affinity datasets, with performance evaluated using root mean square error (RMSE) and correlation coefficients on benchmark datasets.
Experimental Validation of Ligand Binding
Experimental validation of protein-ligand interactions provides the critical foundation for computational method development and verification. Structural biology techniques, particularly X-ray crystallography and cryo-electron microscopy (cryo-EM), enable direct visualization of ligand binding modes and interactions at atomic or near-atomic resolution.
Ligand Validation in Electron Density Maps
The LigPCDS dataset provides a standardized framework for validating ligand binding in electron density maps [6]. This comprehensive dataset contains 244,226 ligand entries derived from X-ray protein crystallography data deposited in the Protein Data Bank. The experimental workflow for creating and validating LigPCDS involves several critical steps. First, a list of valid ligands is filtered and downloaded from the RCSB PDB. Entries are refined using Dimple v2.6.1 in a standardized procedure without added heteroatoms to normalize data quality and highlight ligand blobs in Fo-Fc maps. The 3D images of ligands are derived from their Fo-Fc maps using Gemmi v0.5.8 based on atomic positions of ligand entries. These representations are converted to 3D point clouds with appropriate scaling, background removal, masking, and contouring. Finally, ligand 3D point clouds are labeled pointwise using atomic sphere modeling and designed chemical vocabularies based on atoms and their cyclic structural arrangements [6].
Deep learning models trained on the LigPCDS dataset demonstrate robust performance in semantic segmentation of ligand 3D representations, achieving mean accuracy ranging from 49.7% to 77.4% in terms of Intersection over Union (mIoU) metric and from 62.4% to 87.0% in F1-score (mF1) across different chemical vocabularies [6]. This validation confirms the reliability of the dataset and methodology for interpreting protein ligand chemical structures from experimental data.
High-Quality Dataset Curation
The HiQBind-WF workflow addresses critical challenges in protein-ligand dataset quality that impact validation accuracy [7]. This semi-automated workflow implements multiple curation modules: (1) filtering to reject covalent protein-ligand bonds, rare elements, and severe steric clashes; (2) ligand fixing to ensure correct bond order and protonation states; (3) protein fixing to add missing atoms to binding-related chains; and (4) structure refinement to simultaneously add hydrogens to both proteins and ligands in complex state. Application of HiQBind-WF to existing datasets like PDBbind demonstrates its ability to correct structural imperfections that compromise scoring function accuracy and reliability [7].
Table 2: Key Research Reagent Solutions for Protein-Ligand Interaction Studies
| Reagent/Resource | Type | Primary Function | Key Features | Access Information |
|---|---|---|---|---|
| LigPCDS Dataset [6] | Experimental Dataset | 3D point clouds of protein ligands from X-ray crystallography | 244,226 ligand entries; chemically labeled point clouds | Derived from PDB; Available through publication |
| HiQBind Dataset [7] | Curated Dataset | High-quality protein-ligand structures with binding affinities | >18,000 unique PDB entries; structural artifacts corrected | Open-source workflow and dataset |
| PDBbind [7] | Reference Dataset | Biomolecular complexes with binding affinities | ~19,500 complexes; general/refined/core subsets | http://www.pdbbind-cn.org/ (v2020) |
| BindingDB [7] | Binding Affinity Database | Protein-ligand binding measurements | 2.9 million measurements; 1.3 million compounds | Public database |
| BioLiP [7] | Protein-Ligand Database | Biologically relevant protein-ligand interactions | >900,000 interactions; functional annotations | Public database |
Integration of Computational and Experimental Approaches
The most significant advances in protein-ligand interaction research emerge from the strategic integration of computational predictions with experimental validation. This synergistic approach enables researchers to overcome the limitations inherent in each methodology when used independently.
Signaling Pathways Initiated by Ligand Binding
Ligand binding events typically initiate complex signaling cascades that regulate critical cellular processes [8]. These molecular pathways represent sequences of reactions, often starting with ligand binding to receptors, which subsequently triggers intracellular signaling events. Visualization of these pathways employs specific color semantics, where analogous color palettes (colors adjacent on the color wheel) indicate molecular components that are functionally connected within the same pathway, while color progressions signify the sequential order of molecular interactions [8].
Best Practices for Method Selection
Research objectives should guide the selection of computational and experimental methods for studying protein-ligand interactions. For predicting binding sites for novel ligands, ligand-aware methods like LABind are recommended due to their ability to generalize to unseen ligands [2]. When working with established ligands with known binding sites, affinity prediction methods such as SEGSA_DTA or traditional QSBR approaches may provide sufficient accuracy [4] [5]. For structure-based drug design projects that require atomic-level precision, high-quality curated datasets like HiQBind combined with experimental validation through X-ray crystallography or cryo-EM offer the most reliable approach [7] [6].
The field continues to evolve with emerging trends including the study of ligands targeting intrinsically disordered proteins (IDPs) - challenging therapeutic targets involved in incurable cancers and neurodegenerative disorders [1]. Advanced deep learning models like AlphaFold 3, RosettaFold All-Atom, and molecular diffusion models are increasingly capable of predicting protein-ligand complex structures with accelerating accuracy, potentially revolutionizing virtual screening and de novo drug design in the coming years [1].
The high-resolution visualization of protein-ligand complexes is fundamental to understanding biological function and advancing structure-based drug design. Two primary experimental techniques—X-ray crystallography and cryo-electron microscopy (cryo-EM)—provide atomic-level insights into these interactions, yet they diverge significantly in their methodological approaches, capabilities, and limitations for ligand modeling. While X-ray crystallography has long been the cornerstone of structural biology, offering unparalleled throughput and resolution for crystallizable targets [9], recent breakthroughs in cryo-EM have enabled the determination of complex biomolecules previously inaccessible to crystallization [10]. This guide objectively compares the performance of these techniques for ligand modeling, framed within the broader thesis of validating ligand binding in electron density maps. We present experimental data, detailed methodologies, and practical resources to inform researchers and drug development professionals in selecting and implementing the most appropriate technique for their specific structural biology questions.
Technical Comparison at a Glance
The table below summarizes the core technical characteristics and typical performance metrics of X-ray crystallography and cryo-EM for ligand modeling applications.
Table 1: Key technical characteristics of X-ray crystallography and cryo-EM for ligand modeling
| Characteristic | X-ray Crystallography | Cryo-EM |
|---|---|---|
| Typical Resolution Range | Often 1.5 - 3.0 Å [9] | Often 2.5 - 4.0 Å for ligands, even with 1.5 Å protein resolution [11] |
| Sample State | Crystalline lattice | Vitreous ice (single particles) |
| Temperature Regime | Cryogenic (100 K) standard; Room-temperature emerging [9] [12] | Cryogenic (~100 K) |
| Ligand Introduction Methods | Co-crystallization; Crystal soaking [13] | Incubation with purified protein prior to vitrification |
| Throughput | High (highly automated) [9] | Moderate (rapidly improving) |
| Ideal for Membrane Proteins | Challenging, requires specialized crystallization [10] | Excellent [14] |
| Ability to Trap Intermediates | Yes (via kinetic trapping in crystals) [13] | Limited |
| Radiation Damage Concerns | Mitigated by cryo-cooling; higher at room temperature [9] [12] | Mitigated by low dose imaging and particle averaging |
Experimental Protocols for Ligand Modeling
X-ray Crystallography Workflow
Sample Preparation: Protein-ligand complexes for X-ray crystallography are typically generated via co-crystallization (mixing protein and ligand prior to crystal growth) or soaking (introducing the ligand into pre-grown native crystals) [13]. Soaking is more common for high-throughput applications like fragment-based screening [9].
Data Collection: The standard method involves collecting a single, complete diffraction dataset from one or a few cryo-cooled (100 K) crystals. However, serial crystallography at synchrotrons or XFELs is an emerging method. It merges data from thousands of microcrystals, which is particularly beneficial for room-temperature data collection as it minimizes radiation damage [9] [12].
Ligand Building and Refinement: The initial protein model is fit into the electron density map. The ligand is then modeled into the residual difference density ( Fo - Fc ) in the binding site. Due to conformational heterogeneity, tools like qFit-ligand can be employed. This algorithm uses RDKit's ETKDG method for stochastic conformational sampling and selects a parsimonious ensemble of conformers that best fit the electron density, which is especially useful for flexible ligands and macrocycles [15].
Cryo-EM Workflow
Sample Preparation: The protein is incubated with the ligand in solution, and the complex is then vitrified for data collection. The absence of a crystal lattice can more readily accommodate conformational changes in the protein upon ligand binding [14].
Data Collection & Processing: Thousands of micrographs are collected and processed to reconstruct a 3D density map. A significant challenge is that the global resolution of the map can be high, but the local resolution around the bound ligand is often lower, making ligand identification and modeling difficult [11].
Ligand Building and Refinement: A powerful emerging approach integrates artificial intelligence (AI) with density-guided molecular dynamics (MD) simulations [11] [16].
- AI Prediction: An AI model, such as an AlphaFold3-like model (e.g., Chai-1), predicts the structure of the protein-ligand complex using the protein sequence and the ligand's SMILES string [11].
- Flexible Fitting: The AI-predicted model is then flexibly fit into the experimental cryo-EM map using MD simulations. Additional forces guide the atoms to maximize the cross-correlation between the simulated density of the model and the experimental map, refining both the protein and ligand poses simultaneously [11]. This hybrid approach has been shown to improve ligand model-to-map cross-correlation from 40-71% to 82-95% relative to the deposited structure [11] [16].
Workflow Visualization
The diagrams below illustrate the core methodological workflows for ligand modeling in both techniques, highlighting key differences in approach.
X-ray Crystallography Ligand Modeling
Cryo-EM Ligand Modeling
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful ligand modeling requires a suite of specialized computational tools and reagents. The table below details key solutions for both techniques.
Table 2: Key research reagents and solutions for ligand modeling
| Category | Item / Software | Primary Function | Applicable Technique |
|---|---|---|---|
| Modeling & Refinement | qFit-ligand [15] | Automated modeling of multiconformer ligands into electron density. | X-ray, Cryo-EM |
| RDKit (ETKDG) [15] | Stochastic generation of chemically sensible small molecule conformers. | X-ray, Cryo-EM | |
| GROMACS [11] | Molecular dynamics package for density-guided flexible fitting. | Cryo-EM | |
| AI Prediction | AlphaFold3 / Chai-1 [11] | Predicts protein-ligand complex structure from sequence and SMILES. | Cryo-EM |
| Data Processing | PanDDA [15] | Identifies low-occupancy ligands in X-ray data by subtracting background density. | X-ray (Fragment Screening) |
| Ligand Specification | SMILES String [11] | Standardized line notation for inputting ligand chemistry into modeling software. | X-ray, Cryo-EM |
| Sample Handling | Microporous Fixed-Target Chips [9] | High-throughput serial crystallography at room temperature. | X-ray (SSX) |
Both X-ray crystallography and cryo-EM are powerful techniques for determining protein-ligand structures, yet they offer complementary strengths. X-ray crystallography remains the workhorse for high-throughput studies of crystallizable targets, providing high-resolution data that enables the modeling of subtle ligand conformational heterogeneity [15] [9]. The emergence of room-temperature serial techniques is further enhancing its physiological relevance [12]. In contrast, cryo-EM has revolutionized the study of large complexes and membrane proteins, such as GPCRs and ion channels, which are often recalcitrant to crystallization [14]. Its main challenge for ligand modeling—lower local resolution at the ligand site—is being effectively addressed by innovative hybrid approaches that combine AI-based prediction with experimental density-guided simulation [11] [16]. The choice between techniques depends ultimately on the biological target, the scientific question, and available resources. Future progress will likely be driven by deeper integration of these experimental methods with computational tools, leading to more dynamic and accurate models of molecular recognition.
In the field of structural biology, accurately determining how a small molecule ligand binds to its protein target is fundamental to understanding biological function and for rational drug design. However, this process is susceptible to a range of pitfalls, from ingrained cognitive biases to the technical limitations of experimental data. This guide objectively compares the realities of ligand binding validation against the ideal, providing researchers with the data and methodologies needed to critically assess structural models.
The Impact of Cognitive Bias on Structural Interpretation
Cognitive biases systematically skew the interpretation of experimental data, leading to overconfidence in initial models.
- Confirmation Bias: Scientists naturally tend to favor information that confirms their pre-existing beliefs or hypotheses. In crystallography, this can manifest as building a ligand model to fit weak or ambiguous electron density because it is the expected compound, potentially overlooking discrepancies [17].
- The "Single Structure" Paradigm: A conservative view that insists on "a single dataset, a single structure, a single interpretation" can hinder progress. This mindset may cause researchers to dismiss more complex, multi-state models that better represent the compositional and conformational heterogeneity present in the crystal, especially in fragment-screening campaigns [18].
- The "Flexibilization" of Science: Under external pressures, such as the urgent need for treatments during the COVID-19 pandemic, methodological standards can loosen. This "flexibilization" creates a vicious cycle where low-quality studies produce unreliable data, which in turn generates anecdotal evidence that reinforces pre-existing beliefs [17].
Technical Limitations: The Illusion of High Resolution
Global quality metrics like resolution are often relied upon as a proxy for overall model quality, but they can be dangerously misleading for local ligand interpretation.
Table 1: Prevalence of Ligand Quality Issues in the PDB
| Quality Category | Percentage of Ligands | Description |
|---|---|---|
| Good | 27% | Highly reliable; minimal concerns for use [19]. |
| Dubious | 51% | Not highly reliable; poses minor quality concerns [19]. |
| Bad | 22% | Needs serious attention; unsuitable for applications like drug design [19]. |
A critical analysis of approximately 0.28 million protein-ligand pairs from the PDB reveals that a significant portion of ligands have quality issues [19]. Alarmingly, over half (62.5%) of the ligands classified as "Bad" were determined at a high resolution of 2.5 Å or better [19]. This demonstrates that a high-resolution structure does not guarantee a correct ligand model.
Furthermore, high-resolution studies on Fatty Acid-Binding Proteins (FABPs) uncovered that an estimated 15% of the well-defined ligands had a different chemical composition than expected [20]. These "unexpected ligands" resulted from issues like synthesis side products, degradation, or human error during compound registration. At lower resolutions, these subtle chemical changes can easily go unnoticed, leading to misidentified binding poses that corrupt structure-based design and machine-learning training sets [20].
Experimental Protocols for Robust Validation
To mitigate bias and technical limitations, rigorous and objective validation protocols are essential. The following workflows provide a framework for confident ligand modeling.
Experimental Protocol 1: Validating Ligand Pose and Identity in Cryo-EM Maps
Objective: To accurately identify and build a small molecule ligand into a medium-resolution cryo-EM density map. Background: Cryo-EM is increasingly used for structure-based drug design, but ligand modeling at typical resolutions (worse than 3 Å) is challenging. Traditional crystallographic methods perform poorly at these resolutions [21].
Methodology: The EMERALD-ID method uses a combination of physical forcefields and density agreement to rank ligand identities from a user-provided library [21].
- Input: Provide the cryo-EM density map, a receptor model, and a library of potential ligand identities.
- Docking: The EMERALD tool docks all candidate ligands from the library into the target density map [21].
- Evaluation: A scoring function combines the RosettaGenFF small molecule forcefield (estimating binding affinity) with the density correlation of the docked pose.
- Output: The method ranks the candidate ligands, providing the most probable identity and its conformation. In benchmarks, EMERALD-ID successfully identified the deposited ligand in 44% of instances, and a closely related ligand in 66% of cases [21].
Cryo-EM Ligand Identification Workflow
Experimental Protocol 2: Creating High-Quality Datasets for Computational Studies
Objective: To curate a high-quality, non-covalent protein-ligand dataset for reliable training and validation of scoring functions in drug discovery. Background: Widely used datasets like PDBbind can contain structural artifacts in both proteins and ligands, compromising the accuracy and generalizability of computational models [7].
Methodology: The HiQBind-WF is a semi-automated workflow for data cleaning and structural preparation [7].
- Initial Filtering: Reject covalent binders, ligands with rare elements, and structures with severe steric clashes.
- Ligand Fixing (LigandFixer): Correct bond orders, protonation states, and aromaticity to ensure ligand chemistry is correct.
- Protein Fixing (ProteinFixer): Add missing atoms and residues to all protein chains involved in binding.
- Structure Refinement: Recombine the fixed protein and ligand, then perform a constrained energy minimization to add hydrogens and resolve steric issues in the context of the complex. This workflow systematically addresses common errors, producing a more reliable dataset (HiQBind) for developing drug discovery tools [7].
The Scientist's Toolkit: Essential Research Reagents & Solutions
The following tools and resources are critical for conducting rigorous ligand validation.
Table 2: Key Tools for Ligand Validation and Analysis
| Tool Name | Function | Application Context |
|---|---|---|
| FLEXR-MSA | Unbiased electron-density map comparison of sequence-diverse structures [22]. | X-ray Crystallography |
| VHELIBS | Quantifies local structural quality of ligands and binding site residues [19]. | X-ray Crystallography |
| PDB-EDA | Converts sigma-scaled maps to absolute electron units for physicochemical interpretation [23]. | X-ray Crystallography |
| EMERALD-ID | Docks and evaluates small molecules to determine ligand identity in Cryo-EM maps [21]. | Cryo-Electron Microscopy |
| HiQBind-WF | Semi-automated workflow to create high-quality protein-ligand datasets [7]. | Data Curation for Computational Drug Discovery |
| PanDDA | Identifies low-occupancy ligands by analyzing multiple datasets from screening campaigns [18]. | X-ray Crystallographic Fragment Screening |
Key Takeaways for Researchers
- Trust, but Verify: A high-resolution structure or a PDB code does not guarantee a correct ligand model. Always inspect the ligand's fit to the electron density.
- Embrace Complexity: Move beyond the "single structure" mindset. Consider that your crystal may contain multiple ligand states or conformations, especially in screening experiments.
- Leverage Automated Tools: Use the available computational toolkit to obtain an objective, quantitative assessment of ligand quality. Do not rely solely on visual inspection, which is susceptible to bias.
- Context is Key: The reliability of a ligand model is not just a function of resolution, but also of ligand occupancy, B-factors, and the chemical environment of the binding pocket.
By understanding these pitfalls and adopting rigorous validation protocols, researchers can significantly improve the accuracy of their structural models, leading to more reliable biological insights and a more efficient drug discovery process.
