X-ray Crystallography vs. Cryo-EM: A Structural Biologist's Guide to Choosing the Right Tool
This article provides a comprehensive comparison of X-ray crystallography and cryo-electron microscopy (cryo-EM) for researchers and drug development professionals.
X-ray Crystallography vs. Cryo-EM: A Structural Biologist's Guide to Choosing the Right Tool
Abstract
This article provides a comprehensive comparison of X-ray crystallography and cryo-electron microscopy (cryo-EM) for researchers and drug development professionals. It covers the foundational principles, current methodology, practical troubleshooting, and a direct validation of both techniques. Drawing on the latest data and trends, including the rising contribution of cryo-EM to the PDB, it offers a clear framework for selecting the optimal method based on project goals, sample characteristics, and resource constraints. The scope extends to advanced applications like time-resolved studies and the analysis of membrane proteins and small molecules, concluding with a forward-looking synthesis on the convergent future of structural biology techniques.
The Evolving Landscape of Structural Biology: From X-ray Dominance to the Cryo-EM Revolution
Structural biology relies on powerful techniques to determine the three-dimensional structures of biological macromolecules, with X-ray crystallography and single-particle cryo-electron microscopy (cryo-EM) serving as two pivotal methods. For decades, X-ray crystallography has been the dominant technique for solving atomic-resolution structures, relying on the fundamental principle of Bragg's Law to interpret diffraction patterns from crystalline samples [1] [2]. In contrast, single-particle cryo-EM has emerged more recently as a revolutionary technique that can determine high-resolution structures from non-crystalline, frozen-hydrated samples by computationally analyzing thousands of individual particle images [1] [3].
These two methods are not mutually exclusive but rather highly complementary approaches that together provide a more comprehensive understanding of biological structures and mechanisms [1] [4]. This guide provides a detailed objective comparison of their core principles, methodologies, and applications, specifically framed within the context of structural biology research and drug development. We will examine the theoretical foundations, experimental workflows, technical requirements, and output characteristics of both techniques, enabling researchers to select the most appropriate method for their specific structural biology challenges.
Core Principles and Theoretical Foundations
Bragg's Law in X-ray Crystallography
X-ray crystallography is founded on Bragg's Law, a fundamental principle that describes the condition for constructive interference of X-rays diffracted by crystalline materials. Formulated in 1912 by Sir William Lawrence Bragg and his father Sir William Henry Bragg, this law establishes the relationship between the X-ray wavelength, the distance between atomic planes in the crystal, and the angle of diffraction [5] [6].
The Bragg's Law equation is expressed as: nλ = 2d sinθ, where:
- n is an integer representing the order of the reflection
- λ is the wavelength of the incident X-ray beam
- d is the spacing between consecutive atomic planes in the crystal
- θ is the angle between the incident ray and the scattering planes [5] [7] [6]
In practical terms, Bragg's Law governs the specific angles at which a crystal will produce strong diffraction peaks due to constructive interference when illuminated with X-rays [5]. These "Bragg reflections" occur when X-rays scattering from different crystal planes remain in phase, producing intense spots in the diffraction pattern that can be measured and used to compute the electron density and ultimately determine the atomic structure of the crystallized molecule [7] [6]. The resolution limit in crystallography is directly related to the smallest d-spacing that can be measured, which depends on the maximum diffraction angle (θ) achievable [7].
Single-Particle Analysis in Cryo-EM
Single-particle analysis in cryo-EM operates on fundamentally different principles than X-ray crystallography. Instead of relying on diffraction from crystalline samples, it directly images individual macromolecules using a transmission electron microscope and computationally reconstructs their three-dimensional structure [1] [3].
The core principle involves collecting two-dimensional projection images of thousands to millions of identical, randomly oriented macromolecules preserved in a thin layer of vitreous ice [1] [3]. Through sophisticated image processing algorithms, these particles are identified, aligned, classified, and averaged to generate a three-dimensional electron density map [1]. The reconstruction process relies on the mathematical principles of tomography and the ability to computationally determine the relative orientation of each particle based on common lines in Fourier space [1].
The resolution of the final reconstruction depends on multiple factors including the number of particle images collected, the homogeneity of the sample, the accuracy of particle alignment, the performance of the electron microscope (particularly detector technology and beam coherence), and the effectiveness of computational corrections for microscope aberrations and beam-induced particle movement [1] [8]. Unlike crystallography, where resolution is determined by the highest angle diffraction data, cryo-EM resolution is typically estimated using statistical measures such as the Fourier Shell Correlation (FSC) between independently processed half-maps [8].
Methodological Comparison
Experimental Workflows
The experimental workflows for X-ray crystallography and single-particle cryo-EM differ significantly in their sample preparation, data collection, and data processing stages, each with distinct advantages and challenges.
X-ray Crystallography Workflow:
- Protein Purification: Obtaining highly pure, homogeneous protein samples in sufficient quantities (typically milligrams) [1]
- Crystallization: Screening hundreds to thousands of conditions to obtain well-ordered, diffraction-quality crystals through vapor diffusion, microbatch, or other methods [2]
- Cryo-cooling: Flash-freezing crystals in liquid nitrogen to minimize radiation damage during data collection [2]
- Data Collection: Mounting crystals in the X-ray beam (often at synchrotron sources) and collecting diffraction patterns at various orientations [2]
- Data Processing: Indexing and integrating diffraction spots, merging data from multiple crystals, and determining phases through molecular replacement, anomalous scattering, or other methods [2]
- Model Building and Refinement: Interpreting the electron density map to build an atomic model and iteratively refining it against the diffraction data [2]
Single-Particle Cryo-EM Workflow:
- Sample Preparation: Purifying the macromolecular complex and applying it to specialized grids [3]
- Vitrification: Rapidly plunging the grid into liquid ethane/propane to form a thin layer of vitreous ice that preserves molecules in near-native state [3]
- Screening: Identifying promising samples with optimal particle distribution and ice thickness [3]
- Data Collection: Automatically acquiring thousands to millions of particle images using high-end cryo-electron microscopes [1] [3]
- Image Processing: Computational steps including particle picking, 2D classification, 3D initial model generation, heterogeneous classification, and high-resolution refinement [1]
- Model Building and Refinement: Interpreting the cryo-EM density map to build and refine an atomic model [1]
Technical Requirements and Data Output
The technical requirements, sample characteristics, and data output differ substantially between the two techniques, making each suitable for different types of structural biology problems.
Table 1: Technical Comparison of X-ray Crystallography and Single-Particle Cryo-EM
| Parameter | X-ray Crystallography | Single-Particle Cryo-EM |
|---|---|---|
| Sample Requirement | Highly ordered 3D crystals | Purified macromolecules in solution |
| Sample Amount | 0.1-1 mg (typically larger quantities needed for screening) | <0.1 mg (significantly less material required) [1] |
| Sample State | Molecules constrained in crystal lattice | Near-native state in vitreous ice [1] [4] |
| Molecular Weight Range | No inherent upper limit (small molecules to large complexes) | Typically >50 kDa for high resolution, smaller molecules possible with advanced methods [4] |
| Structural Heterogeneity | Challenging for flexible or dynamic systems | Can resolve multiple conformational states through classification [1] [4] |
| Typical Resolution Range | 1.0-3.5 Å (atomic to near-atomic) | 1.5-4.0 Å for well-behaved samples (near-atomic to sub-nanometer) [1] [8] |
| Membrane Protein Success | Challenging but possible with detergent screening and lipidic cubic phase | Particularly well-suited with many recent successes [4] |
| Data Collection Time | Minutes to hours per dataset | Days to weeks for high-resolution data [3] |
| Key Limiting Factor | Crystal quality and diffraction resolution | Particle homogeneity, microscope performance, computational resources [1] [8] |
Table 2: Statistical Comparison of Structure Determination Methods (Based on PDB Depositions)
| Method | Structures in PDB (2023) | Percentage of Total | Historical Dominance | Trend |
|---|---|---|---|---|
| X-ray Crystallography | ~9,601 | ~66% | ~86% of all structures | Declining proportion but still dominant [2] |
| Cryo-EM | ~4,579 | ~32% | Minimal until 2015 | Rapidly increasing (resolution revolution) [9] [2] |
| NMR | ~272 | ~2% | Consistently small contribution | Stable for small proteins and dynamics [2] |
Comparative Experimental Data
Resolution and Data Quality Metrics
The assessment of resolution and data quality follows different conventions in X-ray crystallography and single-particle cryo-EM, reflecting their different physical principles and data characteristics.
In X-ray crystallography, resolution is traditionally determined by where to truncate the diffraction data based on quality metrics. Key statistics include:
- Signal-to-noise ratio (I/σ(I)): Traditionally truncated when [8]<="" been="" below="" data="" falls="" has="" highest="" in="" include="" li="" questioned="" recommendations="" resolution="" shell,="" standard="" the="" this="" though="" to="" weaker="" with="" σ(i)>="">
- [8]<="" been="" below="" data="" falls="" has="" highest="" in="" include="" li="" questioned="" recommendations="" resolution="" shell,="" standard="" the="" this="" though="" to="" weaker="" with="" σ(i)>="">Rmerge: Measures agreement between multiple measurements of the same reflection, but is inherently flawed as it depends on multiplicity [8] [8]<="" been="" below="" data="" falls="" has="" highest="" in="" include="" li="" questioned="" recommendations="" resolution="" shell,="" standard="" the="" this="" though="" to="" weaker="" with="" σ(i)>="">
- Rmeas: Multiplicity-independent R-factor that provides a more realistic measure of precision [8]
- Rp.i.m.: Precision-indicating merging R-factor for merged reflections [8]
- CC1/2: Pearson's correlation coefficient between random halves of measurements, considered a more reliable quality indicator [8]
The resolution in crystallography represents the smallest lattice spacing (dmin) that can be measured according to Bragg's law, typically reported based on the highest resolution shell where data quality statistics meet certain thresholds [8] [7].
In single-particle cryo-EM, resolution is typically determined using the Fourier Shell Correlation (FSC), which measures the normalized cross-correlation coefficient between two independently determined half-maps in successive resolution shells [8]. The most widely accepted threshold is FSC = 0.143, known as the "gold standard," though the appropriate threshold remains debated [8]. Unlike crystallography, cryo-EM maps often exhibit resolution anisotropy, where resolution varies with direction, and local resolution variations across different regions of the map [8].
Table 3: Resolution Metrics and Quality Indicators
| Quality Measure | X-ray Crystallography | Single-Particle Cryo-EM |
|---|---|---|
| Primary Resolution Metric | Bragg limit (dmin) based on diffraction angles | Fourier Shell Correlation (FSC) between half-maps |
| Standard Threshold | Variable (I/σ, R-factors, CC1/2) | FSC = 0.143 ("gold standard") [8] |
| Typical High Resolution | 0.48 Å (current record) [8] | ~1.2 Å (current record for single-particle) [8] |
| Atomic Resolution Definition | ~1.2 Å or better ("Sheldrick's criterion") [8] | Not strictly defined, but ~1.5-2.0 Å considered near-atomic |
| Key Quality Indicators | I/σ(I), Rmerge, Rmeas, Rp.i.m., CC1/2, completeness, multiplicity | FSC, Q-score, particle orientation distribution, local resolution variations |
| Anisotropy | Can occur in diffraction (anisotropic truncation) | Common in reconstruction (direction-dependent resolution) [8] |
Complementary Applications and Hybrid Approaches
Rather than competing techniques, X-ray crystallography and single-particle cryo-EM increasingly serve complementary roles in structural biology, with hybrid approaches leveraging the strengths of both methods [1] [4].
One powerful application is docking crystallographic structures into cryo-EM maps, where high-resolution atomic models of individual components or homologs are fitted into lower-resolution cryo-EM maps of larger complexes [1]. This approach has been successfully applied to systems such as:
- Ryanodine receptor: The crystal structure of the SPRY2 domain was docked into a 10 Å resolution cryo-EM map, with the docked model showing excellent agreement (2.1 Å RMSD) with later high-resolution cryo-EM structures [1]
- Yeast RNA exosome complex: Docking of crystallographic structures into cryo-EM maps revealed mechanisms of RNA substrate recruitment and processing [1]
- Viral capsids and cytoskeletal filaments: Early applications demonstrating the power of combining crystallographic and EM data [1]
Docking methods include both rigid-body docking (using programs like Situs, EMfit, UCSF Chimera) and flexible docking (using Flex-EM, MDFF, iMODFIT, Rosetta) that can accommodate conformational differences between crystal structures and their counterparts in cryo-EM maps [1].
Conversely, cryo-EM can assist crystallography by providing initial phase information through molecular replacement, helping to solve the notorious "phase problem" in crystallography [4]. Low-to-medium resolution cryo-EM maps can serve as starting models for phasing crystallographic data, particularly for large complexes that are difficult to solve by traditional crystallographic phasing methods [1] [4].
Research Reagent Solutions
Successful structure determination by either method requires specialized reagents and materials optimized for each technique's specific requirements.
Table 4: Essential Research Reagents and Materials
| Reagent/Material | Function | Application in |
|---|---|---|
| Crystallization Screens | Commercial kits with predefined conditions to identify initial crystallization hits | X-ray Crystallography |
| Cryoprotectants | Compounds (glycerol, ethylene glycol) to prevent ice formation during crystal cryo-cooling | X-ray Crystallography |
| Lipidic Cubic Phase (LCP) | Matrix for crystallizing membrane proteins and peptides | X-ray Crystallography |
| EM Grids | Specimen supports (gold or copper with continuous or holey carbon films) for sample application | Cryo-EM |
| Vitrification Systems | Instruments for plunge-freezing samples in ethane/propane for vitreous ice formation | Cryo-EM |
| Direct Electron Detectors | Advanced cameras with high sensitivity and fast readout for recording particle images | Cryo-EM |
| Image Processing Software | Programs (RELION, cryoSPARC, EMAN2) for particle picking, classification, and reconstruction | Cryo-EM |
| Crystallography Data Suites | Software (HKL-2000, XDS, CCP4, PHENIX) for data processing, phasing, and refinement | X-ray Crystallography |
| Model Building Tools | Programs (Coot, O) for interpreting density maps and building atomic models | Both Methods |
X-ray crystallography and single-particle cryo-EM represent two powerful, complementary approaches for determining macromolecular structures, each with distinct advantages and limitations rooted in their fundamental physical principles. X-ray crystallography, governed by Bragg's Law, continues to provide the highest resolution structures for samples that can form well-ordered crystals, remaining the most productive method in terms of total structures determined [2]. Single-particle cryo-EM has undergone a "resolution revolution" that now enables near-atomic resolution for many macromolecular complexes that resist crystallization, particularly membrane proteins, large complexes, and dynamic assemblies [1] [3].
The choice between techniques depends on the specific biological question, sample characteristics, and available resources. For well-behaved proteins that crystallize readily, X-ray crystallography remains the most efficient path to atomic resolution. For large, flexible, or heterogeneous complexes—particularly those difficult to crystallize—single-particle cryo-EM offers unparalleled advantages. Most significantly, these methods are increasingly used together in hybrid approaches that leverage their complementary strengths, such as docking high-resolution crystallographic structures into lower-resolution cryo-EM maps of larger complexes [1] [4].
As both technologies continue to advance, with crystallography pushing toward ever-higher resolutions and cryo-EM expanding its capabilities for smaller proteins and more complex heterogeneous samples, their synergistic application will undoubtedly drive future breakthroughs in understanding biological mechanisms and facilitating structure-based drug design.
Historical Context and the 'Resolution Revolution'
The field of structural biology, dedicated to elucidating the three-dimensional architecture of biological macromolecules, has undergone a profound transformation. For decades, X-ray crystallography stood as the undisputed cornerstone, responsible for the vast majority of structures in the Protein Data Bank (PDB). This landscape shifted dramatically with the advent of a "resolution revolution" in cryo-electron microscopy (cryo-EM), a technological leap that has redefined the possibilities of molecular visualization [10]. This revolution was primarily ignited by the introduction of direct electron detectors, which provided dramatically improved signal-to-noise ratios and enabled accurate correction of beam-induced motion, thereby unlocking near-atomic resolution for previously intractable targets [10]. Understanding the historical context and comparative capabilities of these two powerful techniques is essential for researchers, scientists, and drug development professionals seeking to determine the optimal strategy for their structural inquiries. This guide provides an objective comparison of their performance, supported by experimental data and detailed methodologies.
Historical Dominance of X-ray Crystallography
X-ray crystallography has a long and storied history, originating in the early 20th century following Wilhelm Conrad Röntgen's discovery of X-rays and the subsequent demonstration of X-ray diffraction by crystals by Max von Laue [11]. The development of this technique is firmly rooted in Bragg's Law (nλ = 2dsinϑ), which describes the condition for constructive interference and enables the determination of atomic positions from a crystal's diffraction pattern [11]. Its pivotal role in biology was cemented by the determination of the DNA double helix structure by Watson, Crick, Franklin, and Wilkins [12].
For generations, X-ray crystallography has been the workhorse of structural biology. As of September 2024, it accounts for approximately 84% of all structures deposited in the PDB, a testament to its dominance and reliability [13]. This technique has been instrumental in countless scientific discoveries, from elucidating enzyme mechanisms and solving membrane protein structures to facilitating structure-based drug design, as exemplified by the development of inhibitors for the SARS-CoV-2 main protease [10].
Table 1: Key Historical Milestones in X-ray Crystallography
| Year | Milestone | Key Figures/Study | Significance |
|---|---|---|---|
| 1895 | Discovery of X-rays | Wilhelm Conrad Röntgen | Enabled the development of diffraction-based imaging. |
| 1912 | Demonstration of X-ray diffraction | Max von Laue | Proved crystals could diffract X-rays. |
| 1915 | Development of Bragg's Law | William Henry and William Lawrence Bragg | Provided the foundational equation for analyzing diffraction data. |
| 1953 | Structure of DNA | Watson, Crick, Franklin, Wilkins | Revolutionized understanding of genetic information storage. |
| 1958 | First protein structure (myoglobin) | Kendrew et al. | Opened the door to atomic-level protein visualization [10]. |
| 2011 | β2-adrenergic receptor structure | Rasmussen et al. | Breakthrough in membrane protein crystallography using lipidic cubic phase [10]. |
| 2020 | SARS-CoV-2 main protease structure | Zhang et al. | Critical for antiviral drug discovery during the COVID-19 pandemic [10]. |
The Cryo-EM Resolution Revolution
While cryo-electron microscopy (cryo-EM) has existed for decades, its potential was fully unleashed in the 2010s, an era often termed the "resolution revolution" [12]. This transformation was catalyzed by major technological advancements, most notably the introduction of direct electron detectors [10]. These detectors provided dramatically improved signal-to-noise ratios, accurate electron counting, and rapid frame rates, which enabled the correction of beam-induced motion—a critical barrier to achieving high resolution [10].
A landmark achievement demonstrating this new capability was the determination of the TRPV1 ion channel structure at near-atomic resolution, revealing how this protein detects heat and pain [10]. This structure, which was previously intractable, showcased cryo-EM's power for solving complex membrane protein structures without the need for crystallization. The profound impact of these developments was recognized with the 2017 Nobel Prize in Chemistry for the development of cryo-EM [12].
The growth of the technique has been exponential. From contributing a negligible number of structures in the early 2000s, cryo-EM now accounts for a significant portion of new PDB deposits. In 2023, over 4,500 structures were solved by EM, representing nearly 32% of all releases that year [11]. This trend indicates that cryo-EM is poised to surpass X-ray crystallography as the most used method for determining new structures [9].
Objective Performance Comparison: X-ray Crystallography vs. Cryo-EM
A direct comparison of these two techniques reveals a complementary set of strengths and limitations, which are crucial for selecting the appropriate method for a given project. The following data synthesizes information from industry and academic sources to provide an objective performance comparison.
Table 2: Technical Comparison of X-ray Crystallography and Cryo-EM
| Parameter | X-ray Crystallography | Cryo-EM (Single-Particle) | Supporting Data & Context |
|---|---|---|---|
| Typical Resolution | Atomic (often <1.5 Å) | Near-atomic to Atomic (1.5-4 Å for many structures) [10] | Cryo-EM has not yet reached the physical limits set by radiation damage [9]. |
| Sample Requirement | High-purity, homogeneous, crystallizable protein. Requires 5+ mg at ~10 mg/mL [13]. | High-purity, homogeneous protein. Requires a small amount (≤0.5 mg) at low concentrations (≥0.5 mg/mL) [13]. | Crystallization is the largest hurdle in X-ray crystallography [13]. |
| Sample State | Crystalline solid. | Vitrified solution (frozen-hydrated). | Cryo-EM studies molecules in a near-native state without crystallization [10]. |
| Molecular Weight | No inherent size limit, but crystal quality can be an issue for large complexes [13]. | Ideal for large complexes (>150 kDa), though smaller targets are becoming feasible. | Well-ordered crystals become more difficult to obtain as target size and complexity increase [13]. |
| Throughput | High for established crystal systems; slow and uncertain for novel targets. | Rapid for data collection once grids are optimized; processing is automated but computationally intensive. | X-ray crystallography remains the workhorse for high-throughput structure determination [13]. |
| Membrane Proteins | Challenging; requires special methods like Lipid Cubic Phase (LCP). Successful for many GPCRs [13]. | Excellent; no crystallization needed. Ideal for large, flexible complexes. | The TRPV1 ion channel structure is a prime example of cryo-EM's power for membrane proteins [10]. |
| Time Resolution | Millisecond range with advanced mix-and-quench or XFEL methods [14]. | Currently limited, but development of time-resolved cryo-EM is underway. | Non-photo-initiated mixing methods in crystallography achieve single-millisecond range [14]. |
| PDB Deposition (2023) | ~9,601 structures (66% of total) [11]. | ~4,579 structures (31.7% of total) [11]. | NMR accounted for 272 structures (1.9%) in 2023 [11]. |
Key Research Reagent Solutions
The following table details essential materials and reagents used in these structural biology techniques.
Table 3: Essential Research Reagents and Their Functions
| Reagent / Material | Function | Application in X-ray Crystallography | Application in Cryo-EM |
|---|---|---|---|
| Lipidic Cubic Phase (LCP) | A membrane mimetic matrix for stabilizing membrane proteins. | Used for crystallizing GPCRs and other integral membrane proteins [13]. | Not typically used. |
| Detergents | Solubilizes membrane proteins by mimicking the lipid environment. | Essential for purifying and crystallizing membrane proteins [13]. | Used for purifying membrane proteins prior to grid preparation. |
| Cryo-Protectants | Prevents ice crystal formation during vitrification. | Used for flash-cooling crystals prior to data collection at synchrotrons. | Essential for creating the vitreous ice layer that embeds the sample [12]. |
| Heavy Atom Solutions | Contains atoms with high electron numbers (e.g., Gold, Uranium, Platinum). | Soaked into crystals for experimental phasing (e.g., SAD/MAD) [13]. | Used for negative staining to quickly assess sample quality and distribution (not for high-res). |
| Gold Grids | Sample support for electron microscopy. | Not used. | Standard support film for cryo-EM sample grids. |
| 15N / 13C Isotopes | Stable isotopes incorporated into recombinant proteins. | Not required for standard crystallography. | Not required for standard single-particle cryo-EM. Essential for NMR and solid-state NMR [13]. |
Detailed Experimental Protocols
Protocol 1: Time-Resolved X-ray Crystallography via Mix-and-Quench
This protocol, enabling time resolution in the sub-10 ms range, is used to capture transient enzymatic states [14].
- Crystal Preparation: Grow microcrystals of the target biomolecule (e.g., lysozyme).
- Reaction Initiation (Mixing): Rapidly mix the crystal slurry with a substrate or ligand solution using a specialized instrument. This step initiates the enzymatic reaction.
- Thermal Quenching: After a precise, variable delay (e.g., from 8 ms to seconds), the reaction is halted by rapidly freezing the mixture with a cryogen. This step "traps" the structural state at that specific time point.
- Data Collection: The quenched sample is transferred to a synchrotron beamline for X-ray diffraction data collection. As demonstrated, this can require as few as one crystal per time point [14].
- Data Processing & Analysis: Diffraction data are indexed, integrated, and scaled. Structural models are then built and refined for each time point to create a molecular movie of the reaction.
Protocol 2: Single-Particle Cryo-EM Workflow
This is the standard workflow for determining high-resolution structures from vitrified protein samples [10] [12].
- Sample Vitrification:
- A purified protein solution (at ~0.5-3 mg/mL) is applied to an EM grid.
- The grid is blotted with filter paper to create a thin liquid film.
- The grid is rapidly plunged into a cryogen (typically liquid ethane), freezing the water in a vitreous (non-crystalline) state and embedding the protein particles in a thin layer of ice.
- Data Collection:
- The vitrified grid is loaded into a high-end cryo-electron microscope (e.g., Titan Krios) equipped with a direct electron detector.
- The microscope automatically collects thousands of "micrographs" (images) of the sample, with the electron beam set to a low dose to minimize radiation damage.
- Image Processing:
- Particle Picking: Individual protein particles are automatically identified and extracted from the micrographs.
- 2D Classification: Extracted particles are grouped into classes representing different views of the protein.
- 3D Reconstruction: A preliminary 3D model is generated, and iterative refinements are performed to align all particle images to the model, ultimately reconstructing a high-resolution 3D electron density map.
- Atomic Model Building:
- An atomic model is built into the refined electron density map, either de novo or by fitting and adjusting a known homologous structure.
- The model is stereochemically refined and validated against the map data.
Emerging Trends and Synergistic Integration
The future of structural biology lies not in the supremacy of a single technique, but in their strategic integration and the application of artificial intelligence. A powerful trend is the combination of time-resolved X-ray methods with cryo-EM to visualize molecular movies of biological processes [15]. For instance, mix-and-quench crystallography can capture short-lived intermediates, while cryo-EM can provide detailed snapshots of larger, more complex functional states.
Furthermore, AI and deep learning are revolutionizing both fields. AI-based tools like AlphaFold have demonstrated remarkable accuracy in predicting protein structures from amino acid sequences [10]. These computational models are now being integrated directly into cryo-EM workflows, where they can serve as initial models for refinement, help in interpreting regions of low map quality, and even assess the local quality of a built structure [10] [16]. This AI integration is accelerating the exploration of protein structure-function relationships, directly impacting biomedical research and therapeutic development [10].
The "resolution revolution" in cryo-EM has fundamentally diversified the toolkit available to structural biologists, offering a powerful alternative to the long-dominant technique of X-ray crystallography. While crystallography remains the gold standard for high-throughput atomic-resolution structure determination of proteins that can be crystallized, cryo-EM excels at solving structures of large, flexible, and membrane-embedded complexes that defy crystallization. The choice between them is not a matter of which is universally better, but which is the most appropriate for a specific biological question. The most profound insights will increasingly come from the synergistic use of both methods, augmented by the predictive power of AI, to visualize the dynamic molecular machinery of life in unprecedented detail.
The field of structural biology is powered by three principal experimental techniques for determining the three-dimensional structures of biological macromolecules: X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and electron microscopy (EM), notably cryo-electron microscopy (cryo-EM). For researchers and drug development professionals, understanding the evolving market share and deposition statistics of the Protein Data Bank (PDB) is crucial for making informed decisions about which methodology to employ for their specific projects.
The PDB, the single global archive for three-dimensional structural data of proteins and nucleic acids, provides a clear window into the adoption rates and productivity of these techniques [17]. Historically, X-ray crystallography dominated the landscape, but recent years have witnessed a dramatic shift. The "resolution revolution" in cryo-EM, marked by significant improvements in detector technology and image processing software, has positioned it as a powerful competitor [3] [10]. This guide provides an objective, data-driven comparison of these techniques' current market share based on the latest PDB deposition statistics, offering a snapshot of the modern structural biology toolkit.
Quantitative Analysis of PDB Deposition Trends
The growth in structures released to the PDB has been exponential, reflecting the increasing importance of structural biology in biomedical research. The following table summarizes the total number of structures released annually over recent years, providing context for the methodological shifts discussed later.
Table 1: Total Number of PDB Structures Released Annually (2020-2025)
| Year | Total Number of Structures Released |
|---|---|
| 2020 | 13,982 [18] |
| 2021 | 12,569 [18] |
| 2022 | 14,253 [18] |
| 2023 | 14,447 [18] |
| 2024 | 15,293 [18] |
| 2025 | 15,689 [18] |
A breakdown of the deposition statistics by experimental method reveals a dynamic and shifting landscape. The following table synthesizes the most current data available on the market share of the three primary techniques.
Table 2: Market Share of Primary Structure Determination Techniques in the PDB
| Technique | Historical Context | Recent Annual Share (2023-2024) | Key Strengths |
|---|---|---|---|
| X-ray Crystallography | Dominant method; >86% of all deposited structures [17] | ~66% of new structures in 2023 [17] | Atomic resolution; well-established workflows; high throughput [17] [10] |
| Cryo-EM | Almost negligible in early 2000s [17] | ~31.7% of new structures in 2023 [17]; up to 40% of new deposits by 2023-2024 [17] | No crystallization needed; ideal for large complexes and membrane proteins [3] [19] |
| NMR | Consistently a smaller contributor [17] | ~1.9% of new structures in 2023 [17] | Studies proteins in solution; probes dynamics and flexibility [17] [10] |
Regional Deposition Analysis
Structural biology is a global endeavor, and the geographic distribution of PDB depositions highlights regional research focus and capacity. The data below, sourced from the wwPDB, shows the number of structures deposited by principal investigators based on their geographic location over a recent five-year period.
Table 3: PDB Depositions by Geographic Region (2020-2024)
| Year | North America | Europe | Asia | South America | Australia/New Zealand | Africa |
|---|---|---|---|---|---|---|
| 2020 | 4,757 [18] | 6,167 [18] | 3,379 [18] | 157 [18] | 409 [18] | 9 [18] |
| 2021 | 4,795 [18] | 4,741 [18] | 4,083 [18] | 125 [18] | 491 [18] | 17 [18] |
| 2022 | 5,347 [18] | 5,160 [18] | 4,663 [18] | 87 [18] | 443 [18] | 11 [18] |
| 2023 | 4,670 [18] | 5,669 [18] | 5,297 [18] | 92 [18] | 365 [18] | 11 [18] |
| 2024 | 5,861 [18] | 5,875 [18] | 6,617 [18] | 82 [18] | 388 [18] | 10 [18] |
This data indicates a robust and growing structural biology effort in Asia, which has now surpassed North America and Europe in the number of annual depositions [18]. This growth correlates with significant regional investments in research infrastructure, including cryo-EM facilities [20] [21] [19].
Experimental Protocols for Key Techniques
Protocol for Single-Particle Cryo-EM
Single-particle analysis is the workhorse of modern cryo-EM, allowing for the determination of high-resolution structures from millions of individual particle images [3].
Table 4: Key Research Reagent Solutions in Cryo-EM
| Item | Function |
|---|---|
| Cryo-EM Grids | Tiny mesh grids that hold the vitrified sample for imaging under the electron beam [3]. |
| Vitrification Agents (e.g., liquid ethane) | Used to flash-freeze the aqueous sample so that it forms a glassy (vitreous) ice instead of crystalline ice, which would damage the sample [3]. |
| Direct Electron Detector | A critical camera technology that directly counts incoming electrons with high sensitivity, enabling the "resolution revolution" [10]. |
| Image Processing Software (e.g., RELION, cryoSPARC) | Algorithms used for the complex tasks of 2D classification, 3D reconstruction, and refinement of the final atomic model [3] [19]. |
Workflow Steps:
- Sample Preparation: The purified biological sample is applied to a specially designed grid [3].
- Vitrification: The grid is plunged into a cryogen (like liquid ethane) cooled by liquid nitrogen. This rapid freezing immobilizes the molecules in a thin layer of vitreous ice, preserving their native state [3].
- Data Collection: The vitrified grid is loaded into a cryo-electron microscope (e.g., a Titan Krios or Talos Arctica). The microscope bombards the sample with electrons, and images are collected by a direct electron detector from different orientations [3] [10].
- Image Processing: This computationally intensive step involves several sub-steps:
- Particle Picking: Millions of individual particle images are automatically selected from the micrographs.
- 2D Classification: Particles are grouped into classes representing similar views.
- 3D Reconstruction: A initial model is generated and iteratively refined to produce a high-resolution 3D electron density map.
- Atomic Model Building: Researchers fit and refine an atomic model of the protein or complex into the final electron density map [10].
The following diagram illustrates this complex workflow.
Protocol for X-ray Crystallography
X-ray crystallography remains a dominant method due to its ability to provide atomic-resolution data, though it requires the often-challenging step of growing high-quality crystals [17] [10].
Workflow Steps:
- Crystallization: The purified macromolecule is induced to form a highly ordered crystal through extensive screening of conditions (e.g., pH, precipants) [17].
- Data Collection: A single crystal is exposed to a high-intensity X-ray beam (e.g., from a synchrotron source). The crystal diffracts the X-rays, producing a characteristic pattern of spots on a detector [17].
- Data Processing: The diffraction pattern is processed to determine the amplitude of the diffracted waves. The critical "phase problem" must be solved using methods like molecular replacement or experimental phasing (e.g., SAD/MAD) [17].
- Electron Density Map Calculation: The amplitudes and phases are combined to calculate an electron density map [17].
- Model Building and Refinement: An atomic model is built into the electron density and iteratively refined to improve its fit to the experimental data [17] [10].
The workflow for X-ray crystallography is shown in the diagram below.
The quantitative data from the PDB reveals a structural biology field in the midst of a significant transformation. While X-ray crystallography continues to be the most prolific method in terms of total annual deposits, its historical dominance is steadily being challenged. The meteoric rise of cryo-EM is the most notable trend, with its share of new deposits growing from nearly zero to roughly one-third of all new structures in less than a decade [17]. This shift is a direct result of the technical advancements that constituted the "resolution revolution" [3] [10].
This methodological evolution has profound implications for researchers and drug development professionals. The choice of technique is no longer default but strategic. X-ray crystallography remains unparalleled for high-throughput studies of proteins that readily form crystals and for achieving the very highest resolutions [17] [10]. In contrast, cryo-EM has become the go-to method for visualizing large, flexible, or heterogeneous complexes that have resisted crystallization, such as membrane proteins, ribosomes, and viral capsids [3] [19]. Its ability to capture multiple conformational states within a single sample is particularly valuable for understanding functional mechanisms [10]. NMR spectroscopy, while contributing a smaller fraction of new structures, retains its unique niche in studying protein dynamics and small, soluble proteins in their native, solution-state environment [17] [10].
Looking forward, the integration of artificial intelligence (AI) and machine learning is poised to be the next disruptive force. AI tools like AlphaFold are already revolutionizing computational structure prediction and are being integrated into cryo-EM and crystallography workflows to accelerate model building and refinement [10] [19]. Furthermore, the robust growth in depositions from Asia signals a continued globalization of structural biology, which will likely fuel further innovation and discovery [18]. For the scientific community, this means an increasingly powerful and diverse toolkit to elucidate the molecular mechanisms of life and disease, ultimately accelerating the pace of drug discovery and therapeutic development.
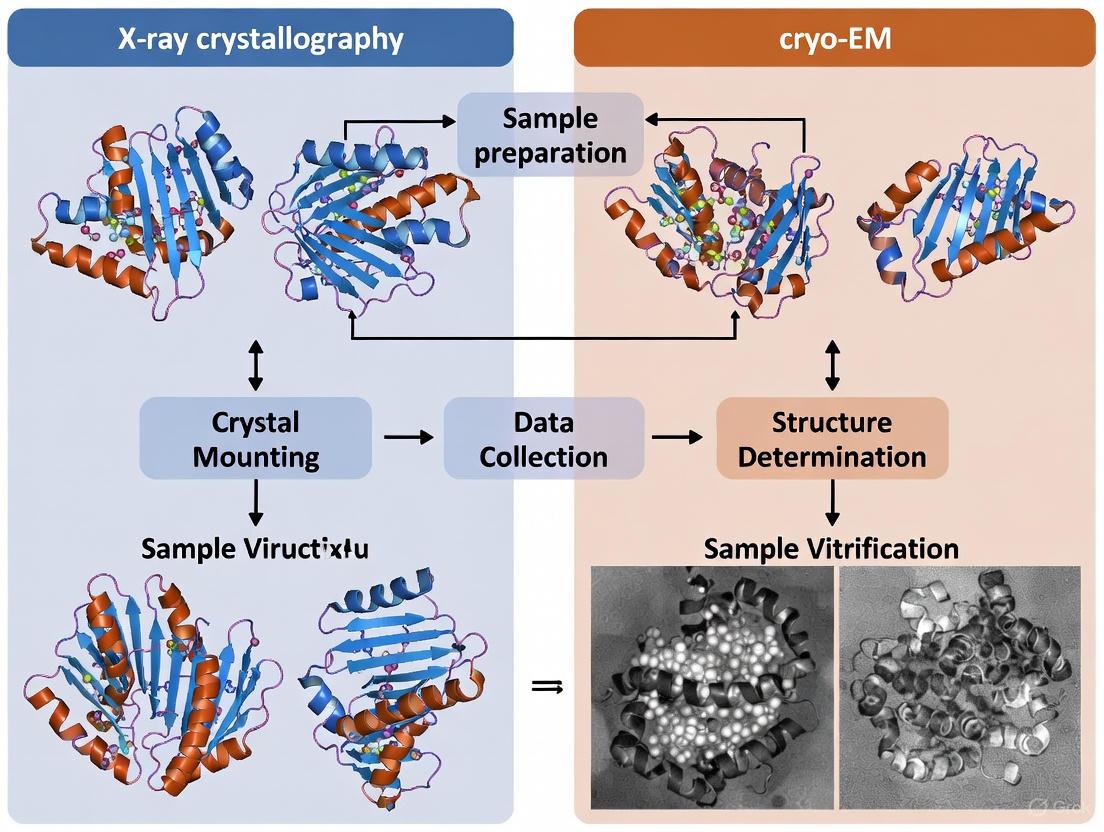
In structural biology, the path to elucidating the three-dimensional architecture of biomolecules hinges critically on sample preparation. This initial phase determines the success of high-resolution techniques like X-ray crystallography and cryo-electron microscopy (cryo-EM). The core methodological divide lies between two distinct preparation philosophies: crystallization, which arranges molecules into a periodic lattice, and vitrification, which flash-freees molecules in a near-native state within amorphous ice [22] [4]. This guide provides a detailed, experimental data-driven comparison of these two foundational approaches, equipping researchers with the knowledge to select and optimize the correct path for their specific biological questions and samples.
The choice between these methods is not merely a technicality; it fundamentally shapes the type of biological information that can be retrieved. Crystallization can yield atomic-resolution snapshots of highly ordered systems, while vitrification excels at capturing the structural heterogeneity and dynamic states of complex assemblies in their native environment [23] [4]. Understanding the requirements, challenges, and opportunities presented by each method is therefore paramount for the efficient allocation of resources and the ultimate success of structural projects.
Crystallization: The Ordered Lattice
Principle and Workflow
Crystallization is the process of inducing a purified macromolecular solution to form a highly ordered, three-dimensional crystal lattice. The principle relies on carefully bringing the sample to a state of supersaturation, typically by gradually removing water or adding precipitants, which drives molecules out of solution and into a growing crystal where they form specific, repeating contacts [24]. A successful crystal acts as an amplifying scaffold, allowing the collective scattering of X-rays to produce a strong, interpretable diffraction pattern [23].
The following workflow outlines the standard steps for a crystallization experiment, from initial sample preparation to data collection.
Key Reagents and Experimental Protocols
Successful crystallization requires meticulous biochemical preparation and the use of specific reagents to drive and control the process [24].
Table 1: Key Research Reagent Solutions for Crystallization
| Reagent Category | Specific Examples | Function in Protocol |
|---|---|---|
| Precipitants | Polyethylene glycol (PEG) of various molecular weights, Ammonium sulfate, 2-methyl-2,4-pentanediol (MPD) | Drives the sample into a supersaturated state by excluding water or competing for solvation, promoting molecular interactions for lattice formation [24]. |
| Buffers | HEPES, Tris, MES (non-phosphate based) | Maintains sample stability at a specific pH, ideally near the protein's isoelectric point (pI) to facilitate crystal contacts [24]. |
| Additives & Salts | Monovalent and divalent salts (e.g., NaCl, MgCl₂), co-factors, substrates | Can stabilize specific conformations, mediate intermolecular contacts, or reduce surface entropy to promote ordered packing [24]. |
| Chemical Reductants | Tris(2-carboxyethyl)phosphine (TCEP), Dithiothreitol (DTT) | Prevents cysteine oxidation over the extended timescale (days to months) of crystal growth, maintaining sample homogeneity [24]. |
| Crystallization Plates & Robots | 96-well sitting-drop or hanging-drop plates, Mosquito/Dragonfly liquid handlers | Enables high-throughput screening of thousands of chemical conditions with nanoliter-volume drops, conserving precious sample [13]. |
A standard vapor diffusion protocol (sitting-drop method) involves:
- Sample Preparation: The target biomolecule is purified to >95% homogeneity and must be monodisperse, as assessed by techniques like size-exclusion chromatography coupled with multi-angle light scattering (SEC-MALS) or dynamic light scattering (DLS) [24]. Typical starting concentrations are 5-20 mg/mL for soluble proteins.
- Drop Setup: Using a liquid-handling robot, nanoliter volumes of the protein sample and crystallization cocktail (precipitant solution) are mixed in a 1:1 to 2:1 ratio on a plate.
- Incubation: The plate is sealed, and the drop is allowed to equilibrate against a larger reservoir of the precipitant solution. Water vapor slowly transfers from the drop to the reservoir, gradually increasing the concentration of both the protein and precipitant in the drop, ideally leading to nucleation and crystal growth [13] [24].
- Optimization: Initial crystal "hits" are optimized by fine-tuning parameters like pH, precipitant concentration, and temperature, or by using techniques like seeding [13].
Technical Requirements and Sample Considerations
Crystallization imposes stringent requirements on sample quality and properties. The following table summarizes the typical prerequisites and the associated challenges, particularly for difficult targets like membrane proteins.
Table 2: Crystallization Sample Requirements and Challenges
| Parameter | Ideal Requirement | Common Challenges & Solutions |
|---|---|---|
| Purity & Homogeneity | >95% pure, monodisperse [24] | Flexible regions or isoforms cause heterogeneity. Solution: Construct engineering using AlphaFold predictions to remove flexible loops [24]. |
| Stability | High, over days to months | Sample degradation during screening. Solution: Addition of stabilizing ligands or substrates to the buffer [24]. |
| Concentration | High (e.g., 5-20 mg/mL for soluble proteins) [13] [25] | Concentration-dependent aggregation. Solution: Pre-crystallization tests to determine the optimal concentration window [24]. |
| Sample Amount | Relatively large (typically >5 mg) [25] | Low-yield expression systems. Solution: Advanced expression systems and miniaturized screening with liquid handlers. |
| Membrane Proteins | Requires detergents or lipidic cubic phase (LCP) [13] | Detergents interfere with crystal contacts. Solution: LCP crystallization provides a more native lipid environment and has been key for GPCR structures [13] [10]. |
Vitrification: Trapping Native State
Principle and Workflow
Vitrification is the rapid freezing of an aqueous sample to form a non-crystalline, glass-like state of ice. This process, which cools the sample to below -150 °C within milliseconds, traps biological particles in a near-native state, preserving their natural conformation and preventing the damaging formation of crystalline ice [22]. In cryo-EM, these vitrified samples are then imaged with an electron beam, and thousands of individual particle images are computationally combined to determine a 3D structure [22] [23].
The workflow for single-particle analysis (SPA) cryo-EM involves several key steps from sample preparation to high-resolution reconstruction.
Key Reagents and Experimental Protocols
The vitrification workflow, while faster than crystallization, requires specialized reagents and equipment to ensure optimal sample preservation [22] [25].
Table 3: Key Research Reagent Solutions for Vitrification
| Reagent Category | Specific Examples | Function in Protocol |
|---|---|---|
| Support Grids | Holey carbon grids (Quantifoil, C-flat), Graphene-based grids (e.g., GraFuture) | Provides a support film with holes where the sample is suspended in a thin layer of vitreous ice. Graphene grids reduce background noise and mitigate preferred orientation [25]. |
| Cryogens | Liquid ethane, liquid ethane/propane mixture | Has high thermal conductivity to enable cooling rates fast enough to bypass water crystal formation and achieve vitrification [22]. |
| Buffer Components | Low-salt buffers, detergents for membrane proteins | Maintains sample stability and monodispersity during grid preparation. Low salt concentrations (<300 mM) are preferred to reduce background noise [25]. |
| Vitrification Robots | Vitrobot (Thermo Fisher), GP2 (Leica) | Automated instruments that control key variables like blotting time, force, and humidity, ensuring reproducible and high-quality vitreous ice [22]. |
A standard plunge-freezing protocol involves:
- Sample Application: A small volume (typically 2-5 µL) of the purified sample at a concentration of ≥ 0.5-2 mg/mL is applied to a glow-discharged EM grid [25].
- Blotting: Excess liquid is removed by pressing filter paper against the grid for a few seconds, leaving a thin film of sample spanning the holes of the support film.
- Plunging: The grid is rapidly plunged into a container of liquid ethane cooled by a surrounding liquid nitrogen dewar. This rapid heat transfer vitrifies the thin film of water [22].
- Storage and Imaging: The vitrified grid is stored in liquid nitrogen and transferred under cryogenic conditions to the electron microscope for data collection.
Technical Requirements and Sample Considerations
Vitrification is less demanding than crystallization in terms of sample amount and order, but it has its own unique set of requirements and challenges related to the behavior of particles in thin ice.
Table 4: Vitrification Sample Requirements and Challenges
| Parameter | Ideal Requirement | Common Challenges & Solutions |
|---|---|---|
| Purity & Homogeneity | ≥ 90% pure [25] | Sample heterogeneity can be addressed computationally. Solution: 2D and 3D classification can separate out different conformational states or complexes from a single dataset [22]. |
| Stability | Short-term (minutes) during grid preparation | Denaturation at the air-water interface. Solution: Use of additives like detergents or specialized grids to shield particles [25]. |
| Concentration | Lower than crystallization (e.g., ≥ 0.5-2 mg/mL) [25] | Finding the right particle density. Solution: Empirical testing of concentration and blotting conditions to achieve a monolayer of well-separated particles. |
| Sample Amount | Minimal (e.g., ~100 µL at 0.5-2 mg/mL) [25] | Often only a few micrograms of protein are consumed per grid, making it suitable for low-yield targets [26]. |
| Particle Size | Optimal for >100 kDa complexes, possible down to ~50 kDa [23] [25] | Small proteins produce weak scattering. Solution: Use of high-end microscopes with direct electron detectors and advanced processing software [10] [25]. |
| Buffer Compatibility | Low salt (≤300 mM), low volatile solvents [25] | Buffer components can form crystalline ice or high background. Solution: Buffer exchange into compatible buffers prior to grid preparation. |
Comparative Analysis: Data-Driven Decision Making
Direct Comparison of Key Parameters
Choosing between crystallization and vitrification requires a pragmatic assessment of sample properties and project goals. The following table provides a side-by-side comparison of quantitative and qualitative metrics derived from experimental data and standard protocols.
Table 5: Direct Comparison of Crystallization and Vitrification for Structural Analysis
| Parameter | X-ray Crystallography (Crystallization) | Single-Particle Cryo-EM (Vitrification) |
|---|---|---|
| Core Requirement | High-quality, ordered 3D crystals | Monodisperse, purified particles in thin ice |
| Typical Sample Amount | >5 mg (soluble protein) [25] | ~0.1-0.2 mg (can be lower) [23] |
| Sample Concentration | High (e.g., >10 mg/mL) [25] | Moderate (e.g., ≥ 2 mg/mL) [25] |
| Molecular Size Suitability | Optimal <100 kDa [23] | Optimal >100 kDa, down to ~50 kDa demonstrated [23] [25] |
| Handling of Flexibility | Poor; flexible regions often disordered or engineered out [24] | Good; can often resolve multiple conformational states from one dataset [23] [4] |
| Typical Timeline | Weeks to months (crystal optimization) [23] | Weeks (faster initial structure) [23] |
| Maximum Resolution | Sub-1.0 Å possible [23] | ~1.4 Å demonstrated, typically 2.5-3.5 Å for complexes [25] |
| Ideal Application | Atomic-level detail of stable, crystallizable molecules | Large, flexible complexes, membrane proteins in near-native states [4] [10] |
Application Scenarios and Method Selection
The comparative advantages of each technique make them uniquely suited for specific biological problems.
Membrane Protein Structural Analysis: For crystallography, membrane proteins often require extensive engineering, detergent screening, or the use of lipidic cubic phase (LCP) methods to form crystals, which can lock the protein into a single conformation [13]. In contrast, vitrification allows membrane proteins to be embedded in lipid nanodiscs or detergent micelles and studied in multiple functional states without the constraints of a crystal lattice, providing insights into mechanisms like gating and ligand binding [23] [10].
Large Protein Complex Studies: Crystallography becomes increasingly challenging with the size and complexity of assemblies, as forming well-ordered crystals is difficult. Vitrification excels here, with no upper size limit, making it the preferred method for massive complexes like ribosomes, viruses, and the nuclear pore complex [23] [10].
Dynamic Structure Visualization: Crystallography provides a single, high-resolution snapshot of the most stable conformation. Vitrification, combined with computational 3D classification, can resolve multiple conformational intermediates from a heterogeneous sample, effectively creating a "movie" of molecular dynamics from a single preparation [23] [4].
The "sample preparation divide" between crystallization and vitrification represents a fundamental fork in the road for structural biologists. Crystallization demands high homogeneity and order to produce a single, atomic-resolution snapshot, while vitrification embraces complexity and flexibility to visualize biomolecules in a near-native state. There is no single "best" method; the choice is dictated by the biological question, the properties of the target molecule, and available resources.
The future of structural biology lies not in the exclusive use of one technique over the other, but in their strategic integration. Medium-resolution cryo-EM maps can provide initial phases for solving crystal structures of challenging targets, a practice known as molecular replacement [4]. Furthermore, the rise of integrative modeling, which combines data from cryo-EM, X-ray crystallography, NMR, and other biophysical techniques, promises a more holistic and dynamic understanding of molecular machines. By mastering both crystallization and vitrification, researchers can leverage the unique strengths of each to illuminate the intricate architecture of life at the atomic level.
Workflow Deep Dive: From Sample to Atomic Model with X-ray and Cryo-EM
X-ray crystallography has long been the cornerstone of structural biology, responsible for determining over 86% of the structures in the Protein Data Bank (PDB) [27]. This dominance is now being challenged by the rapid ascent of cryo-electron microscopy (cryo-EM), which accounted for nearly 40% of new structure deposits by 2023-2024 [27]. Some projections even suggest that single-particle cryo-EM is poised to surpass X-ray crystallography as the most used method for experimentally determining new structures [9]. Despite this shift, X-ray crystallography remains an indispensable tool, especially for obtaining atomic-resolution structures of proteins and protein-ligand complexes, providing critical insights for drug discovery and rational drug design [28] [10].
The fundamental strength of crystallography lies in its ability to reveal molecular structures at atomic resolution, often better than 1.5 Å, enabling the precise visualization of drug-target interactions and enzyme active sites [25]. However, the technique faces a significant bottleneck: the requirement for high-quality, well-ordered crystals, which can be notoriously difficult to obtain for many biologically important targets, particularly membrane proteins and large, flexible complexes [27] [10]. It is within this context of methodological competition and complementarity that we examine the detailed workflow of X-ray crystallography, from crystallization to phasing, while objectively comparing its capabilities and limitations against cryo-EM approaches.
The Crystallization Workflow: From Protein to Crystal
Sample Preparation and Pre-crystallization Assessment
The journey to a high-resolution structure begins with the production of a pure, monodisperse, and stable protein sample. Table 1 outlines the typical sample requirements for successful crystallization. For standard crystallography, protein purity must exceed 95%, with concentrations typically greater than 10 mg/ml and a total sample amount of more than 5 mg [28] [25]. These requirements are generally more stringent than those for cryo-EM Single Particle Analysis (SPA), which can often work with concentrations as low as 2 mg/ml and purity levels around 90% [25].
Table 1: Key Reagents and Materials for Protein Crystallization
| Research Reagent/Material | Function/Purpose in Workflow |
|---|---|
| High-purity protein (>95%) | Ensures homogeneous nucleation and crystal growth; reduces amorphous aggregation. |
| Crystallization screens (e.g., Hampton Research) | Provides thousands of pre-formulated conditions to identify initial crystal hits by varying precipants, pH, and salts [28]. |
| Precipitants (PEG, Ammonium Sulfate) | Drives protein out of solution in a controlled manner to promote crystal formation [28]. |
| Dynamic Light Scattering (DLS) | Assesses sample monodispersity and detects aggregates that interfere with crystallization [28]. |
| Differential Scanning Fluorimetry (DSF) | Determines optimal buffer conditions and protein stability for crystallization [28]. |
| Size-Exclusion Chromatography (SEC) | Removes aggregates and polydisperse populations to improve crystal quality [28]. |
Before crystallization trials, the protein sample undergoes rigorous biophysical characterization. Dynamic light scattering (DLS) assesses monodispersity, while differential scanning fluorimetry (DSF) determines optimal buffer conditions and protein stability [28]. For proteins with suspected flexibility, NMR spectroscopy can be used to verify folding status via HSQC fingerprint analysis, identifying unfolded regions or flexible domains that might prevent crystal formation [28].
Crystallization Strategies and Optimization
The crystallization process itself involves screening hundreds to thousands of conditions to identify initial crystal hits. Commercial screens systematically vary critical parameters including buffer type and pH, ionic strength, salts, and precipitant type and concentration [28]. The most common precipitants are polyethylene glycol (PEG) and ammonium sulfate.