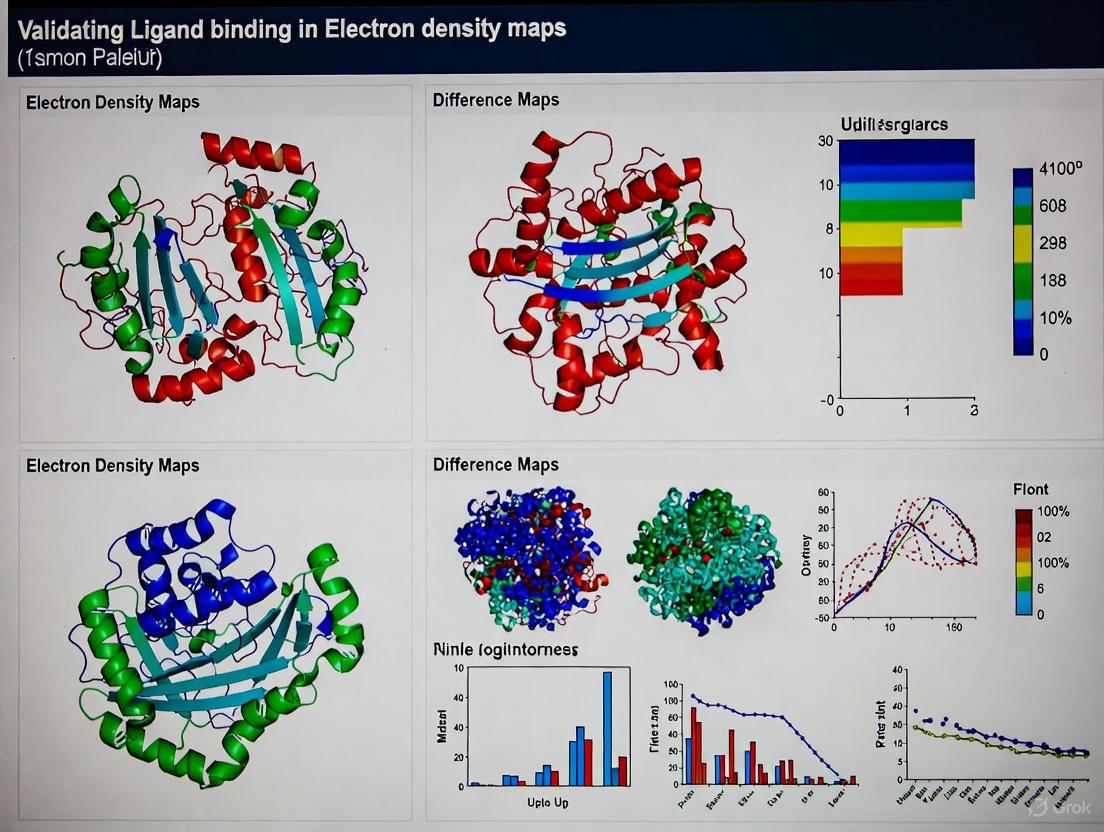
In structural biology, particularly in the context of validating ligand binding in electron density maps, the accuracy and precision of a molecular model are paramount. High-quality structural models serve as the foundation for understanding biological mechanisms and guiding drug development efforts. However, the process of building atomic models into electron density maps, especially those derived from X-ray crystallography or cryo-electron microscopy (cryo-EM), inherently involves interpretation that can be subjective. This challenge is particularly acute for ligand binding sites, which often exhibit lower resolution and higher flexibility than the protein backbone. Without robust, quantitative validation metrics, researchers risk basing scientific conclusions on modeling artifacts or inaccurate atomic placements.
This guide introduces four essential quality metrics—RSCC, RSZD, Q-score, and MolProbity—that provide complementary approaches for assessing model quality. These metrics help researchers move beyond subjective assessment to objective, quantitative evaluation of how well their structural models represent the experimental data and conform to established stereochemical rules. By understanding the strengths and applications of each metric, structural biologists and drug development professionals can make more informed decisions about model quality, particularly when characterizing ligand binding sites that are crucial for structure-based drug design.
Metric Definitions and Theoretical Foundations
Real-Space Correlation Coefficient (RSCC)
The Real-Space Correlation Coefficient (RSCC) quantifies how well the electron density calculated from an atomic model matches the experimentally observed electron density within a specific region of the map. It is defined as the sample correlation coefficient between the observed (ρobs) and calculated (ρcalc) electron density values at all grid points within a defined volume [24]:
RSCC = cov(ρobs, ρcalc) / √[var(ρobs) • var(ρcalc)]
where cov denotes covariance and var denotes variance. The RSCC ranges from 0 to 1, with values closer to 1 indicating better agreement between model and experimental data [25]. In practice, RSCC is typically calculated on a per-residue basis, making it particularly valuable for assessing the fit of individual ligands or specific binding site residues [26]. However, it's important to note that RSCC values are influenced by both model accuracy and precision—they can be affected by factors like atomic displacement parameters (B-factors) and local map quality [24].
Real-Space Z-Difference Score (RSZD)
The Real-Space Z-Difference Score (RSZD) measures the statistical significance of features in the difference electron density map (Fo - Fc), which highlights areas where the model does not adequately explain the experimental data. Unlike RSCC, RSZD focuses specifically on model inaccuracies. The calculation involves analyzing the distribution of difference density values at grid points surrounding the atoms of interest [24]:
RSZD = max(|RSZD-|, RSZD+)
RSZD- and RSZD+ represent Z-scores for negative and positive difference density, respectively, computed by comparing the observed difference density values to the expected distribution. A high RSZD value (typically >3-4) indicates statistically significant unexplained density, suggesting potential modeling errors. The key advantage of RSZD is that it specifically measures model accuracy independently from precision-related parameters like B-factors [24].
Q-score
The Q-score is a metric developed specifically for assessing atom resolvability in cryo-EM maps, though the concept can be extended to crystallographic maps [27]. It quantifies how well individual atoms are resolved by measuring the sharpness of the electron density peaks at atomic positions. The Q-score ranges from 0 to 1, where a value of 1 indicates perfect resolvability of an atom. When averaged across entire models or specific regions, Q-scores correlate strongly with map resolution, providing a local and global measure of map quality [27]. For ligand binding validation, Q-scores are particularly valuable for determining whether the experimental evidence supports the placement of specific ligand atoms.
MolProbity
MolProbity is not a single metric but rather a comprehensive structure validation system that employs multiple criteria to evaluate model quality [28]. Its key components include:
- Clashscore: The number of serious steric overlaps per 1000 atoms, identifying atoms positioned unrealistically close to each other [29] [28]
- Ramachandran analysis: Evaluation of protein backbone torsion angles against preferred conformations [28]
- Rotamer analysis: Assessment of side-chain conformations against preferred rotamer libraries [28]
- Bond length and angle geometry: Comparison of covalent geometry to high-resolution reference data [29]
MolProbity employs all-atom contact analysis, including hydrogen atoms, making it exceptionally sensitive for identifying steric problems and validation outliers [28]. For ligand binding sites, MolProbity can identify clashes between ligands and protein atoms that might indicate incorrect placement.
Comparative Analysis of Quality Metrics
Table 1: Comprehensive Comparison of Structural Validation Metrics
| Metric | Theoretical Range | Optimal Values | Primary Application | Key Strengths | Key Limitations |
|---|---|---|---|---|---|
| RSCC | 0 to 1 | >0.8 (good), >0.9 (excellent) | Local fit to density | Intuitive interpretation; Direct measure of fit to experimental data | Correlated with B-factors; Depends on both accuracy and precision |
| RSZD | 0 to ∞ | <3 (good), <4 (acceptable) | Identifying unmodeled features | Pure measure of model accuracy; Statistical framework | Less intuitive; Requires proper estimation of σ(Δρ) |
| Q-score | 0 to 1 | >0.8 (good), >0.9 (excellent) | Atom resolvability in maps | Direct measure of map quality; Correlates with resolution | More applicable to cryo-EM; Less established for crystallography |
| MolProbity | Varies by component | Clashscore: <10 (good), <20 (acceptable) | Overall model geometry | Comprehensive assessment; All-atom sensitivity | Does not directly assess fit to experimental data |
Table 2: Metric Performance Across Different Resolution Ranges
| Metric | High Resolution (<2.0 Å) | Medium Resolution (2.0-3.0 Å) | Low Resolution (>3.0 Å) | Ligand Validation |
|---|---|---|---|---|
| RSCC | Highly discriminative | Reliable but less sensitive | Affected by ambiguity | Excellent for well-ordered ligands |
| RSZD | Highly sensitive to small errors | Good for identifying major errors | Challenging due to noisy maps | Excellent for identifying incomplete modeling |
| Q-score | Near 1 for most atoms | Declines for side-chain atoms | Limited utility | Directly measures ligand atom evidence |
| MolProbity | Stringent geometry assessment | Essential for guiding modeling | Critical for avoiding over-interpretation | Identifies steric conflicts with protein |
Experimental Protocols and Methodologies
Calculating RSCC and RSZD with EDSTATS
The CCP4 program EDSTATS provides a standardized method for calculating RSCC and RSZD metrics [24]. The recommended protocol is:
Input Preparation: Gather your refined model (PDB format) and either a reflection file (MTZ) or a calculated electron density map (CCP4 format). For ligand-specific analysis, consider creating a separate PDB file containing just the binding site region.
Command Line Execution: Run EDSTATS with the following command structure:
Where MAPIN1 and MAPIN2 are the observed and calculated maps, and XYZIN is the input coordinate file [24].
Result Interpretation: Examine the output file for per-residue statistics. Focus on the ligand and binding site residues. For ligands, RSCC values below 0.8 typically indicate poor fit to density, while RSZD values above 3-4 suggest significant unmodeled features [24].
Implementing MolProbity Validation
MolProbity provides both web-based and command-line interfaces for comprehensive structure validation [28]:
Input Preparation: Prepare your coordinate file in PDB or mmCIF format. Ensure the file contains any ligands or cofactors to be validated.
Web Server Usage:
- Upload your coordinate file to http://molprobity.biochem.duke.edu
- Select "Add H atoms" with automated Asn/Gln/His flip correction enabled
- Choose "Analyze all-atom contacts and geometry" for full validation
- Download results including Clashscore, Ramachandran, and rotamer analysis [28]
Analysis of Results: Pay special attention to clashes between ligands and protein atoms, which may indicate incorrect placement. For binding site residues, check for Ramachandran and rotamer outliers that might suggest strained conformations.
Q-score Calculation for Resolvability Assessment
The Q-score calculation methodology involves [27]:
Input Preparation: Obtain your cryo-EM map (MRC format) and fitted atomic model (PDB format). For ligand binding analysis, extract a submap focused on the binding site.
Software Implementation:
- Use the MapQ plugin for UCSF Chimera, available on GitHub
- Alternatively, implement the published algorithm which compares the experimental map to a simulated ideal map computed from the atomic model
- Calculate Q-scores for individual atoms, then average for residues or ligands
Interpretation: Q-scores below 0.7 for ligand atoms suggest limited resolvability, while scores above 0.8 indicate clear density support. These values should be considered in context with the overall map resolution.
Integrated Workflow for Ligand Binding Validation
Integrated Workflow for Ligand Binding Validation
The recommended workflow begins with an initial model containing the docked ligand. All four validation metrics should be calculated in parallel, as they provide complementary information. The results must be interpreted collectively—for instance, a ligand with acceptable RSCC but poor MolProbity clashscores may be correctly positioned but have steric issues that need resolution. Based on this integrated assessment, researchers decide whether the model requires refinement or can be considered validated.
Essential Research Reagent Solutions
Table 3: Essential Software Tools for Structural Validation
| Tool Name | Primary Function | Application in Ligand Validation | Access Method |
|---|---|---|---|
| CCP4/EDSTATS | RSCC/RSZD calculation | Quantifying ligand fit to density | Command line [24] |
| MolProbity | Comprehensive validation | Identifying steric clashes and geometry issues | Web server/standalone [28] |
| Phenix | Integrated refinement and validation | Model-vs-data analysis during refinement | Command line/GUI [25] |
| UCSF Chimera | Visualization and analysis | Visual integration of validation metrics | GUI [27] |
| MapQ | Q-score calculation | Assessing ligand atom resolvability | Plugin for Chimera [27] |
Robust validation of ligand binding in electron density maps requires a multifaceted approach that combines multiple complementary metrics. RSCC provides a direct measure of how well the model explains the experimental density, RSZD identifies areas of potential omission or error, Q-score assesses the intrinsic resolvability of atoms in the map, and MolProbity ensures proper stereochemistry and absence of steric conflicts. By applying these metrics systematically through integrated workflows, researchers can substantially increase the reliability of their structural models, particularly in the critical context of ligand binding sites that inform drug discovery efforts. As structural biology continues to push toward more challenging targets, including membrane proteins and large complexes, these validation metrics will play an increasingly important role in ensuring the accuracy and biological relevance of structural models.
From Manual Fitting to AI: Modern Methods for Ligand Identification and Modeling
Validating ligand binding in electron density maps represents a critical step in structural biology and structure-based drug design. This process bridges the gap between experimental data and atomic models, ensuring the accuracy of structural interpretations that inform therapeutic development. Traditional computational methods for ligand fitting have long relied on search-and-score algorithms combined with crystallographic refinement, while emerging iterative approaches leverage advanced statistical sampling and deep learning to address complex challenges such as low-occupancy binding and protein flexibility. This guide provides an objective comparison of current methodologies, examining their performance characteristics, experimental protocols, and applicability across different research scenarios. As structural biology increasingly focuses on capturing dynamic processes and transient complexes, the evolution of these tools enables researchers to extract more biological insight from electron density data than ever before.
The landscape of ligand fitting methods spans multiple computational strategies, each with distinct theoretical foundations and workflow integration characteristics. The table below categorizes and compares the fundamental approaches available to researchers.
Table 1: Classification of Ligand Fitting Methodologies
| Method Category | Representative Tools | Core Approach | Typical Input Requirements |
|---|---|---|---|
| Traditional Density Analysis | Ringer, PanDDA | Electron density sampling and statistical comparison | Paired crystallographic datasets (perturbed and native) |
| Automated Model Building | qFit, Phenix-MD, FLEXR | Multi-conformer model building | Single crystal structure with electron density map |
| Iterative Denoising | METEOR | Total variation denoising with negentropy optimization | Difference map with optional reference structure |
| Deep Learning Docking | DiffDock, EquiBind, CWFBind | Geometric deep learning and diffusion models | Protein structure and ligand chemical representation |
| Cross-Map Comparison | FLEXR-MSA | Multiple sequence alignment coupled with density sampling | Multiple structures of sequence-diverse proteins |
The logical relationship between these methodological categories reveals an evolutionary trajectory in the field, from foundational density analysis to integrated learning systems that account for structural flexibility and diversity.
Figure 1: Methodological evolution in ligand fitting, showing the progression from fundamental density analysis to integrated validation approaches.
Performance Comparison and Experimental Data
Quantitative Performance Metrics
Rigorous benchmarking against standardized datasets provides critical insights into the operational performance of ligand fitting methods. The following table summarizes published performance data across multiple tool categories.
Table 2: Quantitative Performance Comparison of Ligand Fitting Approaches
| Tool | Method Type | Success Rate | Resolution Range | Key Strength | Computational Demand |
|---|---|---|---|---|---|
| EMERALD-ID | Cryo-EM ligand identification | 44% exact identification, 66% closely related [21] | Moderate resolution cryo-EM (~3-4Å) | Combines physical forcefield with density agreement | Medium-High |
| LigPCDS Deep Learning | 3D point cloud segmentation | 67.2% top-10 accuracy, 93.6% top-10 recovery [6] | X-ray crystallography | Chemical structure recovery from density | High (GPU required) |
| DiffDock | Deep learning docking | State-of-the-art on PDBBind benchmark [30] | Not resolution-dependent | Blind docking capability | Medium |
| FLEXR-MSA | Electron density comparison | Qualitative improvement in detecting alternative conformations [31] | Medium-high resolution X-ray (<2.5Å) | Identifies hidden differences in sequence-diverse proteins | Medium |
| METEOR | Difference map denoising | Enables detection of previously unresolvable low-occupancy states [32] | Time-resolved crystallography | Reveals low-occupancy populations (<30%) | Low-Medium |
Specialized Capabilities Comparison
Different methodological approaches exhibit distinct strengths depending on the biological context and data quality. The specialized capabilities of each approach highlight their complementary nature in structural biology workflows.
Table 3: Specialized Capabilities Across Method Types
| Method Category | Low-Occupancy Ligand Detection | Handling Protein Flexibility | Sequence-Diverse Comparison | Cryo-EM Compatibility |
|---|---|---|---|---|
| Traditional Density Analysis | Limited (<0.5 occupancy) | Limited to rigid body | Not supported | Limited |
| Automated Model Building | Moderate (0.3-0.5 occupancy) | Side-chain flexibility only | Not supported | Limited |
| Iterative Denoising | Excellent (<0.3 occupancy) [32] | Limited | Not supported | Potential application |
| Deep Learning Docking | Not applicable | Excellent (full flexible docking) [30] | Indirectly through training data | Emerging |
| Cross-Map Comparison | Moderate | Captures conformational landscapes | Excellent (primary purpose) [31] | Not supported |
Experimental Protocols and Workflows
Difference Map Analysis for Low-Occupancy Ligands
The METEOR workflow exemplifies modern approaches to detecting low-occupancy species through optimized difference map calculation [32] [33]. The protocol begins with collection of paired crystallographic datasets—native and derivative—ensuring isomorphism with a maximum recommended RMSD of 0.3 Å for unit cell parameters. Researchers calculate difference structure factors (ΔF = F' - F) and apply negentropy-based weighting to maximize non-Gaussian signal in the resulting map. The core innovation involves total variation denoising, implemented with a regularization parameter optimized by maximizing map negentropy, defined as J(pρ) = H(pgauss) - H(pρ), where H represents differential entropy and pρ is the distribution of map voxel values [32]. This denoising step preserves sharp features while suppressing noise, typically improving signal-to-noise ratio by 30-50% for weak signals. The protocol concludes with phase extrapolation to calculate coefficients for the pure perturbed state, enabling modeling of low-occupancy ligands previously obscured by noise.
Cross-Protein Electron Density Comparison
FLEXR-MSA addresses the challenge of comparing electron densities across proteins with divergent sequences [31]. The methodology begins with multiple sequence alignment of target proteins using standard tools such as Clustal Omega or MAFFT. The aligned sequences guide the mapping of equivalent residues despite different numbering schemes. For each structure, electron density sampling occurs around each residue using five rotameric scans at 10° intervals (0°-360°). The tool calculates electron density values using either 2Fo-Fc or Fo-Fc maps, with sampling focused on the Cβ-Cγ bond for most amino acids. Density comparisons utilize Pearson correlation coefficients between equivalent positions in the sequence alignment, with statistical significance assessed through permutation testing. The output includes visualizations that chart alternative conformations across the protein surface, revealing global changes in conformational landscapes between isoforms or homologs. This approach has proven particularly valuable for studying proteins like HSP90 isoforms, where subtle conformational differences have implications for selective inhibitor design [31].
Cryo-EM Ligand Identification Workflow
EMERALD-ID addresses the growing use of cryo-EM in drug discovery by providing a robust protocol for ligand identification [21]. The process initiates with preparation of a candidate ligand library in SMILES format, which is converted to 3D conformers using tools like Open Babel. The cryo-EM density map and receptor model serve as input, with local resolution estimates informing the docking strategy. The method employs the RosettaGenFF small molecule force field for conformational sampling, generating multiple pose hypotheses for each candidate ligand. Each pose is evaluated using a composite scoring function that combines density correlation (CC) with estimated binding affinity (ΔG). To ensure fair comparison between different-sized ligands, the method applies a linear regression model that normalizes expected density correlation based on ligand size and local map resolution. The final output ranks candidate ligands by a composite score, with the protocol achieving 44% exact identification and 66% close analog identification on benchmarked cryo-EM structures [21].
Successful implementation of ligand validation workflows requires careful selection of computational tools and data resources. The following table catalogs essential components of the modern structural biologist's toolkit for ligand fitting applications.
Table 4: Essential Research Resources for Ligand Fitting and Validation
| Resource Name | Type | Primary Function | Application Context |
|---|---|---|---|
| PDBbind | Curated dataset | Provides protein-ligand structures with binding affinity data | Method training and benchmarking [7] |
| HiQBind-WF | Data processing workflow | Creates high-quality protein-ligand datasets by fixing structural artifacts | Data preparation for analysis [7] |
| LigPCDS | Labeled dataset | 3D point cloud representations of ligands from crystallographic data | Deep learning model training [6] |
| CCD (Chemical Component Dictionary) | Reference database | Standardized chemical descriptions of small molecules | Ligand identity verification [7] |
| RosettaGenFF | Force field | Physical energy function for small molecule conformations | Ligand docking and scoring [21] |
| ESM-2 | Pre-trained model | Protein sequence representation learning | Feature extraction for docking [34] |
The methodological landscape for ligand validation in electron density maps has evolved from rigid docking approaches to sophisticated systems that account for protein flexibility, low-occupancy states, and evolutionary diversity. Traditional methods including difference map analysis and automated model building remain essential for standard crystallographic applications, while iterative denoising approaches like METEOR extend the detectable range to previously inaccessible low-occupancy ligands. Emerging deep learning methods show remarkable performance in flexible docking scenarios but face generalization challenges beyond their training data. Cross-comparison tools such as FLEXR-MSA offer unique capabilities for extracting functional insights from structural variations across protein families. The optimal choice of methodology depends critically on data type, resolution, biological question, and available computational resources. As structural biology continues to push toward more dynamic and complex systems, the integration of these complementary approaches will be essential for fully leveraging structural data in drug discovery applications.
The accurate identification and validation of small-molecule ligands in electron density maps represents a critical challenge in structural biology with profound implications for structure-guided drug design. As noted in recent literature, "the correct identification of ligands is often a vital part of structure-guided drug design" [35]. However, this process remains notoriously susceptible to human bias, as crystallographers may unconsciously fit desired or expected ligands into electron density, sometimes leading to questionable assignments [35] [36]. This challenge is compounded by the fact that ligands are typically modeled manually by chemists or biologists analyzing 3D density maps—a process that is both time-consuming and prone to error, particularly for structures with low resolution or local disorder [35].
Within this context, feature-engineered machine learning tools like CheckMyBlob have emerged as valuable resources to mitigate cognitive bias and standardize ligand identification. Unlike later deep learning approaches that process raw density maps through end-to-end learning, feature-engineered methods rely on carefully designed numerical descriptors to characterize electron density "blobs" [36] [37]. These tools exemplify a significant transition in structural biology informatics, where machine learning supplements human expertise to improve objective interpretation of experimental data. This article examines CheckMyBlob's methodology, performance, and position within the evolving ecosystem of computational tools for ligand validation.
CheckMyBlob: Mechanism and Methodology
CheckMyBlob employs a sophisticated machine learning pipeline that transforms electron density features into ligand predictions. The system operates through a multi-stage process that begins with electron density analysis and culminates in machine learning classification [37].
Electron Density Pre-processing and Blob Detection
The initial phase involves identifying unmodeled fragments of electron density through automated blob detection:
- Input Processing: The system accepts structure files (PDB or mmCIF) and experimental data files (MTZ) containing structure factors [38] [37].
- Blob Identification: CheckMyBlob analyzes all positive electron density peaks within the Fo-Fc difference map, focusing on regions exceeding the 2.8σ isosurface threshold computed with a 0.2 Å grid [35] [37].
- Skeletonization and Merging: To address the challenge of fragmented density, the system detects local maxima and skeletonizes the electron density within each blob's isosurface. Adjacent blobs are combined if the distance between their local maxima or skeleton nodes is less than 2.15 Å [37].
- Polymer Overlap Removal: Any electron density fragments that overlap with the isosurface of already-modeled biopolymer atoms are systematically excised from the blob to focus exclusively on unmodeled regions [37].
Feature Engineering and Machine Learning
Following blob detection, CheckMyBlob employs carefully engineered feature extraction:
- Feature Extraction: Each electron density blob is characterized by 382 numerical descriptors, including Zernike moment invariants, bounding box volume, and features extracted at different contour levels of electron density [36].
- Feature Selection: Through recursive feature selection, this extensive descriptor set is reduced to the 60 most informative features for model training [36].
- Classifier Training: These features train multiple machine learning models, including k-nearest-neighbors, random forests, and gradient boosting machines, with a stacked combination of these methods delivering optimal performance [36]. The model is trained on nearly 700,000 quality-filtered ligand instances from the PDB, clustered into 219 ligand groups based on structural characteristics [35] [37].
The following diagram illustrates CheckMyBlob's workflow from data input to ligand prediction:
Performance Comparison: CheckMyBlob Versus Alternative Approaches
CheckMyBlob's performance must be evaluated against both traditional methods and emerging deep learning alternatives. The following table summarizes key performance metrics across different approaches:
Table 1: Performance Comparison of Ligand Identification Methods
| Method | Approach Type | Top-1 Accuracy | Top-5 Accuracy | Top-10 Accuracy | Applicability |
|---|---|---|---|---|---|
| CheckMyBlob | Feature-engineered ML | 59-71% [36] [37] | 85.2% [36] | 91.3-95% [36] [37] | X-ray crystallography only [35] |
| Deep Learning PointCloud | End-to-end deep learning | Similar to existing ML methods [35] | Not specified | Improved over feature-engineered methods [35] | X-ray & cryo-EM [35] |
| Traditional Iterative Fitting | Iterative fitting of candidate ligands | Lower than ML approaches [35] | Not specified | Not specified | Limited to known candidate ligands [35] |
The performance data reveals CheckMyBlob's particular strength in suggesting plausible ligands, with the correct identification appearing in the top-10 suggestions over 90% of the time [36] [37]. This capability makes it particularly valuable for practical structural biology workflows, where researchers can efficiently evaluate a limited number of candidates rather than searching through entire chemical libraries.
Comparative Limitations and Strengths
Each methodological approach exhibits distinct advantages and limitations:
- CheckMyBlob's Domain Specialization: As a feature-engineered system, CheckMyBlob excels within its domain of X-ray crystallography but cannot be directly applied to cryo-EM density maps without retraining [35]. The developers note that "all the existing ligand prediction approaches are applicable only to X-ray structures" [35].
- Deep Learning Flexibility: Emerging deep learning approaches using 3D point cloud representations demonstrate "similar accuracy to existing machine learning methods for X-ray crystallography while also being applicable to cryoEM density maps" [35]. This represents a significant advantage for multi-modal structural biology.
- Interpretability Trade-offs: Feature-engineered systems like CheckMyBlob offer greater interpretability, as the 60 selected features provide biochemical insight into which electron density characteristics contribute to classification decisions. Deep learning approaches typically function as "black boxes" with limited insight into their decision-making processes [39].
Experimental Protocols and Validation Methodologies
CheckMyBlob Training and Validation Protocol
The experimental methodology underpinning CheckMyBlob's development followed a rigorous validation pipeline:
- Data Curation: 957,855 ligand blobs were initially extracted from PDB structures downloaded as of January 2020. Stringent quality filters were applied, eliminating ligands with resolution > 4.0 Å, RSCC < 0.6, real space Zobs (RSZO) < 1.0, real space Zdiff (RSZD) ≥ 6.0, R factor > 0.3, or occupancy < 0.3 [35].
- Ligand Clustering: Surviving ligands were clustered into groups based on the number of atoms, number of rings, connectivity, chirality, and atomic numbers using RDKit and SMILES/InChI descriptors from the PDB [35].
- Model Validation: Cross-validation experiments assessed performance on 696,887 ligand instances, with additional testing on a separate set of 17,150 ligands gathered after initial training [37].
Performance Assessment Protocol
To ensure fair comparison across methods, standardized assessment metrics must be employed:
- Accuracy Metrics: Top-1, top-5, and top-10 accuracy measurements evaluate how frequently the correct ligand appears within the top suggestions [36] [37].
- Certainty Calibration: CheckMyBlob provides probability estimates with its predictions, and "predictions with higher certainty are in fact very probable, whereas predictions with lower certainty values have a higher chance of being incorrect" [37].
- Cross-Technique Application: When evaluating the deep learning approach, "experiments assessing model performance on 208,896 X-ray crystallography ligands show that the proposed approach has similar accuracy to existing methods while improving in terms of top-10 accuracy" [35].
Structural biologists have access to an expanding toolkit of computational resources for ligand validation. The following table details key solutions available to researchers:
Table 2: Essential Research Reagent Solutions for Ligand Validation
| Tool/Resource | Type | Primary Function | Access Method |
|---|---|---|---|
| CheckMyBlob Web Server | Feature-engineered ML | Identifies and validates ligands in electron density | Web server [38] |
| ChimeraX | Molecular visualization | Interactive model building and visualization | Downloadable software [35] |
| Deep Learning Ligand Classification | Point cloud deep learning | Ligand identification in X-ray and cryo-EM maps | GitHub repository [35] |
| MIC (Metric Ion Classification) | Deep metric learning | Classifies ions and waters in crystal and cryo-EM structures | Open-source package [39] |
| RCSB PDB Ligand Quality Indicators | Quality assessment | Assesses ligand structure quality via composite scores | RCSB.org web portal [40] |
These tools collectively address complementary aspects of the ligand modeling pipeline, from initial identification to final quality assessment. CheckMyBlob serves specifically the ligand identification and validation phase, while tools like MIC specialize in ion/water classification [39], and the RCSB PDB ligand quality indicators provide post-deposition validation [40].
Feature-engineered machine learning tools like CheckMyBlob represent a crucial evolutionary stage in computational structural biology, effectively bridging fully manual ligand modeling and emerging end-to-end deep learning approaches. While deep learning methods demonstrate promising expansion into cryo-EM applications and potentially reduced feature engineering overhead, CheckMyBlob's robust performance within X-ray crystallography—particularly its >90% top-10 accuracy—ensures its continued utility in practical structural biology workflows [36] [37].
The comparative analysis presented here suggests that rather than being rendered obsolete by deep learning, feature-engineered tools like CheckMyBlob will likely maintain specialized roles in contexts where interpretability, speed, and reliability for common ligands are prioritized. As the field advances, the integration of both feature-engineered and deep learning approaches within unified structural biology workbenches promises to further enhance objective ligand validation, ultimately strengthening the foundation for structure-based drug design and functional annotation of biomolecular structures.
In structure-guided drug design, accurately identifying the small-molecule ligands bound to proteins is paramount for understanding function and developing new therapeutics. This process traditionally relies on the manual interpretation of experimental density maps obtained from X-ray crystallography or cryogenic electron microscopy (cryo-EM). However, this interpretation is notoriously challenging and susceptible to human bias, sometimes leading to the incorrect modeling of fictitious compounds [35]. The need for automated, accurate, and objective solutions has never been greater.
While automatic ligand identification methods have existed for X-ray crystallography, they have been largely based on iterative fitting procedures or feature-engineered machine learning, requiring significant manual input and computational time. Furthermore, these existing methods have not been applicable to the growing number of structures determined by cryo-EM [35]. A paradigm shift is underway with the advent of deep learning approaches that treat continuous 3D density maps as discrete 3D point clouds. This end-to-end deep learning framework offers a unified solution for ligand identification across both experimental modalities, demonstrating performance on par with established methods for X-ray data while extending critical capabilities to the cryo-EM field [35] [6].
The Point Cloud Paradigm: From Density Maps to Structural Insights
Fundamental Concepts and Workflow
A 3D point cloud is a dataset representing points in three-dimensional space, typically defined by (x, y, z) coordinates and often augmented with additional features like electron density value or chemical properties. This representation is particularly well-suited for processing by geometric deep learning architectures. The transformation of an experimental density map into a labeled ligand structure involves a multi-step workflow, as illustrated below.
The initial and crucial step involves blob extraction, where the ligand's density is isolated from the map. For X-ray data, this is typically done from the difference electron density map (Fo-Fc map), thresholded at a specific sigma level (e.g., 2.8σ) [35] [6]. The resulting density cluster, or "blob," is then sampled into a 3D point cloud. This point cloud can be processed directly by neural networks to identify the ligand or to perform a semantic segmentation task, where each point in the cloud is assigned a chemical label, effectively building the ligand's atomic structure [6].
Key Research Reagent Solutions
The following table details essential computational tools and datasets that form the foundational "reagent solutions" for this field.
Table 1: Key Research Reagents and Computational Tools for Point Cloud-Based Ligand Identification
| Item Name | Type | Primary Function | Source/Availability |
|---|---|---|---|
| LigPCDS | Dataset | A large-scale, chemically labeled dataset of 3D point clouds of protein ligands derived from X-ray crystallography for training and validation [6]. | Zenodo / Public Datasets |
| CheckMyBlob | Algorithm & Server | A benchmark machine learning method for ligand identification in X-ray maps; used for performance comparison [35]. | Web Server |
| ChimeraX Bundle | Software Plugin | An accompanying tool for the point cloud model, facilitating user-friendly application within a popular structural biology software suite [35]. | GitHub |
| Gemmi | Software Library | A library used for reading crystallographic data and creating 3D point cloud representations of ligands from density maps [6]. | Open Source |
| RDKit | Software Library | Used for cheminformatics tasks, such as clustering ligands into chemically meaningful groups based on descriptors like SMILES and InChI [35]. | Open Source |
Performance Comparison: Point Cloud Models vs. Established Methods
Quantitative Benchmarking on X-ray and Cryo-EM Data
The performance of the deep learning point cloud approach has been rigorously tested against established methods like CheckMyBlob. The following table summarizes key quantitative results from these evaluations.
Table 2: Performance Comparison of Ligand Identification Methods
| Method | Principle | Application Scope | Reported Performance (X-ray) | Reported Performance (cryo-EM) |
|---|---|---|---|---|
| Point Cloud Deep Learning [35] | End-to-end deep learning on 3D point clouds. | X-ray & cryo-EM density maps. | Similar accuracy to CheckMyBlob, with improved top-10 accuracy on 208,896 ligands. | Successful application to 34,671 cryoEM ligands, demonstrating transferability from X-ray training data. |
| CheckMyBlob [35] | Feature-engineered machine learning. | X-ray crystallography only. | Baseline accuracy for comparison. | Not applicable. |
| Iterative Fitting [35] | Trial fitting of candidate ligands. | X-ray crystallography only. | Slower, as requires fitting all candidate ligands. | Not applicable. |
| Semantic Segmentation on Point Clouds [6] | Pointwise labeling of 3D clouds to build chemical structures. | X-ray crystallography (primarily for structure building). | mIoU: 49.7% to 77.4%; F1-score: 62.4% to 87.0% on a stratified dataset (n=78,902). | Not reported. |
The point cloud method achieves comparable accuracy to the state-of-the-art CheckMyBlob for X-ray data but with a significant advantage: it is not limited to a single experimental technique. By training on electron density maps from X-ray crystallography, the model can be directly applied to Coulomb potential maps from cryo-EM, a crucial breakthrough for the field [35]. Experiments on a set of 34,671 cryo-EM ligands confirmed this capability, though the study also highlighted ongoing challenges with standardizing cryo-EM map processing [35].
Architectural Advantages and Limitations
The superior performance of point cloud models stems from several key architectural advantages. They inherently possess translation and rotation invariance, meaning their predictions are not affected by how the ligand is positioned or oriented within the map, a critical feature for robust real-world application [35] [41]. Furthermore, the end-to-end learning paradigm eliminates the need for manual feature engineering, allowing the model to learn the most relevant features directly from the data [35].
Despite these strengths, current limitations exist. The performance on cryo-EM data is contingent upon the quality and standardization of the map processing, which remains a challenge [35]. Moreover, like other data-driven methods, the model's accuracy is dependent on the breadth and quality of its training data, and identifying truly novel ligands (unknown unknowns) remains a complex task [6].
Experimental Protocols and Methodologies
Data Curation and Point Cloud Generation
A critical factor in the success of deep learning models is the creation of high-quality, large-scale datasets. For ligand identification, this involves several standardized steps:
- Data Sourcing and Filtering: Structures are downloaded from the Protein Data Bank (PDB) and filtered based on quality metrics. Common filters include removing structures with resolution worse than 4.0 Å, low real-space correlation coefficient (RSCC), or low ligand occupancy [35] [6].
- Map Calculation and Blob Extraction: For each structure, a difference electron density map (Fo-Fc) is calculated. Ligand "blobs" are extracted by isolating positive density peaks within an isosurface (e.g., 2.8σ) centered on the ligand's atomic coordinates [35] [6].
- Point Cloud Sampling: The density blob is converted into a 3D point cloud. Using tools like Gemmi, the blob is sampled onto a 3D grid with a defined spacing (e.g., 0.2 Å). Each point contains its 3D coordinates and a feature, typically the electron density value at that location [6].
- Chemical Labeling: For semantic segmentation tasks, the point cloud is labeled pointwise. This is often done using atomic sphere modeling, where points within the van der Waals radius of an atom are assigned a label based on a predefined chemical vocabulary (e.g., atom type, membership in an aromatic ring) [6].
Model Architecture and Training
The core innovation lies in the application of neural network architectures designed for 3D point clouds.
As shown in the diagram, the model core often employs rotation-invariant pointnets (e.g., RiConv) to process the point cloud. These layers ensure the model's predictions are not affected by the initial orientation of the ligand in the map, a fundamental requirement for robustness [35]. The network learns to aggregate spatial features from the points and their neighborhoods. The final layers, or "heads," are task-specific: a classification head outputs the identity of the ligand, while a segmentation head assigns a chemical label to each point, effectively building the ligand's structure atom-by-atom [6]. Models are typically trained on hundreds of thousands of ligand instances using standard deep learning optimizers, with a portion of the data held out for validation [35] [6].
The treatment of density maps as 3D point clouds for end-to-end deep learning represents a significant breakthrough in computational structural biology. This paradigm offers a powerful, unified approach for ligand identification that bridges the gap between X-ray crystallography and cryo-EM. By matching the performance of established methods for X-ray data while extending their capabilities to cryo-EM, this technology directly addresses the core thesis of validating ligand binding in electron density maps, reducing reliance on error-prone manual interpretation.