Once initial conditions are identified, extensive optimization follows to improve crystal size and quality. This may involve fine-tuning the pH, precipitant concentration, or adding small molecule additives that enhance crystal packing [28]. For membrane proteins, specialized techniques such as the lipidic cubic phase (LCP) method have been crucial for success, enabling the determination of groundbreaking structures like the β2-adrenergic receptor [10].
The timeline for crystallization screening and optimization typically ranges from 3-6 weeks, though particularly challenging targets may require significantly more time and resources [28]. This represents a key differentiator from cryo-EM, which can often proceed more directly from sample to data collection without the crystallization bottleneck.
Data Collection: Harnessing Synchrotron Radiation
From Crystal to Diffraction Pattern
Once suitable crystals are obtained, they are cryo-cooled in liquid nitrogen to minimize radiation damage during X-ray exposure [28]. The crystals are then exposed to high-intensity X-ray beams, traditionally at synchrotron facilities, though laboratory sources are also used for preliminary characterization.
Synchrotron radiation provides several critical advantages for data collection, as outlined in Table 2. The high brilliance of modern beamlines allows data collection from much smaller crystals than previously possible [28]. The tunable wavelengths enable experimental phasing methods like multiple-wavelength anomalous dispersion (MAD), while the beam focus and stability are essential for collecting high-resolution data [28] [29].
Table 2: Comparative Analysis of X-ray Crystallography and Cryo-EM
| Parameter | X-ray Crystallography | Cryo-EM Single Particle Analysis |
|---|---|---|
| Typical Resolution | Atomic resolution (often better than 1.5 Å) [25] | Near-atomic resolution (typically 1.8-3.5 Å) [25] [10] |
| Sample Requirement | ≥10 mg/ml, >5 mg total, >95% purity [25] | ≥2 mg/ml, ≥100 μL volume, ≥90% purity [25] |
| Key Limiting Step | Crystal growth and quality [27] [10] | Sample preparation and preferred orientation [25] |
| Molecular Weight Range | No upper limit in theory, but crystallization becomes challenging for very large complexes [27] | Optimal for complexes >150 kDa; can go lower with specialized grids [25] |
| Time to Structure (after sample) | Weeks to months (crystallization dependent) [28] | Days to weeks (no crystallization needed) [30] |
| PDB Deposition Share (2023) | ~66% of structures [27] | ~32% of structures and growing [27] |
| Membrane Protein Suitability | Challenging; requires specialized methods like LCP [10] | Excellent; particularly suited for large membrane complexes [10] |
Fourth-generation synchrotrons, such as MAX IV in Sweden, represent the cutting edge in crystallography data collection. These facilities feature multi-bend achromat (MBA) technology that significantly reduces emittance, resulting in increased brightness and coherence of the X-ray beam [29]. This enables faster data collection, opening possibilities for crystallography to be used as a screening method in drug discovery [29].
Emerging Approaches: Serial Crystallography
A significant advancement in data collection methods is the emergence of serial crystallography approaches. This technique involves collecting diffraction patterns from thousands of microcrystals sequentially, which is particularly valuable for systems that only produce microcrystals, such as many membrane proteins [29]. Serial methods also enable time-resolved studies and data collection at room temperature, capturing proteins in more physiological states [31] [29].
Recent work on human cytochrome P450 3A4 (CYP3A4) exemplifies the power of room-temperature serial crystallography. This approach revealed better-defined loops compared to cryo-temperature structures, providing insights into the dynamic properties of this key drug-metabolizing enzyme [31].
Phasing: Solving the Phase Problem in Crystallography
Molecular Replacement and Experimental Phasing
A fundamental challenge in crystallography is the "phase problem" - while diffraction patterns provide intensity information, the phase information is lost during measurement but essential for calculating electron density maps [28]. Two primary approaches address this challenge:
Molecular Replacement (MR) has become the most common phasing method, particularly with the rise of AlphaFold and other AI-based structure prediction tools [29]. MR uses a known homologous structure (generally with sequence identity above 40-50%) as a search model to obtain initial phase information [28] [27]. The availability of accurate computational models has made MR increasingly successful, reducing the need for experimental phasing.
Experimental Phasing methods remain essential when suitable search models are unavailable. The most common approach involves introducing anomalous scatterers, typically by producing selenomethionine (SeMet)-labeled proteins [28]. Techniques like Single-wavelength Anomalous Dispersion (SAD) and Multiple-wavelength Anomalous Dispersion (MAD) exploit the anomalous scattering properties of these incorporated atoms to solve the phase problem [28] [27].
The Growing Role of Computational Methods
The integration of computational methods has dramatically transformed phasing and model building. AlphaFold models are increasingly used as search models for molecular replacement, particularly for targets without close experimental homologs [10]. Furthermore, automated model building and refinement algorithms have significantly accelerated the structure determination process.
Recent advances in automated phase mapping of high-throughput X-ray diffraction data demonstrate how encoding domain-specific knowledge of crystallography, thermodynamics, and materials science into optimization algorithms can solve complex phase analysis challenges [32]. While developed for materials science, these approaches illustrate the growing power of computational methods in crystallographic analysis.
Integrated Workflow and Comparative Visualization
The complete X-ray crystallography workflow integrates multiple steps from gene to structure, with parallel developments in cryo-EM offering complementary approaches for structural biologists. The following diagram illustrates this integrated pathway and its relationship to cryo-EM methodologies.
Integrated Structural Biology Workflows: X-ray Crystallography and Cryo-EM Pathways
X-ray crystallography continues to evolve with advancements in serial crystallography, brighter light sources, and more powerful computational methods [29]. However, its requirement for high-quality crystals remains a significant constraint for many biologically important targets. In contrast, cryo-EM has demonstrated remarkable capabilities for studying large complexes, membrane proteins, and multiple conformational states without crystallization [10].
The most powerful approach for modern structural biology lies in the integration of these complementary techniques. X-ray crystallography provides unparalleled resolution for amenable targets, offering atomic-level details critical for understanding enzyme mechanisms and drug binding [25] [10]. Meanwhile, cryo-EM tackles challenging complexes that defy crystallization, capturing functional states in near-native environments [30] [10]. This methodological synergy, enhanced by AI-based structure prediction, ensures that X-ray crystallography will remain a vital component of the structural biology toolkit, even as cryo-EM continues its exponential growth [9] [10].
For drug development professionals, the choice between techniques should be guided by the specific research question, target properties, and available resources. X-ray crystallography excels for atomic-resolution ligand binding studies when crystals can be obtained, while cryo-EM offers a path forward for intractable targets that have long resisted crystallization efforts. The future of structural biology lies not in the dominance of a single technique, but in the strategic application of multiple complementary methods to illuminate the molecular mechanisms of life and disease.
Structural biology has been transformed by two powerful techniques: cryo-electron microscopy (cryo-EM) and X-ray crystallography. While X-ray crystallography has long been the "gold standard" for determining atomic-resolution structures, cryo-EM has experienced a "resolution revolution" that now enables near-atomic resolution for many biologically significant complexes [4] [10]. These methods are not competing technologies but rather complementary tools that can work together for more complete structural insights [4] [1].
X-ray crystallography requires growing well-ordered three-dimensional crystals and analyzing their diffraction patterns [4] [33]. Its remarkable precision (routinely finer than 2 Å) reveals intimate atomic architecture but faces challenges with crystallization of membrane proteins, large complexes, and dynamic systems [4] [33]. In contrast, cryo-EM studies molecules in near-native states without crystallization by flash-freezing samples in thin ice layers and computationally processing thousands of individual particle images [4] [33]. This flexibility makes it particularly valuable for visualizing large macromolecular complexes, membrane proteins, and multiple conformational states [33].
This guide provides a comprehensive comparison of these techniques, with detailed examination of the cryo-EM single-particle analysis workflow from sample preparation to high-resolution reconstruction, contextualized against traditional crystallographic approaches.
Technical Comparison: Fundamental Principles and Applications
Key Differences in Physical Principles and Output
Table 1: Fundamental methodological differences between Cryo-EM and X-ray Crystallography
| Aspect | Cryo-EM | X-ray Crystallography |
|---|---|---|
| Radiation Source | High-energy electrons [1] | X-ray photons [4] |
| Sample State | Vitrified solution (near-native state) [33] | Well-ordered 3D crystal lattice [4] |
| Primary Interaction | Coulomb potential of atoms [1] | Electron clouds [4] |
| Information Recorded | 2D particle projections [4] | Diffraction pattern (Bragg reflections) [4] |
| Key Challenge | Low signal-to-noise ratio [34] | Phase problem [4] |
| Primary Output | 3D electron density map [35] | Electron density map [4] |
Performance Characteristics and Sample Requirements
Table 2: Practical performance comparison and sample requirements
| Parameter | Cryo-EM | X-ray Crystallography |
|---|---|---|
| Typical Resolution Range | 2.5-4.0 Å [33] | 1.0-2.5 Å (often sub-1Å possible) [33] |
| Optimal Molecular Size | >100 kDa [33] | <100 kDa [33] |
| Sample Amount Required | 0.1-0.2 mg [33] | >2 mg typically [33] |
| Sample Purity Requirements | Moderate heterogeneity acceptable [33] | High homogeneity required [33] |
| Typical Timeline | Weeks [33] | Weeks to months [33] |
| Data Collection Time | Hours to days [33] | Minutes to hours [33] |
| Best Applications | Membrane proteins, large complexes, flexible assemblies [33] | Soluble proteins, small molecules, stable constructs [33] |
The Cryo-EM Single-Particle Analysis Workflow
The cryo-EM single-particle analysis workflow comprises three major stages: grid preparation, imaging, and computational reconstruction. Each stage contributes critically to the final resolution and quality of the determined structure.
Stage 1: Cryo-EM Grid Preparation
Grid preparation, or vitrification, involves flash-freezing aqueous samples to preserve native structure in vitreous ice [36].
Experimental Protocol: Plunge Freezing
Grid Selection and Treatment: TEM grids (typically 300-400 mesh) with holey carbon support films are selected. Grids are cleaned with organic solvents (acetone, ethyl acetate, or chloroform) and treated with glow-discharging to make the surface hydrophilic [35]. For challenging samples, grids may be coated with polylysine or functionalized graphene to improve particle distribution [36].
Sample Application and Blotting: A few microliters of purified sample are applied to the grid. Filter paper is used to blot away excess liquid, leaving an extremely thin aqueous film (typically 10-100 nm thick) [36]. Parameters like blot time, force, and humidity are controlled to optimize ice thickness [36].
Vitrification: The blotted grid is rapidly plunged into liquid ethane cooled by liquid nitrogen. This rapid freezing (~10⁶ K/s) prevents ice crystallization, preserving samples in a vitreous, glass-like state that maintains native structure [35].
Key Challenges and Optimization Strategies:
- Orientation Bias: Particles may adopt limited views due to interactions with the air-water interface. Solutions include adding mild detergents, using supporting films (ultrathin carbon, graphene), or collecting data with a tilted stage [36].
- Particle Partitioning: Particles may fail to enter holes. Optimization strategies include chemical treatments, multiple application rounds, or affinity grids with specialized coatings [36].
- Ice Contamination: Grids must be maintained below -170°C to prevent crystalline ice formation [35].
Stage 2: Cryo-EM Imaging
Imaging occurs in a transmission electron microscope operating at 200-300 kV [35]. Images are collected under low-dose conditions (~1-20 e⁻/Ų) to minimize radiation damage [37].
Experimental Protocol: Data Collection
Microscope Setup: The cryo-holder transfers the vitrified grid into the microscope column. Alignment ensures coherent illumination, and the low-dose system limits electron exposure [35].
Image Acquisition: Micrographs are recorded using direct electron detectors. Movies (typically 20-40 frames) capture beam-induced motion that can be corrected computationally [10]. Multiple micrographs are collected from different grid areas [35].
Contrast Formation Mechanisms:
Biological samples are weak phase objects that produce minimal amplitude contrast. Instead, most usable signal comes from phase contrast, generated by intentional defocusing to create interference between scattered and unscattered electron waves [37]. The weak phase object approximation simplifies computational processing by modeling the exit wave as the sum of unscattered and scattered waves [37].
Stage 3: Image Processing and 3D Reconstruction
This computationally intensive stage transforms 2D projections into 3D density maps [35].
Experimental Protocol: Image Processing Workflow
Pre-processing: Movie frames are motion-corrected to compensate for beam-induced drift [10]. The contrast transfer function (CTF) is estimated and corrected to account for microscope optics [35].
Particle Picking: Hundreds of thousands to millions of individual particle images are extracted from micrographs. Traditional methods use template matching, while modern approaches employ deep learning (e.g., DeepEM convolutional neural networks) for template-free particle selection [34].
2D Classification: Extracted particles are classified into 2D averages to remove non-particle images, contaminants, and poor-quality particles [35].
Initial Model Generation: An initial 3D model is created using random conical tilt, common lines, or model-based approaches [35].
3D Reconstruction: Iterative refinement aligns particles against projections of the 3D model and reconstructs the density map. For symmetric particles like viruses, symmetry constraints (e.g., icosahedral) improve resolution [35].
Validation and Sharpening: Resolution is estimated using gold-standard Fourier shell correlation. Maps are sharpened to correct for attenuation at high resolution [35].
Visual workflow comparing cryo-EM single-particle analysis with X-ray crystallography. The cryo-EM pathway (red) begins with vitrification of purified samples, while crystallography (gray, dashed) requires crystal formation. Both converge at atomic model building.
Integrated Applications in Structural Biology
The complementary strengths of cryo-EM and X-ray crystallography make them powerful when combined. Two integrative approaches are particularly valuable:
Hybrid Modeling: Medium-resolution cryo-EM maps (4-10 Å) provide the overall architecture of complexes, into which high-resolution X-ray structures of individual components can be docked [1]. Software packages like Situs, EMfit, and UCSF Chimera perform rigid-body docking, while Flex-EM and MDFF enable flexible fitting [1]. This approach revealed the architecture of the yeast RNA exosome complex by docking crystal structures of human core complexes into cryo-EM maps [1].
Molecular Replacement for Phasing: Cryo-EM maps can provide initial models for molecular replacement, solving the phase problem in X-ray crystallography [4]. This is particularly valuable for large complexes where traditional phasing methods fail [4].
Table 3: Research reagent solutions for cryo-EM single-particle analysis
| Reagent/Category | Specific Examples | Function and Application |
|---|---|---|
| Grid Materials | Quantifoil grids, C-flat grids, Graphene oxide grids [36] | Support films with patterned holes or continuous surfaces for sample application |
| Grid Treatment | Glow discharge systems (Quorum SC7620), polylysine solution [35] | Modify surface properties to control hydrophobicity/hydrophilicity and particle adhesion |
| Vitrification Systems | FEI Vitrobot, Gatan Cryoplunge [35] | Automated plunge-freezing devices controlling humidity, blotting, and freezing parameters |
| Specialized Buffers | Tris or phosphate buffer (pH 7.0-8.0, 50-100 mM salt) [35] | Maintain biochemical integrity while avoiding crystallization interference |
| Detergents & Additives | Amphipols, fluorinated surfactants [36] | Improve particle distribution and overcome orientation bias at air-water interface |
| Affinity Grid Reagents | Antibodies, streptavidin 2D crystals, functionalized graphene [36] | Enhance specific particle adsorption and concentration on grid surfaces |
Cryo-EM single-particle analysis and X-ray crystallography offer complementary pathways to structural understanding. Cryo-EM excels for large, complex, or dynamic assemblies that resist crystallization, while X-ray crystallography provides unparalleled atomic-level precision for well-behaved samples [4] [33]. The choice between techniques depends on project-specific requirements including sample properties, resolution needs, and available resources [33].
Recent advances continue to blur the boundaries between these methods. Micro-electron diffraction (microED) enables crystallography-style analysis from nanoscale crystals [4], while cryo-EM resolutions continue to improve with direct electron detectors and computational algorithms [10]. Artificial intelligence approaches like DeepEM for particle picking [34] and AlphaFold for model building [10] further enhance both techniques.
The future of structural biology lies not in choosing one method over the other, but in strategically combining cryo-EM, X-ray crystallography, and computational modeling to reveal biological mechanisms at multiple scales. This integrated approach provides unprecedented insights into molecular structure and function, accelerating drug discovery and fundamental biological research.
X-ray crystallography remains a powerful, high-throughput tool for determining the 3D structures of small molecules and their protein targets, playing a critical role in structure-based drug design. This guide compares its performance and applications with cryo-electron microscopy (cryo-EM), providing an objective analysis for research and development professionals.
Core Methodologies and Workflows
The application of X-ray crystallography in drug discovery is underpinned by specific high-throughput methodologies for sample preparation and data collection.
Experimental Protocol: High-Throughput Crystallography for Fragment Screening
Fragment-based drug discovery (FBDD) relies on screening libraries of small molecules (molecular weight 100-250 Da) to identify chemically tractable starting points [38] [39]. The standard workflow involves:
- Protein Crystallization: Using high-throughput vapor-diffusion or batch methods to produce numerous crystals [40]. Automation and standardized screening cocktails enable the processing of large numbers of samples with minimal volume [38] [40].
- Soaking or Co-crystallization: Introducing fragments to pre-grown protein crystals by soaking crystals in solutions containing single fragments or cocktails, or by co-crystallizing the protein with the fragment [38].
- Data Collection: For conventional cryo-crystallography, a single, large crystal is looped and flash-cooled to about -170°C before data collection. For room-temperature serial crystallography, thousands of microcrystals are grown on fixed-target sample holders, and diffraction data from each are merged [41] [42].
- Hit Identification: Electron density maps are analyzed to identify fragments that bind the protein target and to define their binding sites with high precision [38] [39].
Experimental Protocol: Room-Temperature Serial Crystallography
Recent advancements aim to overcome the potential for cryogenic temperatures to mask dynamic states. The HiPhaX (High-throughput Pharmaceutical X-ray screening) instrument at the PETRA III synchrotron exemplifies this [41] [42]:
- On-Chip Crystallization: Crystals are grown directly in microporous compartments of a sample holder.
- Ligand Incubation: Crystallization solution is replaced with fragment solutions.
- Controlled Environment: Data collection occurs at 23°C and 98% relative humidity, maintaining near-physiological conditions.
- Serial Data Collection: The Roadrunner sample delivery system rapidly collects diffraction stills from thousands of microcrystals, minimizing radiation damage [42].
Table 1: Key Performance Metrics from Comparative Studies
| Experimental Parameter | Cryo-Crystallography (100 K) | Room-Temperature Serial Crystallography (296 K) |
|---|---|---|
| Typical Resolution | High (e.g., <1.8 Å for cryo1 dataset [42]) | Comparable to cryo (e.g., ~1.9 Å for RT1/RT2 datasets [42]) |
| Radiation Damage | Minimized by cryo-cooling | Mitigated by spreading dose over thousands of crystals |
| Protein Conformations | May mask functionally relevant states | Can reveal previously hidden conformational states [41] |
| Unit Cell Volume | Smaller | Slightly larger, indicating less structural constraint [42] |
| Ligand Binding Sites | Can identify binders at non-physiological sites | May identify fewer binders, potentially reflecting more physiologically relevant binding [42] |
Comparative Analysis with Cryo-EM
While X-ray crystallography excels in high-throughput screening of small molecules, Cryo-EM has emerged as a powerful complementary technique.
Table 2: X-ray Crystallography vs. Cryo-EM for Structure-Based Drug Discovery
| Feature | X-ray Crystallography | Cryo-Electron Microscopy (Cryo-EM) |
|---|---|---|
| Ideal Sample/Application | Small molecules, fragments, soluble proteins, lead optimization [38] [4] | Large macromolecular complexes, membrane proteins, flexible assemblies [10] [4] |
| Sample Preparation | Requires high-quality, well-ordered crystals [40] | No crystallization needed; samples are flash-frozen in vitreous ice [1] |
| Typical Throughput | High; capable of screening thousands of compounds [38] [41] | Lower; improving but generally not suited for screening large compound libraries [43] |
| Information Obtained | Defines ligand-binding sites with high certainty; precise atomic coordinates [38] | Near-atomic resolution structures of complexes in near-native states; can visualize multiple conformations [10] |
| Key Limitation | Difficulty crystallizing some targets (e.g., membrane proteins); cryo-artifacts [41] [40] | Lower resolution for smaller proteins (<100 kDa); not yet a high-throughput screening tool [4] |
| Data Collection Temp. | Traditionally cryogenic; room-temperature emerging [41] [42] | Always cryogenic [42] |
The techniques are highly complementary. A common integrated approach uses a low-resolution cryo-EM map of a large complex as a framework into which high-resolution X-ray structures of individual components or domains are docked [1] [4]. Conversely, cryo-EM maps can provide initial models to solve the "phase problem" in X-ray crystallography [4].
Diagram 1: Workflow for integrated structural biology in drug discovery.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful high-throughput screening requires specialized materials and reagents.
Table 3: Key Research Reagent Solutions for High-Throughput X-ray Crystallography
| Reagent / Material | Function in the Experimental Process |
|---|---|
| Fragment Libraries (e.g., F2X Entry) | Curated collections of small, low molecular weight compounds (100-250 Da) used to identify initial "hits" by screening against protein crystals [39] [42]. |
| Sparse Matrix & Statistical Screening Cocktails | Pre-formulated solutions (e.g., Jancarik and Kim sparse matrix) that sample chemical space known to promote crystallization, used as a first step in high-throughput pipelines [40]. |
| High-Throughput Crystallization Plates | Specialized multi-well plates (e.g., 24-well format) designed for automated setting of vapor-diffusion or batch crystallization experiments [40]. |
| Fixed-Target Serial Crystallography Chips | Microporous sample holders with multiple compartments that allow for on-chip crystal growth and subsequent high-throughput, room-temperature data collection [41] [42]. |
| Cryo-Protectants | Chemicals (e.g., glycerol, polyethylene glycol) used to prepare crystals for flash-cooling in conventional cryo-crystallography, preventing ice formation [41]. |
The field continues to evolve with automation and integration. The planned PETRA IV synchrotron promises X-rays of unprecedented brilliance, enabling faster, fully automated structural screenings [41]. Furthermore, the combination of highly automated X-ray screening with AI-powered data analysis is poised to substantially accelerate the journey from molecular discovery to medical application [41] [10].
In conclusion, X-ray crystallography is the established, high-throughput method for obtaining precise atomic-level information on small molecules and fragments bound to their protein targets. While cryo-EM provides unparalleled insights for larger, more complex targets, the two techniques are powerfully complementary. The ongoing development of room-temperature serial crystallography ensures that X-ray methods will continue to provide physiologically relevant structural data critical for advancing drug discovery.
Cryo-electron microscopy (cryo-EM) has emerged as a revolutionary technique in structural biology, enabling researchers to visualize biological macromolecules with unprecedented detail. Its unique capabilities are particularly advantageous for studying targets that have long challenged traditional methods, fundamentally expanding the frontiers of biomedical research. This guide objectively compares cryo-EM's performance against X-ray crystallography, with a focused analysis on its ideal applications: large macromolecular complexes, membrane proteins, and proteins exhibiting multiple conformational states.
The Technical Edge: Cryo-EM vs. X-Ray Crystallography
The distinct principles of cryo-EM and X-ray crystallography underlie their differing suitability for various research applications. The following table summarizes their core technical differences.
Table 1: Fundamental Comparison of Cryo-EM and X-ray Crystallography
| Aspect | Cryo-EM | X-ray Crystallography |
|---|---|---|
| Sample State | Flash-frozen in vitreous ice (near-native state) [4] [44] | Crystallized in a well-ordered 3D lattice [4] |
| Probe | High-energy electron beam [4] | X-ray beam [4] |
| Primary Output | 2D projections assembled into a 3D density map [4] | Diffraction pattern of Bragg reflections [4] |
| Key Challenge | Low signal-to-noise for small particles; particle alignment [45] | Requirement for high-quality crystals; phase problem [4] [13] |
| Inherent Strength | Visualizes dynamic, flexible, or heterogeneous samples [4] [44] | Provides extremely high precision for well-ordered structures [4] [44] |
Application 1: Large Macromolecular Complexes
Cryo-EM excels at determining the structures of large, intricate molecular machines without the need for crystallization, which often disrupts their native architecture and inter-subunit interactions.
Experimental Evidence and Performance Data
Cryo-EM has been instrumental in solving structures of massive complexes like ribosomes, viruses, and the nuclear pore complex. A key metric of success is the molecular weight threshold. While X-ray crystallography has no upper size limit in theory, practical difficulties in growing well-ordered crystals for very large complexes are significant [13]. In contrast, cryo-EM faces no such crystallization barrier. The technique has proven highly effective for complexes larger than 100 kDa, and its performance continues to improve with advancements in detectors and software [44].
Table 2: Performance Comparison for Large Complexes
| Criterion | Cryo-EM | X-ray Crystallography |
|---|---|---|
| Typical Size Suitability | Optimal for >100 kDa complexes [44] | Applicable, but crystal quality often degrades with size and complexity [13] |
| Sample Requirement | Minimal sample amount (0.1-0.2 mg typically) [44] | Requires larger sample quantities (>2 mg typically) [44] |
| Key Advantage | Preserves quaternary structure integrity; reveals assembly mechanisms [44] | Can achieve high resolution for stable subcomplexes; precise atomic positioning [44] |
| Representative Achievement | Structure of the nuclear pore complex [10] | Structure of the ribosome [10] |
Detailed Experimental Protocol: Single-Particle Analysis
The primary workflow for studying large complexes is single-particle cryo-EM [44]:
- Purification: The macromolecular complex is purified in solution.
- Vitrification: A small volume of sample is applied to an EM grid and plunge-frozen in liquid ethane, embedding the particles in a thin layer of amorphous ice.
- Data Collection: The grid is transferred to the electron microscope. Thousands to millions of low-dose 2D micrographs are collected, each containing images of individual particles in random orientations.
- Image Processing: Computational algorithms perform motion correction, select particles, and classify them based on orientation and conformational state.
- 3D Reconstruction: 2D particle images are combined to generate an initial 3D reconstruction, which is then iteratively refined to produce a final 3D density map.
- Model Building: An atomic model is built and refined into the experimental density map.
Application 2: Membrane Proteins
Membrane proteins, such as GPCRs and ion channels, are critical drug targets but notoriously difficult to crystallize due to their hydrophobic nature. Cryo-EM allows these proteins to be studied in near-native lipid environments, circumventing the major bottleneck of crystallization [44] [25].
Experimental Evidence and Performance Data
A landmark study published in Nature Communications (2025) showcased cryo-EM's power for membrane proteins by visualizing lysosomal membrane proteins within intact, native lysosomal membranes [46]. The researchers developed a workflow to isolate endolysosomes and used cryo-electron tomography (cryo-ET) with sub-tomogram averaging to refine the structures of key membrane complexes like V-ATPase directly within their native lipid environment [46]. This approach provided insights into their heterogeneous distribution, which would be extremely difficult to capture by crystallography.