The future of this field is bright and points toward greater integration. Promising directions include the incorporation of quantum chemical properties like electron density directly into binding affinity predictions, offering a more fundamental understanding of interaction patterns [42]. Furthermore, the synergy between protein structure prediction tools like AlphaFold and experimental density data is poised to enhance the accuracy and scope of model building, particularly for challenging regions [43] [44]. As these deep learning models mature and become more accessible through user-friendly software integrations, they are set to become an indispensable tool in the arsenal of researchers and drug developers, accelerating the pace of structural biology and drug discovery.
The integration of artificial intelligence-based structure prediction tools into visualization software has revolutionized structural biology, particularly in the validation of ligand binding in electron density maps. UCSF ChimeraX serves as a powerful hub for these tools, allowing researchers to seamlessly transition from prediction to analysis within a single environment. This capability is crucial for drug development professionals who require robust methods to verify ligand-protein interactions and avoid the cognitive biases that can lead to modeling fictitious compounds [45]. The extensible nature of ChimeraX through its "Toolshed" mechanism enables researchers to install specialized bundles that enhance its core functionality, creating customized workflows for specific research needs such as ligand validation [46].
The emergence of tools like AlphaFold, Boltz, and specialized deep learning bundles has transformed how researchers approach structure-guided drug design. These tools provide complementary capabilities—from predicting large protein complexes to estimating small molecule binding affinities—that together form a comprehensive toolkit for experimental validation. This guide objectively compares the performance characteristics, integration pathways, and practical applications of these prediction technologies within ChimeraX workflows, with special emphasis on their utility for ligand binding validation in cryo-EM and X-ray crystallography research.
Comparison of Major Prediction Tools in ChimeraX
ChimeraX integrates multiple structure prediction engines that cater to different research scenarios and computational constraints. The table below summarizes the core tools available for structure prediction within the ChimeraX ecosystem.
Table 1: Core Structure Prediction Tools Available in ChimeraX
| Tool | Developer | Prediction Capabilities | Ligand Support | License |
|---|---|---|---|---|
| AlphaFold | Google DeepMind | Proteins, multimers | Limited (few dozen common ligands via server) | Non-commercial [47] |
| Boltz | MIT/Recursion Pharmaceuticals | Proteins, nucleic acids, modified residues, ligands, ions, solvent | Extensive (arbitrary ligands via CCD codes/SMILES) | Permissive MIT license [48] |
| ColabFold | Academic consortium | Optimized AlphaFold via Google Colab | Similar to AlphaFold | Non-commercial [49] |
| Ligand Recognizer | Karolczak et al. | Ligand identification in density maps | Specialized for ligand identification | Available on GitHub [45] |
Each tool occupies a distinct niche in the research workflow. AlphaFold excels at predicting protein structures and complexes but offers limited ligand support. Boltz provides comprehensive ligand handling capabilities but with more restrictive size limitations. The Ligand Recognizer bundle specializes specifically in identifying ligands within experimental density maps using deep learning, treating density maps as 3D point clouds for classification [45].
Performance Metrics and System Requirements
The practical utility of prediction tools depends heavily on their computational demands and performance characteristics. The following table compares key performance metrics across different hardware configurations.
Table 2: Performance Comparison of Boltz Predictions Across Hardware Platforms (Times in Minutes) [48]
| Assembly (PDB Code) | Tokens | Mac M1 16GB | Mac M1 Max 32GB | Linux Nvidia 4090 | Windows Nvidia 3070 | Intel CPU Only |
|---|---|---|---|---|---|---|
| Small (8rf4) | 129 | 1.2 | 0.8 | 0.3 | 0.8 | 1.1-2.0 |
| Medium (1hho) | 382 | Fail | 1.5 | 0.5 | 2.0 | 8.0-11 |
| Large (9b3h) | 911 | Fail | 10 | 1.1 | 28 | 60-80 |
| Very Large (9gh4) | 1467 | - | 32 | 2.1 | - | 188 |
Tokens represent the number of standard polymer residues plus ligand atoms [48]. Several key observations emerge from this data: GPUs provide significant speed advantages, with high-end Nvidia GPUs performing predictions 10-30x faster than CPU-only configurations [48]. Memory limitations substantially constrain prediction capabilities on personal computers, particularly for larger complexes. The performance characteristics directly influence which tool is appropriate for a given research scenario.
AlphaFold server can handle much larger complexes (up to 5000 residues) but imposes different limitations—it only allows few dozen common ligands and prohibits commercial use [47]. For ligand-focused research, Boltz provides more flexible small-molecule support but with stricter size limitations, creating a practical trade-off that researchers must navigate based on their specific complex size and ligand requirements.
Experimental Protocols and Workflow Integration
Protocol for Ligand Binding Validation Using Multiple Tools
Validating ligand binding requires integrating multiple prediction and analysis tools in a systematic workflow. The following protocol outlines a comprehensive approach:
Initial Structure Prediction: Based on complex size and ligand type, select the appropriate prediction tool. For proteins with uncommon ligands, use Boltz with chemical component dictionary (CCD) codes or SMILES strings to specify ligands [48]. For larger protein-only complexes, use AlphaFold via the ChimeraX ColabFold interface [49].
Confidence Assessment: Evaluate prediction quality using built-in metrics. For AlphaFold, examine pLDDT scores and predicted aligned error (PAE) plots, accessible through the AlphaFold Error Plot tool [49]. For Boltz, analyze the similar coloring scheme and PAE plots to identify low-confidence regions [48].
Experimental Map Fitting: Use ChimeraX fitting tools to align predicted structures with experimental cryo-EM or X-ray density maps. The Volume Viewer tool provides sophisticated fitting capabilities, and extensions like DomainFit can automate domain-level fitting of AI-predicted structures into maps [46].
Ligand Identification Verification: Employ the Ligand Recognizer bundle to automatically identify ligands within density maps using its deep learning approach that treats density maps as 3D point clouds [45]. This provides an unbiased validation method complementary to manual inspection.
Interface Analysis: Utilize ChimeraX's built-in commands and extensions like XMAS for crosslinking data to analyze binding interfaces and validate interaction predictions against experimental evidence [46].
This multi-tool approach leverages the respective strengths of each prediction engine while providing cross-validation through experimental data.
Workflow Visualization
The integration of these tools into a coherent research workflow can be visualized as follows:
Figure 1: Ligand Binding Validation Workflow
This workflow demonstrates how the different prediction tools integrate with experimental data and validation steps to produce rigorously verified ligand binding models. The pathway branches based on complex size and ligand characteristics, then reconverges for structural validation and analysis.
Successful integration of prediction tools requires both software components and practical knowledge of their capabilities and limitations. The table below details key "research reagents" for building effective prediction workflows.
Table 3: Essential Research Reagent Solutions for ChimeraX Prediction Workflows
| Tool/Resource | Function | Access Method | Key Considerations |
|---|---|---|---|
| ChimeraX Toolshed | Extension repository | Tools → More Tools → Toolshed | One-click installation of specialized bundles [46] |
| Boltz Bundle | Structure & affinity prediction | Tools → Structure Prediction → Boltz | Requires 1GB disk space + 3.3GB weights [48] |
| AlphaFold Tool | Database fetch & prediction | Tools → Structure Prediction → AlphaFold | Automatic pLDDT coloring & PAE plots [49] |
| Ligand Recognizer | Deep learning ligand ID | GitHub installation | Treats density as 3D point clouds [45] |
| DeepSSETracer | Secondary structure detection | Toolshed installation | Detects helices & β-sheets in medium-res maps [50] |
| ISOLDE | Interactive refinement | Toolshed installation | Real-time model building with Cryo-ET support [46] |
| Custom Presets | Visualization consistency | Presets menu or .cxc scripts | Standardize display styles for publications [51] |
These tools represent the essential reagents for modern computational structural biology. The ChimeraX Toolshed functions as a centralized distribution point for many extensions, though some specialized tools like Ligand Recognizer require manual installation from GitHub [45]. Custom presets and scriptable workflows ensure reproducibility—a critical concern for publication-quality research.
Beyond the software components, successful prediction workflows require thoughtful experimental design. Researchers should consider prediction time constraints (especially for larger complexes on limited hardware), the trade-offs between local execution (Boltz) and server-based prediction (AlphaFold server), and the importance of confidence metrics for interpreting results. The active development community around ChimeraX means tools are frequently updated, making regular consultation of documentation essential for optimal performance.
The integration of prediction tools into ChimeraX represents a paradigm shift in structural biology, particularly for ligand binding validation. By understanding the performance characteristics, limitations, and complementary strengths of AlphaFold, Boltz, and specialized validation tools, researchers can design efficient workflows that leverage computational predictions to enhance experimental interpretation. The quantitative performance data presented here provides realistic expectations for prediction times across different hardware configurations, enabling informed tool selection based on project requirements and available resources.
The most successful implementations will strategically combine multiple tools—using AlphaFold for large complex frameworks, Boltz for detailed ligand interactions, and specialized bundles like Ligand Recognizer for unbiased validation in experimental maps. This multi-tool approach, coupled with ChimeraX's seamless integration of visualization and analysis, creates a powerful ecosystem for advancing drug development and fundamental structural biology research. As these tools continue to evolve, their increasing accessibility and performance will further democratize high-quality structure prediction, making sophisticated validation workflows available to broader scientific communities.
Navigating Ambiguity: Strategies for Troubleshooting and Optimizing Ligand Models
Accurately identifying and modeling small-molecule ligands in macromolecular structures is a cornerstone of structural biology and structure-based drug design. However, correctly interpreting whether electron density found in a binding site truly represents the soaked or co-crystallized ligand—or merely water, buffer molecules, or noise—is often far from trivial [52]. This challenge intensifies significantly with low-resolution maps, where diminished detail and increased ambiguity can lead to misinterpretation [53] [54]. The proliferation of cryo-electron microscopy (cryo-EM) structures, the majority of which reside at resolutions worse than 10Å, has further amplified the need for robust validation strategies [54]. This guide objectively compares the performance of current computational methods designed to resolve weak density and validate ligand binding, providing scientists with data-driven insights for selecting the right tool for their research.
Performance Comparison of Ligand Modeling Tools
The table below summarizes the quantitative performance metrics of leading tools for ligand modeling in low-resolution density maps.
Table 1: Performance Comparison of Ligand Modeling and Identification Tools
| Tool Name | Methodology | Primary Application | Benchmark Performance | Key Strengths |
|---|---|---|---|---|
| EMERALD [55] | Physical force field (RosettaGenFF) & genetic algorithm with density guidance | Ligand conformation determination in Cryo-EM | 57% replication of deposited models (<1Å RMSD) on 1053 ligands; Performance drops with flexibility & worsening resolution [55] | Integrates full receptor side-chain flexibility; Provides confidence via trajectory convergence |
| EMERALD-ID [21] | Docks candidate libraries & ranks identities using energy and density | Ligand identity determination in Cryo-EM | 44% exact identification; 66% identification of closely related ligands [21] | Discerns plausible misidentifications and omissions in deposited structures |
| CheckMyBlob / Machine Learning (PointNet) [35] | Feature-engineered or deep learning (3D point cloud) on density fragments | Ligand identification in X-ray and Cryo-EM | Similar accuracy to existing methods for X-ray; Improved top-10 accuracy; Applicable to Cryo-EM [35] | Fast analysis; End-to-end deep learning removes need for manual feature engineering |
| Mogul/Geometric Validation [52] | Validates bond lengths/angles against Cambridge Structural Database (CSD) | Ligand geometry quality assessment | Flags ~35% of ligand-binding site pairs as needing attention [53] | Independent, empirical check on ligand stereochemical plausibility |
Experimental Protocols for Method Evaluation
To ensure fair and reproducible comparisons between tools, standardized experimental protocols are essential. The following methodologies are adapted from those used in benchmarking studies.
Protocol for Benchmarking Ligand Conformation Determination (e.g., EMERALD)
1. Dataset Curation:
- Source a set of ligand-bound cryo-EM or X-ray structures from public repositories like the PDB and EMDB.
- Apply quality filters: Include ligands with ≤25 rotatable bonds and maps with a nominal resolution of ≤6 Å. Exclude peptides and ions with coordination bonds [55].
- For cryo-EM, generate difference maps (e.g.,
phenix.real_space_diff_map) by subtracting a ligand-free model from the experimental map to isolate ligand density [35].
2. Computational Docking:
- Run the docking tool (e.g., EMERALD) on the curated dataset. Execute multiple independent trajectories (e.g., three runs) for each ligand to assess convergence [55].
- For each experiment, output a pool of candidate ligand poses with associated scores (e.g., energy, density correlation).
3. Analysis and Success Metrics:
- Calculate the Root-Mean-Square Deviation (RMSD) of non-hydrogen ligand atoms between the top-ranked predicted pose and the deposited reference structure.
- Success Criterion: RMSD < 1.0 Å [55].
- Calculate the density correlation (RSCC) and number of hydrogen bonds for the predicted pose.
- Assess confidence by measuring the convergence of top-ranked models across independent trajectories.
Protocol for Benchmarking Ligand Identity Determination (e.g., EMERALD-ID)
1. Candidate Library Preparation:
- Compile a library of potential ligand identities. This can be a predefined list of common ligands, a custom library of metabolites, or drug fragments [21].
2. Blind Docking and Scoring:
- Input the experimental map, a receptor model, and the candidate library into the identification tool.
- The tool should automatically dock all candidate identities from the library into the target density.
- A regression model that considers ligand size, local resolution, and receptor density fit is used to predict an expected density correlation, allowing for fair cross-comparison between different ligands [21].
3. Analysis and Success Metrics:
- Determine the tool's top-ranked ligand identity for each map.
- Success Criterion: Exact match of the top-ranked identity to the deposited ligand [21].
- Report the percentage of successful identifications across the entire benchmark set.
Workflow Visualization
The following diagram illustrates the logical workflow for validating a ligand in a low-resolution map, integrating the tools and strategies discussed in this guide.
Table 2: Key Research Reagent Solutions for Ligand Validation
| Item | Function in Validation |
|---|---|
| Cryo-EM Density Map (EMDB) | The experimental Coulomb potential map used for initial ligand docking and fitting. |
| Atomic Coordinate Model (PDB) | The structural model of the macromolecule, often requiring a ligand-free version for difference map calculation. |
| Cambridge Structural Database (CSD) | A repository of small-molecule crystal structures used by Mogul to provide expected values for bond lengths and angles, validating ligand geometry [52] [56]. |
| Chemical Component Dictionary (CCD) | The wwPDB's dictionary defining every unique small-molecule ligand in the PDB, crucial for standardizing ligand identities and preparing candidate libraries [56]. |
| Ligand Candidate Library | A user-provided collection of small-molecule structures (e.g., metabolites, drug fragments) used for automated ligand identification by tools like EMERALD-ID [21]. |
| Real Space Correlation Coefficient (RSCC) | A key quantitative metric that measures the agreement between the ligand model and the experimental density, provided in wwPDB validation reports [53] [56]. |
Correcting for Ligand Depletion and Covalent Geometry Outliers
Accurate modeling of protein-ligand interactions is foundational to structure-based drug design, yet two persistent challenges compromise model reliability: ligand depletion (conformational and compositional heterogeneity) and covalent geometry outliers. Ligand depletion occurs when electron density maps fail to fully represent ligand conformational diversity or partial occupancy, leading to incomplete modeling of biologically relevant states [57]. Simultaneously, covalent geometry outliers—deviations from ideal bond lengths, angles, and torsions—introduce structural inaccuracies that misrepresent molecular interactions [58] [59]. Within the broader thesis of validating ligand binding in electron density maps, this guide objectively compares computational methodologies that address these challenges across both crystallographic and cryo-electron microscopy (cryo-EM) structural data. As structural biology increasingly tackles complex therapeutic targets and employs diverse experimental modalities, robust computational correction tools have become indispensable for ensuring the accuracy of structural models that drive drug discovery efforts.
Methodological Comparison of Correction Tools
The computational landscape for addressing ligand modeling errors comprises specialized tools that employ distinct algorithmic strategies. The table below compares four advanced methodologies capable of correcting for ligand depletion and covalent geometry outliers.
Table 1: Comparative Analysis of Ligand Modeling and Correction Tools
| Tool Name | Primary Function | Algorithmic Approach | Experimental Data Compatibility | Key Advantages |
|---|---|---|---|---|
| qFit-ligand [57] | Multiconformer modeling to address conformational heterogeneity | RDKit-based stochastic sampling with ETKDG; Quadratic Programming (QP) and Mixed-Integer QP (MIQP) for parsimonious ensemble selection | X-ray crystallography (including PanDDA maps); cryo-EM density maps | Models complex ligands (e.g., macrocycles); improves Real Space Correlation Coefficient (RSCC) and reduces torsional strain |
| EMERALD-ID [21] | Ligand identity determination and conformation fitting in cryo-EM | RosettaGenFF force field combined with density correlation; linear regression model for identity ranking | Cryo-EM density maps | 44% exact identification success for common ligands; combines physical energy with map agreement |
| HiQBind-WF [7] | Dataset curation and structural correction workflow | Automated pipeline for ligand bond order assignment, protonation state correction, and steric clash resolution | PDB-derived structures with binding affinity data | Corrects covalent geometry errors in existing datasets; ensures chemical validity for scoring function development |
| PanDDA with cluster4x [60] | Identification of low-occupancy "hidden" ligand states in crystallographic screens | Pre-clustering of datasets by structural similarity; differential density analysis (PanDDA) to isolate weak ligand signals | X-ray crystallographic screening datasets | Reveals ~50% more fragment hits; identifies ligands in novel allosteric sites |
Each tool addresses a specific niche within the ligand modeling pipeline. qFit-ligand specializes in modeling conformational heterogeneity, even at partial occupancies, thereby directly addressing ligand depletion by representing multiple states simultaneously [57]. EMERALD-ID confronts the challenge of cryo-EM's typically lower resolution by integrating physical chemistry principles with map fitting to correctly identify ligands and their geometries [21]. HiQBind-WF operates at the data curation level, systematically identifying and rectifying covalent geometry errors across entire datasets [7]. The combined PanDDA/cluster4x approach excels at detecting weakly occupied binding events that conventional refinement misses, uncovering previously hidden ligand states [60].
Quantitative Performance Assessment
Validation metrics provide crucial insights into the practical performance of each correction method. The following table summarizes key quantitative results from independent assessments and benchmark studies.