Table 3: Performance Comparison for Membrane Proteins
| Criterion | Cryo-EM | X-ray Crystallography |
|---|---|---|
| Sample Environment | Studied in detergent or lipid nanodiscs (near-native) [44] [25] | Often requires detergent optimization or lipidic cubic phase (LCP) [13] |
| Key Advantage | Preserves native lipid environment; minimizes protein denaturation [44] | Higher resolution for stable constructs; well-established for some targets [44] |
| Technical Challenge | Requires small sample volumes (≥ 2mg/mL, ≥ 100μL) [25] | Often requires extensive molecular engineering and crystallization screening [4] [13] |
| Representative Achievement | Structures of TRPV1 ion channel and native lysosomal V-ATPase [46] [10] | Structures of β2-adrenergic receptor and other GPCRs using LCP [10] |
Detailed Experimental Protocol: Native Membrane Visualization
The protocol for studying membrane proteins in near-native states, as in the lysosomal study, involves [46]:
- Isolation of Native Organelles: Intact lysosomes are immunopurified from cells using specific antibodies against lysosomal membrane proteins (e.g., TRPML1 or TMEM192), preserving the native membrane architecture.
- Cryo-FIB-SEM Preparation: Cells or isolated organelles are vitrified. Cryo-focused ion beam (cryo-FIB) milling is then used to create thin lamellae (typically 100-300 nm) suitable for electron transparency.
- Cryo-Electron Tomography (Cryo-ET): The lamella is tilted in the electron microscope, collecting a series of 2D images from different angles.
- Tomogram Reconstruction: The tilt series is computationally reconstructed into a 3D tomogram, providing a volumetric view of the cellular context.
- Sub-tomogram Averaging: Identical macromolecular complexes (e.g., V-ATPase particles) are identified, extracted, aligned, and averaged from the tomogram to achieve a higher-resolution structure.
Application 3: Visualizing Multiple Conformations
Many proteins are dynamic machines that sample multiple conformational states to perform their functions. Cryo-EM is uniquely capable of capturing these discrete states from a single sample preparation, providing a molecular movie rather than a single snapshot.
Experimental Evidence and Performance Data
A compelling example is the structural analysis of TcPOP, a vaccine candidate for Chagas disease. Using single-particle cryo-EM, researchers resolved the 3D structure of TcPOP in both open and closed conformations at global resolutions of 3.8 Å and 3.6 Å, respectively [47]. The ability to classify and refine these distinct states from the same dataset was crucial for understanding the enzyme's functional dynamics and for informing immunogen design [47]. In contrast, X-ray crystallography often traps a protein in a single, dominant conformation, potentially missing biologically relevant states.
Table 4: Performance Comparison for Conformational Flexibility
| Criterion | Cryo-EM | X-ray Crystallography |
|---|---|---|
| Conformational Capture | Can capture an ensemble of states from a single sample; reveals transition states [44] | Typically provides a high-resolution snapshot of a single, stable state [44] |
| Data Collection | Hours to days per dataset [44] | Minutes to hours per dataset [44] |
| Key Advantage | Maintains solution-state dynamics; enables time-resolved studies [44] | Provides atomic details of discrete states; temperature-factor analysis can suggest flexibility [44] |
| Representative Achievement | Open/closed states of TcPOP; dynamic ribosomal complexes [47] | Structures of different stable states achieved via crystal packing or ligand binding [44] |
Detailed Experimental Protocol: Conformational State Analysis
The workflow for resolving multiple conformations involves advanced computational classification [47]:
- Heterogeneous Sample Preparation: The protein sample, which contains a mixture of conformational states, is prepared and vitrified as in standard single-particle analysis.
- Initial Processing: Standard steps of motion correction, particle picking, and 2D classification are performed to isolate well-defined particles.
- Heterogeneous 3D Classification: After an initial 3D reconstruction, the dataset of particle images is subjected to 3D classification algorithms without imposing symmetry. This step separates the particles into different groups based on their distinct 3D shapes or conformations.
- Focused Refinement: Each group of particles, now representing a more homogeneous population in a specific conformational state, is refined independently to produce high-resolution 3D maps for each state.
- Model Building and Validation: Atomic models are built and refined for each conformational state.
The Scientist's Toolkit: Key Research Reagent Solutions
Successful cryo-EM studies, especially of challenging targets, often rely on specialized reagents and materials.
Table 5: Essential Research Reagents for Cryo-EM
| Reagent / Material | Function | Application Example |
|---|---|---|
| Nanobody Scaffolds | Bifunctional reagents that bind and dimerize small proteins, increasing their apparent size and facilitating particle alignment [45]. | Imaging proteins under 50 kDa, such as the 14 kDa lysozyme [45]. |
| Graphene Support Grids | Grid coatings that minimize background noise and reduce preferred orientation of particles at the air-water interface [25]. | Improving data quality for low-abundance or difficult-to-bind membrane proteins [25]. |
| Lipid Nanodiscs | Soluble lipid bilayers that provide a native-like environment for purifying and studying membrane proteins [44]. | Structural analysis of GPCRs, ion channels, and transporters [44] [25]. |
| Cryo-FIB/SEM Instrument | Combines a focused ion beam with an SEM to mill thin, electron-transparent lamellae from vitrified cells or tissues [46] [48]. | Preparing samples for cryo-ET to study proteins in their native cellular environment [46]. |
| Direct Electron Detectors | Advanced cameras that provide dramatically improved signal-to-noise and enable motion correction [10]. | Essential for achieving near-atomic resolution in single-particle analysis and cryo-ET [10]. |
Integrated Approaches and Future Directions
The field is increasingly moving towards integrative and complementary approaches. A powerful synergy exists between cryo-EM and X-ray crystallography, where cryo-EM can provide a low-resolution architecture of a large complex into which high-resolution crystal structures of individual components can be docked [4]. Furthermore, the integration of cryo-EM with artificial intelligence (AI) tools like AlphaFold is revolutionizing structure determination. Methods like MICA use multimodal deep learning to combine cryo-EM density maps with AlphaFold3-predicted structures, resulting in more accurate and automated protein structure modeling [49]. This fusion of experimental and computational biology is poised to further accelerate discovery in structural biology.
For researchers seeking to capture molecular movies of biological processes, time-resolved studies represent the frontier of structural biology. Both X-ray crystallography and cryo-electron microscopy (cryo-EM) have developed powerful methodologies to visualize structural changes over time, each with distinct capabilities and applications. This guide provides a detailed comparison of these advanced techniques to inform their use in dynamic structural analysis.
Time-resolved structural biology aims to move beyond static snapshots to capture the sequential structural changes a molecule undergoes during its functional cycle. This is crucial for understanding mechanisms in processes like enzyme catalysis, signal transduction, and ion transport [10].
The fundamental challenge in time-resolved studies lies in initiating a biochemical process synchronously across a molecular population and then capturing structural data at precise time points before the reaction completes. X-ray crystallography and cryo-EM have evolved very different approaches to overcome this challenge, leveraging their respective strengths in resolution and single-particle analysis [4].
Recent technological advances are pushing the boundaries of what's possible in both techniques. For X-ray crystallography, the development of X-ray free-electron lasers (XFELs) has enabled studies on previously inaccessible timescales. In cryo-EM, rapid freezing methods and improved processing algorithms now allow researchers to capture and distinguish multiple conformational states from a single sample [10] [25].
Technical Comparison of Time-Resolved Approaches
The selection between time-resolved X-ray crystallography and cryo-EM involves careful consideration of their technical parameters, as summarized in the table below.
Table 1: Technical Comparison of Time-Resolved Methods
| Parameter | Time-Resolved X-ray Crystallography | Time-Resolved Cryo-EM |
|---|---|---|
| Temporal Resolution | Femtoseconds to milliseconds [10] | Milliseconds to seconds [50] |
| Activation Methods | Light, substrate diffusion, T-jump [10] | Spray mixing, light, substrate diffusion [25] |
| Sample State | Crystal lattice | Vitreous ice [4] |
| Key Enabling Technology | XFELs, microfluidic chips [10] | Rapid freezing, spray plungers [25] |
| Typical Resolution | Atomic (often 1.5-2.5 Å) [50] | Near-atomic to medium (3-8 Å, depending on time point) [50] |
| Data Collection Time | Minutes to hours (per time point) [50] | Hours to days (entire time course) [50] |
| Ideal for | Ultra-fast events, atomic-level dynamics | Slower conformational changes, heterogeneous systems [1] |
Workflow and Methodologies
The experimental pathway for each technique is designed to overcome its unique challenges in capturing dynamic processes.
Table 2: Core Reagent Solutions for Time-Resolved Studies
| Reagent/Tool | Function in Time-Resolved Studies |
|---|---|
| Microfluidic Chips (X-ray) | Enables rapid mixing and precise time-delayed ejection of microcrystals for XFEL studies [10]. |
| Lipidic Cubic Phase (X-ray) | Membrane protein crystallization matrix that can facilitate light-induced or diffusion-initiated reactions [13]. |
| GraFuture Grids (Cryo-EM) | Graphene-based support films that minimize background noise and improve particle distribution for time-resolved samples [25]. |
| Rapid Plunger (Cryo-EM) | Vitrification instrument that can integrate spray mixing for reaction initiation immediately before freezing [25]. |
Diagram 1: Time-resolved structural biology workflows for X-ray crystallography and cryo-EM.
Experimental Protocols and Data Analysis
Time-Resolved Serial Femtosecond Crystallography (TR-SFX)
Principle: TR-SFX uses extremely brief X-ray pulses from X-ray free-electron lasers (XFELs) to probe microcrystals at specific time points after reaction initiation, capturing intermediate states without radiation damage [10].
Detailed Protocol:
- Microcrystal Preparation: Generate high-quality microcrystals (typically 1-5 µm) in their native or substrate-bound state.
- Reaction Initiation: Use one of three primary methods:
- Light Activation: For photosensitive proteins, use a laser pulse to initiate photocycle [10].
- Diffusion-Based: Soak crystals with substrate or use caged compounds that release active molecules upon photolysis.
- Mix-and-Inject: Rapidly mix substrate and crystal streams in a microfluidic device with precise delay before X-ray probing [10].
- Data Collection: Deliver the activated crystal slurry in a liquid jet through the XFEL beam. Each crystal is probed by a single femtosecond X-ray pulse before destruction.
- Data Processing: Apply specialized software to index and merge "hits" from thousands of microcrystals at each time delay to reconstruct a 3D electron density map.
Application Example: This method has been successfully applied to study the catalytic cycle of cytochrome c oxidase, revealing electron and proton transfer mechanisms at various intermediate states [10].
Time-Resolved Cryo-Electron Microscopy
Principle: Time-resolved cryo-EM captures dynamic processes by initiating reactions and vitrifying samples at specific time points, trapping intermediate states for subsequent single-particle analysis [25].
Detailed Protocol:
- Grid Preparation: Apply purified protein sample to cryo-EM grids, typically at concentrations of 2-5 mg/mL [25].
- Reaction Initiation: Employ one of these approaches:
- Spray Mixing: Use a dedicated plunger to mix protein and ligand solutions immediately before blotting and freezing (millisecond resolution).
- Light Activation: For photosystems, illuminate grids before or during the freezing process.
- Diffusion-Based: Incubate grids with substrate for defined periods before freezing.
- Rapid Vitrification: Plunge-freeze the grid into liquid ethane at precise time points after reaction initiation.
- Data Collection: Collect thousands of micrographs using a 300 kV cryo-electron microscope with a direct electron detector [51] [25].
- Heterogeneity Analysis: Use 3D classification algorithms to separate particles into distinct conformational states, effectively reconstructing a temporal trajectory from a single sample.
Application Example: This approach has revealed distinct functional states of membrane proteins like GPCRs and ion channels, showing how they transition between inactive and active conformations [25].
Comparative Strengths and Applications
Resolution and Timescale Capabilities
The choice between techniques often depends on the biological timescale of interest and the required structural detail.
Table 3: Application-Based Method Selection
| Biological Process | Recommended Technique | Rationale |
|---|---|---|
| Enzyme Catalysis | TR-SFX (XFEL) | Atomic resolution needed to visualize bond formation/cleavage; ultrafast timescales [10]. |
| Membrane Protein Activation | Time-Resolved Cryo-EM | Captures large-scale conformational changes; avoids crystal packing constraints [50] [25]. |
| Macromolecular Assembly | Time-Resolved Cryo-EM | Handles size and heterogeneity; reveals assembly pathways [50]. |
| Photosystem Dynamics | TR-SFX (XFEL) | Ideal for light-triggered processes; femtosecond resolution possible [10]. |
Diagram 2: Technique selection guide based on biological process timescales.
Complementary Nature in Structural Biology
Rather than competing technologies, time-resolved X-ray crystallography and cryo-EM serve as complementary tools that can be integrated for a more complete understanding of dynamic processes [1] [4].
Hybrid Approach: Researchers can use time-resolved cryo-EM to identify the major conformational states and then employ TR-SFX to determine atomic-level details of specific intermediates. This strategy combines the breadth of cryo-EM in capturing heterogeneity with the depth of crystallography in revealing atomic mechanics [1].
Phase Problem Solution: In some cases, cryo-EM maps of intermediates can provide initial models for molecular replacement, helping to solve the phase problem in X-ray crystallography [4]. This synergy accelerates the structure determination process for challenging targets.
The field of time-resolved structural biology continues to evolve rapidly. For X-ray crystallography, developments in brighter X-ray sources and faster detectors promise to extend temporal resolution to shorter timescales. In cryo-EM, improvements in rapid freezing technology and AI-driven analysis algorithms are making it possible to resolve finer temporal sampling and smaller molecular targets [10] [25].
The integration of artificial intelligence with both techniques is particularly promising. AI algorithms can better classify heterogeneous particles in cryo-EM and predict likely intermediate states for targeted study by both methods [10]. Additionally, the development of more sophisticated mixing and triggering systems will expand the range of biological processes accessible to time-resolved study.
In conclusion, both time-resolved X-ray crystallography and cryo-EM offer powerful pathways to visualize biomolecular dynamics. TR-SFX with XFELs provides unparalleled temporal and spatial resolution for crystallizable systems, while time-resolved cryo-EM offers unique advantages for studying large, heterogeneous complexes in near-native conditions. The strategic selection between these methods—or their integrated application—provides researchers with a comprehensive toolkit for capturing the molecular movies that underlie biological function.
Overcoming Technical Hurdles: Sample Requirements, Optimization, and Access
X-ray crystallography remains a cornerstone of structural biology, providing unparalleled atomic-level insights into protein function that are indispensable for drug discovery and mechanistic studies. However, its success is fundamentally constrained by a critical bottleneck: the difficulty of obtaining high-quality, well-ordered crystals suitable for diffraction studies. This challenge is particularly pronounced for membrane proteins, flexible complexes, and intrinsically disordered regions that resist crystallization. Despite technological advancements, crystallization remains the primary limiting step, with its unpredictability consuming substantial time and resources in structural biology pipelines. This article examines the crystallization bottleneck within the broader context of methodological competition with cryo-electron microscopy (cryo-EM), comparing their respective capabilities and presenting innovative strategies to overcome crystallization barriers for challenging protein targets.
Understanding the Crystallization Bottleneck
The Fundamental Requirements for Successful Crystallization
Protein crystallization represents a complex process governed by numerous biochemical and physical parameters that must be carefully optimized. Three fundamental requirements must be met for successful crystal formation:
High Purity (>95%): Samples must exhibit exceptional homogeneity, as impurities including oligomerization variants, isoforms, flexible regions, misfolded populations, partial proteolysis products, cysteine oxidation, and deamidation of Asn and Gln residues can disrupt ordered lattice formation [24]. Even when crystals form with impurities, the result is often poor diffraction quality due to a disordered crystal lattice.
Exceptional Stability: Crystals can require days to months to nucleate, necessitating prolonged sample stability maintained through optimized buffer components (typically below ∼25 mM concentration), salts (below 200 mM for sodium chloride), and potentially substrates, ligands, coordinating metals, or reductants [24]. Phosphate buffers should be avoided due to insoluble salt formation.
High Solubility and Homogeneity: The sample must be monodisperse and non-aggregating, with glycerol often required for solubilization but limited to below 5%(v/v) in final crystallization drops [24]. Analytical techniques including dynamic light scattering (DLS), size-exclusion chromatography (SEC), SEC-MALS, and mass photometry provide critical assessment of homogeneity and solubility.
The Phase Diagram and Crystallization Cocktails
Crystallization occurs through careful manipulation of the phase diagram, where chemical cocktails promote crystal formation by modulating protein solubility [24]. These conditions typically include:
- Buffers to control pH, generally within 1-2 pH units of the protein's isoelectric point (pI) to optimize intermolecular interactions.
- Salts that initially enhance stability through electrostatic contacts but at higher concentrations compete for water molecules, forcing proteins to form crystal lattice interactions (salting-out phenomenon).
- Polymers like polyethylene glycols (PEGs) that induce macromolecular crowding and may screen salt-mediated aggregation while potentially reducing entropic motion.
- Additives including 2-methyl-2,4-pentanediol (MPD), cofactors, substrates, non-hydrolyzable substrates, small molecules, partner proteins, and Fab fragments that enhance stability or mediate intermolecular contacts.
Table 1: Common Crystallization Reagents and Their Functions
| Reagent Category | Specific Examples | Primary Function | Considerations |
|---|---|---|---|
| Salts | Ammonium sulfate, Sodium chloride | Reduce solubility via salting-out; mediate interactions | Concentration-dependent effects; some may act as ligands |
| Polymers | PEG 3350, PEG 8000 | Molecular crowding; reduce entropic motion; screen aggregation | Viscosity effects; may serve as cryoprotectants |
| Additives | MPD, ligands, cofactors | Bind hydrophobic regions; enhance stability; mediate contacts | May be essential for ordering flexible regions |
| Buffers | HEPES, Tris, MES | Control pH to optimize surface charges | Keep <25 mM; avoid phosphate buffers |
| Reductants | TCEP, DTT, BME | Prevent cysteine oxidation | Consider half-life relative to crystal growth timescale |
Strategic Approaches to Overcome Crystallization Challenges
Biochemical Optimization Strategies
Construct Design and Engineering
Rational construct design represents the foundational step in overcoming crystallization barriers. AlphaFold has emerged as an invaluable resource for guiding construct design by identifying and eliminating floppy regions that introduce conformational heterogeneity [24]. Strategic approaches include:
- Terminal Truncation: Removing flexible N- or C-terminal regions to enhance protein rigidity and promote ordered packing.
- Surface Engineering: Targeted mutagenesis of surface residues to improve crystal contacts, though validation is essential to ensure structural and functional integrity isn't compromised [24].
- Loop Stabilization: Modifying flexible loops through site-directed mutagenesis or insertion of stabilizing domains.
Affinity Tags and Fusion Partners
Affinity tags serve dual purposes in purification and crystallization enhancement. Beyond improving solubility properties, tags including GST, MBP, and His-tags can function as crystallization chaperones [24]. In challenging cases, fusion partners may provide artificial crystal contacts that facilitate lattice formation, though subsequent tag removal may be necessary for certain targets.
Sample Preparation and Buffer Optimization
Meticulous attention to sample preparation details significantly impacts crystallization success:
- Reductant Selection: Chemical reductants prevent cysteine oxidation but vary substantially in half-life (Table 2). Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) offers superior stability across broad pH ranges compared to dithiothreitol (DTT) or β-mercaptoethanol (BME) [24].
- Stability Assessment: Differential scanning fluorimetry and circular dichroism provide critical data on optimal buffer composition, pH, temperature, and ligand effects.
- Concentration Optimization: Both under-concentration and over-concentration can prevent crystallization success. Pre-crystallization testing using sparse-matrix approaches helps identify appropriate concentration ranges [24].
Table 2: Reductant Stability Under Different pH Conditions
| Chemical Reductant | Solution Half-life (pH 6.5) | Solution Half-life (pH 8.5) | Stability Profile |
|---|---|---|---|
| Dithiothreitol (DTT) | 40 hours | 1.5 hours | pH-dependent degradation |
| β-Mercaptoethanol (BME) | 100 hours | 4.0 hours | Moderate pH sensitivity |
| Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) | >500 hours (pH 1.5-11.1) | >500 hours (pH 1.5-11.1) | Exceptional pH stability |
Physical and Engineering Approaches
Interface-Mediated Crystallization
The strategic use of interfaces has emerged as a powerful approach to promote and modulate nucleation events. Solid/liquid interfaces have gained particular attention due to their ability to control nucleation kinetics [52]. Effective surfaces include:
- Porous Substrates: Provide template effects and increase local protein concentration.
- Functionalized Surfaces: Hydrophobic, charged, or chemically modified substrates that promote molecular alignment.
- Nanoparticles and DNA Assemblies: Offer precisely tunable surfaces for nucleation enhancement.
These interfaces operate through both electrostatic and non-electrostatic mechanisms, influencing early nucleation events and subsequent crystal growth [52].
Scaffold-Assisted Crystallization
Natural and engineered protein scaffolds provide pre-organized templates that facilitate crystal lattice formation:
- Natural Crystalline Scaffolds: Certain proteins possess intrinsic sequence features promoting spontaneous crystal formation, which can be leveraged through fusion strategies to promote crystallization of challenging targets [53].
- Engineered Assemblies: Symmetrical scaffolds including DARPins and coiled-coil modules create ordered environments that support crystal formation [54].
- In-Cell Biocrystallization: Harnessing cellular machinery for intracellular crystal formation represents an emerging approach with potential for stable protein formulation and direct structural analysis [53].
Computational and Predictive Methods
AI-Driven Crystal Packing Prediction
The integration of artificial intelligence with structural modeling has transformed crystal packing prediction. The MASCL approach combines AlphaFold with symmetrical docking to simulate crystal packing, introducing PackQ as a stringent metric for evaluating packing quality [55]. In benchmark tests, this methodology successfully predicted packing interfaces for 26.8%-30.1% of targets within top 100 models, with success rates improving to 57.9% when initial dimeric assemblies were successfully predicted [55].
Physicochemical Descriptor Analysis
AAI-PatchBag represents an innovative patch-based method that utilizes physicochemical descriptors to assess molecular interface similarity [55]. Compared to conventional condition-searching strategies like sequence alignment and structure superposition, this approach significantly reduces the number of trials required to identify potential crystallization conditions, as demonstrated in lysozyme crystallization studies [55].
Crystallization Workflow and Experimental Design
High-Throughput Screening Methodologies
Given the vast chemical space of potential crystallization conditions, efficient screening approaches are essential:
- Sparse-Matrix Screening: Empirically designed condition sets that efficiently sample diverse chemical space [24].
- Incomplete Factorial Approaches: Systematically explore parameter combinations to identify critical crystallization factors.
- Homology-Guided Screening: Leveraging crystallization conditions from structurally related proteins deposited in the PDB.
- Additive Screening: Targeted screening of cofactors, substrates, and small molecules that may enhance stability or promote specific interactions.
The likelihood of crystallization success increases substantially with the number of conditions tested, making high-throughput approaches particularly valuable [24].
Comparative Analysis: X-ray Crystallography vs. Cryo-EM
Technical and Methodological Comparisons
The emergence of cryo-EM as a dominant structural biology technique has transformed the landscape, offering distinct advantages and limitations compared to X-ray crystallography:
Table 3: Comparative Analysis of X-ray Crystallography and Cryo-EM
| Parameter | X-ray Crystallography | Cryo-EM (Single Particle) |
|---|---|---|
| Sample Requirement | High-quality crystals; highly homogeneous, concentrated samples | Vitrified solution; lower concentration possible; tolerance for heterogeneity |
| Size Limitations | No upper size limit; minimal practical size ~10 kDa | Theoretical limit ~38 kDa; practical minimum ~50 kDa without scaffolds |
| Resolution Range | Typically 1.5-3.0 Å (atomic) | Typically 2.5-4.5 Å (near-atomic to atomic) |
| Throughput | Months to years (crystallization bottleneck) | Days to weeks (after sample optimization) |
| Membrane Proteins | Challenging; requires detergents/nanodiscs; LCP methods helpful | Excellent; native-like environments; major recent advances |
| Dynamic/Flexible Regions | Poorly resolved; often missing electron density | Better tolerated; specialized processing can resolve conformations |
| Intrinsic Disorder | Highly challenging; typically unresolved | Moderate success with advanced processing [56] |
| Scaffolding Requirements | Occasionally for difficult targets | Frequently essential for small proteins (<100 kDa) [54] |
Practical Applications in Drug Discovery
Both techniques have demonstrated significant impact in structure-based drug design, with complementary strengths:
X-ray Crystallography: Remains unparalleled for elucidating precise atomic interactions between drugs and targets, as demonstrated in the structure determination of the SARS-CoV-2 main protease with inhibitor nirmatrelvir [10]. Provides critical information for optimizing small molecule interactions within binding pockets.
Cryo-EM: Excels for large complexes and membrane proteins difficult to crystallize, such as G protein-coupled receptors (GPCRs) and ion channels. Enables visualization of drug binding in near-native environments, as demonstrated with kRasG12C structures at 3.7 Å resolution with bound inhibitor MRTX849 and GDP [54].
Case Studies: Integrated Approaches for Challenging Targets
Membrane Protein Crystallization
Membrane proteins represent particularly challenging targets for crystallization due to their inherent instability in solution. Successful strategies often combine multiple approaches:
- Detergent Screening: Identification of optimal detergents that maintain stability while allowing crystal contacts.
- Lipidic Cubic Phase (LCP) Crystallization: Provides a membrane-mimetic environment that has enabled structure determination of numerous GPCRs and transporters [10].
- Antibody Fragment Complexation: Using Fab fragments to increase hydrophilic surface area and provide crystal contacts.
Cryo-EM Scaffolding Strategies for Small Proteins
For proteins below 50 kDa, cryo-EM faces significant technical challenges due to limited signal-to-noise ratios and difficulties in particle alignment [54]. Multiple scaffolding approaches have been developed to overcome these limitations:
- Coiled-Coil Fusion Strategies: As demonstrated with kRasG12C-APH2 fusions, these provide rigid, symmetric assemblies that enable high-resolution reconstruction (3.7 Å) [54].
- DARPin Cages: Engineered protein cages that encapsulate target proteins in ordered symmetric arrays, though requiring extensive optimization [54].
- Megabodies and Legobodies: Nanobody-based scaffolds with engineered extensions that increase particle size while maintaining binding specificity.