Table 2: Experimental Performance Metrics of Correction Tools
| Tool | Validation Context | Reported Performance Improvement | Key Validation Metrics |
|---|---|---|---|
| qFit-ligand [57] | Redeposited PDB structures with multiconformer ligands | Improved fit to electron density; Reduced torsional strain | Real Space Correlation Coefficient (RSCC), Electron Density Support for Individual Atoms (EDIA), ligand strain energy |
| EMERALD-ID [21] | Benchmark on deposited cryo-EM maps with known ligands | 44% exact ligand identification; 66% closely related identification | Success rate in top-1 and top-5 identification; density correlation (Q-score) |
| PanDDA/cluster4x [60] | Reanalysis of PTP1B fragment screen | 65 new fragment hits identified (+46% increase) | Real Space Correlation Coefficient (RSCC) improvement in event maps; number of new valid binding sites discovered |
| wwPDB Validation [58] [59] | Community-wide standards for ligand geometry | Identification of outliers in bond lengths, angles, and torsions | MolProbity scores, Clashscore, Ramachandran outliers, rotamer outliers |
The quantitative data reveals distinct performance profiles. qFit-ligand demonstrates measurable improvements in both electron density fit and chemical geometry, directly addressing both ligand depletion and covalent geometry issues [57]. EMERALD-ID's 44% success rate in cryo-EM ligand identification, while modest in absolute terms, represents a significant advancement for a domain where existing methods perform poorly at resolutions worse than 3Å [21]. The PanDDA/cluster4x approach provides the most dramatic quantitative evidence, increasing identified fragment hits by approximately 50% in a real-world application, powerfully demonstrating its capability to correct for ligand depletion in crystallographic screens [60].
Experimental Protocols for Implementation
Protocol for Multiconformer Modeling with qFit-ligand
The qFit-ligand workflow requires specific inputs and produces validated multiconformer models through these methodical steps [57]:
- Input Preparation: Provide a starting protein-ligand complex (PDBx/mmCIF format), experimental data (CCP4 map or MTZ structure factors), and the ligand's SMILES string for bond order assignment.
- Conformer Generation: Using RDKit's ETKDG algorithm, stochastically sample the ligand's conformational space (5,000-7,000 conformations) with distance geometry and knowledge-based potentials from the Cambridge Structural Database.
- Pocket-Compatibility Filtering: Apply specialized sampling functions (unconstrained, fixed terminal atoms, blob search) to bias conformer generation toward structures compatible with the binding site geometry.
- Ensemble Optimization: Use Quadratic Programming (QP) and Mixed-Integer QP (MIQP) to select a parsimonious set of conformers (maximum 3 for X-ray, 2 for cryo-EM) and their occupancies that best explain the experimental density.
- Output and Validation: The algorithm outputs a multiconformer ligand model embedded in the unaltered structural context, which should be validated using RSCC, EDIA, and torsional strain metrics.
Protocol for Ligand Identification in Cryo-EM with EMERALD-ID
The EMERALD-ID method addresses identity and conformation uncertainties in moderate-resolution cryo-EM data through this multi-stage process [21]:
- Input Specification: Supply a cryo-EM density map, a receptor model, and a custom ligand library of candidate identities in SMILES format.
- Systematic Docking: Use the EMERALD framework to dock all candidate ligands from the library into the target density, generating multiple pose hypotheses for each identity.
- Scoring and Normalization: For each pose, calculate a combined score incorporating the RosettaGenFF force field energy and the density correlation (Q-score). Normalize density correlation by ligand size and local map resolution to enable fair cross-comparison.
- Identity Ranking: Rank all candidate ligands by their normalized scores, with the top-ranking identity representing the most probable identification.
- Validation and Diagnostics: Examine the correlation between the best-fitting pose and the local density, and check for consistency with the chemical environment of the binding pocket (e.g., hydrogen bonding, hydrophobic contacts).
Protocol for Detecting Hidden Ligands with PanDDA and cluster4x
This specialized protocol reveals low-occupancy fragment binders missed by conventional crystallographic analysis [60]:
- Dataset Pre-clustering: Apply cluster4x to group crystallographic datasets from a fragment screen based on reciprocal space structure factors and Cα displacements, identifying structurally isomorphic clusters.
- Event Map Calculation: For each cluster, perform PanDDA analysis to compute a statistical model of the background density and identify significant positive density features ("events") corresponding to potential ligand binding.
- Model Building into Event Maps: Build and refine ligand models into the PanDDA event maps, which amplify the signal from low-occupancy binders by subtracting the averaged background density.
- Validation: Compare the Real Space Correlation Coefficient (RSCC) of the ligand model in the traditional 2mFo-DFc map versus the PanDDA event map to confirm improved fit.
- Deposition: Publicly deposit the newly identified fragment-bound structures with comprehensive validation reports to enable community access and verification.
Figure 1: Decision workflow for selecting the appropriate correction methodology based on the specific ligand modeling problem encountered.
Successful implementation of ligand correction methodologies requires both computational tools and curated data resources. The following table details essential components of the structural biologist's toolkit for addressing ligand depletion and geometry outliers.
Table 3: Essential Research Resources for Ligand Modeling and Validation
| Resource Name | Type | Primary Function | Application Context |
|---|---|---|---|
| RDKit [57] | Software Library | Chemical informatics and conformer generation (ETKDG) | Provides stochastic sampling of ligand conformational space in qFit-ligand |
| CCD (Chemical Component Dictionary) [58] [7] | Reference Database | Standardized chemical definitions for ligands | Source of ideal bond length/angle parameters for geometry validation |
| MolProbity [59] | Validation Suite | All-atom contact analysis and geometry validation | Identification of covalent geometry outliers (clashes, rotamer outliers) |
| wwPDB Validation Server [58] [59] | Web Service | Comprehensive structure validation pre-deposition | Generation of validation reports highlighting ligand geometry issues |
| PDBbind [7] | Curated Dataset | Protein-ligand complexes with binding affinity data | Benchmark for scoring functions; source of structures for correction |
| BioLiP [7] | Annotated Database | Biologically relevant protein-ligand interactions | Source of functional annotations and binding affinity data |
| EMDB (Electron Microscopy Data Bank) [58] [21] | Public Repository | Experimental cryo-EM density maps | Benchmarking and testing of cryo-EM ligand modeling methods |
These resources collectively provide the foundation for rigorous ligand modeling, validation, and correction. RDKit's ETKDG conformer generator is particularly notable for its integration into multiple correction tools, including qFit-ligand, where it enables sampling of low-energy conformations [57]. The wwPDB validation services and MolProbity implement community-standard geometry validation, providing the critical benchmarks against which correction tools must demonstrate improvement [58] [59]. Public data repositories like PDBbind and EMDB supply the essential experimental data required for both method development and independent benchmarking [7] [21].
Integrated Workflow for Comprehensive Ligand Validation
Addressing complex ligand modeling challenges often requires integrating multiple tools in a sequential workflow. The most effective approach begins with identifying the specific nature of the modeling problem through systematic validation, then applying specialized tools in an appropriate sequence, and concluding with comprehensive validation to ensure all issues have been resolved. The decision workflow (Figure 1) provides a logical pathway for tool selection based on problem type, emphasizing that these methodologies are often complementary rather than mutually exclusive. For instance, a structure with both ligand depletion and geometry issues might benefit from sequential application of qFit-ligand followed by geometry correction using HiQBind-WF principles. The integration of these computational methods represents a significant advancement in the broader thesis of ligand binding validation, moving from passive identification of problems to active computational correction, thereby enhancing the reliability of structural models for drug discovery applications.
In structural biology, particularly in cryogenic electron microscopy (cryo-EM), the accuracy of an atomic model relative to its experimental density map is paramount. This model-to-map fit validates the structural interpretation, especially when studying ligand binding sites crucial for understanding biological function and drug development. Quantitative metrics like Q-scores and real-space correlation coefficients (RSCC) have emerged as essential tools for objectively measuring this fit [61] [62]. Q-scores specifically measure the resolvability of individual atoms in a cryo-EM map by comparing the map's density profile around an atom to an ideal reference Gaussian, with scores ranging from 0 (no resolvability) to 1 (perfectly resolved) [63]. Real-space correlation, conversely, evaluates the agreement between the experimental density and a density map simulated from the atomic model [61]. This guide objectively compares the performance of contemporary software tools and methodologies for optimizing these critical validation metrics, providing researchers with data-driven insights for improving model quality.
Comparative Analysis of Modeling and Validation Software
The landscape of software for cryo-EM model building, refinement, and validation is diverse, with tools developed from both crystallographic and cryo-EM-specific traditions [64]. The following analysis compares their performance based on community-wide assessments and benchmark studies.
Table 1: Software Tools for Model Building, Refinement, and Validation
| Tool Name | Primary Function | Key Features | Performance Data |
|---|---|---|---|
| COOT [64] | Interactive model building | GUI for manual building, real-space refinement, rotamer fitting | Widely used for manual correction of backbone traces and side-chain placement [64]. |
| Phenix [61] | Comprehensive suite | Automated model building, real-space refinement, validation tools | Real-space refinement improves Fit-to-Map scores; Map-Model FSC and RSCC are key metrics [61]. |
| Rosetta [64] [21] | De novo modeling | Model building using structure prediction, energy functions | Successful de novo interpretation at 3.1-4.8 Å resolution; used for ligand identification (EMERALD-ID) [64] [21]. |
| Q-score [63] [62] | Validation metric | Quantifies atom-level resolvability in cryo-EM maps | Correlates strongly with map resolution (R²=0.70); ~0.5 for resolved side chains, ~0.2 for resolved helices [62]. |
| EMRinger [61] | Validation metric | Assesses side-chain fit by density near C-β atoms | Score values improve with increasing map resolution; clusters with Q-score in validation analyses [61]. |
The 2019 EMDataResource Model Challenge provided critical insights into the performance of various modeling approaches and the metrics used to evaluate them [61]. A key finding was that no single validation metric is sufficient; a combination of Q-score, EMRinger, and real-space correlation is necessary for a complete assessment [61]. The challenge also revealed that model quality is highly reproducible when different groups model the same map, but common errors include peptide-bond flips, local sequence misalignment, and failure to model bound ligands [61].
Table 2: Benchmark Performance of Ligand Identification Tools in Cryo-EM
| Method | Approach | Reported Success Rate | Key Application Context |
|---|---|---|---|
| EMERALD-ID [21] | Docking with RosettaGenFF force field and density correlation | 44% exact identification; 66% for closely related ligands | Identifies small molecules from custom libraries (e.g., metabolites, drug fragments). |
| Phenix Ligand Identification [21] | Density map correlations and shape features | Lower accuracy than EMERALD-ID at resolutions worse than 3 Å | Developed primarily for crystallographic data. |
| MinkLoc3Dv2 [21] | Deep learning for ligand classification | Provides ligand classes but does not generate conformations | Limited to identification without conformational modeling. |
For ligand modeling, EMERALD-ID demonstrates superior performance by combining physical forcefields with density agreement, effectively identifying mismodeled ligands in deposited structures [21]. This highlights the importance of using energy-based functions alongside density fit for accurate ligand placement and identity assignment.
Experimental Protocols for Optimizing Model-to-Map Fit
Workflow for Integrated Model Building and Validation
The following diagram illustrates a robust, iterative workflow for building and validating a cryo-EM model, emphasizing steps that directly improve Q-scores and real-space correlation.
Protocol for De Novo Model Building and Refinement
Objective: Construct an atomic model with optimal fit to the cryo-EM density map, maximizing Q-scores and real-space correlation while maintaining proper stereochemistry.
Materials and Input Data:
- Cryo-EM density map (in .mrc or .map format)
- Amino acid and/or nucleic acid sequence of the macromolecule
- Computational Tools: Software suite (e.g., Phenix, Coot, Rosetta) installed on a high-performance computing cluster or workstation.
Methodology:
Map Segmentation and Preparation:
- If the map contains multiple subunits, use segmentation tools like Segger to isolate the region of interest [64]. Note that automatic segmentation can be error-prone with intertwined subunits.
- Inspect the map in a visualization program (e.g., ChimeraX, Coot) to assess local resolution variations and overall quality.
Initial Model Building:
- For high-resolution maps (better than ~3.5 Å), use automated tools like Phenix autobuild or Buccaneer to generate an initial backbone trace and place side chains [64].
- For medium-resolution maps (~3.5-4.5 Å), employ a Rosetta-based de novo approach, which uses predicted backbone conformations and Monte Carlo sampling to register sequence in ambiguous density [64].
- For lower-resolution maps where only secondary structures are visible, use tools like SSEhunter and Gorgon to identify and place helices and sheets, then determine topology via graph matching [64].
Iterative Real-Space Refinement:
- Perform multiple cycles of real-space refinement using Phenix or Coot [64] [61].
- Refinement parameters should optimally balance the fit-to-map term with strong stereochemical restraints (bond lengths, angles, dihedrals) and, if applicable, secondary structure restraints to prevent overfitting [61].
- If the resolution permits, consider refining individual atomic B-factors, which can modestly improve Fit-to-Map scores [61].
Ligand and Cofactor Modeling:
- For identified binding pockets, use EMERALD-ID to dock and evaluate small-molecule conformations from a custom library [21].
- The tool combines the RosettaGenFF small molecule force field with density correlation from the EMERALD fitting method to rank ligand identities and their poses [21].
- Ensure all tightly bound ligands (e.g., NADH, metal ions) are included, as their omission can lead to local backbone mistracing [61].
Protocol for Quantitative Model Validation
Objective: Systematically quantify the model's accuracy using Q-scores, real-space correlation, and other validation metrics.
Materials: The initial and refined atomic models in PDB format, and the corresponding cryo-EM map.
Methodology:
Calculate Global and Local Q-scores:
- Use the Q-score implementation in Phenix or EMDB validation tools [62].
- The Q-score calculation involves generating an atomic map profile by averaging map values at radial distances (0-2.0 Å) from an atom's position, considering only points closer to this atom than any other. This profile is then correlated against a reference Gaussian function (centered at 0, width σ=0.6 Å) using a normalized cross-correlation formula [63].
- Analyze the Q-scores averaged over the entire model, per chain, and per residue. Compare the global average to the expected value for the map's resolution using the established statistical model: Q-scores typically decrease from ~1.0 at 1 Å resolution to ~0.3 at 5 Å resolution [62].
- Identify atoms and residues with significantly low Q-scores (e.g., below 0.3 at 3 Å resolution) for targeted re-examination and refinement [62].
Assess Real-Space Correlation (RSCC):
- Calculate the RSCC for the entire model and for individual residues using Phenix or TEMPy [61].
- This metric correlates the experimental map with a map simulated from the model. For local analysis, it is typically computed within a model-based mask.
- Residues with RSCC in the lowest 1% should not be trusted, while those in the lowest 5% should be treated with caution [65].
Comprehensive Multi-Metric Validation:
- Run a full validation report using the wwPDB Validation Server, which incorporates Q-scores, RSCC, EMRinger, and geometry scores [61] [65].
- Use MolProbity to evaluate stereochemical quality (Clashscore, Rotamer outliers, Ramachandran outliers) [61].
- Employ CaBLAM to check for errors in protein backbone conformation, such as misoriented peptide bonds, which are common in medium-resolution models [61].
The Scientist's Toolkit: Essential Research Reagents and Software
Table 3: Key Research Reagent Solutions for Cryo-EM Model Optimization
| Item Name | Category | Function/Benefit |
|---|---|---|
| Phenix Software Suite | Software | Integrated platform for macromolecular structure determination, featuring tools for model building, refinement, and validation [61]. |
| COOT | Software | Interactive molecular graphics tool for model building and manipulation, ideal for manual correction of atomic models [64]. |
| Rosetta Software Suite | Software | Platform for comparative and de novo structure prediction, includes modules for cryo-EM model building and ligand docking [64] [21]. |
| ChimeraX / PyMOL | Software | Molecular visualization systems for exploratory analysis and creating high-quality renderings of maps and models [66]. |
| MolProbity | Web Service/Software | System for validating the stereochemical quality of macromolecular structures [61]. |
| wwPDB Validation Server | Web Service | Provides comprehensive validation reports for experimental structures prior to deposition, incorporating multiple metrics [65]. |
| EMDB Map Distribution | Data Resource | Public repository for electron microscopy density maps, enabling access to experimental data for validation and re-analysis [66]. |
Optimizing model-to-map fit in cryo-EM is a multifaceted process that relies on iterative refinement guided by robust quantitative metrics. The data demonstrates that Q-scores provide a reliable, atom-level measure of resolvability that correlates strongly with global resolution [63] [62], while real-space correlation coefficients are essential for evaluating local fit [61] [65]. The most successful outcomes are achieved not by relying on a single tool or metric, but by leveraging a synergistic workflow: using Rosetta or Phenix for de novo building, Phenix and Coot for refinement, and EMERALD-ID for ligand modeling, all under the rigorous scrutiny of a multi-metric validation framework (Q-score, RSCC, EMRinger, MolProbity). For researchers focused on ligand binding, this integrated approach is critical for producing models that are not only structurally sound but also functionally informative, thereby providing a reliable foundation for mechanistic insights and drug discovery.
Cryogenic Electron Microscopy (cryo-EM) has revolutionized structural biology by enabling the determination of biomolecular structures that are challenging to resolve using conventional methods such as X-ray crystallography [43]. However, the rapid expansion of cryo-EM applications, particularly in drug discovery where accurately modeling ligand-protein interactions is crucial, has exposed significant challenges in standardizing map processing and quality assessment. The inherent limitations of cryo-EM, including variable resolution, noise, and the potential for conformational changes during sample vitrification, necessitate robust validation frameworks to ensure the reliability of atomic models built from density maps [67].
The process of interpreting cryo-EM maps remains challenging, especially at lower resolutions or in regions of local flexibility. While artificial intelligence (AI) has dramatically transformed structure modeling from cryo-EM density maps, the integration of these powerful tools requires standardized validation to prevent over-interpretation [43] [68]. This guide objectively compares current methodologies for cryo-EM map processing and quality assessment, providing researchers with experimental protocols and data-driven comparisons to standardize their workflow, with special emphasis on validating ligand binding.
Core Challenges in Cryo-EM Map Interpretation
Resolution Variability and Model Building
A fundamental challenge in cryo-EM stems from the variability in map resolution, which directly dictates the appropriate modeling strategy. Methods for structure modeling are typically classified into two categories based on resolution thresholds:
- De novo modeling (up to ~5 Å resolution): At resolutions up to approximately 5 Å, deep learning techniques can identify amino acids, nucleotides, and atomic positions, enabling full-atom structure modeling directly from the map [43]. Tools like DeepTracer, ModelAngelo, and MICA fall into this category.
- Structure-fitting methods (worse than 5 Å resolution): At resolutions worse than 5 Å, identifying key structural features becomes challenging. Modeling must therefore rely on fitting existing atomic structures or predicted models into the lower-resolution density [43]. Methods such as SITUS, UCSF Chimera's
fitmap, and VESPER are designed for this regime.
This empirical 5 Å cutoff is critical for standardization, as applying de novo methods to low-resolution maps can lead to over-interpretation and modeling errors.
Ligand Building at Intermediate Resolutions
A specific and critical challenge in drug development is the accurate modeling of small molecule ligands. Often, even when the surrounding protein is resolved to high resolution, the ligand density may be of significantly lower quality (e.g., 3–3.5 Å while the protein is at 1.5 Å) [69]. This complicates automated model building, as traditional methods from X-ray crystallography, which rely on well-defined topological features in high-resolution density, see their accuracy drop substantially below 3 Å resolution [55]. Furthermore, sample preparation for cryo-EM can sometimes induce conformational changes during blotting and vitrification, meaning the final map may not represent the native solution state [67]. This highlights the need for independent validation tools.
Comparative Analysis of Cryo-EM Tools and Methods
AI-Driven De Novo Modeling Tools for High-Resolution Maps
For high-resolution maps (<4 Å), AI-driven tools have automated the process of building protein backbone and side-chain atoms. The table below summarizes key features and performance metrics of state-of-the-art tools.
Table 1: Comparison of Automated De Novo Cryo-EM Structure Modeling Tools
| Tool | Key Technology | Input Requirements | Strengths | Reported Performance (TM-score) | Reference |
|---|---|---|---|---|---|
| MICA | Multimodal deep learning integrating cryo-EM maps and AlphaFold3 structures | Cryo-EM map, protein sequence(s) | High accuracy and completeness; robust to protein size and map resolution | 0.93 (average on high-res maps) | [70] |
| ModelAngelo | 3D-CNN for Cα detection; GNNs for chain tracing; HMM for sequence alignment | Cryo-EM map, protein sequence(s) | Combines map features with protein language models (ESM) | Lower than MICA | [43] [70] |
| EModelX(+AF) | 3D U-Net for atom/amino acid detection; sequence-guided threading with AlphaFold2 | Cryo-EM map, protein sequence(s) | Uses AF2 structures to refine and fill unmodeled gaps | Lower than MICA | [70] |
| DeepTracer | Four U-Nets for atom, secondary structure, amino acid, and backbone detection | Cryo-EM map, protein sequence(s) | Early deep learning tool with a user-friendly web server | N/A | [43] |
| Cryo2Struct | 3D transformer for atom/amino acid detection; HMM for chain tracing | Cryo-EM map (ab initio, no templates) | Fully automated without relying on predicted or homologous templates | N/A | [43] [70] |
Experimental Data and Protocol: The performance of MICA was evaluated on the Cryo2StructData test dataset (resolution range: 2.05 Å to 3.9 Å). The protocol involves:
- Input: Feeding the cryo-EM density map and AlphaFold3-predicted structures for the protein chains into a multi-task encoder-decoder network with a Feature Pyramid Network (FPN).
- Prediction: The network simultaneously predicts backbone atoms, Cα atoms, and amino acid types.
- Tracing & Refinement: An initial backbone model is built from the predictions, unmodeled gaps are filled using AF3 structural information, and the final model is refined against the density map using
phenix.real_space_refine[70].
Specialized Tools for Ligand Building and Validation
Accurately placing small molecules in medium-resolution cryo-EM maps is paramount for drug discovery. The following table compares tools designed for this specific task.
Table 2: Comparison of Ligand Building and Validation Tools for Cryo-EM
| Tool | Approach | Input Requirements | Key Metrics | Reported Performance | Reference |
|---|---|---|---|---|---|
| EMERALD | Density-guided genetic algorithm docking with RosettaGenFF force field | Cryo-EM map, protein structure, ligand specification | Ligand RMSD, density correlation, hydrogen bonds | 57% of ligands replicate deposited model within 1 Å RMSD; 16% confidently find alternate conformations | [55] |
| Chai-1 + Flexible Fitting | AI-predicted complex (via SMILES) + density-guided molecular dynamics | Protein sequence, ligand SMILES, cryo-EM map | Model-to-map cross-correlation (CC), protein-ligand interaction energy | Improved CC from 40-71% to 82-95% vs. deposited structures for 10 pharmaceutically relevant targets | [69] |
| AUSAXS | Independent validation by comparing SAXS profile from EM map with solution SAXS data | Cryo-EM map, experimental SAXS data | Goodness-of-fit (reduced χ²) | Identifies discrepancies between vitrified and solution states | [67] |
Experimental Protocol for EMERALD:
- Initial Placement: The target cryo-EM map is processed into a pseudo-atomic skeleton. The ligand is placed at the skeleton's center.
- Genetic Algorithm Docking: A population of 100 ligand conformers, with flexible surrounding protein side chains, is iteratively optimized. The scoring function combines RosettaGenFF energy and density correlation.
- Refinement and Analysis: The top poses are refined, and confidence is assessed through convergence across multiple independent trajectories [55].
Quality Assessment and Validation Tools
Validation is a critical step to ensure model quality and prevent overfitting.
Table 3: Comparison of Quality Assessment and Validation Methods
| Method | Type | Core Principle | What it Validates | Reference |
|---|---|---|---|---|
| Half-Map Validation | Internal consistency | Compares two independent reconstructions from random halves of the particle data | Prevents overfitting; assesses global map quality | [71] |
| DAQ | AI-Based QA | Deep learning model that learns local density features to assess residue-level quality | Identifies local model errors in regions of low resolution | [72] |
| Independent Particle Set (BioEM) | External validation | Monitors map probability evolution over a control particle set not used in refinement | Assesses overall map quality and overfitting | [71] |
| AUSAXS | External/Independent | Compares SAXS curve from the EM map dummy model with experimental solution SAXS data | Validates that the map represents the solution conformation | [67] |
Essential Research Reagent Solutions
A standardized cryo-EM workflow relies on a suite of computational tools and data resources. The following table details key "research reagents" for processing, modeling, and validation.
Table 4: Essential Research Reagent Solutions for Cryo-EM Workflows
| Item Name | Function/Brief Explanation | Example Tools / Databases |
|---|---|---|
| Deep Learning Denoising Models | Enhances interpretability of intermediate-resolution maps by reducing noise and boosting local contrast. | DeepEMhancer, CryoSAMU [68] |
| AI-Based Model Building Suites | Automates the construction of atomic models from cryo-EM density maps. | MICA, ModelAngelo, DeepTracer, EModelX [43] [70] |
| Ligand Docking Pipelines | Automatically determines small molecule ligand structures guided by cryo-EM density. | EMERALD [55] |
| Quality Assessment (QA) Metrics | Assesses the quality of a built model based on map-model agreement and protein stereochemistry. | DAQ, DAQ-Refine [72] |
| Independent Validation Datasets | Provides an external standard to validate the map without relying on the refined atomic model. | Control particle sets [71], SAXS data [67] |
| Standardized Benchmark Datasets | Used for training and benchmarking AI models for map enhancement and model building. | Labeled dataset for cryo-EM map enhancement [73] |
Visualizing Workflows and Relationships
Cryo-EM Processing and Validation Workflow
The following diagram illustrates a robust, standardized workflow for cryo-EM processing, model building, and validation, integrating the tools and methods discussed.
Cryo-EM Processing and Validation Workflow
Ligand Validation Logic Pathway
For drug development projects, confirming ligand placement is critical. This diagram outlines a decision pathway for validating and interpreting ligand density.
Ligand Validation Logic Pathway
The field of cryo-EM is transitioning from a technique focused on structure determination to one capable of discovery-driven science, particularly in drug design. Standardization of map processing and quality assessment is the cornerstone of this transition. As demonstrated by the tools and data in this guide, the integration of AI with cryo-EM is powerful but must be tempered with rigorous, multi-faceted validation. The most robust workflows combine internal checks (half-maps, geometry) with external validation (independent particles, SAXS) and specialized AI tools for model building (MICA) and ligand placement (EMERALD). By adopting these standardized protocols and critical assessment frameworks, researchers can confidently generate and interpret cryo-EM structures, ensuring that insights into protein function and ligand binding are built upon a foundation of reliable, reproducible data.
Beyond the Model: A Comparative Guide to Validation Metrics and Benchmarking
Accurate modeling of ligands within their macromolecular environment is fundamentally important to structural biology and drug discovery, as these small molecules can substantially influence larger-scale structure and biological functions [58]. As the number of novel ligands in cryo-EM-derived structures continues to increase rapidly, investigating how best to validate them has become crucial to ensure optimal modeled ligand quality using various measures such as fit of model-to-map, geometry scores, and local interactions with surrounding protein or nucleic acid components [58]. The EMDataResource Ligand Model Challenge brought together the international research community to assess the reliability and reproducibility of modeling ligands bound to protein and protein/nucleic-acid complexes in cryogenic electron microscopy (cryo-EM) maps determined at near-atomic resolution (1.9-2.5 Å) [74]. This challenge revealed that a composite rather than a single score was needed to assess macromolecule+ligand model quality effectively [75], leading to the development and refinement of the validation metrics explored in this guide.
The EMDataResource challenge identified four key validation categories that provide complementary assessment capabilities for ligand modeling in cryo-EM structures. The table below summarizes the core purpose, measurement focus, and key strengths of each primary metric discussed in this guide.
Table 1: Core Ligand Validation Metrics Overview
| Metric | Primary Purpose | Measurement Focus | Key Strengths |
|---|---|---|---|
| Q-score | Map-model fit assessment | Local map quality around atoms | Quantifies density fit; applicable to cryo-EM; wwPDB validation standard |
| LIVQ | Ligand+environment integration | Density fit of ligand AND surrounding residues | Contextual assessment; identifies environmental mismatches |
| Pharmacophore Modeling | Binding site interaction quality | Energetic favorability of ligand-environment interactions | Time-tested energetic measure; functional group optimization |
| Probescore | Atomic contact analysis | Specific all-atom contacts (H-bond, clash, van der Waals) | Identifies specific interaction types; quantifies contact quality |
Deep Dive into Individual Metrics
Q-score: Quantifying Map-Model Fit
Q-score has emerged as a fundamental metric for evaluating how well atomic models fit their experimental cryo-EM density maps. Inspired and introduced by the 2019 EMDataResource Model Metrics Challenge, Q-score has now been adopted by the wwPDB Validation System used at deposition as well as in the detailed validation report [58].
The Q-score algorithm operates by calculating the local map quality around each atom. It assesses the density distribution within a sphere around atomic positions, providing a quantitative measure between 0 (no fit) and 1 (perfect fit). This metric is particularly valuable because it offers a standardized approach to evaluate map-model fit specifically designed for cryo-EM applications, addressing a critical need in the field as cryo-EM structures continue to increase in both number and resolution [58].
LIVQ Scores: Contextual Ligand+Environment Assessment
The LIVQ (Ligand+Immediate enVironment Q scores) metric was introduced through the EMDataResource Ligand Challenge to address a crucial gap in ligand validation [74] [75]. Traditional metrics that focus solely on the ligand itself often miss critical contextual information about whether the ligand is placed in a chemically reasonable environment.
LIVQ scores measure the density fit of both the ligand and the surrounding protein or nucleic acid residues within its binding site [75]. This dual focus is particularly important because a ligand might appear to have reasonable density fit in isolation, but if the surrounding environment shows poor density quality, the entire ligand placement becomes questionable. The LIVQ approach recognizes that ligand binding requires complementary density for both the ligand and its binding site, providing a more holistic assessment of ligand placement validity.
Pharmacophore Modeling: Energetic Validation
Pharmacophore modeling provides an optimized and time-tested energetic measure for evaluating how well a binding site would interact with a specific ligand [75]. A pharmacophore represents the ensemble of steric and electronic features necessary to ensure optimal supramolecular interactions with a specific biological target and to trigger (or block) its biological response [76].
In the context of ligand validation, pharmacophore modeling abstracts the key molecular recognition elements into features such as hydrogen bond donors/acceptors, charged groups, hydrophobic regions, and aromatic rings. These features are used to assess whether the modeled ligand makes chemically sensible interactions with its environment [76]. The EMDataResource challenge specifically recommended pharmacophore modeling as one of several independent metrics that should be applied to evaluate ligand binding site interactions [74]. If the ligand model shows only weak interaction with its environment as assessed by pharmacophore modeling, the model likely requires refinement [75].
Probescore: Atomic Contact Analysis
Probescore complements other metrics by providing detailed analysis of specific all-atom contacts between the ligand and its binding environment [75]. This metric both quantifies and identifies specific interactions including hydrogen bonds, steric clashes, and van der Waals interactions [74].
The strength of Probescore lies in its ability to detect specific problematic interactions that might be missed by more global metrics. For example, it can identify individual atomic clashes that might strain the model or highlight missing favorable interactions that would be expected in a biologically relevant binding mode. When used in combination with other metrics, Probescore provides the atomic-resolution insight needed to refine and validate ligand models with high precision.
Experimental Protocols and Workflows
EMDataResource Challenge Design
The EMDataResource Ligand Challenge established a rigorous experimental framework for evaluating ligand modeling approaches and validation metrics. The challenge design selected three cryo-EM map targets based on specific criteria: recently published with resolution better than 3 Å, maps released in the Electron Microscopy Databank (EMDB), associated coordinates in the Protein Data Bank (PDB), presence of diverse small molecules (ligands, water, metal ions, detergent, and/or lipid), and current topical relevance [58].
The specific targets included:
- Target 1: 1.9 Å E. coli β-Galactosidase (β-Gal) in complex with inhibitor 2-phenylethyl 1-thio-beta-D-galactopyranoside (PETG) with PDB Chemical Composition Dictionary (CCD) id PTQ, EMDB map entry EMD-7770, PDB reference model 6CVM
- Target 2: 2.5 Å SARS-CoV-2 RNA-dependent RNA polymerase (RNAP) with the pharmacologically active, nucleotide form of the prodrug remdesivir (CCD id F86) covalently-bound to RNA, EMD-30210, PDB reference model 7BV2
- Target 3: 2.1 Å SARS-CoV-2 Open Reading Frame 3a (ORF3a) putative ion channel in complex with 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine phospholipid (CCD id PEE), EMD-22898, PDB reference model 7KJR
Seventeen independent research groups participated, submitting sixty-one models with supporting workflow details, creating a robust dataset for evaluating validation metrics [58] [74].
Recommended Validation Workflow
Based on the challenge outcomes, the following integrated workflow for validating cryo-EM structures of liganded macromolecules is recommended:
Research Reagent Solutions
Table 2: Essential Research Tools for Ligand Validation
| Tool/Category | Specific Examples | Primary Function |
|---|---|---|
| Modeling Software | Coot, Rosetta, Chimera, ChimeraX, Phenix | Ligand placement and refinement |
| Validation Suites | MolProbity, wwPDB Validation Server | Comprehensive structure validation |
| Geometry Analysis | CaBLAM, EMRinger, Q-score | Conformational and map-fit assessment |
| Ligand Restraint Generation | Phenix eLBOW, CCP4 AceDRG, Open Babel | Chemical parameter generation |
| Interaction Analysis | Probescore, Pharmacophore Modeling (LigandScout) | Binding site contact evaluation |
| Specialized Metrics | LIVQ scores, DNATCO, Sugar pucker analysis | Domain-specific validation |
Comparative Performance Data
The EMDataResource challenge provided quantitative insights into how different validation metrics perform across various ligand types and resolution ranges. While specific numerical results for each metric across all submissions are extensive, several key patterns emerged from the analysis.
Table 3: Metric Performance Across Challenge Targets
| Metric Category | Target 1 (β-Galactosidase) | Target 2 (RNA Polymerase) | Target 3 (ORF3a Lipid) |
|---|---|---|---|
| Q-score Range | 0.6-0.8 (high resolution advantage) | 0.4-0.7 (nucleic acid complexity) | 0.3-0.6 (membrane protein challenges) |
| Geometry Outliers | Minimal (well-defined small molecule) | Moderate (nucleotide analog complexity) | Significant (flexible lipid chains) |
| Contact Scores | Consistent favorable interactions | Variable protein-RNA-ligand interfaces | Challenging membrane environment |
| Key Strength | Excellent small molecule validation | Covalent ligand assessment | Membrane context evaluation |
The comparative analysis revealed that no single metric could comprehensively assess ligand model quality across all targets. The highest-quality submissions consistently showed strong performance across multiple complementary metrics rather than excelling in just one dimension [58] [74]. RNA-protein complexes (Target 2) presented particular challenges for ligand validation due to the complexity of balancing protein-ligand, RNA-ligand, and protein-RNA interactions simultaneously.
Best Practice Recommendations
Based on the comprehensive analysis from the EMDataResource Ligand Challenge and recent advances in validation methodologies, the following best practices are recommended for researchers validating ligand binding in electron density maps:
Implement Multi-Metric Validation Strategies Relying on a single validation metric is insufficient for comprehensive ligand assessment. Composite scoring using Q-score, LIVQ, pharmacophore modeling, and Probescore provides complementary insights that collectively ensure robust validation [75].
Contextualize Ligand Quality Within Macromolecular Environment Ligands cannot be validated in isolation. The EMDataResource challenge demonstrated that the quality of the surrounding macromolecular model significantly impacts ligand validation results. LIVQ scores specifically address this by evaluating both ligand and immediate environment [74].
Standardize Geometric Checks Across Experimental Methods Apply rigorous geometric validation standards comparable to those used in crystallography, including MolProbity evaluation with CaBLAM and clashscore for proteins, and specialized checks like sugar pucker and DNATCO conformational analysis for nucleic acid components [75].
Correlate Local Density Quality with Interaction Patterns Strong density fit (Q-score) should correlate with chemically sensible interaction patterns (Probescore, pharmacophore modeling). Discordance between these metrics often indicates problematic modeling that requires reinvestigation.
Future Directions and Community Standards Future cryo-EM model challenges should incorporate automated checks and immediate author feedback similar to established CASP and CAPRI events in the prediction community [75]. The rapidly growing number of RNA-containing cryo-EM structures suggests future validation efforts should focus particularly on RNA-ligand complexes and associated ion identification.
The Validator's Toolkit for assessing ligand binding in cryo-EM structures has evolved significantly through community-wide efforts like the EMDataResource Ligand Challenge. The integrated application of Q-score, LIVQ, pharmacophore modeling, and Probescore provides a robust, multi-faceted approach to ligand validation that addresses map-model fit, local environmental context, energetic favorability, and specific atomic interactions. As cryo-EM continues to advance into near-atomic resolution for increasingly complex macromolecular systems, these complementary validation metrics will play an essential role in ensuring the reliability and reproducibility of structural models, ultimately supporting more accurate structure-based drug design and mechanistic understanding of biological processes.
Accurate modeling of small-molecule ligands within macromolecular complexes is fundamental to structural biology, with critical implications for understanding biological mechanisms and structure-based drug discovery. As cryo-electron microscopy (cryo-EM) has emerged as a mainstream method for determining high-resolution structures of macromolecular complexes, the need for robust validation methods for ligand modeling has become increasingly pressing. The EMDataResource 2021 Ligand Model Challenge was established to address this need by systematically evaluating the reliability and reproducibility of modeling ligands in near-atomic resolution cryo-EM maps [77] [75]. This community-wide assessment revealed that no single validation metric could adequately assess ligand model quality, leading to the recommendation for a composite validation approach that integrates multiple complementary measures [77] [74]. This article analyzes the outcomes of this landmark challenge and synthesizes its implications for validation practices in structural biology.