Research Reagent Solutions for Structural Biology
Table 4: Essential Research Reagents for Structural Biology Applications
| Reagent Category | Specific Examples | Primary Application | Key Considerations |
|---|---|---|---|
| Crystallization Screens | Hampton Research Screens, MemGold | Initial crystallization condition screening | Sparse-matrix, grid screening, and specialized membrane protein screens available |
| Detergents | DDM, LMNG, OG, Cymal | Membrane protein solubilization and stabilization | Critical for membrane proteins; impact crystal quality and diffraction |
| Fusion Tags | His-tag, GST, MBP, SUMO | Solubility enhancement and purification | May influence crystallization; removal often necessary |
| Scaffold Proteins | DARPins, APH2 coiled-coil, BRIL | Cryo-EM particle size enhancement and stabilization | Require optimization for each target; may influence conformational states |
| Nanobodies | Anti-APH2 nanobodies, VHH libraries | Conformational stabilization and complex formation | High affinity and stability; accessible epitopes may be limited |
| Additive Libraries | Hampton Additive Screen | Crystallization optimization | Identify specific compounds improving crystal quality |
| Cryoprotectants | Glycerol, ethylene glycol, sucrose | Crystal and vitreous ice preservation | Must be optimized for each crystal system |
Future Perspectives and Emerging Technologies
Integrative Structural Biology Approaches
The future of structural biology lies in integrative approaches that combine multiple methodologies to overcome individual technique limitations:
- Hybrid Modeling: Combining cryo-EM density maps with AlphaFold predictions to resolve flexible regions and validate models [10] [57].
- Time-Resolved Studies: Serial crystallography and time-resolved cryo-EM enabling visualization of dynamic processes and intermediate states.
- In Situ Structural Biology: Cryo-electron tomography advancing toward cellular context structural determination, bridging the resolution gap between cellular and molecular scales [57].
AI and Machine Learning Revolution
Artificial intelligence continues to transform structural biology through multiple avenues:
- Structure Prediction Integration: AlphaFold predictions guide construct design and identify structured domains for crystallization [24].
- Crystallization Condition Prediction: Machine learning algorithms trained on PDB crystallization data increasingly inform condition selection [24].
- Dynamic Region Analysis: Tools like the CLTC hybrid model (combining CNN, LSTM, Transformer, and CRF architectures) predict flexible regions challenging for both crystallography and cryo-EM [56].
Methodological Convergence
The historical competition between X-ray crystallography and cryo-EM is evolving toward methodological complementarity:
- Microcrystal Electron Diffraction (MicroED): Bridges the gap between crystallography and cryo-EM by enabling structure determination from nanocrystals [58].
- Serial Femtosecond Crystallography (SFX): Using X-ray free-electron lasers to collect data from microcrystals before radiation damage occurs [52].
- Cellular Crystallography: Emerging approaches leveraging natural crystalline scaffolds for in cellulo crystal formation [53].
The crystallization bottleneck, while persistent, is being addressed through innovative multidisciplinary strategies that combine biochemical, physical, and computational approaches. The integration of AI-driven prediction tools, advanced scaffolding methodologies, and interface-mediated crystallization has substantially improved success rates for challenging targets. Rather than representing competing technologies, X-ray crystallography and cryo-EM have evolved complementary roles in the structural biology toolkit, each with distinct advantages for specific biological questions. The future of structural biology lies in methodological integration, leveraging the atomic precision of crystallography with the compositional and conformational flexibility of cryo-EM to illuminate biological mechanisms and accelerate therapeutic development. As these technologies continue to converge and advance, the crystallization bottleneck will progressively diminish, opening new frontiers in our understanding of complex biological systems.
While cryogenic electron microscopy (cryo-EM) has revolutionized structural biology by enabling the determination of high-resolution structures of biological macromolecules without crystallization, two persistent challenges frequently compromise data quality: preferred orientation and air-water interface (AWI) effects. These phenomena are interconnected sample preparation issues that can introduce significant resolution anisotropy, reconstruction artifacts, or even complete loss of structural information for certain domains [59] [60].
Preferred orientation occurs when protein particles adopt a specific, non-random alignment on the cryo-EM grid, rather than distributing themselves in uniformly random orientations [61] [62]. Much like viewing milk jugs from only one angle where the handle remains invisible, this preferential alignment prevents researchers from capturing all the necessary views to reconstruct a complete three-dimensional structure [60]. The AWI presents a particularly damaging environment where proteins can become partially denatured, dissociate into subunits, or accumulate at the interface before freezing [59] [63]. Understanding and mitigating these challenges is crucial for obtaining high-resolution structures that accurately represent the native state of biological molecules.
Understanding the Fundamental Challenges
The Preferred Orientation Problem
In single-particle cryo-EM, the three-dimensional structure of a biomolecule is reconstructed from thousands of two-dimensional projection images taken from different orientations [61]. The fundamental assumption is that the molecules are randomly oriented in the vitreous ice. However, this ideal is frequently not met. Proteins, particularly those with elongated shapes (rods) or flat surfaces (discs), often orient with their long axis parallel to the grid [60]. Furthermore, hydrophobic patches on the protein surface can cause them to stick to the hydrophobic AWI, leading to a highly imbalanced distribution of particle views [62] [59].
The consequences of preferred orientation are severe. When significant orientation bias exists, certain views of the particle are over-represented while others are missing entirely. This leads to resolution anisotropy in the reconstructed density map, where some regions are well-resolved while others appear blurred or are completely absent [61] [62]. In extreme cases, the missing views make accurate three-dimensional reconstruction impossible, as critical structural information is lost [60]. Computational methods struggle with this imbalance because signals from particles in non-preferred views are frequently overshadowed by those from dominant preferred views during refinement processes [62].
The Air-Water Interface Threat
The AWI represents a potentially destructive environment for biological samples during cryo-EM grid preparation. When the sample solution is applied to the grid and blotted to form a thin liquid film, proteins are exposed to the interface before vitrification. This interface is chemically disruptive because of its high surface energy and hydrophobic nature [63]. Exposure to the AWI can lead to subunit dissociation, partial denaturation, and preferred orientation of particles [59].
The problem is particularly acute for membrane proteins and complex assemblies with both hydrophobic and hydrophilic surfaces. A 2021 study on the KtrA protein demonstrated that extensive AWI exposure resulted in complete lack of density for the C-lobe domains, which could only be restored by addressing both AWI exposure and molecular flexibility [59]. Traditionally, more than 99% of collected images might be unusable due to AWI-induced damage, representing a massive inefficiency in data collection [63].
Table 1: Key Differences in Sample Preparation Challenges Between Cryo-EM and X-ray Crystallography
| Challenge Factor | Cryo-EM | X-ray Crystallography |
|---|---|---|
| Primary Sample Hurdle | AWI exposure & preferred orientation | Crystal formation & quality |
| Sample State | Near-native, frozen-hydrated state [64] | Highly ordered crystalline lattice [64] |
| Sample Amount | Minimal volume (0.1-0.2 mg typically) [23] | Larger quantities (>2 mg typically) [23] |
| Flexibility Handling | Can capture multiple conformational states [23] | Requires rigid structure for well-ordered crystals [23] |
| Membrane Protein Suitability | Ideal for membrane proteins in near-native environments [23] | Challenging; requires detergent optimization or lipidic cubic phase [13] |
Experimental Solutions and Mitigation Strategies
Sample Preparation Innovations
Addressing the Air-Water Interface
Multiple innovative approaches have been developed to minimize AWI damage:
High-Speed Droplet Vitrification: A groundbreaking method developed by Russo's group at the MRC Laboratory of Molecular Biology sprays microscopic droplets of protein solution at high speed (approaching 100 m/s) onto cryogenically cooled grids. The droplets flatten and freeze in under 10 microseconds, locking proteins in place before they can diffuse to the AWI. This technique has been validated with apoferritin, yielding a 2.7 Å resolution reconstruction without structural degradation [63].
Reduced Plunge Time: Modifying standard blotting techniques to minimize the time between blotting and vitrification significantly reduces AWI exposure. The KtrA study demonstrated that reducing plunge times from 6 seconds to ≤100 milliseconds notably improved density for the vulnerable C-lobe domains [59].
Surfactants and Additives: Including surfactants or other additives just before vitrification can modulate protein-AWI interactions. These compounds form a protective layer that shields proteins from the denaturing interface [60].
Alternative Grid Supports: Utilizing graphene supports or other functionalized grids can equalize particle pose distribution by providing a more favorable surface than the AWI [62].
Overcoming Preferred Orientation
No single solution universally addresses preferred orientation, necessitating a multi-pronged approach:
Grid Treatment: Chemically modifying the grid surface to induce alternative orientations can help achieve a more balanced particle distribution [61].
Biomolecule Modifications: Engineering the protein surface characteristics or selecting different protein/complex constructs can reduce surface stickiness. Adding ligands to reduce flexibility has also proven effective, as demonstrated in the KtrA study where cyclic di-AMP binding combined with fast plunge time eliminated preferred orientation [59].
Tilt Collection Strategy: Collecting data with the grid tilted at an angle (typically 10-40°) relative to the electron beam provides different viewing perspectives, helping to fill in missing orientations. However, this approach introduces drawbacks including reduced image acquisition efficiency and increased beam-induced movement [62].
Diagram 1: Experimental workflow for tackling Cryo-EM sample preparation challenges, showing key problems and mitigation strategies.
Computational Solutions
When sample optimization alone is insufficient, computational methods offer powerful alternatives for addressing preferred orientation:
cryoPROS Framework: This computational framework, introduced in 2025, corrects misalignment caused by preferred orientation by co-refining raw particles with computationally generated auxiliary particles. A deep generative model synthesizes these auxiliary particles with evenly distributed orientations in a self-supervised manner, creating a more balanced combined dataset that significantly improves pose estimation accuracy. CryoPROS has achieved near-atomic resolution with untilted datasets that previously could not be solved [62].
Manifold Learning Analysis: Researchers have proposed a measure based on manifold learning to identify when cryo-EM data comes from biomolecules adopting preferred orientations. By analyzing the geometry of manifolds projected onto low-dimensional spaces, this method can detect orientation bias before attempting reconstruction, potentially saving significant computational time [61].
Extended Data Collection: Advances in direct electron detectors (Gatan K3, TFS Falcon 4) now support high-throughput image acquisition, making larger particle sets available for processing. By collecting massive datasets, researchers increase the probability of capturing rare, non-preferred views that can fill critical gaps in the reconstruction [60].
Table 2: Quantitative Comparison of Solution Efficacy for Cryo-EM Challenges
| Solution Strategy | Technical Approach | Reported Efficacy | Key Limitations |
|---|---|---|---|
| High-Speed Droplet Vitrification [63] | Prevents AWI contact via microsecond freezing | 2.7 Å resolution achieved with apoferritin; eliminates >99% data loss | Challenging ice thickness control |
| cryoPROS Computational Framework [62] | Generative AI creates auxiliary particles for co-refinement | Near-atomic resolution from untilted HA-trimer data | Requires substantial computational resources |
| Fast Plunge + Ligand Combination [59] | 100ms vitrification with cyclic di-AMP | Restored missing C-lobe density in KtrA | Condition-specific optimization needed |
| Grid Tilting [62] | Collects data at 10-40° tilt angles | Improves directional isotropy | Increased noise & beam-induced movement |
Comparative Analysis with X-ray Crystallography
Fundamental Differences in Sample Handling
X-ray crystallography approaches sample preparation from a fundamentally different perspective than cryo-EM. Rather than seeking to preserve molecules in a near-native state, crystallography requires the formation of a highly ordered, repeating crystalline lattice [4] [64]. This process involves bringing protein molecules out of solution at a controlled rate that promotes crystal growth rather than precipitation [13].
The primary advantage of crystallography lies in its atomic-resolution precision, routinely achieving resolutions finer than 2 Å, which reveals intimate architectural details of molecules including how atoms connect to form chemical bonds and exactly where drug molecules dock with their targets [23] [65]. However, the crystallization process presents the largest hurdle in X-ray crystallography, particularly for membrane proteins, flexible complexes, and dynamic assemblies that resist crystallization [4] [13].
For membrane proteins, crystallography often requires extensive molecular engineering, including removal of flexible regions or domains to reduce overall flexibility and increase stable crystal contacts. Integral membrane proteins pose a specific challenge as they typically require detergents or nanodiscs for purification, with the membrane mimetic generally reducing potential crystal contact sites [13].
Complementary Strengths in Structural Biology
Rather than competing technologies, cryo-EM and X-ray crystallography serve as highly complementary tools [4]. One of the most powerful synergies between these methods involves solving structures through an integrated approach:
Cryo-EM can generate an initial low- to medium-resolution 3D map of a large protein complex, providing the overall architecture. X-ray crystallographic data of individual components can then be docked into the cryo-EM map, revealing high-resolution details of specific domains or subunits [4].
Conversely, cryo-EM can assist X-ray crystallography by helping to solve the phase problem through molecular replacement. Cryo-EM maps can provide an initial model for phasing, enabling researchers to solve crystal structures at higher resolution [4].
The distinct physical principles underlying the two techniques mean they provide slightly different structural information, allowing researchers to study molecules in different states or conformations for a more holistic view of biological structure and function [4].
Diagram 2: Complementary workflow between Cryo-EM and X-ray Crystallography for integrated structural biology.
Essential Research Reagent Solutions
Successful structural biology research requires specific reagents and materials to address the challenges discussed in this guide. The following table details key resources mentioned in recent literature.
Table 3: Essential Research Reagents and Materials for Addressing Cryo-EM Challenges
| Reagent/Material | Primary Function | Application Example |
|---|---|---|
| Cyclic di-AMP (CDA) [59] | Ligand that reduces flexibility and stabilizes specific conformations | Restored C-lobe density in KtrA when combined with fast plunge freezing |
| Surfactants/Detergents [60] | Modulate protein-air-water interface interactions to prevent denaturation | Protective layer shielding proteins from denaturing interface during grid preparation |
| Graphene Grid Supports [62] | Provide alternative surface chemistry to reduce AWI interactions | Equalize particle pose distribution for more balanced orientation sampling |
| Liquid Ethane Cryogen [63] | Medium for high-speed vitrification to preserve native structure | Used in droplet spray method for microsecond freezing of samples |
| Apoferritin Control Sample [63] | Well-characterized standard for validating new methods and equipment | Achieved 2.7 Å resolution in validation of high-speed spraying technique |
Preferred orientation and air-water interface effects represent significant but addressable challenges in cryo-EM sample preparation. Through innovative techniques ranging from high-speed vitrification and optimized grid preparation to advanced computational methods like cryoPROS, researchers now have multiple strategies to overcome these hurdles. The complementary nature of cryo-EM and X-ray crystallography continues to provide a powerful integrated approach to structural biology, with each method compensating for the limitations of the other. As both technologies advance, their synergistic application will undoubtedly drive future breakthroughs in understanding complex biological systems and facilitating structure-based drug design.
Structural biology relies on several powerful techniques to determine the three-dimensional structures of biological macromolecules, with X-ray crystallography and cryo-electron microscopy (cryo-EM) standing as the most prominent methods. X-ray crystallography has served as the dominant technique for decades, accounting for approximately 84% of structures in the Protein Data Bank (PDB) [13]. This method requires growing high-quality crystals of the target molecule, which are then exposed to X-rays to produce diffraction patterns. The analysis of these patterns reveals the electron density map from which an atomic model is built [66]. The technique's requirement for well-ordered crystals has historically limited its application for certain challenging targets, such as membrane proteins and large, flexible complexes [4].
In contrast, cryo-EM has emerged as a transformative technology, particularly over the past decade, experiencing what has been termed a "resolution revolution" [9] [4]. This technique involves flash-freezing purified protein samples in a thin layer of vitreous ice, preserving them in a near-native state. Electron micrographs of these frozen-hydrated particles are collected and computationally processed to reconstruct a three-dimensional structure [25] [67]. A significant advantage of cryo-EM is that it does not require crystallization, making it uniquely suited for studying large macromolecular complexes, membrane proteins, and dynamic systems that have proven refractory to crystallization [4] [68]. The selection between these methods often hinges on the specific sample properties and research goals, with sample requirements representing a critical factor in determining the most appropriate and feasible approach.
Comparative Analysis of Sample Requirements
The successful determination of a protein structure is profoundly influenced by the quality and preparation of the sample itself. The requirements for purity, concentration, and buffer conditions differ significantly between X-ray crystallography and cryo-EM, often making one technique more accessible than the other for a given biological sample.
Sample Purity and Homogeneity
X-ray crystallography demands exceptionally high sample purity and homogeneity, typically greater than 95% [13] [25]. This stringent requirement is necessary to promote the formation of a well-ordered, single crystal lattice. Even minor heterogeneity in the sample, such as the presence of protein aggregates, contaminants, or conformational flexibility, can prevent crystallization or result in poorly diffracting crystals. For membrane proteins, which are notoriously difficult to crystallize, additional steps like truncating flexible regions or engineering stability are often required to obtain crystals [13].
Cryo-EM is more tolerant of moderate sample heterogeneity, with purity requirements generally around 90% or higher [25]. While a pure sample is still ideal, cryo-EM can often resolve structures from samples that contain multiple conformational states or minor impurities. This is because the single-particle analysis computational algorithms can classify and separate different particle views and conformations during processing [67]. This tolerance for heterogeneity is a key advantage for studying dynamic complexes that exist in multiple states in solution.
Sample Concentration and Volume
The amount of sample required is another area of significant divergence between the two techniques, largely due to their different preparation workflows.
X-ray crystallography typically requires high protein concentrations, often in the range of 10 mg/mL or higher, with a total sample amount of at least 5 mg [13] [25]. For more complex experiments, such as studying antigen-antibody complexes, the total amount required can be even greater (e.g., >10 mg) [25]. This high consumption is due to the extensive screening and optimization needed to find initial crystallization conditions and then grow large, high-quality crystals suitable for diffraction experiments.
Cryo-EM requires a much lower total sample consumption. A typical single-particle analysis (SPA) experiment needs a protein concentration of ≥ 2 mg/mL in a volume of ≥ 100 μL, translating to a total requirement of about 0.2-0.5 mg of protein [25]. This minimal sample requirement makes cryo-EM particularly valuable for studying proteins that are difficult to express and purify in large quantities.
Buffer Conditions and Molecular Characteristics
Buffer composition and the intrinsic properties of the target molecule also influence the choice of method.
For X-ray crystallography, the buffer should be optimized to promote crystal contacts. Phosphate buffers are generally avoided as they can crystallize in the presence of divalent cations, leading to false positives in crystallization screens [13]. The technique is most successfully applied to soluble, stable proteins with rigid structures. While there is no strict size limit, very large complexes can present increased difficulties in obtaining well-ordered crystals [13].
For Cryo-EM, the buffer should have a low concentration of organic solvents (like glycerol) and salt ion concentrations typically ≤ 300 mM [25]. It is also recommended to send a separate 50-100 mL aliquot of the buffer for sample concentration exploration during grid preparation [25]. Cryo-EM excels with large molecules (optimal >100 kDa) and is ideally suited for membrane proteins, flexible complexes, and targets that are unstable over the long time periods often required for crystallization trials [67].
Table 1: Direct Comparison of Sample Requirements for X-ray Crystallography and Cryo-EM SPA
| Requirement | X-ray Crystallography | Cryo-EM (Single Particle Analysis) |
|---|---|---|
| Purity | >95% (High homogeneity essential) [13] [25] | ≥90% (Moderate heterogeneity tolerated) [25] |
| Concentration | >10 mg/mL [13] [25] | ≥2 mg/mL [25] |
| Total Sample Amount | >5 mg (Soluble proteins); >10 mg (Complexes) [25] | ~0.2 mg (≥100 μL at 2 mg/mL) [25] |
| Buffer/Solution | Avoid phosphate buffers [13] | Low organic solvent; Salt ≤300 mM [25] |
| Ideal Molecular Size | No strict limit, but smaller, rigid proteins are favorable [13] | Optimal >100 kDa [67] |
| Key Sample Property | Monodisperse, stable, crystallizable | Stable in vitreous ice for data collection |
Experimental Workflows and Protocols
The journey from a purified protein sample to a refined structural model involves distinct and critical steps for both X-ray crystallography and cryo-EM. Understanding these workflows is essential for planning and executing a successful structure determination project.
X-ray Crystallography Workflow
The following diagram outlines the key stages in structure determination by X-ray crystallography:
Crystallization is the most significant bottleneck in X-ray crystallography. The process involves creating supersaturated conditions where the protein slowly comes out of solution to form an ordered crystal lattice. This requires extensive screening of countless conditions varying precipitant, buffer, pH, and temperature [13] [66]. For initial crystal hits, optimization is performed to improve crystal size and diffraction quality. Membrane proteins often require specialized methods like lipidic cubic phase (LCP) crystallization to mimic their native environment [13].
Data Collection occurs at synchrotron facilities, which provide intense, tunable X-ray beams. A single crystal is exposed to the X-ray beam, and the resulting diffraction pattern is captured on a detector. A complete dataset consists of hundreds of images collected as the crystal is rotated [13] [66].
Data Processing and Phase Determination involves several computational steps. The diffraction spots on the images are indexed, integrated, and scaled to produce a set of structure factors containing amplitude information. A major hurdle, known as the "phase problem," is that the phase information is lost during data collection. This is typically solved by Molecular Replacement (MR), which uses a similar existing structure as a search model, or by experimental methods like Single-wavelength Anomalous Dispersion (SAD) or Multi-wavelength Anomalous Dispersion (MAD), which involve collecting data from crystals containing heavy atoms (e.g., selenium) [13] [66].
Model Building and Refinement is an iterative process. An initial atomic model is built into the experimental electron density map. This model is then refined by adjusting atomic positions to improve the agreement with the observed diffraction data while respecting standard chemical geometry [13] [66].
Cryo-EM Single Particle Analysis Workflow
The following diagram illustrates the standard workflow for structure determination by cryo-EM single particle analysis:
Grid Preparation and Vitrification is a critical first step. A small volume (~3-4 µL) of the purified sample is applied to an EM grid, blotted with filter paper to create a thin film, and then rapidly plunged into a cryogen (like liquid ethane) to freeze it into a glass-like (vitreous) ice layer. This process preserves the native structure of the particles [25] [67]. Challenges like preferential particle orientation or air-water interface disruption can be mitigated using specialized grids, such as graphene-based supports [25].
Data Collection is performed on a high-end transmission electron microscope operating at 200-300 kV. Modern automated microscopes equipped with direct electron detectors can collect thousands of micrograph movies in a single session, which may take from hours to days [25] [67].
Image Processing and 3D Reconstruction is a computationally intensive, multi-step process. The collected movies are first motion-corrected to account for beam-induced movement and the Contrast Transfer Function (CTF) of the microscope is estimated [67]. Individual protein particles are then automatically picked from the micrographs. These particle images are subjected to 2D classification to remove junk particles and generate clean class averages. An initial 3D model is generated and then iteratively refined. A powerful feature of cryo-EM is 3D classification, which can separate different conformational states or compositional heterogeneity within the dataset, allowing for the determination of multiple structures from a single sample [67].
Model Building and Refinement is the final stage. For a high-resolution map (typically better than ~3.5 Å), an atomic model can be built de novo or by docking and refining a known homologous structure. The model is refined against the cryo-EM map to produce the final atomic coordinates [67].
Research Reagent Solutions and Materials
Successful structure determination relies on a suite of specialized reagents, instruments, and software. The following table details key solutions used in the featured experimental workflows.
Table 2: Essential Research Reagents and Materials for Structural Biology
| Item | Function/Application | Relevant Technique |
|---|---|---|
| Crystallization Screens | Commercial suites of pre-dispensed solutions to empirically find initial crystal formation conditions. | X-ray Crystallography [13] |
| Lipidic Cubic Phase (LCP) Materials | A monolein-based lipid matrix used to crystallize membrane proteins in a membrane-mimetic environment. | X-ray Crystallography [13] |
| Selenomethionine (Se-Met) | An amino acid used to produce proteins with incorporated selenium atoms for experimental phasing via SAD/MAD. | X-ray Crystallography [13] |
| Cryo-EM Grids (e.g., Graphene) | Supports for vitrified samples. Graphene grids reduce background noise and mitigate preferred orientation. | Cryo-EM [25] |
| Nanobodies / DARPins | Engineered binding proteins used to increase the effective size and stability of small or flexible target proteins. | Cryo-EM [54] |
| Volta Phase Plate (VPP) | An EM component that enhances image contrast, particularly beneficial for very small particles. | Cryo-EM [54] |
| Synchrotron Beamtime | Access to a high-intensity X-ray source for diffraction data collection. | X-ray Crystallography [13] [66] |
| 300kV Cryo-Electron Microscope | High-end microscope capable of generating high-resolution images of vitrified samples. | Cryo-EM [25] |
Case Studies and Experimental Data
Case Study 1: Cryo-EM of a Small Protein Target (kRasG12C)
Background: The protein kRas is a critically important oncogenic target in cancer research, but its small size (~19 kDa) places it well below the traditional practical size limit for single-particle cryo-EM analysis [54].
Experimental Protocol: To overcome the size limitation, researchers employed a fusion strategy. The kRasG12C protein was fused to a coiled-coil motif (APH2) using a continuous alpha-helical linker. This fusion was designed to form a stable dimer, effectively doubling the molecular weight and providing a larger, more rigid complex. Furthermore, the APH2 motif was targeted by high-affinity nanobodies, adding further size and rigidity to the complex for improved particle alignment during image processing [54].
Results and Data: This engineered approach enabled the determination of a 3.7 Å resolution cryo-EM structure of kRasG12C. The map was of sufficient quality to clearly visualize the bound inhibitor drug MRTX849 and a GDP molecule within the active site. This case study demonstrates a practical and generalizable methodology for applying cryo-EM to small protein targets, which constitute a large proportion of the proteome and include many drug targets [54].
Case Study 2: High-Throughput Cryo-EM for Drug Discovery on TRPML1
Background: TRPML1 is a lysosomal ion channel, an integral membrane protein that is typically challenging to study. Access to multiple high-resolution structures of protein-ligand complexes is a prerequisite for efficient structure-based drug design [43].
Experimental Protocol: Researchers applied a high-throughput cryo-EM approach to determine the structures of TRPML1 in complex with ten different chemically diverse modulators, including both agonists and antagonists. This involved purifying the protein-ligand complexes, preparing cryo-grids for each, and systematically collecting and processing high-resolution datasets [43].
Results and Data: The study successfully yielded a series of high-resolution structures that provided profound insights into the structure-function relationships of the ligands. The structural data revealed the mechanistic basis for channel pore opening induced by agonists and closing induced by antagonists. The depth of structural information generated established a foundation for conducting iterative cycles of structure-based design to develop improved modulators of TRPML1, showcasing cryo-EM's transformative potential in drug discovery for targets refractory to X-ray crystallography [43].
For researchers in structural biology and drug development, selecting the right high-end instrumentation is a critical strategic decision. This guide provides a detailed, objective comparison between two pivotal technologies: synchrotrons for X-ray crystallography and high-end cryo-electron microscopes (cryo-EM) for single-particle analysis, focusing on their instrumentation, access models, and application in modern research.
Technical Specifications and Performance Metrics
The core instrumentation for these techniques differs significantly in its operation, capabilities, and physical requirements.
Table 1: Core Instrumentation Comparison
| Feature | Synchrotron (for X-ray Crystallography) | High-End Cryo-Electron Microscope |
|---|---|---|
| Principle | Particle accelerator generating high-brightness X-rays [13] | High-voltage electron microscope with cryogenic sample stage [69] |
| Key Components | Storage ring, insertion devices, beamlines, detectors [70] | Electron gun (e.g., X-FEG), objective lens, cryo-holder, direct electron detector (e.g., Falcon C) [69] |
| Typical Operating Energy | High-energy X-rays (e.g., 5-20 keV) | 100 kV, 200 kV, or 300 kV electron beams [69] |
| Resolution Range | Routine sub-1.5 Å to atomic (<1.0 Å) [71] | ~2.5 Å to 4.0 Å for typical single-particle analysis [71] |
| Sample Environment | Cryo-cooled crystals (typically ~100 K) [13] | Vitreous ice at cryogenic temperatures [72] |
| Data Output | Diffraction pattern (spot intensities) [13] | 2D micrographs of individual particles [37] |
Recent advancements have made 100 kV cryo-TEMs, like the Tundra Cryo-TEM, viable for high-resolution work on symmetrical molecules, featuring hardware improvements such as an extreme brightness field emission gun (X-FEG) and a short focal length objective lens to reduce aberrations [69]. For synchrotrons, modern low-emittance sources provide high-flux microfocus X-ray beams ideal for microcrystals [70].
Access Models and Operational Workflows
Gaining access to these facilities and conducting experiments involves distinct pathways and procedures, which are summarized in the diagram below.
Synchrotron Access
Access to synchrotron beamlines is primarily grant-based. Researchers typically submit proposals for beamtime, which are assessed competitively based on scientific merit [73]. Successful applicants are allocated a time slot on a specific beamline. Data collection is highly automated; users often ship their crystals to the facility, where robotic sample changers mount crystals and collect diffraction data, sometimes with remote user supervision [70] [74]. Facilities like Diamond Light Source offer highly automated beamlines that allow researchers to rapidly collect diffraction data from large numbers of crystals, significantly accelerating drug discovery pipelines [74]. Training courses, such as the RapiData course at SSRL, are available to educate scientists on optimal data collection and processing techniques [73].
Cryo-EM Access
Access to high-end cryo-EM is often managed through centralized national facilities or institutional core facilities. The process usually involves booking instrument time through the facility's scheduling system [72]. Users either send purified protein samples to the facility for grid preparation and data collection, or they may prepare grids themselves and operate the microscope during their scheduled session, often with staff assistance. The data collection process itself can be highly automated, with systems screening grids and collecting thousands of micrographs unattended [72]. This "democratization" of cryo-EM has increased access, though the demand for time on these instruments remains very high.
Experimental Protocols and Methodologies
The journey from a purified protein to a 3D structure differs fundamentally between the two techniques.
Synchrotron X-ray Crystallography Workflow
The core workflow for macromolecular crystallography at a synchrotron involves several defined stages, from crystal preparation to final model refinement.
- Crystallization: The protein is concentrated and subjected to a wide range of conditions to induce the formation of well-ordered crystals. This is often the major bottleneck and can take weeks to months [13] [71].
- Crystal Preparation & Data Collection: A single crystal is harvested, cryo-cooled, and mounted in the X-ray beam. The crystal is rotated to collect a complete diffraction dataset, a process that can take minutes to hours [13] [70].
- Data Processing & Structure Solution: The diffraction patterns are processed to determine the intensities and phases of the reflections (the "phase problem"). AI-powered models from AlphaFold have dramatically simplified phasing via molecular replacement [75]. An atomic model is then built and refined against the experimental data [13].
Cryo-EM Single-Particle Analysis Workflow
The single-particle analysis workflow for cryo-EM focuses on preparing and imaging individual protein particles suspended in a thin layer of ice.
- Grid Preparation (Vitrification): A small volume of purified sample is applied to an EM grid, blotted to form a thin film, and rapidly plunged into a cryogen. This "vitrifies" the water, preserving the protein particles in a near-native state within a thin layer of amorphous ice [72] [71].
- Data Collection: The vitrified grid is loaded into the microscope. Under low-dose conditions to minimize beam damage, thousands of micrographs are collected automatically. Each micrograph contains 2D projections of thousands of individual protein particles in random orientations [72] [71].
- Image Processing & 3D Reconstruction: This is a computationally intensive process. It involves correcting for beam-induced motion, estimating the microscope's contrast transfer function, and picking individual particles. These particles are then classified and used to reconstruct a 3D density map, into which an atomic model can be built [71].
Essential Research Reagents and Materials
Successful experiments rely on specialized consumables and reagents unique to each technique.
Table 2: Key Research Reagents and Consumables
| Item | Technique | Function |
|---|---|---|
| Crystallization Plates/Screens | X-ray Crystallography | Pre-formulated chemical matrices to identify initial crystal growth conditions [13]. |
| Cryoprotectants | X-ray Crystallography | Chemicals (e.g., glycerol, ethylene glycol) added to prevent ice formation during crystal cryo-cooling [13]. |
| EM Grids | Cryo-EM | Small metal meshes (e.g., gold or copper) that support the vitreous ice sample film [72]. |
| Detergents / Amphipols / Nanodiscs | Cryo-EM | Membrane mimetics used to solubilize and stabilize membrane proteins in a native-like lipid environment [72]. |
| Phase Plates | Cryo-EM | Devices that introduce a phase shift to the electron beam to enhance image contrast, though their use is not yet universal [37]. |
The choice between synchrotron-based X-ray crystallography and high-end cryo-EM is not a matter of one being superior to the other, but rather which is the most appropriate tool for a specific research question.
- Choose Synchrotron X-ray Crystallography when your target can be crystallized and your goal requires the highest possible atomic resolution for precise ligand binding or mechanistic studies, particularly for soluble proteins and smaller complexes [13] [75] [71].
- Choose High-End Cryo-EM when studying large, flexible complexes, membrane proteins in a lipid environment, or heterogeneous samples that are recalcitrant to crystallization. It is the preferred method for capturing multiple conformational states [72] [71].
In modern drug discovery, these techniques are increasingly used in a complementary fashion, with each providing unique and valuable structural insights that accelerate the development of new therapeutics [75] [71].
Single-particle cryo-electron microscopy (cryo-EM) has revolutionized structural biology by enabling the visualization of biological macromolecules at near-atomic resolution. However, a significant challenge remains: the technique has traditionally struggled with proteins smaller than 100 kDa, particularly those in the 30-40 kDa range that represent the median size of eukaryotic and prokaryotic proteins [76]. This size limitation exists because smaller molecules provide weaker signal-to-noise ratios and present difficulties in particle alignment during image processing [76] [77]. To overcome these challenges, researchers have developed innovative strategies employing scaffolds and fusion proteins that effectively increase the size and symmetry of target complexes, enabling high-resolution structure determination of previously intractable targets. These advanced approaches are transforming structural biology and expanding the horizons of cryo-EM applications in drug discovery and basic research.
Table 1: Key Challenges for Small Protein Cryo-EM and Scaffolding Solutions
| Challenge | Impact on Small Protein Cryo-EM | Scaffolding Solution |
|---|---|---|
| Molecular Size | Weaker signal, difficult particle picking | Increases complex size to >100 kDa [76] |
| Preferred Orientation | Incomplete 3D reconstruction | Symmetric scaffolds (octahedral, etc.) mitigate orientation bias [76] |
| Structural Flexibility | Blurred reconstructions | Rigid attachments stabilize conformation [77] |
| Particle Alignment | Inaccurate alignment during processing | Large, distinct fiducial markers enable precise alignment [77] |
Scaffold-Based Strategies for Cryo-EM Structure Determination
RNA Scaffolding for Nucleic Acid Structures
The structural determination of protein-free RNAs has historically presented considerable difficulties, with most attempts yielding only low to moderate resolution. These challenges are compounded for small RNAs, as cryo-EM is inherently more difficult for lower molecular weight macromolecules [78]. A breakthrough scaffold-enabled approach has been developed using group II introns as fusion partners for target RNAs.
This technology involves fusing small RNAs to a group II intron scaffold, which yields high-resolution structures of the appended RNA. Researchers demonstrated this strategy by determining the structures of an 86-nucleotide thiamine pyrophosphate (TPP) riboswitch aptamer domain and a 210-nucleotide raiA bacterial non-coding RNA. The scaffolding approach allowed visualization of the riboswitch ligand binding pocket at 2.5 Å resolution, enabling precise modeling of the thiamine pyrophosphate ligand [78]. The ideal RNA scaffold exhibits specific properties: (1) minimal grid orientation preference, (2) high solubility, (3) structure resistant to denaturation, (4) molecular mass >100 kDa, and (5) a bridging region that can accommodate target RNAs [78].
Comparison with earlier scaffold attempts reveals significant improvements. Previous approaches using the Tetrahymena group I intron, RNA origami, or the ribosome as scaffolds typically achieved only ~5 Å resolution for target RNAs, which does not allow discrimination of individual nucleotides and makes precise modeling impossible [78]. The new group II intron scaffold represents a substantial advancement, enabling de novo modeling of entire RNA structures.
Protein Scaffolds for Small Protein Targets
For protein targets, a highly effective scaffold strategy has been developed that securely anchors proteins of interest to a robust, symmetric base via a selective adapter. The most efficacious constructs feature a designed ankyrin-repeat protein (DARPin) rigidly linked to an octahedral human apoferritin via a helical linker [76]. This design creates a large, highly symmetric scaffold of approximately 1 MDa, which significantly mitigates the prevalent challenge of preferred particle orientation in cryo-EM and reduces demands on image collection and data processing.
By utilizing these DARPin-apoferritin scaffolds, researchers have achieved near-atomic-resolution cryo-EM structures of green fluorescent protein (GFP) and maltose-binding protein (MBP), reaching resolutions of 3.47 Å for GFP and 4.0 Å for MBP [76]. The modular design allows adaptation of new DARPins through minor amino-acid-sequence modifications, enabling the binding and visualization of a diverse array of proteins. The key advantage of this system lies in its combination of large size, high symmetry, and modularity, presenting a versatile solution that breaks through the size constraints traditionally limiting single-particle cryo-EM.
Fusion Protein Strategies for Membrane Proteins and GPCRs
Fusion Protein Design Principles
G protein-coupled receptors (GPCRs) and other small integral membrane proteins present particular challenges for structural studies due to their relatively small sizes and structural dynamics. Applying cryo-EM to GPCRs without signaling proteins remains challenging because most receptors lack structural features in their soluble domains to facilitate image alignment [77]. Fusion protein strategies have emerged as a powerful solution to this problem.
The general approach involves inserting a fusion protein between transmembrane helices 5 and 6 of GPCRs, replacing the third intracellular loop (ICL3) [77]. This strategy has been highly successful in GPCR crystallography, where the inserted fusion protein can mediate crystal contacts. For cryo-EM applications, the design must ensure rigid attachment of the fusion protein to serve as an effective fiducial marker for particle alignment. Research indicates that a Fab fragment tightly bound to a rigidly attached BRIL (apocytochrome b562 RIL) domain is necessary to serve as a sufficient fiducial marker for single-particle image alignment of GPCRs [77].
Figure 1: Fusion Protein Strategy Workflow for GPCR Cryo-EM
Experimental Applications and Outcomes
Through systematic exploration of fusion protein designs, researchers have determined structures of antagonist-bound A2A adenosine receptor at 3.4 Å resolution and unliganded Smoothened at 3.7 Å resolution [77]. These successes provide important design guidelines for cryo-EM interrogation of other GPCRs and small integral membrane proteins.
A critical finding from these studies is that a BRIL domain alone (MW ~10 kDa), even when rigidly attached to a GPCR, does not provide sufficient features to enable particle alignment. Only when an anti-BRIL Fab fragment was added to enlarge the fiducial marker was successful particle alignment achieved, yielding a reconstruction with global resolution of ~3.4 Å [77]. Further focused refinement improved the resolution of the transmembrane domain to 3.2 Å, sufficient for detailed model building and visualization of drug binding.
The research also revealed that attachment geometry significantly impacts outcomes. A single-helix BRIL connection proved insufficient for high-resolution reconstruction, indicating that rigid attachment with two extended helices (connecting TM5 and TM6 of the receptor to the N- and C-terminus of BRIL, respectively) is necessary for successful particle alignment [77]. These findings establish clear design principles for fusion strategies in cryo-EM studies of membrane proteins.
Table 2: Comparison of Scaffold and Fusion Protein Strategies
| Characteristic | RNA Scaffold (Group II Intron) | Protein Scaffold (DARPin-Apoferritin) | GPCR Fusion (BRIL-Fab) |
|---|---|---|---|
| Target Class | Protein-free RNAs [78] | Small soluble proteins (e.g., GFP, MBP) [76] | Membrane proteins/GPCRs [77] |
| Scaffold Size | 400-850 nucleotides [78] | ~1 MDa [76] | ~10 kDa BRIL + Fab [77] |
| Symmetry | Asymmetric | Octahedral [76] | Asymmetric |
| Achieved Resolution | 2.5 Å (core) [78] | 3.47 Å (GFP) [76] | 3.2-3.4 Å (TM domain) [77] |
| Key Innovation | Bridging region for RNA attachment | Modular DARPin adapter [76] | Rigid double-helix attachment [77] |
| Typical Sample Amount | Not specified | 0.1-0.2 mg [79] | Not specified |
Experimental Protocols and Methodologies
Protocol: DARPin-Apoferritin Scaffold Preparation
The DARPin-apoferritin scaffold system involves a well-defined preparation protocol. Constructs are engineered by fusing the DARPin to the N-terminus of apoferritin and incorporating an α-helical element at both the N-terminus of the DARPin and the C-terminus of apoferritin, thereby enhancing scaffold stability [76]. Most constructs are cloned into the pET-28a vector for expression.
The experimental workflow proceeds as follows:
- The DARPin-apoferritin scaffold and target proteins (e.g., GFP, MBP) are individually expressed heterologously in SHuffle T7 Escherichia coli strain
- Expression is induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside at 16°C during overnight incubation
- Following cell lysis using a French press, the supernatant is purified using a HisTrap HP column
- Proteins are eluted with an imidazole gradient and buffer-exchanged into binding buffer (20 mM Tris-HCl, 50 mM NaCl pH 8.0)
- The scaffold and target proteins are mixed in a 1:3 molecular ratio and incubated at 4°C overnight
- The complex undergoes size-exclusion chromatography using a Superose 6 Increase 10/300 GL column
- Complex peak fractions are collected, concentrated to 1 mg ml−1, and characterized by SDS–PAGE, static light scattering, and negative-stain EM [76]
Protocol: GPCR Fusion with BRIL and Fab
For GPCR fusion strategies, the experimental approach involves:
- Engineering a BRIL fusion into the ICL3 region of the GPCR between transmembrane helices 5 and 6
- Ensuring rigid attachment through two continuous helices connecting TM5 and TM6 of the receptor to the N- and C-terminus of BRIL, respectively
- Purifying the GPCR-BRIL fusion construct in L-MNG/CHS detergent with appropriate ligand
- Adding an anti-BRIL Fab fragment to enlarge the fiducial marker
- Utilizing the Fab-bound complex for grid preparation and data collection [77]
The critical quality control step involves verifying rigid attachment through structural analysis, as a single-helix BRIL connection has been shown to be insufficient for high-resolution reconstruction [77].
Comparative Analysis with X-ray Crystallography
Technical and Operational Considerations
The choice between cryo-EM and X-ray crystallography for structural determination depends on multiple factors, including target characteristics, project requirements, and available resources. Each technique offers distinct advantages and limitations that must be considered in experimental design.
Figure 2: Comparative Workflows: Cryo-EM vs. X-ray Crystallography
Table 3: Method Selection Guide: Cryo-EM vs. X-ray Crystallography
| Factor | Cryo-EM | X-ray Crystallography |
|---|---|---|
| Optimal Molecular Size | >100 kDa (scaffolds enable smaller targets) [76] [79] | <100 kDa [79] |
| Sample Amount | 0.1-0.2 mg [79] | >2 mg typically [79] |
| Resolution Range | Typically 2.5-4.0 Å [79] | Up to 1.0 Å possible [79] |
| Membrane Protein Studies | Excellent in lipid environments [80] | Requires detergent optimization [79] |
| Structural Dynamics | Captures multiple conformations [79] | Crystal packing constraints [78] |
| Typical Timeline | Weeks typically [79] | Weeks to months [79] |
| Data Collection | Hours to days [79] | Minutes to hours [79] |
| Sample Preparation | Vitrification optimization [79] | Crystal growth & optimization [79] |
Application-Based Method Selection
The comparative advantages of each technique make them suitable for different research applications. Cryo-EM excels in analyzing large molecular complexes, studying proteins in their native states, capturing multiple conformational states, and examining challenging targets like membrane proteins [79]. The scaffold and fusion protein approaches detailed in this review significantly expand these advantages to include smaller proteins that were previously inaccessible to cryo-EM analysis.
X-ray crystallography remains superior for obtaining atomic resolution structures, high precision for small molecules, and established data processing pipelines [79]. However, the constraining crystal lattice can suppress dynamic movements and favor compact states, as demonstrated by comparative studies of group II introns where cryo-EM revealed a 90° swinging action of the branch-site helix that was not observed in crystal structures [78].
For drug discovery applications, cryo-EM enables visualization of drug binding sites and analysis of conformational changes in large drug targets, while X-ray crystallography provides ultra-high resolution ligand binding information and established fragment screening capabilities [79] [81]. The integration of scaffold strategies with cryo-EM is particularly valuable for structure-based drug design on targets refractory to crystallization, such as integral membrane proteins [43].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of scaffold and fusion protein strategies for cryo-EM requires specific reagents and materials. The following table details key research reagent solutions essential for these advanced structural biology approaches.
Table 4: Essential Research Reagents for Scaffold-Based Cryo-EM
| Reagent/Material | Function/Application | Examples/Specifications |
|---|---|---|
| Group II Intron Scaffold | RNA scaffolding platform | High solubility, >100 kDa, minimal orientation preference [78] |
| DARPin-Apoferritin Scaffold | Protein scaffolding platform | ~1 MDa, octahedral symmetry, modular DARPin adapter [76] |
| BRIL Fusion Protein | GPCR fusion partner | Apocytochrome b562 RIL, enables rigid attachment [77] |
| Anti-BRIL Fab | Fiducial marker for alignment | Binds BRIL domain, enlarges particle size [77] |
| HisTrap HP Column | Protein purification | Cytiva, purification of His-tagged constructs [76] |
| Superose 6 Increase Column | Size-exclusion chromatography | Cytiva, complex separation and analysis [76] |
| L-MNG/CHS Detergent | Membrane protein purification | Maintains stability of GPCRs during purification [77] |
| Direct Electron Detector | Cryo-EM data collection | Improved signal-to-noise, rapid frame rates [10] |
Scaffold and fusion protein strategies represent transformative advancements in cryo-EM methodology, effectively breaking through the traditional size limitations that have constrained structural studies of small proteins, RNAs, and membrane proteins. The development of RNA scaffolds using group II introns, protein scaffolds employing DARPin-apoferritin systems, and fusion approaches for GPCRs has enabled high-resolution structure determination of targets previously considered intractable to cryo-EM analysis. These approaches leverage several key principles: increasing overall complex size to improve particle alignment, incorporating symmetry to mitigate orientation bias, and ensuring rigid attachments to stabilize complexes for high-resolution reconstruction. As these methodologies continue to evolve and become more widely adopted, they promise to expand the frontiers of structural biology, enabling researchers to visualize increasingly challenging biological targets and accelerating drug discovery efforts against previously inaccessible protein classes.
Head-to-Head Comparison: Resolution, Cost, Throughput, and Data Validation
For researchers, scientists, and drug development professionals, selecting the appropriate structural biology technique is crucial for obtaining meaningful and reliable structural information. The ongoing comparison between X-ray crystallography and cryo-electron microscopy (cryo-EM) represents a central theme in modern structural biology, with each method offering distinct advantages and limitations in terms of resolution, accuracy, and applicability to real-world research scenarios [82]. The choice between these techniques significantly impacts the interpretation of biological mechanisms and the rational design of therapeutic compounds.
This guide provides an objective comparison of the performance of X-ray crystallography and cryo-EM, focusing on their achievable limits of resolution and accuracy across various experimental scenarios. We present supporting experimental data and detailed methodologies to enable informed decision-making for your specific research requirements in structural biology and drug development.
Quantitative Comparison of Resolution and Accuracy
The following tables summarize the key performance metrics and technical considerations for X-ray crystallography and cryo-EM, based on current experimental data and statistical measures.
Table 1: Achievable Resolution Limits and Statistical Measures
| Parameter | X-ray Crystallography | Single-Particle Cryo-EM |
|---|---|---|
| Current Absolute Resolution Record | 0.48 Å [8] | 1.54 Å (single-particle) [8] |
| Typical High Resolution | 1.5-2.5 Å [13] | 2.5-3.5 Å [83] |
| Resolution Determination Method | Bragg's law; Signal-to-noise ([8]<="" r-factors="" rmeas,="" rp.i.m.)="" td="" σ(i)>),=""> | Fourier Shell Correlation (FSC=0.143 "gold standard") [8] |
| Effective Resolution Concept | Accounts for anisotropy and data incompleteness [8] | Varying resolution across different regions of the map [8] |
| Atomic Resolution Definition | ~1.2 Å or better ("Sheldrick's criterion") [8] | Not strictly defined, but near-atomic considered ~2 Å or better [8] |
Table 2: Accuracy, Model Quality, and Technical Considerations
| Parameter | X-ray Crystallography | Single-Particle Cryo-EM |
|---|---|---|
| Primary Output | Electron density map [13] | Coulomb potential map [84] |
| Key Accuracy Feature | Distinguishes individual atoms at high resolution [8] | Potential to identify specific ions and protonation states [84] |
| Model Quality Assessment | R-factors (Rwork, Rfree), geometry validation [13] | Map-model correlation, geometry validation [85] |
| Radiation Damage Concerns | Crystal decay during X-ray exposure [82] | Beam-induced sample movement & damage [84] [8] |
| Conformational Heterogeneity | Multiconformer modeling possible at high resolution [85] | Direct visualization of multiple states from heterogeneous samples [83] |
Experimental Protocols for Structure Determination
X-ray Crystallography Workflow
The process of structure determination via X-ray crystallography follows a well-established pipeline with distinct critical stages:
Sample Preparation and Crystallization: The target protein is purified to homogeneity, and crystallization screens are performed to identify conditions that yield well-ordered, diffraction-quality crystals. This remains the major bottleneck [83] [13]. For membrane proteins, lipidic cubic phase (LCP) crystallization has been particularly successful [13].
Data Collection: Crystals are exposed to high-energy X-rays, typically at a synchrotron beamline. The crystal diffracts the X-rays, producing a pattern of discrete spots (reflections) on a detector [13] [8].
Data Processing: The diffraction images are processed to index the spots, measure their intensities, and determine the crystal's symmetry. This yields a set of structure factor amplitudes [13] [8].
Phasing: The phase information, which is lost during data collection (the "phase problem"), is determined using methods like molecular replacement (using a similar known structure), or experimental techniques such as SAD/MAD [13] [8].
Model Building and Refinement: An atomic model is built into the experimental electron density map and iteratively refined to improve its agreement with the diffraction data and ideal geometry [13].
The following diagram illustrates this workflow:
X-ray Crystallography Workflow
Single-Particle Cryo-EM Workflow
The single-particle cryo-EM methodology involves a different set of specialized steps to determine structures from individual protein particles:
Vitrification: The purified protein sample is applied to an EM grid and rapidly plunged into a cryogen (like liquid ethane) to create a thin layer of vitreous ice, preserving the native structure of the particles in a frozen-hydrated state [83].
Data Acquisition: The vitrified grid is loaded into a transmission electron microscope operating at 200 or 300 kV. Thousands to millions of low-dose images (micrographs) are automatically collected using a direct electron detector [83] [10].
Particle Picking: Individual protein particles are identified and extracted from the micrographs [83].
2D Classification and 3D Reconstruction: The extracted particle images are classified into 2D averages to remove poor-quality particles. Subsequently, an initial 3D model is generated and refined through iterative cycles of 3D classification and refinement to produce a final 3D Coulomb potential map [83].
Model Building and Refinement: An atomic model is built into the cryo-EM map, often using computational tools and/or integrating AI-based predictions, and refined against the map [10] [85].
The following diagram illustrates this workflow:
Single-Particle Cryo-EM Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Successful structure determination relies on specialized reagents and instrumentation. The following table outlines essential materials and their functions for both techniques.
Table 3: Essential Research Reagents and Instrumentation
| Item | Function | Primary Application |
|---|---|---|
| Direct Electron Detectors | High-sensitivity cameras that enable motion correction and dramatically improve signal-to-noise ratio [10]. | Cryo-EM |
| Synchrotron Beamline | Source of high-intensity, tunable X-rays necessary for collecting high-resolution diffraction data [13]. | X-ray Crystallography |
| Lipidic Cubic Phase (LCP) | A membrane-mimetic matrix used for crystallizing membrane proteins like GPCRs [13]. | X-ray Crystallography |
| Plunge Freezer | Instrument for rapid vitrification of samples in ethane/propane to form vitreous ice [83]. | Cryo-EM |
| Selenomethionine | Used for creating heavy-atom derivatives for experimental phasing via SAD/MAD [82]. | X-ray Crystallography |
| qFit Software | Automated computational tool for building multiconformer models into high-resolution density maps [85]. | X-ray Crystallography & Cryo-EM |
Real-World Application Scenarios
Application in Drug Discovery
Both techniques provide critical structural information for drug development, but their applications often differ based on the target characteristics:
X-ray Crystallography has been a cornerstone for structure-based drug design, providing atomic-resolution information on protein-ligand complexes. It is routinely used for fragment screening, where small molecules are soaked into crystals to identify binding events [13]. The high-resolution data allows medicinal chemists to precisely visualize interactions between a drug candidate and its target, enabling rational design of more potent and specific compounds [83]. For example, the structure of the SARS-CoV-2 main protease (Mpro) with inhibitors was solved by crystallography, directly guiding antiviral drug development [10].
Cryo-EM is particularly impactful for studying large drug targets that are difficult to crystallize, such as G protein-coupled receptors (GPCRs) in complex with their signaling partners, or large viral machinery [83] [10]. It can investigate a broad spectrum of drug-target interactions and dynamic conformational states that might be trapped in a crystal lattice [83]. This technique is increasingly used to visualize the binding of small molecules to large complexes, expanding the druggable proteome.
Studying Dynamic Processes and Flexibility
Understanding biomolecular function often requires insight into dynamics and conformational flexibility:
Cryo-EM excels at probing systems with inherent conformational heterogeneity. Through advanced image analysis and classification, it can resolve multiple conformational or compositional states from a single sample preparation [83] [82]. This ability to deconvolute structural heterogeneity from a mixture of particles is a distinct advantage for studying flexible assemblies.
X-ray Crystallography can access dynamic information through time-resolved studies. Using rapid mixing and thermal quenching, this approach can capture structural states with sub-10 millisecond time resolution, enabling the visualization of intermediate states in enzymatic reactions [86]. Furthermore, at high resolution, conformational flexibility can be investigated by building multiconformer models using tools like qFit, which reveals alternative side-chain and backbone conformations within the crystal [85].