Challenge Design and Experimental Framework
The Ligand Model Challenge employed a rigorous blind assessment design where participating research groups submitted models for predefined cryo-EM targets without access to reference structures. The challenge incorporated three carefully selected cryo-EM map targets determined at near-atomic resolution (1.9-2.5 Å), representing diverse biological systems with current relevance [77] [58]:
Table 1: EMDataResource Ligand Challenge Targets
| Target | Biological System | Ligand | Resolution | EMDB ID | Reference PDB |
|---|---|---|---|---|---|
| Target 1 | E. coli beta-galactosidase | PETG inhibitor | 1.9 Å | EMD-7770 | 6CVM |
| Target 2 | SARS-CoV-2 RNA-dependent RNA polymerase | Remdesivir (F86) | 2.5 Å | EMD-30210 | 7BV2 |
| Target 3 | SARS-CoV-2 ORF3a ion channel | Phospholipid (PEE) | 2.1 Å | EMD-22898 | 7KJR |
The challenge attracted seventeen independent research groups who submitted a total of sixty-one models, each with detailed workflow documentation [58]. Participant methodologies spanned diverse approaches to both polymer and ligand modeling, with some groups performing ab initio modeling while others optimized existing models. Ligand restraint generation utilized various software solutions including Phenix eLBOW, Open Babel, CCP4 AceDRG, and molecular dynamics force fields [58].
Key Findings: The Imperative for Composite Validation
Analysis of the submitted models revealed substantial variation in ligand modeling quality across participants. The assessment employed multiple validation dimensions including local map quality, model-to-map fit, geometry, energetics, and contact scores [77]. Critically, the challenge demonstrated that:
- No single metric correlated sufficiently with model accuracy across all targets and modeling scenarios
- Different metrics captured complementary aspects of model quality, with each having strengths and limitations
- Model quality depended significantly on both ligand placement and the accuracy of the surrounding macromolecular environment [77] [74]
These findings directly contradicted the assumption that any individual validation metric could serve as a definitive measure of ligand model quality, highlighting the necessity of a composite approach.
The Composite Validation Framework: Recommended Metrics and Methods
Based on their comprehensive analysis, the challenge organizers recommended a multi-faceted validation framework that assesses quality at multiple levels:
Macromolecular Model Validation
The foundation of accurate ligand modeling begins with proper validation of the surrounding macromolecular structure. Recommended checks include [75] [74]:
- Standard covalent geometry validation (bond lengths, angles)
- MolProbity evaluation including CaBLAM and clashscore analysis
- Nucleic acid-specific checks for sugar pucker and DNATCO conformational analysis
- Model-map fit assessment using EM Ringer, Q-score, and FSC
Ligand-Specific Validation
Ligands require specialized validation beyond standard protein checks [75] [74]:
- Covalent geometry measures (bond lengths, angles, planarity, chirality) per wwPDB standards
- Density fit assessment using cryo-EM appropriate metrics like Q-score
- Occupancy evaluation considering both absolute density strength and relative to surroundings
- Completeness assessment identifying missing atoms or ions
Binding Site Interaction Analysis
The detailed interaction between ligand and binding site requires particularly careful assessment using multiple independent metrics [75] [74]:
- Pharmacophore modeling to evaluate energetic compatibility with the binding site
- LIVQ scores (Ligand+Immediate enVironment Q scores) measuring density fit of both ligand and surrounding residues
- Probescore analysis quantifying all-atom contacts including H-bond, clash, and van der Waals interactions
Composite Ligand Validation Workflow
Comparative Performance of Validation Metrics
The challenge provided unique insights into the strengths and limitations of individual validation metrics when applied to cryo-EM ligand models:
Table 2: Validation Metrics and Their Applications
| Metric Category | Specific Metrics | Strengths | Limitations | Optimal Use Case |
|---|---|---|---|---|
| Geometry Validation | Mogul RMSZ, MolProbity | Identifies stereochemical errors, standardized benchmarks | May not detect correct geometry in poor density, size-dependent RMSZ | Initial quality screening |
| Density Fit | Q-score, EM Ringer | Direct measure of experimental support, resolution-dependent | Does not assess chemical plausibility, affected by flexibility | Placement confidence assessment |
| Energetic Validation | RosettaGenFF, Pharmacophore | Evaluates physical plausibility, interaction quality | Computationally intensive, force field limitations | Binding mode validation |
| Contact Analysis | Probescore, LIVQ | Specific interaction mapping, environment consideration | Requires accurate surroundings, may miss global issues | Binding site refinement |
Advanced Methodologies: EMERALD and Automated Ligand Placement
Concurrent with the Ligand Challenge, methodological advances have emerged to address the complexities of cryo-EM ligand modeling. The EMERALD (EM Maps ERoded for Automatic Ligand Docking) method exemplifies this progress by combining cryo-EM density with the RosettaGenFF force field to enable robust ligand modeling without user intervention [55].
The EMERALD methodology employs a sophisticated multi-step process:
- Density-guided initial placement using pseudo-atomic skeletonization of cryo-EM maps
- Genetic algorithm optimization maintaining a population of 100 conformations
- Iterative refinement balancing density correlation and physical energy functions
- Convergence assessment across multiple independent trajectories to estimate confidence [55]
Validation across 1,053 ligands from the EMDB demonstrated that EMERALD recapitulated deposited models in 57% of cases, confidently identified alternate conformations in 16% of cases, and produced incorrect placements in only 5% of cases [55]. In several instances where EMERALD proposed alternate conformations differing from deposited models, subsequent comparison with high-resolution crystal structures validated the EMERALD placements [55].
Table 3: Research Reagent Solutions for Cryo-EM Ligand Validation
| Tool/Resource | Type | Function | Access |
|---|---|---|---|
| MolProbity | Software Suite | Macromolecular geometry validation, clashscores | Web server/standalone |
| Phenix eLBOW | Restraint Generator | Ligand coordinate and restraint generation | Integrated in Phenix |
| CCP4 AceDRG | Restraint Generator | Chemical geometry dictionary generation | Integrated in CCP4 |
| EM Ringer | Map-Metric | Sidechain-model consistency assessment | Standalone/Web |
| Q-score | Map-Metric | Model-to-map fit quantification | Integrated in wwPDB |
| EMERALD | Automated Docking | Ligand placement in cryo-EM maps | Rosetta-based |
| Probescore | Contact Analyzer | All-atom interaction quantification | Integrated in MolProbity |
Implementation Protocol: Best Practices for Structural Biologists
Based on the challenge findings, researchers should implement the following workflow for comprehensive ligand validation:
Experimental Design Phase
- Target resolution consideration: Ensure local resolution around binding site supports ligand modeling (generally ≤3.0 Å)
- Restraint preparation: Generate accurate chemical restraints before refinement using eLBOW, AceDRG, or similar tools
- Control modeling: Include known ligands or inhibitors as positive controls when feasible
Modeling and Refinement Phase
- Macromolecular context first: Refine protein/nucleic acid environment before introducing ligands
- Multiple placement trials: Experiment with alternate ligand conformations and orientations
- Occupancy refinement: Adjust occupancy parameters when density suggests partial binding
Validation Phase
- Systematic metric application: Apply all recommended validation categories rather than selective checking
- Comparative assessment: Evaluate metrics against both global averages and local binding site context
- Outlier investigation: Carefully examine any geometric or energetic outliers rather than automatically dismissing them
Future Directions and Community Recommendations
The Ligand Challenge outcomes have prompted several forward-looking recommendations for the structural biology community [75] [74]:
- Standardized challenge framework: Future cryo-EM model challenges should follow the established formats of CASP and CAPRI competitions
- Automated validation pipelines: Development of integrated validation systems providing immediate author feedback
- RNA-focused challenges: Given the increasing number of RNA-containing structures, future challenges should specifically address RNA-ligand validation
- Validation metric refinement: Continued development and testing of improved metrics, particularly for intermediate-resolution maps
The EMDataResource Ligand Challenge has fundamentally advanced our understanding of ligand validation in cryo-EM structural biology. Its most significant contribution lies in demonstrating that composite assessment using multiple complementary metrics surpasses any single validation measure for evaluating ligand model quality. The recommended framework—encompassing macromolecular geometry, ligand-specific checks, and detailed binding site analysis—provides a robust methodology for researchers across structural biology and drug discovery. As cryo-EM continues to evolve as a dominant structural method, these validation principles will ensure the reliability of ligand-containing structures that drive mechanistic understanding and therapeutic development.
Validating ligand binding in electron density maps represents a critical challenge in structural biology and drug discovery. Accurately identifying small molecules and quantifying their interactions with target proteins is fundamental to understanding biological function and guiding therapeutic design. This review provides a comparative analysis of three dominant computational approaches—traditional geometry-based methods, traditional machine learning (ML), and deep learning (DL)—for modeling protein-ligand interactions and interpreting electron density data. As structural data from cryo-electron microscopy (cryo-EM) and X-ray crystallography continues to grow exponentially, benchmarking the performance and generalizability of these methods becomes increasingly important for the research community.
The following table summarizes the representative methods from each category and their key performance metrics as reported in recent literature.
Table 1: Performance Comparison of Ligand Binding Validation Methods
| Method Category | Representative Methods | Key Performance Metrics | Reported Results | Primary Applications |
|---|---|---|---|---|
| Geometry / Physics-Based | EMERALD-ID [21], CheckMyBlob [35] | Ligand Identification Rate (Exact/Related) | 44% exact, 66% related ligands identified [21] | Ligand identification in cryo-EM & X-ray maps |
| Traditional Machine Learning | P2Rank [2], DeepPocket [2] | AUPR (Binding Site Prediction) | ~0.47 (DS1), ~0.32 (DS3) [2] | Binding site residue prediction |
| Deep Learning (Structure-Based) | CORDIAL [78], LumiNet [79], LABind [2] | ROC AUC (Affinity), PCC (ABFE), AUPR (Binding Sites) | Maintains ROC AUC on novel families [78], PCC=0.85 (CASF-2016) [79], AUPR=0.72 (DS1) [2] | Affinity ranking, ABFE calculation, ligand-aware binding site prediction |
| Deep Learning (Density-Based) | MICA [70], Rotation-Invariant PointNets [35] | TM-score (Structure Modeling), Top-1/Top-10 Accuracy (Ligand ID) | TM-score=0.93 [70], Performance on par with ML for cryo-EM [35] | Protein structure modeling from maps, Ligand identification |
Detailed Experimental Protocols and Validation
A critical differentiator among modern methods is the rigor of their validation strategies, which directly impacts the reliability of reported performance metrics.
Validation of Generalizability
A significant challenge for ML and DL models is performance degradation on novel targets. To assess this, stringent benchmarks have been developed:
- Leave-Superfamily-Out (LSO) Validation: This protocol, utilized for frameworks like CORDIAL, involves withholding entire protein homologous superfamilies from the training data. It simulates real-world discovery scenarios where models encounter proteins with novel architectures and implicitly associated chemical scaffolds [78].
- Cross-Dataset Evaluation: Models are trained on one data source (e.g., high-quality X-ray crystallography ligands) and tested on another (e.g., cryo-EM ligands). This tests robustness to differences in data processing and map quality [35].
- Unseen Ligand Prediction: Methods like LABind are specifically evaluated on their ability to predict binding sites for small molecules and ions not present in the training dataset, a key requirement for prospective drug discovery [2].
Key Experimental Workflows
The experimental protocols for benchmarking can be categorized into several major workflows, which are visualized in the diagram below.
Performance on Specific Tasks
Binding Affinity Prediction
Accurate prediction of protein-ligand binding affinity is crucial for virtual screening. CORDIAL, an interaction-only DL framework, was designed to overcome the generalizability problem of structure-centric models. It focuses exclusively on the physicochemical properties of the protein-ligand interface by creating interaction radial distribution functions from distance-dependent cross-correlations [78]. Under LSO validation, CORDIAL uniquely maintained predictive performance and calibration, whereas the performance of 3D-CNN and GNN models degraded significantly [78].
In a different approach, LumiNet bridges physics-based models and black-box algorithms. It uses a subgraph transformer and geometric neural networks to map atomic pair structures into key physical parameters of non-bonded interactions in classical force fields for absolute binding free energy (ABFE) calculation [79]. It achieved a Pearson Correlation Coefficient (PCC) of 0.85 on the CASF-2016 benchmark and demonstrated the ability to rival the accuracy of Free Energy Perturbation (FEP+) in some tests with a speed improvement of several orders of magnitude [79].
Ligand and Binding Site Identification
For identifying small molecules in cryo-EM data, the geometry/physics-based method EMERALD-ID combines a physical forcefield with density agreement to rank ligand identities. When benchmarked on deposited cryo-EM maps, it successfully identified the exact deposited ligand 44% of the time, and a closely related ligand 66% of the time [21].
For predicting binding sites, the deep learning method LABind utilizes a graph transformer and a cross-attention mechanism to learn distinct binding characteristics between proteins and ligands in a ligand-aware manner [2]. It significantly outperformed other multi-ligand-oriented methods (e.g., P2Rank, DeepSurf) on benchmark datasets, achieving an AUPR of 0.72 on the DS1 dataset. Its explicit modeling of ligand properties enables it to generalize to predicting binding sites for unseen ligands [2].
Protein Structure Modeling from Maps
Deep learning has enabled high-accuracy, automated protein structure determination from cryo-EM density maps. MICA introduces a multimodal deep learning approach that integrates cryo-EM density maps with AlphaFold3-predicted structures at both the input and output levels [70]. This integration compensates for limitations in each individual modality, such as low-resolution regions in maps or incorrect predictions in AF3 structures. MICA significantly outperformed other state-of-the-art methods like ModelAngelo and EModelX(+AF), building high-accuracy models with an average TM-score of 0.93 from high-resolution maps [70].
Successful implementation of the methods discussed relies on a foundation of key datasets, software tools, and computational resources.
Table 2: Key Research Reagents and Computational Resources
| Resource Name | Type | Primary Function | Relevance to Validation |
|---|---|---|---|
| PDBbind [80] | Curated Dataset | Provides comprehensive set of protein-ligand complexes with binding affinity data. | Standard benchmark for training and testing affinity prediction models. |
| CATH Database [78] | Protein Classification | Hierarchical classification of protein domains into superfamilies. | Enables rigorous Leave-Superfamily-Out (LSO) validation protocols. |
| CASF-2016 Benchmark [79] | Benchmarking Suite | Standardized set of complexes for evaluating scoring power, docking power, ranking power, and screening power. | Core benchmark for assessing binding affinity prediction accuracy (e.g., used by LumiNet). |
| CheckMyBlob [35] | Web Server / Tool | Statistical and machine learning-based ligand identification and validation for X-ray crystallography. | Established baseline for comparing new deep learning ligand identification methods. |
| Phenix Software Suite [21] [70] | Software Platform | Integrated toolbox for macromolecular structure determination. | Used for tasks like real-space refinement and difference map calculation in workflows like EMERALD-ID and MICA. |
| AlphaFold3 (AF3) [70] | AI Model | Predicts 3D structures of proteins and biomolecular complexes from sequence. | Provides prior structural knowledge that can be integrated with experimental maps in methods like MICA. |
| RosettaGenFF [21] | Force Field | Physics-based energy function for small molecules and proteins. | Used for conformational sampling and energy evaluation in tools like EMERALD-ID. |
Integrated Workflow for Ligand Binding Validation
The synergy between different method categories is leading to more powerful and reliable integrated workflows. The following diagram illustrates how geometry-based, physical, and deep learning principles can be combined.
This integrated approach, as exemplified by LumiNet [79] and CORDIAL [78], leverages the powerful pattern recognition of deep learning while grounding predictions in well-established physical laws. The DL module learns to extract structural features and map them into key physical parameters, which are then used within a physics-based scoring function or framework to generate a final, interpretable prediction. This strategy enhances generalizability and provides researchers with actionable insights, such as identifying critical atom pairs contributing to binding.
The benchmarking data reveals a nuanced landscape. Geometry and physics-based methods provide robust, interpretable tools for specific tasks like ligand identification, often without requiring extensive training data. Traditional machine learning methods offer a solid baseline but may be surpassed by more complex deep learning architectures. Deep learning models, particularly those employing 3D convolutions, graph networks, and multimodal integration, are setting new performance standards in structure modeling, affinity prediction, and binding site identification.
A critical finding is that a model's performance on a standard test set can be a poor indicator of its real-world utility. The ability to generalize to novel protein families and unseen ligands, as demonstrated by CORDIAL and LABind, is the true benchmark of a robust method. The most promising future direction lies in hybrid approaches that combine the data-driven power of deep learning with the fundamental constraints of physical laws, creating models that are not only accurate but also generalizable and interpretable for researchers in drug discovery and structural biology.
The accurate identification of protein-ligand binding sites is a fundamental challenge in structural biology and computer-aided drug discovery. Over the past three decades, more than 50 computational methods have been developed to predict binding sites from protein structures, representing a paradigm shift from geometry-based approaches to modern machine learning techniques [81] [82]. The evaluation of these methods has traditionally relied on benchmark datasets such as sc-PDB, PDBbind, binding MOAD, COACH420, and HOLO4K [82]. However, these established datasets often include 1:1 protein-ligand complexes or consider asymmetric units, which may not accurately represent biologically relevant binding interfaces [81] [82].
The introduction of the LIGYSIS dataset addresses critical limitations in previous benchmarking resources by aggregating biologically relevant unique protein-ligand interfaces across biological units of multiple structures from the same protein [81] [83]. This approach represents a significant advancement for method evaluation, as the asymmetric unit (the smallest portion of a crystal structure that can reproduce the complete unit cell) often does not correspond to the true biological assembly and can introduce artificial crystal contacts or redundant protein-ligand interfaces [82]. The LIGYSIS dataset comprises approximately 30,000 proteins with bound ligands, with a human subset of 3,448 proteins serving as a robust benchmark for objective assessment of prediction capabilities [82].
This article examines how LIGYSIS enables more accurate evaluation of ligand binding site prediction methods and provides a comprehensive comparison of computational tools within this improved validation framework.
The LIGYSIS Advantage: Dataset Design and Innovations
Fundamental Improvements Over Previous Datasets
LIGYSIS introduces several key innovations that address fundamental limitations in previous datasets. Unlike earlier resources that typically include 1:1 protein-ligand complexes or consider asymmetric units, LIGYSIS aggregates biologically relevant protein-ligand interfaces across different biological assemblies of the same protein deposited in the PDBe [81] [83]. This approach consistently considers biological units, which is critical for any analysis investigating molecular interactions at the residue or atomistic level [82].