X-ray crystallography and cryo-EM are powerful, complementary techniques in the structural biologist's arsenal. The choice between them depends heavily on the specific biological question and the nature of the target macromolecule.
X-ray crystallography remains the technique of choice for obtaining the most precise atomic coordinates of proteins, particularly those under a few hundred kDa, and is exceptionally well-suited for high-throughput ligand screening. Its main challenge remains the crystallization bottleneck.
Cryo-EM has revolutionized the study of large, flexible macromolecular complexes and membrane proteins that have resisted crystallization efforts. Its strength lies in visualizing structural heterogeneity and solving structures without the need for crystals.
For researchers in drug development, the decision framework should consider the target size, flexibility, and the required resolution for informed decision-making. As both technologies continue to advance—with crystallography pushing toward faster time resolution and cryo-EM toward higher resolution and automation—their synergistic application will continue to drive discoveries in basic biology and therapeutic development.
Throughput and Cost-Benefit Analysis for Drug Discovery Pipelines
The selection of structural biology techniques is a pivotal strategic decision in drug discovery, directly impacting a project's cost, timeline, and probability of success. For researchers and drug development professionals, the choice between established workhorses like X-ray crystallography and the transformative capabilities of cryo-electron microscopy (cryo-EM) involves careful consideration of throughput, cost, and technical applicability. This guide provides an objective, data-driven comparison of these two methods, framing them within the context of modern drug discovery pipelines. We evaluate performance metrics, detail experimental protocols, and analyze cost-benefit ratios to inform strategic decision-making in R&D.
Market Context and Adoption Trends
The structural biology landscape is dynamically evolving, with both X-ray crystallography and cryo-EM occupying significant and sometimes overlapping niches. Understanding their market presence and growth trajectories provides context for their current capabilities and future potential.
X-ray crystallography maintains a strong position, with its global market valued at approximately US$1.8 billion in 2025 and projected to grow at a Compound Annual Growth Rate (CAGR) of 7.6% to reach US$3.0 billion by 2032 [87] [88]. Its dominance is particularly evident in the pharmaceutical sector, which accounts for over 45% of its application share [87]. This sustained growth is fueled by the technique's entrenched role in structure-based drug design, continuous technological advancements in automation and AI integration, and its established infrastructure in both industry and academia [87] [88].
In contrast, the market for cryo-EM structure analysis services, while smaller, is expanding at a more accelerated pace. It is estimated at USD 1.30 billion in 2025 and is expected to grow at a remarkable CAGR of 9.8% to USD 2.51 billion by 2032 [20]. This rapid growth follows the "resolution revolution" that began around 2013, triggered by the introduction of direct electron detectors [9] [89]. Surveys indicate that about 65% of pharmaceutical companies now incorporate cryo-EM data in their drug discovery pipelines, a dramatic increase from just 20% five years ago [89]. The demand is so high that commercial service providers report waiting periods of 3-6 months for project slots [89].
Table 1: Global Market Overview and Key Growth Drivers
| Feature | X-ray Crystallography | Cryo-Electron Microscopy (cryo-EM) |
|---|---|---|
| Market Size (2025) | ~US$ 1.8 Billion [87] [88] | ~USD 1.30 Billion [20] |
| Projected Market (2032) | ~US$ 3.0 Billion [87] [88] | ~USD 2.51 Billion [20] |
| Projected CAGR | 7.6% [87] [88] | 9.8% [20] |
| Key Growth Drivers | - Automation & AI Integration- Demand in Pharmaceutical Drug Design- Established Infrastructure [87] [88] | - "Resolution Revolution"- Demand for Membrane Protein Structures- Pharmaceutical R&D Investments [9] [89] [20] |
| Pharmaceutical Sector Adoption | >45% application share [87] | ~65% of companies use it (up from 20% in 5 years) [89] |
Technical Comparison and Performance Metrics
The fundamental differences between X-ray crystallography and cryo-EM lead to distinct performance profiles, making each technique suitable for different types of biological problems in the drug discovery pipeline.
Methodological Workflows
The journey from a purified protein to a resolved structure differs significantly between the two techniques. The following diagrams illustrate the core workflows for X-ray crystallography and cryo-EM single-particle analysis (SPA), highlighting key bottlenecks and decision points.
Quantitative Performance Comparison
A direct comparison of key performance metrics reveals the complementary strengths and weaknesses of each technique, which are crucial for project planning.
X-ray crystallography excels in producing very high-resolution structures, often at resolutions better than 1.5 Å, allowing for the precise visualization of atoms, water molecules, and inhibitors in a binding pocket [10]. Its sample throughput can be high once a robust crystallization system is established, especially with modern automated systems. However, its most significant limitation is the crystallization bottleneck itself. Many biologically critical targets, such as membrane proteins and large flexible complexes, are notoriously difficult or impossible to crystallize. The method also provides limited native information on conformational dynamics, as the protein is locked in a crystalline state.
Cryo-EM has revolutionized the study of targets that resisted crystallization. It can handle a vast range of molecular weights, from large complexes (>200 kDa) down to smaller proteins (~60 kDa) with specialized techniques [9] [10]. A key advantage is its ability to resolve multiple conformational states from a single sample, providing dynamic insights into molecular mechanisms [10]. The primary technical challenge lies in sample preparation, where achieving a thin, homogeneous layer of vitreous ice without preferred particle orientation requires significant expertise and optimization. Furthermore, while resolutions of 2-3 Å are now routine for well-behaved samples, the technique can struggle with inherent structural flexibility, leading to blurred regions in maps.
Table 2: Technical Performance and Application Scope
| Parameter | X-ray Crystallography | Cryo-Electron Microscopy (cryo-EM) |
|---|---|---|
| Typical Resolution Range | ≤ 2.0 Å (Often ≤ 1.5 Å) [10] | 2.0 - 4.0 Å (Routine near-atomic) [9] [10] |
| Optimal Size Range | Versatile, but limited by crystal formation | ≥ 60 kDa (ideally > 200 kDa), techniques improving for smaller proteins [9] [10] |
| Sample Throughput | High (after crystallization is achieved) | Moderate to Low (Data collection & processing is time-consuming) |
| Key Technical Bottleneck | Protein Crystallization [10] | Sample Preparation (Ice Quality, Particle Distribution) [89] |
| Handling Flexibility/ Dynamics | Limited (static snapshot in crystal lattice) | Excellent (can resolve multiple conformations) [10] |
| Ideal Application Scope | - Soluble proteins amenable to crystallization- Atomic-level drug binding details- High-throughput screening of analogs | - Membrane proteins (GPCRs, ion channels) [89] [10]- Large, flexible complexes (ribosomes, filaments) [10]- Structurally heterogeneous samples |
Throughput and Economic Analysis
The economic viability of a technique is a critical factor in resource-constrained R&D environments. The cost structures for X-ray crystallography and cryo-EM differ significantly in both capital investment and operational expenses.
Cost-Benefit and Infrastructure Considerations
X-ray crystallography benefits from a mature market and relatively lower entry costs. A high-end diffractometer system represents a substantial investment but is typically below the cost of a high-end cryo-EM microscope. Furthermore, the widespread availability of benchtop crystallography instruments has democratized access for many labs. The well-established and often automated workflows contribute to lower operational costs and higher sample throughput for suitable targets, making it a cost-effective workhorse for many applications [87] [88].
Establishing a cryo-EM facility, however, requires a massive capital outlay. A single high-end cryo-EM instrument (e.g., a 300 kV microscope) typically costs between $5-7 million, with supporting equipment, facility modifications for vibration and magnetic isolation, and computational infrastructure potentially doubling the total investment [89]. Annual maintenance contracts alone can exceed $300,000 per instrument, not including salaries for highly specialized staff [89]. To distribute these high costs, collaborative models such as multi-institutional partnerships, regional shared facilities, and fee-for-service cores have become common [89].
Service Market and Operational Throughput
The vibrant service market for both techniques provides an alternative to in-house infrastructure, offering valuable insights into their practical cost and throughput.
Cryo-EM service costs for a full structure determination typically range from $50,000 to $200,000 per project, depending on the target's complexity and the desired resolution [89]. The reported 3-6 month waiting periods at commercial providers indicate very high demand and limited throughput relative to that demand [89]. A major operational challenge is the massive data generation, often exceeding 10 terabytes per week, which necessitates substantial investment in high-performance computing for data processing, storage, and analysis [89].
While specific service pricing for X-ray crystallography is less highlighted in the search results, its longer history has fostered a mature and competitive service market. The integration of automation and AI in crystallography workflows is focused on enhancing efficiency, accuracy, and throughput, particularly for drug screening applications [87] [88].
Table 3: Economic and Operational Comparison
| Factor | X-ray Crystallography | Cryo-Electron Microscopy (cryo-EM) |
|---|---|---|
| Instrument Cost (Capital) | High, but generally lower than cryo-EM [87] | $5 - 7 million per high-end instrument [89] |
| Additional Infrastructure Cost | Moderate (X-ray room, robotics) | Very High (Vibration isolation, EM shielding, computing) [89] |
| Typical Service Cost per Project | Mature, competitive market (Specific price not listed) | $50,000 - $200,000 [89] |
| Operational Cost & Maintenance | High (X-ray sources, consumables) | Very High (>$300,000/year maintenance + specialized staff) [89] |
| Data Management Needs | Moderate | Very High (>10 TB/week, HPC required) [89] |
| Access Model Trends | In-house instruments, core facilities, CROs | Shared regional facilities, core labs, service providers [89] |
Experimental Protocols and Reagent Solutions
Reproducibility is the cornerstone of reliable science. This section outlines generalized protocols for key experiments and lists essential research reagents, providing a practical foundation for researchers to plan and execute their structural biology studies.
Key Experimental Protocols
Protocol 1: High-Throughput Crystallization Screening for Ligand Binding Studies (X-ray Crystallography)
This protocol is designed to efficiently identify initial crystallization conditions for a target protein, a critical first step in structure-based drug design.
- Protein Preparation: Purify the target protein to high homogeneity (>95%) and concentrate it to a suitable level (typically 5-20 mg/mL, target-dependent) in a stable buffer.
- Ligand Soaking or Co-crystallization:
- Soaking: Grow apo-protein crystals. Then, transfer a crystal into a stabilizing solution containing a high concentration of the ligand/drug candidate for a defined period (minutes to days).
- Co-crystallization: Mix the purified protein directly with the ligand prior to setting up crystallization trials and incubate to ensure binding.
- Initial Screening: Use commercial sparse-matrix screens (e.g., from Hampton Research, Molecular Dimensions) in 96-well or 384-well format. Employ an automated liquid handling system to set up sitting-drop vapor diffusion trials by mixing nanoliter volumes of protein-ligand complex with reservoir solution.
- Crystal Optimization: Identify "hits" from the initial screen. Systematically optimize conditions around the hit by varying pH, precipitant concentration, salt type, and temperature to improve crystal size and diffraction quality.
- Data Collection & Analysis: Cryo-protect the optimized crystal and flash-cool it in liquid nitrogen. Collect X-ray diffraction data at a synchrotron or home source. Solve the structure by molecular replacement (using a known homologous structure) and refine the model with the ligand included [10].
Protocol 2: Single-Particle Analysis of a Protein-Ligand Complex (Cryo-EM)
This protocol describes the process of determining the structure of a stabilized protein-ligand complex without the need for crystallization.
- Complex Formation and Validation: Incubate the purified protein with a saturating concentration of the ligand. Validate complex formation and stability using a biophysical method such as Size Exclusion Chromatography (SEC) or Native Mass Spectrometry.
- Grid Preparation: Use a vitrification robot (e.g., Vitrobot, CP3) to apply 3-4 µL of the sample to a freshly plasma-cleaned EM grid. Blot away excess liquid to form a thin film and rapidly plunge-freeze the grid into a cryogen (typically liquid ethane) to create vitreous ice.
- Screening and Data Collection: Initially screen grids on the microscope to assess ice thickness, particle concentration and distribution. For a high-resolution dataset, collect thousands of micrograph movies at a high defocus range (e.g., -0.8 to -2.5 µm) on a 300 kV microscope equipped with a direct electron detector, using a total electron dose of ~40-60 e⁻/Ų [10] [90].
- Data Processing:
- Pre-processing: Perform beam-induced motion correction and estimate the contrast transfer function (CTF) for each micrograph.
- Particle Picking: Use template-based or AI-driven picking (e.g., in CryoSPARC, RELION) to extract millions of particle images.
- 2D Classification: Generate 2D class averages to remove junk particles and assess sample homogeneity.
- 3D Reconstruction: Generate an initial model ab initio or using a known structure as an initial reference, followed by 3D classification to isolate structurally homogeneous subsets. Refine a final 3D map using homogeneous particle sets.
- Model Building and Refinement: If the resolution is sufficient (~3.5 Å or better), build an atomic model into the density map de novo or by docking and flexibly fitting a known structure. Refine the model against the map, validating the fit and the density for the bound ligand [10].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Reagents and Solutions for Structural Biology
| Item | Function/Description | Key Considerations |
|---|---|---|
| Crystallization Screening Kits | Commercial suites of solutions (e.g., from Hampton Research, Molecular Dimensions) that screen a broad range of chemical conditions to induce crystallization [88]. | Sparse-matrix screens are efficient for initial hits. Optimization screens are then used to refine conditions. |
| Cryo-EM Grids | Specimen supports, typically gold or copper grids with a holy carbon film, onto which the sample is applied for vitrification. | Grid quality and surface properties (hydrophilicity) are critical for achieving uniform ice thickness and particle distribution [89]. |
| Direct Electron Detectors | Advanced cameras (e.g., from Gatan, Dectris) that directly detect electrons with high sensitivity and fast readout, enabling motion correction and are fundamental to the "resolution revolution" [10]. | Essential for high-resolution cryo-EM. Specifications like detective quantum efficiency (DQE) and frame rate are key performance metrics. |
| Vitrification Robots | Automated instruments (e.g., Thermo Fisher Vitrobot, Leica GP) that standardize and reproduce the process of plunge-freezing samples [89]. | Critical for minimizing human error and variability in sample preparation, a major bottleneck in cryo-EM. |
| Lipidic Cubic Phase (LCP) Materials | A lipid-based matrix used for crystallizing membrane proteins, which mimics their native lipid environment [10]. | A key breakthrough for obtaining high-resolution structures of GPCRs and other challenging membrane targets. |
Integrated Analysis and Future Outlook
The choice between X-ray crystallography and cryo-EM is not a simple declaration of a superior technology, but a strategic decision based on the specific target, project goals, and available resources.
X-ray crystallography remains the gold standard for obtaining the highest-resolution structures of targets that readily form crystals. It is exceptionally powerful for iterative drug optimization cycles where atomic-level detail of ligand binding is required. Its higher throughput and lower operational costs, once crystallization is mastered, make it a highly efficient and cost-effective tool for many drug discovery programs, particularly for soluble enzyme targets [87] [10].
Cryo-EM has broken fundamental barriers in structural biology, making formerly "undruggable" targets accessible. Its primary strategic value lies in solving structures of large complexes, membrane proteins, and dynamic systems without the need for crystallization. This comes at the cost of lower throughput, significantly higher infrastructure investment, and complex data management [9] [89] [10]. It is the method of choice when the target itself precludes the use of crystallography.
The future lies in integration and synergy. AI-driven structure prediction tools like AlphaFold are already being integrated into both X-ray and cryo-EM workflows, aiding in model building and map interpretation [10]. The combination of computational prediction, cryo-EM for large complexes, and crystallography for high-resolution ligand screening represents a powerful, multi-pronged approach. Furthermore, advancements in automation, AI-integrated data processing, and the development of more affordable and compact cryo-EM systems are poised to continue improving the accessibility, throughput, and cost-effectiveness of both techniques, further solidifying their central role in accelerating drug discovery [9] [87] [89].
The central challenge in structural biology is not only determining the three-dimensional architecture of biological macromolecules but also understanding their dynamic nature. For decades, X-ray crystallography has served as the traditional workhorse, providing exceptionally detailed static snapshots of molecular structures [13]. In contrast, cryo-electron microscopy (cryo-EM) has emerged as a powerful technique that captures macromolecules in a near-native state, preserving their inherent flexibility and conformational diversity [91] [92]. This guide provides an objective comparison of how these two foundational techniques approach the critical task of capturing molecular dynamics, offering researchers a framework for selecting the appropriate method for their structural studies.
Core Principles and Workflows
The fundamental difference in how these techniques capture molecular states stems from their distinct sample preparation and data collection methods.
X-ray Crystallography: The Structured Approach
X-ray crystallography requires biomolecules to be packed into a highly ordered crystal lattice. When X-rays pass through these crystals, they diffract, producing patterns that can be transformed into electron density maps [4] [13]. The workflow involves:
- Crystallization: Inducing the protein to form ordered crystals, often the major bottleneck [13].
- Data Collection: Measuring diffraction patterns, typically at synchrotron facilities [13].
- Phase Solving: Overcoming the "phase problem" to interpret diffraction data [13].
- Model Building and Refinement: Fitting an atomic model into the electron density [91].
This process provides a single, static conformation of the molecule as it exists in the crystal lattice, which may not represent its physiological state.
Cryo-EM: The Native-State Approach
Cryo-EM images biological molecules that have been rapidly frozen in vitreous ice, preserving their solution-state structure [92] [22]. The workflow consists of:
- Vitrification: Plunge-freezing the sample in cryogens to form amorphous ice [22].
- Data Acquisition: Collecting 2D projection images of individual particles in an electron microscope [22].
- Image Processing: Using computational algorithms to align and classify particles [91].
- 3D Reconstruction: Generating a 3D density map from the 2D projections [22].
- Model Building and Refinement: Fitting and refining atomic coordinates into the density map [91].
This technique can capture multiple conformational states present in the sample, providing direct insight into molecular dynamics and flexibility.
Comparative Analysis: Capturing Molecular Dynamics
The following table summarizes the key differences between the two techniques in the context of structural flexibility and dynamics.
Table 1: Technique Comparison for Capturing Dynamics and Flexibility
| Aspect | X-ray Crystallography | Cryo-Electron Microscopy |
|---|---|---|
| Sample State | Fixed in crystal lattice [4] | Near-native, frozen-hydrated state [92] [4] |
| Conformational Information | Typically single, homogeneous conformation [4] | Potential to resolve multiple conformations through classification [91] [43] |
| Dynamic Range | Limited; crystal packing may restrict motions [4] | Broader; can capture flexible regions and large-scale movements [91] |
| Ideal for Studying | Stable conformations, atomic-level detail of static states [4] [13] | Flexible complexes, molecular machines, membrane proteins [91] [43] |
| Key Limitation for Dynamics | May capture non-physiological conformations; difficult for transient states [4] | Lower signal-to-noise for highly flexible regions may limit resolution [91] |
Experimental Data and Case Studies
Cryo-EM Reveals Functional Conformations of TRPML1
A 2025 study on the lysosomal ion channel TRPML1 exemplifies cryo-EM's power for capturing dynamics in drug discovery. Researchers determined high-resolution structures of TRPML1 bound to ten different modulators [43]. The experimental protocol involved:
- Sample Preparation: Purifying TRPML1 and incubating with agonists or antagonists.
- Grid Preparation: Vitrifying samples on cryo-EM grids.
- Data Collection: Imaging particles and processing datasets for each ligand condition.
- 3D Reconstruction: Generating density maps for each TRPML1-ligand complex.
Results: The study revealed that agonists and antagonists induced distinct channel conformations—open and closed pore states, respectively [43]. This provided direct structural evidence of ligand-induced gating mechanisms, demonstrating cryo-EM's ability to visualize functionally relevant, dynamic changes in a membrane protein that is challenging to crystallize.
Advancing Small Protein Analysis with Cryo-EM
Traditional cryo-EM faced limitations with proteins below 50 kDa due to low signal-to-noise. A 2025 study on the oncogenic protein kRasG12C (19 kDa) demonstrated an innovative scaffold-based solution [54]. The methodology involved:
- Fusion Strategy: Fusing kRasG12C to a coiled-coil motif (APH2) recognized by nanobodies.
- Complex Formation: Creating a larger, symmetric assembly suitable for cryo-EM analysis.
- Data Processing: Achieving a 3.7 Å resolution map.
Results: The structure clearly showed kRasG12C bound to both the inhibitor drug MRTX849 and GDP, proving that strategic scaffolding extends cryo-EM's reach to small, medically relevant proteins while preserving ligand-binding details [54].
The Scientist's Toolkit: Essential Research Reagents
Successful structural studies require specialized reagents and materials. The following table outlines key solutions for both techniques, with a focus on supporting the analysis of dynamics.
Table 2: Key Research Reagent Solutions for Structural Studies
| Reagent / Material | Function | Application Context |
|---|---|---|
| Lipidic Cubic Phase (LCP) Materials | Membrane mimetic environment for crystallizing membrane proteins like GPCRs [13]. | X-ray Crystallography |
| Crystallization Screening Kits | Sparse matrix screens to identify initial crystallization conditions [13]. | X-ray Crystallography |
| Direct Electron Detectors | High-sensitivity cameras that enable high-resolution single-particle analysis [91] [92]. | Cryo-EM |
| Scaffold Proteins (e.g., DARPins, Nanobodies) | Rigid binding partners or fusion modules to increase effective particle size and stabilize specific conformations [92] [54]. | Cryo-EM (esp. for small proteins) |
| Cryo-EM Grids | Specialized grids (e.g., gold or copper with holy carbon film) for sample application and vitrification [92]. | Cryo-EM |
Workflow and Data Integration
The following diagram illustrates the fundamental workflows for both techniques, highlighting the key steps where the handling of molecular dynamics diverges.
The Emerging Role of Artificial Intelligence
The integration of Artificial Intelligence (AI) is transforming structural biology, particularly in cryo-EM. Deep learning methods like MICA demonstrate that integrating cryo-EM density maps with AlphaFold3-predicted structures at the input level significantly improves modeling accuracy and completeness [49]. This AI-integration is especially valuable for interpreting regions of flexibility or lower resolution in experimental maps, potentially revealing dynamic features that are otherwise challenging to resolve.
The choice between X-ray crystallography and cryo-EM for studying molecular dynamics is not a matter of superiority but of strategic application. X-ray crystallography remains unparalleled for providing ultra-high-resolution static snapshots of crystallizable, stable macromolecules, offering a crucial baseline for understanding atomic interactions. Cryo-EM excels at visualizing structural flexibility and multiple native-state conformations of large complexes, membrane proteins, and dynamic assemblies without the constraint of crystal packing.
For researchers focused on drug discovery targeting dynamic processes or flexible proteins, cryo-EM offers a powerful path to visualize ligand-induced conformational changes. Meanwhile, X-ray crystallography continues to provide the atomic-level precision needed for understanding detailed mechanistic interactions. As both technologies advance, particularly through AI integration, their complementary use will continue to push the boundaries of our understanding of molecular machines in motion.
The resolution of biomolecular structures is fundamental to advancing our understanding of biology and accelerating drug discovery. For decades, techniques like X-ray crystallography and the more recent cryo-electron microscopy (cryo-EM) have been the pillars of experimental structural biology. The emergence of sophisticated artificial intelligence (AI) tools, most notably AlphaFold, has introduced a powerful new paradigm. This guide objectively examines the evolving role of AI, exploring whether it serves as a complement to experimental methods or a potential replacement, framed within the ongoing comparison of X-ray crystallography and cryo-EM.
The Evolving Landscape of Structural Biology Methods
The three primary techniques for determining macromolecular structures are X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and electron microscopy (EM). According to the Protein Data Bank (PDB), while X-ray crystallography remains the dominant technique, its annual proportion has been declining as the contribution of EM, particularly cryo-EM, has surged to account for nearly a third of new deposits by 2023 [93]. Each method possesses distinct strengths and limitations, summarized in the table below.
Table 1: Comparison of Major Structural Biology Techniques
| Feature | X-ray Crystallography | NMR Spectroscopy | Cryo-Electron Microscopy (Cryo-EM) |
|---|---|---|---|
| Typical Resolution | Atomic (0.5 - 3.0 Å) | Atomic (for smaller structures) | Near-atomic to Atomic (2.0 - 4.5+ Å) |
| Sample State | Crystalline solid | Solution | Vitrified ice (frozen-hydrated) |
| Key Advantage | High-resolution, gold standard | Studies dynamics in solution | No crystallization needed; great for large complexes |
| Key Limitation | Requires high-quality crystals | Limited to smaller proteins/complexes | Resolution can be heterogeneous |
| Sample Throughput | Medium to Low (crystallization bottleneck) | Low | Medium to High |
| Impact of AI | Model building and refinement | Data interpretation and structure calculation | Significant, from map interpretation to model building |
The Rise of AI and AlphaFold in Structure Prediction
AI, particularly deep learning, has revolutionized computational biology. AlphaFold, an AI system developed by DeepMind, can predict a protein's 3D structure from its amino acid sequence with accuracy competitive with experimental methods [94] [95]. By mid-2025, systems like AlphaFold 3 have expanded capabilities to model protein-ligand complexes, nucleic acids, and other biomolecules [10]. These tools have made structural information more accessible, providing models for millions of proteins whose structures were previously unknown.
However, AI predictions are not infallible. They can struggle with proteins that undergo conformational changes, have intrinsic disorder, or are part of novel complexes not well-represented in training data [96] [95]. This is where the synergy with experimental methods becomes critical.
AI as a Complementary Tool: Enhancing Experimental Workflows
The prevailing evidence strongly indicates that AI currently functions as a powerful complement to, rather than a replacement for, experimental data. AI tools are being integrated into experimental workflows to solve specific challenges, significantly accelerating and enhancing structure determination.
Interpreting and Modeling from Low-Resolution Cryo-EM Maps
A major challenge in cryo-EM is building accurate atomic models from low-resolution maps (worse than ~4 Å). Traditional de novo modeling tools see a dramatic drop in efficacy at these resolutions [95]. New AI-driven protocols are designed to address this gap.
Experimental Protocol: DeepTracer-LowResEnhance This innovative framework integrates AlphaFold's sequence-based predictions directly into the map interpretation process [95].
- Input: A low-resolution cryo-EM density map and the corresponding protein amino acid sequence.
- AlphaFold Integration: AlphaFold is run on the sequence to generate a structural prediction.
- Deep Learning Refinement: A convolutional neural network (CNN) uses features from the AlphaFold model to enhance and "sharpen" the low-resolution cryo-EM map.
- Model Building: The enhanced map is fed into a map-to-model tool (like DeepTracer) to build the final all-atom model.
- Validation: The final model is validated against the original experimental map to ensure a proper fit.
This method has demonstrated substantial improvements, achieving an average TM-score improvement of 3.53x over baseline predictions on low-resolution datasets [95].
Table 2: Performance of AI-Ehanced vs. Traditional Methods on Low-Resolution Cryo-EM Maps
| Method | Average TM-score (Maps <4 Å) | Key Advantage | Limitation |
|---|---|---|---|
| Baseline DeepTracer | Low (Baseline) | Fully automated | Efficacy drops significantly at low resolution |
| Traditional Sharpening (e.g., Phenix) | Moderate | Widely adopted; model-free | Can introduce artifacts; limited improvement |
| DeepTracer-LowResEnhance (AI) | High (3.53x improvement) | Integrates sequence-prediction; superior accuracy | Limited effectiveness on already high-res maps (<4 Å) |
Modeling Alternative Conformational States and Ligand Binding
Proteins are dynamic machines, and understanding different functional states is crucial for drug design. A common scenario is having a high-resolution structure in one state but only a low-resolution cryo-EM map in another, alternative state. A combined AI-simulation protocol addresses this [96]:
Experimental Protocol: AlphaFold2-Ensemble with Density-Guided Simulation
- Generate Conformational Ensemble: Instead of a single prediction, AlphaFold2 is run multiple times with stochastic subsampling of the multiple sequence alignment (MSA) to generate a diverse ensemble of potential structures [96].
- Cluster Models: The thousands of generated models are filtered and clustered (e.g., via k-means) to identify a manageable set of representative starting structures.
- Flexible Fitting with Simulation: Each representative model is rigidly aligned to the target cryo-EM density and then subjected to density-guided molecular dynamics (MD) simulations. This flexible fitting process biases the model to better match the experimental map while maintaining physically realistic geometry.
- Model Selection: The final model is selected based on a compound score balancing the fit to the map (cross-correlation) and model quality (GOAP score).
This approach has successfully resolved state-dependent differences, such as helix bending in GPCRs and substantial domain rearrangements in transporters, where starting from a single known-state structure failed [96].
The workflow for modeling a protein's alternative state using AI-generated ensembles and cryo-EM data can be visualized as follows:
Placing and Validating Ligands and Small Molecules
Resolving the atomic details of protein-ligand interactions is key for drug discovery, but ligand density in cryo-EM maps is often poor. AI is now assisting in this precise task [97].
- Experimental Protocol: AI-Predicted Pose with Density-Guided Refinement
- AI Prediction: An AlphaFold3-like model (e.g., Chai-1) is used to predict the structure of the protein-ligand complex. The input is the protein sequence and the ligand's SMILES string (a standardized notation for chemical structure).
- Initial Placement: The best AI-predicted complex is rigidly aligned into the experimental cryo-EM map.
- Simulation-Based Refinement: The complex undergoes density-guided MD simulations, where forces drive the protein and ligand atoms to better fit the experimental density without artificial restraints. This step is crucial when the AI prediction is not perfect.
- Validation: The refined model is validated by calculating the model-to-map cross-correlation. This pipeline has been shown to improve cross-correlation from 40-71% to 82-95% relative to the deposited structure for various kinases, GPCRs, and transporters [97].
Furthermore, specialized deep learning tools like MIC (Metric Ion Classification) have been developed to correctly identify ions and water molecules in cryo-EM and crystal structures—a task that is notoriously difficult from experimental data alone. MIC uses interaction fingerprints and a deep metric learning model to classify spherical density features, achieving superior accuracy over empirical methods and helping to correct mis-modeled sites in the PDB [98].
Research Reagent Solutions for Integrated AI-Experimental Workflows
The following table details key computational and experimental "reagents" essential for the workflows described in this guide.
Table 3: Essential Research Reagents and Tools for AI-Enhanced Structural Biology
| Item / Solution | Function / Purpose | Example Use Case |
|---|---|---|
| AlphaFold (or similar model) | Predicts protein 3D structure from amino acid sequence. | Generating initial models or conformational ensembles for cryo-EM fitting [96] [95]. |
| Cryo-EM Density Map | Experimental 3D reconstruction of the macromolecule. | Serves as the experimental constraint for model validation and refinement [97] [96]. |
| Molecular Dynamics (MD) Software | Simulates physical movements of atoms and molecules over time. | Performing density-guided flexible fitting of models into experimental maps [97] [96]. |
| DeepTracer-LowResEnhance | Specialized AI tool for building models from low-resolution maps. | Interpreting cryo-EM maps with resolution worse than 4 Å [95]. |
| MIC (Metric Ion Classification) | Deep learning tool for identifying ions/water in structures. | Validating and correcting the identity of bound ions and waters in PDB models [98]. |
| Ligand SMILES String | Standardized notation for inputting ligand chemistry into AI models. | Specifying the small molecule for protein-ligand complex prediction with tools like AlphaFold3/Chai-1 [97]. |
The question of whether AI replaces or complements experimental data has a clear answer based on current evidence: AI is a powerful complement. It has not rendered experimental methods obsolete but has instead integrated with them to create more powerful and efficient hybrid workflows.
- X-ray crystallography remains a high-resolution gold standard.
- Cryo-EM has become the leading technique for large, flexible complexes that are difficult to crystallize.
AI, exemplified by AlphaFold, enhances both by solving specific bottlenecks: interpreting low-resolution data, modeling conformational dynamics, and accurately placing ligands and ions. The future of structural biology lies not in choosing between computation or experiment, but in strategically integrating both to accelerate the pace of scientific discovery and drug development.
Selecting the right technique for determining a macromolecular structure is a critical first step in any structural biology project. This guide provides an objective, data-driven comparison between X-ray crystallography and cryo-electron microscopy (cryo-EM), framing the choice within a clear decision matrix to help researchers and drug development professionals select the optimal method for their specific needs.
Quantitative Comparison of Technique Capabilities
The choice between X-ray crystallography and cryo-EM hinges on the properties of the target macromolecule and the desired structural information. The table below summarizes the core performance characteristics of each technique.
Table 1: Key Technique Comparison for Structure Determination
| Criterion | X-ray Crystallography | Single-Particle Cryo-EM |
|---|---|---|
| Typical Resolution | Atomic-level (typically high) | Near-atomic to atomic (increasingly common) |
| Sample State | Crystalline solid | Vitrified solution (near-native) |
| Molecular Weight | Versatile, but limited for very large complexes | Ideal for large complexes (>~50 kDa) |
| Throughput | High for well-behaved targets | Rapidly increasing, enabling high-throughput studies [43] |
| Membrane Protein Utility | Challenging, requires specialized crystallization (e.g., LCP) [10] | Highly effective; a major strength [10] [43] |
| Dynamic/Time-Resolved Studies | Yes (e.g., mix-and-quench, serial crystallography) [14] | Emerging, but more challenging |
| Context of Structure | Purified, crystal-packed environment | Purified, single particles in ice |
| PDB Deposit Share (2023) | ~66% [99] | ~32% [99] |
Detailed Experimental Protocols
Understanding the standard workflows for each technique is essential for evaluating their feasibility for a given project.
Protocol for X-ray Crystallography
The process of structure determination by X-ray crystallography follows a well-established pathway [99].
- Crystallization: The purified target macromolecule is induced to form a highly ordered crystal. This is often the most significant bottleneck and may require extensive screening of thousands of conditions.
- Data Collection: A single crystal is exposed to an intense X-ray beam. As the X-rays pass through the crystal, they diffract, producing a pattern of spots on a detector.
- Data Processing: The diffraction pattern is processed computationally. The key challenge is solving the "phase problem" to convert the pattern into an electron density map. Methods include Molecular Replacement (using a known similar structure) or experimental phasing like SAD/MAD [99].
- Model Building and Refinement: An atomic model is built into the electron density map. This model is then iteratively refined to improve its fit to the experimental data and to ensure proper stereochemistry [99].
Time-Resolved Variation: For capturing intermediate states, a "mix-and-quench" approach can be used. The crystal is rapidly mixed with a substrate or ligand solution, and the reaction is stopped after a defined delay (as short as milliseconds) by thermal quenching. This allows for the trapping of short-lived structural states [14].
Protocol for Single-Particle Cryo-EM
The cryo-EM workflow, while requiring less sample preparation than crystallization, involves sophisticated instrumentation and computation [100] [10].
- Vitrification: A solution of the purified macromolecule is applied to a grid and rapidly plunged into a cryogen (like liquid ethane). This "flash-freezing" creates a layer of vitreous (non-crystalline) ice, trapping the particles in a near-native state.
- Data Collection: The vitrified grid is loaded into a transmission electron microscope. An electron beam passes through the sample, and a direct electron detector captures thousands to millions of 2D projection images of individual, randomly oriented particles.
- Image Processing: This is a computationally intensive, multi-step process:
- Particle Picking: Individual particle images are automatically selected from the micrographs.
- 2D Classification: Particles are grouped into classes representing similar views.
- 3D Reconstruction: A initial low-resolution 3D model is generated and then iteratively refined against the particle images to produce a high-resolution 3D electron density map.
- Model Building and Refinement: Similar to crystallography, an atomic model is built and refined into the cryo-EM density map. For high-resolution maps, automated tools like qFit can be used to model conformational heterogeneity directly from the data [85].
Technique Selection Pathways
The following decision diagram synthesizes the quantitative and qualitative data into a step-by-step selection guide.
Research Reagent and Material Solutions
Successful structure determination relies on key reagents and instruments. The following table details essential items for both techniques.
Table 2: Essential Research Reagents and Materials
| Item | Technique | Function Description |
|---|---|---|
| Lipidic Cubic Phase (LCP) | X-ray Crystallography | A membrane-mimetic matrix used for crystallizing membrane proteins like GPCRs [10]. |
| Microcrystals | X-ray Crystallography | Tiny crystals used in Serial Femtosecond Crystallography (SFX) at XFELs to study dynamics and avoid radiation damage [10]. |
| Direct Electron Detector | Cryo-EM | A critical camera technology that provides high signal-to-noise and enables motion correction, underpinning the "resolution revolution" [10]. |
| Cryo-Electron Tomography (Cryo-ET) | Cryo-EM | An advanced modality that constructs 3D images of flash-frozen cells or tissues, visualizing macromolecules in their native cellular context [12]. |
| qFit Software | Both (High-Res) | An automated computational tool that builds multiconformer models into high-resolution density maps, revealing conformational heterogeneity [85]. |
| Titan Krios Microscope | Cryo-EM | A high-end electron microscope with a cold field emission source, representing one of the highest-resolution setups available [12]. |
Conclusion
X-ray crystallography and cryo-EM are not mutually exclusive but rather powerful, complementary tools in the structural biologist's arsenal. The choice is project-dependent: X-ray remains the workhorse for high-resolution, high-throughput structure determination of crystallizable targets, while cryo-EM excels for large, flexible complexes and membrane proteins in near-native states. The future lies in convergence—combining these techniques with X-ray free-electron lasers, next-generation synchrotrons like PETRA IV, and AI-driven analysis to capture molecular movies of biological processes in real-time. This integrated approach will profoundly accelerate drug discovery, enable the rational design of therapeutics targeting transient states, and deepen our understanding of life's fundamental mechanisms at an atomic level.
References
- 1. How cryo‐electron microscopy and X‐ray crystallography ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5192981/]
- 2. Comparison of X-ray Crystallography, NMR and EM [https://www.creative-biostructure.com/comparison-of-crystallography-nmr-and-em_6.htm?srsltid=AfmBOoqeXgDMXDmhZ3-034nKqpn9YgJJbKXcA_5adEYkkwMzt3NollFM]
- 3. Microscopic Magnificence [https://www.utsouthwestern.edu/ctplus/stories/2025/cryo-em-research.html]
- 4. JEOL USA blog | Cryo-EM vs. X-ray Crystallography [https://www.jeolusa.com/NEWS-EVENTS/Blog/cryo-em-vs-x-ray-crystallography]
- 5. Bragg's law [https://en.wikipedia.org/wiki/Bragg%27s_law]
- 6. X-ray diffraction, Bragg's law and Laue equation [https://eng.libretexts.org/Bookshelves/Materials_Science/Supplemental_Modules_(Materials_Science)/Electronic_Properties/X-ray_diffraction_Bragg's_law_and_Laue_equation]
- 7. Bragg's Law - an overview [https://www.sciencedirect.com/topics/medicine-and-dentistry/braggs-law]
- 8. The Resolution in X-ray Crystallography and Single ... [https://www.mdpi.com/2073-4352/10/7/580]
- 9. Extending the reach of single-particle cryoEM [https://www.sciencedirect.com/science/article/pii/S0959440X25000235]
- 10. High-resolution protein modeling through Cryo-EM and AI [https://pmc.ncbi.nlm.nih.gov/articles/PMC12590954/]
- 11. Comparison of X-ray Crystallography, NMR and EM [https://www.creative-biostructure.com/comparison-of-crystallography-nmr-and-em_6.htm?srsltid=AfmBOoqMw-vv44p92skBpYAPGhzHpKMXurHfdL0wN83RoqzQUSWA1ECZ]
- 12. Capitalizing on the resolution revolution in structural and ... [https://www.stjude.org/research/progress/2025/capitalizing-on-the-resolution-revolution-in-structural-and-cell-biology-with-cryo-electron-tomography.html]
- 13. Comparison Of Structural Techniques: X-ray, NMR, Cryo-EM [https://peakproteins.com/a-comparison-of-the-structural-techniques-used-at-sygnature-discovery-x-ray-crystallography-nmr-and-cryo-em/]
- 14. Instrumentation and methods for efficient time-resolved X-ray ... [https://pubmed.ncbi.nlm.nih.gov/40277177/]
- 15. Research team sheds light on the future potential of X-ray ... [https://desy.de/e18959/e18960/e20311/index_eng.html]
- 16. AI-based quality assessment methods for protein structure ... [https://www.sciencedirect.com/science/article/pii/S2665928X25000017]
- 17. Comparison of X-ray Crystallography, NMR and EM [https://www.creative-biostructure.com/comparison-of-crystallography-nmr-and-em_6.htm?srsltid=AfmBOop-57Akzts0D7hoM1wTNYzlm4CJnflhyVBUuaKOsqVfbXApiguJ]
- 18. wwPDB: Deposition Statistics [https://www.wwpdb.org/stats/deposition]
- 19. Cryo Electron Microscopy Market Size Analysis [https://media.market.us/cryo-electron-microscopy-market-news/]
- 20. Cryo-EM Structure Analysis Services Market Share, 2025- ... [https://www.coherentmarketinsights.com/industry-reports/cryo-em-structure-analysis-services-market]
- 21. Global Cryo-Electron Microscopy Market Size, Share, and ... [https://www.databridgemarketresearch.com/reports/global-cryo-electron-microscopy-market?srsltid=AfmBOorCU2RTyWvm7s65-4PNtGrOqvG6Uj2LOAZ_oV6zK4KOP7Q-c9yL]
- 22. Electron Microscopy: Principle, Strengths... | Technology Networks Cryo [https://www.technologynetworks.com/analysis/articles/cryo-electron-microscopy-principle-strengths-limitations-and-applications-377080]
- 23. - X - ray : Which is... - Creative Biostructure Crystallography vs Cryo EM [https://www.creative-biostructure.com/resource-cryo-em-vs-xray-crystallography-comparison.htm]
- 24. Preparing for successful protein crystallization experiments [https://journals.iucr.org/f/issues/2025/07/00/oq5003/]
- 25. Cryo-EM vs. X-Ray: Which Structural Method Is Best? [https://shuimubio.com/blogs/cryo-em-vs-x-ray-which-structural-method-is-best]
- 26. Sample delivery methods for protein X-ray crystallography ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12594859/]
- 27. Comparison of X-ray Crystallography, NMR and EM [https://www.creative-biostructure.com/comparison-of-crystallography-nmr-and-em_6.htm?srsltid=AfmBOoqqiGBHK7jYRE75EvvQoFzTWrAlyHHo8i28XoQc6tyKcxO-JxFU]
- 28. Protein Crystallography Services [https://www.saromics.com/x-ray-crystallography-services/]
- 29. Status and perspective of protein crystallography at the first ... [https://journals.iucr.org/paper?sze5001]
- 30. A large-scale curated and filterable dataset for cryo-EM ... [https://www.nature.com/articles/s41597-025-05179-2]
- 31. Microcrystallization and room-temperature serial ... [https://www.sciencedirect.com/science/article/pii/S0003986125001328]
- 32. Automated phase mapping of high-throughput X-ray ... [https://www.nature.com/articles/s41524-025-01837-6]
- 33. X-ray Crystallography vs Cryo-EM: Which is Best for Your ... [https://www.creative-biostructure.com/resource-cryo-em-vs-xray-crystallography-comparison.htm?srsltid=AfmBOor4ojVj0Xyw_zqMRp7I1jgXIB4DwIGSb2sqocbel6XzX0uO8ttd]
- 34. A deep convolutional neural network approach to single- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5521087/]
- 35. Single Particle Cryo-electron Microscopy and 3-D ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4020923/]
- 36. Chapter 2: Cryo-EM Grid Preparation [https://cryoem101.org/chapter-2/]
- 37. Contrast in Cryo-EM [https://guide.cryosparc.com/cryo-em-foundations/image-formation/contrast-in-cryo-em]
- 38. High-throughput X-ray crystallography for drug discovery [https://www.sciencedirect.com/science/article/abs/pii/S1471489204001225]
- 39. X-ray crystallography: Assessment and validation of protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3138648/]
- 40. Lessons from high-throughput protein crystallization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3637395/]
- 41. DESY scientists advance drug development with room- ... [https://desy.de/desy_latest_news/2025/hiphax/index_eng.html]
- 42. Room-temperature X-ray fragment screening with serial ... [https://www.nature.com/articles/s41467-025-64918-6]
- 43. High throughput cryo-EM provides structural understanding ... [https://www.sciencedirect.com/science/article/pii/S0969212625001923]
- 44. X-ray Crystallography vs Cryo-EM: Which is Best for Your ... [https://www.creative-biostructure.com/resource-cryo-em-vs-xray-crystallography-comparison.htm?srsltid=AfmBOoqaDqiTpFaaaWb0jfziRpc1BklUyOaLiDuBMDBSdJDToqt8J8SY]
- 45. Smallest protein structure imaged with cryo-EM [https://www.diamond.ac.uk/Home/News/LatestNews/2025/New-nanobody-scaffold-unlocks-cryo-EM-imaging-of-the-smallest-proteins.html]
- 46. Visualization of lysosomal membrane proteins by cryo ... [https://www.nature.com/articles/s41467-025-64314-0]
- 47. Cryo-EM led analysis of open and closed conformations ... [https://www.nature.com/articles/s41467-025-62068-3]
- 48. Cryo-electron microscopy (2025) [https://www.sciencedirect.com/journal/current-opinion-in-structural-biology/special-issue/1094R30CRCM]
- 49. Multimodal deep learning integration of cryo-EM and ... [https://www.nature.com/articles/s42004-025-01718-5]
- 50. X-ray Crystallography vs Cryo-EM: Which is Best for Your ... [https://www.creative-biostructure.com/resource-cryo-em-vs-xray-crystallography-comparison.htm?srsltid=AfmBOoqA_1lwmgvYeO2GR1mjmB0C9VNsNX5KaH9SJFGVH59s6Hj1UDrm]
- 51. High-resolution cryo-EM performance comparison of two ... [https://www.sciencedirect.com/science/article/pii/S1047847722000752]
- 52. Solid/liquid interface induced protein crystallization [https://www.sciencedirect.com/science/article/abs/pii/S096089742500018X]
- 53. Biocrystallization: harnessing natural scaffolds for stable ... [https://www.sciencedirect.com/science/article/pii/S1871678425001062]
- 54. Structural determination of small proteins by cryo-EM using ... [https://www.nature.com/articles/s41598-025-20608-3]
- 55. Using AlphaFold and Symmetrical Docking to Predict ... [https://pubmed.ncbi.nlm.nih.gov/40401365/]
- 56. Exploring the Bottleneck in Cryo-EM Dynamic Disorder ... [https://www.mdpi.com/2673-4125/5/3/39]
- 57. Specialty grand challenges in theoretical modeling, ... [https://www.frontiersin.org/journals/chemical-biology/articles/10.3389/fchbi.2025.1635423/full]
- 58. Comparison of X-ray Crystallography, NMR and EM [https://www.creative-biostructure.com/comparison-of-crystallography-nmr-and-em_6.htm?srsltid=AfmBOop-W0mtYXsQQzmUtThj-sKMK3UsxsfnPQJmeKLEGO-c_2m4iP-h]
- 59. Untangling the effects of flexibility and the AWI in cryoEM ... [https://www.sciencedirect.com/science/article/pii/S1047847725000413]
- 60. Solve Preferred Orientation in Cryo-EM [https://www.nanoimagingservices.com/about/blog/solving-the-challenges-of-preferred-orientation-in-cryo-em-sample-preparation]
- 61. A measure for the identification of preferred particle ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8116199/]
- 62. CryoPROS: Correcting misalignment caused by preferred ... [https://www.nature.com/articles/s41467-025-59797-w]
- 63. A new method to prepare cryo-EM samples avoids protein ... [https://www2.mrc-lmb.cam.ac.uk/a-new-method-to-prepare-cryo-em-samples-avoids-protein-damage-during-freezing/]
- 64. Three Pillars: Cryo-EM, X-Ray & NMR Compared [https://shuimubio.com/blogs/three-pillars-cryo-em-x-ray-nmr-compared]
- 65. Protein structure determination techniques: X - ray .... Crystallography vs [https://penprofile.com/protein-structure-determination-techniques-x-ray-crystallography-vs-cryo-electron-microscopy/]
- 66. Comparison of X-ray Crystallography, NMR and EM [https://www.creative-biostructure.com/comparison-of-crystallography-nmr-and-em_6.htm?srsltid=AfmBOoondSPigoJCLDE3rxQVlobwRc8ZfDIifl2ah_xAsq0uDnOCCGxC]
- 67. X-ray Crystallography vs Cryo-EM: Which is Best for Your ... [https://www.creative-biostructure.com/resource-cryo-em-vs-xray-crystallography-comparison.htm?srsltid=AfmBOoqosPALJ9wLoUP4HjnXd5Rf8oiHJSBmLaFWBXBywBisH4ofg-Ks]
- 68. Comparing Analytical Techniques for Structural Biology [https://www.nanoimagingservices.com/about/blog/unraveling-the-mysteries-of-the-molecular-world-structural-biology-techniques]
- 69. Advances in high-resolution cryo-EM at 100 kV [https://www.thermofisher.com/blog/atomic-resolution/advances-in-100-kv-high-resolution-cryo-em-single-particle-analysis/]
- 70. Useful experimental aspects of small-wedge synchrotron ... [https://journals.iucr.org/d/issues/2025/01/00/wa5149/]
- 71. X-ray Crystallography vs Cryo-EM: Which is Best for Your ... [https://www.creative-biostructure.com/resource-cryo-em-vs-xray-crystallography-comparison.htm?srsltid=AfmBOoq5wA9oZdSqSNPmGRemt1FmEmoHKdxngfQWwZzrbDLIJhBCRiyH]
- 72. Better, Faster, Cheaper: Recent Advances in Cryo–Electron ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10393189/]
- 73. About RapiData 2025 at SSRL - Stanford University [https://www-ssrl.slac.stanford.edu/rapidata/rapidata-2025/about.html]
- 74. A comprehensive approach to X-ray crystallography for ... [https://www.sciencedirect.com/science/article/pii/S1740674920300214]
- 75. The current role and evolution of X-ray crystallography in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10620067/]
- 76. A large, general and modular DARPin–apoferritin scaffold ... [https://journals.iucr.org/m/issues/2025/03/00/eh5021/]
- 77. Fusion protein strategies for cryo-EM study of G ... [https://www.nature.com/articles/s41467-022-32125-2]
- 78. Scaffold-enabled high-resolution cryo-EM structure ... [https://www.nature.com/articles/s41467-024-55699-5]
- 79. X-ray Crystallography vs Cryo-EM: Which is Best for Your ... [https://www.creative-biostructure.com/resource-cryo-em-vs-xray-crystallography-comparison.htm?srsltid=AfmBOopDD8wlqlU14p7aUYnX2e0Pth7uvHJKwuJnOSljS3DW8m5cnwHv]
- 80. Single-particle cryogenic electron microscopy structure ... [https://www.sciencedirect.com/science/article/abs/pii/S0959440X2500065X]
- 81. Cryo-electron microscopy-based drug design [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2024.1342179/full]
- 82. X-rays in the Cryo-EM Era: Structural Biology's Dynamic Future [https://pmc.ncbi.nlm.nih.gov/articles/PMC5999524/]
- 83. Cryo-electron microscopy and X-ray crystallography [https://pmc.ncbi.nlm.nih.gov/articles/PMC5379166/]
- 84. Exploiting the full potential of cryo-EM maps [https://www.sciencedirect.com/science/article/pii/S2667160324000012]
- 85. Automated Multiconformer Model Building for X- ray ... [https://elifesciences.org/reviewed-preprints/90606]
- 86. Instrumentation and methods for efficient time-resolved X- ... [https://journals.iucr.org/paper?min5001]
- 87. X-Ray Crystallography Market Forecast 2025 - 2032 [https://www.persistencemarketresearch.com/market-research/xray-crystallography-market.asp]
- 88. X-ray Crystallography Market to Expand at 7.6% CAGR, ... [https://www.openpr.com/news/4197017/x-ray-crystallography-market-to-expand-at-7-6-cagr-reaching]
- 89. Cost-Benefit Analysis Of Establishing A Cryo-EM Facility [https://eureka.patsnap.com/report-cost-benefit-analysis-of-establishing-a-cryo-em-facility]
- 90. Cost-benefit analysis of cryogenic electron tomography ... [https://www.sciencedirect.com/science/article/abs/pii/S0969212624004982]
- 91. Frontiers | High- resolution protein modeling through Cryo - EM and AI... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2025.1688455/full]
- 92. Cryogenic electron microscopy - Wikipedia [https://en.wikipedia.org/wiki/Cryogenic_electron_microscopy]
- 93. Comparison of X-ray Crystallography, NMR and EM [https://www.creative-biostructure.com/comparison-of-crystallography-nmr-and-em_6.htm?srsltid=AfmBOorl_Jt1dIZXcS9Kkh32cKyVttbW8pXa1UxpXSGvR2u6KxLW4ZFT]
- 94. Transformative Role of Artificial Intelligence in Drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12406033/]
- 95. Integrating deep learning and AlphaFold for enhanced ... [https://www.sciencedirect.com/science/article/abs/pii/S1476927125001549]
- 96. Modeling cryo-EM structures in alternative states with ... [https://www.nature.com/articles/s42004-025-01751-4]
- 97. Cryo-EM ligand building using AlphaFold3-like model and ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1013367]
- 98. MIC: A deep learning tool for assigning ions and waters in ... [https://www.nature.com/articles/s41467-025-61315-x]
- 99. Comparison of X-ray Crystallography, NMR and EM [https://www.creative-biostructure.com/comparison-of-crystallography-nmr-and-em_6.htm?srsltid=AfmBOooxa0p8x6m5vwY3ZjFS9uTLSnLuKNTZVx3FfXGBOxHuxrZ6ttoJ]
- 100. CryoEM milestones and future directions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12585528/]