The distinction between asymmetric units and biological units is crucial for accurate binding site characterization. The asymmetric unit represents the smallest portion of a crystal structure that can reproduce the complete unit cell through symmetry operations, but it may not correspond to the biologically functional assembly. The biological unit constitutes the actual macromolecular assembly that functions in biological contexts, which may comprise one, multiple copies, or a portion of the asymmetric unit [82]. This distinction is illustrated by PDB: 1JQY, present in the HOLO4K dataset, where the asymmetric unit contains three copies of a homo-pentamer, while the biological unit consists of a single pentamer [82]. Analyzing the asymmetric unit can therefore introduce artificial crystal contacts that do not represent genuine biological interactions.
Dataset Composition and Accessibility
The full LIGYSIS dataset encompasses approximately 65,000 protein-ligand binding sites across 25,000 proteins, representing one of the most comprehensive resources available [83] [84]. The publicly accessible LIGYSIS-web platform provides free access to this database without login requirements, featuring a Python Flask web application with a JavaScript frontend that integrates the 3Dmol.js structure viewer with dynamic tables and Chart.js graphs for interactive analysis [83]. Researchers can upload their own structures in PDB or mmCIF format for analysis, visualization, and download, significantly enhancing the utility of the resource for practical research applications [83].
LIGYSIS defines binding sites by clustering ligands using their protein interaction fingerprints to identify unique binding sites, as described by Utgés et al. [82]. This methodology ensures the removal of redundant protein-ligand interfaces, providing a more accurate representation of biologically relevant binding sites compared to previous datasets.
Comprehensive Performance Benchmarking on LIGYSIS
Experimental Design and Evaluation Metrics
The most extensive benchmark study to date, conducted by Utgés et al., evaluated 13 original ligand binding site prediction methods and 15 variants against the human subset of LIGYSIS using 10 different metrics [81] [82]. This analysis represents the largest benchmark in the field in terms of dataset size (2,775 structures), number of methods compared (28 total), and metrics employed [82]. The study prioritized open-source, peer-reviewed methods that are easy to install, ensuring reproducibility and practical utility for researchers [82].
The benchmark included methods spanning 30 years of research development, focusing on the latest machine learning-based approaches such as VN-EGNN, IF-SitePred, GrASP, PUResNet, and DeepPocket, while also including established methods like P2Rank, PRANK, and fpocket, and earlier geometry-based techniques including PocketFinder, Ligsite, and Surfnet [82]. The researchers proposed top-N+2 recall as a universal benchmark metric for ligand binding site prediction, addressing variations in the number of binding sites per protein and providing a standardized evaluation framework [81].
Table 1: Performance Comparison of Ligand Binding Site Prediction Methods on LIGYSIS
| Method | Type | Recall (%) | Precision Improvement with Re-scoring | Key Features |
|---|---|---|---|---|
| fpocketPRANK | Hybrid (Geometry + ML) | 60 | N/A | fpocket predictions re-scored by PRANK |
| DeepPocket | Machine Learning | 60 | N/A | Convolutional neural networks on grid voxels |
| IF-SitePred | Machine Learning | 39 | 14% recall improvement | ESM-IF1 embeddings with LightGBM models |
| Surfnet | Geometry-based | N/A | 30% precision improvement | Molecular surface geometry analysis |
| P2Rank | Machine Learning | N/A | N/A | Random forest on solvent accessible surface points |
| VN-EGNN | Machine Learning | N/A | N/A | Virtual nodes with equivariant graph neural networks |
Key Findings and Performance Trends
The benchmark results revealed substantial performance variations among methods. Re-scoring of fpocket predictions by PRANK and DeepPocket displayed the highest recall at 60%, while IF-SitePred showed the lowest recall at 39% [81] [82]. The study demonstrated the detrimental effect of redundant binding site prediction on performance metrics and highlighted the beneficial impact of stronger pocket scoring schemes, with improvements up to 14% in recall for IF-SitePred and 30% in precision for Surfnet through appropriate re-scoring approaches [81].
The research also highlighted how method design influences performance characteristics. Machine learning methods employed diverse architectural strategies: VN-EGNN combines virtual nodes with equivariant graph neural networks using ESM-2 embeddings [82]; IF-SitePred utilizes ESM-IF1 embeddings with 40 different LightGBM models [82]; GrASP employs graph attention networks for semantic segmentation on surface protein atoms [82]; PUResNet combines deep residual and convolutional neural networks on grid voxels [82]; and DeepPocket exploits convolutional neural networks to re-score and extract pocket shapes from fpocket candidates [82].
Experimental Protocols and Methodologies
Benchmarking Methodology
The comparative evaluation executed all methods with their standard settings to replicate real-world usage conditions [82]. Methods were assessed based on multiple performance metrics including recall, precision, F1-score, and the proposed top-N+2 recall [81]. The benchmark specifically analyzed factors such as the number of predicted ligand sites, their size, shape, proximity, overlap, and redundancy relative to the LIGYSIS reference dataset [82].
The experimental workflow involved:
- Protein Structure Preparation: Utilizing biological units from LIGYSIS instead of asymmetric units to avoid artificial crystal contacts [82]
- Binding Site Prediction: Running all methods with default parameters on the curated structures
- Performance Quantification: Comparing predictions against experimentally validated binding sites from LIGYSIS
- Re-scoring Analysis: Applying alternative scoring schemes to evaluate performance improvements [81]
LIGYSIS Dataset Construction
The LIGYSIS dataset was constructed through a meticulous process:
- Collection of protein-ligand complexes for 3,448 human proteins [82]
- Identification of biologically relevant protein-ligand interactions according to BioLiP criteria [82]
- Consideration of interactions across PISA-defined biological assemblies of multiple entries deposited in the PDBe [82]
- Clustering of ligands using protein interaction fingerprints to identify unique binding sites [82]
- Removal of redundant protein-ligand interfaces to create a non-redundant benchmark set
Table 2: Essential Research Reagents and Computational Resources
| Resource Name | Type | Function in Research | Access Information |
|---|---|---|---|
| LIGYSIS-web | Database | Hosts 65,000 protein-ligand binding sites for analysis | https://www.compbio.dundee.ac.uk/ligysis/ |
| LIGYSIS (Dataset) | Benchmark | Curated reference dataset for method evaluation | 30,000 proteins with bound ligands |
| BioLiP | Database | Provides biologically relevant protein-ligand interactions | Used for defining relevant interactions |
| PDBe | Database | Source of protein structures and biological assemblies | https://www.ebi.ac.uk/pdbe/ |
| PISA | Software | Defines biological units from crystal structures | Integrated in LIGYSIS pipeline |
Visualization of Workflows and Relationships
LIGYSIS Dataset Construction Workflow
Method Benchmarking Pipeline
Impact of Re-scoring on Method Performance
Implications for Drug Discovery and Structural Biology
The introduction of LIGYSIS and comprehensive benchmarking studies have significant implications for drug discovery and structural biology research. The demonstrated impact of non-redundant datasets on method evaluation underscores the importance of using biologically relevant benchmarks for developing and validating computational tools [81] [82].
For drug discovery applications, accurate binding site prediction is crucial for understanding protein function and modulating therapeutic targets [81] [82]. The benchmark results provide guidance for selecting appropriate methods based on performance characteristics, with hybrid approaches like fpocketPRANK and DeepPocket showing particularly strong recall rates [81]. The significant performance improvements achievable through re-scoring strategies (up to 30% precision and 14% recall enhancements) highlight the importance of robust scoring functions in binding site prediction pipelines [81].
The field continues to evolve with emerging challenges, including the application of binding site prediction methods to cryo-EM structures where determining ligand conformation and identity remains challenging at typical resolutions [21]. Tools like EMERALD-ID address this gap by combining physical forcefields with density agreement to rank ligand identities in cryo-EM data, achieving 44% success in identifying deposited ligands [21]. Continued development and benchmarking of methods on comprehensive, non-redundant datasets like LIGYSIS will be essential for advancing structural biology applications in drug discovery.
The introduction of the LIGYSIS dataset represents a significant advancement in the evaluation of ligand binding site prediction methods. By aggregating biologically relevant protein-ligand interfaces across biological units and removing redundant interactions, LIGYSIS provides a more accurate benchmark than previous datasets reliant on asymmetric units or 1:1 protein-ligand complexes [81] [82].
Comprehensive benchmarking on LIGYSIS has revealed substantial performance variations among methods, with re-scoring approaches demonstrating significant improvements in recall and precision [81]. The recommendation for open-source sharing of both methods and benchmarks promotes reproducibility and continued advancement in the field [81]. As structural biology continues to evolve with increasing numbers of cryo-EM structures and computational methods, robust non-redundant datasets like LIGYSIS will remain essential for validating ligand binding site prediction tools and advancing drug discovery research.
Conclusion
The reliable validation of ligands in electron density maps is paramount for deriving meaningful biological insights and making informed decisions in drug discovery. As this article has outlined, a multi-faceted approach is necessary, combining a solid understanding of foundational principles with the application of modern AI-driven methods, rigorous troubleshooting of model quality, and adherence to comprehensive, multi-metric validation standards. The field is rapidly evolving, with future directions pointing towards increased standardization in cryo-EM processing, the development of more sophisticated deep learning architectures, and the community-wide adoption of benchmarks like LIGYSIS and challenge-driven best practices. By embracing these tools and methodologies, structural biologists can significantly enhance the accuracy of their models, thereby accelerating the development of new therapeutics.
References
- 1. Protein–Ligand Interactions: Recent Advances in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12525376/]
- 2. LABind: identifying protein binding ligand-aware sites via ... [https://www.nature.com/articles/s41467-025-62899-0]
- 3. Computational modeling of protein–ligand interactions [https://www.sciencedirect.com/science/article/pii/S0959440X25001708]
- 4. Protein–ligand binding affinity prediction with edge ... [https://www.sciencedirect.com/science/article/pii/S2589004222021654]
- 5. The Development of Quantitative Structure-Binding Affinity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2773514/]
- 6. Labeled dataset of X-ray protein ligand images in 3D point ... [https://www.nature.com/articles/s41597-025-06002-8]
- 7. A workflow to create a high-quality protein–ligand binding ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d4dd00357h]
- 8. Considering best practices in color palettes for molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9377702/]
- 9. Room-temperature X-ray fragment screening with serial ... [https://www.nature.com/articles/s41467-025-64918-6]
- 10. High-resolution protein modeling through Cryo-EM and AI [https://pmc.ncbi.nlm.nih.gov/articles/PMC12590954/]
- 11. Cryo-EM ligand building using AlphaFold3-like model and ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1013367]
- 12. Perspective: Structure determination of protein-ligand ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2023.1113762/full]
- 13. X-ray crystallography of protein-ligand interactions [https://pubmed.ncbi.nlm.nih.gov/15939997/]
- 14. High throughput cryo-EM provides structural understanding ... [https://www.sciencedirect.com/science/article/pii/S0969212625001923]
- 15. Expanding Automated Multiconformer Ligand Modeling to ... [https://elifesciences.org/reviewed-preprints/103797v1]
- 16. Cryo-EM ligand building using AlphaFold3-like model and ... [https://pubmed.ncbi.nlm.nih.gov/40788932/]
- 17. Flexibilization of Science, Cognitive Biases, and the COVID ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7449982/]
- 18. Of problems and opportunities—How to treat ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9424839/]
- 19. 'All That Glitters Is Not Gold': High-Resolution Crystal ... [https://www.mdpi.com/1422-0067/22/13/6830]
- 20. A high-resolution data set of fatty acid-binding protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12315585/]
- 21. Automated identification of small molecules in cryoelectron ... [https://www.sciencedirect.com/science/article/abs/pii/S0969212625002503]
- 22. FLEXR-MSA: electron-density map comparisons of sequence ... [https://journals.iucr.org/m/issues/2025/02/00/jt5079/]
- 23. A chemical interpretation of protein electron density maps in ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0236894]
- 24. EDSTATS — CCP4 documentation [https://www.ccp4.ac.uk/html/edstats.html]
- 25. Real Space Correlation Coefficient [https://www.blopig.com/blog/2020/07/real-space-correlation-coefficient/]
- 26. Machine learning to estimate the local quality of protein ... [https://www.nature.com/articles/s41598-021-02948-y]
- 27. Measurement of atom resolvability in cryo-EM maps with Q - scores [https://www.nature.com/articles/s41592-020-0731-1?error=cookies_not_supported&code=e7b17eef-e435-4b13-bb42-4017b6213381]
- 28. MolProbity: all-atom structure validation for macromolecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2803126/]
- 29. Validation Options - MolProbity Help [http://molprobity.manchester.ac.uk/help/validation_options/validation_options.html]
- 30. Beyond rigid docking: deep learning approaches for fully ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12406700/]
- 31. FLEXR-MSA: electron-density map comparisons of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11878447/]
- 32. Denoising and iterative phase recovery reveal low- ... [https://www.nature.com/articles/s42003-025-09031-6]
- 33. Denoising and iterative phase recovery reveal low-occupancy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12644873/]
- 34. Geometry-Awareness for Fast and Accurate Protein-Ligand ... [https://arxiv.org/html/2508.09499v1]
- 35. Ligand Identification in CryoEM and X-ray Maps Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11383698/]
- 36. Check My Blob | Oxford Protein Informatics Group [https://www.blopig.com/blog/2018/11/check-my-blob/]
- 37. About - CheckMyBlob [https://checkmyblob.bioreproducibility.org/server/about/]
- 38. CheckMyBlob | Identify ligands in electron density using ... [https://checkmyblob.bioreproducibility.org/]
- 39. MIC: A deep learning tool for assigning ions and waters in ... [https://www.nature.com/articles/s41467-025-61315-x]
- 40. Simplified quality assessment for small-molecule ligands in ... [https://www.sciencedirect.com/science/article/pii/S0969212621003713]
- 41. A Point Cloud Graph Neural Network for Protein–Ligand ... [https://www.mdpi.com/1422-0067/25/17/9280]
- 42. Tokenizing Electron Cloud in Protein-Ligand Interaction ... [https://arxiv.org/html/2505.19014v2]
- 43. Advancing structure modeling from cryo-EM maps with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12224893/]
- 44. Deep learning for reconstructing protein structures from ... [https://www.sciencedirect.com/science/article/pii/S0959440X23000106]
- 45. Ligand identification in CryoEM and X-ray maps using ... [https://pubmed.ncbi.nlm.nih.gov/39700427]
- 46. UCSF ChimeraX: Tools for structure building and analysis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10588335/]
- 47. Practical structure prediction with AlphaFold 3, Boltz 2 ... [https://www.rbvi.ucsf.edu/chimerax/data/pncc-workshop-sep2025/pncc_boltz.html]
- 48. Making Boltz Structure Predictions in ChimeraX [https://www.rbvi.ucsf.edu/chimerax/data/boltz-apr2025/boltz_help.html]
- 49. Tool: AlphaFold [https://www.cgl.ucsf.edu/chimerax/docs/user/tools/alphafold.html]
- 50. A Tool for Segmentation of Secondary Structures in 3D ... [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2021.710119/full]
- 51. ChimeraX tutorial: Analysing AlphaFold Predictions and More [https://www.cgl.ucsf.edu/chimerax/data/vorlander-jan2025/ChimeraX_tutorial.html]
- 52. Validation of ligands in macromolecular structures ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5947763/]
- 53. High-Resolution Crystal Structures of Ligand-Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8268033/]
- 54. Validation methods for low-resolution fitting of atomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4543555/]
- 55. Automatic and accurate ligand structure determination ... [https://www.nature.com/articles/s41467-023-36732-5]
- 56. Ligand Structure Quality in PDB Structures [https://www.rcsb.org/docs/general-help/ligand-structure-quality-in-pdb-structures]
- 57. Expanding automated multiconformer ligand modeling to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12208665/]
- 58. Outcomes of the EMDataResource Cryo-EM Ligand ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10854310/]
- 59. User guide to the wwPDB EM validation reports [http://www.wwpdb.org/validation/2017/EMValidationReportHelp]
- 60. An expanded trove of fragment-bound structures for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11316629/]
- 61. Cryo-EM model validation recommendations based on ... [https://www.nature.com/articles/s41592-020-01051-w]
- 62. (IUCr) Q-score as a reliability measure for protein, nucleic acid ... [https://journals.iucr.org/d/issues/2025/08/00/ic5125/index.html]
- 63. Measurement of Atom Resolvability in CryoEM Maps with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7446556/]
- 64. Tools for Model Building and Optimization into Near-Atomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5103630/]
- 65. Assessing the Quality of 3D Structures [https://www.rcsb.org/docs/general-help/assessing-the-quality-of-3d-structures]
- 66. Electron Density Maps and Coefficient Files [https://www.rcsb.org/docs/general-help/electron-density-maps-and-coefficient-files]
- 67. Validation of electron-microscopy maps using solution ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11220840/]
- 68. From Density to Atoms: Deep Learning Tools Advancing ... [https://neurosnap.ai/blog/post/from-density-to-atoms-deep-learning-tools-advancing-cryo-em/68cf0028c0e73282471b6a75]
- 69. Cryo-EM ligand building using AlphaFold3-like model and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12364316/]
- 70. Multimodal deep learning integration of cryo-EM and ... [https://www.nature.com/articles/s42004-025-01718-5]
- 71. Validation tests for cryo-EM maps using an independent ... [https://www.sciencedirect.com/science/article/pii/S2590152420300143]
- 72. AI-based quality assessment methods for protein structure ... [https://www.sciencedirect.com/science/article/pii/S2665928X25000017]
- 73. A labeled dataset for AI-based cryo-EM map enhancement [https://www.sciencedirect.com/science/article/pii/S2001037025002570]
- 74. Paper Published: Cryo-EM Ligand Modeling Challenge [https://www.rcsb.org/news/6679793750bece1b815a1450]
- 75. Cryo-EM Ligand Modeling Challenge Results & ... [https://www.emdataresource.org/news/ligand-challenge-publication.html]
- 76. Pharmacophore modeling: advances, limitations, and ... [https://www.dovepress.com/pharmacophore-modeling-advances-limitations-and-current-utility-in-dru-peer-reviewed-fulltext-article-JRLCR]
- 77. Outcomes of the EMDataResource cryo-EM Ligand ... [https://www.nature.com/articles/s41592-024-02321-7]
- 78. A generalizable deep learning framework for structure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12557521/]
- 79. Robust protein–ligand interaction modeling through ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc07405j]
- 80. Binding Affinity Prediction: From Conventional to Machine ... [https://arxiv.org/html/2410.00709v2]
- 81. Comparative evaluation of methods for the prediction ... [https://pubmed.ncbi.nlm.nih.gov/39529176/]
- 82. Comparative evaluation of methods for the prediction of ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00923-z]
- 83. LIGYSIS-web: a resource for the analysis of protein-ligand ... [https://pubmed.ncbi.nlm.nih.gov/40377089/]
- 84. PDBe tools for an in‐depth analysis of small molecules ... [https://www.semanticscholar.org/paper/PDBe-tools-for-an-in%E2%80%90depth-analysis-of-small-in-the-Choudhary-Kunnakkattu/3a94af9930686aa8df19e916f7a93d7b6e394fcc]